
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transport and programmed release of nanoscale cargo from cells by using NETosis†
Daniel
Meyer
a,
Saba
Telele
a,
Anna
Zelená
b,
Alice J.
Gillen
 c,
Alessandra
Antonucci
c,
Elsa
Neubert
d,
Robert
Nißler
a,
Florian A.
Mann
a,
Luise
Erpenbeck
d,
Ardemis A.
Boghossian
c,
Sarah
Köster
b and
Sebastian
Kruss
*a
c,
Alessandra
Antonucci
c,
Elsa
Neubert
d,
Robert
Nißler
a,
Florian A.
Mann
a,
Luise
Erpenbeck
d,
Ardemis A.
Boghossian
c,
Sarah
Köster
b and
Sebastian
Kruss
*a
aInstitute of Physical Chemistry, Göttingen University, 37077 Göttingen, Germany. E-mail: skruss@uni-goettingen.de
bInstitute of X-Ray Physics, Göttingen University, 37077 Göttingen, Germany
cÉcole Polytechnique Fédérale de Lausanne (EPFL), Lausanne, Switzerland
dDepartment of Dermatology, Venerology and Allergology, University Medical Center, Göttingen University, 37075 Göttingen, Germany
First published on 3rd April 2020
Abstract
Cells can take up nanoscale materials, which has important implications for understanding cellular functions, biocompatibility as well as biomedical applications. Controlled uptake, transport and triggered release of nanoscale cargo is one of the great challenges in biomedical applications of nanomaterials. Here, we study how human immune cells (neutrophilic granulocytes, neutrophils) take up nanomaterials and program them to release this cargo after a certain time period. For this purpose, we let neutrophils phagocytose DNA-functionalized single-walled carbon nanotubes (SWCNTs) in vitro that fluoresce in the near infrared (980 nm) and serve as sensors for small molecules. Cells still migrate, follow chemical gradients and respond to inflammatory signals after uptake of the cargo. To program release, we make use of neutrophil extracellular trap formation (NETosis), a novel cell death mechanism that leads to chromatin swelling, subsequent rupture of the cellular membrane and release of the cell's whole content. By using the process of NETosis, we can program the time point of cargo release via the initial concentration of stimuli such as phorbol 12-myristate-13-acetate (PMA) or lipopolysaccharide (LPS). At intermediate stimulation, cells continue to migrate, follow gradients and surface cues for around 30 minutes and up to several hundred micrometers until they stop and release the SWCNTs. The transported and released SWCNT sensors are still functional as shown by subsequent detection of the neurotransmitter dopamine and reactive oxygen species (H2O2). In summary, we hijack a biological process (NETosis) and demonstrate how neutrophils transport and release functional nanomaterials.
1. Introduction
Targeted delivery of (nano)materials and pharmaceuticals is one of the great challenges in biomedicine.1 Encapsulation of drugs in colloidal structures such as liposomes or polymeric micelles has been studied thoroughly over the years. These approaches demonstrated great success in delivery of anti-cancer agents or antimicrobials.1–3 Nanomaterials such as nanoparticles,4 carbon nanotubes5 or nanobots6 offer several benefits for such applications due to their optoelectronic properties, tunable surface chemistry and ability to infiltrate cellular membranes.5 Additionally, they can be imparted with useful functions. For example, single-walled carbon nanotubes (SWCNTs) are known for their near infrared (nIR) fluorescence and serve as versatile building blocks for optical nanosensors.7–10 Their surface can be chemically tailored with DNA, peptides, lipids, nanobodies or viruses to sense biologically relevant signaling molecules with high spatiotemporal resolution.11–18 Thus, such nanomaterials are attractive candidates for biomedical applications. It is also known that nanomaterials such as SWCNTs can be taken up by living cells depending on their size.19 Furthermore, surface functionalization plays an important role and determines uptake and retention inside cells.20From a different perspective, the uptake of nanomaterials by cells can be used as a concept for cargo delivery or transport.21,22 One example is engineered bacteria that are programmed for lysis in vivo, resulting in the delivery of cytotoxic agents and a potential way to tackle cancer.23 Another example is the binding and transport of cargo molecules by surface-modified red blood cells, which form long-living, biocompatible hybrid carriers.24,25 Neutrophilic granulocytes (neutrophils) are the most abundant type of white blood cells. They are interesting candidates for cargo delivery because they are able to take up materials (phago-/endocytosis),26 sense and migrate along chemical gradients (chemotaxis) to inflammatory sites27,28 or cross dense borders, such as the blood–brain barrier.29,30 The abilities of neutrophils have been used to take up silica particles to follow E. coli gradients and paclitaxel-containing liposomes for cancer treatment.29,31
In general, neutrophils have been discussed in different ways for cargo transport and delivery.29,30,32 However, so far it was not possible to program cargo release. Another function of neutrophils is neutrophil extracellular trap (NET) formation (NETosis), a defense strategy and novel type of cell death.33,34 During NETosis, the cell's chromatin is chemically modified, which leads to its expansion and ultimately the rupture of the cellular membrane and the release of their cytosolic content (Fig. 1).35 Neubert et al. showed that this process consists of different phases, including a first active phase in which the cell remains fully functional.35 NETosis is triggered by different (chemical) stimuli, such as phorbol 12-myristate-13-acetate (PMA) or lipopolysaccharides (LPS), that activate signaling pathways that include the enzymes protein-arginine deiminase 4 (PAD4),36 neutrophil elastase (NE) or myeloperoxidase (MPO).33,37–40 In summary, neutrophils possess several functions that are highly interesting for cargo delivery of nanoscale materials.
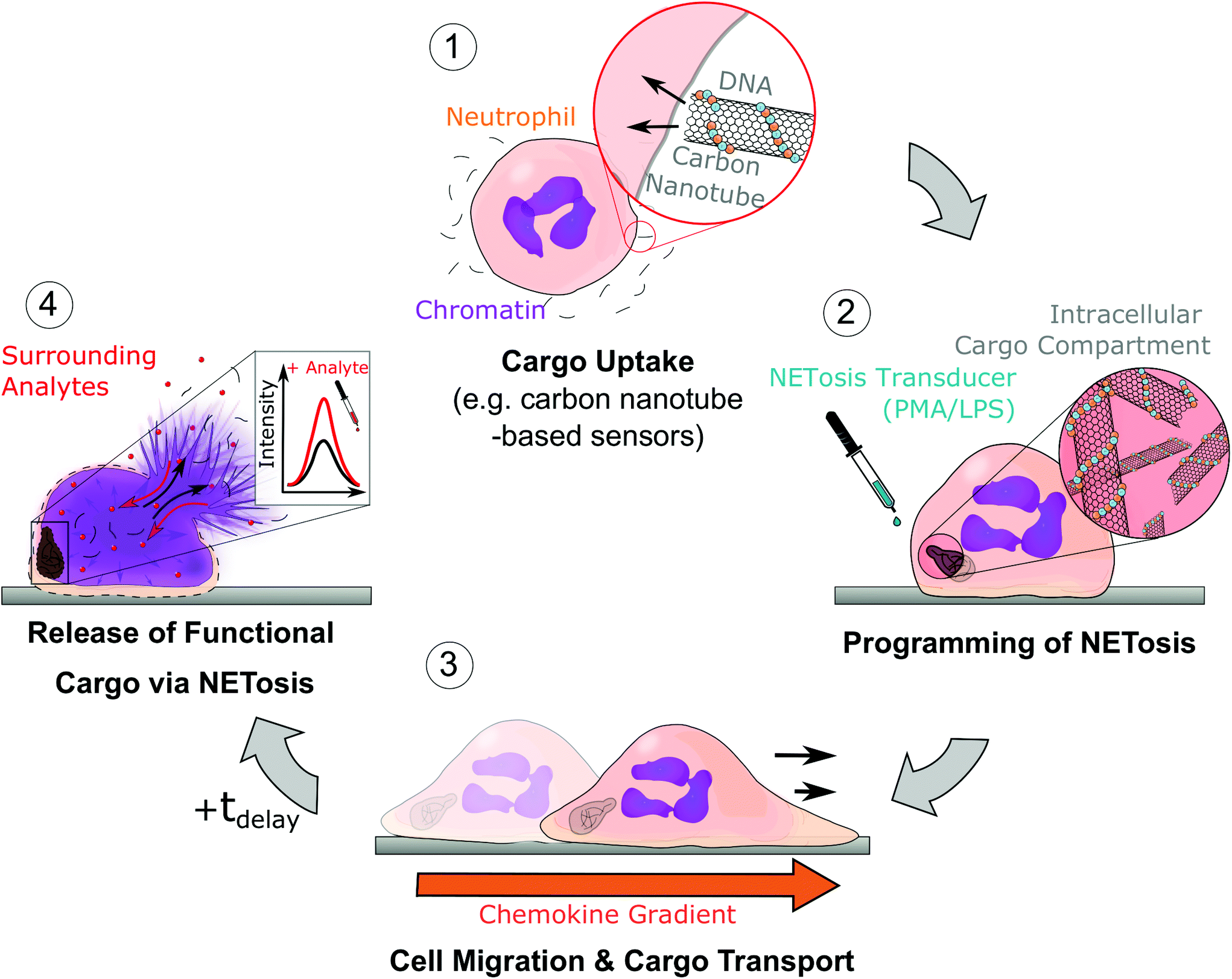 | ||
| Fig. 1 Schematic of uptake, transport and programmed release of cargo by neutrophils; (1) neutrophils take up a nanomaterial such as DNA-coated single-walled carbon nanotube (SWCNT) sensors. (2) Afterwards, NETosis is chemically induced (with LPS or PMA), which determines the timepoint of NETosis and membrane rupture. During the first phase of NETosis, cells are still functional and able to migrate and follow inflammatory signals (3). (4) Finally, at the end of the NETotic process (tdelay), the cell membrane ruptures and releases the cargo into the extracellular space. In the case of SWCNT sensors (as cargo), they sense analytes at the new location via their near infrared fluorescence signal. | ||
Here, we make use of these functions and demonstrate uptake, transport and programmed release of functional nanosensors. We show that neutrophils take up carbon nanotube-based nIR fluorescent sensors as cargo, transport them and release them again via NETosis. Importantly, we quantify time and length scales of this process and showcase that the cargo is fully functional after delivery by detecting small molecules with the transported nanosensors.
2. Results and discussion
2.1 Uptake and release of carbon nanotubes by neutrophils
Neutrophils use phagocytosis to destroy foreign objects.26,41 Therefore, neutrophils should also be able to take up nanomaterials. In this study, we chose SWCNTs as a model cargo to make use of their unique nIR fluorescent sensing properties. Neutrophils readily took up DNA-functionalized (GT)15-(6,5)-SWCNTs (Fig. 2a and b). The nIR signal (red) of the uptaken SWCNTs is localized in a region of the neutrophil outside the nucleus (blue), and this compartment often remained in the rear of the cells during migration (Fig. 2a, ESI Movie 1†). | ||
| Fig. 2 Uptake of nanomaterial cargo by neutrophils; (a) neutrophils take up (GT)15-SWCNT nanosensors, are still fully functional and able to migrate. Phase contrast (top) and fluorescence images of chromatin (blue, Hoechst 33342 staining) and nIR fluorescent SWCNTs (red). SWCNTs appear to be located at the rear of the cell. Scale bar = 10 μm. (b) SWCNT uptake measured by nIR fluorescence intensity increases inside the cells. Uptake takes place within minutes and saturates after 15 minutes. Mean ± SEM, N = 2 donors, n > 60 cells for each time point. (c) The SWCNT fluorescence signal changes during NETosis. Both SWCNT area (blue, circular data points) as well as intensity (black, circular data points) decrease during the process. Chromatin area (blue, triangular data points) increases as expected from NETosis. Mean ± SEM, N = 3 donors, n > 30 cells. (d) Time course of NETosis in different (GT)15-SWCNT-loaded and activated neutrophils show cell rounding and chromatin decondensation. SWCNTs (>900 nm, red), chromatin (blue), cell membrane (green), phase contrast (grey). SWCNTs appeared to be compressed/pushed by the expanding chromatin to the outer cell membrane in the final phase of NETosis. Scale bar = 10 μm. | ||
It is most likely that this compartment is the phagosome, as it is often found in the actomyosin-rich rear of polarized neutrophils.42 Similarly, streptavidin-functionalized SWCNTs in HL60 cells, a model cell line for primary neutrophils, were found in a similar location.43 The nuclei (blue), on the other side, remained rather at the middle/front of the cell during migration as previously described.44 Uptake of SWCNTs increased with concentration and incubation time as evidenced by the nIR fluorescent signal inside the cells (Fig. 2b). After around 15 min, uptake reached a plateau (Fig. 2b). For low SWCNT concentrations (0.1 nM), cells showed normal behavior while at higher concentrations (>1 nM), we sometimes observed cell agglomerates (ESI Fig. S1a†). For this reason, we used 0.1 nM SWCNT for uptake and for all following experiments. Cells that took up SWCNTs were still able to perform NETosis after stimulation with 100 nM PMA and demonstrated the well-documented time course of chromatin decondensation and subsequent cell rupture (Fig. 2c). Surprisingly, the distribution of the SWCNT cargo changed during NET formation (Fig. 2d).
The size of the intracellular SWCNT-containing compartment did not change in early phases. In contrast, in later stages of NETosis, the compartment with the SWCNTs shrank parallel to chromatin expansion (Fig. 2d, ESI Fig. S2 and Movie 2†), which could be explained by an increasing intracellular pressure by chromatin swelling that compresses this compartment.35 This finding also explains why SWCNTs ended up in close proximity to the cell membrane. These observations are in agreement with measurements taken with a recently developed nIR fluorescence confocal microscope.45 The improved resolution in the z-direction provides additional insights into the location of SWCNTs inside neutrophils before and after activation (ESI Fig. S3†). SWCNTs are distributed in the whole cell but accumulate in the phagosome. Over the time course of NETosis, the SWCNTs are pushed closer to the cell wall, as seen from wide-field microscopy. After membrane rupture, the SWCNT signal is found in all areas around the cell as expected.
Additionally, the SWCNT fluorescence intensity decreased during the time course of NETosis, which could be attributed to several processes within the cell. During migration, no significant changes in nIR fluorescence intensity were observed (ESI Fig. S4†). Changes in the pH were reported to cause SWCNT fluorescence quenching.46 Similarly, SWCNTs that get closer to each other during the process could quench themselves. Degradation of SWCNTs by myeloperoxidase (MPO) or neutrophil elastase (NE) is also well known, but unlikely as it occurs over longer time scales (hours to days).47,48
2.2 Functionality of cargo-loaded neutrophils
Uptake and release of the cargo is a necessary step for a delivery system. However, it was unclear if DNA-SWCNT-loaded cells activated to perform NETosis were still functional and able to migrate for a considerable amount of time. Therefore, live-cell imaging of neutrophils exposed to different types and concentrations of NETosis-inducing compounds (LPS and PMA) was performed in the next step. For this purpose, the random movement of the most motile cells (n = 30) for each condition and blood donor (N = 3 independent donors) were tracked. Exemplary tracks are shown in Fig. 3a and trajectories for all conditions are shown in ESI Fig. S5.†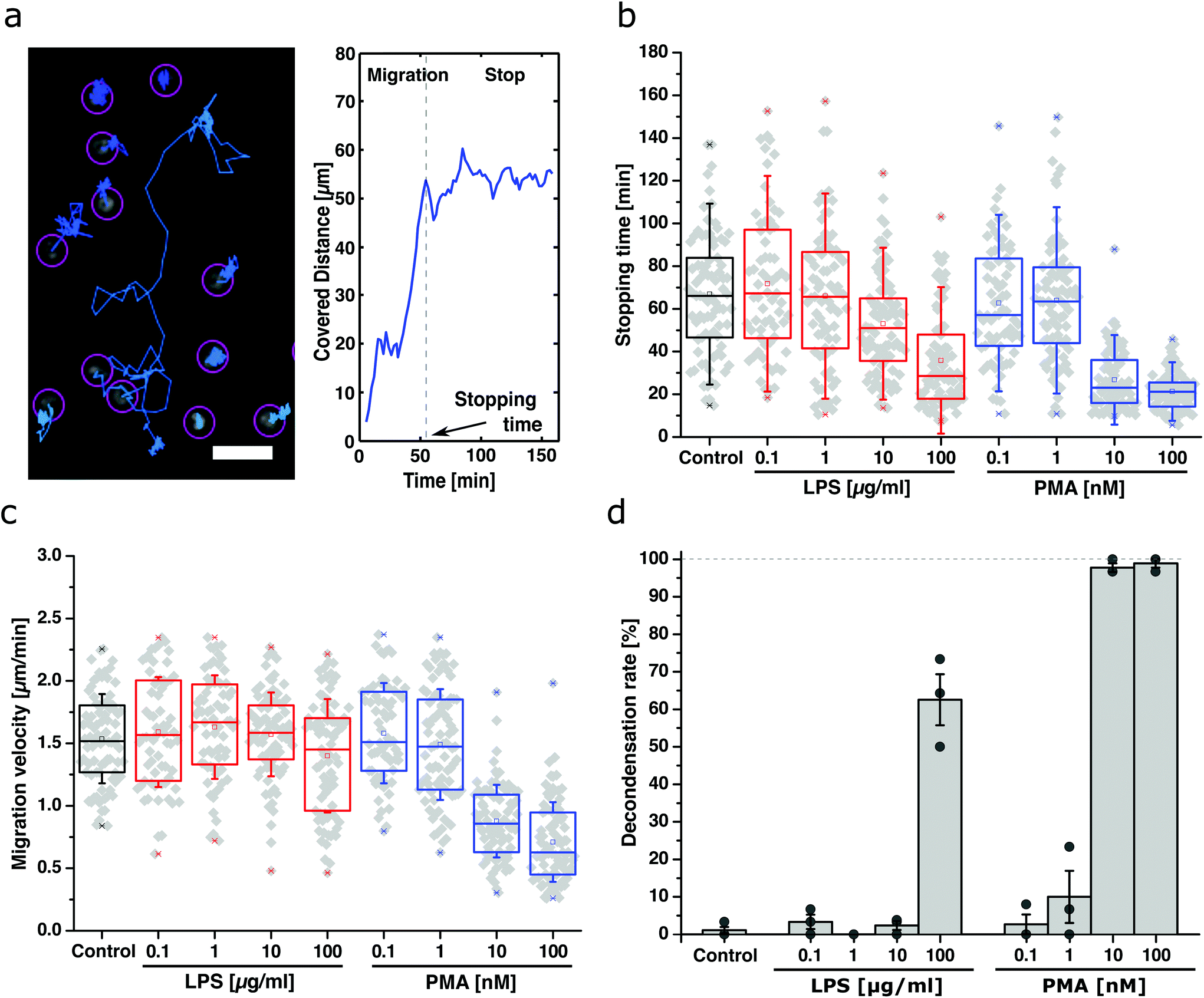 | ||
| Fig. 3 Migratory properties of SWCNT-loaded and NETosis-programmed neutrophils; (a) typical trajectories (left) and the (absolute) covered distance of a neutrophil (right). Without inflammatory gradient, cells randomly move in all directions until they stop due to the onset of the second phase of NETosis. Scale bar = 10 μm. (b) and (c) Time until the cells stop moving (stopping time) and migration velocity of activated neutrophils for different activation conditions. Increasing the concentration of LPS (red) decreases stopping time linearly. For PMA (blue) there was practically no movement above a certain concentration. Likewise, LPS does not influence the migration velocity whereas PMA slows them down at higher concentrations. N = 3 donors, n > 60 cells. Boxplot shows box line = 25–75% percentile, cross = mean, dot = min/max, error bars = SD. (d) Decondensation (NETosis) rates of neutrophils from (b)/(c) 160 minutes after activation. Very low amounts of LPS and PMA did not trigger NETosis. In contrast, higher values (100 μg mL−1 LPS, 10–100 nM PMA) lead to massive chromatin decondensation/NETosis. Data: mean ± SEM, N = 3, n > 60 cells. Nucleus stained with Hoechst 33342. | ||
The results show that the time for the neutrophils to reach a stationary phase (stopping time) decreased with increasing concentration of the activator for both LPS and PMA as expected (Fig. 3b). The migration velocity stayed relatively constant for all LPS concentrations (Fig. 3c) but drastically decreased with increasing PMA concentration (≥10 nM). Furthermore, NETosis (indicated by chromatin decondensation) was assessed 160 min after activation and showed a dose-dependent probability for cells to become NETotic (Fig. 3d).
Together, these results show that low activator concentrations (0.1–10 μg mL−1 LPS and 0.1–1 nM PMA) do not trigger high NETosis rates but maintained the neutrophil's motility. On the contrary, too high concentrations (10–100 nM PMA) resulted in high decondensation rates but also inhibited cell migration completely. Only for certain concentrations (100 μg mL−1 LPS and 1 nM PMA), cells were still able to migrate and perform NETosis during the time course of the experiment (decondensation images are shown in ESI Fig. S6,† example of cell migration behavior in ESI Movie 3†). In summary, we identified 100 μg mL−1 LPS as an optimal concentration to guarantee both migratory capabilities and efficient cargo release via NETosis.
2.3 Transport of nanoscale cargo via migration of neutrophils
To further investigate whether SWCNTs inside neutrophils affect their migration, a gradient migration assay (under agarose migration) was performed with cargo-loaded and unloaded cells.49 This assay mimics an in vivo scenario in which cargo-loaded neutrophils are supposed to follow inflammatory cues and finally release their cargo at the inflammatory site. Both experiments were performed in commonly used fetal calve serum (FCS) environments at low (0.5%, Fig. 4 and ESI Fig. S7†), as well as high concentrations (20%, ESI Fig. S8†) and showed (ESI Movie 4†) that cells react to chemokine gradients of interleukin-8 (IL8). In addition, cells were again exposed to different amounts of activators (0.1–10 nM PMA & 1–100 μg mL−1 LPS) and their migration distance was quantified after three hours of consecutive movement. In agreement with Fig. 3, increasing LPS concentrations decreased motility/covered distances. In contrast, addition of PMA showed either the same behavior as control samples (0.1–1 nM) or no migration at all (10 nM), implying an “all or nothing” behavior for PMA-induced NETosis pathways (Fig. 4c). This result is in agreement with a recent study that shows that PMA-induced NETosis does not require adhesion or mechanical input at all.50 Interestingly, cells migrated over longer distances at higher serum concentration (20% vs. 0.5%) conditions, which highlights that this approach could also work in vivo (ESI Fig. S7–S9 and Tables T1/T2†). Cargo-loaded and stimulated (LPS, 100 μg mL−1) cells migrated between 145 ± 44 μm (0.5% FCS) and 478 ± 162 μm (20% FCS).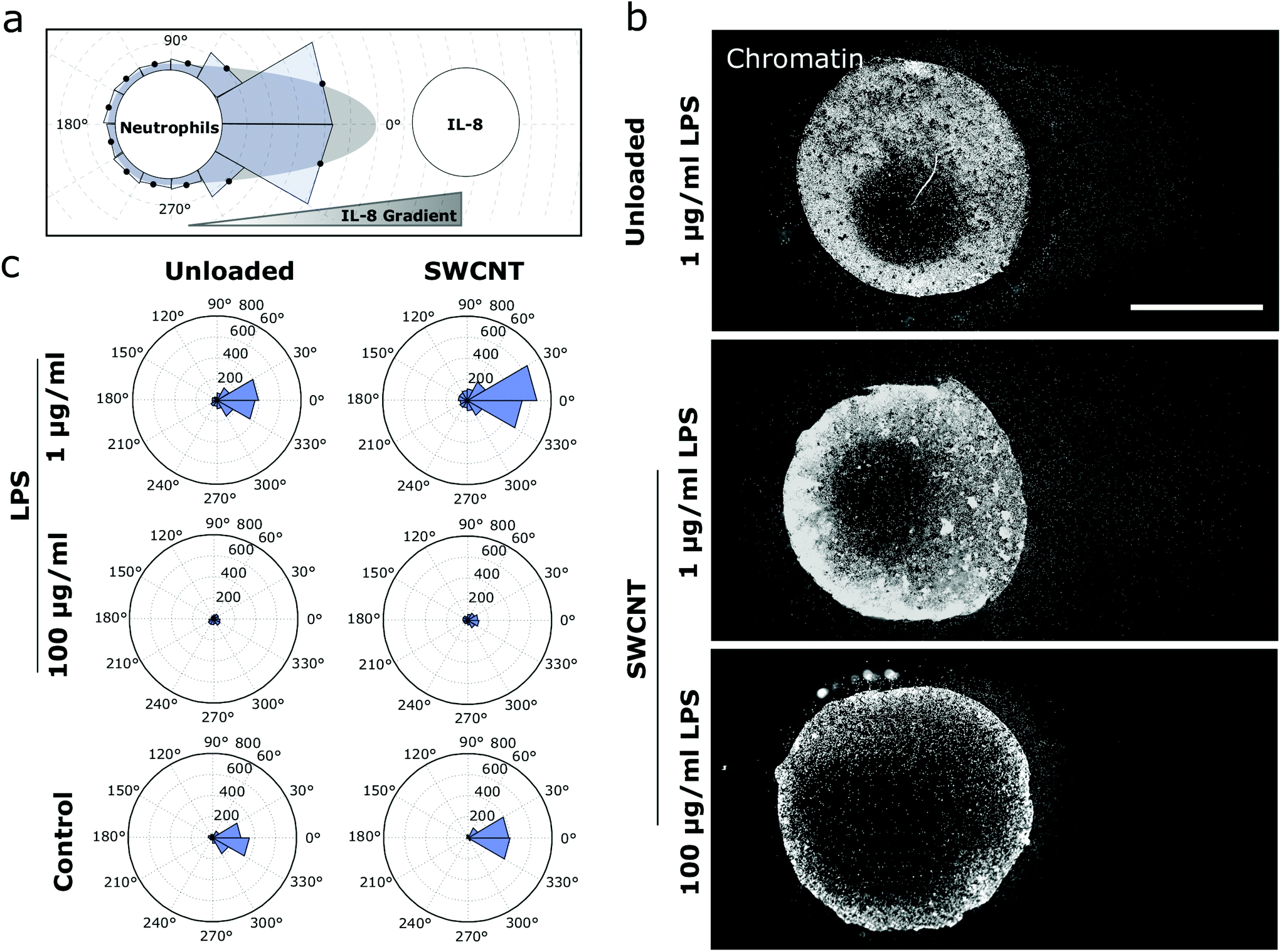 | ||
| Fig. 4 Collective migration and nanoscale cargo transport by activated neutrophils; (a) design of the migration experiment: neutrophils programmed to perform NETosis migrate along an IL-8 gradient (under-agarose assay). Then the distance to the frontier of the leading cells is quantified. (b) The concentration of the NETosis activator affects the period during which the cells still migrate and the onset of NETosis. Images show nuclei of neutrophils (chromatin stained by Hoechst 33342) after 3 h of migration. Scale bar = 500 μm. (c) Mean radial migration plots for different activators of NETosis and SWCNT uptake. Higher activator concentrations reduce the distance migrated by the cells. Interestingly, SWCNT-loaded cells travel around 30% further than the control cells after 3 h. | ||
2.4 Programmed release of functional nanosensors
In the final step, we performed functionality tests of the SWCNT cargo in inactivated and ruptured cells to investigate whether the specific abilities of the internalized material remain intact throughout NETosis. SWCNTs are useful nIR fluorescent building blocks of nanosensors for novel applications, and their selectivity depends on the specific surface functionalization.8,51 We used (GT)15-(6,5)-SWCNTs because they change their fluorescence in the presence of the neurotransmitter dopamine and are therefore powerful sensors.52–55 Such sensors have been used to image dopamine release from cells with high spatiotemporal resolution.56 As a second functional nanomaterial, we employed hemin-aptamer-functionalized SWCNTs,57,58 which are known to decrease their fluorescence in the presence of H2O2.57–59 In both cases, SWCNTs were taken up by neutrophils and their responses were measured via consecutive nIR imaging either while they were carried by non-activated cells or after NETotic membrane rupture. Here, the addition of 100 nM dopamine led to an instantaneous increase of the sensors’ intensity for disrupted cells. In comparison, in non-damaged cells, dopamine is not expected to enter the cell, and indeed, such cells showed no sensor response to dopamine (Fig. 5a and ESI Movies 5/6†). This result indicates a successful release and accessibility of the cargo after NETosis and full functionality of the dopamine nanosensors after release. In contrast, 100 μM H2O2 addition decreased the nIR signaling for both intact and NETotic cells (Fig. 5b and ESI Movies 7/8†), which can be explained by the diffusion of H2O2 through the cellular membrane.60,61 Interestingly, we were also able to locate differences in the sensors’ uptake behavior depending on the associated surface functionalization. While (GT)15-(6,5)-SWCNTs appeared most often in larger, intracellular structures, hemin-aptamer-SWCNTs were found closer to the cell membrane in smaller agglomerates (Fig. 5a/b and ESI Fig. S10a†). Nevertheless, both sensor types were functional after cargo transport and rupture, as evidenced by the same fluorescence response performance prior to cellular uptake (ESI Fig. S10b†) and control experiments (ESI Fig. S10c/d†). | ||
| Fig. 5 Release of functional nanomaterials at target sites; (a) cells programmed for NETosis release functional cargo such as (GT)15-(6,5)-SWCNTs that serve as nIR sensors for the neurotransmitter dopamine (by intensity increase). SWCNTs inside unactivated cells (grey line) showed no fluorescence changes to 100 nM dopamine. In contrast, the area around cells that underwent NETosis showed a sensor response (black line). Scale bar = 5 μm, chromatin stained with Hoechst 33342. (b) Other cargo such as hemin-aptamer-SWCNTs (H202 sensor) responded (intensity decrease) to 100 μM H202 both in unactivated (grey) and ruptured cells because H202 diffuses through cell membranes. (c) (GT)15-(6,5)-SWCNT-loaded neutrophils find adhesive cues and adhere to them. Here, fibrinogen was patterned on glass (top). After NETosis, chromatin (and cargo) is found in those locations (bottom). (d) nIR-colocalization of the (GT)15-(6,5)-SWCNTs transported by neutrophils and the adhesive cues. Sensor positions correlated with the pattern (white lines), but the soft chromatin (including the sensors) quickly diffuses away and therefore the pattern does not remain stable. Sensors were still functional and respond to dopamine (100 nM). Scale bar = 100 μm. Chromatin stained with Hoechst 33342. | ||
Finally, we also demonstrated the transport and release of the functionalized nanosensors to specific target sites. For this purpose, fibrinogen was patterned on glass surfaces and SWCNT-loaded, activated neutrophils were allowed to migrate over the coated area, resulting in an alignment of the cells along the fibrinogen pattern (Fig. 5c). Again, the immobilized sensors were still functional after cell rupture on the pattern and showed an instant response to 100 nM dopamine (Fig. 5d and ESI Movie 9†). Thus, neutrophils can be programmed to take up such a nanoscale cargo, transport it to specific sites and release it in a functional state.
Of course, SWCNTs can vary in length, functionalization and chirality. It is known that those properties affect uptake and retention in cells.20 Therefore, one could envision to tailor the nanoscale cargo for specific uptake/retention kinetics. In this study, functionalized SWCNTs have been used as sensors for two important signaling molecules (dopamine, H2O2). However, sensors for many other interesting molecules have been developed and could be transported by this approach.11,12,15–18,62 For example, recently, a SWCNT-based sensor for another important neurotransmitter (serotonin) has been introduced.13 Another application is to use SWCNTs as mechanical sensors. In this context, SWCNTs have been used to study movements in cells and the extracellular space in brain tissue.63,64 Therefore, the cargo transport approach presented in this work could also be extended to bring SWCNT sensors into specific in vivo locations and explore the local mechanical properties.
3. Conclusion
Over the years, much effort has been put forward to study uptake and release of nanomaterials by cells. Here, we demonstrated a novel approach that makes use of particular and unique functions of neutrophils including phagocytosis, migration and NET formation. As we show, neutrophils take up nIR fluorescent SWCNT sensors and precise chemical activation determines how long the cells migrate and the time point of cargo release. In this process, the internalized nanoscale cargo remains functional at all times and is protected from most extracellular influences. This new type of transport-and-release mechanism might be of great benefit for various in vivo biomedical applications as it combines the biocompatibility and targeting capabilities of cells and a tool to program the time scale of release. For example, one could envision isolation of neutrophils from a patient, loading with functional nanomaterials and reinjection for programmed release. This approach would benefit from using the patient's own cells. Furthermore, circulating neutrophils would most likely accumulate in inflammatory sites because the vessel surfaces present surface cues that lead to enhanced adhesion.65,66 Consequently, inflammatory sites, for example around a tumor, are targeted, which would reduce side effects from drugs. Besides the potential for in vivo drug delivery, this work also emphasizes the utility of SWCNTs as versatile chemical sensors that can be transported inside cells and are highly stable. In conclusion, we present a concept for nanoscale cargo transport and delivery by programming immune cells via NETosis.4. Experimental
4.1 Cell isolation of human neutrophils
Neutrophils were isolated from human venous blood of healthy donors. The study itself was approved by the ethics committee of the university medical center in Göttingen (chairman Prof. Jürgen Brockmöller). A fully informed consent of all donors was obtained after being informed about possible consequences of the study and procedure. Consent could be withdrawn at any time during the study. The study and all experiments were carried out in compliance with all relevant laws and the guidelines of Göttingen University as well as the Göttingen University Medical Center. Isolation of human neutrophils was performed according to a standard protocol.67 In brief, fresh blood of healthy donors was collected with S-Monovettes KE 7.5 mL (Sarstedt) and layered gently on top of a Histopaque 1119 solution (ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). After a first centrifugation step (1100g for 21 minutes), the transparent third, as well as the pink fourth layer, were collected and washed with HBSS (w/o Ca2+/Mg2+, Thermo Fisher Scientific). Cells were then again centrifuged for 10 minutes at 400g and the resulting pellet was resuspended in HBSS before layered on top of a phosphate buffered percoll (GE Healthcare) gradient with concentrations of 85, 80, 75, 70 and 65%. After a third centrifugation (1100g, 22 minutes), neutrophils were extracted by collecting half of the 70% and 80% layer as well as the entire 75% layer and washed once with HBSS. The remaining cell pellet was then resuspended in 1 mL HBSS, counted and lastly diluted to the needed concentration of the experiment. As a culture medium, RPMI 1640 (Lonza) with the addition of 10 mM HEPES and 0.5% fetal calf serum (FCS, Sigma-Aldrich) was used if not stated otherwise. Cellular identity was furthermore confirmed by a standard cytospin assay (Cytospin 2 Zentrifuge, Shannon) and a subsequent Diff-Quick staining (Medion Diagnostics). Cell purity had to exceed a 95% threshold to be used for any experiment.
:1). After a first centrifugation step (1100g for 21 minutes), the transparent third, as well as the pink fourth layer, were collected and washed with HBSS (w/o Ca2+/Mg2+, Thermo Fisher Scientific). Cells were then again centrifuged for 10 minutes at 400g and the resulting pellet was resuspended in HBSS before layered on top of a phosphate buffered percoll (GE Healthcare) gradient with concentrations of 85, 80, 75, 70 and 65%. After a third centrifugation (1100g, 22 minutes), neutrophils were extracted by collecting half of the 70% and 80% layer as well as the entire 75% layer and washed once with HBSS. The remaining cell pellet was then resuspended in 1 mL HBSS, counted and lastly diluted to the needed concentration of the experiment. As a culture medium, RPMI 1640 (Lonza) with the addition of 10 mM HEPES and 0.5% fetal calf serum (FCS, Sigma-Aldrich) was used if not stated otherwise. Cellular identity was furthermore confirmed by a standard cytospin assay (Cytospin 2 Zentrifuge, Shannon) and a subsequent Diff-Quick staining (Medion Diagnostics). Cell purity had to exceed a 95% threshold to be used for any experiment.
4.2 SWCNT modification with ssDNA
Surface modification of SWCNT was performed as described previously.52,54 Briefly, 125 μL ssDNA solution (2 mg mL−1 stock in phosphate buffered saline (PBS)) (Sigma Aldrich) and 125 μL (6,5 chirality)-enriched SWCNTs (Sigma Aldrich, Product no. 773735) (2 mg mL−1 stock in PBS) were placed for tip sonication (15 min/30% Amplitude, Fisher Scientific TM Model 120 Sonic Dismembrator) in an ice bath. The obtained dispersion was centrifuged 2 × 30 min/RT/16100g, followed by removal of the excess ssDNA using a Vivaspin 500 MWCO-filter (100000 Da cut-off). The sequence of the hemin-binding aptamer was (5′-AGT GTG AAA TAT CTA AAC TAA ATG TGG AGG GTG GGA CGG GAA GAA GTT TAT TTT TCA CAC T-3′).57,59
4.3 SWCNT uptake
To increase uptake rates, around 400000 cells were resuspended in 200 μL RPMI 1640 medium in a standard 1.5 mL Eppendorf tube and mixed 1:1 with a SWCNT solution (0.2 nM if not stated otherwise) that was diluted in RPMI 1640 medium. Cells were then incubated at 37 °C, 5% CO2 for 20 minutes, centrifuged once at 600g (5 minutes) and washed extensively with RPMI 1640 medium before seeding on the desired substrate.
The uptake analysis, shown in Fig. 2b, was performed likewise. Neutrophils were incubated, placed in a commonly used μ-slide 8 well chamber slide (ibidi, 75000 cells per well and 200 μL RPMI 1640 medium) and fixated using 4% paraformaldehyde (PFA) (Sigma Aldrich, CAS 30525-89-4) for 1 hour after letting the cells adhere for another 30 minutes at 37 °C, 5% CO2. Fixed samples were then placed in a custom-build nIR microscope composed of an Olympus IX-51 housing (Olympus), regular fluorescence (X-Cite Series 120 Q lamp, EXFO) and white light illumination (TH4-200 lamp, Olympus), a 561 nm laser (Jive 500, Cobolt) and two cameras (Zyla 4.2 sCMOS, Andor and Xeva-1.7-320, Xenics) with a 900 nm long-pass filter (FEL0900, Thorlabs) in front to visualize SWCNT excitation as well as phase contrast and fluorescence in parallel. For imaging, a 20× objective (MPLFLN20X, Olympus) was chosen. For each condition, four images at each side of a well were recorded in phase contrast and nIR-mode (100 mW laser power, 500 ms exposure time) using the Zyla camera and saved in separate 16-bit files for subsequent analysis.
Measuring the intensity of SWCNTs inside the cells was then performed using ImageJ's thresholding system (v3.52i). nIR images were tuned to 8-bit depths, cropped into 20 × 20 μm areas containing only the respective SWCNTs and thresholded via a common MinError thresholding algorithm to calculate a mean intensity. SWCNT spots that could not be correlated with a cell area in the corresponding phase contrast image, as well as sensors that were found in cell agglomerations, were excluded from the analysis. The remaining data were averaged (weighted mean of all experiments) and normalized over the corresponding background value (Ibr = 1) to calculate a normalized mean intensity for each condition.
4.4 Live cell imaging of SWCNT during NETosis
1 mL of RPMI medium containing 200000 (GT)15-(6,5)-SWCNT-loaded cells and a 1 μg mL−1 concentration of Hoechst 33342 (Cat. H1399, Thermo Fisher Scientific) was poured on a glass-bottom Petri dish (Cat. 150680, Thermo Fisher Scientific) and incubated to enable sufficient cell adhesion.
Subsequently, the sample was placed into a preheated incubation system (37 °C) (Cat. 11922, ibidi), on top of the custom-build nIR microscope mentioned in the previous section. Using a 100× oil objective (UPLSAPO 100XO, Olympus) consecutively, phase contrast as well as chromatin (DAPI) and nIR images were taken manually from the chosen sample position every ten minutes for 150 minutes after addition of 100 nM phorbol myristate acetate (PMA) (Sigma-Aldrich) to the cell sample. SWCNTs were excited by a common fluorescence lamp in combination with a built-in 561 nm filter cube (F48-553, AHF Analysentechnik), and excitation powers and exposure times were kept constant to ensure data comparability.
Analysis of chromatin and SWCNT area as well as SWCNT intensity was similar to the uptake study described before.
4.5 Live cell imaging of activated neutrophil migration
To record the migration behavior of activated neutrophils, around 200 μL of RPMI 1640 medium containing 75000 untreated cells and 1 μg mL−1 Hoechst 33342 stain were incubated in 8 well μ-slides (ibidi) for 20 minutes. Subsequently, the μ-slides were incorporated in pre-heated ibidi heating chambers (37 °C, 5% CO2, 90% humidity) on top of Olympus IX-81 microscopes. Using integrated XY-stages, 20× objectives (UCPLFLN 20×, Olympus) and corresponding phase contrast and DAPI channels (CBH white light lamp, U-HGLGPS fluorescence lamp, Olympus and 86-370-OLY DAPI filter-cube), a suitable position within each well was chosen and saved using the implemented steering software (Cell Sense, v. 2.1, Olympus). Subsequently, 200 μL of RPMI medium containing a distinct concentration of PMA (0.2, 2, 20, 200 nM) or lipopolysaccharide from Pseudomonas aeruginosa (LPS) (0.2, 2, 20, 200 μg mL−1) (Sigma-Aldrich) were added to a well in a blinded fashion and phase contrast as well as DAPI channels were recorded every two minutes for 160 minutes to image the cell movement.
4.6 Migration analysis of preactivated neutrophils
Cell tracking was performed by ImageJ's TrackMate plugin. Briefly, all chromatin images gained by the image process were combined to form a z-stack and implemented into the plugin. Segmentation was then performed using the Laplacian of Gaussian (LoG) detector module with an estimated blob diameter of 20 pixels (6.5 μm) and a 5 pixel (1.6 μm) threshold. As a result, all cell trajectories within a stack could be traced back by using a subsequent simple LAP tracker model with a maximal linking and gap-closing distance of 50 pixels (16 μm) and a maximal frame gap of two frames. All trajectories were then further analyzed via a custom-built MATLAB code (v. Matlab 2014a) which was able to calculate the traveled distance d for every cell and frame i according to the formula | (1) |
Subsequently, for each condition, 30 cells with the highest average distance values of the data set were further analyzed to detect the migration behavior of the most motile neutrophils within the environment. The stopping time of the migratory phase was calculated by plotting the distance-plots, likewise in a blinded fashion, together with the respective cell trajectory and manually searching for a time point of stagnation within the data set for each cell. Cells that did not show a clear change of moving patterns were excluded from the data set. The cell velocity of the migratory phase was then calculated by averaging the cell speed
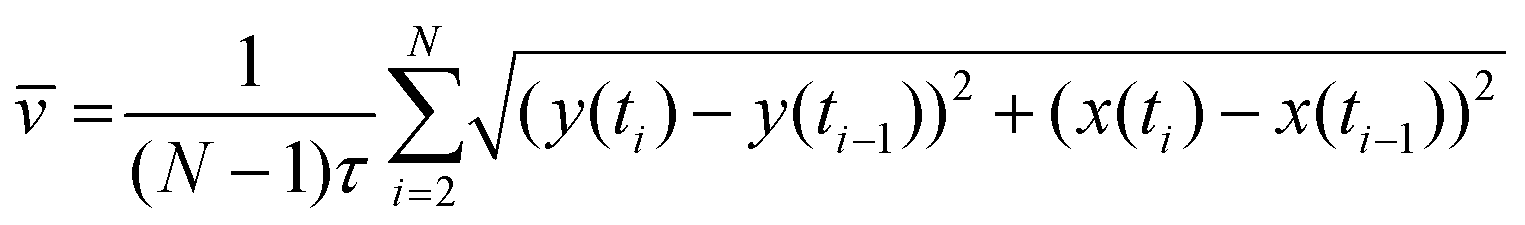 | (2) |
4.7 Decondensation rate analysis
Counting decondensed and intact/globular shaped nuclei was performed according to existing protocols.35,68 Briefly, chromatin images of the recorded positions were taken after 180 minutes and analyzed via ImageJ's Cell Counter plugin. Nuclei that appeared in their known, compressed shape were counted and defined as intact/condensed whereas nuclei that showed increased, roundish chromatin distributions defined a basis for the decondensed/NETotic state. Cells were counted and the number of decondensed cells was divided by the total number of cells to generate a relative decondensation value.4.8 Migration in a gradient
Under-agarose assays were performed to measure cell migration of SWCNT-loaded neutrophils in a gradient. Gels were manufactured according to the protocol of B. Heit et al.49 A HBSS/RPMI 1640 solution was prepared by mixing 5 mL HBSS (w/o Ca2+, Mg2+, Thermo Fisher Scientific) and 10 mL RPMI containing 0.75% FCS (Merck) in a common 50 mL Eppendorf tube and heated up to 68 °C using a common water bath. Meanwhile, 0.24 g ultra-pure agarose (Roth) was added to a vial containing 5 mL of milliQ and the solution was vortexed extensively in order to suspend the agarose homogeneously. The latter was subsequently heated up until boiling by the use of a common Bunsen burner and quickly vortexed three times. The HBSS/RPMI solution was added to the agarose and 3 mL of each mixture was evenly distributed on a plastic-bottom Petri dish (cat. 81156, ibidi). Agarose gels were then allowed to solidify at room temperature and samples were stored overnight at 4 °C with the dish lid covered in dust-free, milliQ-saturated cloth to avoid gel draining. Shortly before the cell experiment, two wells with a diameter of 3 mm and a distance of 2.2 mm were punched in each gel using a dermal biopsy punch (cat. KBP-48101, kai medical), and remaining agarose within each of the wells was extensively removed by vacuum aspiration. Lastly, gels were equilibrated with RPMI 1640 medium for one hour (37 °C, 5% CO2), and the supernatant medium was again removed by vacuum aspiration shortly before cell loading.For the latter, around 100000 neutrophils ((GT)15-(6,5)-SWCNTs loaded or without any pre-treatment) were poured in one of the prepared agarose wells using 5 μL of medium plus 1.6 μM Hoechst 33342 solution and were allowed to equilibrate for 20 minutes at 37 °C, 5% CO2. Subsequently, 10 μL of a 0.1 μM IL-8 solution was poured into the second well while 5 μL of the respective NETosis activator concentration (LPS: 200, 20, 2 μg mL−1, PMA: 20, 2, 0.2 nM or medium as control) was added to the cell well at the same time. All samples were stored for three hours in an incubator (37 °C, 5% CO2) and imaged afterwards using the aforementioned IX-81 microscopic setup in combination with a 4× objective (UPLFLN 4×, Olympus). Afterward, cells were fixated by adding 2% PFA in PBS after carefully removing the gels with a scalpel and tweezers, washed twice with PBS, stored at 4 °C overnight and nIR-imaged by the aid of our aforementioned, customized nIR setup the next day.
For image analysis, single images of each sample were stitched using ImageJ's plugin MosaicJ. Fused images were background corrected, and the existing cell distributions outside of the well were divided radially into 30-degree sections (see ESI Fig. S7†) in which the distance of the leading cell edge was manually analyzed using ImageJ's line tool. Resulting distances were averaged over all experiments and plotted as radial bar plots shown in Fig. 4 or ESI Fig. S7/8 or listed in ESI Tables T1/2.†
4.9 nIR imaging of sensors
Cells for SWCNT functionality tests were prepared similarly to those used for live-cell imaging. Likewise, recordings of the sensor response in intact or ruptured neutrophils were performed with the custom-built nIR setup mentioned above in combination with an integrated ibidi heating chamber (37 °C) and the 100× oil objective. SWCNTs for each condition were excited by a 561 nm laser (200 mW) and recorded with a frame time of 2 or 5 seconds per image for 4 (dopamine addition) or 10 minutes (H2O2 addition), respectively, during which, at around the 1 minute mark, 500 μL of the particular analyte (300 nM dopamine, 300 μM H2O2, both in medium) was added. Subsequent exportation of image files, intensity analysis of single SWCNT areas or data handling was performed as described in the live cell imaging section.4.10 Cell patterning
Cell patterns on substrates shown in Fig. 5c were achieved by light-induced fibrinogen printing controlled by a PRIMO micropatterning machine (alvéole). Briefly, 18 × 18 mm glass coverslips (Fisher Scientific) were washed with 75% ethanol twice and plasma-treated for five minutes to clean and improve the hydrophilicity of the sample. Subsequently, PDMS stencils with a circular well (r = 2 mm) in the center were pressed onto the glass and filled with 0.1 mg mL−1 PLL-g-PEG (Sigma) in PBS for one hour to guarantee a homogeneous passivation layer. Next, the as-prepared sample was fixed on the respective PRIMO setup, calibrated according to the manufacturers’ instructions (microscope specs: Olympus IX83 with an UCPlanFL N 20× objective and an IX3-SSU stage) and filled with 5 μL of the photoactive reagent PLPP (1×, alvéole). Local radiation of the PLPP-bound PEG substrate using the patterning software LEONARDO (alvéole) then led to protein degradation within the illuminated spots and enabled deposition of a second substrate protein. Samples were washed three times with PBS, coated with Alexa-488-labeled fibrinogen solution (10 μL of a 50 μg mL−1 solution in PBS, Sigma) for twenty minutes and stored at 4 °C after an additional three washing steps. Patterns were normally used the next day. However, pattern degradation was not visible within one week after printing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the state of Lower Saxony (life@nano) and the German Research Foundation (DFG grant KR 4242/4-1 and ER 723/2-1). We thank Andreas Janshoff and Claudia Steinem for fruitful discussions and support. We are grateful for fruitful discussions with members of the collaborative research center SFB 937 funded by the DFG. We thank the IMPRS PBCS for a PhD student fellowship (D. M.).References
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- T. Nakanishi, S. Fukushima, K. Okamoto, M. Suzuki, Y. Matsumura, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, J. Controlled Release, 2001, 74, 295–302 CrossRef CAS PubMed.
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, J. D. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674–679 CrossRef CAS PubMed.
- Y. Saadeh and D. Vyas, Am. J. Rob. Surg., 2014, 1, 4–11 CrossRef PubMed.
- M. J. O'Connell, S. M. Bachilo, C. B. Huffman, V. C. Moore, M. S. Strano, E. H. Haroz, K. L. Rialon, P. J. Boul, W. H. Noon, C. Kittrell, J. Ma, R. H. Hauge, R. B. Weisman and R. E. Smalley, Science, 2002, 297, 593–596 CrossRef PubMed.
- S. Kruss, A. J. Hilmer, J. Zhang, N. F. Reuel, B. Mu and M. S. Strano, Adv. Drug Delivery Rev., 2013, 65, 1933–1950 CrossRef CAS PubMed.
- E. Polo and S. Kruss, Anal. Bioanal. Chem., 2016, 408, 2727–2741 CrossRef CAS PubMed.
- G. Hong, S. Diao, A. L. Antaris and H. Dai, Chem. Rev., 2015, 115, 10816–10906 CrossRef CAS PubMed.
- F. A. Mann, N. Herrmann, D. Meyer and S. Kruss, Sensors, 2017, 17(7), E1521 CrossRef PubMed.
- J. D. Harvey, P. V. Jena, H. A. Baker, G. H. Zerze, R. M. Williams, T. V. Galassi, D. Roxbury, J. Mittal and D. A. Heller, Nat. Biomed. Eng., 2017, 1, 1–43 CrossRef PubMed.
- M. Dinarvand, E. Neubert, D. Meyer, G. Selvaggio, F. A. Mann, L. Erpenbeck and S. Kruss, Nano Lett., 2019, 19(9), 6604–6611 CrossRef CAS PubMed.
- F. A. Mann, J. Horlebein, N. F. Meyer, D. Meyer, F. Thomas and S. Kruss, Chem. – Eur. J., 2018, 24, 12241–12245 CrossRef CAS PubMed.
- G. Bisker, J. Dong, H. D. Park, N. M. Iverson, J. Ahn, J. T. Nelson, M. P. Landry, S. Kruss and M. S. Strano, Nat. Commun., 2016, 7, 1–14 Search PubMed.
- V. Zubkovs, N. Schuergers, B. Lambert, E. Ahunbay and A. A. Boghossian, Small, 2017, 13, 1701654 CrossRef PubMed.
- F. A. Mann, Z. Lv, J. Grosshans, F. Opazo and S. Kruss, Angew. Chem., Int. Ed., 2019, 58(33), 11469–11473 CrossRef CAS PubMed.
- N. M. Bardhan, D. Ghosh and A. M. Belcher, Nat. Commun., 2014, 5, 1–11 Search PubMed.
- H. Jin, D. A. Heller, R. Sharma and M. S. Strano, ACS Nano, 2009, 3, 149–158 CrossRef CAS PubMed.
- M. Gravely, M. M. Safaee and D. Roxbury, Nano Lett., 2019, 19, 6203–6212 CrossRef CAS PubMed.
- V. Agrahari, V. Agrahari and A. K. Mitra, Expert Opin. Drug Delivery, 2017, 14, 285–289 CrossRef PubMed.
- A. Khazi-Syed, M. T. Hasan, E. Campbell, R. Gonzalez-Rodriguez and A. V. Naumov, Nanomaterials, 2019, 9, 1685 CrossRef CAS PubMed.
- M. O. Din, T. Danino, A. Prindle, M. Skalak, J. Selimkhanov, K. Allen, E. Julio, E. Atolia, L. S. Tsimring, S. N. Bhatia and J. Hasty, Nature, 2016, 536, 81–85 CrossRef CAS PubMed.
- C. H. Villa, A. C. Anselmo, S. Mitragotri and V. Muzykantov, Adv. Drug Delivery Rev., 2016, 106, 88–103 CrossRef CAS PubMed.
- J. Shi, L. Kundrat, N. Pishesha, A. Bilate, C. Theile, T. Maruyama, S. K. Dougan, H. L. Ploegh and H. F. Lodish, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10131–10136 CrossRef CAS PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef CAS PubMed.
- R. David, Nat. Rev. Mol. Cell Biol., 2013, 14, 547–547 Search PubMed.
- F. Wang, Cold Spring Harbor Perspect. Biol., 2009, 1, a002980 Search PubMed.
- J. Xue, Z. Zhao, L. Zhang, L. Xue, S. Shen, Y. Wen, Z. Wei, L. Wang, L. Kong, H. Sun, Q. Ping, R. Mo and C. Zhang, Nat. Nanotechnol., 2017, 12, 692–700 CrossRef CAS PubMed.
- D. Chu, X. Dong, X. Shi, C. Zhang and Z. Wang, Adv. Mater., 2018, 30, 1706245 CrossRef PubMed.
- J. Shao, M. Xuan, H. Zhang, X. Lin, Z. Wu and Q. He, Angew. Chem., Int. Ed., 2017, 56, 12935–12939 CrossRef CAS PubMed.
- D. Chu, J. Gao and Z. Wang, ACS Nano, 2015, 9, 11800–11811 CrossRef CAS PubMed.
- V. Brinkmann, V. Reichard, C. Goosmann, B. Fauler, Y. Uhlemann, D. Weiss, Y. Weinrauch and A. Zychlinsky, Science, 2004, 303, 1532–1535 CrossRef CAS PubMed.
- T. A. Fuchs, U. Abed, C. Goosmann, R. Hurwitz, I. Schulze, V. Wahn, Y. Weinrauch, V. Brinkmann and A. Zychlinsky, J. Cell Biol., 2007, 176, 231–241 CrossRef CAS PubMed.
- E. Neubert, D. Meyer, F. Rocca, G. Günay, A. Kwaczala-Tessmann, J. Grandke, S. Senger-Sander, C. Geisler, A. Egner, M. P. Schön, L. Erpenbeck and S. Kruss, Nat. Commun., 2018, 9, 3767 CrossRef PubMed.
- S. R. Clark, A. C. Ma, S. A. Tavener, B. McDonald, Z. Goodarzi, M. M. Kelly, K. D. Patel, S. Chakrabarti, E. McAvoy, G. D. Sinclair, E. M. Keys, E. Allen-Vercoe, R. DeVinney, C. J. Doig, F. H. Y. Green and P. Kubes, Nat. Med., 2007, 13, 463–469 CrossRef CAS PubMed.
- E. F. Kenny, A. Herzig, R. Krüger, A. Muth, S. Mondal, P. R. Thompson, V. Brinkmann, H. von Bernuth and A. Zychlinsky, eLife DOI:10.7554/eLife.24437.
- V. Brinkmann, J. Innate Immun., 2018, 10, 414–421 CrossRef CAS PubMed.
- C. M. de Bont, W. J. H. Koopman, W. C. Boelens and G. J. M. Pruijn, Biochim. Biophys. Acta, Mol. Cell Res., 2018, 1865, 1621–1629 CrossRef CAS PubMed.
- H. R. Thiam, S. L. Wong, R. Qiu, M. Kittisopikul, A. Vahabikashi, A. E. Goldman, R. D. Goldman, D. D. Wagner and C. M. Waterman, bioRxiv, 2019, 663427.
- C. Rosales, Front. Physiol., 2018, 9, 1–17 Search PubMed.
- T. Mitchell, A. Lo, M. R. Logan, P. Lacy and G. Eitzen, Am. J. Physiol.: Cell Physiol., 2008, 295, C1354–C1365 CrossRef CAS PubMed.
- N. W. S. Kam, T. C. Jessop, P. A. Wender and H. Dai, J. Am. Chem. Soc., 2004, 126(22), 6850–6851 CrossRef CAS PubMed.
- H. R. Manley, M. C. Keightley and G. J. Lieschke, Front. Immunol., 2018, 9, 2867 CrossRef CAS PubMed.
- V. Zubkovs, A. Antonucci, N. Schuergers, B. Lambert, A. Latini, R. Ceccarelli, A. Santinelli, A. Rogov, D. Ciepielewski and A. A. Boghossian, Sci. Rep., 2018, 8, 13770 CrossRef PubMed.
- N. Mohammadshafie, Tuning the Photoluminescence of DNA-Wrapped Carbon Nanotubes by pH, 2019, DOI: 10.26434/CHEMRXIV.8362103.V1.
- K. A. Gogick, S. Petoud, W. A. Saidi, B. A. Barth, Y. Zhao, G. P. Kotchey, C. F. Chiu and A. Star, J. Am. Chem. Soc., 2013, 135, 13356–13364 CrossRef PubMed.
- C. Farrera, K. Bhattacharya, B. Lazzaretto, F. T. Andón, K. Hultenby, G. P. Kotchey, A. Star and B. Fadeel, Nanoscale, 2014, 6, 6974 RSC.
- B. Heit and P. Kubes, Sci. Signaling, 2003, 2003(170), pl5 CrossRef PubMed.
- L. Erpenbeck, A. L. Gruhn, G. Kudryasheva, G. Günay, D. Meyer, J. Busse, E. Neubert, M. P. Schön, F. Rehfeldt and S. Kruss, Front. Immunol., 2019, 10, 508366 CrossRef PubMed.
- J. P. Giraldo, H. Wu, G. M. Newkirk and S. Kruss, Nat. Nanotechnol., 2019, 14, 541–553 CrossRef CAS PubMed.
- S. Kruss, M. P. Landry, E. Vander Ende, B. M. A. Lima, N. F. Reuel, J. Zhang, J. Nelson, B. Mu, A. Hilmer and M. Strano, J. Am. Chem. Soc., 2014, 136, 713–724 CrossRef CAS PubMed.
- E. Polo and S. Kruss, J. Phys. Chem. C, 2016, 120(5), 3061–3070 CrossRef CAS.
- R. Nißler, F. A. Mann, P. Chaturvedi, J. Horlebein, D. Meyer, L. Vuković and S. Kruss, J. Phys. Chem. C, 2019, 123, 4837–4847 CrossRef.
- R. Nißler, F. A. Mann, H. Preiß, G. Selvaggio, N. Herrmann and S. Kruss, Nanoscale, 2019, 11, 11159–11166 RSC.
- S. Kruss, D. P. Salem, L. Vuković, B. Lima, E. Vander Ende, E. S. Boyden and M. S. Strano, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1789–1794 CrossRef CAS PubMed.
- J. Pan, H. Zhang, T.-G. G. Cha, H. Chen, J. H. Choi, T.-G. G. Cha, H. Zhang, J. Pan and H. Chen, Anal. Chem., 2013, 85, 8391–8396 CrossRef CAS PubMed.
- H. Atsumi and A. M. Belcher, ACS Nano, 2018, 12, 7986–7995 CrossRef CAS PubMed.
- H. Wu, R. Nißler, V. Morris, N. Herrmann, P. Hu, S.-J. Jeon, S. Kruss and J. P. Giraldo, Nano Lett., 2020 DOI:10.1021/acs.nanolett.9b05159.
- J.-H. Kim, C. R. Patra, J. R. Arkalgud, A. A. Boghossian, J. Zhang, J.-H. Han, N. F. Reuel, J.-H. Ahn, D. Mukhopadhyay and M. S. Strano, ACS Nano, 2011, 5, 7848–7857 CrossRef CAS PubMed.
- Y. Ohno and J. I. Gallin, J. Biol. Chem., 1985, 260, 8438–8446 CAS.
- N. F. Reuel, B. Grassbaugh, S. Kruss, J. Z. Mundy, C. Opel, A. O. Ogunniyi, K. Egodage, R. Wahl, B. Helk, J. Zhang, Z. I. Kalcioglu, K. Tvrdy, D. O. Bellisario, B. Mu, S. S. Blake, K. J. Van Vliet, J. C. Love, K. D. Wittrup and M. S. Strano, ACS Nano, 2013, 7, 7472–7482 CrossRef CAS PubMed.
- N. Fakhri, A. D. Wessel, C. Willms, M. Pasquali, D. R. Klopfenstein, F. C. MacKintosh and C. F. Schmidt, Science, 2014, 344, 1031–1035 CrossRef CAS PubMed.
- A. G. Godin, J. A. Varela, Z. Gao, N. Danné, J. P. Dupuis, B. Lounis, L. Groc and L. Cognet, Nat. Nanotechnol., 2017, 12, 238–243 CrossRef CAS PubMed.
- S. Kruss, L. Erpenbeck, K. Amschler, T. A. Mundinger, H. Boehm, H.-J. Helms, T. Friede, R. K. Andrews, M. P. Schön and J. P. Spatz, ACS Nano, 2013, 7, 9984–9996 CrossRef CAS PubMed.
- S. Kruss, L. Erpenbeck, M. P. Schön and J. P. Spatz, Lab Chip, 2012, 12, 3285 RSC.
- A. Mantovani, M. A. Cassatella, C. Costantini and S. Jaillon, Nat. Rev. Immunol., 2011, 11, 519–531 CrossRef CAS PubMed.
- S. L. Wong, M. Demers, K. Martinod, M. Gallant, Y. Wang, A. B. Goldfine, C. R. Kahn and D. D. Wagner, Nat. Med., 2015, 21(7), 815–819 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information given about SWCNT uptake, migration and NETosis behavior of activated cells as well as cargo functionality and properties. See DOI: 10.1039/d0nr00864h |
| This journal is © The Royal Society of Chemistry 2020 |