
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-directed arrangement of soft synthetic compartments and their behavior in vitro and in vivo†
Juan
Liu‡§
a,
Ioana
Craciun‡
a,
Andrea
Belluati
 a,
Dalin
Wu
a,
Sandro
Sieber
b,
Tomaz
Einfalt
b,
Dominik
Witzigmann¶
b,
Mohamed
Chami
c,
Jörg
Huwyler
b and
Cornelia G.
Palivan
*a
a,
Dalin
Wu
a,
Sandro
Sieber
b,
Tomaz
Einfalt
b,
Dominik
Witzigmann¶
b,
Mohamed
Chami
c,
Jörg
Huwyler
b and
Cornelia G.
Palivan
*a
aDepartment of Chemistry, University of Basel, Mattenstrasse 24a, Basel-4058, Switzerland. E-mail: cornelia.palivan@unibas.ch
bDivision of Pharmaceutical Technology, Department of Pharmaceutical Sciences, University of Basel, Klingelbergstrasse 50, Basel-4056, Switzerland
cBioEM lab, Biozentrum, University of Basel, Mattenstrasse 26, Basel-4058, Switzerland
First published on 20th April 2020
Abstract
DNA has been widely used as a key tether to promote self-organization of super-assemblies with emergent properties. However, control of this process is still challenging for compartment assemblies and to date the resulting assemblies have unstable membranes precluding in vitro and in vivo testing. Here we present our approach to overcome these limitations, by manipulating molecular factors such as compartment membrane composition and DNA surface density, thereby controlling the size and stability of the resulting DNA-linked compartment clusters. The soft, flexible character of the polymer membrane and low number of ssDNA remaining exposed after cluster formation determine the interaction of these clusters with the cell surface. These clusters exhibit in vivo stability and lack of toxicity in a zebrafish model. To display the breadth of therapeutic applications attainable with our system, we encapsulated the medically established enzyme laccase within the inner compartment and demonstrated its activity within the clustered compartments. Most importantly, these clusters can interact selectively with different cell lines, opening a new strategy to modify and expand cellular functions by attaching such pre-organized soft DNA-mediated compartment clusters on cell surfaces for cell engineering or therapeutic applications.
1. Introduction
The unique features of DNA enable the organization of nanostructures into larger superstructures and macroscopic superlattices with well-defined orientations depending on their intrinsic architecture.1,2 Current knowledge and experiences with DNA-organized supramolecular structures revolve around exploiting DNA-linked rigid nanostructures,3,4 for example gold nanoparticles.5,6 The rigidity of particles imparts directionality to hybridization interactions and the high-density of DNA strands on the surfaces act as ‘valencies’ to manipulate the stereo-organization precisely, either when DNA chains or DNA origami direct the organization,7–9 or when steric hindrance induces formation of core-satellite structures.5,7,10,11A completely different framework of interactions is induced when the nanostructure is deformable, and molecules can laterally diffuse inside the assembly, causing a constant fluctuation in exposed molecular groups bearing the DNA. Thus, the unique properties of the resulting organization open up a new field of soft DNA-linked compartment assemblies. Only recently have soft nanocompartments, especially liposomes and polymersomes, been exploited for DNA-mediated organization12,13 due to the preconception that their inherent flexibility interferes with the creation of defined structures,14 such as superlattices assembled by rigid nanoparticles. For example, in the case of liposomes, DNA-mediated assembly led to their precipitation from solution or membrane fusion due to their low stability.13,15,16 In the case of polymersome based sub-micrometer sized assemblies, only one example has been reported12 with no in vitro or in vivo testing, thus insights into the behavior of such soft materials in a biological environment are still lacking. Nevertheless, nanocompartments have unique advantages due to their intrinsic architecture, hollow spheres with hydrophilic cavities surrounded by a membranous structure possessing hydrophobic cores. They enable the efficient loading of various hydrophilic and hydrophobic therapeutic agents ranging from small molecular weight drugs to DNA and RNA for drug delivery,17–21 nanoparticles for catalytic properties,22,23 and various enzymes for the development of artificial organelles24 functional in vivo.25 While both liposomes and polymersomes allow tailoring for biocompatibility, stimuli-responsiveness and biodegradability, polymersomes are an improved alternative as they exhibit higher mechanical stability compared to their counterpart, liposomes.24,26–28
There are several examples of sub-micrometer sized nanoparticle assemblies, however, only a few were proposed for cellular interactions and phototherapies.5,10,29,30 The biological application of such nanoparticle assemblies remains challenging with most focusing on DNA intercalating molecules loaded non-covalently in the hybridized DNA strands for their delivery.5,30,31 Furthermore such nanoparticle assemblies undergo fast disassembly in vitro and in vivo,10,30 implemented as a strategy for clearance.10
Here, we present soft DNA-linked polymersome clusters as a new type of compartment-assembly able to support efficient bio-applications with the unique advantage of simultaneously allowing the encapsulation of catalytic compounds and their localization at desired locations controlled by their interaction with cells. DNA-linked polymersomes represent a complementary platform to DNA–nanoparticle systems with a hierarchic self-organization that has been manipulated by fine-tuning the polymer membrane composition, the surface density of ssDNA and polymersome concentration (Scheme 1). As the soft polymersome clusters differ greatly with respect to other DNA-mediated assemblies, we methodically probe their behavior in vitro and in vivo to determine their suitability for future applications. We present how such soft DNA-mediated compartment assemblies interact with different cell lines resulting in binding selectivity towards epithelial cells via scavenger receptors. Notably, for the first time the realization of cellular membrane attachment of DNA-linked super-assemblies is achieved. These clusters are non-toxic and their characteristic behavior is also maintained in vivo. Attachment of such clusters to the cell surface could prove momentous, as it would provide a universal and innocuous approach for non-genetic therapeutic cell engineering32,33 but also detoxification of xenotoxins by encapsulating enzymes within the inner compartment. Following along this line, we encapsulated laccase as an extensively studied enzyme with multiple applications34,35 within the clustered polymersomes and demonstrated the catalytic potential of the clusters to prevent internalization of harmful compounds inducing apoptosis. In contrast to rigid DNA-linked nanoparticles and assemblies that offer limited bioactivity after endocytosis, we reveal that both the deformability of the polymersome's membrane and the lateral diffusion of the polymer chains within the membrane contribute to their surprising stability and determine the clusters’ destiny in biologically relevant environments. The multiple compartments with both aqueous cavities and hydrophobic boundaries benefit the integration of active biomolecules with diverse chemical properties into one nanoplatform, providing combined and synchronized therapeutic and diagnostic solutions. All these features make them truly unique soft compartment assemblies with high potential for bio-applications, such as regenerative medicine or cell engineering to restore, replace or integrate new cellular functions.
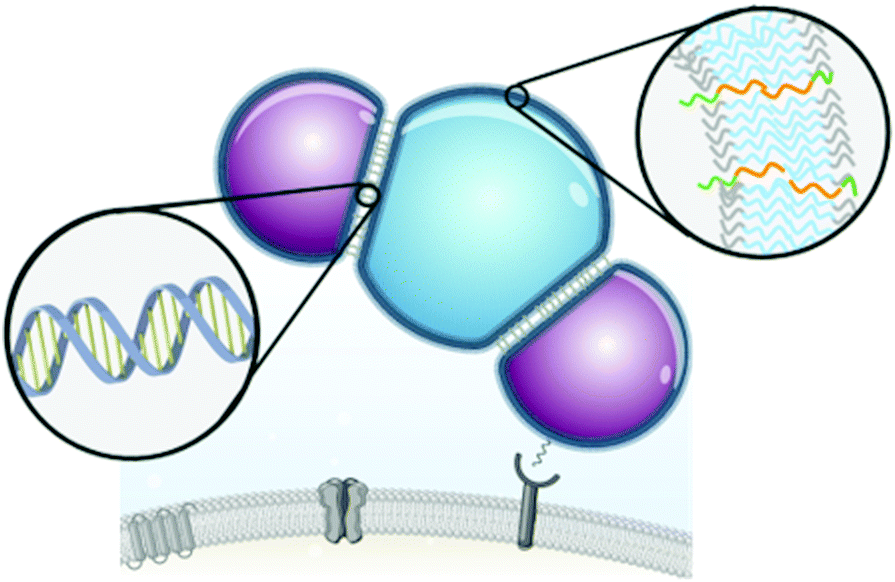 | ||
| Scheme 1 Synthetic nanocompartments (polymersomes, shown in blue and magenta) assembled from amphiphilic diblock copolymers (right insert depicting the polymeric membrane) are interconnected using DNA hybridization as the driving force for their self-organization into clusters (left insert highlighting the hybridized DNA that locks in place the polymersomes). Such soft flexible assemblies selectively interact with the scavenger receptors on the cell membranes in vitro and in vivo leading to attachment to the cell surface. | ||
2. Results and discussion
2.1. Assembly of ssDNA functionalized polymersomes and DNA-linked polymersome clusters
In order to construct stable sub-micrometer sized polymersome clusters in a controllable manner, we carefully considered three key molecular factors: (i) the chemical nature and morphology of the polymersomes’ membrane, (ii) the DNA density and distribution on the polymersomes’ surface, and (iii) the concentration of polymersomes.12 For polymersome formation, we selected the diblock copolymer poly(dimethylsiloxane)-block-poly(2-methyloxazoline) (PDMS-PMOXA) because of its low toxicity and biocompatibility.36,37 As previously shown, the combination of PMOXA-PDMS-PMOXA triblock copolymer with azide-functionalized diblock PDMS-PMOXA copolymer, bearing a longer PMOXA block, forms polymersomes with exposed azide functional groups on the polymersomes’ surface.12 We then linked dibenzocyclooctyne (DBCO)-functionalized ssDNA (DBCO-ssDNA) to the polymersomes and promoted their zipping by DNA hybridization.12 Here we changed the approach and synthesized only diblock copolymers, PDMS62-PMOXA13 and PDMS71-PMOXA25-OEG3-N3, with similar PDMS block lengths aimed at circumventing hydrophobic mismatch and phase separation between the two polymers, consequently favoring formation of polymersomes (Fig. 1A, Fig. S1, and Table S1†). Switching to this new block copolymer combination facilitated an increase in PDMS71-PMOXA25-OEG3-N3 content to 20 mol% without perturbing the self-assembly (Fig. S2†), thus obtaining polymersomes with average apparent hydrodynamic diameter (DH) of 190 ± 30 nm (s.e.m, n = 3) as obtained by dynamic light scattering (DLS, Fig. S4 and Table S2†). | ||
| Fig. 1 Principles and preparation of DNA-linked polymersome clusters. (A) Schematic representation of assembled ssDNA–polymersomes, chemical structures of the two block copolymers used and the sequence of the two complementary ssDNAs. (B) Schematic representation of the cluster assembly process where the polymersomes first link together via DNA hybridization, followed by migration of non-hybridized ssDNAs and subsequent hybridization to form a DNA bridge area in between the polymersomes. (C) Self-organization of ssDNA–polymersomes into clusters reported as the change in their apparent DH as a function of time for 0.5 mol% (orange and light orange), 1 mol% (magenta and light magenta), 5 mol% (red and light red), 10 mol% (green and light green) and 20 mol% (blue and light blue) azide-functionalized diblock copolymers. The results represent two independent measurements shown on the graph as hues of the same color. (D) The distribution of the average apparent DH of: ssDNAa-polymersomes (200 nm ± 30 nm, s.e.m, n = 3,) comprised of 20 mol% azide-functionalized diblock copolymers (blue) and of the corresponding DNA-linked polymersome clusters (440 nm ± 100 nm, s.e.m, n = 3) (red). (E–F) Transmission electron microscopy (TEM) and cryo-TEM micrographs of the DNA-linked polymersome clusters P20-ab. Scale bars are 500 nm (E) and 100 nm (F). | ||
As mentioned above, key molecular factors that affect the process of hierarchic self-organization of polymersomes into clusters are both the number of ssDNAs per polymersome and concentration of ssDNA–polymersomes. To modulate the apparent DH of DNA-linked polymersome clusters, we kept the concentration of complementary ssDNA–polymersomes constant (0.2 mg mL−1) and varied only the number of ssDNAs per polymersome. We linked DBCO-ssDNAs onto the polymersomes’ surface by strain-promoted azide–alkyne cycloaddition (SPAAC) reaction between DBCO-ssDNAs and polymersomes with exposed N3 groups. By varying the N3 content per polymersome, we varied the number of ssDNAs per polymersome from 1 ± 1 to 289 ± 72, as measured by fluorescence correlation spectroscopy (FCS, Fig. S3, see ESI† for details). The perikinetic clustering (solely driven by diffusion) of complementary ssDNA–polymersomes, at equal mass ratios and 37 °C, was investigated by monitoring the change in apparent DH over time (Fig. 1B and C). The hybridization of complementary ssDNA on the polymersome surface resulted in a rapid increase in the apparent DH until reaching a plateau with a defined range, where controlled cluster formation occurs. Self-organization into sub-micrometer sized clusters takes place only for complementary ssDNA–polymersomes with a number of linked ssDNAa/b ranging from 34 ± 33 to 188 ± 58 and 8 ± 5 to 111 ± 29 (Fig. S3 and S4†), corresponding to 1 to 20 mol% of azide-functionalized diblock content, respectively (Fig. 1E and F). Lowering the amount of linked ssDNA per polymersome does not sustain cluster formation, while a higher number of ssDNA per polymersome resulted in uncontrollable aggregation, as observed by the instantaneous formation of large clusters with an apparent DH > 1 μm.
Ultimately, we obtained a series of polymersome clusters with a size distribution corresponding to a lognormal distribution function (statistical analysis, ESI†) and an average apparent DH from 280 ± 80 nm to 440 ± 100 nm (s.e.m., n = 3, Fig. 1D and Fig. S4†). The distribution of the number of polymersomes/cluster for polymersomes with 1 to 20 mol% azide-functionalized diblock content was fitted with a log-normal distribution function as well (statistical analysis, ESI†). The clusters are composed on average of 2.5 ± 1.3 to 3.4 ± 1.6 polymersomes per cluster with a non-bound polymersome fraction lower than 5%, determined from transmission electron microscopy (TEM, Fig. S4†). In contrast with ssDNA-coupled rigid nanoparticles, ssDNA–polymersomes are advantageous when forming sub-micrometer sized clusters in a controllable manner as the ssDNA-bearing block copolymers migrate through the membrane and are then locked in a bridging area between linked polymersomes (Fig. 1B and F).12,38 The formation of this three-dimensional bridging area with an area regulated by the number of dsDNAs holding the polymersomes together and opening dependent on the length of dsDNA, rather than a ‘spot’, functions as a barrier thus sheltering the dsDNA from exposure to the environment. The migration of ssDNA to the bridging area also decreases the ssDNA density in the non-bridging area and eventually terminates the clustering process. Therefore, the formation of sub-micrometer sized clusters is kinetically controlled by using mechanically stable polymersomes with a relatively low ssDNA density on the surface to prevent aggregation or rupture.
To further probe the stability of the bridging region, we incubated the clusters in presence of DNase I, the most prevalent endonuclease in serum (Fig. S5†). We observed that polymersome clusters with an apparent DH above 300 nm remain clustered in presence of 1 μg mL−1 of DNase I. However, no clusters were formed when the ssDNA–polymersomes were pre-treated with 1 μg mL−1 of DNase I, suggesting the DNA strands on single ssDNA–polymersomes have been digested. Narrowing the bridging area between polymersomes from 7.1 ± 0.5 nm to 3.5 ± 0.3 nm by reducing the length of the ssDNA from 22 bp to 11 bp effectively protects the polymersome clusters against DNase I hydrolysis (Fig. S5†). The clusters linked by 22 bp DNA disassembled to single polymersomes at a DNase I concentration of 10 μg mL−1, while clusters linked by 11 bp DNA preserved their clustered structures.
2.2. Examination of DNA-linked polymersome clusters’ behavior in vitro
As the polymersome clusters we designed differ greatly with respect to other DNA-mediated assemblies, and no other soft compartment assemblies were studied in vitro, it was fundamental to study their interactions in vitro and determine their potential for biomedical applications. Under physiological conditions, e.g. in cell medium, the colloidal stability of DNA functionalized materials might be affected due to interactions between proteins and the DNA.39 Thus, we first examined the stability of the 22 bp ssDNA–polymersome clusters in cell medium (Fig. S6A†). The clusters remained stable up to 10 hours due to the strong interactions between complementary ssDNAs located in the sheltered bridging region (Fig. S6A†).Prior to determining if there are selective interactions between the clusters and epithelial cells, we evaluated the toxicity of the ssDNA polymersomes and DNA-linked clusters using an MTS assay (Fig. S6E†). No toxicity was obtained up to 40 μg mL−1. Subsequently, we examined their interactions with three cell lines, i.e. HeLa, HEK293T and U87-MG, to determine if the DNA-linked polymersome clusters are stable, internalized or have any inherent selectivity towards a specific cell line. We incubated cells in the presence of low concentrations (13 μg mL−1) of unmodified polymersomes, ssDNA–polymersomes, 400 nm sized small clusters and 1000 nm large clusters (see Methods for the preparation of large clusters). In polymersomes functionalized with 22 bp ssDNAa we encapsulated a fluorescent dye DY-633 (DY-633-P20-a) and in polymersomes functionalized with the complementary 22 bp ssDNAb ATTO 488 dye (ATTO-488-P20-b) as small molecule mimics and for visualization via confocal laser scanning microscopy (CLSM, Fig. S6C and D†). After 24 h, we thoroughly washed the cells to remove unbound polymersomes or clusters and stained the cellular membrane using the CellMask™ orange plasma membrane stain, prior to imaging. DY-633-P20-a as well as both small and large polymersome clusters accumulated on the cell surface predominantly at cell junctions of HEK293T and HeLa cells (Fig. 2A, Fig. S7A–C† for transmission channels, Fig. S8A–J and S9A–C† for repeat experiments). Using flow cytometry, we confirmed the presence of clusters, most likely still attached to the cell surface; however, the precise location cannot be determined using this technique (Fig. S10A and B†). Unmodified polymersomes showed no binding and were not internalized (Fig. S8B and G†). Furthermore, the binding interaction between the clusters with the cell membrane appears to be more favorable than the one between single ssDNA–polymersomes and cells, since rigorous washing steps prior to imaging did not result in detachment and there appear to be more clusters than single ssDNA–polymersomes retained on the cell membrane as shown by the fluorescence intensity (Fig. 2). A local accumulation of clusters on the cell surface is key feature that will allow their future application for biomedical applications. In addition, polymersome clusters have the advantages, opposed to non-clustered or ssDNA–polymersomes to provide segregated spaces at the nanoscale supporting simultaneous functionality of separate compounds and simultaneous co-localization of such comportments to improve their overall efficiency. As observed in the merged image, DY-633-P20-a overlaps with ATTO-488-P20-b, to a larger extent as compared to the control polymersomes bearing non-complementary ssDNA (Fig. 2B), indicating that the clusters are intact and did not dissociate over the 24 h incubation period. The large 1000 nm clusters appear to form a network like structure that expands to cover large portions of the cell outer membrane (Fig. 2C) suggesting an active rearrangement of the clusters in vitro. Binding and accumulation of the clusters to the cell surface occurred within the first hour of incubation with no precipitation or accumulation visible in regions where the cells were not present, thus further demonstrating the high affinity of the clusters towards the cell surface (Fig. S11†). Binding to the cell surface is unique as most reported DNA coupled nanostructures and assemblies are internalized within cells and do not form networks on the surface.5,10,29,30 This behavior is again an exceptional emerging property of using soft DNA-linked polymersomes bearing a low DNA surface density. This leads to a low adhesion energy for full wrapping of cells through receptor binding and thus might not be sufficient to activate endocytosis.40 A second factor contributing to this unique behavior is that ssDNA–polymersomes and the DNA-linked polymersome clusters are much larger and more deformable compared to rigid gold nanoparticles where the stiffness of the surface promotes cell membrane engulfment.41 In stark contrast, our clusters spread along the cell membrane without significant membrane deformation, thereby limiting the internalization processes.
 | ||
| Fig. 2 Interaction of DNA-linked polymersome clusters with HEK293T and U87-MG cells. (A) CLSM micrographs showing attachment to surface of HEK293T cells of ssDNA polymersomes loaded with DY-633 (DY-633-P20-a, magenta), 400 nm small clusters and 1000 nm large clusters composed of ssDNAa-polymersomes loaded with DY-633 (DY-633-P20-a magenta) and ssDNAb-polymersomes loaded with ATTO 488 (ATTO-488-P20-b, cyan). Scale bar = 10 μm. (B) 3D z-stack reconstruction showing 400 nm clusters on the cell surface (left) and non-complementary ssDNA–polymersomes (right). Scale bar = 20 μm. (C) Z-Stack of large polymersome clusters forming a network on the surface of HEK293T cells. Scale bar = 20 μm. (D) CLSM micrographs showing uptake in U87-MG cells of ssDNA polymersomes loaded with DY-633 (DY-633-P20-a, magenta), 400 nm small clusters and 1000 nm large clusters composed of ssDNAa-polymersomes loaded with DY-633 (DY-633-P20-a, magenta) and ssDNAb-polymersomes loaded with ATTO 488 (ATTO-488-P20-b, cyan). Scale bar = 10 μm. The experiments were performed in cell media containing 10% fetal calf serum. The cell membrane was stained using CellMaskTM orange plasma membrane stain prior to imaging. | ||
Next, we examined the interaction of unmodified polymersomes, ssDNA–polymersomes and the small and large clusters with U87-MG glioblastoma cells, known to have a higher endocytotic potential and favor uptake of larger sized particles.42,43 Interestingly, we observed limited internalization of both the 400 nm clusters and to a lesser extent of the larger clusters (Fig. 2D, Fig. S7D–F, S8K–P and S9D–E†). DY-633-P20-a and ATTO-488-P20-b polymersomes from within the cluster colocalize, as seen also from the individual CLSM z-stack slices (Fig. S8P†) indicating that the clusters preserve their integrity following uptake, as re-clustering within the cell is improbable (Fig. 2D). In the case of ssDNA–polymersomes and unmodified polymersomes it was difficult to determine the extent of uptake or attachment by CLSM due to their scarcity; however, analysis of the flow cytometry data suggests that both were internalized or in the case of ssDNA possibly attached to the cell surface (Fig. S10C†). As mentioned above, the precise location cannot be determined via flow cytometry, however unmodified polymersomes cannot attach to the cell surface and have been shown to be readily uptaken by cells.25,44 The observed uptake via CLSM of the small and large clusters is in agreement with the fact that U87-MG cell line preferentially uptakes larger particles (Fig. S10C†).42,43
2.3. Mechanism of DNA-linked polymersome cluster attachment to cells
To better understand the in vitro behavior and specific cellular interaction of the polymersome clusters, we explored the binding mechanism of the ssDNA–polymersomes and clusters with HEK293T cells, as model cell line. Patel et al. showed that DNA-functionalized nanoparticles are internalized via binding to scavenger receptors.45 To test whether our DNA-polymersome clusters also bind to this class of receptors, we performed competitive inhibition experiments. We preincubated the cells with increasing concentrations of two scavenger receptor ligands (i.e. polyinosinic acid (PolyI) and Fucoidan) for 1 hour prior to addition of ssDNA–polymersomes, small (400 nm) and large (1000 nm) clusters. As the concentration of the inhibitors increases, surface binding of the ssDNA–polymersomes, both small and large clusters, decreased in a dose responsive fashion as compared to controls (Fig. 3A, B depicting trend of large clusters and Fig. S12† transmission channel, Fig. S13† for complete analysis). At the highest concentration of PolyI, binding of the clusters was almost fully inhibited and to a lesser extent in the case of the Fucoidan inhibitor. These results indicate that specific binding to epithelial cells is indeed mediated through attachment of negatively charged polymersome clusters to scavenger receptors. Of note, although DNA migration occurs during the polymersome clustering, approximately 50% of ssDNA on the polymersomes’ surface remains non-hybridized. The percentage of non-hybridized ssDNA was determined by fluorescent resonance energy transfer analysis (FRET, Fig. S6B†). Excess amount of ssDNAa-Cy3 and polymersomes linked with ssDNAa-Cy3 (P20-b-Cy3) were incubated with polymersomes linked with ssDNAb-Cy5 (P20-b-Cy5), respectively. The percentage of hybridized ssDNA, when forming clusters, was calculated by dividing the fluorescence intensity of P20-b-Cy5 hybridized with P20-b-Cy3 by the value when P20-b-Cy5 is hybridized with an excess amount of ssDNAa-Cy3. 50% of non-hybridized ssDNA on the polymersomes’ surface is also indicated by the increase in ζ-potential from −11.00 ± 0.04 mV for DY-633-P20-a and −10.60 ± 0.02 mV for ATTO-488-P20-b to −5.47 ± 0.02 mV for the clusters (s.e.m., n = 3, Table S4†). Therefore, the non-hybridized ssDNA on the clusters contributes to their cellular interactions. | ||
| Fig. 3 Effect of scavenger receptor inhibitors on cluster cellular interaction. (A) CLSM micrographs of 1000 nm large DNA-linked polymersome clusters composed of ssDNAa-polymersomes loaded with DY-633 (DY-633-P20a, magenta) and ssDNAb-polymersomes loaded with ATTO-488 (ATTO-488-P20b, cyan) in presence of increasing concentration of PolyI, a scavenger receptor inhibitor (0 to 50 μg mL−1, final concentration). Addition of PolyI completely inhibits cell binding of the DNA-linked polymersome clusters even at the lowest concentration tested. (B) CLSM micrographs of the 1000 nm large DNA-linked polymersome clusters in presence of increasing concentration of Fucoidan, a scavenger receptor inhibitor (0 to 50 μg mL−1, final concentration). Scale bar 10 μm. | ||
2.4. Development of catalytically active DNA-linked polymersome clusters
Capitalizing on the fact that our clusters stably attach to the cell surface, we subsequently focused on developing the clusters for potential detoxification applications in the blood circulation. Polymersome clusters attached to the cell surface of blood vessels and loaded with catalytically active enzymes can be applied as exogenous tool kits that simultaneously protect the encapsulated cargo from degradation or deactivation and can be used for the elimination of circulating toxic chemicals. One solution we envision resides in taking a true and tested enzyme, fungal laccase, and encapsulate it within the polymersomes. Laccases have been extensively used for bioremediation applications, as they are known to act on multiple xenotoxic compounds, such as pesticides and insecticides,34 anti-inflammatory drugs with a high incidence of misuse and overdose, as for example Acetaminophen and aspirin,46 phenolic compounds,47,48 polychlorinated biphenyls (PCBs) and polycyclic aromatic hydrocarbons (PAH).49 While actions to reduce the exposure to such toxins have been taken, microplastics, ubiquitously present in the environment are now making their way up the food chain and represent a new source of concern as they leach out PCBs, phenolic substances and various other endocrine disrupting compounds.50 Phenolic substances are main chemical pollutants in soil and water and their production is tremendous: for example, the annual output of biphenyl A alone is up to 4.69 million tons.51 Luckily laccases can also degrade such substrates relieving the toxic burden.52Here as a proof of concept we encapsulate laccase in polymersomes permeabilized by inserting the outer membrane protein F (OmpF) into the membrane. We confirm that upon encapsulation, either in single, unmodified polymersomes (Lac-Polymersomes) or in DNA-clustered Lac-Polymersomes, laccase maintains its activity and converts the substrate Amplex Ultra Red to the fluorescent product resorufin (Fig. 4). At the same concentration, both unmodified and clustered Lac-Polymersomes show 40% of the activity of free enzymes, whereas almost no activity can be detected from control Lac-Polymersomes with no OmpF because their membrane is not permeable for small molecules. In human serum, the DNA-clustered Lac-Polymersomes truly exhibit their advantages, displaying an increase in activity up to 80% of the activity of free laccase in the same medium (Fig. 4). Previously, we have shown that encapsulation not only helps maintain enzymatic activity in complex medium, but also increases the stability of the enzymes and protects them from degradation.44 In serum, free laccases were partially degraded while the ones inside the polymersomes and clusters were protected. However, in serum trace amounts of heme-containing proteins lead to further oxidation of Amplex Ultra Red, causing a higher background than observed in PBS. Nevertheless, by combining the robust DNA-cluster system that attaches to the cell surface with the catalytic capability of these clusters, we can utilize our system as extracellular detoxification patches. In addition, the clusters provide a better spatial localization than single-polymersomes, especially supported by their specific interactions with the cells. With future optimization, our enzyme loaded networks could become a first line of protection for farmers under constant exposure to pesticides and insecticides, for patients with increased levels of xenobiotics in their blood or for those with increased levels of microplastics in their system.
 | ||
| Fig. 4 Catalytic activity of DNA-polymersome clusters containing laccase. (A) Schematic representation of DNA-linked polymersome cluster with catalytic activity by encapsulation of laccase (Lac) and permeabilization of the polymer membrane by insertion of OmpF. The encapsulated enzyme laccase converts in situ Amplex Ultra Red to the fluorescent product resorufin. (B) Reaction efficiency of the free laccase, non-ssDNA modified polymersomes containing encapsulated laccase (Naked Lac-Polymersomes), DNA-clustered Lac-Polymersomes equipped with OmpF, and control Lac-Polymersomes that are not permeabilized (without OmpF). Reaction efficiency was determined by measuring the production of resorufin (Ex. λ = 529 nm, Em. λ = 600 nm; n = 3) both in PBS (orange) and in human serum (green). | ||
2.5. Examination of DNA-linked polymersome cluster behavior in vivo
We finally examined the in vivo behavior of ssDNA–polymersome clusters (i.e. biodistribution, cluster stability) using the zebrafish as a vertebrate screening model. Recently, it has been shown that the zebrafish model is a straightforward tool to study the systemic circulation and biodistribution of nanoparticles53 and the stability as well as enzymatic activity of artificial organelles.25 Therefore, we investigated mixtures of non-complementary ssDNA–polymersomes encapsulating either sulforhodamine B (SRB) or DY-633, as small molecule mimics, and small and large polymersome clusters in transgenic kdrl:EGFP zebrafish embryos, which express EGFP in their vasculature endothelial cells. We injected all nanostructures intravenously into the blood circulation via the duct of Cuvier (Fig. 5A and Fig. S6F, G† – characterization of clusters with encapsulated SRB and DY-633 used in this in vivo study) and analyzed the in vivo biodistribution at specific time points (0.5, 4, and 24 h) using CLSM (Fig. 5B). Interestingly, polymersome mixtures as well as both small and large clusters showed a distinct binding pattern in the caudal vein region already after 30 min. After 24 hours all types of polymersomes remained attached to the posterior caudal vein (PCV, Fig. 5B). This is an interesting observation, since this region has been identified as a model to assess the interaction of anionic nanoparticles with scavenger receptors.54 Thus, both ssDNA–polymersomes and the polymersome clusters were also recognized in vivo by scavenger receptor expressing cells, confirming our in vitro results.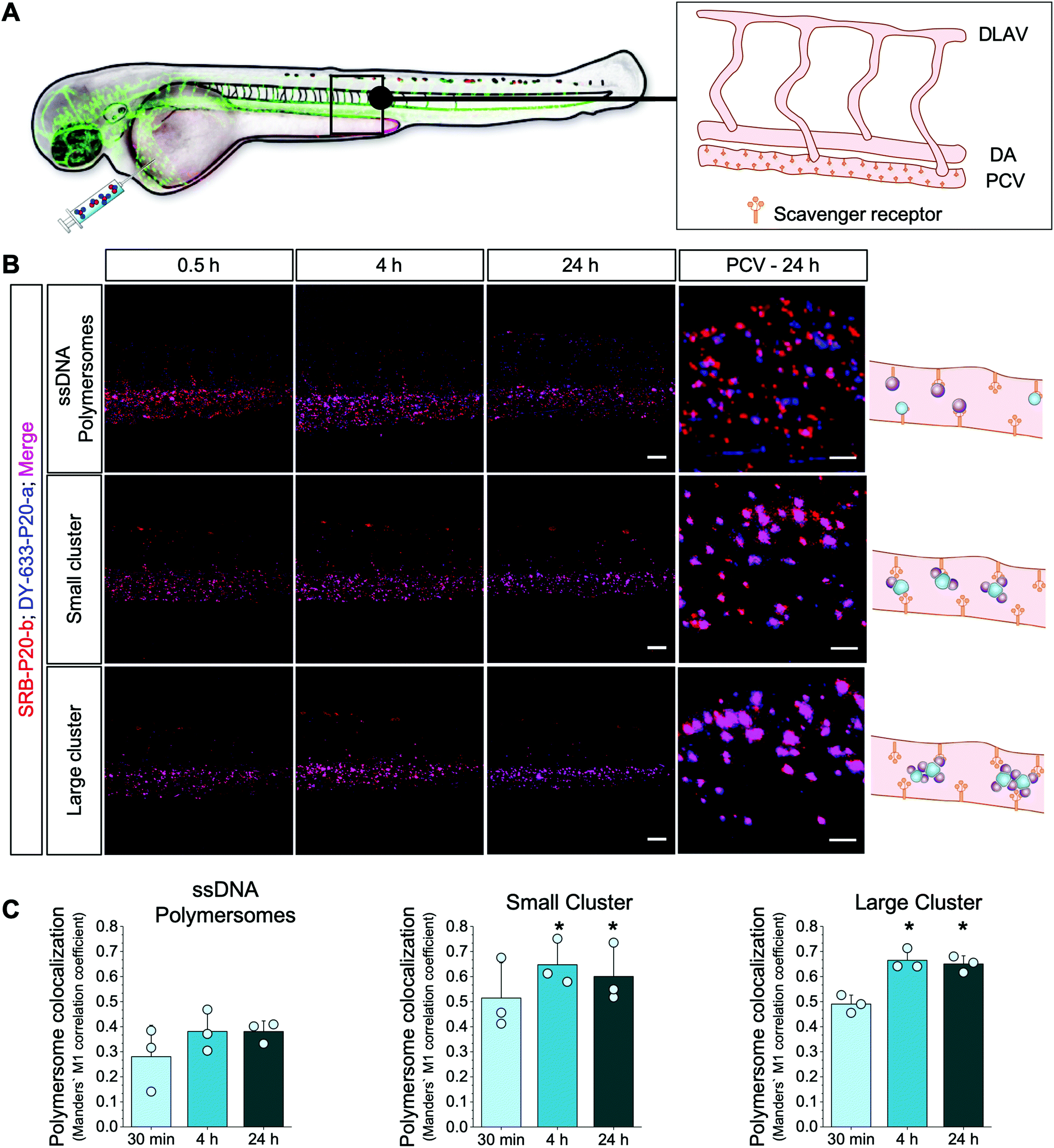 | ||
| Fig. 5 Biodistribution and stability of DNA-linked polymersome clusters in vivo. (A) Schematic representation of anatomical regions in the zebrafish model (DLAV = dorsal longitudinal anastomotic vessel, DA = dorsal aorta, PCV = posterior cardinal vein lined with scavenger receptors). Non-coupling ssDNA polymersomes (SRB-P20-a and Dy-633-P20-a), small clusters and large clusters were injected into transgenic zebrafish embryos. CLSM micrographs from the tail region were taken at specific time points after intravenous injection. Auto-fluorescence of pigment cells (bright red spots) are visible in the dorsal region. (B) Time-dependent analysis of biodistribution of non-coupling ssDNA polymersomes or preformed DNA-linked polymersome clusters (small clusters – 300 nm and big clusters – 500 nm) at 0.5, 4 and 24 hours (left). Scale bars = 50 μm. Schematic presentation and biodistribution analysis of non-coupling polymersomes, small clusters and large clusters in PCV. Representative high magnification images are shown (right). Scale bar = 10 μm. (C) Colocalization analysis of DY-633-P20-a and SRB-P20-b modified polymersomes, small clusters (average size 300 nm) and larger clusters (average size 500 nm) in vivo based on Manders’ M1 colocalization coefficient. Statistical analysis was performed by one-way ANOVA with a Bonferroni post hoc test. * p < 0.05 vs. non-coupling polymersomes at the same time point (means ± s.e.m., n = 3). | ||
To assess the in vivo stability of non-complementary ssDNA polymersomes, small and large clusters, we performed a colocalization analysis of DY-633-P20-a and SRB-P20-b bearing polymersome signals (Fig. 5C). In the case of non-complementary polymersome mixtures, both polymersome types showed a strong binding to the same scavenger receptor expressing regions, however with very low colocalization at all time points (30 min: 0.28 ± 0.07; 4 h: 0.38 ± 0.05; 24 h: 0.38 ± 0.02, mean ± s.e.m., n = 3). Thus, non-complementary polymersomes do not assemble into clusters in blood circulation but bind to the same target region. In sharp contrast, preformed polymersome clusters exhibited a significantly higher colocalization of DY-633-P20-a and SRB-P20-b signals at binding sites in vivo (Fig. 5B and C). Manders’ M1 colocalization coefficients 24 h post injection were 0.60 ± 0.07 and 0.65 ± 0.02 (mean ± s.e.m., n = 3) for small and large polymersome clusters, respectively. Thus, the multiple factors that influence the disassembly of DNA-linked nanomaterials such as serum proteins, enzymes or shear force did not affect the polymersome clusters’ integrity over time, confirming their stability even under in vivo conditions.
3. Conclusions
In summary, we have developed soft DNA-linked assemblies based on hierarchical organization of polymersomes that show specific interactions with cell surfaces with exceptional stability in vitro and in vivo thus strengthening our vision of developing them for biological applications. We demonstrated the ability to control the size of DNA-linked polymersome clusters by varying the ssDNA surface density. In vitro and in vivo behavior of the clusters are a direct result of the inherent deformability and membrane fluidity of the polymersomes, where a fraction of the ssDNAs can migrate to the protective bridging regions, while the remaining unbound ssDNA fraction can interact with scavenger receptors on the surface of epithelial cells. Finally, we also demonstrated that a medically relevant enzyme can be encapsulated within the DNA-polymersome clusters and maintains its activity. Polymersome clusters have the following advantages as compared to non-clustered or ss-DNA-polymersomes: (i) they allow segregated nano-spaces where different reactions can take place simultaneously, (ii) support co-localization of polymersomes inducing an increase in their overall functionality, (iii) tend to have better binding to the cell surface and (iv) accumulate more on the cell surface, thus supporting an improved functionality. In addition, polymersome clusters are physiochemically stable in the physiological environment and strongly attach to scavenger receptor-expressing cells thus making them ideal candidates to serve as nano-implants for sustainable and long-term therapies. The unique in vitro and in vivo properties and robustness propel this system towards the development of protective detoxification networks. More importantly, the global enzyme replacement treatment market is expected to grow 6.5% per year and generate more than $13.7 billion each year by 2028.55 Our new approach and results could be extended to the enzyme replacement market by replacing the encapsulated enzyme to protect and deliver a broad context of therapeutic enzymes to selective cells and complement cell functions in blood circuits.4. Materials and methods
See the ESI† for additional details. As a reporting standard has been developed to strengthen the quality and reproducibility of published research in the field of bio-nano science,56 we provide information of materials including synthesis and characterization, size distribution, shape, dimensions, aggregation, zeta potentials, density (in culture), enzyme loading efficiency, labelling efficiency, stability in biological fluids, as well as biological characterization including toxicity, 3D-mapping of materials on cell surfaces for selective targeting quantification, mechanism of selective targeting, material distribution in Zebra fish, and statistics for data analysis.4.1. Materials
Hydroxylethoxypropyl terminated polydimethylsiloxane was purchased from ABCR GmbH (AB 116675), Germany. The oligonucleotides with DBCO modification on the 5′-end and the ones with both DBCO modification on the 5′-end and ATTO 488/Cy5 modification on the 3′-end were purchased from IBA GmbH, Germany. DBCO-OEG4-ATTO 488 was purchased from Jena Bioscience, Germany. The fluorescent probes ATTO 488 and DY-633 were purchased from ATTO-TEC GmbH, Germany, and Dyomics GmbH, Germany, respectively. CHCl3 in HPLC-grade quality for GPC elution were obtained from Scharlab, Spain. All other chemicals were purchased from Sigma-Aldrich in the highest available grade and used as received if not stated otherwise.4.2. Block copolymers
PDMS62-PMOXA13 was synthesized according to the published procedure.57 PDMS62-PMOXA13-OH: 1H NMR (400 MHz, δ, CDCl3): 0 ppm (m, –Si(CH3)2), 0.54 ppm (m, –SiCH2), 0.88 ppm (t, –CH3), 1.31 ppm (m, –CH2–CH2–), 1.62 ppm (m, –SiCH2–CH2–CH2O–), 2.08–2.21 ppm (m, CH3–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 3.40–3.60 ppm (m, –CH2–O–CH2–CH2–N–CH2–CH2–), 3.75 ppm (t, –CH2OH).
O), 3.40–3.60 ppm (m, –CH2–O–CH2–CH2–N–CH2–CH2–), 3.75 ppm (t, –CH2OH).
For the synthesis of N3-functionalized polymer, PDMS65-PMOXA32 (400 mg, 54.0 μmol) was dissolved in 5 mL anhydrous CHCl3, following the addition of 14-azido-3,6,9,12-tetraoxatetradecanoic acid solution (0.5 M in tert-butyl methyl ether, 2.6 eq.), N,N′-dicyclohexylcarbodiimide (DCC, 2.6 eq.), triethylamine (5.2 eq.) and 4-dimethylaminopyridine (0.2 eq.) under Ar flow. The reaction flask was placed in an ice bath at 0 °C, warmed to room temperature gradually and reacted further for 2 days. The crude azide-functionalized diblock copolymers were purified by dialysis (MWCO 3.5 kDa) in H2O/EtOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 V/V). 1H NMR (400 MHz, CDCl3): 0 ppm (m, –Si(CH3)2), 0.54 ppm (m, –SiCH2), 0.88 ppm (t, –CH3), 1.31 ppm (m, –CH2–CH2–), 1.62 ppm (m, –SiCH2–CH2–CH2O–), 2.08–2.21 ppm (m, CH3–CO), 3.40–3.60 ppm (m, –CH2–O–CH2–CH2–N–CH2–), 3.65 ppm (s, –O–CH2–CH2–O–CH2–CH2–O–CH2–CH2–O–CH2–).
:6 V/V). 1H NMR (400 MHz, CDCl3): 0 ppm (m, –Si(CH3)2), 0.54 ppm (m, –SiCH2), 0.88 ppm (t, –CH3), 1.31 ppm (m, –CH2–CH2–), 1.62 ppm (m, –SiCH2–CH2–CH2O–), 2.08–2.21 ppm (m, CH3–CO), 3.40–3.60 ppm (m, –CH2–O–CH2–CH2–N–CH2–), 3.65 ppm (s, –O–CH2–CH2–O–CH2–CH2–O–CH2–CH2–O–CH2–).
4.3. ssDNA–polymersomes
To visualize the polymersomes by CLSM, 0.2 mM ATTO 488, 0.2 mM DY-633 or 0.2 mM SRB was dissolved in 1 mL PBS and used for the film rehydration instead of PBS to prepare the polymersomes P20. Dye-encapsulated polymersomes were extruded as mentioned above and purified from non-encapsulated fluorescent probes by size exclusion chromatography (SEC, Sepharose 4B column; 8 cm length). The fraction of individual non-encapsulated fluorescent probes was collected and the concentration was determined by measuring the maximal UV-vis absorbance. The maximal molar extinction coefficient (εmax) of ATTO 488 (λmax = 500 nm) DY-633 (λmax = 637 nm) and SRB (λmax = 550 nm) is 9 × 104, 17 × 104 and 11 × 104 M−1 cm−1, respectively. The encapsulation efficiency of ATTO 488, DY-633 and SRB for P20 is subsequently calculated as 10.4%, 24.7% and 18.7%, respectively (eqn (1)).
 | (1) |
4.4. DNA-linked polymersome clusters
DNase I digestion. 0.2 mg mL−1 of P1-ab, P5-ab, P10-ab and P20-ab polymersome clusters were diluted in PBS to reach a concentration of 0.025 mg mL−1, 0.05 mg mL−1 and 0.1 mg mL−1. The diluted polymersome cluster solutions were further treated with various concentrations of DNase I (0.1 μg mL−1, 1 μg mL−1 and 10 μg mL−1, final volume 400 μL) in presence of 5 mM of the cofactor MgCl2. All samples were incubated at 37 °C for 1 day followed by DLS measurements for the size analysis. For the control experiment, 0.1 mg mL−1 of P20-a and P20-b were pre-treated with 1 μg mL−1 of DNase I at 37 °C for 24 h, respectively, and mixed at 37 °C for another 24 h, following up with characterization of TEM. In order to understand the influence of DNA length on the nuclease resistance of polymersome clusters, polymersomes were linked with complementary 11 bp ssDNA, respectively, and purified as aforementioned. The sequences of 11 bp ssDNA are 5′-DBCO-CCT CGC TCT GC-ATTO 488-3′ and 5′-DBCO-GCA GAG CGA GG-ATTO 643N-3′, respectively. The DNA density on the polymersome’ membrane was carefully controlled in order to obtain the polymersome clusters with a size similar to P20-ab. 0.025 mg mL−1, 0.05 mg mL−1 and 0.1 mg mL−1 of polymersome clusters linked by 11 bp DNAs was treated with DNase I at 0.1 μg mL−1, 1 μg mL−1 and 10 μg mL−1 at 37 °C for 1 day, respectively. The samples were incubated followed by characterization as aforementioned for polymersome clusters.
4.5. In vitro studies of the behavior of the DNA-linked polymersome clusters
000 single cells for each sample were analyzed, and data processed using Flow Jo VX software (TreeStar, Ashland, OR).
 with intensity measured as arbitrary fluorescent units.
with intensity measured as arbitrary fluorescent units.
4.6. In vivo studies of the behavior of the DNA-linked polymersome clusters
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
We gratefully acknowledge the financial support provided by the Swiss National Science Foundation (SNSF), the National Centre of Competence in Research Molecular Systems Engineering and the University of Basel. DW is supported by an SNSF Early Postdoc Mobility Fellowship (#174975).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Prof. W. Meier (University of Basel) for useful discussions regarding the polymer synthesis and Prof. Dr M. Affolter (University of Basel) for supporting zebrafish breeding. CP thanks Dr H. Palivan for statistics analysis and discussions on the relevance of fitting models. R. M. Subasic and D. Hughes are acknowledged for useful discussions and reading the manuscript.References
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- R. Merindol, S. Loescher, A. Samanta and A. Walther, Nat. Nanotechnol., 2018, 13, 730–738 CrossRef CAS PubMed.
- N. Liu and T. Liedl, Chem. Rev., 2018, 118, 3032–3053 CrossRef CAS PubMed.
- B. Roark, J. A. Tan, A. Ivanina, M. Chandler, J. Castaneda, H. S. Kim, S. Jawahar, M. Viard, S. Talic, K. L. Wustholz, Y. G. Yingling, M. Jones and K. A. Afonin, ACS Sens., 2016, 1, 1295–1300 CrossRef CAS PubMed.
- V. Raeesi, L. Y. T. Chou and W. C. W. Chan, Adv. Mater., 2016, 28, 8511–8518 CrossRef CAS PubMed.
- N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- R. Schreiber, J. Do, E.-M. Roller, T. Zhang, V. J. Schüller, P. C. Nickels, J. Feldmann and T. Liedl, Nat. Nanotechnol., 2014, 9, 74–78 CrossRef CAS PubMed.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- M. G. Warner and J. E. Hutchison, Nat. Mater., 2003, 2, 272–277 CrossRef CAS PubMed.
- L. Y. T. Chou, K. Zagorovsky and W. C. W. Chan, Nat. Nanotechnol., 2014, 9, 148–155 CrossRef CAS PubMed.
- R. C. Mucic, J. J. Storhoff, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 12674–12675 CrossRef CAS.
- J. Liu, V. Postupalenko, S. Lörcher, D. Wu, M. Chami, W. Meier and C. G. Palivan, Nano Lett., 2016, 16, 7128–7136 CrossRef CAS PubMed.
- U. Jakobsen, A. C. Simonsen and S. Vogel, J. Am. Chem. Soc., 2008, 130, 10462–10463 CrossRef CAS PubMed.
- A. Czogalla, H. G. Franquelim and P. Schwille, Biophys. J., 2016, 110, 1698–1707 CrossRef CAS PubMed.
- P. A. Beales and T. K. Vanderlick, J. Phys. Chem. A, 2007, 111, 12372–12380 CrossRef CAS PubMed.
- N. Dave and J. Liu, ACS Nano, 2011, 5, 1304–1312 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS PubMed.
- M. Rasoulianboroujeni, G. Kupgan, F. Moghadam, M. Tahriri, A. Boughdachi, P. Khoshkenar, J. J. Ambrose, N. Kiaie, D. Vashaee, J. D. Ramsey and L. Tayebi, Mater. Sci. Eng., C, 2017, 75, 191–197 CrossRef CAS PubMed.
- Y. Xia, J. Tian and X. Chen, Biomaterials, 2016, 79, 56–68 CrossRef CAS PubMed.
- F. Wang, J. Gao, J. Xiao and J. Du, Nano Lett., 2018, 18, 5562–5568 CrossRef CAS PubMed.
- A. Wijaya and K. Hamad-Schifferli, Langmuir, 2007, 23, 9546–9550 CrossRef CAS PubMed.
- M. Spulber, P. Baumann, J. Liu and C. G. Palivan, Nanoscale, 2015, 7, 1411–1423 RSC.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- T. Einfalt, D. Witzigmann, C. Edlinger, S. Sieber, R. Goers, A. Najer, M. Spulber, O. Onaca-Fischer, J. Huwyler and C. G. Palivan, Nat. Commun., 2018, 9, 1127–1138 CrossRef CAS PubMed.
- H. Che and J. C. M. van Hest, J. Mater. Chem. B, 2016, 4, 4632–4647 RSC.
- G.-Y. Liu, C.-J. Chen and J. Ji, Soft Matter, 2012, 8, 8811–8821 RSC.
- J. Gaitzsch, X. Huang and B. Voit, Chem. Rev., 2016, 116, 1053–1093 CrossRef CAS PubMed.
- S. Ohta, D. Glancy and W. C. W. Chan, Science, 2016, 351, 841–845 CrossRef CAS PubMed.
- J. Kim, Y. M. Lee, Y. Kang and W. J. Kim, ACS Nano, 2014, 8, 9358–9367 CrossRef CAS PubMed.
- H. Li, X. Zhou, D. Yao and H. Liang, Chem. Commun., 2018, 54, 3520–3523 RSC.
- M. T. Stephan, J. J. Moon, S. H. Um, A. Bershteyn and D. J. Irvine, Nat. Med., 2010, 16, 1035–1041 CrossRef CAS PubMed.
- C. M. Csizmar, J. R. Petersburg and C. R. Wagner, Cell Chem. Biol., 2018, 25, 931–940 CrossRef CAS PubMed.
- D. M. Mate and M. Alcalde, Microb. Biotechnol., 2017, 10, 1457–1467 CrossRef CAS PubMed.
- A. D. Moreno, D. Ibarra, M. E. Eugenio and E. Tomás-Pejó, J. Chem. Technol. Biotechnol., 2020, 95, 481–494 CrossRef CAS.
- K. Kiene, S. H. Schenk, F. Porta, A. Ernst, D. Witzigmann, P. Grossen and J. Huwyler, Eur. J. Pharm. Biopharm., 2017, 119, 322–332 CrossRef CAS PubMed.
- A. Najer, D. Wu, A. Bieri, F. Brand, C. G. Palivan, H.-P. Beck and W. Meier, ACS Nano, 2014, 8, 12560–12571 CrossRef CAS PubMed.
- Q. Luo, Z. Shi, Y. Zhang, X.-J. Chen, S.-Y. Han, T. Baumgart, D. M. Chenoweth and S.-J. Park, J. Am. Chem. Soc., 2016, 138, 10157–10162 CrossRef CAS PubMed.
- C. H. J. Choi, L. Hao, S. P. Narayan, E. Auyeung and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7625–7630 CrossRef CAS PubMed.
- X. Yi and H. Gao, Soft Matter, 2015, 11, 1107–1115 RSC.
- X. Yi, X. Shi and H. Gao, Phys. Rev. Lett., 2011, 107, 098101–098105 CrossRef PubMed.
- J. Yue, T. J. Feliciano, W. Li, A. Lee and T. W. Odom, Bioconjugate Chem., 2017, 28, 1791–1800 CrossRef CAS PubMed.
- J. Yan, C. Hu, P. Wang, B. Zhao, X. Ouyang, J. Zhou, R. Liu, D. He, C. Fan and S. Song, Angew. Chem., Int. Ed., 2015, 54, 2431–2435 CrossRef CAS PubMed.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19, 4023–4033 CrossRef CAS PubMed.
- P. C. Patel, D. A. Giljohann, W. L. Daniel, D. Zheng, A. E. Prigodich and C. A. Mirkin, Bioconjugate Chem., 2010, 21, 2250–2256 CrossRef CAS PubMed.
- J. Yang, W. Li, T. B. Ng, X. Deng, J. Lin and X. Ye, Front. Microbiol., 2017, 8, 832–856 CrossRef PubMed.
- Z. Asadgol, H. Forootanfar, S. Rezaei, A. H. Mahvi and M. A. Faramarzi, J. Environ. Health Sci. Eng., 2014, 12, 93 CrossRef PubMed.
- J. M. Bollag, K. L. Shuttleworth and D. H. Anderson, Appl. Environ. Microbiol., 1988, 54, 3086–3091 CrossRef CAS PubMed.
- Shraddha, R. Shekher, S. Sehgal, M. Kamthania and A. Kumar, Enzyme Res., 2011, 2011, 217861 CAS.
- M. Smith, D. C. Love, C. M. Rochman and R. A. Neff, Curr. Environ. Health Rep., 2018, 5, 375–386 CrossRef CAS PubMed.
- L. Zhao, Q. Wu and A. Ma, IOP Conf. Ser. Earth Environ. Sci., 2018, 111, 012024 CrossRef.
- G. Macellaro, C. Pezzella, P. Cicatiello, G. Sannia and A. Piscitelli, BioMed Res. Int., 2014, 2014, 614038 Search PubMed.
- S. Sieber, P. Grossen, P. Detampel, S. Siegfried, D. Witzigmann and J. Huwyler, J. Controlled Release, 2017, 264, 180–191 CrossRef CAS PubMed.
- F. Campbell, F. L. Bos, S. Sieber, G. Arias-Alpizar, B. E. Koch, J. Huwyler, A. Kros and J. Bussmann, ACS Nano, 2018, 12, 2138–2150 CrossRef CAS PubMed.
- H. Tiwari and P. Mhaisekar, Enzyme Replacement Therapy Market - Global Industry Analysis, Size and Forecast, 2018 to 2028, https://www.futuremarketinsights.com/reports/enzyme-replacement-therapy-market, (accessed July 16, 2019).
- M. Faria, M. Björnmalm, K. J. Thurecht, S. J. Kent, R. G. Parton, M. Kavallaris, A. P. R. Johnston, J. J. Gooding, S. R. Corrie, B. J. Boyd, P. Thordarson, A. K. Whittaker, M. M. Stevens, C. A. Prestidge, C. J. H. Porter, W. J. Parak, T. P. Davis, E. J. Crampin and F. Caruso, Nat. Nanotechnol., 2018, 13, 777 CrossRef CAS PubMed.
- D. Wu, M. Spulber, F. Itel, M. Chami, T. Pfohl, C. G. Palivan and W. Meier, Macromolecules, 2014, 47, 5060–5069 CrossRef CAS.
- T. Wang, Y. Xiang, X. Liu, W. Chen and Y. Hu, Talanta, 2017, 162, 143–150 CrossRef CAS PubMed.
- S. Sieber, S. Siegrist, S. Schwarz, F. Porta, S. H. Schenk and J. Huwyler, Macromol. Biosci., 2017, 17, 1700015–1700024 CrossRef PubMed.
- B. M. Weinstein, D. L. Stemple, W. Driever and M. C. Fishman, Nat. Med., 1995, 1, 1143–1147 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr00361a |
| ‡ These authors contributed equally to this work. |
| § Present address: Chemistry Research Laboratory, Department of Chemistry, University of Oxford, 12 Mansfield Road, OX1 3TA, Oxford, UK. |
| ¶ Present address: Department of Biochemistry and Molecular Biology, University of British Columbia, 2350 Health Sciences Mall, Vancouver, British Columbia-V6T 1Z3, Canada. |
| This journal is © The Royal Society of Chemistry 2020 |