
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Platinum nanoparticles stabilized by N-heterocyclic thiones. Synthesis and catalytic activity in mono- and di-hydroboration of alkynes†
Leonardo C.
Moraes
ab,
Rute C.
Figueiredo
ac,
Juan P.
Espinós
d,
Florencia
Vattier
d,
Antonio
Franconetti
 e,
Carlos
Jaime
e,
Bertrand
Lacroix
fg,
Javier
Rojo
a,
Patricia
Lara
*ab and
Salvador
Conejero
*ab
e,
Carlos
Jaime
e,
Bertrand
Lacroix
fg,
Javier
Rojo
a,
Patricia
Lara
*ab and
Salvador
Conejero
*ab
aInstituto de Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, C/Américo Vespucio 49, 41092, Seville, Spain. E-mail: sconejero@iiq.csic.es; patricia@iiq.csic.es
bCSIC and Universidad de Sevilla, Departamento de Química Inorgánica, Centro de Innovación en Química Avanzada (ORFEO-CINQA), Spain
cUniversidade Federal de Ouro Preto, Departamento de Química, Instituto de Ciências Exatas e Biológicas, Rua Costa Sena, 171, Centro, 35400-000, Ouro Preto, Minas Gerais, Brazil
dNanotechnology on Surfaces Laboratory, Instituto de Ciencia de Materiales de Sevilla (ICMS), CSIC – Universidad de Sevilla, c/Américo Vespucio 49, 41092 Sevilla, Spain
eDepartament de Química, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Spain
fDepartment of Materials Science and Metallurgical Engineering, and Inorganic Chemistry, University of Cádiz, Spain
gIMEYMAT, Institute of Research on Electron Microscopy and Materials of the University of Cádiz, Spain
First published on 17th March 2020
Abstract
N-Heterocyclic Thiones (NHT) proved to be efficient ligands for the stabilization of small platinum nanoparticles (1.3–1.7 nm), synthesized by decomposition of [Pt(dba)2], under a H2 atmosphere, in the presence of variable sub-stoichiometric amounts of the NHT. Full characterization by means of TEM, HR-TEM, NMR, ICP, TGA and XPS have been carried out, providing information about the nature of the metal nanoparticles and the interaction of the NHT ligands to the metal surface. Importantly, DFT calculations indicate that some NHT ligands interact with the metal through the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the imidazole fragment in addition to the sulfur atom, thus providing additional stabilization to the nanoparticles. According to XPS, TGA and ICP techniques, the surface coverage by the ligand increases by decreasing the size of the substituents on the nitrogen atom. The platinum nanoparticles have been used as catalyst in the hydroboration of alkynes. The most active system is that with a less covered surface area lacking an interaction of the ligand by means of the CC double bond. This catalyst hydroborates alkynes with excellent selectivities towards the monoborylated anti-Markovnikov product (vinyl-boronate) when one equiv. of borane is used. Very interestingly, aliphatic alkynes undergo a second hydroborylation process leading to the corresponding 1,1- and 1,2-diboroylated species with good selectivities towards the former.
C double bond of the imidazole fragment in addition to the sulfur atom, thus providing additional stabilization to the nanoparticles. According to XPS, TGA and ICP techniques, the surface coverage by the ligand increases by decreasing the size of the substituents on the nitrogen atom. The platinum nanoparticles have been used as catalyst in the hydroboration of alkynes. The most active system is that with a less covered surface area lacking an interaction of the ligand by means of the CC double bond. This catalyst hydroborates alkynes with excellent selectivities towards the monoborylated anti-Markovnikov product (vinyl-boronate) when one equiv. of borane is used. Very interestingly, aliphatic alkynes undergo a second hydroborylation process leading to the corresponding 1,1- and 1,2-diboroylated species with good selectivities towards the former.
Introduction
Metal nanoparticles (NPs) are nowadays well recognized materials with potential applications in different research areas spanning from electronic devices to catalysis.1 Most specifically, platinum nanoparticles have played a major role in important catalytic processes such as the oxygen reduction reaction,2 oxidation of CO3 or hydrosilylation4 to name a few. The small size of the metal nanoparticles improves its chemical reactivity or even their selectivity.5 The shape, size and ligand coating of the NPs dictate its catalytic behavior and thus most of the research groups have focused their attention on these parameters to control the activity and selectivity of the processes.6 With respect to the stabilizer of the metal nanoparticle, most of the ligands used for unsupported platinum systems are based on thiols,7 phosphines8 and amines,9 ligands that bind the metal strongly enough to prevent agglomeration processes. This is in stark contrast to the acquired knowledge that we have in molecular chemistry, for which a vast library of ligands have been used over the years to tune the properties of their corresponding platinum complexes. In recent years, some efforts have been made to transfer this knowledge to the realm of the metal nanoparticles, with some relevant illustrative examples in the use of the now prevalent N-heterocyclic carbenes (still under-developed in the field of metal nanoparticles),10 zwitterionic amidinates11 or other type of reactive (non-innocent) carbenes and vinylidenes.12 These ligands modify the properties of the surface of the metal nanoparticle both electronically and sterically, and thus open the door for controlling their stability and catalytic performance. Very recently, we have succeeded in introducing N-heterocyclic thiones (NHTs) as ligand stabilizers for gold NPs.13 This success was due, in part, to the presence of a sulfur atom on the ligand that, beside forming a strong interaction with the gold surface, can adopt a resonant form with a negative charge on it that confers good coordinating properties to the NHT and a thiolate-like behavior (Fig. 1). In addition, some of the advantages of these types of ligands are found on the simplicity of their synthesis, the easiness of tuning their steric environment and their tolerance to air and protic solvents, which facilitates considerably their handling or their use during the synthesis of the metal NPs.13 Platinum has also an excellent affinity to sulfur based ligands,7c–e with the paradigmatic example on the thiolate based platinum nanoparticles,7 for which extensive utilization has been reported. Therefore, we have considered that NHTs could also be excellent ligand stabilizers for platinum nanoparticles. Moreover, non-supported platinum nanoparticles have been barely used in hydroboration reaction of alkynes,10c,14 a process that lead to the formation of C–B bonds, which are starting materials for a myriad of organic transformations.15 There have been some reports on the use of metal nanoparticles for the hydroboration of alkynes. However, the boron source for such transformations are based on diboron reagents, which usually requires the use of external additives and have the inconvenient of losing half of the boron source, and consequently the generation of large amounts of waste.16 In this sense, the search for catalytic systems that can incorporate into an unsaturated organic fragment (alkynes) two boryl units through diborylation reactions is gaining relevance, since such organoboron compounds can be further poly-functionalized. Moreover, diborylation processes have been considerably less explored, although some important contributions have recently appeared in the literature by using homogeneous catalysts.17 The large surface area found in metal (platinum) nanoparticles might be relevant to achieve a sufficient active catalyst able to diborylate alkynes. | ||
| Fig. 1 Top: Resonant structure of N-heterocyclic thiones (NHTs). Bottom: N-Heterocyclic thiones used in this work. | ||
In this contribution, we wish to report the synthesis of well-defined, small platinum nanoparticles stabilized by N-heterocyclic thiones, their characterization by different methods (including TEM, HR-TEM, XPS, NMR, ICP and TGA) and their use for the hydroboration of alkynes with hydroboranes. Additionally, DFT calculations have provided some clues about the coordination mode of the NHT to the surface of the NPs. We have found that some of these platinum nanoparticles are very efficient in the hydroboration of alkynes, leading to either mono- or diborylated species.
Results and discussion
Synthesis and characterization of platinum-NHT nanoparticles
We have considered the use of different types of N-heterocyclic thiones with different substituents on the nitrogen atom, either aromatic or alkylic, the latter with different lengths to improve the stability of the platinum nanoparticle (see Fig. 1). We have used the organometallic method developed by Chaudret et al. for the synthesis of the metal nanoparticles,18 using as starting material the Pt(0) complex bis(dibenzylideneacetone)platinum, [Pt(dba)2], in the presence of variable amounts of the NHT ligands under a H2 atmosphere (see ESI;†Scheme 1). Employing this methodology, we can get well-controlled metallic nanostructures in terms of shape, size dispersion, organization and surface state. The amount of ligand employed as stabilizer plays a key role in the characteristics of the nanostructures. Particularly, in the organometallic approach, a bottom-up method, individual Pt nuclei are present in the reaction media. In order to avoid the formation of traditional organometallic complexes, sub stoichiometric amounts of stabilizing ligands (typically between 0.2 to 0.5 equiv.) must be employed (see below). Purification of the Pt NPs can be easily achieved through successive washing with pentane at low temperature (see ESI† for details). Under these reaction conditions, well defined platinum NPs can be obtained depending on the structure and the amount of the NHT present during decomposition of the platinum starting material. Thus, methyl substituents at the nitrogen atom of the NHT (NHTMe) don't seem to be sufficiently bulky to form stable Pt NPs, neither using 0.2 nor 0.5 equiv. of this ligand per [Pt(dba)2]. However, increasing the steric bulk by changing the methyl groups by iso-propyl fragments has a great impact on the formation of well-defined platinum NPs. Nevertheless, the amount of ligand present during decomposition of the platinum metal complex is very important to avoid the formation of agglomerates. Thus, 0.5 equiv. of NHTiPr are at least needed to produce Pt NPs of mean-size 1.5(0.2) nm (Fig. 2) with a narrow distribution, whereas 0.2 equiv. lead to large amounts of agglomerates according to TEM images (see ESI, Fig. S16†). Similar behavior has been observed for ligands NHTC6, NHTC14, and the aryl substituted NHTMes for which 0.5 equiv. are required to generate stable, well dispersed Pt NPs (NP mean-size 1.3(0.2)–1.5(0.2) nm, Fig. 3), with smaller amounts leading to platinum black. Increasing the length of the alkylic chain such as in NHTC18 is beneficial, since the decomposition process can be successfully achieved either with 0.2 and 0.5 equiv. of this ligand. | ||
| Scheme 1 Reaction conditions for the synthesis of the platinum nanoparticles. | ||
 | ||
| Fig. 2 TEM image of platinum nanoparticles PtNP-NHTiPr synthesized from Pt(dba)2 and 0.5 equiv. of NHTiPr. | ||
 | ||
| Fig. 3 TEM images and size distribution of platinum NPs: (a) PtNP- NHTC6,(b) PtNP- NHTC14,(c) PtNP-NHTMes and (d) PtNP-NHTC18. | ||
Finally, the very bulky NHTIPr is not able to stabilize the nanoparticles at any concentration of the ligand. As will be discussed below, it is likely that the bulkiness of this ligand precludes an efficient coordination to the metal surface making this capping ligand inefficient towards stabilization of the metal nanoparticles.
These Pt colloids are rather soluble in organic solvents such as THF and benzene, allowing recording their 1H NMR spectra. As expected from previous results on gold-NHT NPs,13 the very broad signals showed in these spectra are consistent with the presence of platinum nanoparticles stabilized with organic ligands (see ESI, Fig. S2, S5, S8, S11, S14†).19 The broadening of the signals is even more important when the proton atoms are closer to the surface of the metal nanoparticle, to the point that, in some cases, the signals can be lost in the base line. This seem to be the case for most of the colloids, for which no signals due to the back-bone of the imidazolyl fragment or the N–CH2 are clearly discernible in their proton NMR spectra. This is indicating that such fragments are likely in very close proximity to the surface of the metal nanoparticle (see below).
High Resolution Transmission Electron Microscopy (HR-TEM) provides evidence for the crystallinity of the platinum NPs, with interplanar distances characteristic of (111) and (002) planes in the face-centered cubic structure (Fig. 4(a)–(d)). The EDX spectra recorded across various NPs of sample PtNHTC6 using the scanning TEM (STEM) mode shows unequivocally three peaks at 2.1, 9.4 and 11.1 keV characteristic of platinum (besides the corresponding peaks of copper, carbon and Si arising from the grid and the specimen holder) (see Fig. 4(e)). The metal content was analyzed by Inductive Coupled Plasma (ICP) showing and array of values going from 31.65% (NHTC18) to 68.66% (NHTMes) fitting very well with the values obtained for the content of organic material by TGA (see ESI† for details).
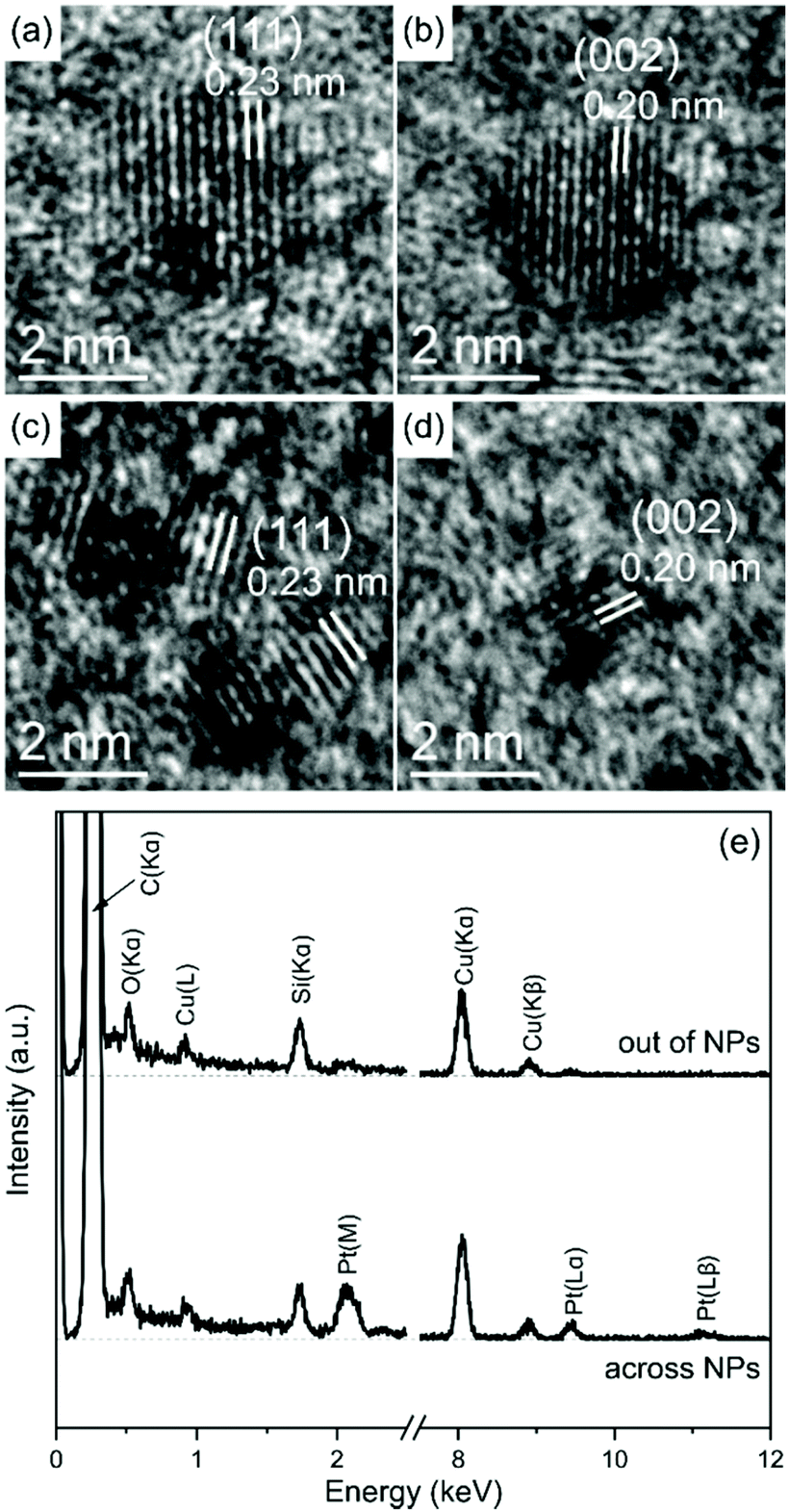 | ||
| Fig. 4 HR-TEM images of platinum NPs: (a) and (b) PtNP-NHTMes, (c) and (d) PtNP-NHTC6. (e) EDX spectra recorded across various NPs and across the support only (for reference). | ||
X-ray Photoelectron Spectroscopy (XPS) provided evidence for the coordination of the NHT to the surface of the platinum nanoparticle. In all the cases, the intensities (signal areas) of the signals in the spectra have been normalized by the relative intensity of the N 1s peak. The spectral regions of the Pt 4f, N 1s and S 2p peaks are depicted in Fig. 5.
 | ||
| Fig. 5 Detailed X-ray photoelectron spectral regions for the main signals of the present elements in the four studied samples: Pt 4f, N 1s and S 2p. | ||
The N 1s spectra for the four samples consist of a single, very symmetrical and narrow signal (fwhm = 1.7–1.9 eV) which indicates that a single chemical nitrogenated species are present in all the cases. The Pt 4f photoemission signal is a doublet, consisting of two energetic levels, Pt 4f7/2 and Pt 4f5/2, which are separated by 3.34 eV by electron spin–orbit interactions, with an intensity ratio of 0.75. For all the samples, the binding energy of the main peak, Pt 4f7/2, is in the range 71.3–70.9 eV, which are close to the binding energy of bulky metallic platinum (71.2 eV), and far away from the binding energy for Pt(II) or Pt(IV) compounds (in the range 73.6–76.3 eV).20 In this sense, it must be noted that the electronic structure of nanometric particles can be strongly affected by their particle size and by the interaction with the environment. In particular, the Pt 4f7/2 binding energy for nanometric Pt particles increases when the particle size decreases when supported on “inert” substrates (for instance, up to 0.75 eV when supported on graphite).21 Consequently, it can be assumed that platinum is present in metallic state for all the samples, while the small differences in their binding energy (≤0.46 eV) values could be caused by differences in the mean particle sizes or by interaction with the particular organic ligand. In the S 2p spectral region, an asymmetric signal is always found, consisting of a main peak at ∼162.2 ± 0.2 eV with a long tail at the high binding energy side. The intensity, width and shape of this tail slightly differing among the samples (see discussion below).
The relative intensities of the signals provide us with additional pieces of information concerning the surface composition of the samples (Table 1). Importantly, the intensities (N/S atomic ratios) of the global S 2p spectra are quite similar for all samples.
| Pt4f | N | S | N/S ratio | Pt/S ratio | Pt/N ratio | |
|---|---|---|---|---|---|---|
| %At | %At | %At | ||||
| PtNP-NHTiPr | 6.55 | 6.41 | 3.43 | 1.87 | 1.91 | 1.02 |
| PtNP-NHTMes | 9.39 | 4.73 | 2.55 | 1.85 | 3.68 | 1.99 |
| PtNP-NHTC6 | 7.82 | 4.94 | 2.50 | 1.98 | 3.13 | 1.58 |
| PtN-PNHTC14 | 3.80 | 3.20 | 1.58 | 2.03 | 2.41 | 1.19 |
A remarkable feature is that the N/S ratios are close to 2, consistent with the composition of the NHT ligands. Additionally, the relative values of the Pt/N ratios range from 1.02 to 1.99. These differences might be related to two factors: the adsorption of the organic molecule and the mean size of the platinum nanoparticles. With respect to the former, higher coverage degree of the surface and higher packing density of the adsorbate molecules, lead to lower Pt/N ratio. Concerning the particle size, for Pt particles with diameters lower than 3 nm, (which is the Pt 4f photoelectrons analysis depth),22 the lower the mean size, the lower the Pt/N ratio (the Pt/N ratio will not be affected for particles diameters larger than 3 nm). Consequently, a lower Pt/N ratio could indicate a higher surface coverage of the Pt particles by the adsorbate or a smaller Pt particle diameter. However, since the mean sizes of the Pt nanoparticles, as determined by TEM, is in a very close range for all the studied samples, between 1.3 and 1.7 nm, it is reasonable to assume that the Pt/N atomic ratio is mainly determined by the adsorption coverage. Therefore, the Pt-NHTiPr are highly covered by the ligand (Pt/N ratio = 1.02) whereas Pt-NHTMes are less covered, in agreement with the higher steric hindrance exerted by the mesityl groups in comparison to the iso-propyl fragments. Nevertheless, the case for platinum nanoparticles PtNP-NHTC6 and PtNP-NHTC14 might be, in principle, counterintuitive. The longer length of the C14 side-chains should give rise to a more sterically hindered environment compared to the C6 alkyl chains. However, the Pt/N ratio observed (Table 1) suggest that the platinum nanoparticles have a higher content of NHTC14 ligands than the NHTC6. It is not entirely understood the reasons underlying to this effect, but one possible explanation could be that the NHTC14 ligands are more packed around the surface as a consequence of lateral packing interactions of the alkyl chains, whereas the alkyl arms in NHTC6 are less ordered, spreading randomly, leading to a less covered surface.23 In fact, according to theoretical calculations the interaction of two C14 chains is favored by ca. 5 kcal mol−1 (ΔEbind = −3.3 vs. −8.1 kcal mol−1 for C6/C6 and C14/C14, respectively), supporting a preference for a more ordered structure.
The S 2p signals have been deconvoluted and fitted with three components (three doublets as shown in Fig. 6; see Experimental section for details). The energy position and the width of the main component is well defined for the four samples; the peak S 2p3/2 is located at 161.8 ± 0.3 eV and its width is relatively low (fwhm = 1.9 ± 0.1 eV). However, the energy positions and widths of the second and third component are poorly defined due to instrumental noise: the peak S 2p3/2 is located at 164.3 ± 0.5 eV and at 166.9 ± 0.5 eV for the second and third component, respectively, while their widths are around 20% wider (fwhm = 2.25 ± 0.15 eV) than that of the main component. Nonetheless, the proportions of the three components vary slightly from one sample to another. The minor component, with a proportion around 10% (with a high binding energy), is very likely related to some that oxidized sulfur species (sulphates, sulphites, …), and it is a regular by-product in the study of adsorbed thiols on metal surfaces. It is worth comparing energy positions of free and coordinated thiones to that observed in the platinum nanoparticles reported herein. The S 2p signal from a free thiourea is located at ∼162.4 ± 0.1 eV, while coordination to metal cations (such as Co(II), Ni(II), Cu(I), Re(IV)) through its sulfur atom leads to small positive S 2p chemical shifts (between +0.3 and +0.6 eV).24 On the contrary, small negative chemical shifts can be expected for coordination of thiourea molecules to metal particles as previously reported on thiourea absorbed on polycrystalline platinum.25 Consequently, the main component in the S 2p spectra observed for PtNPs-NHT is due to chemisorbed molecules to platinum atoms.
 | ||
| Fig. 6 Experimental and fitted S 2p X-ray photoelectron spectral region for the four studied samples PtNp-BHTC6, PtNP-NHTiPr, PtNP-NHTMes and PtNP-NHTC14. | ||
Theoretical calculations: DFT and DPD
In order to understand the role of NHT on the stabilization of PtNPs, theoretical calculations have been carried out using two approaches. Firstly, DFT calculations have been applied at the CAM-B3LYP-D3(BJ)/LanL2DZ level of theory to study the feasible coordination modes of different NHTs to PtNPs. In this context, the analysis of molecular electrostatic potential (MEP) surface of Pt10 subnanocluster reveals positive (+26 kcal mol−1) and negative (−12 kcal mol−1) regions on the surface highlighting the donor–acceptor ability of Pt nanoparticles thus enabling interactions with different moieties (Fig. 7). Expectedly, the most negative electrostatic potential is found for the thione (ca. −30 kcal mol−1) moiety whereas the positive electrostatic potential (ca. +11 kcal mol−1) is located in the CC bond region. Therefore, both functional groups should have the capability to interact favourably with the PtNP surface. In fact, Pt-NHTiPr and Pt-NHTC5 (as a model for a lineal alkylic NHT) exhibit large adsorption energies (ΔEads of −132.5 and −130.2 kcal mol−1, respectively) showing thione (RS–Pt = 2.42 Å) and CC (RC–Pt = 2.12 Å) groups chemisorbed onto the surface (Fig. 7). Even though the complexation of NHTMes and NHTiPr are also equally favoured due to the presence of several Pt⋯H–C and Pt⋯π non-covalent interactions,26 a different geometrical situation arises. NHTMes stabilizes Pt nanoparticles through chemisorption of the thione moiety whereas the CC bond of the imidazolyl fragment is pointing outward far away from the Pt10 cluster. However, we roughly estimate a weaker coordination based on the directionality of Pt–S bond (φCS–Pt = 116° vs. 96° in NHTC5). This situation ends up with loss of complexation observed for NHTiPr (RS–Pt = 2.44 Å, φCS–Pt = 117°) in agreement with experimental results about the stabilization of Pt nanoparticles with this NHT. At this point, we have used Dissipative Particles Dynamics (DPD) simulations as second approach to get further insight about the stabilization of Pt nanoparticles in THF by means of NHT ligands. Since we have correlated soft-repulsive parameters with DFT-based binding energies to build our coarse grained model (Fig. S18, ESI†),27 this second approach is an efficient way to transfer the atomistic features to mesoscopic simulations. In fact, this kind of calculations encompasses different phenomena such as cooperativity and competitiveness between ligands as well as aggregation and stability over the time (temperature fixed at 23.5 °C) in a periodic environment.
 | ||
| Fig. 7 (a) MEP of Pt nanoparticles plotted onto the 0.001 a.u. isodensity surface; (b) optimized structures for the interaction between Pt10 cluster and different NHT ligands at CAM-B3LYP-D3(BJ)/LanL2DZ level. Energies are given in kcal mol−1 and distances in Å. | ||
Regarding the model, we have considered the stabilization of one Pt nanoparticle (formed by 100 beads) and lineal NHTC14 (50 molecules, 12 beads each one). In addition, Pt nanoparticle has a computed diameter of 1.6 nm in the range of experimental findings. After the simulation time (900 ns), PtNP is surrounded by ca. 20 NHT molecules (Fig. 8a). Radial distribution function (RDF) analysis reveals that both S and N/CC moieties are located on the Pt surface although there is a subtle but clear preference for S beads, and therefore, thione groups. Nevertheless, the calculated distance from mPtNP is around 0.2 nm, in agreement with RS–Pt (and RC–Pt) distance aforementioned in DFT calculations. An analogous simulation has been carried out to study the stabilization of one PtNP by hindered NHTMes (50 molecules, 10 beads each one). Interestingly, this system is stabilized much earlier than NHTC14 along the simulation time (125 vs. 450 ns, respectively). Despite being stabilized faster, the nanoparticle surface is covered by only 10% of NHTMes molecules (Fig. 8b). In this case, the outcomes of the analysis of RD functions provide differences between distances of S and N/CC beads. In fact, the S beads are on the Pt NP surface while beads for mesityl groups are further away. In addition, the calculated distances of N/CC beads from mPtNP is even further away (around 1.07 nm; 0.27 nm from the surface) as expected for hindered NHTMes molecules.
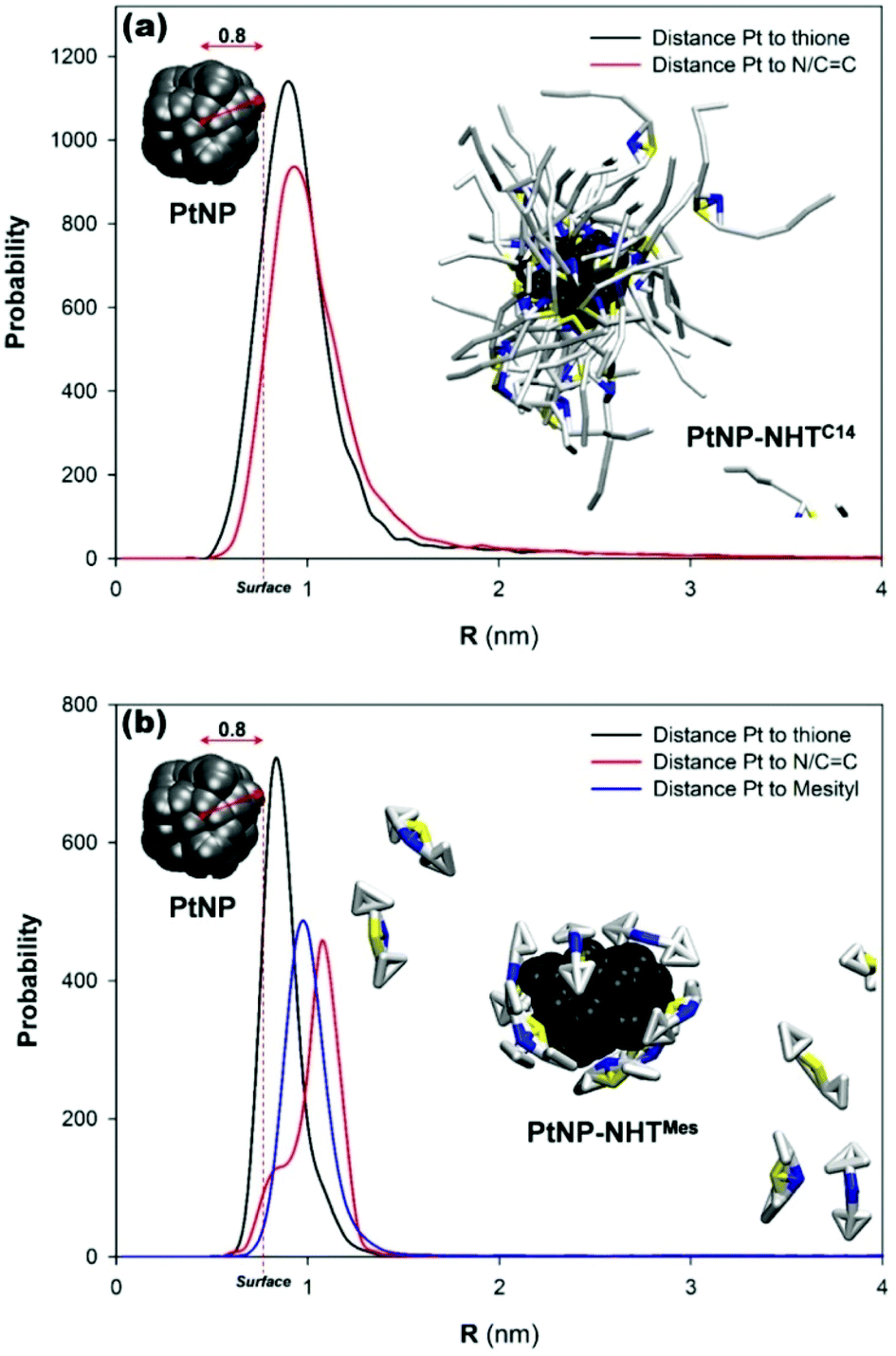 | ||
| Fig. 8 (a) Radial distribution function for the distance between the center of mass and different functional groups of NHTC14. Results from the simulations of one stabilized PtNP at 900 ns is also shown; (b) radial distribution function for the distance between the center of mass and different functional groups of NHTMes and final snapshot of the stabilization of one PtNP taken at 900 ns. Color codes: Pt in black, carbon in gray, sulfur in yellow, and the CC/N beads in blue; THF beads have been removed for clarity. | ||
Catalytic activity in hydroboration of alkynes
With the well-defined Pt nanoparticles in hand, we have tested their catalytic activity in the hydroboration of alkynes. First, we have used as a model reaction the hydroboration of phenylacetylene with a slight excess of pinacolborane,28 using 5% of the PtNP colloid in benzene-d6 at 80 °C (Scheme 2). The platinum colloids PtNP-NHTiPr, PtNP-NHTMes, PtNP-NHTC6, PtNP-NHTC14 and PtNP-NHTC18 have been evaluated in the process, and the results are collected in Table 2. | ||
| Scheme 2 Hydroboration of phenylacetylene catalyzed by different PtNp-NHTX colloids. | ||
| Entry | Catalyst | Time (h) | Conversion (%) | Selectivity (2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a:4a:5a) :3a:4a:5a) |
|---|---|---|---|---|
| a Reaction conditions: 0.35 mmol of phenylacetylene (1 M in C6D6), 0.36 mmol of HBpin, 5% PtNP-NHTX, 80 °C. b 20% of styrene is formed. c 5% of styrene is formed. d 15% of styrene is formed. e 14% of styrene is formed. | ||||
| 1 | PtNP-NHTiPr | 15 | 20 | 50:10:20:0b |
| 2 | PtNP-NHTMes | 4 | >99 | 79:6:5:5c |
| 3 | PtNP-NHTC6 | 19 | 34 | 72:2:11:<1d |
| 4 | PtNP-NHTC14 | 20 | 44 | 66:3:10:7e |
| 5 | PtNP-NHTC18 | 20 | <5% | –c |
In all cases, both the mono- and di-borylated species 2a–5a have been detected in variable amounts. However, the best system in terms of conversion, selectivity and time appeared to be the colloid PtNP-NHTMes, for which full conversion is achieved in only 4 h with an excellent selectivity towards the monoborylated, anti-Markovnikov trans-isomer 2a (Table 2, entry 2). On the other hand colloids, PtNP-NHTiPr, PtNP-NHTC6 and PtNP-NHTC14 exhibited a rather similar behaviour, but conversions below 45% are observed after 15–20 h (Table 2, entries 1, 3 and 4). The platinum nanoparticles, PtNP-NHTC18 did not show any catalytic activity. A possible explanation for the different performance of the alkyl-substituted NHT colloids compared to the very active PtNP-NHTMes might be related to a different coordination of the NHT to the surface of the platinum nanoparticle. As described above, the N-heterocyclic thiones substituted with alkyl chains seems to bind the surface using both the sulfur atom and the CC double bond, with a flat disposition of the heterocyclic ring towards the metal surface. However, the approach of the CC double bond of the mesityl derivative NHTMes is hampered by the aromatic rings, leading to coordination of the NHT exclusively through the S atom. This bonding situation might have two possible effects; first, the metal surface is more exposed to the incoming substrates, and second the NHT ligand is less effectively anchored to the surface facilitating its detachment to undergo exchange with the substrates. These effects together with the fact that, according to XPS (see above), the colloid PtNP-NHTMes is less covered by ligands, are likely responsible for the enhanced catalytic activity of PtNP-NHTMes in comparison to the other nanoparticles due to the presence of more active sites.
At this point it's worth mentioning that the morphology and size of the metal nanoparticles are retained after the catalytic process, as shown in the TEM image of Fig. 9 obtained from the content of the crude reaction mixture solutions after hydroboration of phenylacetylene with pinacolborane. This analysis revealed the presence of individual nanoparticles with a similar mean size than the starting material (1.7(0.3) nm; Fig. 9).
 | ||
| Fig. 9 TEM images of the colloid PtNP-NHTMes after a catalytic reaction. | ||
Once established the most efficient catalytic system, several reaction conditions were tested. The solvent, temperature and catalyst loadings were modified and the results are summarized in Table 3. The catalyst loading has an influence on the rate of the reaction but only marginally on the selectivity. On the other hand, lower catalyst loadings (1 and 2%, entries 5–8) lead to good selectivities towards the monoborylated product 2a, but slightly longer times are required to achieve full conversion. In this sense an increase of the reaction temperature to 80 °C is clearly beneficial, reducing about 2 h the reaction time (entries 5–7 vs. 6–8), and increasing slightly the monoborylated species 2a to levels even higher than those values found using 5% of catalyst loadings at the same temperature (see Table 2, entry 2). As will be discussed below, the better selectivity observed under lower catalyst loadings is likely related to changes in the active catalyst species during the course of the reaction.
| Entry | Catalyst loading (%) | Solv. | T (°C) | Time (h) | Conv. (%) | Selectivity (2a:3a:4a:5a)a |
|---|---|---|---|---|---|---|
| a In all cases minor amounts of styrene (less than 2%) was observed. | ||||||
| 1 | 5.0 | C6D6 | 60 | 12 | >99 | 79:2:2:16 |
| 2 | 5.0 | C6D6 | 70 | 4 | >99 | 76:3:4:11 |
| 3 | 3.0 | C6D6 | 60 | 12 | >99 | 84:2:2:10 |
| 4 | 3.0 | C6D6 | 70 | 4 | >99 | 87:2:2:8 |
| 5 | 2.0 | C6D6 | 70 | 6 | >99 | 86:2:2:8 |
| 6 | 2.0 | C6D6 | 80 | 4 | >99 | 88:3:1:6 |
| 7 | 1.0 | C6D6 | 70 | 8 | >99 | 87:1:1:10 |
| 8 | 1.0 | C6D6 | 80 | 6.5 | >99 | 88:2:1:7 |
| 9 | 1.0 | THF d8 | 80 | 15 | 79 | 89:2:0:6 |
| 10 | 1.0 | CD3CN | 80 | 24 | >99 | 83:<1:<1:16 |
The effect of the solvent has been also explored with tetrahydrofurane-d8 or acetonitrile-d3 (Table 3, entries 9 and 10).
However, the results were in all cases poorer in terms of activity, selectivity or rate. Therefore, coordinating solvents such as THF or acetonitrile, inhibit to some extent the catalytic process according to their ligand binding properties.
The scope of the reaction has been explored by using, initially, terminal and internal aromatic alkynes bearing electron-donor and withdrawing groups under the optimized reaction conditions (1% cat., 80 °C, benzene-d6). The conversion and selectivity has been determined by means of NMR and/or GC-MS. The monoborylated products (E isomers in the case of terminal alkynes and Z for internal) have been obtained in all cases as the major product in reaction times going from 4 to 60 h (Table 4). The presence of electron–donor or withdrawing groups has an effect on the reaction rate but not significantly on the selectivity (small amounts of diborylated species were detected). In all cases the major product is the one arising from a syn addition of the borane in an anti-Markovnikov manner. The yield of products formed through the anti- or the Markovnikov additions is, typically, below 7%.
a
| a Yields are given for the inseparable mixture of compounds 2–5. |
|---|

|
The reaction using electron donor groups in the para position of the aromatic ring took place considerably faster than with electron-withdrawing fragments (Table 4, alkynes 1b,cvs.1d,e). Nevertheless, the effect of groups in the ortho position was very diverse and difficult to rationalize. For example, the presence of F and CF3 groups in the ortho position (Table 4, alkynes 1h,i) decreases and increases the rate of the reaction, respectively, in comparison with their para substituted counterparts (alkynes 1d,e). With respect to the selectivity of the process, the ortho substituents slightly favour the amount of the monoborylated products. The borylation process has been extended to both aromatic and aliphatic internal alkynes. Besides obtaining full conversion in all cases, the reactions were considerably faster than those of the terminal alkynes, taking place in as short as 1 h (Table 4, alkynes 1j–l). Although excellent selectivities are obtained for diphenylacetylene and 3-hexyne, the asymmetrical 1-phenyl-1-propyne generates mixtures of isomers 2l and 2l′ in a 1:3.7 ratio together with a poorer selectivity with respect to the diborylated isomer (Scheme 3).
 | ||
| Scheme 3 Hydroboration of 1-phenyl-1-propyne catalyzed by PtNP-NHTMes. | ||
At this stage, it is important to remark that the amount of the diborylated species formed in the process, generated in principle from the monoborylated derivatives, is not increased when an excess (2 or more equiv.) of pinacolborane is used, even after prolonged periods of heating (see Table S3 in ESI†). Likewise, increasing the catalyst loading or the temperature of the reaction has nearly no effect in the mono-/diborylated ratio.
As for aromatic systems, terminal aliphatic alkynes can be efficiently hydroborated, but as inferred from the data shown in Table 5, the selectivity of the process is slightly poorer, in which the diborylated species are produced in amounts ranging from 4 to 13% of the total amount. In fact, at variance to the results obtained for aromatic alkynes, when the reaction is carried out with an excess (3 equiv.) of pinacolborane, full conversion to the diborylated derivatives is produced, generated as mixtures of the 1,2 and 1,1dihydroboration reaction (Scheme 4 and Table 6). The ratio of these isomers depends strongly on the bulkiness of the alkyl moiety at the 3 position of the alkyne. Thus, the less hindered systems (Table 6, alkynes 1p–r) lead to an approximately 2:1 ratio in favour to the 1,1 disubstituted alkylborane, whereas the presence of a bulkier group such as cyclopropyl, cyclohexyl or iso-propyl, increase the selectivity up to 100% (alkynes 1m–o). Interestingly, under these reaction conditions (excess of borane) no 1,2-diborylated species is formed for the cyclopropylacetylene 1m, in stark contrast with the reaction of this alkyne with 1.2 equiv. of borane, a reaction that produces small amounts of the 1,2-diborylated derivative 5m with no signs of 6m being detected. We do not have an explanation to this contradictory result, but a possible explanation might be found in a different active surface catalyst, when the reaction is carried out in the presence of an excess of pinacolborane, or in a different active catalyst that is only present at early stages of the reaction. This latter assumption might also explain the formation of small amounts of 1,2-diborylated species in the reaction of aromatic alkynes with pinacolborane that, as stated above, do not increase with either time or in the presence of an excess of the borane.
 | ||
| Scheme 4 Dihydroboration of terminal alkyl-alkynes with an excess of pinacolborane. | ||
a
| a Yields are given for the inseparable mixture of compounds 2–5. |
|---|

|
a
| a Yields are given for the inseparable mixture of compounds 5 and 6. |
|---|
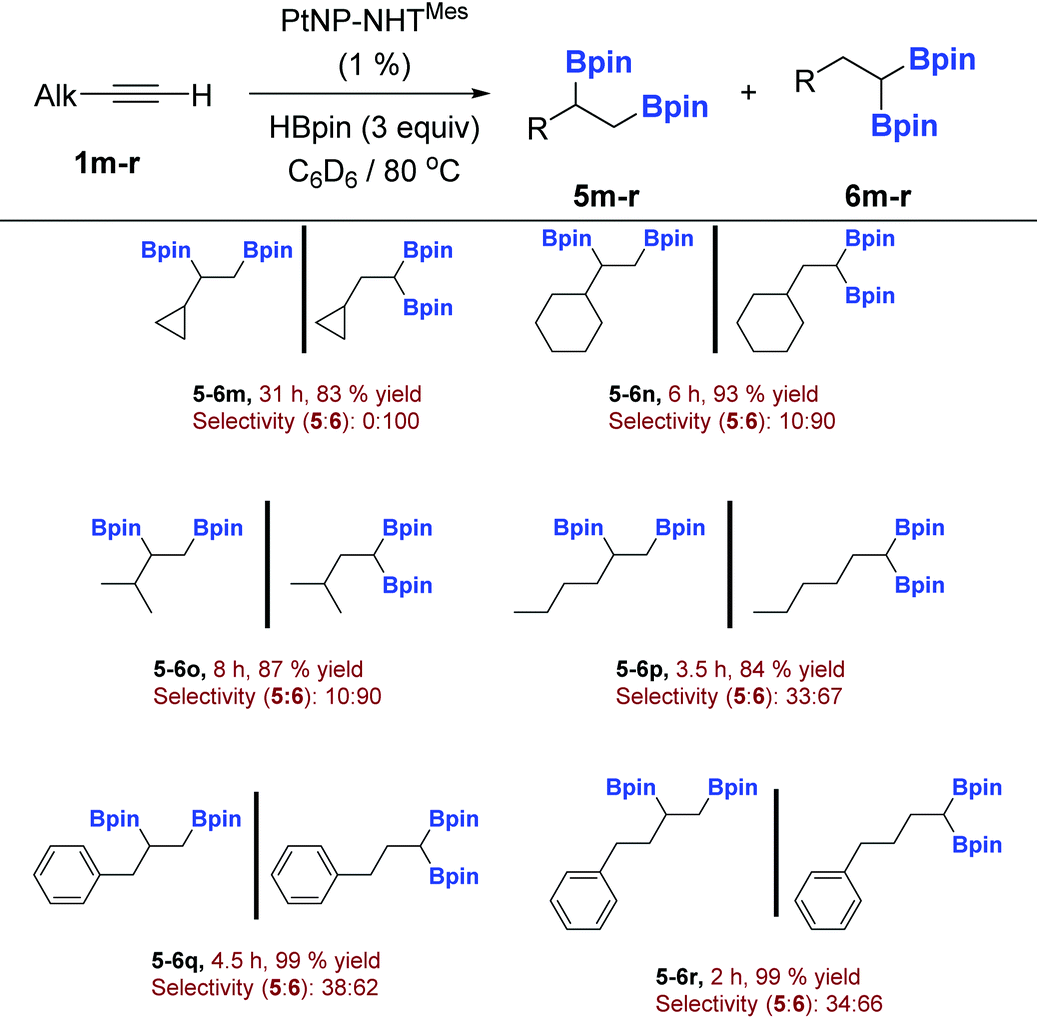
|
Although the hydroboration of alkynes leading to vinylboronates (monoborylation) has been previously studied using NHC-stabilized Pt nanoparticles,10c this is the first time that diborylation products have been obtained using platinum nanoparticles.
Conclusions
Readily available, air stable N-heterocyclic thiones (NHTs), are excellent ligands for the synthesis and stabilization of well-defined, small platinum nanoparticles. The surface coverage can be controlled through the modification of the steric environment on the nitrogen atoms of the NHT ligands. XPS clearly indicate that the NHT is bound to the metal through the sulphur atom but, in addition, according to DFT calculations, further stabilization through coordination of the CC double bond of the imidazole moiety is present when relatively small substituents are present on the nitrogen atoms. Larger substituents, such as mesityl, disfavours this type of interaction, likely due to steric repulsions of the flanking mesityl groups with the metal surface. These metal nanoparticles are active in the catalytic hydroboration of terminal and internal alkynes and, interestingly, the most active one is PtNP-NHTMes containing the smallest amount of ligand per surface area and lacking the interaction of the CC double bond with the metal surface. The metal nanoparticles PtNP-NHTMes showed an excellent performance in the hydroboration of alkynes in terms of selectivity (leading to the anti-Markonikov vinyl-boronate products) and activity when one equivalent of borane is used but, additionally, the vinyl-boronate products can be further hydroborated to the 1,1- and 1,2-diborylated alkanes when aliphatic alkynes are used as starting materials. The selectivity of this latter process can be directed towards the 1,1-diborylated species by increasing the steric environment of the alkyne at the 3 position.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support (FEDER contribution) from the MINECO (Projects CTQ2016-76267-P, CTQ2016-80814-R and CTQ2016-81797-REDC) and the Junta de Andalucía (Project FQM-2126) is gratefully acknowledged. R. C. F thanks Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-CsF) and Fundación Carolina for financial support. L. C. M. thanks the CAPES Foundation, Ministry of Education of Brazil (CAPES-CsF 13465-13-9) for a doctoral grant. B. L. acknowledges the “Talent Attraction Program” of the University of Cádiz for supporting his research (contract code E-11-2017-0117214). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).Notes and references
- (a) C. Amiens, D. Ciuculescu-Pradines and K. Philippot, Coord. Chem. Rev., 2016, 308, 409–432 CrossRef CAS; (b) M. Zahmakıran and S. Özkar, Nanoscale, 2011, 3, 3462–3481 RSC; (c) Metal NPs: Preparation, Characterization and Applications, ed. D. L. Feldheim and A. F. Colby Jr, Marcel Dekker, New York, 2002 Search PubMed; (d) The chemistry of Nanomaterials; Synthesis, Properties and Applications, ed. N. R. Rao, A. Müller and A. K. Cheetham, C. Wiley-VCH, Weinheim, 2004, vol. 1 and 2 Search PubMed; (e) Handbook of Nanoparticles, ed. M. Aliofkhazraei, Springer, 2016 Search PubMed.
- (a) L. Wang, A. Holewinski and C. Wang, ACS Catal., 2018, 8, 9388–9398 CrossRef CAS; (b) M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed.
- (a) S. B. Vendelbo, C. F. Elkjær, H. Falsig, I. Puspitasari, P. Dona, L. Mele, B. Morana, B. J. Nelissen, R. van Rijn, J. F. Creemer, P. J. Kooyman and S. Helveg, Nat. Mater., 2016, 13, 884–890 CrossRef PubMed; (b) R. M. Arán-Ais, F. J. Vidal-Iglesias, M. J. S. Farias, J. Solla-Gullón, V. Montiel, E. Herrero and J. M. Feliua, J. Electroanal. Chem., 2017, 793, 126–136 CrossRef.
- T. Galeandro-Diamant, M.-L. Zanota, R. Sayah, L. Veyre, C. Nikitine, C. de Bellefon, S. Marrot, V. Meille and C. Thieuleux, Chem. Commun., 2015, 51, 16194–16196 RSC.
- (a) D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) Nanoparticles and Catalysis, ed. P. Serp and K. Philippot, Wiley-VCH, Weinheim, 2013 Search PubMed.
- (a) L. Bai, X. Wang, Q. Chen, Y. Ye, H. Zheng, J. Guo, Y. Yin and C. Gao, Angew. Chem., Int. Ed., 2016, 55, 15656–15661 CrossRef CAS PubMed; (b) W. Zang, G. Li, L. Wangab and X. Zhangab, Catal. Sci. Technol., 2015, 5, 2532–2553 RSC; (c) Y. Dai, Y. Wang, B. Liu and Y. Yang, Small, 2015, 3, 268–289 CrossRef PubMed; (d) F. Zaera, ChemSusChem, 2013, 6, 1797–1820 CrossRef CAS PubMed; (e) K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS; (f) K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- (a) S. Tricard, O. Said-Aizpuru, D. Bouzouita, S. Usmani, A. Gillet, M. Tassé, R. Poteau, G. Viau, P. Demont, J. Carreya and B. Chaudret, Mater. Horiz., 2017, 4, 487–492 RSC; (b) P. Wanda, E. Kratzer, U. Heiz, M. Cokoja and M. Tschurl, Catal. Commun., 2017, 100, 85–88 CrossRef; (c) K. A. San and Y.-S. Shon, Nanomaterials, 2018, 8, 346 CrossRef PubMed; (d) K. A. San, V. Chen and Y.-S. Shon, ACS Appl. Mater. Interfaces, 2017, 9, 9823–9832 CrossRef CAS PubMed; (e) F. Raynal, A. Etcheberry, S. Cavaliere, V. Noël and H. Perez, Appl. Surf. Sci., 2006, 252, 2422–2431 CrossRef CAS; (f) S. E. Eklund and D. E. Cliffel, Langmuir, 2004, 20, 6012–6018 CrossRef CAS PubMed; (g) J. Yang, J. Y. Lee, T. C. Deivaraj and H.-P. Too, Langmuir, 2003, 19, 10361–10365 CrossRef CAS; (h) F. Dassenoy, K. Philippot, T. O. Ely, C. Amiens, P. Lecante, E. Snoeck, A. Mosset, M.-J. Casanove and B. Chaudret, New J. Chem., 1998, 703–711 RSC.
- P. Wand, J. D. Bartl, U. Heiz, M. Tschurl and M. Cokoja, J. Colloid Interface Sci., 2016, 478, 72–80 CrossRef CAS PubMed.
- (a) I. Schrader, J. Warneke, J. Backenköhler and S. Kunz, J. Am. Chem. Soc., 2015, 137, 905–912 CrossRef CAS PubMed; (b) E. Morsbach, M. Nesselberger, J. Warneke, P. Harz, M. Arenz, M. Bäumera and S. Kunz, New J. Chem., 2015, 39, 2557–2564 RSC; (c) J. H. Ryu, S. S. Han, D. H. Kim, G. Henkelman and H. M. Lee, ACS Nano, 2011, 5, 8515–8522 CrossRef CAS PubMed; (d) M. Cabié, S. Giorgio, C. R. Henry, M. R. Axet, K. Philippot and B. Chaudret, J. Phys. Chem. C, 2010, 114, 2160–2163 CrossRef; (e) E. Ramirez, L. Eradès, K. Philippot, P. Lecante and B. Chaudret, Adv. Funct. Mater., 2007, 17, 2219–2228 CrossRef CAS; (f) J. Yang, J. Y. Lee and H.-P. Too, Anal. Chim. Acta, 2006, 571, 206–210 CrossRef CAS PubMed; (g) J. Yang, T. C. Deivaraj, H.-P. Too and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 2181–2185 CrossRef CAS.
- (a) Y. Zeng, T. Zhang, M. R. Narouz, C. M. Crudden and P. H. McBreen, Chem. Commun., 2018, 54, 12527–12530 RSC; (b) J. M. Asensio, S. Tricard, Y. Coppel, R. Andrés, B. Chaudret and E. de Jesús, Chem. – Eur. J., 2017, 23, 13435–13444 CrossRef CAS PubMed; (c) L. M. Martínez-Prieto, L. Rakers, A. M. López-Vinasco, I. Cano, Y. Coppel, K. Philippot, F. Glorius, B. Chaudret and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2017, 23, 12779–12786 CrossRef PubMed; (d) E. A. Baquero, S. Tricard, J. C. Flores, E. de Jesús and B. Chaudret, Angew. Chem., Int. Ed., 2014, 53, 13220–13224 CrossRef CAS PubMed; (e) P. Lara, A. Suárez, V. Collière, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87–90 CrossRef CAS.
- L. M. Martínez-Prieto, I. Cano, A. Márquez, E. A. Baquero, S. Tricard, L. Cusinato, I. del Rosal, R. Poteau, Y. Coppel, K. Philippot, B. Chaudret, J. Cámpora and P. W. N. M. van Leeuwen, Chem. Sci., 2017, 8, 2931–2941 RSC.
- P. Hu, L. Chen, X. Kang and S. Chen, Acc. Chem. Res., 2016, 49, 2251–2260 CrossRef CAS PubMed.
- L. C. Moraes, B. Lacroix, R. C. Figueiredo, P. Lara, J. Rojo and S. Conejero, Dalton Trans., 2017, 46, 8367–8371 RSC.
- Y. Zhua and N. S. Hosmaneb, Coord. Chem. Rev., 2015, 293–294, 357–367 CrossRef.
- Synthesis and Applications of Organoboron Compounds, in Topics in Organometallic Chemistry, ed. E. Fernández and A. Whiting, Springer, Heidelberg, 2015 Search PubMed.
- (a) H.-Y. Tsai, M. Madasu and M. H. Huang, Chem. – Eur. J., 2019, 25, 1300–1303 CrossRef CAS PubMed; (b) S. Thoka, M. Madasu, C.-F. Hsia, S.-Y. Liu and M. H. Huang, Chem. – Asian J., 2017, 12, 2318–2322 CrossRef CAS PubMed; (c) A. Khan, A. M. Asiri, S. A. Kosa, H. Garcia and A. Grirrane, J. Catal., 2015, 329, 401–412 CrossRef CAS; J. Zhao, Z. Niu, H. Fu and Y. Li, Chem. Commun., 2014, 50, 2058–2060 Search PubMed; (d) S. Rawat and B. Sreedhar, Synlett, 2014, 25, 1132–1136 CrossRef.
- (a) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC; (b) S. Lee, D. Li and J. Yun, Chem. – Asian J., 2014, 9, 2440–2443 CrossRef CAS PubMed; (c) Z. Zuo and Z. Huang, Org. Chem. Front., 2016, 3, 434–438 RSC; (d) J. Takaya and N. Iwasawa, ACS Catal., 2012, 2, 1993–2006 CrossRef CAS.
- (a) P. Lara, K. Philippot and B. Chaudret, ChemCatChem, 2013, 5, 28–45 CrossRef CAS; (b) C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulanticac and K. Philippot, New J. Chem., 2013, 37, 3374–3401 RSC; (c) K. Philipppot and B. Chaudret, Organometallic Derived-I: Metals, Colloids and Nanoparticles, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and M. P. Mingos, Elsevier, Amsterdam, vol. 12, 2007 Search PubMed.
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS.
- D. Briggs and M. P. Seah, Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy. Apendix 5, Wiley, Chichester, 1983, p. 621 Search PubMed.
- M. G. Mason, Phys. Rev. B, 1983, 27, 748–762 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 2011, 43, 689–713 CrossRef CAS.
- (a) J. Kubackova, I. Izquierdo-Lorenzo, D. Jancura, P. Miskovskybcd and S. Sanchez-Cortes, Phys. Chem. Chem. Phys., 2014, 16, 11461–11470 RSC; (b) S. T. Marshall, M. O'Brien, B. Oetter, A. Corpuz, R. M. Richards, D. K. Schwartz and J. W. Medlin, Nat. Mater., 2010, 9, 853–858 CrossRef CAS PubMed; (c) L. Guerrini, I. Izquierdo-Lorenzo, J. V. Garcia-Ramos, C. Domingo and S. Sanchez-Cortes, Phys. Chem. Chem. Phys., 2009, 11, 7363–7371 RSC.
- V. Srinivasan and R. A. Walton, Inorg. Chim. Acta, 1977, 25, L85–L86 CrossRef CAS.
- M. Quinet, F. Lallemand, L. Ricq, J. Y. Hihn and P. Delobelle, Surf. Coat. Technol., 2010, 204, 3108–3117 CrossRef CAS.
- J. Cordón, G. Jiménez-Osés, J. M. López-de-Luzuriaga and M. Monge, Nat. Commun., 2017, 8, 1657 CrossRef PubMed.
- A. Franconetti, J. M. Carnerero, R. Prado-Gotor and F. Cabrera-Escribano, Carbohydr. Polym., 2019, 207, 806–814 CrossRef CAS PubMed.
- A slight excess of pinacolborane was necessary in all cases to achieve full conversion of the alkyne. Reactions carried out with only 1 equiv. of the borane resulted in incomplete conversions with similar selectivities for both the mono- and diborylated species.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of PtNP-NHTs. Catalytic procedures. Spectroscopic data of boron compounds. Computational methods. See DOI: 10.1039/d0nr00251h |
| This journal is © The Royal Society of Chemistry 2020 |