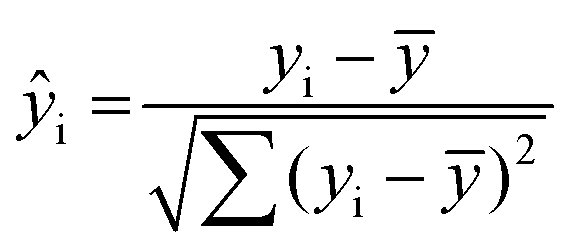DOI:
10.1039/D0NR00227E
(Paper)
Nanoscale, 2020,
12, 10226-10239
Circumvention of common labelling artefacts using secondary nanobodies†
Received
8th January 2020
, Accepted 17th April 2020
First published on 24th April 2020
Abstract
A standard procedure to study cellular elements is via immunostaining followed by optical imaging. This methodology typically requires target-specific primary antibodies (1.Abs), which are revealed by secondary antibodies (2.Abs). Unfortunately, the antibody bivalency, polyclonality, and large size can result in a series of artifacts. Alternatively, small, monovalent probes, such as single-domain antibodies (nanobodies) have been suggested to minimize these limitations. The discovery and validation of nanobodies against specific targets are challenging, thus only a minimal amount of them are currently available. Here, we used STED, DNA-PAINT, and light-sheet microscopy, to demonstrate that secondary nanobodies (1) increase localization accuracy compared to 2.Abs; (2) allow direct pre-mixing with 1.Abs before staining, reducing experimental time, and enabling the use of multiple 1.Abs from the same species; (3) penetrate thick tissues more efficiently; and (4) avoid probe-induced clustering of target molecules observed with conventional 2.Abs in living or poorly fixed samples. Altogether, we show how secondary nanobodies are a valuable alternative to 2.Abs.
Introduction
Standard immunodetection approaches use typically a primary antibody (1.Ab) which binds the protein of interest (POI) and a secondary antibody (2.Ab) that binds to the 1.Ab and carries a detection element. In fluorescent microscopy techniques, the detection element is a fluorophore1,2 or a single strand of DNA. The latter is used in DNA Point Accumulation for Imaging in Nanoscale Topography (DNA-PAINT), a single molecule localization microscopy technique reaching <5 nm resolution by transiently binding of single stranded DNA bearing a fluorophore to their complementary strand on the target of interest.3 The complex formed by the primary antibody and the secondary antibody (1.Ab–2.Ab) is widely used because it is a cost effective and flexible approach since only the 2.Abs need to be coupled to the detection element. However, the use of this complex carries some relevant limitations. First, the 1.Ab–2.Ab can measure up to 30 nm, leading to a large distance between the targeted molecule and the detection element, causing the so called “linkage” or “displacement” error.4 While this might not influence the results in some applications (e.g. epifluorescence, ELISA or FACS), it is of major relevance for super-resolution microscopy techniques where the localization precision can be as high as 1 nm.5 The linkage error can be reduced by using directly labelled small affinity probes like camelid single domain antibodies (sdAbs) also known as nanobodies (Nbs),4,6 affibodies,7 aptamers8,9 or affimers,10 which all have sizes below 3 nm. Unfortunately, such small probes exist only for a handful of targets11 due to their rather laborious selection and validation process, while conventional 1.Abs are readily available for a large number of POIs. An alternative to the standard 2.Abs was recently developed: monovalent recombinant secondary nanobodies (2.Nbs).12 Secondly, the large size of the 1.Ab–2.Ab complex makes them to perform poorly in crowded cellular environments or when the epitopes are abundant and densely arranged. In this respect, smaller probes such as aptamers or nanobodies are more efficient in the detection of the POI.8,13,14 Moreover, sample penetration of full antibodies is a problem when staining thick biological specimen such as tissues, biopsies or whole organisms.13,15 For the optimal labelling of these thick samples, protocols have been established, but they are often laborious and require time-consuming incubations of weeks16 or need artefact-prone epitope retrieval protocols.17 Smaller probes are expected to shorten some of these long incubations. Third, in a multiplex immunostaining, i.e. when multiple targets are stained in the same sample, scientists are typically constrained to use 1.Abs coming from different species. This is because, standard immunostaining needs to be done in a sequential manner: first 1.Abs are incubated on the sample, washed off and only then 2.Abs are incubated. Therefore, 1.Abs should be raised in different species and 2.Abs should recognize one species specifically, limiting the choice of antibodies for a multiplexing staining. It has been shown that by pre-mixing 1.Abs with 2.Nbs in a tube prior staining, one could circumvent this species limitation and use on a sample 1.Abs from the same species.12 Finally, conventional antibodies used commonly for immunodetections are bivalent binders, i.e. each antibody molecule can bind two POIs/epitopes simultaneously. In the case of polyclonal antibodies, they are not only bivalent binders, but they also contain an unknown number of different antibodies able to bind the POI. This is the case of the vast majority of 2.Abs used for detection of 1.Ab in immunofluorescence applications. The bivalency and polyclonality of 2.Abs combined have been proposed as characteristics that induce clustering of the POI and their interactors, which can have a strong impact in the conclusions obtained from such experiments.18,19 The use of monovalent secondary probes should minimize the potential of secondary probe induced clustering effects.
In this work we tested and thoroughly validated the use of 2.Nbs for several microscopy applications. We first confirmed that the usage of 2.Nbs decreases linkage error by using them in STED microscopy and DNA-PAINT. We then exploited the ability of these probes to allow the simultaneous use of several 1.Abs from the same species by using them in Exchange-PAINT multiplexed super-resolution microscopy. This technique enables to image a virtually infinite number of targets in high resolution in the same sample.20,21 Additionally, we observed that pre-mixing 1.Ab and 2.Nb can save time in staining thick biological samples imaged under light-sheet microscopy, ensuring also a better sample penetration and homogenous staining. Finally, we systematically compared the probe-induced clustering of the target protein either using directly-labelled monovalent probes, like affibodies and single Fab’ fragments, and conventional 1.Abs detected by polyclonal and bivalent 2.Abs or by monovalent 2.Nbs. We observed that 2.Nbs drastically reduced the clustering of the target in both live and fixed sample. This makes 2.Nbs a real alternative to conventional 2.Abs by minimizing experimental time, expanding the multiplexing ability of immunostainings, improving the tagging precision and signal linearity, and finally avoiding the probe-induced clustering artefacts.
Results
Secondary nanobodies provide higher staining accuracy than secondary antibodies
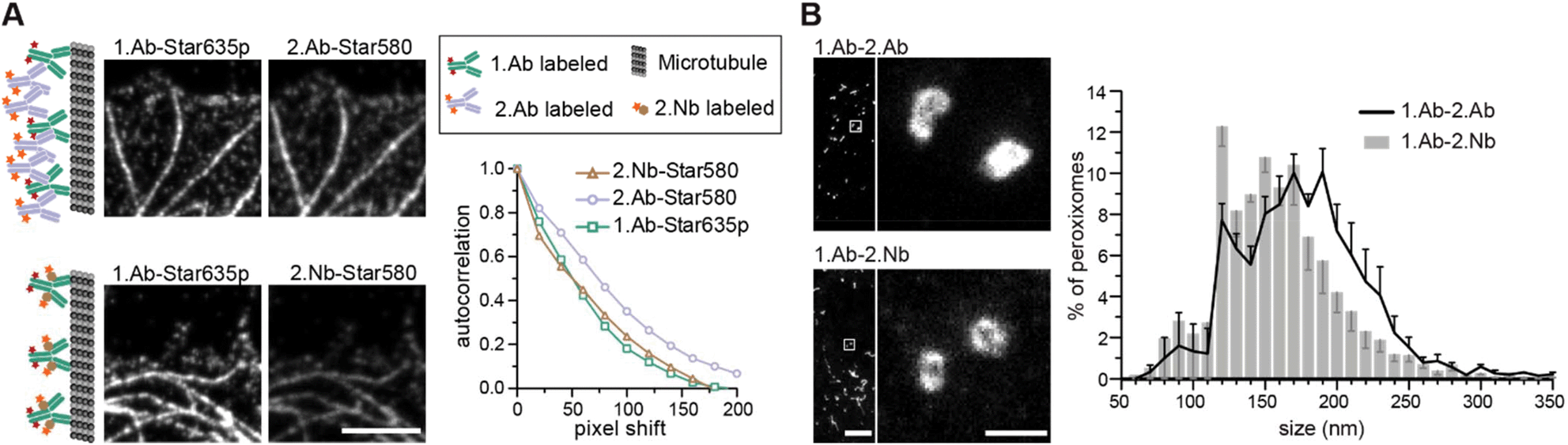
First, we investigated the accuracy of 2.Abs or 2.Nbs in revealing their 1.Ab target. To do so, we imaged (using a two-color STED microscopy setup) COS-7 cells fixed with cold methanol to ensure a strong immobilisation of the POI,22 then stained with a monoclonal 1.Ab anti-alpha tubulin directly conjugated to the fluorAbberiorStar635P. The primary antibody was subsequently recognized by either a polyclonal 2.Ab or a monovalent 2.Nb, both carrying the fluorophore AbberiorStar580. An autocorrelation analysis was performed on these images to evaluate the staining accuracy of the secondary probes by comparing them with the directly labelled primaries. Initially, the autocorrelation of the images with the fluorescent 1.Ab provided an idea of the distribution or density of the 1.Ab on microtubule filaments. The autocorrelation curve obtained from the signal of the 2.Nb followed the tendency of the autocorrelation obtained for the anti-alpha tubulin primary antibody, which proposes that the 2.Nb signal accurately follows the fluorescent signal from the 1.Ab. In contrast, when performing the same analysis on the staining performed with the polyclonal 2.Abs, the correlation curve was shifted to the right. This suggests that the 2.Ab inaccurately reveals the location of the 1.Ab anti-alpha tubulin (Fig. 1A). To confirm this, we decided to analyse paraformaldehyde (PFA) fixed peroxisomes within primary hippocampal neurons. We compared the diameter of these small organelles when imaged with STED microscopy after using a 1.Ab anti-pmp70 (a peroxisomal membrane protein) revealed by a 2.Ab or a 2.Nb.
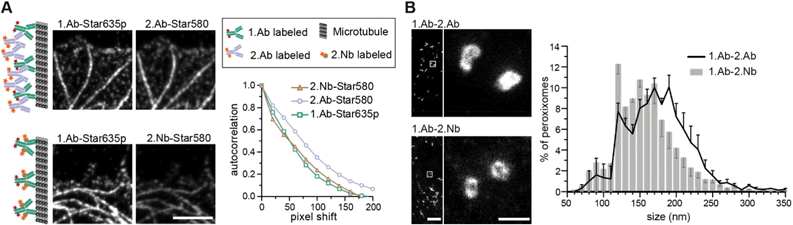 |
| | Fig. 1 2.Nbs minimize the linkage error caused by 2.Abs and increase detection accuracy. (A) Two color STED imaging of microtubules stained with 1.Ab directly labelled with AbberiorStar635P dye and secondary reagents (either 2.Ab or 2.Nb) labelled with AbberiorStar580. Fixation conditions were the same for both conditions (methanol fixation, see Methods). Example images and schematic representation of the experimental procedure. Scale bar 2.5 μm. Autocorrelation analysis on signal obtained from either the 1.Ab or the secondary probe microtubules. N = 51 line profiles for 2.Nb, N = 70 for 2.Ab and N = 121 for 1.Ab. One-way ANOVA p = 1.061 × 10−6F = 14.58 followed by post hoc Bonferroni tests indicates that the 2.Ab is different with p < 0.01 from the 1.Ab and 2.Nb which themselves are indistinguishable. (B) Primary hippocampal neurons were fixed for 30 minutes with 4% PFA, and stained against the peroxisome protein (pmp70) with 1.Ab–2.Ab or 1.Ab–2.Nb. Exemplary STED images. Scale bars 10 μm (overview) and 100 nm (zoom). Size distribution analysis of peroxisomes. N = 3109 peroxisomes were analysed when stained with 2.Nb and N = 3020 stained with 2.Ab. Error bars correspond to the standard error of the mean from 4 independent experiments. Paired t-test shows that the apparent size of peroxisomes stained with 2.Nb is on average smaller with p < 0.01 compared to the one stained with 2.Ab. | |
We observed a significant shift to smaller diameters of 1.Ab–2.Nb labelled organelles after comparing 3020 peroxisomes stained with 1.Ab–2.Ab and 3109 peroxisomes stained with 1.Ab–2.Nb (Fig. 1B). To evaluate more precisely if the 2.Nbs decrease the linkage error, we needed a technique providing higher spatial resolution. For this purpose, we used DNA-PAINT that has achieved sub-10 nm resolution.3 As DNA-PAINT uses affinity reagents attached to short DNA oligonucleotides, we coupled the 2.Nbs site-specifically to a single stranded DNA oligo (termed docking strand) as described previously.23 We performed an assay which has been used as gold standard in the field to assess linkage error.24 We immunostained the microtubule network of fibroblast cells with a monoclonal 1.Ab against alpha tubulin, and detected it by either a 2.Ab or a 2.Nb coupled to docking strands (Fig. 2). After analysing the diameter of ∼80 microtubules for each condition, we obtained an average diameter of 61.3 ± 13.2 nm (mean ± SD) when using the 2.Ab and 38.3 ± 9.34 nm when using a 2.Nb (Fig. 2G and I). A similar gain in precision was observed previously using dSTORM.12 Additionally, we performed an autocorrelation analysis on single microtubules to corroborate their difference in size, and we observed a significantly faster loss in autocorrelation for microtubules stained with 2.Nb (Fig. 2H).
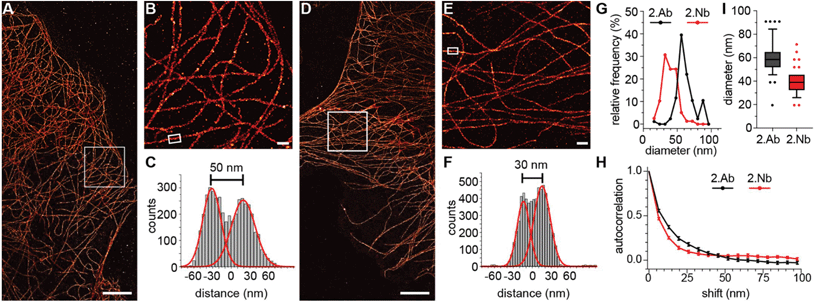 |
| | Fig. 2 DNA-PAINT imaging using secondary antibodies or secondary nanobodies shows differences in microtubule's diameter size. (A) Overview DNA-PAINT image of COS-7 cells fixed with cold methanol and stained with 1.Ab targeting alpha tubulin and 2.Ab coupled to a DNA-PAINT docking strand. (B) Zoom-in image of the region highlighted in A. (C) Cross-sectional histogram example of the region highlighted in B, showing a microtubule filament diameter of ∼50 nm. (D) Overview DNA-PAINT image of alpha tubulin stained with 1.Ab and 2.Nb. (E) Zoom-in image of the region highlighted in D. (F) Cross-sectional histogram example of the region highlighted in E, showing a microtubule filament diameter of ∼30 nm. Scale bars: 5 μm (A and D), 500 nm (B and E). (G) Histogram analysis for the microtubules diameter distribution (86 cross-sections for 2.Ab and 78 for 2.Nb). (I) Box & Whiskers graph of G. Whiskers represent 10 & 90 percentiles. Two-tailed unpaired t-test results in p < 0.0001 (****). (H) Autocorrelation analyses with N = 57 line profiles for each condition; 2.Nb display a significantly faster loss of autocorrelation than 2.Abs (Mann Whitney test p < 0.0001). | |
Premixing secondary nanobodies bypasses the primary antibody animal-species limitations
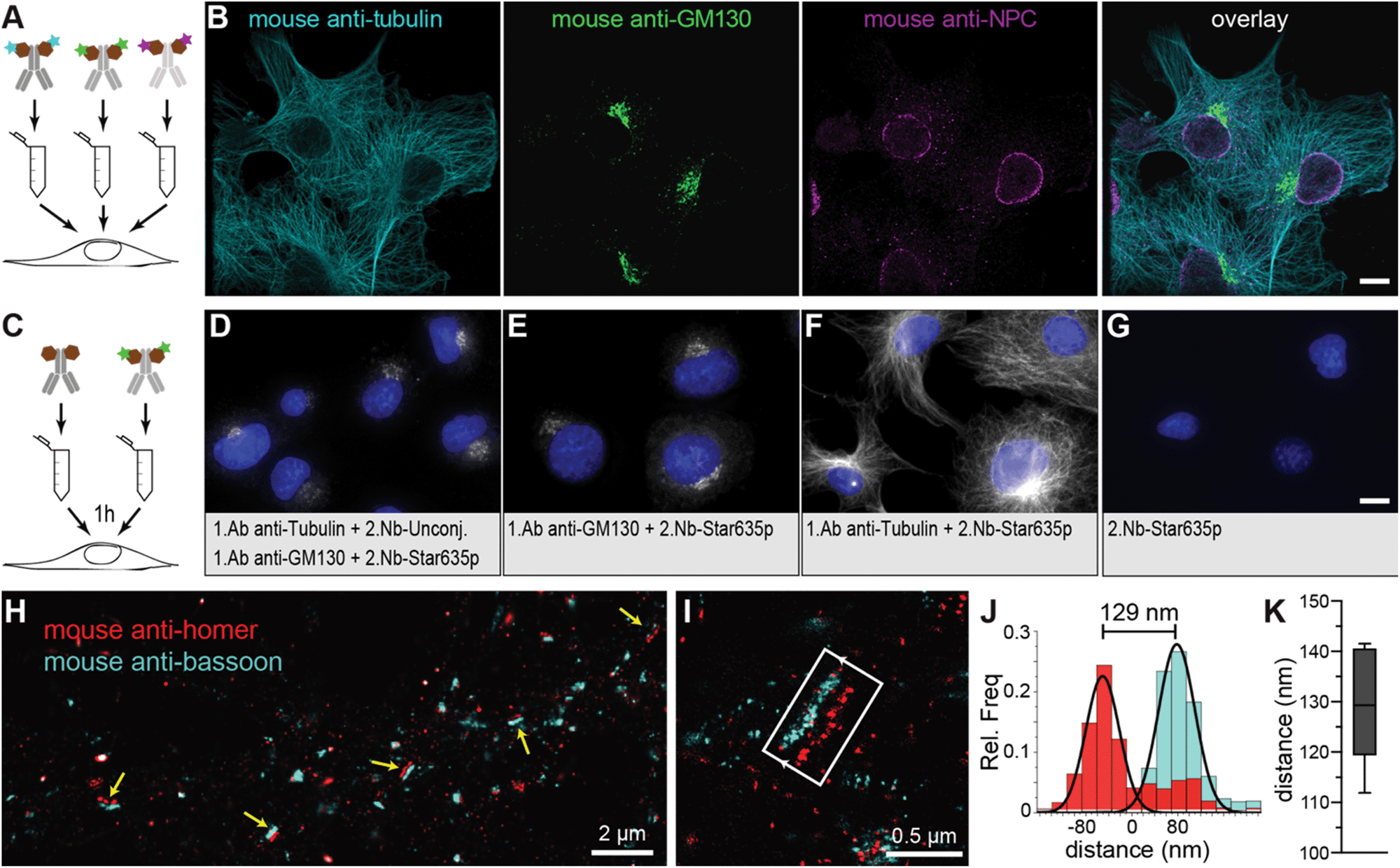
In standard immunoassays, 1.Abs are first incubated with the sample followed by washes to remove unbound excess of primary antibodies. Only at this point, labelled-2.Abs are incubated for a period of time with the sample followed by washes to eliminate the non-bound excess of 2.Abs before imaging the specimen. Pre-mixing the 1.Ab with the 2.Ab before adding them to the sample would shorten protocols and save considerable amount of time and costs (e.g. in clinical pathology laboratories). However, this is not possible due to the polyclonality and bivalency of 2.Abs that result in agglutination or aggregation of the 1.Abs–2.Abs complexes and thus in a failure to stain the intended target in the sample (ESI Fig. 1†). If the secondary probe binds to the 1.Ab in a monovalent fashion, pre-mixing primary and secondary probes would be possible. The pre-mixing of 2.Nbs with a mouse monoclonal 1.Ab anti-alpha tubulin for 15 minutes resulted in properly stained filaments (ESI Fig. 1†) and single bands detected in a fluorescent Western blot assay (ESI Fig. 2A†). Bypassing this limitation by pre-mixing with monovalent secondary probes open a new possibility in immunoassays, i.e. it allows to use several 1.Abs against different targets raised in the same species. To detect two or more POIs it was typically required that each 1.Ab comes from a different animal (e.g. mouse, rabbit and chicken for the detection of 3 POIs on the same specimen). This strict requisite is necessary to ensure the indirect detection of the POIs with species-specific 2.Abs. This restriction provides a limitation for the choice of 1.Ab and it can reduce the multiplexing capability of immunoassays. Here we chose three different monoclonal 1.Abs raised all in mouse directed against alpha-tubulin, GM130 (Golgi), and FXFG repeats in nucleoporins (nuclear pore complex; NPC). Each was pre-mixed with anti-mouse kappa-light chain 2.Nbs carrying different fluorophores (Fig. 3A and B). COS-7 cells were imaged under scanning confocal microscopy, and they clearly displayed the three stained structures (microtubules, Golgi and NPC) with minimal background and negligible cross-talk between the channels. This multiplexing assay requires that the 2.Nb stays bound to its primary antibody without swapping to another primary during the staining incubation, which would result in the cross-contamination of signals. To asses this potential problem, we performed a control experiment where mouse 1.Ab anti-GM130 was pre-mixed with excess of fluorescently labelled 2.Nb anti mouse antibody (2.Nb-Star635p). In parallel, mouse 1.Ab anti-alpha tubulin was premixed with excess of non-fluorescent 2.Nb anti mouse antibody. Finally, these individual pre-mixtures were added simultaneously to methanol-fixed cells and were incubated for 1 hour, 3 hours or overnight. We were not able to observe the distinctive microtubule pattern in any of the conditions, which demonstrates that no cross-contamination from the fluorescent 2.Nbs pre-mixed with the 1.Ab anti-GM130 onto the 1.Ab anti-tubulin occurred (Fig. 3C–G and ESI Fig. 3†). Similarly, this pre-mixing capability could be applied for the detection of 2 different POIs in fluorescent Western blots assays (ESI Fig. 2B†). The option of pre-mixing primary antibodies with secondary nanobody is ideal for techniques that allow the detection of multiple targets (multiplexing). Therefore, we turned once again to DNA-PAINT, this time we used an extension termed Exchange-PAINT that can, in theory, image an unlimited number of POIs on the same sample with a few nanometer precision.20,23 We stained primary hippocampal neurons with two mouse monoclonal 1.Abs and each was pre-mixed with 2.Nbs conjugated to DNA docking strands with orthogonal sequences. We performed Exchange-PAINT on synapses stained against bassoon, a protein highly enriched at the pre-synaptic active zone,25 and the scaffold protein homer that is concentrated at the post-synaptic density26 (Fig. 3H and I). Notably, we obtained a super-resolved view of single neuronal synapses using two primary antibodies from the same species. We measured a distance of 129.5 ± 10.9 nm (Fig. 3J and K) between bassoon (pre-synaptic) and homer (post-synaptic), reproducing previous results obtained with other advanced microscopy techniques such as dSTORM27 and X10 expansion microscopy.28 The presence of localisations of homer in the pre-synaptic area is most likely due to unspecific background staining caused by the respective 1.Ab (ESI Fig. 4†).
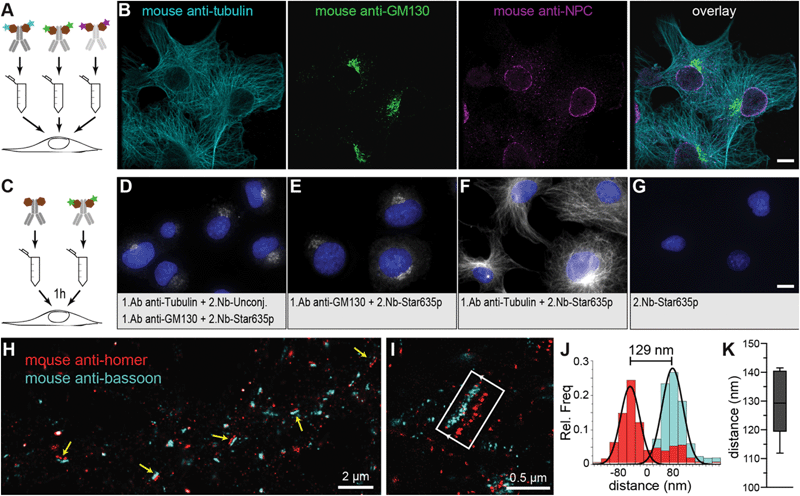 |
| | Fig. 3 Pre-mixing 1.Abs with 2.Nbs allows to use same animal-species 1.Ab for several target proteins on the same sample. (A) Scheme of pre-mixing: different 1.Abs were pre-mixed with 2.Nbs each carrying different fluorophores and subsequently incubated on cells. (B) Example of confocal images performed on COS-7 cells stained with the pre-mixing methods. Cyan: Mouse anti-tubulin 1.Ab pre-mixed with 2.Nb-CF633. Green: mouse anti-GM130 1.Ab pre-mixed with 2.Nb-Alexa488. Magenta: mouse anti-NPC 1.Ab pre-mixed with 2.Nb-Alexa546. Scale bar represents 10 μm. (C) Scheme of the experimental procedure: a mouse monoclonal anti-tubulin antibody was pre-mixed with unconjugated 2.Nbs. A monoclonal anti-GM130 antibody was pre-mixed with 2.Nb conjugated to Star635p. (D) Epifluorescence example image of cells co-incubated with both mixtures simultaneously for 1 h. (E) Control where only the anti-GM130 premixed with 2.Nb-Star635p was used. (F) Control sample stained only with anti-Tubulin premixed with 2.Nb-Star635p. (G) Control where 2.Nb conjugated to Star635p was used without 1.Ab. All images displayed in D–G are equally scaled, for direct comparison. Scale bar represents 10 μm. (H) Exchange-PAINT overview image of primary rat hippocampal neurons. Yellow arrows indicate evident mature synapses where the pre-synaptic active zone (mouse 1.Ab anti-bassoon) and post-synaptic density (mouse 1.Ab anti-homer) are in front of each other. (I) Higher magnification of a selected synapse where a synaptic cleft is recognized. (J) Exemplary histogram analysis of the selected synapses displaying the length of the synaptic cleft. (K) Distance analysis of 8 different synapses averaging a mean of 129.5 ± 10.9 nm (mean ± SD). Graph shows the data as Box & Whiskers representing 10 & 90 percentiles. | |
Secondary nanobodies enhance sample penetration in shorter incubation time
We used the time advantage of pre-mixing 1.Abs with the 2.Nbs to stain a complex thick sample that requires long incubation with the probes to ensure proper sample staining. We used cochleae extracted from three weeks old mice and stained parvalbumin-α, a calcium buffering protein present in inner hair cells and type I spiral ganglion neurons (Fig. 4). We compared how long the 1.Ab–2.Nb and 1.Ab–2.Ab needed to be incubated to result in a homogenous staining throughout the sample. In order to image the entire volume, we used light-sheet microscopy after decalcification and clearing. Two cochleae obtained from the same animal were stained either with 1.Abs and sequentially with 2.Abs or with 1.Abs pre-mixed with 2.Nbs for comparable amount of time. The cochleae stained with 1.Ab–2.Ab for a total of 6 days (3 days 1.Ab, 3 days 2.Ab) showed insufficient penetration of the staining, with signals accumulated in the outer bone surface and in the edges exposed to the solution (Fig. 4A). The cochlea stained with the same antibody for 14 days (7 days 1.Ab, 7 days 2.Ab) showed a better staining performance, revealing hair cells and neurons. However, the ganglion displayed a staining gradient with stronger signals on the edges, indicating insufficient detection of target molecules deep in the tissue (Fig. 4A, area depicted with white discontinuous lines). On the other hand, the cochleae stained with pre-mixed 1.Ab–2.Nb for 6 and 14 days revealed a homogenous staining of neurons in an analogue area. No apparent difference in term of signal homogeneity between the two incubation times was observed (Fig. 4B, area depicted with white discontinuous lines). A custom written analysis quantifying the signal intensity throughout the ganglion of the cochleae (Fig. 4C and ESI Fig. 5, 6†) showed how the signal coming from cochleae stained with 1.Ab–2.Nb displayed a plateau phase meaning homogenous staining, while the ones stained with 1.Ab–2.Ab displayed a peak showing a gradual staining from the distal to the central portion of the ganglion.
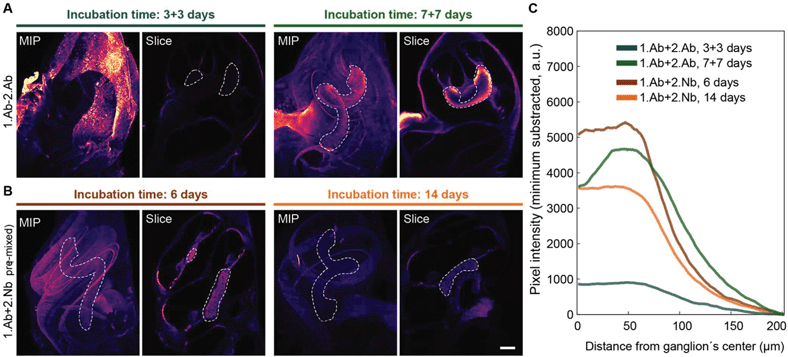 |
| | Fig. 4 Pre-mixing decreases the incubation time necessary to obtain homogenous staining of the cochlea. Mice intact cochleae were stained with a parvalbumin-α antibody, either pre-mixed with 2.Nb (A) or sequentially incubated with 2.Ab (B) for the time indicated. In each panel the maximal intensity z-projection (MIP) and an exemplary light-sheet microscopy slice of an intact cochlea (Slice) are depicted. Scale bar: 200 μm. Ganglion outlined by dotted line. (C) Mean pixel line profile from radii crossing the ganglion distributed along the centerline of the ganglion. See ESI Fig. 5† for schematic analysis explanation and ESI Fig. 6† for raw data. N = 2 cochlea per condition. Note: The plateau profile depicted by the samples stained with 2.Nb, as opposed to the relatively pronounced peak profile in the samples stained with 2.Ab for 7 days or to the flat profile in the samples stained for 3 + 3 days. | |
Secondary nanobodies reduce the probe-induced clusters of target proteins on living cells
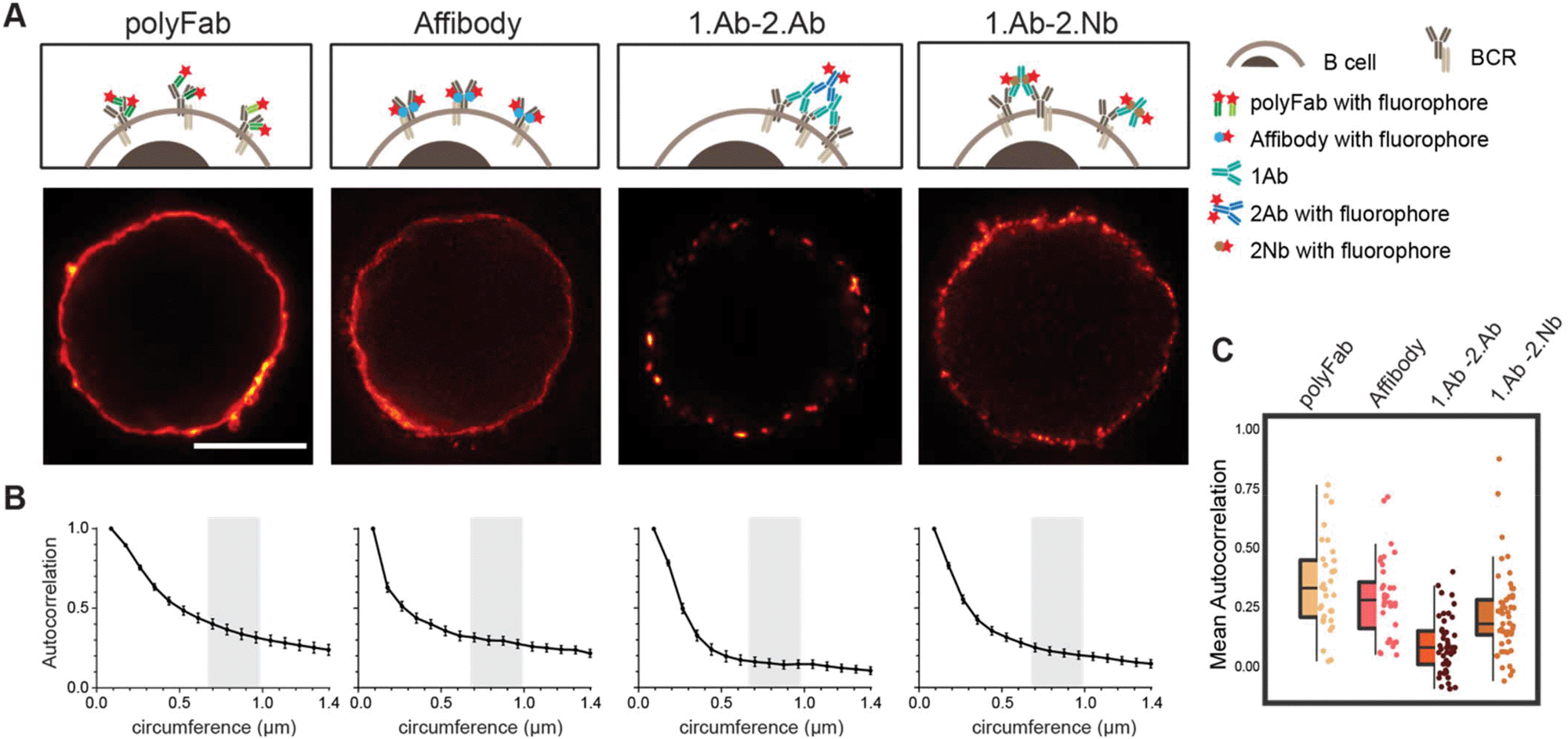
To test if the 2.Nbs have an effect in the probe-induced clustering of target molecules, we decided to analyse the surface distribution of IgM containing B cell receptors (IgM-BCRs) on a human B cell line (Ramos cells). This cellular model allows simple visual inspection and numerical analysis because the POIs are evenly distributed in the cellular surface of these resting B cells.29 Cells were first stained and then chemically fixed with aldehydes to be imaged under stimulation emission depletion (STED) microscopy. Initially, the surface IgM-BCRs on living Ramos cells were stained using fluorescent-monovalent probes: a monoclonal affibody29 or a polyclonal single Fab fragment (polyFab) (Fig. 5). In this case a smooth continuous plasma membrane signal from the surface IgM-BCRs distribution was observed at the optically sliced equator of the cells. However, when cells were stained using a 1.Ab–2.Ab, a sparse clustered signal was clearly identified (Fig. 5). Finally, we tested if the 2.Nbs elicit a similar clustering effect observed using 1.Ab–2.Ab detection system. Interestingly, a considerably milder effect was observed when using the same 1.Ab detected by a 2.Nbs, partially rescuing the distribution pattern observed when stained with the monovalent affibody or polyFab that bind directly to the IgM-BCRs (Fig. 5). These results suggest that although the bivalency of the monoclonal 1.Ab still deviates slightly from the signal distribution obtained with fluorescent monovalent primary probes, the major cluster-inducing element is contributed by the conventional 2.Abs. A Pearson's autocorrelation analysis30 was used to quantify the probe-induced clustering. The custom-written analysis consists of collecting the STED image intensity along the membrane and correlating it to itself for different rotation angles. We then plotted the autocorrelation curves, which start with a perfect correlation (r = 1) at zero rotation and decrease at higher rotations (Fig. 5B, with rotation angle converted to corresponding membrane distance). The major empirical effect between the different conditions was observed at membrane distances between 0.7 to 1 μm. Therefore, the correlations measured throughout this interval were then averaged with the value obtained from each cell corresponding to a spot on the scatter plot (Fig. 5C). With this method we determined an average autocorrelation of 0.34 ± 0.19 (mean ± SD) for cells stained with the monovalent polyFab and 0.29 ± 0.16 for cells stained with the monovalent affibody, while it was only 0.07 ± 0.12 for the cells stained with monoclonal 1.Ab and polyclonal 2.Ab. Interestingly, this effect was not only evident using super-resolution microscopy, but it was also observed in diffraction limited scanning confocal microscopy images (ESI Fig. 7†). As expected by the more continuous pattern observed, the average autocorrelation of cells stained with 1.Ab–2.Nb was 0.21 ± 0.17 indicating a significant decrease (rescue) of the probe-induced clustering artefact caused by the polyclonal 2.Ab (Fig. 5).
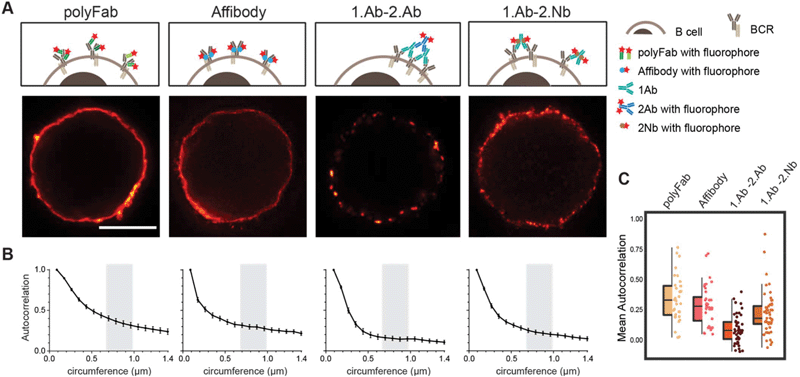 |
| | Fig. 5 Live staining of IgM-BCRs on Ramos cells shows different pattern according to the probe used. (A) STED images of a B cell stained with fluorescent polyclonal single Fab’ fragment (polyFab); affibody; primary antibody revealed by a fluorescent secondary antibody (1.Ab–2.Ab); primary antibody revealed by a secondary nanobody (1.Ab–2.Nb). All fluorescent probes were conjugated to AbberiorStar635P fluorophores. Scale bars = 5 μm. (B) Autocorrelation analysis along the circumference of cells. We analysed three independent experiments with N ≥ 10 cells for each conditions. (C) Box-dot plots show the average autocorrelation from 0.7 μm to 1.0 μm circumference (grey zones shown in the graphs in B). Boxes show the interquartile range (IQR). Lines signify medians, and whiskers extend to 1.5 times the IQR. Lower box represents higher clustering. The autocorrelation observed in 1.Ab–2.Ab differs from the monovalent probes (polyFab and affibody) by p ≤ 0.0001 and from 1.Ab–2.Nb by p ≤ 0.001. P values were calculated with one-way ANOVA followed by Tukey Multiple Comparison Test. See ESI Table 5† for full statistics. | |
Probes induce clusters of target proteins in aldehyde-fixed cells
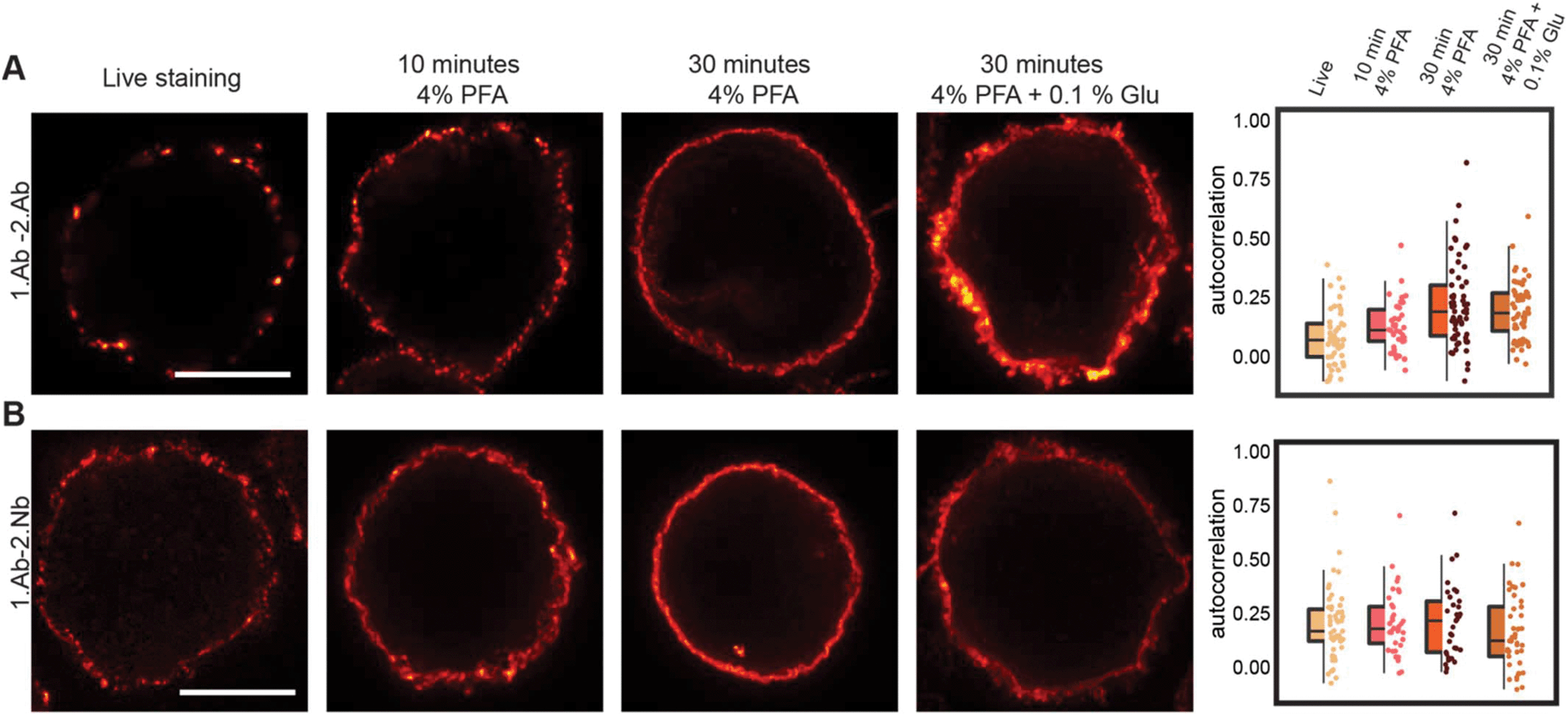
It has been noticed that conventional fixation times with 4% PFA does not necessarily prevent protein movement.22 Also, other variables like blocking reagents and temperature need to be taken into consideration and tested case-by-case depending on the imaged target.31 A more efficient fixative such as glutaraldehyde (GLU) could be used, but it generates unwanted autofluorescence and only few affinity molecules bind their target epitopes after GLU crosslinking. A recently described di-aldehyde alternative that seems to alleviate some of these problems caused by PFA and GLU is glyoxal.32 However, glyoxal implementation is very recent and the vast majority of researchers still use PFA-fixation for conventional immunofluorescence. Therefore, we tested and compared the probe-induced clustering after exposing the Ramos cells for 10 and 30 minutes with 4% PFA or 30 minutes with a combination of 4% PFA and 0.1% GLU (Fig. 6 and ESI Fig. 8†). We compared these fixative conditions and live staining using the classical 1.Ab–2.Ab complexes or the 1.Ab–2.Nb imaged under STED microscopy. Our observations suggest that applying 4% PFA for 10 minutes is not enough to avoid the artefactual formation of clusters induced by 1.Ab–2.Ab (autocorrelation of 0.14 ± 0.11 not significantly different from the live staining condition 0.07 ± 0.12). However, 4% PFA fixation for 30 minutes seems to be sufficient to rescue to a great degree the clustering artefact caused by the 2.Ab (0.23 ± 0.18 different from the live staining condition with p value p ≤ 0.001; see also Fig. 2A). Using the 2.Nbs had no significant change between live, 10 or 30 minutes of fixation with 4% PFA (0.21 ± 0.17, 0.20 ± 0.17 and 0.21 ± 0.18 respectively; Fig. 6B). As expected, similar non-clustering effects are observed for a primary monovalent probe like the monovalent polyFab directed against human IgM-BCRs (ESI Fig. 8†). In addition, when observing the staining pattern created by the combination of 4% PFA and 0.1% GLU for 30 minutes, the stained rim of the cells is not a thin layer as observed by PFA fixation, but it displays a texture-like surface. From studies in electron microscopy, it is expected that GLU fixation results in a better ultrastructure preservation. Due to the uneven texture-like surface when fixing with PFA and GLU, and therefore a reduced homogeneity at the investigated spatial scale, the Pearson's correlation analysis has the tendency to paradoxically display a slightly lower correlation (Fig. 6, boxplot).
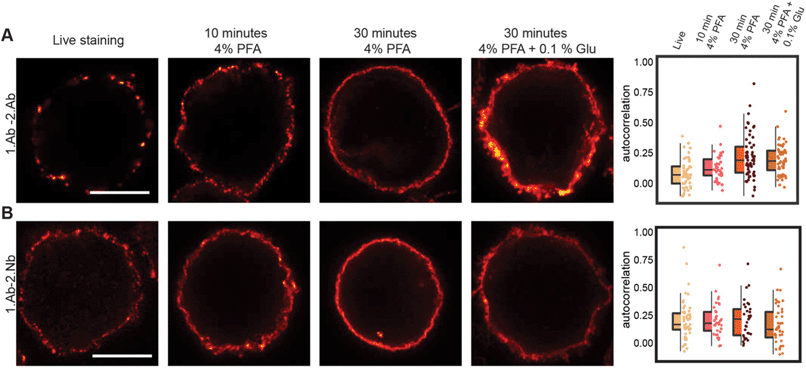 |
| | Fig. 6 Probe-induced clustering on aldehyde fixed B cells. (A) and (B) STED images showing the effect of fixation on clustering induced by 1.Ab–2.Ab or 1.Ab–2.Nb. Scale bars represent 5 μm. Box-dot plots show the average autocorrelation from 0.7 μm to 1.0 μm circumference (grey zones shown in the ESI Fig. 9†). Boxes show the interquartile range (IQR). Lines signify medians, and whiskers extend to 1.5 times the IQR. Lower box represents higher clustering. Autocorrelation curves are detailed in ESI Fig. 9 and statistics in ESI Table 5.† | |
Discussion
In this study we have systematically studied how secondary nanobodies detecting primary antibodies can be used to overcome several limitations and artefacts caused by conventional polyclonal secondary antibodies. Additionally, problems with polyclonal secondary antibodies have been repeatedly attributed to the poor reproducibility provided by polyclonal serums, which can be highly heterogeneous.37 Therefore, we propose that using recombinantly produced monoclonal and monovalent secondary reagents, like the nanobodies characterized here, not only eliminates the ethically controversial use of animals for conventional 2.Abs (e.g. from donkey, goat, sheep, etc.), but importantly also minimizes artefacts, linkage errors and increases the reproducibility of biomedical experiments.
Smaller size of the secondary probe decreases linkage error and increases staining accuracy
Although it has been already demonstrated that small primary affinity probes are able to minimize the linkage error,4,8,12 there was just one indication performed in dSTORM that secondary nanobodies can increase the labelling precision.12 Here we show, in addition to the resolution improvement by using 2.Nbs on DNA-PAINT and STED microscopy, that bulky secondary antibodies not accurately represent the distribution of the primaries, due to a combination of their polyclonal nature and large size (Fig. 1A). We show that the 1.Abs directly labelled with a fluorophore decorates microtubules with a certain periodicity that can be followed when revealed with a 2.Nb but not with a conventional 2.Ab, suggesting that the 2.Ab blurs the localization of the 1.Ab. This inaccuracy of the polyclonal 2.Abs can have major consequences in one of the main application of fluorescence microscopy, namely co-localization studies.
Pre-mixing overcomes the species limitation for multiplexing microscopy
Mixing the primary and the secondary reagents prior to incubating them with the sample (pre-mixing) is a desired feature as it saves experimental time. This cannot be performed with conventional bivalent polyclonal 2.Abs (ESI Fig. 1†), however, pre-mixing has been shown to work when using monovalent binders against 1.Abs.12 This feature eliminates the animal-species limitation of the primaries when detecting two or more POIs. We first showed that it is possible to use 2 mouse 1.Abs in a simpler Western-blot assay (ESI Fig. 2†), and then we tried 3 different mouse 1.Abs in immunofluorescence under conventional scanning confocal microscopy. Nevertheless, pre-mixing needs to be carefully tested and well validated for every application and for each set of 1.Abs, since the 2.Nbs are not covalently bound to the 1.Abs. However, our control experiments (Fig. 3C and ESI Fig. 3†), suggest that no significant movement between primaries can be observed if incubation with pre-mixed primary and secondary nanobodies is for 1 and 3 hours, even overnight incubations did not show signs of cross-contamination when using two primaries of the same species. To ensure the permanence of the secondary nanobody on its primary antibody in demanding applications like DNA-PAINT, we decided to perform a short post fixation between the applications of the different pre-mixed pairs. Here we showcased the proof-of-principle of pre-mixing with 2.Nbs and multiplexing using Exchange-PAINT super-resolution microscopy. We determined the distance between the pre- and post-synapses with high accuracy (Fig. 3), and we obtained average synaptic cleft distances comparable to values from other methods.27 Exchange-PAINT not only provides high spatial resolution, but it also eliminates the limit on the number of POIs that can be imaged in the same sample. This makes the combination of pre-mixing using 2.Nbs with Exchange-PAINT a very powerful approach for multiplexing.
Pre-mixing shortens experimental time and allows a better penetration of probes in thick tissue
Immunostaining protocols of complex thick tissue samples typically require days to weeks.16,17 This is because it takes time for the antibody to penetrate into the tissue, for thoroughly washing unbound binders, and also because the 1.Abs and 2.Abs have to be performed sequentially. This may be a problem even in cell monolayers, where it has been suggested that primary and conventional secondary antibodies have difficulties penetrating in crowded areas and revealing all epitopes.8,13
Pre-mixing the primary antibodies and the secondary nanobodies reduces the experimental time of immunostainings and becomes a very important time-saver when used in samples that require long staining protocols. Here we used cleared mouse cochlea imaged with light sheet microscopy to compare the staining pattern of primaries pre-mixed with 2.Nbs or using conventional secondary antibodies. Our observation suggest that pre-mixing shorten the conventional protocol by at least half the time (i.e. 6 days of staining; Fig. 4). We did not test shorter times for pre-mixing, but the fact that no clear difference in intensity or signal distribution between pre-mixed stainings for 6 or 14 days were observed, suggests that optimal incubation time might be even shorter.
Antigen clustering on cells rescued by the use of secondary nanobodies
Our results on probe-induced clustering of the POI show strong indications of conventional polyclonal 2.Abs as the major clustering element. We first demonstrated that these probe-induced artefacts can be minimized using monovalent probes as secondary reagents (e.g. 2.Nbs). The staining of living Ramos cells show how the distribution of BCRs at the plasma membrane went from a smooth to a clustered pattern when using 1.Ab–2.Ab, which was rescued by the use of 2.Nb (Fig. 6). This results suggests that monoclonal bivalent 1.Ab has some minor effect on probe-induced clustering compared to the major clusters of the POI observed when using polyclonal 2.Ab. Importantly, this probe-induced clustering artefact could also be observed when short aldehyde fixation was performed. Sample fixation with 4% paraformaldehyde for 10 minutes is a widespread practice in biology laboratories, but seems to be insufficient to fully immobilize cellular elements, in agreement with previous work.22,32 We complemented the previous observations by demonstrating that polyclonal 2.Abs drastically enhance the clustering of the POIs if samples are poorly fixed (Fig. 6). The artificial aggregation of POIs even after chemical fixation can lead to several misleading conclusions when studying for example co-localization of two or more POIs, poly-molecular arrangements or if molecular mechanisms are interpreted after imaging analysis.
Conclusion
Small, monovalent, and monoclonal probes specific to the endogenous targets are clearly the ideal probes to reveal POIs. Unfortunately, their availability is limited to a handful of targets. On the other hand, a large amount of well validated monoclonal antibodies is available. Our data suggests that the localization of primary antibodies with recombinant secondary nanobodies or probably other small monovalent binder such as protein A,33 can minimize the probe-induced clustering of targets, increase the localization accuracy in super-resolution microscopy, lower steric hindrance for detecting more target molecules, enhance the sample penetration, remove the species-limitation by pre-mixing allowing high multiplexing capabilities, and finally, increase the reproducibility of results with no needs of animals.
Materials and methods
Cell culture
In standard cells were cultured at 37 °C, 5% CO2 in a humified incubator. The human Burkitt lymphoma B cell lines DG75 and COS-7 fibroblast were obtained from the Leibniz Institute DSMZ—German Collection of Microorganisms and Cell Culture (DSMZ Braunschweig, Germany). For maintenance, cell lines were kept on Petri dishes. For experiments cells were plated on poly-L-lysine (PLL)-coated coverslips. DG75 cells were splitted every 3 days using fresh complete medium (RPMI medium supplemented with 10% Fetal Bovine Serum (FBS), 4 mM L-glutamine and 100 U ml−1 penicillin and streptomycin). COS-7 fibroblast cells were cultured in complete Dulbecco's MEM with the addition of 10% FBS, 4 mM L-glutamine, 0.6% penicillin and streptomycin. A549 cells (ATCC, Cat. No. CRL-1651) were maintained in DMEM (Thermo-Fisher Scientific, Cat. No. 10566016), supplemented with 10% Fetal Bovine Serum (Thermo-Fisher Scientific, Cat. No. 10500-064) and 1% Penicillin/Streptomycin (Thermo-Fisher Scientific, Cat. No. 15140-122). Rat primary hippocampal neuron cultures were prepared as described before by Opazo et al.30 In brief, the brains of P1–2 were extracted and placed in cold HBSS (Thermo-Fisher, Waltham, Massachusetts, USA). The hippocampi were extracted and placed in a solution containing 10 mL DMEM (Thermo Fisher), 1.6 mM cystein, 1 mM CaCl2, 0.5 mM ethylenediaminetetraacetic acid (EDTA), 25 units of papain per mL of solution, with CO2 bubbling, at 37 °C for 1 h. The solution was removed and the hippocampi were incubated in 10% FBS-DMEM, 73 μM albumin for 15 minutes. The hippocampi were triturated using a 10 mL pipette in complete-neurobasal medium [Neurobasal A (Thermo Fisher), containing 2% B27 (Thermo Fisher) and 1% Glutamax-I (Thermo Fisher). Neurons were plated (12-well plate) on poly-L-lysin-hydrochloride (Sigma-Aldrich, St Louis, Missouri, United States) coated coverslips in plating medium (500 mL MEM, 50 mL horse serum, 5 mL glutamine, 330 mg glucose. After 2 h the plating medium was replaced with 1.25 ml neurobasal-A Medium.
Staining of BCRs
For the staining of BCRs on living cells the staining was performed on ice to avoid the internalization of BCRs. Cells (∼200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per sample) were pelleted by centrifuging at 1000g, resuspended in 50 μL of ice-cold complete medium (see above) containing the investigated affinity probe (see ESI Table 1†) and incubated for 10 minutes on ice. Cells were centrifuged at 400g at 4 °C in a table top centrifuge and the excess of probe was removed. Cells were washed by resuspension in 1 ml of ice-cold Dulbecco's Phosphate Buffered Saline (DPBS) followed by incubation on ice for 3 minutes and centrifugation at 400g at 4 °C. The washing step was repeated 3 times to remove most of the excess of the fluorescent probes. When a secondary probe was used (see ESI Table 1†), the cells were further incubated with 50 μL ice-cold complete medium containing the secondary reagent and incubated for another 30 minutes on ice (staining controls without secondary probes were left for the same time on DPBS only). Washing was performed as described for the primary probe. After staining, cells were resuspended in 1 ml of cold DPBS and transferred to a 12 well plate (containing PLL coated coverslips). The plate was centrifuged at 500 rpm for 5 minutes at 4 °C. The DPBS was carefully discarded and cells were fixed with 1 mL of 4% paraformaldehyde and 0.1% GLU in PBS for 10 minutes on ice followed by 30 minutes at room temperature. The fixative was removed and quenched by adding 1 mL of 0.1 M Glycine in DPBS and incubated at room temperature for additional 20 minutes. Finally, cells were rinsed with 1 mL DPBS and mounted on a glass slide using Mowiol (6 g glycerol, 6 ml deionized water, 12 ml 0.2 M Tris buffer pH 8.5, 2.4 g Mowiol 4–88, Merck Millipore). The staining of BCRs of fixed Ramos cells, around 200000 cells per sample were pelleted by centrifuging at 1000g, resuspended in 1 mL DPBS and transferred to a single well on a 12 well plate containing PLL coated coverslips. The cells were let to sediment on the coverslips at 37 °C for 1 h. DPBS was removed and cells were fixed with one of the following conditions: 10 minutes with 4% PFA, 30 minutes with 4% PFA or 30 minutes with 4% PFA and 0.1% GLU. For all fixation conditions the first 5 minutes incubation were performed on ice and the remaining fixation time at room temperature. After fixation, the quenching of reactive aldehydes was performed as described above. Cells were finally rinsed and staining was done in 1 mL DPBS containing the different probes. After staining, cells were washed 3 times with DPBS for 5 minutes at room temperature and coverslips were mounted in Mowiol.
000 cells per sample) were pelleted by centrifuging at 1000g, resuspended in 50 μL of ice-cold complete medium (see above) containing the investigated affinity probe (see ESI Table 1†) and incubated for 10 minutes on ice. Cells were centrifuged at 400g at 4 °C in a table top centrifuge and the excess of probe was removed. Cells were washed by resuspension in 1 ml of ice-cold Dulbecco's Phosphate Buffered Saline (DPBS) followed by incubation on ice for 3 minutes and centrifugation at 400g at 4 °C. The washing step was repeated 3 times to remove most of the excess of the fluorescent probes. When a secondary probe was used (see ESI Table 1†), the cells were further incubated with 50 μL ice-cold complete medium containing the secondary reagent and incubated for another 30 minutes on ice (staining controls without secondary probes were left for the same time on DPBS only). Washing was performed as described for the primary probe. After staining, cells were resuspended in 1 ml of cold DPBS and transferred to a 12 well plate (containing PLL coated coverslips). The plate was centrifuged at 500 rpm for 5 minutes at 4 °C. The DPBS was carefully discarded and cells were fixed with 1 mL of 4% paraformaldehyde and 0.1% GLU in PBS for 10 minutes on ice followed by 30 minutes at room temperature. The fixative was removed and quenched by adding 1 mL of 0.1 M Glycine in DPBS and incubated at room temperature for additional 20 minutes. Finally, cells were rinsed with 1 mL DPBS and mounted on a glass slide using Mowiol (6 g glycerol, 6 ml deionized water, 12 ml 0.2 M Tris buffer pH 8.5, 2.4 g Mowiol 4–88, Merck Millipore). The staining of BCRs of fixed Ramos cells, around 200000 cells per sample were pelleted by centrifuging at 1000g, resuspended in 1 mL DPBS and transferred to a single well on a 12 well plate containing PLL coated coverslips. The cells were let to sediment on the coverslips at 37 °C for 1 h. DPBS was removed and cells were fixed with one of the following conditions: 10 minutes with 4% PFA, 30 minutes with 4% PFA or 30 minutes with 4% PFA and 0.1% GLU. For all fixation conditions the first 5 minutes incubation were performed on ice and the remaining fixation time at room temperature. After fixation, the quenching of reactive aldehydes was performed as described above. Cells were finally rinsed and staining was done in 1 mL DPBS containing the different probes. After staining, cells were washed 3 times with DPBS for 5 minutes at room temperature and coverslips were mounted in Mowiol.
Imaging and analysis of BCRs
Cells were imaged with a multicolor confocal STED microscope (Abberior Instruments GmbH, Göttingen, Germany) described below. Imaging was performed using a 640 nm excitation laser and a 775 nm depletion laser. The final raw STED images were obtained after the summation of 3 successive scans. STED images of cells were analysed using custom written MATLAB scripts (MATLAB Release 2014b, The MathWorks, Inc., Natick, Massachusetts, United States). For each cell center, the radii of two circles were manually adjusted so that the area between the circles contained all of the cell membrane. From this area, pixels were grouped by their angle to the cell center (in 360 bins of 1°) and maximum-projected to obtain the angle-dependent intensity ŷi along the membrane. The self-similarity of this function was then assessed by calculating its normalized autocorrelation:| |
ai = ![[scr F, script letter F]](https://www.rsc.org/images/entities/char_e141.gif) −1{(ŷi) × conj((ŷi))} −1{(ŷi) × conj((ŷi))}
| (1) |
using the normalized intensity| |
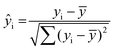 | (2) |
the mean value ŷ, the complex conjugate conj and the fast Fourier transform F. It gives a measure of how similar the intensity of two points are on the membrane depending on their angular distance. As the effect of different labelling homogeneities was best observed at a range of 8–12°, the autocorrelation from this area was then averaged for each cell (and translated to the perimeter in μm in the figures by approximating the cell diameters to 10 μm).
Peroxisome size
Primary neurons from rat hippocampi were fixed with 4% PFA 30 minutes at 4 °C followed by 30 minutes at room temperature. The neurons were incubated in a blocking and permeabilizing solutions containing 5% bovine serum albumin (BSA) and 0.1% Triton X-100 for 20 minutes at room temperature. The rabbit polyclonal anti Pmp70 antibody (Abcam, Cat. No.: ab85550) was added on the cells in a 1:300 dilution in PBS containing 2.5% BSA 0.05% for 1 h at room temperature. The cells were washed 3 time for 10 minutes each in PBS and incubated with either secondary goat anti rabbit conjugated to AbberiorStar635P (Abberior GmbH, Cat. No.: 2-0012-007-2) or the 2.Nb FluoTag-X2 anti rabbit also conjugated to AbberiorStar635P (NanoTag Biotechnologies, Cat. No.: N1002) and diluted to 1:200 and 1:100 respectively in 2.5% BSA, 0.05% Triton X-100 for 1 h at room temperature. The cells were washed 3 times for 10 minutes in PBS and finally mounted in Mowiol. The peroxisomes on neurons were imaged with the STED setup described above using a 640 nm excitation laser and 775 nm depletion laser. For determining the peroxisome diameter, the images were filtered using a bandpass filter, in MATLAB, to remove background noise, and peroxisome regions of interest were identified using an empiric threshold. The smallest ellipse diameter that fitted each peroxisome region of interest was then obtained by using the self-written MATLAB routine.
Autocorrelation on microtubule stainings
COS-7 cells were fixed with −20 °C pre-cooled methanol for 20 minutes at −20 °C. Methanol was removed and cells were blocked with 3% BSA for 20 minutes at room temperature. The cells were incubated with primary mouse monoclonal antibody anti-alpha tubulin (SySy, Cat. No.: 302211) directly coupled to AbberiorStar635p fluorophore and diluted at 1:25 in 1.5% BSA for 1 h at room temperature. The cells were washed 3 times, 5 minutes each with PBS. Cells were then incubated with either secondary nanobody FluoTag-X2 anti mouse IgG kLC conjugated to AbberiorStar580 (NanoTag Biotechnologies GmbH, Cat. No.: N1202) or secondary full antibody anti mouse coupled to AbberiorStar580 (Abberior, Cat. No.: 2-0002-005-1) diluted at 1:100 in 1.5% BSA for 1 h at room temperature. Finally, cells were washed as described above and mounted in Mowiol. Images of microtubules were taken using the Abberior Expert line STED system. A 640 nm excitation laser and 775 nm depletion laser were used for imaging the 1.Ab (AbberiorStar635P) while a 561 nm excitation laser and 775 nm depletion laser were used for imaging the fluorophore on the secondary probes (AbberiorStar580). The correlation of the STED signal provided by the secondary probe to the primary probe was analysed as follows. Lines were drawn following the stained microtubules using a self-written routine in MATLAB. The Pearsońs correlation between the directly labelled 1.Ab and the secondary probes were measured at the drawn lines. The autocorrelation of the signal from the 1.Ab was used as control.
Pre-mixing experiment Immunostaining
COS-7 cells were fixed in −20 °C pre-cooled methanol for 20 minutes at −20 °C. The cells were blocked by addition of 3% BSA in PBS for 30 minutes at room temperature. In the meantime, the 1.Abs were pre-mixed for 30 minutes with two molar excess of fluorescently-labelled 2.Nbs in PBS containing 1.5% of BSA (see ESI Table 2†). The pre-mixed complexes were then incubated on the fixed cells sequentially. In between each round of pre-mixed complex, the cells were washed 3 times for 5 minutes each with PBS and post fixed with 4% PFA for 10 minutes. The excess of fixative was quenched with 0.1 M glycine in PBS for 10 minutes. The cells were mounted in Mowiol and imaged using a multicolor laser scanning confocal microscope (the STED system described before). For the immunostaining of Fig. 3D–G and ESI Fig. 3,† COS-7 cells were stained and blocked as described above. The 1.Abs were premixed with approximately two-fold molar excess of the 2.Nbs (either fluorescently labelled or unconjugated) for 30 minutes. The premixed complexes were in this case incubated simultaneously on the cells for the amount of time indicated. The cells were then washed three times for 5 min with PBS. Hoechst was added and rinsed after 5 minutes before they were mounted in Mowiol and imaged with STED microscopy.
Pre-mixing experiment Western Blot
A confluent plate of COS-7 cells was briefly washed with ice-cold PBS before lysing the cells on the plate sitting on ice with pre-chilled Lysis buffer (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.5% IgePAL, 0.5% Sodium deoxycholate and freshly added DNAse, 1 mM PMSF and protease inhibitor cocktail (Roche). Cells were scrapped and passed through a syringe with needle gauge 26 several times avoiding foam. After max. speed centrifugation at 4 °C in a table-top centrifuge for 15 minutes. Supernatant was taken and mixed with 2× loading dye (50 mM Tris-HCl, 4% sodium dodecyl sulfate (SDS), 0.01% Serva Blue G, 12% glycerol, pH 6.8, 50 mM DTT) and heated at 95 °C for 10 minutes. Boiled samples were then loaded in 10% SDS-PAGE. Proteins in the gel were then transferred to a nitrocellulose membrane in wet trans-blot cell (Biorad). The membranes were blocked in blocking buffer (5% Nonfat Dried Milk in PBS + 0.1% Tween-20) for 1 hour at room temperature. 1.Abs were pre-mixed with the corresponding fluorescent 2.Nb for 10 minutes and then added together on the blocked nitrocellulose membranes for 60 minutes at room temperature. Membranes were washed 5 times with large volumes of PBS for 5 minutes each and read with a LiCor Sytem Odyssey Clx.
Conjugating secondary nanobodies to ssDNA or fluorophores
Secondary nanobodies (obtained from NanoTag Biotechnologies GmbH, see ESI Table 1†) were coupled to docking oligonucleotide strands (Biomers GmbH, Ulm, Germany) functionalized with an azide group at the 5′-end and an Atto488 fluorophore at the 3′-end following the protocol described by Sograte-Idrissi et al.23 In brief, the nanobody containing an extra C-terminal cysteine was reduced with 5 mM TCEP (Sigma-Aldrich, Cat. No. C4706) for 2 h on ice. TCEP was removed via 10 kDa molecular weight cut-off (MWCO) Amicon spin filters (Merck, Cat. No. UFC500324) and the nanobody was coupled through maleimide conjugation chemistry to a maleimide-DBCO crosslinker (Sigma-Aldrich, Cat. No. 760668). After removal of excess crosslinker through 10 kDa MWCO Amicon spin filters, the nanobody was coupled to the docking oligo containing an azide group at its 5′-end (Biomers) through a strain promoted azide–alkyne cycloaddition reaction. To avoid background signal, the excess of docking oligo was removed by a size exclusion chromatography column (Superdex® Increase 75, GE Healthcare) on an ÄKTA pure 25 system (GE Healthcare). The docking strand sequences were obtained from Agasti et al.20 and can be found in ESI Table 2.†
Nanobodies bearing ectopic cysteines were first reduced with 10 mM of TCEP for 1–2 h. After removing TCEP with a Nap5 column (GE Healthcare), reduced nanobodies were immediately exposed to ∼3 molar excess of maleimide-functionalized fluorophore (e.g. CF633, Alexa488, Alexa546, Star635p) for 2 hours. Subsequently, the excess of dye was removed using a size exclusion chromatography column (Superdex® Increase 75, GE Healthcare) on an ÄKTA pure 25 system (GE Healthcare).
DNA-coupling of antibody
Donkey anti-mouse secondary antibody (Jackson Immunosearch, Cat. No. 715-005-151) was labelled with a DNA strand via a DBCO-sulfo-NHS ester linker according to the protocol as previously described.3 Briefly, primary amines of the antibody were reacted with a DBCO-sulfo-NHS ester cross-linker (Jena Bioscience, Cat. No. CLK-A124-10) for two hours at 4 °C. Unreacted cross linker was then removed using a Zeba desalting column (40 kDa MWCO, Thermo-Fisher Scientific, Cat. No. 87766). The antibody-DBCO conjugate was then attached to a DNA strand functionalized with an azide group at the 5′-end via copper-free click chemistry. Excess DNA-strands were removed using 100 kDa MWCO Amicon spin filters (Merck Millipore, Cat. No. UFC510096). Docking strand sequences were obtained from Agasti et al.20 and can be found in ESI Table 2.†
Stainings for DNA-PAINT
Cells for DNA PAINT imaging were plated on an 8-well chamber coverglass II (Sarstedt, Cat. No.: 94.6190.802 or ibidi, Cat. No. 80827 ibidi, Cat. No. 80827) and grown overnight. The next day, cells were fixed. COS-7 cells were fixed with pre-cooled methanol for 20 minutes at −20 °C. The cells were then blocked with 3% (w/v) BSA for 20 minutes at room temperature and incubated with a primary mouse monoclonal anti-alpha tubulin antibody directly labelled with Atto647N (SySy, Cat. No.: 302 211) and diluted 1:25 in 1.5% BSA for 1 h at room temperature. Unbound 1.Ab was removed by washing the cells 3 times with PBS for 5 minutes each. They were then incubated with the 2.Nb or 2.Ab coupled to DNA-PAINT docking sequences. The cells were washed 3 times for 5 minutes with PBS.
Rat primary hippocampal neuron, were fixed by adding 4% PFA for 30 minutes on ice and 4% PFA for 30 additional minutes at room temperature. The neurons were blocked and permeabilized with 3% (w/v) BSA + 0.1% (v/v) Triton X-100 for 20 minutes at room temperature. The mouse monoclonal anti Bassoon (Enzo, Cat. No.: ADI-VAM-PS003-F) and the mouse monoclonal anti Homer (SySy, Cat. No.: 1600111) were pre-mixed in a 1:5 molar ratio with 2.Nb anti mouse coupled to P1 (5′-TTATACATCTATTTT-Atto488-3′) and P5 (5′-TTTCAATGTATTTTT-Atto488-3′) respectively. The pre-mixed anti Homer 1.Ab and its 2.Nb were added on the cells for 1 h with slow orbital shaking. The cells were then washed 3× 5 minutes each with PBS and 1× 5 minutes with PBS supplemented with 0.1 M NaCl. The 1.Ab–2Nb. complex were briefly fixed by adding 4% PFA for 5 minutes. The fixative was removed and the remained quenched with 0.1 M glycine for 5 minutes. The pre-mixed anti Bassoon 1.Ab with its 2.Nb was added to the cells for 1 h at room temperature, and post-fixed and quenched as before. For drift correction purposes, cells were incubated with a 1:10 dilution of 90 nm gold particles (cytodiagnostics, Cat. No. G-90-100) for 10 minutes, rinsed 4× with PBS and stored at 4 °C until imaging was performed.
DNA-PAINT imaging
The correspondent imager strand to the DNA-PAINT docking sites used on the nanobodies (ESI Table 3†), were equipped with a Cy3b fluorophore at their 3′-end. Imager strands were diluted in PBS supplemented with 500 mM NaCl and 1× Trolox (Sigma-Aldrich, Cat. No. 238813-1G). Imager strands were used at concentrations between 0.5 nM and 2 nM to optimize the number of binding events per time (see ESI Table 4†). The focal plane was found by searching in the 488 nm channel. Cells were then imaged in the 561 nm channel with a 100–200 ms exposure time per frame for 30.000–60.000 frames. When exchange of imager was performed, the chamber was washed 10 times with PBS supplemented with 0.5 M NaCl until no residual blinking was observed anymore. The reconstruction of the raw data and the drift correction with cross correlation and gold particles as fiducial markers was performed with Picasso Sotware.3 Microtubule filament sizes were measured via exported regions and Gaussian fits in Origin on the localizations. Images were acquired as described below and raw data movies were reconstructed with the Picasso software suite. Drift correction and multicolor alignment was performed via redundant cross-correlation and 90 nm gold particles as fiducial markers. The Picasso software suite was also used to detect the localisation frequency events in ESI Fig. 4A.†
Cochlear staining
Mice C75Bl6/J of 3 weeks of age were euthanized by decapitation. Cochleae were harvested and fixed in 4% PFA for 45 minutes at room temperature. Afterwards, they were processed following the cochlea-adapted version of the iDISCO+protocol (Keppeler and Duque-Afonso et al., in preparation). Briefly, they were decalcified in 10% EDTA in PBS, pH 8, for 2 days and treated with 25% N,N,N′,N′-tetrakis(2-hydroxypropyl)ethylenediamine in PBS for another 2 days, in order to remove endogenous fluorescence34 at room temperature under constant rotation. The samples underwent the methanol-free pre-treatment of the iDISCO+protocol,35 followed by the regular procedure for immunostaining and clearing. The pre-treatment consisted in subsequent incubations at 37 °C under constant shaking of the following solutions: 0.2% Triton X-100 in PBS (2 × 1 h), 0.2% Triton X-100/20% DMSO in PBS (1 day), 0.1% Triton X-100/20% DMSO/0.1% Tween-20/0.1% Deoxycholate/0.1% IGEPAL CA-630 in PBS (1 day), Triton X-100 in PBS (2 × 1 h). The immunostaining continued at 37 °C, under constant shaking, with the incubation of the tissue in a Permeabilization solution (0.16%TritonX-100/20%DMSO/2.3% Glycine (0.3M) in PBS, 2 days) and in a Blocking solution (0.16% TritonX-100/10%DMSO/3%BSA in PBS, 2 days). The 1.Ab (Guinea Pig antiserum anti-parvalbumin- α, 195 004, Synaptic System) was pre-mixed with the 2.Nb (Nanobody anti-guinea pig Alexa546) using a molar ratio of 1:3 or 45 min, under constant rotation, at room temperature. The PTwH buffer contained 0.2% Tween-20/0.001% Heparin in PBS. The primary antibody was diluted in a solution containing 5%DMSO/1.5%BSA in PTwH with a concentration of 1:300. The 2.Ab (Goat-Anti Guinea pig 568, Invitrogen, A11075, 1:500) and the 1.Ab pre-mixed with the 2.Nb were diluted in a solution containing only 1.5%BSA in PTwH. The sample were incubated in 4 different ways (37 °C, under shaking): (1) 6 days and (2) 14 days in the solution containing the 1.Ab premixed with the 2.Nb, (3) 3 days and (4) 7 days with the 1.Ab followed by a washing step of 1 day in PTwH at room temperature and the incubation of the 2.Ab for 3 and 7 days respectively. Before the clearing procedure, the samples were washed in PTwH for 1 day at room temperature. Finally, samples were dehydrated in an increasing methanol dilution series (20, 40, 60, 80, 100 and 100% Methanol in ddH2O, one hour each), incubated in 66% Dicloromethane/33% Methanol for 3 hours plus two consecutive incubation in 100% DCM for 15 minutes each for lipid extraction, and immerse in Dibenzylether, as a refractive index matching solution.
Cochlear probe penetration quantification
The original stack was resampled by a factor of 2.15 × 2.15 × 2 and converted to 8-bits in FIJI.36 Then, the ganglion was coarsely segmented manually with TrakEM237 and imported to 3DSlicer.38,39 There, a median filter with a kernel of 10 × 10×1 pixel was applied and the resulting image was threshold segmented, converted to a 3D closed surface or mesh and stored as a .stl file, as it is the input format needed for the following step. Centerlines of the ganglion were then calculated using the vmtkcenterline function of the open source software VMTK (the Vascular Modelling Toolkit, Orobix Srl) and then imported to MATLAB for further analysis. For every sample, the mesh, centerline and raw stack were imported to MATLAB. The centerline was fitted using spline interpolation and 100 position equally spaced were retrieved. In each of these positions, 14 radii of 200 μm were positioned, 6 orthogonal to the rest. The chosen orientation was parallel to the apical-basal axis formed by the most apical and most basal coordinate of the centerline. Those radii that were inside of the mesh, checked by the function inpolyhedron, or outside of the original image space, were removed. Radii were mapped in the image space and the pixel values in their coordinates were used to obtain the line profiles. The minimum of each profiles was subtracted for each to have a comparable baseline.
Microscopy setups
Fluorescent imaging of ESI Fig. 1† was done with Nikon inverted epifluorescence microscope. The microscope was equipped with an HBO 100 W lamp and an IXON X3897 Andor Camera. For all samples, a 60× Plan apochromat oil immersion objective (NA 1.4) was used (from Nikon). Images were obtained using the image acquisition software NiS-Elements AR (Nikon). STED microscopy images were obtained using STED Expert line microscope (Abberior Instruments, Göttingen, Germany) composed of a IX83 inverted microscope (Olympus, Hamburg, Germany) with a UPLSAPO 100× 1.4 NA oil immersion objective (Olympus). Confocal images were obtained from the same setup without using the STED depletion laser. DNA-PAINT imaging was carried out on an inverted Nikon Eclipse Ti microscope (Nikon Instruments) with the Perfect Focus System, applying an objective-type TIRF configuration with an oil-immersion objective (Apo SR TIRF 100×, NA 1.49, Oil). Two lasers were used for excitation: 561nm (200 mW, Coherent Sapphire) or 488 nm (200 mW, Toptica iBeam smart). The laser beam was passed through a clean-up filter (ZET488/10× or ZET561/10×, Chroma Technology) and coupled into the microscope objective using a beam splitter (ZT488rdc or ZT561rdc, Chroma Technology). Fluorescence light was spectrally filtered with two emission filters (ET525/50m and ET500lp for 488 nm excitation and ET600/50 and ET575lp for 561 nm excitation, Chroma Technology) and imaged on a sCMOS camera (Andor Zyla 4.2) without further magnification, resulting in an effective pixel size of 130 nm after 2 × 2 binning. Camera Readout Sensitivity was set to 16-bit, Readout Bandwidth to 540 MHz. Light-sheet images of the cochleae were done using a light-sheet microscope (LaVision Biotec Ultramicroscope II). The laser power was constant for all the samples except for the sample incubated with 1.Ab–2.Ab for 14 days, which was 6.75 times lower (13.5% vs. 2%). The stacks were acquired with a total zoom of 8× (2× MVPLAPO Objective and 4× Optic Zoom microscope body), a step size of 3 μm, with a light-sheet of 30% width and a thickness of 5 μm (NA: 0.148, unidirectional illumination and 11–12 steps of dynamic horizontal focus. The images were imported to FIJI36 for calculating the maximum intensity projection image and to generate the RGB tif files with a mpl-magma look-up-table.
Conflicts of interest
F.O. and S.O.R. are funders shareholders of NanoTag Biotechnologies. S.O.R. received compensation as consultant of NanoTag Biotechnologies. All the other authors declare no competing interests.
Acknowledgements
We thank Niklas Engels for providing Ramos cells. We thank Riccardo Testolin for the help in designing the hybrid plot graphs. We thank Eugenio F. Fornasiero, Sebastian Jaehne and Sven Truckenbrodt for reading and commenting the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) through Cluster of Excellence Nanoscale Microscopy and Molecular Physiology of the Brain (CNMPB) to F.O. and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2067/1-390729940 to S. O. R. and T. M. T. S. and S. S. acknowledge support from the DFG through the Graduate School of Quantitative Biosciences Munich (QBM). This work was further supported by the DFG through the Emmy Noether Program (DFG JU 2957/1-1), the SFB 1032 (Nanoagents for spatiotemporal control of molecular and cellular reactions, Project A11), the ERC through an ERC Starting Grant (MolMap, grant agreement no. 680241), the Max Planck Society, the Max Planck Foundation, and the Center for Nanoscience (CeNS) to R. J.
Notes and references
- A. H. Coons, H. J. Creech, R. N. Jones and E. Berliner, The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody, J. Immunol., 1942, 45(3), 159–170 CAS.
- R. P. Haugland, Coupling of Monoclonal Antibodies with Fluorophores, in Monoclonal Antibody Protocols 205–222, Humana Press, 1995, DOI:10.1385/0-89603-308-2:205.
- J. Schnitzbauer, M. T. Strauss, T. Schlichthaerle, F. Schueder and R. Jungmann, Super-resolution microscopy with DNA-PAINT, Nat. Protocol., 2017, 12, 198–1228 CrossRef PubMed.
- J. Ries, C. Kaplan, E. Platonova, H. Eghlidi and H. Ewers, A simple, versatile method for GFP-based super-resolution microscopy via nanobodies, Nat. Methods, 2012, 9, 582–584 CrossRef CAS PubMed.
- Y. Eilers, H. Ta, K. C. Gwosch, F. Balzarotti and S. W. Hell, MINFLUX monitors rapid molecular jumps with superior spatiotemporal resolution, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6117–6122 CrossRef CAS PubMed.
- T. Pleiner, et al., Nanobodies: site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation, eLife, 2015, 4, e11349 CrossRef PubMed.
- D. B. Peckys, U. Korf and N. de Jonge, Local variations of HER2 dimerization in breast cancer cells discovered by correlative fluorescence and liquid electron microscopy, Sci. Adv., 2015, 1, e1500165 CrossRef PubMed.
- F. Opazo, et al., Aptamers as potential tools for super-resolution microscopy, Nat. Methods, 2012, 9, 938–939 CrossRef CAS PubMed.
- S. Strauss, et al., Modified aptamers enable quantitative sub-10-nm cellular DNA-PAINT imaging, Nat. Methods, 2018, 15, 685–688 CrossRef CAS PubMed.
- T. Schlichthaerle, et al., Site-Specific Labeling of Affimers for DNA-PAINT Microscopy, Angew. Chem., Int. Ed., 2018, 57, 11060–11063 CrossRef CAS PubMed.
- R. Bedford, et al., Alternative reagents to antibodies in imaging applications, Biophys. Rev., 2017, 9, 299–308 CrossRef CAS PubMed.
- T. Pleiner, M. Bates and D. Görlich, A toolbox of anti-mouse and anti-rabbit IgG secondary nanobodies, J. Cell Biol., 2018, 217, 1143–1154 CrossRef CAS PubMed.
- M. Maidorn, S. O. Rizzoli and F. Opazo, Tools and limitations to study the molecular composition of synapses by fluorescence microscopy, Biochem. J., 2016, 473, 3385–3399 CrossRef CAS PubMed.
- M. A. Gomes de Castro, C. Höbartner and F. Opazo, Aptamers provide superior stainings of cellular receptors studied under super-resolution microscopy, PLoS One, 2017, 12, e0173050 CrossRef PubMed.
- T. Fang, et al., Nanobody immunostaining for correlated light and electron microscopy with preservation of ultrastructure, Nat. Methods, 2018, 15, 1029–1032 CrossRef CAS PubMed.
- L. Manning and C. Q. Doe, Immunofluorescent antibody staining of intact Drosophila larvae, Nat. Protoc., 2017, 12, 1–14 CrossRef CAS PubMed.
- T. Ezaki, Antigen retrieval on formaldehyde-fixed paraffin sections: its potential drawbacks and optimization for double immunostaining, Micron, 2000, 31, 639–649 CrossRef CAS PubMed.
- S. Mayor, K. G. Rothberg and F. R. Maxfield, Sequestration of GPI-anchored proteins in caveolae triggered by cross-linking, Science, 1994, 264, 1948–1951 CrossRef CAS PubMed.
- S. Spiegel, S. Kassis, M. Wilchek and P. H. Fishman, Direct visualization of redistribution and capping of fluorescent gangliosides on lymphocytes, J. Cell Biol., 1984, 99, 1575–1581 CrossRef CAS PubMed.
- S. S. Agasti, et al., DNA-barcoded labeling probes for highly multiplexed Exchange-PAINT imaging, Chem. Sci., 2017, 8, 3080–3091 RSC.
- O. K. Wade, et al., 124-Color Super-resolution Imaging by Engineering DNA-PAINT Blinking Kinetics, Nano Lett., 2019, 19(4), 2641–2646 CrossRef CAS PubMed.
- K. A. K. Tanaka, et al., Membrane molecules mobile even after chemical fixation, Nat. Methods, 2010, 7, 865–866 CrossRef CAS PubMed.
- S. Sograte-Idrissi, et al., Nanobody
Detection of Standard Fluorescent Proteins Enables Multi-Target DNA-PAINT with High Resolution and Minimal Displacement Errors, Cells, 2019, 8, 48 CrossRef CAS PubMed.
- A. Auer, et al., Nanometer-scale Multiplexed Super-Resolution Imaging with an Economic 3D-DNA-PAINT Microscope, ChemPhysChem, 2018, 19, 3024–3034 CrossRef CAS PubMed.
- K. Richter, et al., Presynaptic cytomatrix protein bassoon is localized at both excitatory and inhibitory synapses of rat brain, J. Comp. Neurol., 1999, 408, 437–448 CrossRef CAS PubMed.
- J.-H. Tao-Cheng, S. Thein, Y. Yang, T. S. Reese and P. E. Gallant, Homer is concentrated at the postsynaptic density and does not redistribute after acute synaptic stimulation, Neuroscience, 2014, 266, 80–90 CrossRef CAS PubMed.
- A. Dani, B. Huang, J. Bergan, C. Dulac and X. Zhuang, Super-resolution Imaging of Chemical Synapses in the Brain, Neuron, 2010, 68, 843–856 CrossRef CAS PubMed.
- S. Truckenbrodt, et al., X10 expansion microscopy enables 25-nm resolution on conventional microscopes, EMBO Rep., 2018, 19, e45836 CrossRef PubMed.
- M. A. Gomes de Castro, et al., Differential organization of tonic and chronic B cell antigen receptors in the plasma membrane, Nat. Commun., 2019, 10, 820 CrossRef CAS PubMed.
- F. Opazo, et al., Limited Intermixing of Synaptic Vesicle Components upon Vesicle Recycling, Traffic, 2010, 11, 800–812 CrossRef CAS PubMed.
- P. M. Pereira, et al., Fix Your Membrane Receptor Imaging: Actin Cytoskeleton and CD4 Membrane Organization Disruption by Chemical Fixation, Front. Immunol., 2019, 10, 675 CrossRef CAS PubMed.
- K. N. Richter, et al., Glyoxal as an alternative fixative to formaldehyde in immunostaining and super-resolution microscopy, EMBO J., 2018, 37, 139–159 CrossRef CAS PubMed.
- T. Schlichthaerle, M. Ganji, A. Auer, O. K. Wade and R. Jungmann, Bacterial-derived antibody binders as small adapters for DNA-PAINT microscopy, ChemBioChem, 2019, 20, 1032–1038 CrossRef CAS PubMed.
- A. Greenbaum, et al., Bone CLARITY: Clearing, imaging, and computational analysis of osteoprogenitors within intact bone marrow, Sci. Transl. Med., 2017, 9, eaah6518 CrossRef PubMed.
- N. Renier, et al., Mapping of Brain Activity by Automated Volume Analysis of Immediate Early Genes, Cell, 2016, 165, 1789–1802 CrossRef CAS PubMed.
- J. Schindelin, et al., Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- A. Cardona, et al., TrakEM2 Software for Neural Circuit Reconstruction, PLoS One, 2012, 7, e38011 CrossRef CAS PubMed.
- R. Kikinis, S. D. Pieper and K. G. Vosburgh, 3D Slicer: A Platform for Subject-Specific Image Analysis, Visualization, and Clinical Support, in Intraoperative Imaging and Image-Guided Therapy, Springer New York, 2014, pp. 277–289, DOI:10.1007/978-1-4614-7657-3_19.
- A. Fedorov, et al., 3D Slicer as an image computing platform for the Quantitative Imaging Network, Magn. Reson. Imaging, 2012, 30, 1323–1341 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: File 3. See DOI: 10.1039/d0nr00227e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  abc,
Thomas Schlichthaerlede,
Carlos J. Duque-Afonso
abc,
Thomas Schlichthaerlede,
Carlos J. Duque-Afonso