
Switchable reinforced streptavidin†
 ‡a
Steffen M.
Sedlak
‡a
and
‡a
Steffen M.
Sedlak
‡a
and
Abstract
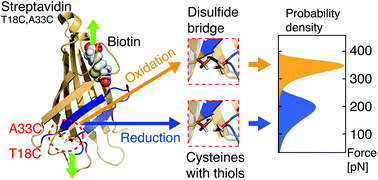
The complex of the small molecule biotin and the homotetrameric protein streptavidin is key to a broad range of biotechnological applications. Therefore, the behavior of this extraordinarily high-affinity interaction under mechanical force is intensively studied by single-molecule force spectroscopy. Recently, steered molecular dynamics simulations have identified a low force pathway for the dissociation of biotin from streptavidin, which involves partial unfolding of the N-terminal β-sheet structure of monovalent streptavidin's functional subunit. Based on these results, we now introduced two mutations (T18C,A33C) in the functional subunit of monovalent streptavidin to establish a switchable connection (disulfide bridge) between the first two β-strands to prevent this unfolding. In atomic force microscopy-based single-molecule force spectroscopy experiments, we observed unbinding forces of about 350 pN (at a force-loading rate of 10 nN s−1) for pulling a single biotin out of an N-terminally anchored monovalent streptavidin binding pocket – about 1.5-fold higher compared with what has been reported for N-terminal force loading of native monovalent streptavidin. Upon addition of a reducing agent, the unbinding forces dropped back to 200 pN, as the disulfide bridge was destroyed. Switching from reducing to oxidizing buffer conditions, the inverse effect was observed. Our work illustrates how the mechanics of a receptor–ligand system can be tuned by engineering the receptor protein far off the ligand-binding pocket.
- This article is part of the themed collection: Editor’s Choice: Recent breakthroughs in nanobiotechnology
 Please wait while we load your content...
Please wait while we load your content...

