
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoclusters as electrocatalysts: size, ligands, heteroatom doping, and charge dependences†
Bharat
Kumar‡§
a,
Tokuhisa
Kawawaki§
 ab,
Nobuyuki
Shimizu
a,
Yukari
Imai
a,
Daiki
Suzuki
a,
Sakiat
Hossain
a,
Lakshmi V.
Nair
a and
Yuichi
Negishi
*ab
ab,
Nobuyuki
Shimizu
a,
Yukari
Imai
a,
Daiki
Suzuki
a,
Sakiat
Hossain
a,
Lakshmi V.
Nair
a and
Yuichi
Negishi
*ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.kagu.tus.ac.jp
bPhotocatalysis International Research Center, Tokyo University of Science, 2641 Yamazaki, Noda, Chiba 278-8510, Japan
First published on 4th March 2020
Abstract
To establish an ultimate energy conversion system consisting of a water-splitting photocatalyst and a fuel cell, it is necessary to further increase the efficiencies of the hydrogen evolution reaction (HER), the oxygen evolution reaction (OER), and the oxygen reduction reaction (ORR). Recently, it was demonstrated that thiolate (SR)-protected gold clusters, Aun(SR)m, and their related alloy clusters can serve as model catalysts for these three reactions. However, as the previous data have been obtained under different experimental conditions, it is difficult to use them to gain a deep understanding of the means to attain higher activity in these reactions. Herein, we measured the HER, OER, and ORR activities of Aun(SR)m and alloy clusters containing different numbers of constituent atoms, ligand functional groups, and heteroatom species under identical experimental conditions. We obtained a comprehensive set of results that illustrates the effect of each parameter on the activities of the three reactions. Comparison of the series of results revealed that decreasing the number of constituent atoms in the cluster, decreasing the thickness of the ligand layer, and substituting Au with Pd improve the activities in all reactions. Taking the stability of the cluster into consideration, [Au24Pd(PET)18]0 (PET = 2-phenylethanethiolate) can be considered as a metal cluster with high potential as an HER, OER, and ORR catalyst. These findings are expected to provide clear design guidelines for the development of highly active HER, OER, and ORR catalysts using Aun(SR)m and related alloy clusters, which would allow realization of an ultimate energy conversion system.
1. Introduction
The development of an ultimate energy conversion system that is able to generate hydrogen (H2) from water (H2O) using a photocatalyst and then use the H2 in a fuel cell to generate electricity is of great interest.1 Using such a system, it would be possible to obtain energy only from light and water and circulate the energy medium (H2) (Scheme S1†). However, to realize such a system, it is essential to further improve the reaction efficiencies of the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), which are half reactions of the water-splitting reaction, and the hydrogen oxidation reaction (HOR) and oxygen reduction reaction (ORR), which are the reverse reactions of the HER and OER, respectively. To improve the reactivity per unit volume, it is important to increase the number of active sites and the rate of these reactions. Catalyst miniaturization is an effective method to achieve the former. The latter is strongly affected by the adsorption and desorption energies of the reactive molecules on the catalyst surface. Therefore, in recent years, there has been intensive research on controlling these energies by using alloy catalysts.1Thiolate (SR)-protected gold clusters (Aun(SR)m)2–19 are size-controllable at the atomic level and have high stability in both solution and solid states. Au in these clusters can be partially substituted with silver (Ag), copper (Cu), platinum (Pt), palladium (Pd), cadmium (Cd), and mercury (Hg). Aun(SR)m and its alloy clusters exhibit catalytic activities in numerous reactions such as carbon monoxide oxidation,20 alcohol oxidation,21 and epoxidation.22,23 Because the geometrical structure of Aun(SR)m and its alloy clusters can be determined by single-crystal structural analysis,24 a deep understanding of the correlation between the geometrical structure and the catalytic activity is expected to be obtained for these clusters. For these reasons, Aun(SR)m and its alloy clusters have drawn considerable attention as model catalysts in recent years, and their catalytic activity has been intensively studied by many research groups.23
Recently, it has been demonstrated that Au and its related alloy clusters also exhibit catalytic activities in the HER,25–32 OER,33,34 and ORR.35–44 Consequently, these clusters are also expected to be model catalysts in the above-mentioned energy conversion system. If we can develop a deep understanding of the effective activation of Aun(SR)m and its related alloy clusters through catalyst studies, it may be possible to develop HER, OER, and ORR catalysts using Aun(SR)m and alloy clusters with performance which surpasses that of conventional catalysts. However, because the current data on catalytic activity were obtained under different experimental conditions (e.g., electrode preparation methods and measurement conditions), it is difficult to quantitatively compare the effects of each parameter of Aun(SR)m and its alloy clusters on catalytic activity, which in turn makes it difficult to obtain clear design guidelines to achieve effective activation.
In this study, we measured the HER, OER, and ORR activities of Aun(SR)m and its alloy clusters containing different numbers of constituent atoms, ligand functional groups, and heteroatom species under the same experimental conditions to address the above-mentioned problem (Scheme 1). Accordingly, we systematically clarified the effect of each parameter on the HER, OER, and ORR activities of the cluster catalysts.
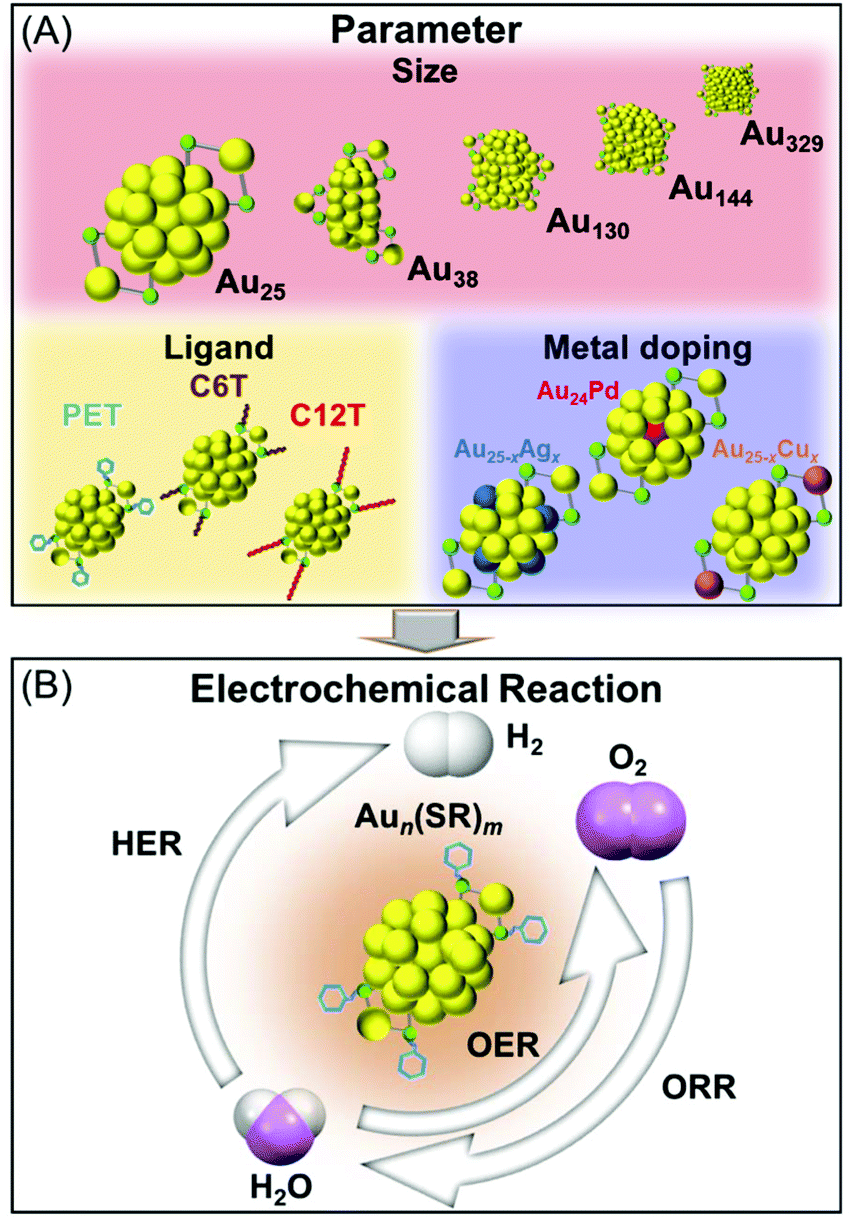 | ||
| Scheme 1 Schematics of the (A) parameters and (B) electrochemical reactions investigated in this study. | ||
2. Results and discussion
2-Phenylethanethiolate (PET) is one of the most frequently used ligands in the synthesis of Aun(SR)m clusters.45–47 Therefore, Aun(PET)m clusters were the primary focus of this study. [Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0 (Fig. S1†) were used to elucidate the dependence of catalytic activity on the number of constituent atoms. To evaluate the dependence of catalytic activity on ligand functional groups, [Au25(PET)18]0, hexanethiolate (C6T)-protected [Au25(C6T)18]0, and dodecanethiolate (C12T)-protected [Au25(C12T)18]0 were used (Fig. S1(a) and S2†). In addition, [Au25(PET)18]0, [Au20.5Ag4.5(PET)18]0, [Au23.7Cu1.3(PET)18]0, and [Au24Pd(PET)18]0 were used to investigate how the heteroatom species affect the catalytic activity (Fig. S3†). [Au25(PET)18]0 and [Au25(PET)18]− were used to evaluate the dependence of catalytic activity on the charge state of the cluster. Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry and optical absorption spectroscopy confirmed that all eleven types of clusters (Table 1) were synthesized with high purity (Fig. S4–S11†). After loading an identical amount (0.5 mg) of each of these clusters on electrodes, the HER, OER, and ORR characteristics of each cluster were evaluated using electrochemical measurements (see section 4 and Fig. S12† for details).| Cluster |
M
w![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
N (×1017)b | N surf. (×1017)c |
|---|---|---|---|
| a Molecular weight. b Number of metal atoms in 0.5 mg of the cluster. c Number of surface atoms (Fig. S1†) in 0.5 mg of the cluster. d This molecular weight includes that of the counter cation (tetraoctylammonium ion). | |||
| [Au25(PET)18]0 | 7394.2 | 10.2 | 4.89 |
| [Au38(PET)24]0 | 10778.1 |
10.6 | 5.87 |
| [Au130(PET)50]0 | 32466.7 |
12.1 | 4.64 |
| [Au144(PET)60]0 | 36596.5 |
11.8 | 4.94 |
| [Au329(PET)84]0 | 76328.5 |
13.0 | 5.68 |
| [Au25(C6T)18]0 | 7034.3 | 10.7 | 5.14 |
| [Au25(C12T)18]0 | 8549.2 | 8.8 | 4.23 |
| [Au20.5Ag4.5(PET)18]0 | 6993.2 | 10.8 | 5.17 |
| [Au23.7Cu1.3(PET)18]0 | 7220.7 | 10.4 | 5.00 |
| [Au24Pd(PET)18]0 | 7303.6 | 10.3 | 4.95 |
| [Au25(PET)18]− | 7861.2d | 9.6 | 4.60 |
2.1. Hydrogen evolution reaction for water splitting
The HER is a half reaction of water splitting. In this reaction, protons (H+) first form bonds with metal atoms on the cluster surface via the Volmer–Heyrovsky or Volmer–Tafel mechanism. H2 is then formed on the metal surface and subsequently desorbed. Under acidic conditions, the HER is expressed as follows:1| Volmer reaction: Au + H+ + e− → Au–H | (1) |
| Heyrovsky reaction: Au–H + H+ + e− → Au + H2 | (2) |
| Tafel reaction: 2Au–H → 2Au + H2 | (3) |
Fig. 1(a) shows linear sweep HER voltammograms for [Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0. The horizontal axis of each voltammogram is the voltage applied to the cluster-supported electrode versus the reversible hydrogen electrode (RHE). At the same applied voltage, the current density increased continuously as the number of constituent atoms in the cluster decreased (Table 2). Fig. 1(b) compares the mass activities of the clusters at an applied voltage of −0.7 V vs. RHE. With decreasing number of constituent atoms, the current density increased, and [Au25(PET)18]0 (−17.8 mA cm−2) exhibited a mass activity which was 223% that of [Au329(PET)84]0 (−8.0 mA cm−2) (Table 2). These results indicate that decreasing the number of constituent atoms in the cluster catalyst induces an increase in HER activity. In the case of bulk Au, it has been shown that Au binds weakly to H+ and the HER does not progress easily.1 In contrast, small Aun(SR)m clusters have an electronic/geometrical structure different from that of bulk Au.48 Because of this different electronic/geometrical structure, Aun(SR)m clusters form stronger bonds with H+ than Au and thus exhibit higher HER activity than Au.27 To understand the reason for the improved activity with decreasing number of constituent atoms, we calculated the number of Au atoms in 0.5 mg of each cluster and the number of surface atoms actually bonded to the reactants (Fig. S13† and Table 1). It was found that these values did not change much regardless of the number of constituent atoms in the cluster. This indicates that the dependence of catalytic activity on the number of constituent atoms observed in Fig. 1(b) cannot be explained by the change in the number of surface atoms with cluster size. In general, as the number of constituent atoms in the Aun(SR)m clusters decreases, Au–H bonds are formed more easily on the surface of the metal core.27 It is considered that because a similar change in the electronic structure occurs as the number of constituent atoms decreases, the catalytic activity depends on the number of constituent atoms, as is observed in Fig. 1(b).
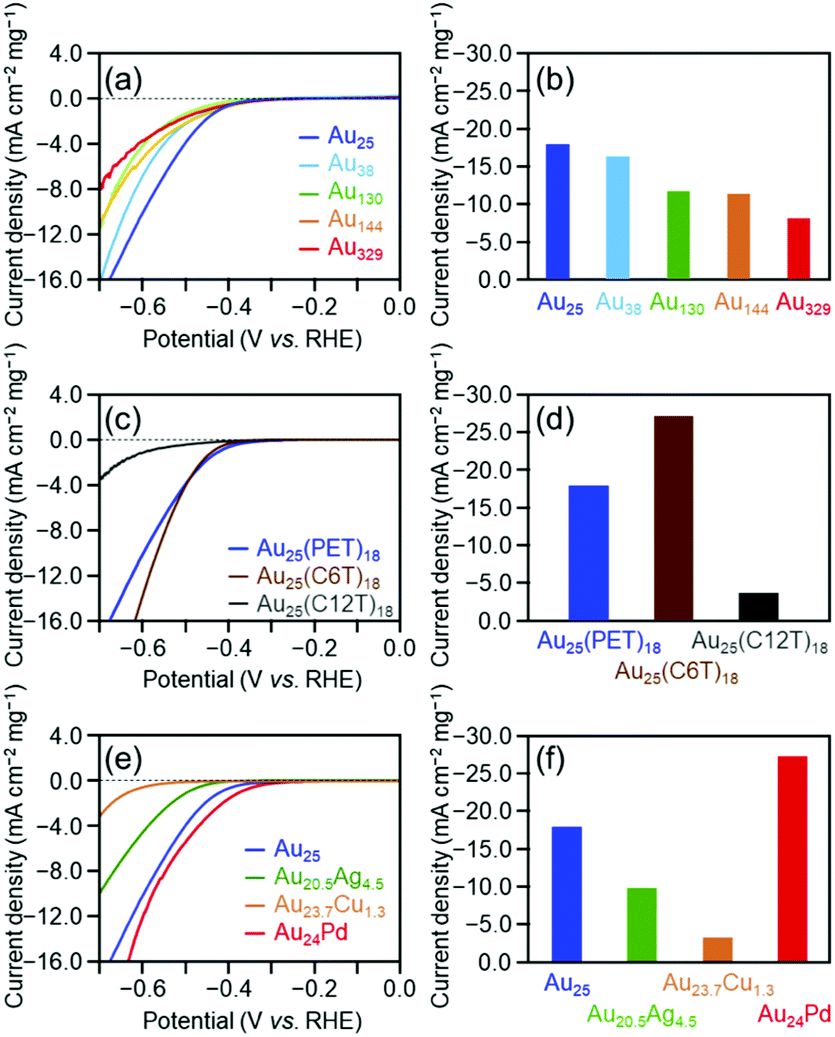 | ||
| Fig. 1 HER voltammograms of Aun(SR)m clusters with different (a) sizes ([Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0), (c) ligands ([Au25(PET)18]0, [Au25(C6T)18]0, and [Au25(C12T)18]0), and (e) doped heteroatoms ([Au25(PET)18]0, [Au20.5Ag4.5(PET)18]0, [Au23.7Cu1.3(PET)18]0, and [Au24Pd(PET)18]0) in 0.2 M HClO4 saturated with N2 (pH = 0.7). Mass HER activity of gold clusters with different (b) sizes, (d) ligands, and (f) doped heteroatoms at −0.7 V vs. RHE evaluated from (a), (c), and (e), respectively. | ||
| Cluster | Onset potentiala (V) | Current densityb (mA cm−2 mg−1) | Normalized current densityc (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HER | OER | ORR | HER | OER | ORR | HER | OER | ORR | |
| a These values were estimated from Fig. 1(a), (c), and (e), 3(a), (c), and (e) and 5(a), (c), and (e). b The values for the HER, OER, and ORR were estimated at −0.7, 2.2, and −0.2 V vs. RHE respectively. c These values are normalized to that of [Au25(PET)18]0 (Fig. 7). | |||||||||
| [Au25(PET)18]0 | −0.33 | 1.90 | 0.24 | −17.8 | 8.55 | −0.85 | 100 | 100 | 100 |
| [Au38(PET)24]0 | −0.33 | 1.90 | 0.24 | −16.3 | 5.63 | −0.49 | 91.3 | 65.9 | 57.0 |
| [Au130(PET)50]0 | −0.37 | 2.00 | 0.19 | −11.6 | 4.52 | −0.44 | 65.0 | 52.9 | 51.2 |
| [Au144(PET)60]0 | −0.31 | 2.03 | 0.15 | −11.3 | 3.56 | −0.30 | 63.2 | 41.6 | 35.3 |
| [Au329(PET)84]0 | −0.31 | 2.02 | 0.20 | −8.0 | 3.19 | −0.14 | 44.8 | 37.3 | 16.8 |
| [Au25(C6T)18]0 | −0.37 | 1.86 | 0.18 | −27.0 | 8.95 | −0.91 | 151.4 | 104.7 | 106.8 |
| [Au25(C12T)18]0 | −0.42 | 2.16 | 0.17 | −3.6 | 1.23 | −0.54 | 20.4 | 14.4 | 63.7 |
| [Au20.5Ag4.5(PET)18]0 | −0.42 | 1.94 | 0.20 | −9.9 | 7.39 | −0.78 | 55.7 | 86.5 | 91.4 |
| [Au23.7Cu1.3(PET)18]0 | −0.53 | 1.90 | 0.15 | −3.2 | 8.65 | −0.75 | 18.0 | 101.2 | 88.4 |
| [Au24Pd(PET)18]0 | −0.29 | 1.92 | 0.24 | −27.1 | 9.47 | −0.95 | 151.7 | 110.8 | 111.8 |
| [Au25(PET)18]− | −0.19 | 1.79 | 0.23 | −26.0 | 10.31 | −1.13 | 145.7 | 120.6 | 133.4 |
As described above, the HER activity of the Aun(SR)m clusters increases as the number of constituent atoms decreases. Furthermore, mass spectrometry revealed that [Au25(PET)18]0 maintained its chemical composition after the electrochemical experiment with twenty scans (Fig. 2). Therefore, we used [Au25(PET)18]0 (Fig. S1(a)†), which showed the highest activity in the above-mentioned experiments, as the reference to examine the catalytic activity in subsequent experiments. Fig. 1(c) shows the linear sweep voltammograms of [Au25(PET)18]0, [Au25(C6T)18]0, and [Au25(C12T)18]0. [Au25(C12T)18]0 showed lower activity than [Au25(PET)18]0. For example, at an applied voltage of −0.7 V vs. RHE, [Au25(PET)18]0 showed a mass activity of −17.8 mA cm−2, whereas that of [Au25(C12T)18]0 was only −3.6 mA cm−2 (Fig. 1(d)). In all our experiments, the weight of the metal cluster was set to 0.5 mg. Because PET (Mw = 137.2) has a lower molecular weight than C12T (Mw = 201.4), the number of metal atoms in 0.5 mg of [Au25(PET)18]0 (20.4 × 1017) was 1.16 times greater than that of [Au25(C12T)18]0 (17.6 × 1017) (Table 1). However, the activity of the former was 4.89 times higher than that of the latter (Fig. 1(d)). Therefore, the difference in activity cannot be explained solely by the difference in the number of metal atoms in these clusters. C12T has a longer hydrocarbon moiety than PET (Fig. S2†). Thus, it is inferred that when [Au25(C12T)18]0 was used as the catalyst, a thicker insulating layer than that formed with [Au25(PET)18]0 was formed between the electrode and metal core. The thicker insulating layer in turn made the electron transfer between the electrode and metal core more difficult, and consequently, [Au25(C12T)18]0 showed lower HER activity than [Au25(PET)18]0. On the other hand, [Au25(C6T)18]0 showed higher activity than [Au25(PET)18]0. For example, at an applied voltage of −0.7 V vs. RHE, [Au25(PET)18]0 showed a mass activity of −17.8 mA cm−2, whereas that of [Au25(C6T)18]0 was only −27.0 mA cm−2 (Fig. 1(d)). This difference of activity also cannot be explained solely by the difference in the number of metal atoms in these clusters (Table 1). C6T has a little longer hydrocarbon moiety than PET (Fig. S2†). However, since the ligand in [Au25(PET)18]0 includes the phenyl group and thereby π–π interaction should occur between the ligands in [Au25(PET)18]0, the interaction between ligands should be lower in [Au25(C6T)18]0 than in [Au25(PET)18]0. Thus, it is assumed that the ligand layer in [Au25(C6T)18]0 is more liquid-like and thereby thinner on the electrode than that in [Au25(PET)18]0. This seems to be the main reason why [Au25(C6T)18]0 showed higher HER activity than [Au25(PET)18]0.
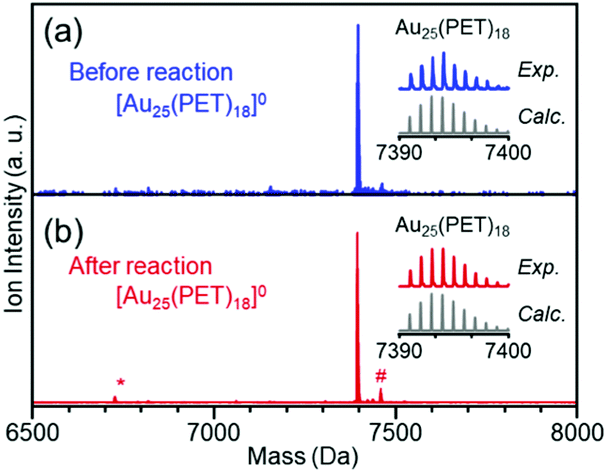 | ||
| Fig. 2 MALDI mass spectra of [Au25(PET)18]0 (a) before and (b) after the HER experiment with twenty scans. The peaks indicated by asterisk (*) and hash (#) symbols are assigned to Au23(PET)16 and Au25(PET)18S2, respectively, which are caused by laser irradiation. | ||
Fig. 1(e) illustrates the linear voltammograms of the HER for [Au25(PET)18]0, [Au20.5Ag4.5(PET)18]0, [Au23.7Cu1.3(PET)18]0, and [Au24Pd(PET)18]0. Based on these voltammograms, the HER activity of the catalysts shows the following order from highest to lowest: [Au24Pd(PET)18]0 > [Au25(PET)18]0 > [Au20.5Ag4.5(PET)18]0 > [Au23.7Cu1.3(PET)18]0 (Fig. 1(f)). Regarding the HER of the Pd-substituted cluster, Lee and co-workers conducted studies on [Au24Pd(C6T)18]0.25 Their studies showed that [Au24Pd(C6T)18]0 exhibits higher HER activity than [Au25(C6T)18]0. According to the density functional theory (DFT) calculations reported by Jiang et al.,27 when the central Au atom of [Au25(C1T)18]0 (C1T = methanethiolate) is substituted with Pd, the electronic structure changes, and Au on the metal surface can form a bond with H+ more easily than in the case of the unsubstituted cluster. For example, they estimated the adsorption energies of the first and second H+ to be −2.15 and −2.16, respectively, for [Au24Pd(C1T)18]0, whereas those to be −2.04 and −1.25, respectively, for [Au25(C1T)18]0. Although the Pd-substituted cluster used in our study contains ligand functional groups (PET) different from those investigated in previous studies (C6T and C1T), it can be considered that [Au24Pd(PET)18]0 showed higher HER activity than [Au25(PET)18]0 because a similar change also occurred between [Au24Pd(PET)18]0 and [Au25(PET)18]0. The effect of Ag or Cu substitution in the Aun(SR)m cluster on HER activity was determined experimentally for the first time.49 Jiang and colleagues performed theoretical calculations and predicted that Ag substitution weakens the bond between the metal surface and H+.27 It is considered that because of this effect, [Au20.5Ag4.5(PET)18]0 showed lower HER activity than [Au25(PET)18]0. Unlike Pd and Ag, Cu is considered to substitute Au not in the metal core, but in the Au2(SR)3 staple covering the metal core (Fig. S3(c)†).50 However, the Cu substitution at such locations also has some effect on the orbital energy of the metal core.51 It is assumed that the change in the electronic structure of the metal core caused by Cu substitution makes it difficult to perform the HER reaction on the surface of the metal core, leading to the observed decrease of the HER activity for [Au23.7Cu1.3(PET)18]0. Overall, the results obtained in this study are in good agreement with the theoretical predictions by Jiang et al.27
From these experiments, it became clear that efficient ways to increase the HER activity include the following: (i) decreasing the number of constituent atoms, (ii) using a shorter ligand, and (iii) inducing strong metal–H bond formation via substitution with a heteroatom. Based on these results, it is assumed that effective methods to improve the HER activity include, but are not limited to, using cluster catalysts with fewer constituent atoms than [Au25(PET)18]0 and making the ligand shorter than PET. However, these methods are not necessarily effective from the viewpoint of the stability of the catalyst. It has been reported that an Aun(SR)m cluster with fewer constituent atoms than [Au25(SR)18]0 shows lower stability than [Au25(SR)18]0 in solution.52 In addition, there are no reports of the precise synthesis of Pd-substituted products for such clusters.53 Thus, it is considered that small Pd-substituted clusters have low stability. In terms of the ligand, the clusters used in this study could be precisely synthesized even when an SR with a shorter hydrocarbon chain is used.54–56 However, such clusters are generally less stable in solution than those containing a PET ligand.57 We also examined the effect of the charge state on the HER activity of the clusters using [Au25(PET)18]0 and [Au25(PET)18]−. The results revealed that although the negative charge temporarily improved the activity (Fig. S14†), the negative ion was oxidized during the potential sweep (Fig. S11(c)†). Taking all these points into consideration, it can be said that [Au24Pd(PET)18]0 is a metal cluster showing high potential for use as an HER catalyst.
2.2. Oxygen evolution reactions for water splitting
The OER is also a half reaction of water splitting. The OER is a multistage four-electron reaction that proceeds through different routes depending on the OER intermediates (O, OH, OOH, etc.). Because the measurements in this study were performed under acidic conditions, the OER probably proceeds according to the following route:1| Au + H2O → Au–OH + H+ + e− | (4) |
| Au–OH + OH− → Au–O + H2O + e− | (5) |
| 2Au–O → 2Au + O2 | (6) |
| Au–O + H2O → Au–OOH + H+ + e− | (7) |
| Au–OOH → Au + O2 + H+ + e− | (8) |
Fig. 3(a) shows the linear sweep voltammograms for the OER of [Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0. Similar to the HER activity, the OER activity increased continuously as the number of constituent atoms decreased (Fig. 3(b)). A previous study reported that the electronic structure of Au clusters becomes discrete as the number of constituent atoms decreases, which facilitates the adsorption of hydroxyl ions (OH−) onto the Au cluster surface.35 It can be considered that the dependence of the OER activity on the number of constituent atoms observed in this study is also strongly influenced by the change in the adsorption ability of OH− caused by the change in the electronic structure with the number of constituent atoms.
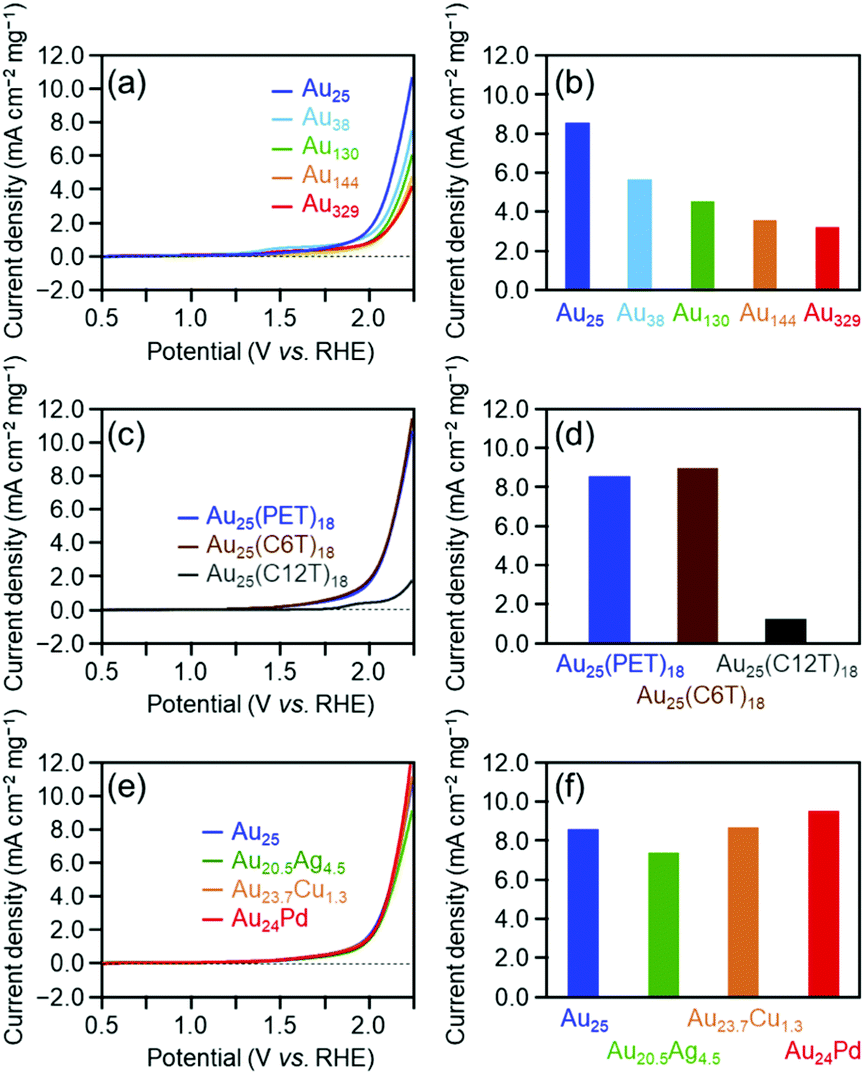 | ||
| Fig. 3 OER voltammograms of Aun(SR)m clusters with different (a) sizes ([Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0), (c) ligands ([Au25(PET)18]0, [Au25(C6T)18]0, and [Au25(C12T)18]0), and (e) doped heteroatoms ([Au25(PET)18]0, [Au20.5Ag4.5(PET)18]0, [Au23.7Cu1.3(PET)18]0, and [Au24Pd(PET)18]0) in 0.2 M HClO4 saturated with N2 (pH = 0.7). Mass OER activity of gold clusters with different (b) sizes, (d) ligands, and (f) doped heteroatoms at +2.2 V vs. RHE evaluated from (a), (c), and (e), respectively. | ||
Fig. 3(c) shows the linear sweep voltammograms for the OER of [Au25(PET)18]0, [Au25(C6T)18]0, and [Au25(C12T)18]0. These voltammograms indicate that [Au25(C12T)18]0 has lower activity than [Au25(PET)18]0, whereas [Au25(C6T)18]0 has a little higher activity than [Au25(PET)18]0 (Fig. 3(d)). As mentioned above, the ligands of the Aun(SR)m cluster act as an insulating layer between the electrode and the metal core.58 Thus, it can be considered that these tendencies are also mainly caused by the difference in the thickness of the ligand layer.
Fig. 3(e) shows the OER voltammograms of [Au25(PET)18]0, [Au20.5Ag4.5(PET)18]0, [Au23.7Cu1.3(PET)18]0, and [Au24Pd(PET)18]0. The order of the OER activity of the clusters from highest to lowest was [Au24Pd(PET)18]0 > [Au23.7Cu1.3(PET)18]0 > [Au25(PET)18]0 > [Au20.5Ag4.5(PET)18]0 (Fig. 3(f)). In general, the activity of an electrochemical reaction of Aun(SR)m and related alloy clusters is strongly influenced by the binding force between the cluster and reactants, which depends on the electronic structure of the cluster.27 It is interpreted that Pd and Cu substitution induces an electronic structure suitable for the OER, whereas Ag substitution generates unfavorable electronic structures for the OER. DFT calculations are expected to reveal the origin of this dependence in detail in the future.27
Overall, the effect of each parameter on catalyst activity in the HER and OER is generally similar; the difference in the effect between two reactions was only the Cu doping effect. Because these two reactions have different mechanisms (eqn (1)–(8)), the factors that induce changes in the catalytic activity are probably different from each other. However, interestingly, the effect of each parameter on the catalyst activity in each reaction is overall similar. Durability tests demonstrated that [Au25(PET)18]0 and [Au24Pd(PET)18]0 can maintain their chemical compositions (Fig. S15†) and thereby their activities even after twenty sweeps (Fig. 4). Based on these results, it can be said that [Au24Pd(PET)18]0 shows considerable promise as an OER catalyst.
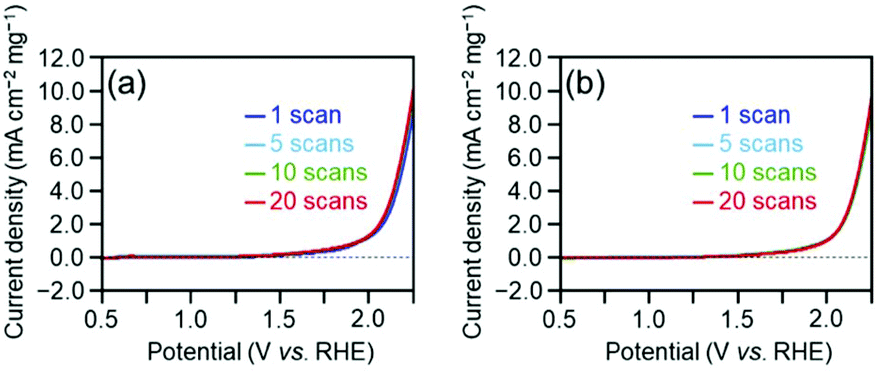 | ||
| Fig. 4 Durability of (a) [Au25(PET)18]0 and (b) [Au24Pd(PET)18]0 in the OER. | ||
2.3. Oxygen reduction reactions for fuel cells
Two types of fuel cells have been developed to date that use alcohol or H2 as the fuel. In H2 fuel cells, the reactions are the HOR and ORR. The HOR is the reverse reaction of the HER and is a one-electron reaction. Therefore, the reaction mechanism is simple and catalysts active in the HER are often also active in the HOR. The ORR is the reverse reaction of the OER and is a four- or two-electron reaction. Because the ORR follows a complicated reaction pathway, a catalyst active in the OER is not necessarily active in the ORR. The ORR pathway in an acidic electrolyte can be described as follows:1| O2 + 4H+ + 4e− → 2H2O | (9) |
| O2 + 2H+ + 2e− → H2O2 | (10) |
| H2O2 + 2H+ + 2e− → 2H2O | (11) |
Fig. 5 shows the dependence of the ORR activity of the clusters on the number of constituent atoms, ligand functional groups, and heteroelement species. Overall, these dependencies are very similar to those obtained for the OER activity (Fig. 3). This indicates that in the case of Aun(SR)m and related alloy clusters, clusters which are highly active for the OER also show high activity for the ORR.
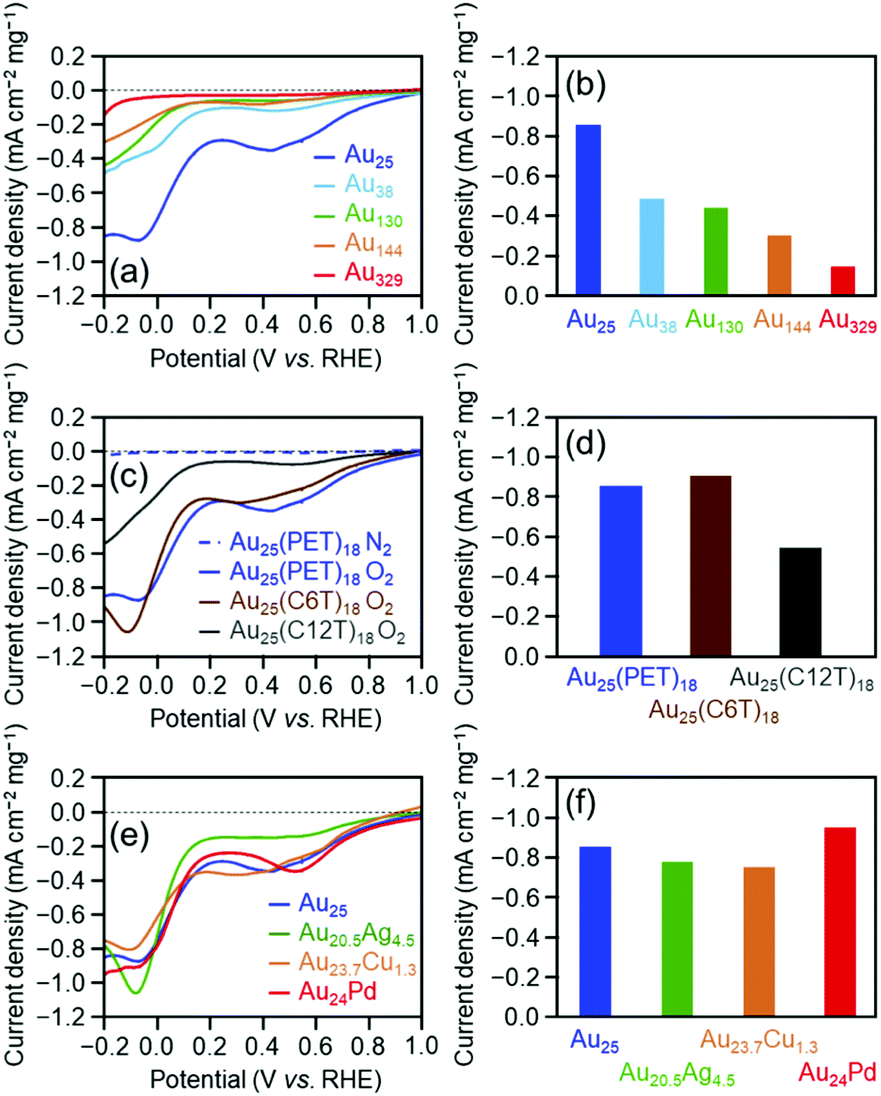 | ||
| Fig. 5 ORR voltammogram of Aun(SR)m clusters with different (a) sizes ([Au25(PET)18]0, [Au38(PET)24]0, [Au130(PET)50]0, [Au144(PET)60]0, and [Au329(PET)84]0), (c) ligands ([Au25(PET)18]0, [Au25(C6T)18]0, and [Au25(C12T)18]0) and (e) doped heteroatoms ([Au25(PET)18]0, [Au20.5Ag4.5(PET)18]0, [Au23.7Cu1.3(PET)18]0, and [Au24Pd(PET)18]0) in 0.2 M HClO4 saturated with O2 or N2 (pH = 0.7). Mass ORR activity of gold clusters with different (b) sizes, (d) ligands, and (f) doped heteroatoms at −0.2 V vs. RHE evaluated from (a), (c), and (e), respectively. | ||
Regarding the dependence of catalytic activity on the number of constituent atoms in the cluster, as the case of the OER, it can be explained that the difference in the adsorption force between Au and oxygen on the metal core surface mainly causes the tendency seen in Fig. 5(a) and (b). Actually, previous studies have shown that with the decreasing core size of Au nanoclusters, the d-bands become narrowed and shift towards the Fermi level.59,60 This finding suggests that smaller Au clusters are energetically more favorable for O2 adsorption. For the dependence of catalytic activity on the ligand functional group, it is assumed that the difference in the distance between the electrode and metal core mainly causes the trend in Fig. 5(c) and (d), similar to the case for the HER or OER. With respect to the effect of substitution with different elements on the catalytic activity, the tendency seen in Fig. 5(e) and (f) seems to occur because Pd substitution gives an electronic structure suitable for the ORR, whereas Ag and Cu substitution generates unfavorable electronic structures, similar to the case of the OER. This Cu doping effect is different from the case of the OER reaction. To gain a deep understanding of this reason, it is anticipated that the geometrical structure of [Au23.7Cu1.3(PET)18]0 would be determined by single crystal X-ray diffraction analysis.
We also evaluated the reaction activity of [Au25(PET)18]0 and [Au24Pd(PET)18]0 using a rotating ring disc electrode (RRDE). The results showed that the four-electron reduction reaction was dominant under acidic conditions in the range of −0.25–0.05 V vs. RHE (Fig. 6(a) and (b)). For [Au25(C12T)18]0, it has been reported that the two-electron reduction occurs with high selectivity under alkaline conditions.38 Our results strongly suggest that Aun(SR)m and related alloy clusters undergo different ORR pathways depending on whether the conditions are acidic or alkaline.
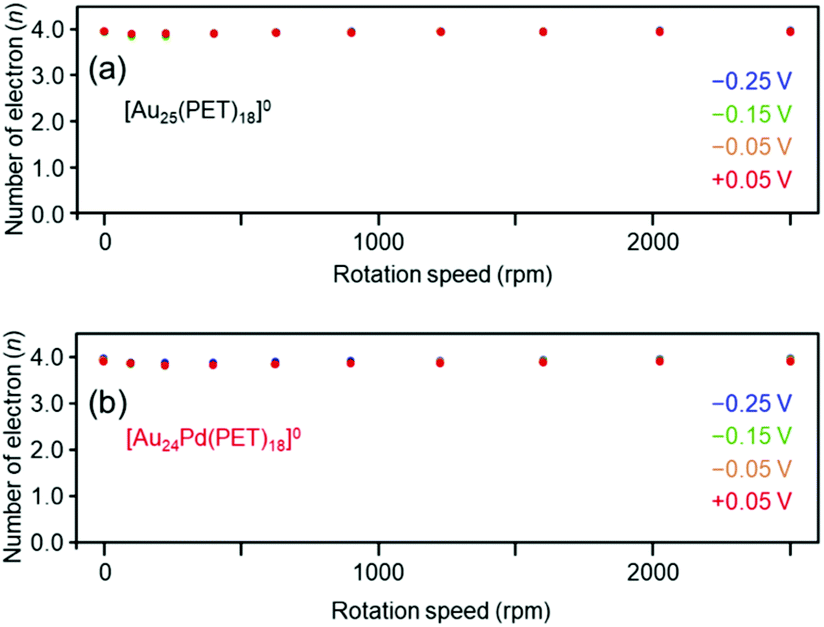 | ||
| Fig. 6 Number of electrons transferred vs. the rotation speed at different potentials (vs. RHE) obtained by RRDE measurements for (a) [Au25(PET)18]0 and (b) [Au24Pd(PET)18]0. | ||
3. Conclusions
The Aun(SR)m and the related clusters are very specific because one material has several different functions. However, in previous studies, each work studied only one reaction for Aun(SR)m clusters. In this study, we systematically examined the effects of different parameters of Aun(SR)m and related alloy clusters on their HER, OER, and ORR activities using the same apparatus (Fig. 7). The results of this study are summarized as follows:(1) Comprehensive results that can serve as a library revealing the effect of each parameter on the activities of these three reactions were obtained.
(2) Comparing the series of data revealed that decreasing the number of constituent atoms in the cluster, decreasing the thickness of the ligand layer, and Pd substitution increase the catalytic activity of the clusters in all three reactions despite the mechanisms of these reactions being different from each other.
(3) Taking both activity and stability into consideration, it was found that [Au24Pd(PET)18]0 is a metal cluster with high potential in all reactions.
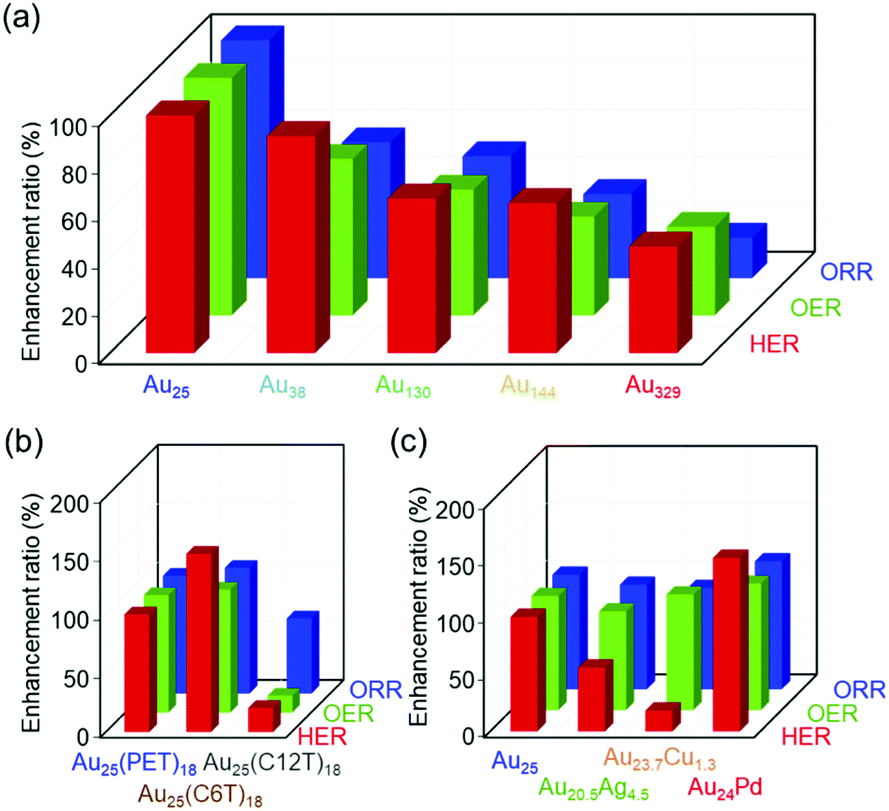 | ||
| Fig. 7 Enhancement ratio of the HER, OER, and ORR activities for gold clusters with different (a) sizes, (b) ligands, and (c) metal substitutions compared with the case for [Au25(PET)18]0. | ||
These findings are expected to lead to the development of highly active HER, OER and ORR catalysts, which in turn will lead to design guidelines to realize ultimate energy conversion systems.
At the same time, more ingenuity is required to construct catalysts with even higher activity using Aun(SR)m and related alloy clusters. Based on the report by Lee et al. on [Au24Pt(SR)18]0 (SR = C6T or C1T),30 substituting the central atom of [Au25(PET)18]0 with Pt is one way to increase its catalytic activity. If multiple Au in [Au25(PET)18]0 could be substituted with Pd, even higher catalytic activity is expected to be achieved by Pd substitution. In addition, we used three types of hydrophobic SR clusters (PET, C6T, and C12T) as ligands in this study. However, it may be necessary to use a hydrophilic SR as the ligand to further improve the activity in each reaction (HER, OER, or ORR) by facilitating the approach of the reactants (H+, H2O, O, OH, OOH, O2etc.) to the cluster in each reaction. Thus, hydrophilic SR clusters29 are expected to be extensively used to produce HER, OER, and ORR catalysts possessing even higher activities. At present, the activities of Aun(SR)m and related alloy clusters are only around 10% of that of the commercial catalysts, e.g. Pt/C. The difference in the sample preparation method seems to be strongly related to this reason. In the case of commercial Pt/C, Pt nanoparticles are loaded on the carbon to increase the number of surface Pt atoms. This electrode preparation method has been already well established. On the other hand, in the study of Aun(SR)m and related clusters, the clusters are typically just loaded on the electrode with Nafion. Thus, for Aun(SR)m and related clusters, there seems to be still a large room for improving the activity. Future studies are expected to attempt not only the improvement of the activity of each cluster but also the improvement of the electrode preparation methods to make the most of each cluster's activity.
4. Experimental
4.1. Chemicals
All chemicals were commercially obtained and used without further purification. Hydrogen tetrachloroaurate tetrahydrate (HAuCl4·4H2O) was purchased from Tanaka Kikinzoku. Palladium(II) sodium chloride trihydrate (Na2PdCl4·3H2O), copper(II) chloride dihydrate (CuCl2·2H2O), 1-dodecanethiol, glutathione, and tetraoctylammonium bromide ([(C8H17)4N]Br) were obtained from FUJIFILM Wako Pure Chemical. 2-Phenylethanethiol and Nafion® perfluorinated resin solution (5 wt% dispersion, contains 45% water) were obtained from Aldrich. Methanol, acetonitrile, toluene, acetone, tetrahydrofuran (THF), ethanol, dichloromethane, perchloric acid (60% aqueous solution), triethylamine and silver nitrate (AgNO3) were obtained from Kanto Chemical. We purchased sodium tetrahydroborate (NaBH4), 1-hexanethiol and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene] malononitrile (DCTB) from Tokyo Chemical Industry. Pt/C (EC-10-PTC) was purchased from Toyo Co. Polishing alumina and diamond solutions were obtained from BAS. Deionized water with a resistivity of >18 MΩ cm was used in all experimental work.4.2. Synthesis
[Au25(PET)18]− was synthesized by a method previously reported in the literature61 with slight modifications. First, HAuCl4·4H2O (0.76 mmol) was dissolved in THF (30 mL) containing [(C8H17)4N]Br (0.76 mmol) at room temperature. After stirring for 15 min, 2-phenylethanethiol (4.7 mmol) was added to the solution and the resulting mixture was stirred for 30 min. Then, a cold aqueous solution of NaBH4 (8.7 mmol, 5.8 mL) was rapidly added to the stirred solution. After stirring overnight at room temperature, THF was removed by evaporation. The remaining precipitate was washed with methanol to remove excess thiol and other by-products. [Au25(PET)18][(C8H17)4N] was extracted from the precipitate using acetonitrile.
[Au25(PET)18][(C8H17)4N] was oxidized by stirring with silica gel using dichloromethane as the eluent, similar to a reported procedure.62 The obtained [Au25(PET)18]0 was further purified and then used for characterization and other measurements.
[Au25(C6T)18]− was synthesized by a reported method with slight modifications.66 First, HAuCl4·4H2O (0.75 mmol) was dissolved in THF (30 mL) containing [(C8H17)4N]Br (0.76 mmol) at room temperature. After stirring for 15 min, 1-hexanethiol (4.07 mmol) was added and then the resulting mixture was stirred for 30 min. A cold aqueous solution of NaBH4 (8.7 mmol, 5.8 mL) was rapidly added and then the resulting mixture was stirred at room temperature overnight. THF was removed by evaporation and then the remaining precipitate was washed with a water/methanol (= 1:1) solvent mixture to remove excess thiol and other by-products. The [Au25(C6T)18][(C8H17)4N] product was extracted from the dried sample using acetonitrile.
The oxidation of [Au25(C6T)18][(C8H17)4N] was performed by stirring [Au25(C6T)18][(C8H17)4N] with silica gel using dichloromethane as the eluent, analogous to the oxidation of [Au25(PET)18]−. The obtained [Au25(C6T)18]0 was purified before characterization and use in electrochemical measurements.
[Au25(C12T)18]− was synthesized by a reported method with slight modifications.67 First, HAuCl4·4H2O (0.75 mmol) was dissolved in THF (30 mL) containing [(C8H17)4N]Br (0.76 mmol) at room temperature. After stirring for 15 min, 1-dodecanethiol (4.07 mmol) was added and then the resulting mixture was stirred for 30 min. A cold aqueous solution of NaBH4 (8.7 mmol, 5.8 mL) was rapidly added and then the resulting mixture was stirred at room temperature overnight. THF was removed by evaporation and then the remaining precipitate was washed with methanol to remove excess thiol and other by-products. The [Au25(C12T)18][(C8H17)4N] product was extracted from the dried sample using a mixture of acetone and acetonitrile (4:1).
The oxidation of [Au25(C12T)18][(C8H17)4N] was performed by stirring [Au25(C12T)18][(C8H17)4N] with silica gel using dichloromethane as the eluent, analogous to the oxidation of [Au25(PET)18]−. The obtained [Au25(C12T)18]0 was purified before characterization and use in electrochemical measurements.
[Au20.5Ag4.5(PET)18]0 was synthesized by a reported metal exchange method.68 Ag(I)–(PET) complexes were synthesized as follows. First, 2-phenylethanethiol (4.52 mmol) and triethylamine (2 mL) were added to ethanol (7 mL) containing AgNO3 (0.21 mmol). After stirring for 30 min at room temperature, the resulting precipitate was washed with methanol and then water to remove the unreacted AgNO3 and excess thiol. [Au25(PET)18]0 (0.5 μmol) was dissolved in CH2Cl2 (1 mL) and Ag(I)–(PET) (25 μmol) was added. After stirring for 50 or 70 s, the resulting suspension was filtered through filter paper with 0.2 μm pores. The product was washed with methanol, ethanol, and finally water to remove the excess Ag(I)–(PET) and by-products. Finally, [Au20.5Ag4.5(PET)18]0 was extracted from the dried sample using dichloromethane.
[Au23.7Cu1.3(PET)18]0 was synthesized by a reported method68 with slight modifications. Cu(II)–(PET)2 was synthesized as follows. First, 2-phenylethanethiol (4.52 mmol) and triethylamine (2 mL) were added to ethanol (7 mL) containing CuCl2·2H2O (0.21 mmol). After stirring for 30 min at room temperature, the resulting precipitate was washed with methanol and then water to remove the unreacted CuCl2 and excess thiol. [Au25(PET)18]0 (8 mg) and Cu(II)–(PET)2 (0.007 mmol, 2.5 mg) were dissolved in THF (5 mL). After 30 s., NaBH4 (0.09 mmol, 3.5 mg) was rapidly added. The resulting solution was stirred for 30 min at 0 °C. THF was removed by evaporation and then the remaining precipitate was washed with water to remove excess NaBH4. Finally, [Au23.7Cu1.3(PET)18]0 was extracted from the dried sample using acetonitrile to remove other by-products.
[Au24Pd(PET)18]0 was synthesized by a method similar to that in the literature.69 First, [Au24Pd(C12T)18]0 was synthesized using the method previously reported by our group.69 Specifically, HAuCl4·4H2O (0.54 mmol) and Na2PdCl4·3H2O (0.21 mmol) in water (150 mL) were added to toluene (150 mL) containing [(C8H17)4N]Br (0.85 mmol) and then stirred at room temperature for 30 min. The aqueous layer was removed and 1-dodecanethiol (9.0 mmol) was added. The resulting solution was stirred for 30 min at room temperature and then for 30 min at 0 °C. A cold aqueous solution of NaBH4 (7.5 mmol, 150 mL) was rapidly added. The solution was stirred for 3 h at room temperature. Toluene was removed by evaporation and the remaining precipitate was washed with methanol to remove excess thiol and other by-products. The product was extracted using toluene and then dried. Subsequently, all the ligands of [Au24Pd(C12T)18]0 were replaced with PET by reacting [Au24Pd(C12T)18]0 with 2-phenylethanethiol in dichloromethane. This was accomplished by repeating the ligand-exchange reaction and removal of excess 2-phenylethanethiol and by-products using a mixture of water and methanol (1:4) several times.
4.3. Characterization
MALDI mass spectra were collected using a spiral time-of-flight mass spectrometer (JEOL Ltd, JMS-S3000) with a semiconductor laser (wavelength: 349 nm) and DCTB as the matrix. The cluster-to-matrix ratio was set to 1:1000.
UV-Vis absorption spectra of the clusters were recorded in toluene at ambient temperature with a spectrometer (JASCO, V-630).
Electrocatalytic experiments were carried out using an electrochemical workstation (ALS/HCH Instruments Electrochemical Analyzer Model 610 D + SWV). The HER, OER, and ORR were studied using an Ag/AgCl reference electrode, a Pt coil counter electrode, and a rotating-disc glassy carbon (0.196 cm2) working electrode. The working electrode was polished with alumina paste and diamond paste, and subsequently ultrasonicated in distilled water. An electrocatalyst paste solution was made by thoroughly mixing each cluster, toluene, and Nafion® with a ratio of 1 mg:10 μL:10 μL with a micropipette. The paste (10 μL) was placed on the working electrode and then dried for 30 min. The amount of each cluster on the working electrode was weighed (weight of the metal cluster electrocatalyst = weight of the electrode with the metal cluster electrocatalyst − weight of the blank electrode; the weight was measured three times and the repeatability was checked in each experiment). The same weighing balance was used for all working electrodes to avoid error caused by the balance. A schematic diagram of the preparation of working electrodes with cluster catalysts is presented in Fig. S12.† All three types of electrodes were placed in 0.2 M HClO4 solution (pH = 0.7). Linear sweep voltammetry was carried out at a scan rate of 0.025 V s−1 in the potential range of 0 to −1.0 vs. Ag/AgCl in HER studies, whereas a potential range of 0 to 2.0 V vs. Ag/AgCl was used for the OER study. The ORR was carried out from 1.0 to −0.5 V vs. Ag/AgCl. N2 gas flow was maintained during the HER and OER measurements, whereas in ORR measurements, N2 and O2 gas flow was maintained at a constant flow rate. The ORR measurements were also carried out at different rotation speeds to calculate the number of electrons transferred during the ORR process using the Koutecky–Levich equation.35 During these electrocatalytic measurements, contamination of Pt, which makes up the electrode, in the thiolate-protected metal clusters was not observed (Fig. S15†).
4.4. Analysis
The average number of the substituted atoms (x′) of Au25−x′Mx′(PET)18 (M = Ag or Cu) was calculated using the following equation,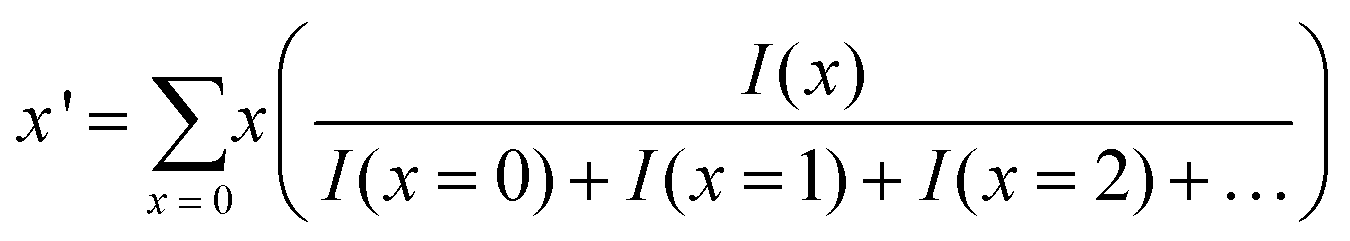 | (12) |
Here, x is the number of substituted metal atoms and I(x) is the ion intensity (height) of MxAu25−x(PET)18 (M = Ag or Cu) with each x, which was estimated from mass spectral data (Fig. S8(b), (c), and (e)†).
The number of electrons transferred (n) in the ORR (Fig. 6) was quantified using eqn (13) based on the RRDE voltammograms measured at different rotation speeds:
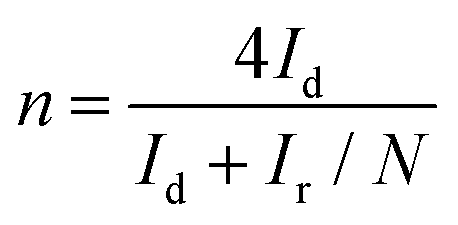 | (13) |
Here, Id is the disk current, Ir is the ring current, and N is the RRDE collection efficiency (= 0.37).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mr Keisuke Nishido, Ms Juri Maekawa, and Mr Atsuya Harasawa for technical assistance. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant number JP16H04099 and 16K21402), Scientific Research on Innovative Areas “Coordination Asymmetry” (grant number 17H05385), and Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion” (grant number 18H05178). Funding from the Takahashi Industrial and Economic Research Foundation, the Futaba Electronics Memorial Foundation, the Iwatani Naoji Foundation, and the Asahi Glass Foundation is also gratefully acknowledged.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- H. Qian, M. Zhu, Z. Wu and R. Jin, Acc. Chem. Res., 2012, 45, 1470–1479 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS PubMed.
- R. L. Whetten, H.-C. Weissker, J. J. Pelayo, S. M. Mullins, X. López-Lozano and I. L. Garzón, Acc. Chem. Res., 2019, 52, 34–43 CrossRef CAS PubMed.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed.
- Y. Pei, P. Wang, Z. Ma and L. Xiong, Acc. Chem. Res., 2019, 52, 23–33 CrossRef CAS PubMed.
- T. P. Bigioni, R. L. Whetten and Ö. Dag, J. Phys. Chem. B, 2000, 104, 6983–6986 CrossRef CAS.
- C. M. Aikens, Acc. Chem. Res., 2018, 51, 3065–3073 CrossRef CAS PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- Q. Tang, G. Hu, V. Fung and D.-E. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- M. Haruta, Faraday Discuss., 2011, 152, 11–32 RSC.
- H. Tsunoyama, N. Ichikuni, H. Sakurai and T. Tsukuda, J. Am. Chem. Soc., 2009, 131, 7086–7093 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- T. Higaki, Q. Li, M. Zhou, S. Zhao, Y. Li, S. Li and R. Jin, Acc. Chem. Res., 2018, 51, 2764–2773 CrossRef CAS PubMed.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-E. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- D. Eguchi, M. Sakamoto and T. Teranishi, Chem. Sci., 2018, 9, 261–265 RSC.
- G. Hu, Q. Tang, D. Lee, Z. Wu and D.-E. Jiang, Chem. Mater., 2017, 29, 4840–4847 CrossRef CAS.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- K. Kwak, W. Choi, Q. Tang, D.-E. Jiang and D. Lee, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-E. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- X. Zhao, P. Gao, Y. Yan, X. Li, Y. Xing, H. Li, Z. Peng, J. Yang and J. Zeng, J. Mater. Chem. A, 2017, 5, 20202–20207 RSC.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed.
- H. Yin, H. Tang, D. Wang, Y. Gao and Z. Tang, ACS Nano, 2012, 6, 8288–8297 CrossRef CAS.
- C. Jeyabharathi, S. S. Kumar, G. V. M. Kiruthika and K. L. N. Phani, Angew. Chem., Int. Ed., 2010, 49, 2925–2928 CrossRef CAS PubMed.
- Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 RSC.
- D. R. Kauffman, D. Alfonso, C. Matranga, P. Ohodnicki, X. Deng, R. C. Siva, C. Zeng and R. Jin, Chem. Sci., 2014, 5, 3151–3157 RSC.
- T. C. Jones, L. Sumner, G. Ramakrishna, M. B. Hatshan, A. Abuhagr, S. Chakraborty and A. Dass, J. Phys. Chem. C, 2018, 122, 17726–17737 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed.
- K. Kwak, U. P. Azad, W. Choi, K. Pyo, M. Jang and D. Lee, ChemElectroChem, 2016, 3, 1253–1260 CrossRef CAS.
- L. Wang, Z. Tang, W. Yan, H. Yang, Q. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 20635–20641 CrossRef CAS PubMed.
- L. Sumner, N. A. Sakthivel, H. Schrock, K. Artyushkova, A. Dass and S. Chakraborty, J. Phys. Chem. C, 2018, 122, 24809–24817 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- T. Tsukuda and H. Häkkinen, Protected Metal Clusters: From Fundamentals to Applications, Elsevier B.V., Amsterdam, The Netherlands, 2015 Search PubMed.
- X. L. Du, X. L. Wang, Y. H. Li, Y. L. Wang, J. J. Zhao, L. J. Fang, L. R. Zheng, H. Tong and H. G. Yang, Chem. Commun., 2017, 53, 9402–9405 RSC.
- S. Yamazoe, W. Kurashige, K. Nobusada, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2014, 118, 25284–25290 CrossRef CAS.
- Y. Negishi, K. Munakata, W. Ohgake and K. Nobusada, J. Phys. Chem. Lett., 2012, 3, 2209–2214 CrossRef CAS PubMed.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- Y. Niihori, D. Shima, K. Yoshida, K. Hamada, L. V. Nair, S. Hossain, W. Kurashige and Y. Negishi, Nanoscale, 2018, 10, 1641–1649 RSC.
- M. S. Devadas, S. Bairu, H. Qian, E. Sinn, R. Jin and G. Ramakrishna, J. Phys. Chem. Lett., 2011, 2, 2752–2758 CrossRef CAS.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS PubMed.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS PubMed.
- J. Jung, S. Kang and Y.-K. Han, Nanoscale, 2012, 4, 4206–4210 RSC.
- K. Maeda, N. Okabayashi, S. Kano, S. Takeshita, D. Tanaka, M. Sakamoto, T. Teranishi and Y. Majima, ACS Nano, 2012, 6, 2798–2803 CrossRef CAS PubMed.
- J. A. van Bokhoven and J. T. Miller, J. Phys. Chem. C, 2007, 111, 9245–9249 CrossRef CAS.
- N. S. Phala and E. van Steen, Gold Bull., 2007, 40, 150–153 CrossRef CAS.
- H. Qian, C. Liu and R. Jin, Sci. China: Chem., 2012, 55, 2359–2365 CrossRef CAS.
- M. A. Tofanelli, K. Salorinne, T. W. Ni, S. Malola, B. Newell, B. Phillips, H. Häkkinen and C. J. Ackerson, Chem. Sci., 2016, 7, 1882–1890 RSC.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- C. Kumara and A. Dass, Anal. Chem., 2014, 86, 4227–4232 CrossRef CAS PubMed.
- H. Qian and R. Jin, Chem. Mater., 2011, 23, 2209–2217 CrossRef CAS.
- K. Kwak, Q. Tang, M. Kim, D.-E. Jiang and D. Lee, J. Am. Chem. Soc., 2015, 137, 10833–10840 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malona, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed.
- Y. Niihori, Y. Koyama, S. Watanabe, S. Hashimoto, S. Hossain, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 4930–4934 CrossRef CAS PubMed.
- Y. Niihori, M. Matsuzaki, C. Uchida and Y. Negishi, Nanoscale, 2014, 6, 7889–7896 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Geometrical structure of each cluster, MALDI mass spectra, UV-vis spectra, schematic of the proposed energy conversion system, additional linear sweep voltammograms of the products. See DOI: 10.1039/d0nr00702a |
| ‡ Present address: Department of Chemistry, M V College Buxar, Veer Kunwar Singh University, Ara, Bihar 802101, India. |
| § These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |