
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal atomic layer deposition of gold nanoparticles: controlled growth and size selection for photocatalysis†
Fatemeh S. M.
Hashemi
 *a,
Fabio
Grillo
ab,
Vikram R.
Ravikumar
a,
Dominik
Benz
a,
Ankit
Shekhar
a,
Matthew B. E.
Griffiths
c,
Seán T.
Barry
c and
J. Ruud
van Ommen
a
*a,
Fabio
Grillo
ab,
Vikram R.
Ravikumar
a,
Dominik
Benz
a,
Ankit
Shekhar
a,
Matthew B. E.
Griffiths
c,
Seán T.
Barry
c and
J. Ruud
van Ommen
a
aDepartment of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: fhashemi@alumni.stanford.edu
bDepartment of Materials, ETH Zurich, Vladimir Prelog Weg 5, 8093 Zürich, Switzerland
cDepartment of Chemistry, Carleton University, 1125 Colonel By Dr, Ottawa, ON K1S 5B6, Canada
First published on 1st April 2020
Abstract
Gold nanoparticles have been extensively studied for their applications in catalysis. For Au nanoparticles to be catalytically active, controlling the particle size is crucial. Here we present a low temperature (105 °C) thermal atomic layer deposition approach for depositing gold nanoparticles on TiO2 with controlled size and loading using trimethylphosphino-trimethylgold(III) and two co-reactants (ozone and water) in a fluidized bed reactor. We show that the exposure time of the precursors is a variable that can be used to decouple the Au particle size from the loading. Longer exposures of ozone narrow the particle size distribution, while longer exposures of water broaden it. By studying the photocatalytic activity of Au/TiO2 nanocomposites, we show how the ability to control particle size and loading independently can be used not only to enhance performance but also to investigate structure–property relationships. This study provides insights into the mechanism underlying the formation and evolution of Au nanoparticles prepared for the first time via vapor phase atomic layer deposition. Employing a vapor deposition technique for the synthesis of Au/TiO2 nanocomposites eliminates the shortcomings of conventional liquid-based processes opening up the possibility of highly controlled synthesis of materials at large scale.
Introduction
Gold was long thought to be far less catalytically active than other transition metals. Yet studies in the late 80s showed that Au nanoparticles supported on metal oxides such as TiO2 exhibits an extraordinary high activity towards various catalytic oxidation and reduction reactions, such as low-temperature combustion, partial oxidation of hydrocarbons, hydrogenation of unsaturated hydrocarbons, and reduction of nitrogen oxides.1–10 The catalytic properties of Au nanoparticles originate from quantum, electronic, and geometric finite-size effects, and are therefore strongly related to the particle size.3,11 For example, Au nanoparticles supported on TiO2 exhibit a high activity towards the oxidation of CO only when their size is within an optimal range of 2–3 nm.3 Furthermore, Au nanoparticles can extend the absorption of TiO2 in the visible range to an extent that depends on their size thanks to plasmon resonance, that is, the resonant oscillation of free electrons induced by incident light.12,13 While the sensitivity of the structure–property relationship of Au nanoparticles can add a new dimension to the design of catalysts, it also poses a challenge. This is because practical applications often require large amounts of material, and fabricating supported nanoparticles with a controlled size in a scalable fashion remains challenging.14–16 Conventional techniques for the synthesis of supported Au nanoparticles typically rely on liquid-phase processes.17–20 While these methods have been extensively studied and optimized, their practical applicability still faces some challenges. These include polydispersity of produced nanoparticles, impurities in the final product arising from residual solvents, poor control over the composition in large batches, and high sensitivity to operating conditions.20,21 These factors can significantly affect the activity, selectivity, and stability of the catalyst.15,22 An alternative solvent-free method with precise control over the particle size and composition at large-scale would circumvent these shortcomings. Furthermore, vapor based methods for the deposition of Au nanoparticles would enable their integration into the industrial processing of nanostructures, opening up the possibility of using Au nanoparticles in emerging applications.Atomic layer deposition (ALD) is a vapor-phase deposition technique for the synthesis of supported nanoparticles as it boasts high precursor utilization efficiency, low degree of contamination of the final product, and an excellent degree of control over both the metal loading and the particle size.16,23–29 While there exist over seventy ALD processes for the deposition of noble metals, only limited success with regards to Au has been reported.10 Two successful studies on ALD of Au include a plasma process at 120 °C using (trimethylphosphino)trimethylgold(III) as metal precursor; and a thermal process at 180 °C, with dimethylgold(III)(diethylthiocarbamato) as metal precursor.30,31 In both studies, the deposition was carried out on flat substrates under vacuum conditions. While such processes are certainly promising for a number of applications, plasma and vacuum processes are not readily scalable to large quantities of high-surface-area supports, which are relevant for catalytic applications.28 In this work, we demonstrate a synthesis method for scalable fabrication of supported Au nanoparticles on TiO2via thermal atmospheric-pressure ALD. This ALD process uses trimethylphosphino-trimethyl gold(III) as the metal precursor, and ozone and water as co-reactants, at low temperatures (105 °C). We show that it is possible to control the broadness of the particle size distribution while retaining a given metal loading by tuning the exposure of the co-reactants. Interestingly, ozone and water have opposing effects on the size distribution: long ozone exposures narrow the size distribution, whereas long water exposures broaden it. Finally, by leveraging our independent control over loading and particle size we explore the structure sensitivity of the Au/TiO2 nanocomposites against the photocatalytic decomposition of a model dye. In doing so, we show a threefold enhancement in the photocatalytic activity of commercial titania P25. The photocatalytic enhancement of Au/P25 achieved by the ALD deposited Au nanoparticles is superior to the previously reported Au/P25 nanocomposites prepared via colloidal synthesis.32 By boasting an independent control over particle size and metal loading, our thermal atmospheric-pressure ALD process can open new possibilities for the application of supported Au nanoparticle in large scale catalytic processes.52
Experimental section
Aeroxide P25 titanium oxide particles from Evonik Industries with a mean diameter of 32.7 nm, a specific area of 52.4 m2 g−1 were used as the support material for the deposition of Au. The nanoparticles were treated with oxygen (99.999 vol%) for 10 min followed by nitrogen (99.999 vol%) for 10 min at 105 °C before the Au ALD experiments. The deposition experiments were carried out in a custom-built vibration-assisted fluidized bed ALD reactor operating at atmospheric pressure described elsewhere.33 The ALD reaction chamber was a 25 mm I.D. glass column into which 2 g of titania P25 NPs were loaded for each ALD run. The metal precursor bubbler and the line connecting the bubbler to the reactor were heated to 85 °C and 95 °C, respectively. All the reactants were transferred to the reaction chamber with a nitrogen flow rate of 0.5 lit min−1. An infra-red lamp was used to heat and maintain the reactor at 105 °C. Trimethylphosphino-trimethyl gold(III) was synthesized as described previously.30The photocatalytic tests were based on the degradation of rhodamine B. First, 30 mg of catalyst material was dispersed into a solution of rhodamine B (30 ml, 16 mg l−1, in DI water). Then sodium polyphosphate (0.33 ml, 100 g l−1 in DI water) was added to improve the dispersion of the particles. The dispersion was sonicated for 10 min and stirred in the dark for 20 min to reach the adsorption–desorption equilibrium. This procedure is adapted from previous studies on the photocatalytic activity of TiO2.34 It should be noted that sodium polyphosphate may also graft to the surface of Au NPs and affect their electronic properties. Investigating the additional effects of the dispersing agent is subject to further research. Then the samples were top-illuminated in an Atlas SUNTEST XXL equipped with three Xenon lights to ensure homogeneous light distribution (45 W m−2). The reactor (irradiation surface 11.3 cm2) was cooled using a water bath at 20 °C while the dispersion was continuously stirred during the test using a magnetic stirrer operating at 700 rpm. After distinct time intervals, samples of 1 ml were taken, centrifuged and the light absorption at 629 nm was measured using a HACH LANGE DR5000 UV/Vis Spectrometer to follow the degradation of rhodamine B with time. The apparent kinetic constant of the degradation reaction was estimated by assuming first-order reaction kinetics.
To determine Au and P loading (wt%) on powders, Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) analyses were performed in PerkinElmer Optima 5300DV Optical emission spectrometer. About 25 to 50 mg of samples were used for each ICP-OES measurement. X-ray Photoelectron Spectroscopy (XPS) samples were analyzed in a Thermo Scientific K-Alpha X-ray spectrometer. The X-ray source was a monochromatic AlKa source with a beam energy of 1486.7 eV and a spot size of 400 μm. The obtained Au 4f, P 2p, O 1s, C 1s, and Ti 2p spectra were processed in CasaXPS. Diffuse Reflectance Infrared Fourier-Transform Spectroscopy (DRIFTS) measurements were made using a NICOLET 8700 spectrometer in diffuse reflectance mode with a range of 500–4000 cm−1 (128 scans).
For Transmission Electron Microscopy (TEM) measurements multiple samples were taken from the 2 g batch. The NPs were suspended in a droplet of ethanol which was then cast on a copper TEM grid. The TEM micrographs were obtained by a JEOL JEM 1400TEM operating at 120 kV. Many points were imaged on the TEM grid. Number based particle size distributions were obtained from image analysis of the micrographs. The projected area of each particle (Api) was measured manually using ImageJ. Then the equivalent particle diameter was estimated as  for ∼100–600 NPs from 15 to 40 micrographs taken at different locations of the TEM grid and at different magnifications. The measured diameters are displayed as binned scatter plots and box-and-whisker diagrams. In the scatter plots all the measured diameters are grouped into bins whose width and height indicate different size ranges and their relative frequency, respectively. The box-and-whisker diagrams represent different statistical descriptors of the populations: the whiskers indicate the minimum and maximum observed diameter, the lower and upper side of the box indicate the first to the third quartile of the population, respectively, and the diamond inside the box indicates the mean diameter. Diffuse Reflectance-Ultraviolet/Visible Spectroscopy (DRS) measurements were performed on a PerkinElmer Lambda 900 spectrometer in diffuse reflectance mode at a range of 250 to 800 nm using an integration sphere. Barium sulfate (BaSO4) was used as a blank standard.
for ∼100–600 NPs from 15 to 40 micrographs taken at different locations of the TEM grid and at different magnifications. The measured diameters are displayed as binned scatter plots and box-and-whisker diagrams. In the scatter plots all the measured diameters are grouped into bins whose width and height indicate different size ranges and their relative frequency, respectively. The box-and-whisker diagrams represent different statistical descriptors of the populations: the whiskers indicate the minimum and maximum observed diameter, the lower and upper side of the box indicate the first to the third quartile of the population, respectively, and the diamond inside the box indicates the mean diameter. Diffuse Reflectance-Ultraviolet/Visible Spectroscopy (DRS) measurements were performed on a PerkinElmer Lambda 900 spectrometer in diffuse reflectance mode at a range of 250 to 800 nm using an integration sphere. Barium sulfate (BaSO4) was used as a blank standard.
Results and discussion
As described by Griffiths et al., trimethylphosphino-trimethyl gold(III) vapor pressure follows a Antoine equation (ln(p) = 0.059T − 1.65).30 We estimate the molar flux of precursor to the reactor to be about 2.6 e−4 mol h−1, assuming the partial pressure of the Au precursor in the gas stream to be equal to its vapor pressure at the bubbler temperature: 28.9 Pa at 85 °C. For titania nanoparticles with the surface area of 50 m2 g−1, a long Au dosage time (>60 min) is required to saturate the titania surface groups. Thus for demonstrating the saturation behavior of this precursor we only perform 1 ALD cycle. To achieve thermal ALD of Au on gram-scale batches of high-surface-area powders we adapted the plasma process reported by Griffiths et al., which consists of ABC cycles comprising the sequence of pulses: Au precursor/oxygen plasma/water. In our process, the oxygen plasma step is replaced by exposure to 1 bar of ozone. The effect of the exposure to ozone and water is studied by performing more cycles (3 to 9) where short exposures of the Au precursor were used. As we will discuss below, within the cycle range explored here (1–9), the deposition process results in the formation of nanoparticles rather than films. In particular, we show that both the Au loading and the size distribution of the nanoparticles can be controlled even when running the chemisorption of the Au precursor in a sub-saturation regime.The self-limiting behavior of the precursor chemisorption was probed by measuring the Au loading (wt%) on the TiO2 nanopowders after exposure times in the range of 5–150 min. The Au loading was measured with ICP-OES (Fig. 1a). For all saturation experiments, ozone and water exposures were kept at 10 min and 30 s, respectively. The Au loading progressively increases up to exposure times of about 60 min, after which it plateaus and remains virtually constant at around 2.7 wt% within a time window of about one hour, i.e. for a total exposure of 120 min. This demonstrates the self-limiting nature of the deposition process. Upon prolonging the exposure time up to 150 min, well past the minimum saturation time, higher Au loadings were observed (ESI Fig. 1†). At such long exposures, it is not surprising that parasitic chemical vapor deposition (CVD) components might manifest themselves. In fact, although precursor is known to decompose at about 120 °C and the bulk temperature of the reactor is kept constant at 105 °C, after the surface reactions have terminated, unreacted precursor can still be expected to decompose at a very slow rate, which over long time scales (∼30 min) can add up to noticeable amounts of precursor being decomposed. Furthermore, local hot spots occurring sporadically across the reactors can also contribute to a small CVD component. Nonetheless, a self-limiting deposition is retained over a broad time window (∼1 hour), attesting the robustness of this ALD process. A loading of 2.7 wt% after one cycle corresponds to a growth per cycle of ∼1.6 Au atoms per nm2 and thus to ∼0.1 Au(111) monolayer (1.387 × 1015 atoms per cm2). This figure is very much in line with what has been reported for a similar process: Pt ALD on P25, where ΔnPt after one cycle at saturation is about ∼1 at nm−2.29,33 The deposition of less than a monolayer per cycle further attests to the ALD-nature of the precursor chemisorption on TiO2.35 Furthermore, we estimate about 0.57 g of Au to be fed to the reactor in 60 min (saturation time), this amount results in Au to TiO2 weight fraction of 2.8 wt% which is very close to the experimentally obtained saturation value (2.7 wt%). This attests to the near-perfect efficiency in utilizing the dosed precursor, which has been previously shown for the ALD processes performed in a fluidized bed reactor.24
 | ||
| Fig. 1 Self-limiting behavior of the Au precursor chemisorption. (a) Evolution of the Au loading on TiO2 nanoparticles after one cycle with increasing exposure time. The ozone and water exposure times were: 10 min, and 30 s, respectively, and the reactor temperature was kept at 105 °C (the dashed red line is drawn to guide the eye). (b) FTIR spectra acquired at 120 °C on the Au/TiO2 nanocomposites obtained after 1 ALD cycle with different Au precursor exposure times. | ||
A detailed analysis of the TiO2 surface functional groups by FTIR measurements reveals an interesting trend in the evolution of surface functional groups. Upon longer Au precursor exposures, the Ti–OH peak decreases while a –CH peak emerges. The –CH peak is originated from the precursor's –CH3 ligands and the organic contaminants. Furthermore, the H–O–H bending peak related to adsorbed water also decreases with an increase in the Au precursor exposure (Fig. 1b).
To gain further insights into the growth process, we repeated the saturation experiments by performing 5 ALD cycles. This allowed us to study the aggregation of the as-deposited Au atoms into nanoparticles in more detail, as lower cycle numbers result in nanoparticles too small to be observed with TEM. In particular, we studied the evolution of the loading and the size distribution of the nanoparticles by exposing the TiO2 nanopowders to the Au precursor for pulse times in the range of 5–40 min (i.e., sub-saturating doses). Similar to the saturation experiments, we kept the ozone and water exposures at 10 min and 30 s, respectively (Fig. 2).
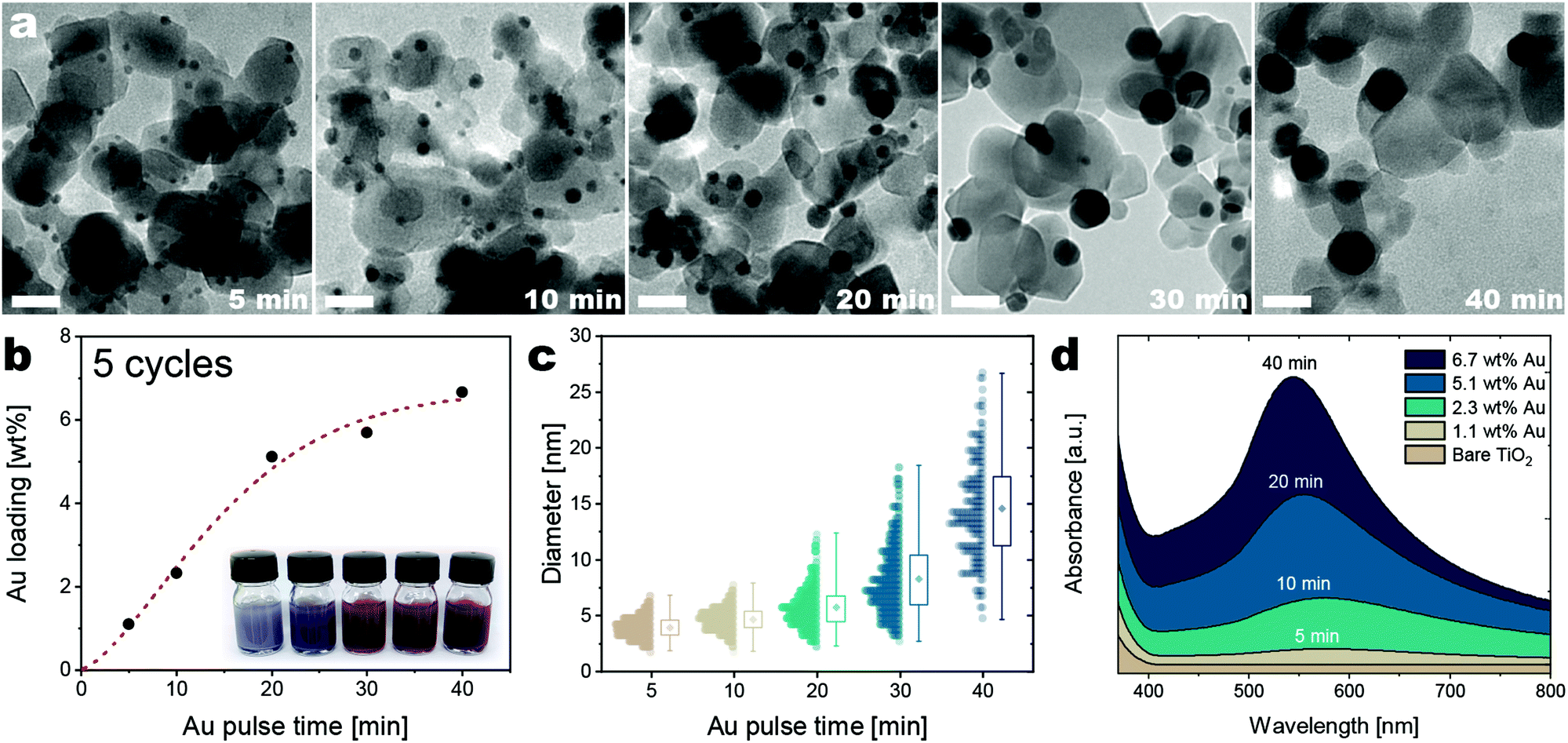 | ||
| Fig. 2 Evolution of the morphology, Au loading, and absorbance in the visible spectrum of the Au/TiO2 obtained after 5 cycles with different precursor exposure times. (a) TEM micrographs of the Au/TiO2 nanocomposites (scale bar 20 nm), (b) ICP results Au loading as the Au exposure time increases with corresponding photos of colloidal suspensions of the Au/TiO2 nanocomposites (the dashed red line is drawn to guide the eye), (c) size distribution of the ALD-grown Au nanoparticles, (d) absorbance of the Au/TiO2 nanocomposites as obtained via DRS measurements. | ||
Image analysis of TEM micrographs together with elemental analysis shows that an increase in Au loading with increasing Au precursor exposure time is accompanied by a higher degree of metal aggregation, as attested by the increase in particle size, the broadening of the size distribution, and the decreased number of particles (Fig. 2). This points to the fact that sintering mechanisms, such as nanoparticle diffusion and coalescence or Ostwald ripening, play an important role during the growth process.27,36 This is not surprising because the high surface energy of noble metals typically translates into a poor wetting of metal oxide surfaces, and in particular of titania.37–40 Increasing the Au precursor exposure time from 5 to 40 min results in the formation of fewer but larger particles, as the average diameter grows from ∼4 nm to ∼15 nm, while the span of the size distribution (the difference between the maximum and the minimum observed diameter) grows from ∼5 to ∼21 nm (Fig. 2a and c). Diffuse Reflectance-Ultraviolet/Visible Spectroscopy (DRS) was employed to characterize the absorption properties of Au nanoparticles at different Au precursor dose times (Fig. 2d). The absorption spectra show the emergence and evolution of the Plasmon resonance as the Au precursor dosage time is increased. With the growth of larger particles at higher loadings of Au, the absorption band narrows, the plasmon resonance shifts to lower wavelengths (from 576 to 536 nm), and the intensity of the plasmon resonance wavelength increases from 0.2 to 1.7 (Fig. 2d). For Au nanoparticles smaller than 20 nm, the blue shift and narrowing of the absorption are attributed to the increase in particle size.41–43 The intensification of the absorption is correlated with the increase in Au loading and particle size.13,44 The DRS results, therefore, confirm the quantum confinement effects emerging in the ALD-grown nanoparticles and demonstrates the ability to tune them.
As synthesizing nanoparticles with an average size below 10 nm is essential for the catalytic applications of Au/TiO2 nanocomposites, we focused on tuning the size distribution and the Au loading by tuning the exposures of the counter reactants while using short Au precursor exposures. First, we studied the effect of varying the ozone exposure from 30 s to 10 min, while keeping the precursor and water exposure time constant at 10 min and 30 s, respectively. The elemental analysis confirms that the Au loading on P25 nanoparticles is virtually unaffected by varying the ozone exposure such a time window (Fig. 3b). Nonetheless, the ozone exposure time has a dramatic effect on the size distribution of the ALD-grown nanoparticles, in that both the average size and the width of the size distribution are drastically reduced from 9.35 nm (std. dev. of 3.3 nm) to 4.68 nm (std. dev. of 1.0 nm) when the ozone exposure time is increased from 0.5 to 10 min (Fig. 3a and d). Furthermore, the long tail that is observed at shorter ozone exposures gradually disappears with increasing exposure times. The fact that the loading is virtually unaffected by increasing ozone exposures, while the size distribution varies dramatically, suggests that the ozone changes the chemical affinity between Au and titania, and thus its degree of wetting and propensity to aggregate. XPS results confirm that the ozone affects the chemistry of the deposited Au as a significant decrease in Au(III) fraction is observed as the ozone exposure is increased (Fig. 3c). Since ozone participates in the combustion reactions of the organic ligands remaining after the precursor chemisorption, an exposure to ozone is expected to promote the reduction of the deposited Au. At the same time, prolonged ozone exposures may also induce the formation of a thin oxide layer on the surface of the nanoparticles that promote their stability.37,45,46
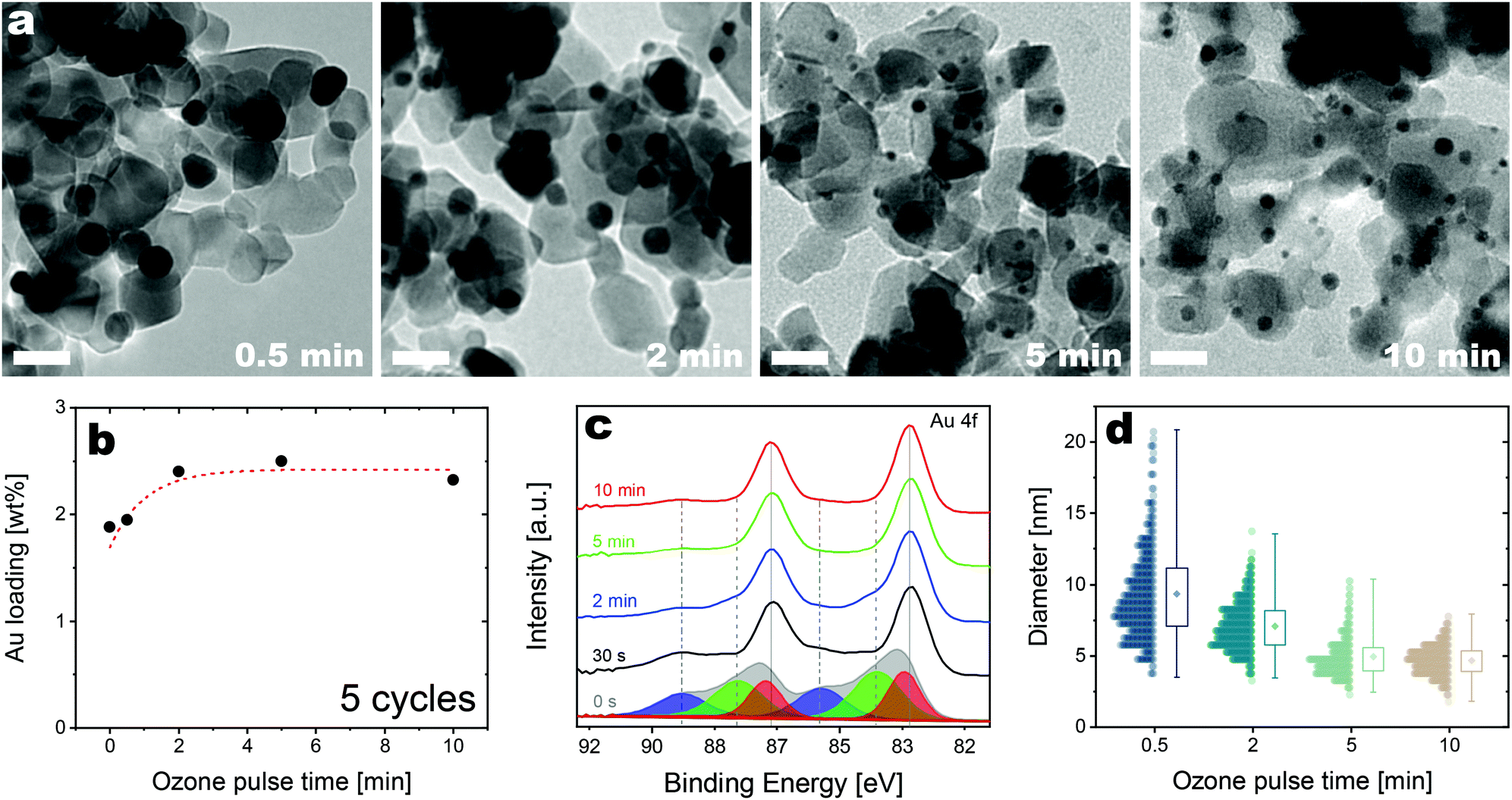 | ||
| Fig. 3 Evolution of the morphology, Au loading, and chemical state of the Au/TiO2 nanocomposites obtained after 5 cycles with different ozone exposure times. (a) TEM micrographs of the Au/TiO2 nanocomposites (scale bar: 20 nm), (b) evolution of the Au loading with the ozone exposure time (the dashed red line is drawn to guide the eye), (c) evolution of the chemical state of the ALD-grown Au as captured by the Au 4f region of XPS spectra. [Fitted peaks in 0 s ozone curve show different oxidation states of Au (red Au(0), green Au(I) and blue Au(III)), (d) evolution of the size distribution of the ALD-grown Au nanoparticles with increasing ozone exposure times.] | ||
Next, the effect of water exposure was studied by keeping Au and ozone exposure at 10 min and varying the water exposure from 0 to 2 min. An analysis of the elemental composition by XPS and ICP shows no appreciable changes in the loading of Au on titania nanoparticles upon longer water exposures (Fig. 4e and ESI Fig. 2†). Nonetheless, longer water exposures broaden the size distribution of the Au nanoparticles (Fig. 4a–d and f). These results suggest that water promotes surface mobility, and thus the propensity of adatoms and nanoparticles to diffuse and coalesce, while not affecting the amount of precursor that can chemisorb in every cycle. Griffiths et al. performed a ternary Au ALD process on Si wafers using water as the second counter reactant to eliminate the phosphorus impurity.30 Water would react with the phosphorous impurities producing phosphoric acid, which more readily desorbs from the growth surface. However, in our case, ICP-OES and XPS show no changes in the phosphorous content upon increasing the water exposure time (ESI Fig. 2†). The ineffectiveness of water exposure in removing P can be attributed to the differences in the ALD environment and support material used here compared to the study by Griffiths et al. When the ALD process is performed in vacuum and on flat substrates with a surface area of ∼cm2, several seconds of water exposure suffice to hydrolyze the phosphorus to phosphoric acid and purge the latter from the reactor. However, in our experiments, the high surface area of the powders (∼100 m2) and the use of atmospheric pressure might hinder the removal of phosphorous. In fact, the removal of low-vapor-pressure substances, such as phosphoric acid, can be particularly slow in powders compared with flat substrates because of two phenomena: (1) capillary condensation, and (2) the fact that molecules that desorb from one particle can adsorb onto other particles before they can escape from the reactor.47 In particular, the latter is exacerbated by operating the reactor at relatively high pressure compared with the vacuum reactors used by Griffiths et al.30 The effective removal of phosphorus in the form of phosphoric acid would probably require much longer water exposures, which would be cumbersome as they tend to disrupt the fluidization behavior of the powders. Hence, the fabrication of phosphorus-free Au/TiO2 nanocomposites via fluidized bed reactors operating at atmospheric pressure might require more aggressive treatments. The complete removal of P would allow understanding the role of P in catalytic properties of our nanocomposites and is subject to further research.
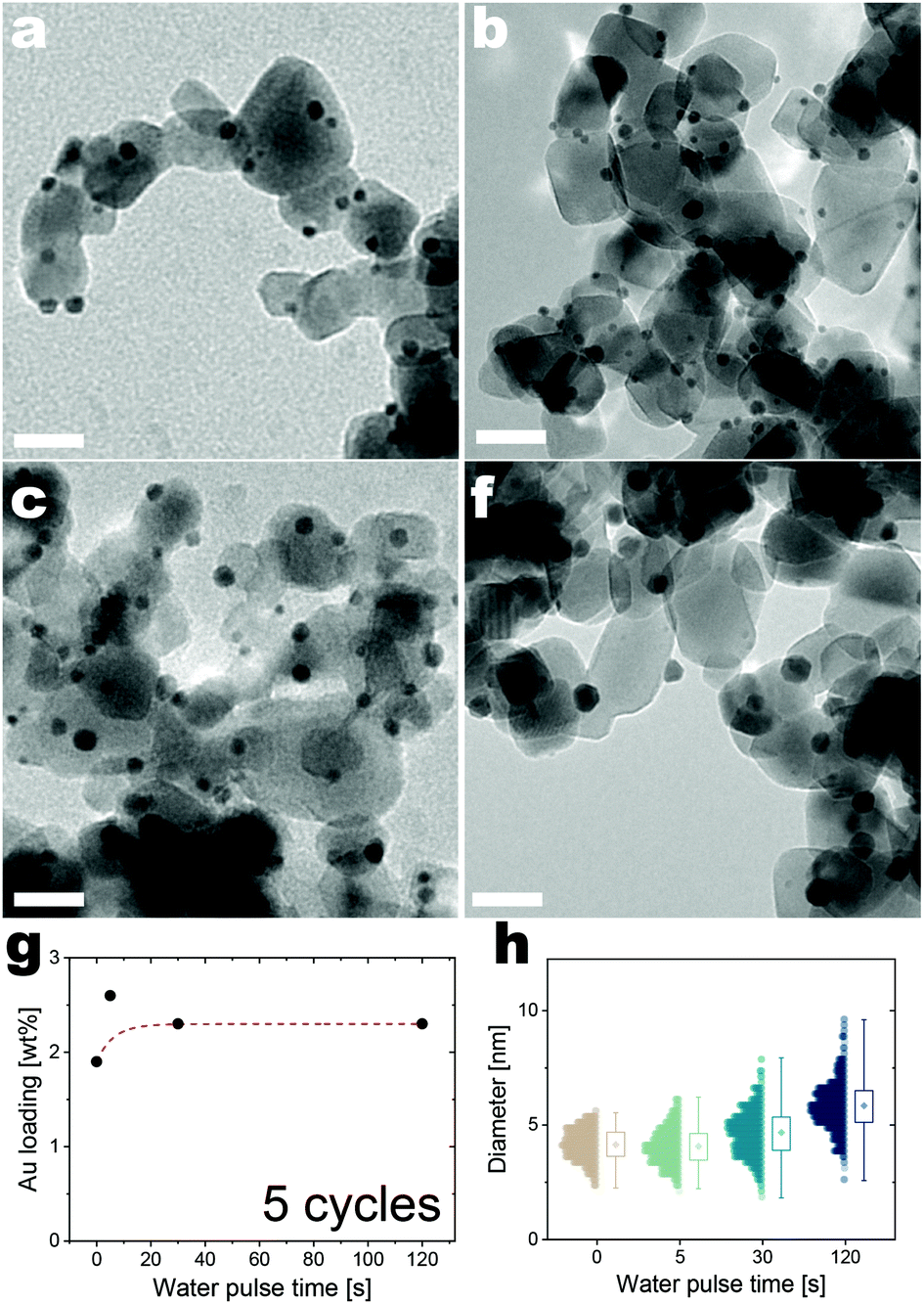 | ||
| Fig. 4 Evolution of the morphology and Au loading of the Au/TiO2 nanocomposites obtained after 5 cycles with different water exposure times. (a–d) TEM micrographs of the Au/TiO2 nanocomposites (scale bar 20 nm), (e) evolution of the Au loading (the dashed red line is drawn to guide the eye), (f) size distribution of the ALD-grown Au nanoparticles at different water exposure times. | ||
Next, the effect of varying the number of ALD cycles was studied using the following sequence of Au–O3–H2O exposure times: 10 min–10 min–30 s. A 10 min nitrogen purge was applied after each precursor/co-reactant exposure. The Au loading shows linear evolution with the number of cycles in the range of 1 to 9 cycles (Fig. 5d). Increasing the number of cycles results in a slight increase in the average diameter of the ALD-grown nanoparticles, while the span of size distribution remains relatively constant (Fig. 5a–c and e). This is not surprising because a precursor exposure time of 10 min translates into a growth per cycle of only ∼0.26 at nm−2, which is 0.02 Au(111) ML. As a result, increasing the number of cycles from 3 to 9 would result in a total increase of ∼1.6 at nm−2, which in itself cannot result in an appreciable change in particle size. Hence, this clearly illustrates how the use of long ozone exposures in conjunction with short precursor exposures mitigates nanoparticle sintering in each ALD cycle, and thus lends itself to the fine-tuning of the size of the ALD-grown nanoparticles down to the sub-nanometer level. The plasmon resonance peak measured from the samples with a varying number of cycles shifts towards lower wavelengths (578 to 544 nm) as the number of Au ALD cycles increases (Fig. 5f). A stronger and narrower absorption band is also observed as a result of increasing Au loading upon increasing the number of cycles. As the Au particle size variation from the 3 cycles to 5 cycles experiments is less than one nanometer (4.5 to 4.7 nm), the detected shift in the resonance peak wavelength is almost negligible (∼8 nm shift). On the other hand, the Au loading has a 40% increase from 3 to 5 cycles. This substantial change in the Au loading explains the enhanced resonance intensity and narrowing down of the plasmon resonance peak.
 | ||
| Fig. 5 Effect of varying the number of cycles using long ozone exposures and short precursor exposures. (a–c) TEM micrographs of the Au/TiO2 nanocomposites (scale bar 20 nm), (d) evolution of the Au loading with the number of cycles (the dashed red line is drawn to guide the eye), (e) size distribution of the ALD-grown Au nanoparticles (f) Absorbance of the Au/TiO2 nanocomposites as obtained via DRS measurements. | ||
To further demonstrate the ability of our process to control the size distribution of the ALD-grown nanoparticles at a given Au loading, we tested the photocatalytic activity of the Au/TiO2 nanocomposites towards the degradation of a model dye: rhodamine B. Incorporating titania with Au nanoparticles has been reported to increase its photocatalytic activity towards various reactions, including the oxidation of organic molecules and water splitting.13,48–51 Several mechanisms have been proposed in the literature to explain such an enhancement. For example, Au nanoparticles can extend the absorption of titania in the visible region because of plasmon resonance, and they can mediate surface charge recombination rates. In general, the effect of Au nanoparticles varies across different reactions and strongly depends on both the loading and the particle size. Although understanding the exact mechanism behind the enhanced photocatalytic activity of Au/TiO2 towards the decomposition of rhodamine B is beyond the scope of this work, we do show how the precision of our process can be used to decouple the effect of loading and particle size, and thus lead to optimal photocatalysts.
First, we tested the photocatalytic activity of the nanocomposites obtained after five cycles using five different precursor exposures (Fig. 6a), which translate into Au loadings that vary in the range ∼1–7 wt% and particle sizes in the range ∼5–15 nm. On these samples, the Au loading and the particle size are not decoupled as higher loadings correspond to larger particle sizes. As shown in Fig. 6a, higher loadings, and thus larger particle sizes have a non-monotonic effect on the photocatalytic activity of the nanocomposites, as quantified in terms of the apparent rate constant (kapp) extracted from the decay over time of the rhodamine B concentration upon illumination (ESI Fig. 3†). The rate constant increases threefold upon increasing the Au loading to ∼5 wt% compared with bare TiO2. Yet increasing the loading further sees a gradual drop in kapp. Although a maximum exists, loading and particle size are coupled and so it is not clear whether this trend is to be attributed to the loading or the particle size. This exemplifies one of the common problems encountered in fundamental studies into the catalytic activity of nanostructured materials.
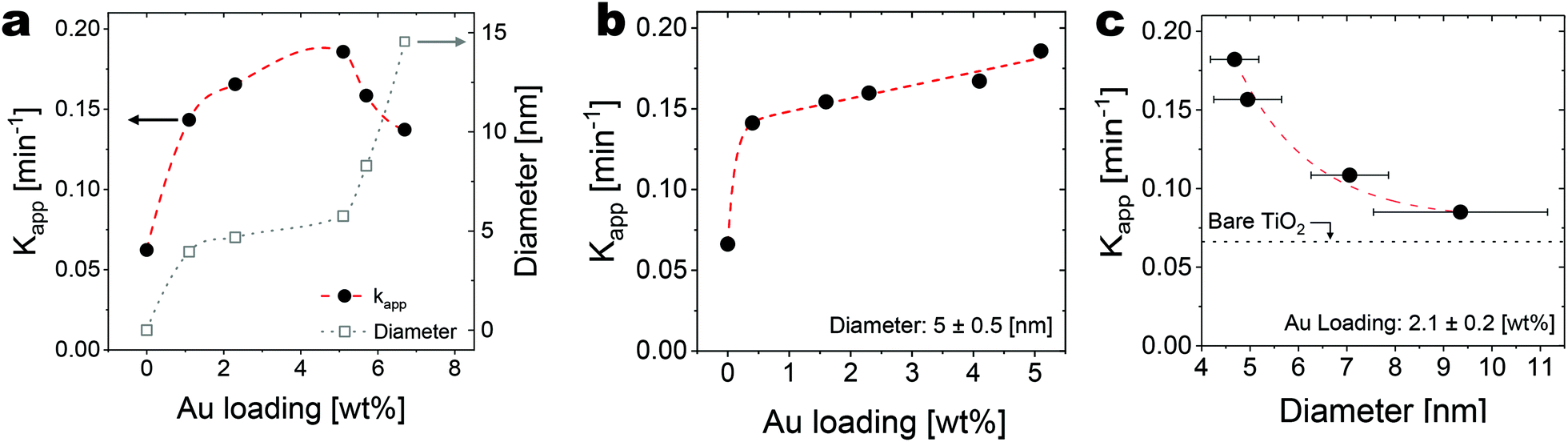 | ||
| Fig. 6 Photocatalytic activity of the Au/TiO2 nanocomposites towards the decomposition of rhodamine B with (a) varying both Au loading and particle size, (b) fixed particle size (5 nm) and varying Au loading, (c) fixed Au loading (2%) and increasing particle size (error bars: Standard deviation of the particle size distribution). | ||
To illustrate how our process can be used to gain insights into the structure-dependent properties of Au/TiO2 nanocomposites, we studied the photocatalytic activity of samples obtained by decoupling the Au loading from the particle size. Fig. 6b shows that for given particle size (5 nm), kapp increases roughly linearly with the Au loading in the range ∼0.5 to 5 wt%. On the other hand, Fig. 6c shows that at a given loading (∼2.1 wt%), kapp rapidly decreases with increasing particle size (in the range ∼4–10 nm), and approaches the value obtained for the control case with bare TiO2 for a particle size around 10 nm and larger. A similar trend was reported in the seminal work by Haruta,2 where they show that the catalytic activity of Au nanoparticles towards CO oxidation rapidly decreases with increasing particle size, and virtually vanishes beyond 10 nm. While the deposition of Au nanoparticles with a size smaller than 5 nm was not explored in this work, our proposed method provides a systematic approach to tune the Au nanoparticles size and loading and can be used to generate small nanoparticles (<5 nm) whose catalytic properties can be further investigated.
Conclusions
We demonstrated ALD of Au on gram-scale batches of titania nanopowders using a thermal process employing a fluidized bed reactor operated at atmospheric pressure. We performed ALD at the relatively low temperature of 105 °C using trimethylphosphino-trimethyl gold(III) as the metal precursor and ozone and water as co-reactants. This process leads to the formation of supported Au nanoparticles, with the number of cycles controlling the Au loading. We found that by varying the exposure time of the co-reactants, it is possible to control the broadness of the size distribution of the Au nanoparticles independently from the Au loading. In particular, ozone and water exposures have opposite effects: long ozone exposures suppress nanoparticle sintering and thus result in narrow size distributions, whereas long water exposures lead to broad ones. By leveraging the ability of the process to control the particle size at given loading, we could explore the structure–property relationship of the ALD-grown Au/TiO2 in the photocatalytic degradation of rhodamine B. We could achieve up to a threefold increase in the photocatalytic activity of titania by increasing the Au loading in the range 0–5 wt%, while maintaining a constant nanoparticle size of ∼5 nm. Conversely, increasing the nanoparticle size from 5 to 9 nm at the given loading of ∼2.1 wt% resulted in a decrease of the photocatalytic activity which gradually approached that of bare TiO2. This excellent control over the size and loading of Au nanoparticles ultimately enabled the enhanced photocatalytic activity which is superior to the previously reported Au/P25 nanocomposites prepared via colloidal synthesis. This process opens new avenues not only for the scalable fabrication of Au/TiO2 nanocomposites, but also for investigating the structure–property relationship of supported Au nanoparticles.Conflicts of interest
J. R. van Ommen has a financial interest in Delft IMP.Acknowledgements
This research is supported by the TU Delft | Global Initiative, a program of the Delft University of Technology to boost Science and Technology for Global Development.References
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS.
- F. Moreau, G. C. Bond and A. O. Taylor, J. Catal., 2005, 231, 105–114 CrossRef CAS.
- A. Corma, P. Serna and H. García, J. Am. Chem. Soc., 2007, 129, 6358–6359 CrossRef CAS.
- M. Hosseini, S. Siffert, H. L. Tidahy, R. Cousin, J. F. Lamonier, A. Aboukais, A. Vantomme, M. Roussel and B. L. Su, Catal. Today, 2007, 122, 391–396 CrossRef CAS.
- T. Barakat, J. C. Rooke, M. Franco, R. Cousin, J.-F. Lamonier, J.-M. Giraudon, B.-L. Su and S. Siffert, Eur. J. Inorg. Chem., 2012, 2012, 2812–2818 CrossRef CAS.
- M. Hosseini, T. Barakat, R. Cousin, A. Aboukais, B. L. Su, G. De Weireld and S. Siffert, Appl. Catal., B, 2012, 111, 218–224 CrossRef.
- A. Dollinger, L. Stolch, Y. Luo, M. Beck, C. H. Strobel, M. Hagner, S. Dilger, M. Bein, S. Polarz, G. F. Gantefoer, Y.-D. Kim and S. Proch, Phys. Chem. Chem. Phys., 2014, 16, 11017–11023 RSC.
- J. Hämäläinen, M. Ritala and M. Leskelä, Chem. Mater., 2014, 26, 786–801 CrossRef.
- L. Li, A. H. Larsen, N. A. Romero, V. A. Morozov, C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Norskov, J. Phys. Chem. Lett., 2013, 4, 222–226 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- M. Cargnello, Chem. Mater., 2019, 31, 576–596 CrossRef CAS.
- B. R. Cuenya and F. Behafarid, Surf. Sci. Rep., 2015, 70, 135–187 CrossRef.
- H. Van Bui, F. Grillo, S. S. Kulkarni, R. Bevaart, N. Van Thang, B. van der Linden, J. A. Moulijn, M. Makkee, M. T. Kreutzer and J. R. van Ommen, Nanoscale, 2017, 9, 10802–10810 RSC.
- L. Zang, W. Macyk, C. Lange, W. F. Maier, C. Antonius, D. Meissner and H. Kisch, Chem. – Eur. J., 2000, 6, 379–384 CrossRef CAS.
- D. Li, J. T. McCann, M. Gratt and Y. Xia, Chem. Phys. Lett., 2004, 394, 387–391 CrossRef CAS.
- P. N. Amaniampong, K. Li, X. Jia, B. Wang, A. Borgna and Y. Yang, ChemCatChem, 2014, 6, 2105–2114 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- B. J. O'Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef.
- J. E. Mondloch, E. Bayram and R. G. Finke, J. Mol. Catal. A: Chem., 2012, 355, 1–38 CrossRef CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS.
- F. Grillo, M. T. Kreutzer and J. R. van Ommen, Chem. Eng. J., 2015, 268, 384–398 CrossRef CAS.
- A. J. M. Mackus, M. J. Weber, N. F. W. Thissen, D. Garcia-Alonso, R. H. J. Vervuurt, S. Assali, A. A. Bol, M. A. Verheijen and W. M. M. Kessels, Nanotechnology, 2016, 27(3), 034001 CrossRef PubMed.
- J. Dendooven, R. K. Ramachandran, E. Solano, M. Kurttepeli, L. Geerts, G. Heremans, J. Rongé, M. M. Minjauw, T. Dobbelaere, K. Devloo-Casier, J. A. Martens, A. Vantomme, S. Bals, G. Portale, A. Coati and C. Detavernier, Nat. Commun., 2017, 8, 1074 CrossRef PubMed.
- F. Grillo, H. Van Bui, J. A. Moulijn, M. T. Kreutzer and J. R. van Ommen, J. Phys. Chem. Lett., 2017, 8, 975–983 CrossRef CAS PubMed.
- H. Van Bui, F. Grillo and J. R. van Ommen, Chem. Commun., 2017, 53, 45–71 RSC.
- F. Grillo, H. Van Bui, D. La Zara, A. A. I. Aarnink, A. Y. Kovalgin, P. Kooyman, M. T. Kreutzer and J. R. van Ommen, Small, 2018, 14(29), 1800765 CrossRef PubMed.
- M. B. E. Griffiths, P. J. Pallister, D. J. Mandia and S. T. Barry, Chem. Mater., 2016, 28, 44–46 CrossRef CAS.
- M. Mäkelä, T. Hatanpää, K. Mizohata, J. Räisänen, M. Ritala and M. Leskelä, Chem. Mater., 2017, 29, 6130–6136 CrossRef.
- Z. Bian, T. Tachikawa, P. Zhang, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 458–465 CrossRef CAS PubMed.
- A. Goulas and J. Ruud van Ommen, J. Mater. Chem. A, 2013, 1, 4647–4650 RSC.
- B. A. van Driel, P. J. Kooyman, K. J. van den Berg, A. Schmidt-Ott and J. Dik, Microchem. J., 2016, 126, 162–171 CrossRef CAS.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 121301 CrossRef.
- F. Grillo, J. A. Moulijn, M. T. Kreutzer and J. R. van Ommen, Catal. Today, 2018, 316, 51–61 CrossRef CAS.
- E. Ruckenstein, J. Cryst. Growth, 1979, 47, 666–670 CrossRef CAS.
- M. J. J. Jak, C. Konstapel, A. van Kreuningen, J. Verhoeven and J. W. M. Frenken, Surf. Sci., 2000, 457, 295–310 CrossRef CAS.
- A. La Torre, M. D. Gimenez-Lopez, M. W. Fay, G. A. Rance, W. A. Solomonsz, T. W. Chamberlain, P. D. Brown and A. N. Khlobystov, ACS Nano, 2012, 6, 2000–2007 CrossRef CAS PubMed.
- G. R. Bell, P. M. Dawson, P. A. Pandey, N. R. Wilson and P. A. Mulheran, APL Mater., 2014, 2, 012109 CrossRef.
- E. Cottancin, G. Celep, J. Lermé, M. Pellarin, J. R. Huntzinger, J. L. Vialle and M. Broyer, Theor. Chem. Acc., 2006, 116, 514–523 Search PubMed.
- Y. Fleger and M. Rosenbluh, Int. J. Opt., 2009, 2009, 5 Search PubMed.
- S. Peng, J. M. McMahon, G. C. Schatz, S. K. Gray and Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14530 CrossRef CAS PubMed.
- E. D. Martínez, C. Boissiére, D. Grosso, C. Sanchez, H. Troiani and Galo J. A. A. Soler-Illia, J. Phys. Chem. C, 2014, 118, 13137–13151 CrossRef.
- E. Solano, J. Dendooven, R. K. Ramachandran, K. V. Van de Kerckhove, T. Dobbelaere, D. Hermida-Merino and C. Detavernier, Nanoscale, 2017, 9, 13159–13170 RSC.
- G. Goula, G. Botzolaki, A. Osatiashtiani, C. M. A. Parlett, G. Kyriakou, R. M. Lambert and I. V. Yentekakis, Catalysts, 2019, 9, 541 CrossRef CAS.
- T. Onn, R. Küngas, P. Fornasiero, K. Huang and R. J. I. Gorte, Inorganics, 2018, 6, 34 CrossRef.
- X. Z. Li and F. B. Li, Environ. Sci. Technol., 2001, 35, 2381–2387 CrossRef CAS PubMed.
- I. Paramasivam, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 71–75 CrossRef CAS.
- O. Rosseler, M. V. Shankar, M. K.-L. Du, L. Schmidlin, N. Keller and V. Keller, J. Catal., 2010, 269, 179–190 CrossRef CAS.
- P. Sangpour, F. Hashemi and A. Z. Moshfegh, J. Phys. Chem. C, 2010, 114, 13955–13961 CrossRef CAS.
- J. R. van Ommen and A. Goulas, Mater. Today Chem., 2019, 14, 100183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr01092h |
| This journal is © The Royal Society of Chemistry 2020 |