
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoparticle rearrangement under stress in networks of cellulose nanofibrils using in situ SAXS during tensile testing†
J.
Engström‡
 ab,
A. M.
Jimenez‡
c and
E.
Malmström
*ab
ab,
A. M.
Jimenez‡
c and
E.
Malmström
*ab
aKTH Royal Institute of Technology, School of Engineering Sciences in Chemistry, Biotechnology and Health, Department of Fibre and Polymer Technology, Sweden. E-mail: mavem@kth.se
bWallenberg Wood Science Centre, Teknikringen 56-58, SE-100 44 Stockholm, Sweden
cDepartment of Chemical Engineering, Columbia University, 10027 New York, NY, USA
First published on 25th February 2020
Abstract
This study aims to describe and evaluate the mechanism for increased strain-at-break of composites made of cellulose nanofibrils (CNFs) reinforced with nanoscopic latex particles (<200 nm) stabilized by a cationic polyelectrolyte as corona. The applied latex nanoparticles (NPs), synthesized by polymerization-induced self-assembly (PISA), are composed of a neutral core polymer, either poly(butyl methacrylate) (PBMA) or poly(methyl methacrylate) (PMMA). At room temperature, PBMA is close to its glass transition (Tg), while PMMA is below its Tg. Nanocomposites with 75 wt% CNFs and 25 wt% NPs were analyzed using in situ small angle X-ray scattering during tensile testing, monitoring the structural evolution of the NPs under strain. The scattering of the spherical PMMA NPs, which do not coalesce like the PBMA NPs, shows changes to the organization of the NPs in the CNF-network. The observations are corroborated by cross-sectional transmission and scanning electron microscopy. No distinct change from spherical to ellipsoidal shape is evidenced for the PMMA NP cores during tensile strain. Changes in anisotropic scattering produced by the three-dimensional NP structure appear to be very different between nanocomposites loaded with PMMA or PBMA NPs, contrasting commonly described two-dimensional CNF networks. The discrete PMMA NPs can reorganize within the CNF–NP double network under strain, resulting in maintained strength and increased strain-at-break. Increasing the humidity (20, 50 and 80% RH) during in situ measurements further emphasizes this effect in the PMMA composite, relative to the PBMA composite and CNF reference films. The onset of deformation occurs at strain values beyond the fracture of the more brittle films, indicating the effect of secondary nanoscale interaction available only for the PMMA composite, extending the plastic deformation and increasing the ductility. These results provide key insights into the deformation mechanism occurring during tensile testing in the CNF composites loaded with PMMA NPs.
Introduction
There is a growing interest in cellulose nanofibrils (CNFs) for material applications with the increasing urge to replace fossil resources for plastic materials with bio-based counterparts. Despite CNFs’ promising properties as nanomaterials, with individual fibrils having estimated elastic moduli above 130 GPa, networks of CNFs inherently come with significant challenges.1–3 When produced with carboxyl groups on the surface (such as TEMPO-oxidation4 or carboxymethylation5), the CNFs hydrophilic character can provide good strainability in the wet state, similar to nanocomposites composed of polysaccharides from algae and Ca2+.6 In their dried state, these CNF networks can form very strong and stiff films,7–10 with relatively brittle properties (strain-at-break <20% at 50% RH). Promising approaches have been reported for improving CNF-type composite materials (including oxidized species from TEMPO- or periodate oxidation, and choice of counter ions), however, these materials remain significantly more brittle than many conventional thermoplastic materials used for packaging and food containers (such as high density polyethylene, polypropylene and polylactic acid) which commonly show strainability greater than 50%.10,11Attempts have been made to use polymeric nanoparticle additives in networks of CNFs, though often only small improvements can be observed, especially when targeting high loadings of CNFs (>50 wt%).12–14 Our previous study on nanocomposites based on CNFs using a tailored latex-system (PISA-latexes) was a breakthrough in this regard, when an unexpected increase in strainability was observed.15 PISA-latexes are particularly interesting in structure–property studies as their core–shell morphology is stabilized by a covalently bonded hydrophilic corona polymer to result in stable colloidal dispersions and eliminating the risk of migrating stabilizers once dried. The controlled radical polymerization synthesis provides unprecedented possibilities to fine-tune the chemical composition and properties of the corona and the core, respectively, and their resulting compatibility with the matrix of CNFs. This was demonstrated in our previous work, where nanocomposite films of CNFs with 25 wt% of a PDMAEMA-b-PMMADP1410 latex (D ≈ 100 nm) resulted in almost two times the strain-at-break compared to using PDMAEMA-b-PBMADP1410 latex and the CNF reference. This result, showing reduced brittleness upon addition of NPs with a brittle core-polymer (PMMA), indicates that nanoscale interactions between CNF-surfaces and the NPs are more important than their individually combined properties. At CNF-loadings of 75 wt% the CNFs form a distinct percolating network with notable directionality.15 The rigid PMMA NPs, compared to the softer PBMA NPs, maintained its spherical shape after drying at room temperature, and seemed to be able to interact with the CNFs in the nanocomposite in a way that allowed for increased strain-at-break. These promising results did not come with any in-depth understanding of the toughening mechanism. A stick-and-slip mechanism between CNFs and latex NPs was postulated, with only the decreasing mechanical properties as supporting evidence. In order to gain a more fundamental understanding on how double networks can result in enhanced toughness, it is necessary to utilize a non-destructive technique that can provide environmentally controlled nanoscale structural analysis; hence our focus on small angle X-ray scattering (SAXS) in this study. Combined with the possibility to perform tensile testing in situ during the analysis makes it an even more useful characterization tool for elucidating the underlying mechanism behind the strain enhancement induced by the nanoscopic latex particles, especially when used in combination with microscopy techniques.16–18 The structural evolution of nanoparticles has previously been analyzed with SAXS, for example, tensile testing of latex films caused the spherical latex nanoparticles to become more ellipsoidal.19 However, despite analysis of composites of inorganic nanoparticles in cellulose networks and polymer matrices, the use of CNFs as the composite matrix and their interactions with tailored PISA-latexes, especially during the straining-process, has, to the best of our knowledge, not been studied.16,20
The structural evolution of PISA-latex NPs in the CNF network, and the subsequent toughening, was monitored with SAXS during in situ tensile testing by analyzing the anisotropy in scattering, as well as the direct change in the form and structure factors of the NPs. Results indicate that PMMA NPs re-organize under strain, like that of the PBMA NPs, but that the maintained rigid NPs allow for a secondary structural change. Building from the previous hypothesis, latexes acting with a stick-and-slip mechanism, or structural reorganization of NPs and CNFs, seems more probable than a nanoscale change of the NP cores from spherical to ellipsoidal. This implies that the CNFs are forced to untangle during strain, and if all three dimensions of the film have fibrils in the percolating network, the presence of the PMMA-latex allows the film to become more flexible.
Experimental section
Materials
2-Dimethylaminoethyl methacrylate (DMAEMA, Aldrich, 98%), methyl methacrylate (MMA, Acros, 99%), n-butyl methacrylate (nBMA, Sigma Aldrich, 99%) 2,2′-azobis(2-methylpropion-amidine)dihydrochloride (AIBA, Aldrich, 97%), hydrochloric acid (HCl, VWR Prolabo, 35 wt%, technical grade), 1,3,5-trioxane (Aldrich, ≥99%), sodium chloride (VWR), potassium chloride (Merck), sodium chlorite (puriss p.a.), sodium hypochlorite (14% solution), 2,2,6,6-tetramethyl-1-piperidineyloxy (TEMPO), all purchased from Sigma Aldrich, and sodium bromide (Alfa Aesar, 99+%) were used as received. Water used was either deionized or Milli-Q water. 4-Cyano-4-thiothiopropylsulfanyl pentanoic acid (CTPPA) was synthesized according to literature procedure.21–23The pulp used for the preparation of cellulose nanofibrils (CNFs) was a never-dried dissolving pulp (60% Norwegian spruce and 40% Scots Pine) kindly supplied by Domsjö Aditya Birla AB, Domsjö, Sweden. The preparation of CNFs was conducted using TEMPO-oxidation at pH 6.8 in phosphate buffer to ease the liberation of the fibrils from the fibres, according to a method from Saito et al.4,24 to reach a charge density of ∼600 μeq g−1, as measured by conductometric titration.25 After TEMPO-oxidation the oxidized fibres were passed through a high-pressure homogenizer (Microfluidizer M-110EH, Microfluidics Corp) by two passes through 400 μm and 200 μm chambers at 1000 bar and four passes through 200 μm and 100 μm at 1650 bar, similarly to the method used by Cervin et al.26 to produce a gel of 0.89 wt% dry content.
Instrumentation and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm was used for 20 minutes.
000 magnification on 7.8 mm working distance. The samples were mounted on a metal stub with carbon tape and coated with a 5 nm layer of Pt/Pd with a Cressington 208HR sputter coater.
000 rpm was used for 20 minutes.
000 magnification on 7.8 mm working distance. The samples were mounted on a metal stub with carbon tape and coated with a 5 nm layer of Pt/Pd with a Cressington 208HR sputter coater.
Preparation of NPs (PISA-latexes)
Nanoparticles (NPs) with PMMA or PBMA in the core were obtained in water by RAFT-mediated surfactant-free emulsion polymerization using polymerization-induced self-assembly (PISA), according to previously reported procedures by employing a pre-formed PDMAEMA-based macroRAFT agent.15,28Preparation of CNF–NP nanocomposite films
A gel of CNFs (∼33.7 g) at 0.89 wt%, as produced after homogenization, was dispersed in Milli-Q (116 mL added and total volume ∼150 mL, CNF content ∼0.2 wt%) with an ultra-turrax at 10000 rpm for 20 min in a 500 mL flask. The dispersion of CNFs was degassed for 30 min under vacuum to remove any air bubbles. The NP dispersion of either PMMA37, PMMA100, PnBMA32 or PnBMA82 (3 g L−1 in 5 mM sodium phosphate buffer) were added dropwise to the CNF dispersion under magnetic stirring. The mixtures had a total dry weight of 400 mg, a CNF content of 75 wt% and latex NPs content of 25 wt%. The final dispersion was left under slow magnetic stirring for 1 h to allow latex adsorption to the CNFs’ surfaces, where after the dispersion was vacuum filtered through a glass filter funnel (7.2 cm in diameter) using a 0.65 μm PVDF membrane, DVPP Millipore, USA,7,29 with a targeted thickness of around 80 μm and grammage of 100 g m−2. After filtration, the films were left to dry for at least 24 hours in a Petri dish in the fume hood prior to being stored in a conditioned room (23 °C, 50% RH, min 24 h) prior to further characterizations. The sample notation of resulting composite films is CNF followed by the NPs used as filler and its size assessed by DLS in subscript; CNF-PMMAxx, e.g. CNF-PBMA32.
A reference film with only CNFs was also prepared, denoted as CNF reference. This film was prepared as described above with the sole addition of 33 mL of 5 mM sodium phosphate buffer since no NPs were added, before the film formation.
Results and discussion
Mechanical properties of CNF nanocomposites comparing ex situ and in situ
The herein investigated NPs (PISA-latexes), Table 1, have been thoroughly analyzed in previous studies.15,28 Nanocomposites were prepared by adding the NPs (PISA-latex) dropwise to a dispersion of TEMPO-oxidised CNFs where after films (7.2 cm ∅) were formed by filtration. Four nanocomposites were prepared (Fig. 2), all having the same ratio between NPs and CNF (25:75 wt%.) A reference film of only CNF was also prepared. Cross-sectional SEM images, together with a schematic illustration of the formed nanocomposites are shown in Fig. 1.
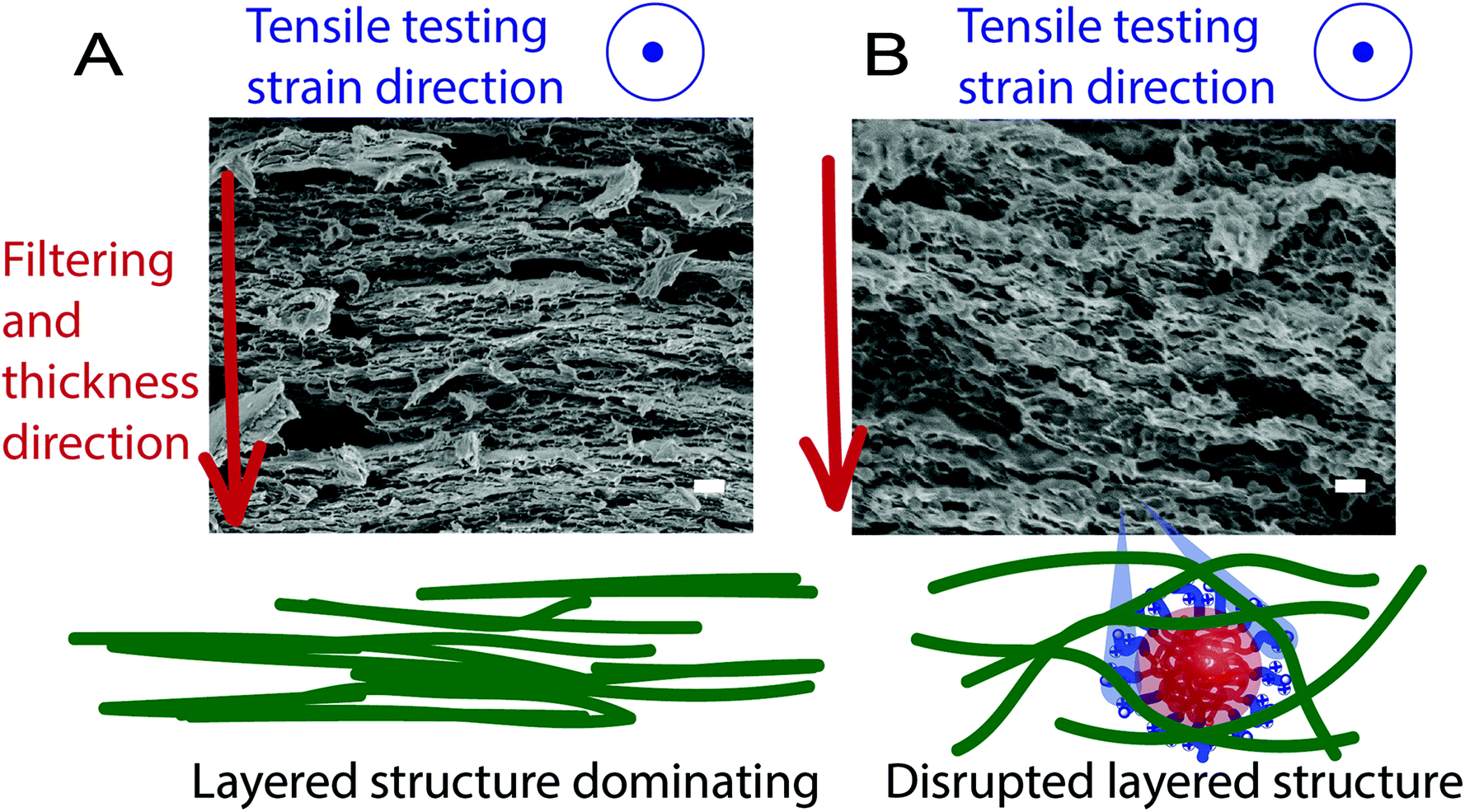 | ||
| Fig. 1 SEM-images of the cross-section of (A) CNF reference and (B) CNF-PMMA100 composites created by cryo-fracture. Scale bars are 300 nm. Included are the schematic illustrations of the nanostructures comparing CNF and CNF-PMMA100 to show the formation of layered structures and the tensile direction. | ||
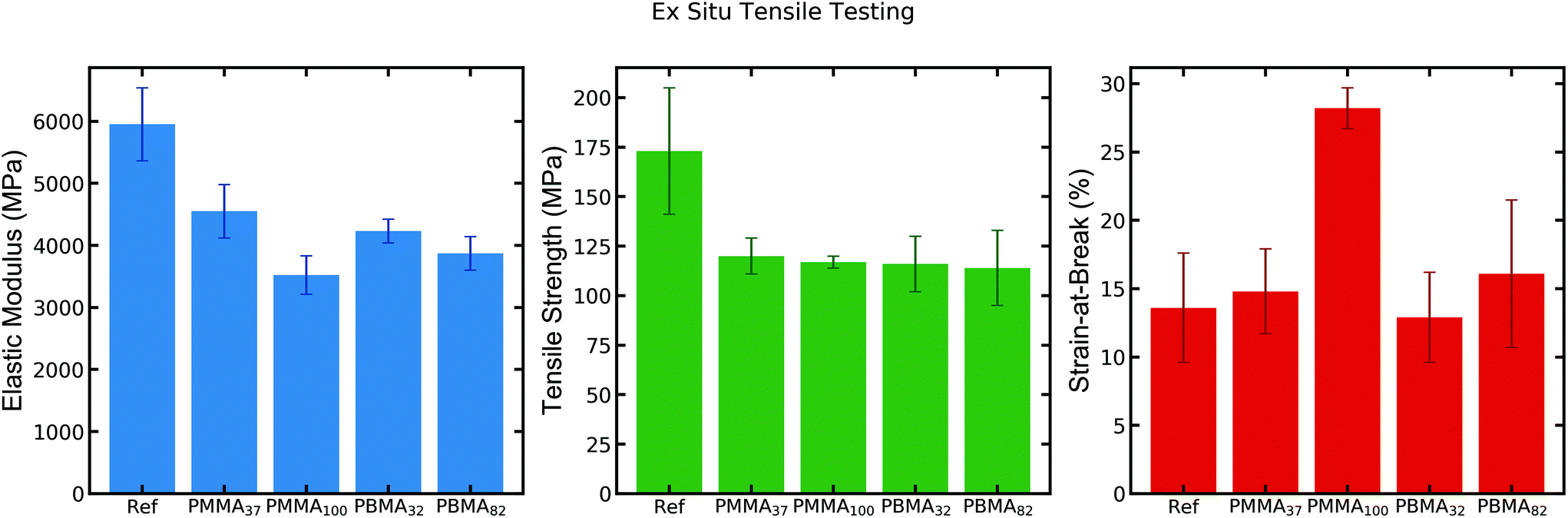 | ||
| Fig. 2 Ex situ tensile testing (10% strain rate) of CNF reference and CNF-latex nanocomposites at 50% RH.15 | ||
The resulting mechanical properties from ex situ tensile testing show a consistent drop in tensile strength and elastic modulus (25–35%) for the latex-containing nanocomposites as compared to the neat CNF-film (Fig. 2). The strain-at-break is largely unaffected for most samples, except for that of CNF-PMMA100, which exhibits roughly twice the strain-at-break compared to the CNF reference film. This observation shows that the addition of the latex PMMA100 imparts a toughening effect into the nanocomposite. Despite the small decrease in strength of this sample (which could offset the toughness value), it demonstrates a significantly enhanced strain-at-break at 50% RH.
To further investigate the toughening induced by PMMA100 we used high resolution synchrotron small-angle X-ray scattering (SAXS) at the CMS beamline at NSLS-II while performing in situ tensile testing on a Linkam tensile stage. The SAXS data is discussed in more depth, vide infra, but the in situ tensile testing during the SAXS analysis was first compared with the ex situ results to verify that no large deviation in material properties could be observed. To increase the temporal resolution of the in situ measurement, a strain rate of 1% was implemented. One set of tensile testing at 10% strain-rate was also performed in situ for comparison. Both the 1% and 10% strain-rate resulted in minor deviations in the resulting mechanical properties relative to that of the ex situ 10% strain measurements performed on an Instron instrument. Since it is well known that networks of CNFs are very sensitive to moisture and swell at high humidity, measurements were performed at a variety of RH-values.3,30 The Linkam stage provided a sealed sample chamber in which the humidity could be controlled with an inlet flow of combined dry and saturated nitrogen with regulated flows. The resulting humidity control provided a tuneable test environment between 20–80% RH at room temperature. As observed for the samples measured in situ at 50% RH in Fig. 3, the results follow closely with the ex situ measurements and show great control of the strain and measurement of strength, focusing specifically on the two larger latexes, PBMA82 and PMMA100.
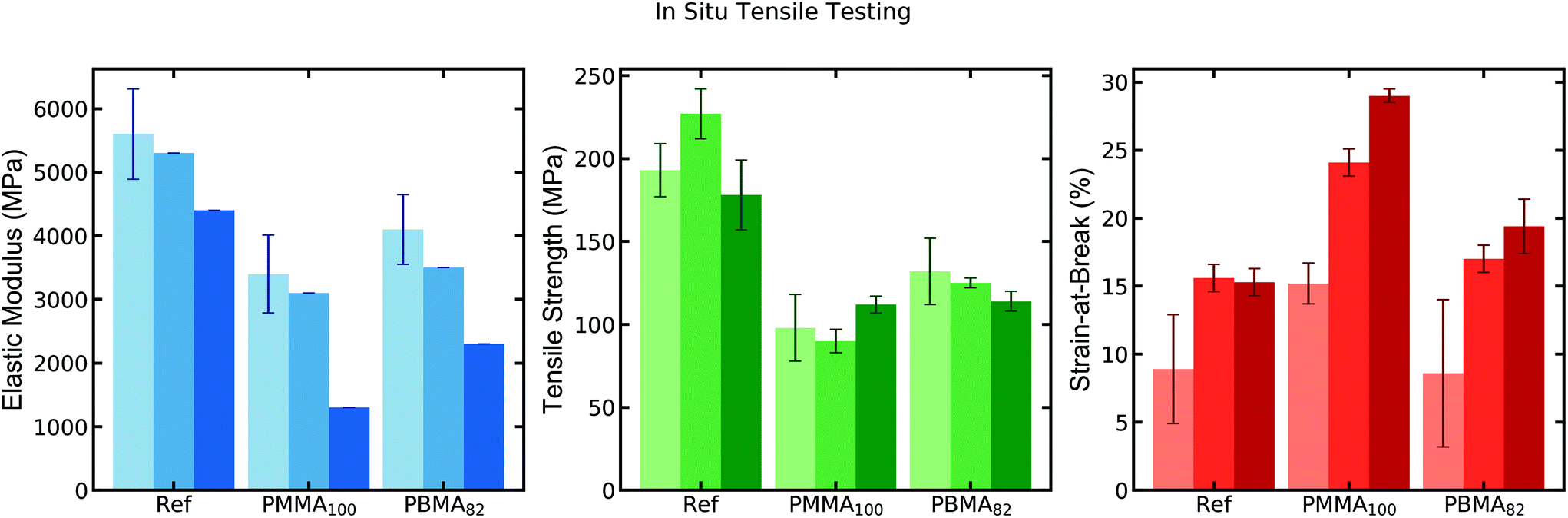 | ||
| Fig. 3 In situ tensile testing (1% strain rate) of CNF reference and CNF–NP nanocomposites at 20% (light shade), 50% (medium shade) and 80% (dark shade) RH. (Samples analyzed at 50 and 80% RH consist of only two repeated measurements due to limited time for analysis at the beamline.) | ||
While having relatively little effect on the strength of the samples, the humidity noticeably increases the strainability of all the samples. At 80% RH, the strength of the samples begins to change, decreasing for CNF reference due to the plasticizing effect of the water, whereas the latex-filled nanocomposites attain a higher strength from the prolonged plastic deformation (larger strain-at-break). At lower RH (20%) the materials become much more brittle and few samples reach the same maximum values of strength due to the shortened plastic deformation, with fractures occurring around roughly half the strain observed at 50% RH. It is worth noting that the strength increases for some of the nanocomposites with the lower humidity. Focusing on the strainability of the most interesting sample, CNF-PMMA100, the strain-at-break increases from 15% to 24% when moving from 20% to 50% RH, though always significantly higher than the other samples.
The mechanical properties, Fig. 2 and 3, together with the images in Fig. 1, indicate the importance of the bottom-up design to achieve more strainable nanomaterials of CNFs (herein CNFs content of >75 wt%) in the dry state. Based on the microscopy images, three-dimensional entanglements of CNFs must be formed rather than the otherwise reported two-dimensional sheets in the tensile direction (Fig. 1). With the formation of a 3D-network utilizing these specific NPs where size and core stiffness matters, the CNFs can interact over the whole thickness direction, thus withstanding larger strain. The layered structure as observed for the CNF reference, Fig. 1A, could indicate that the strain divides into two steps: (i) initial stiff elastic region (E-modulus) with large stress transfer and (ii) the plastic deformation where the CNF layers slips before fracture. Step (ii) would be governed by the secondary interactions and adhesion between the CNFs, with both hydrogen bonding and van der Waals interactions present. These interactions and the resulting mechanical properties of the CNF networks would also be affected by parameters such as hemicellulose content and porosity, but they all together explain why water and humidity decrease the strength and stiffness; by interacting and weakening the strong interactions between the CNFs and the layers within the structure.7 Seemingly, the layers form during filtration of the CNFs and the drying of an otherwise dense CNF material, both are processes in need of further investigations, and there are structural artefacts that can be made simply by the method of fracture formation using either a scissor or blade cut, tensile testing and cryo-fractures. In our previous study, it was shown that the swelling of CNF films, only dried at room temperature, results in cavities between the layered planes.30 Walther et al. also presented a corroborating image of the layered structure from film formation and discussed the effect of the weakening of the network's internal bonding with water.30,31 Hence, the placement of strongly adhering NPs (PDMAEMA corona) between such layered structures, with the formation of bundles of CNFs along the thickness direction, would allow for a more flexible material with prolonged plastic deformation, Fig. 2 and 3. The assumption that the NPs maintain their spherical core shape during this assembly, from wet to dry state, appears to aid in maintaining the 3D network (Fig. 1), drastically increasing the strain-at-break. The effect is pronounced mostly for the NPs with PMMA cores, in contrast to previous studies where low Tg latexes gave an increase in ductility, thus supporting that the interactions of the CNF and the NP corona play a large role.13
Owing to the monodisperse nature of the NPs (Table 1) and the high loading (25 wt%), the structural integrity of the NPs makes them perfect visualizing probes, allowing for analysis at nanoscale using TEM and in situ SAXS. The analyses were performed for the CNF reference and the nanocomposites containing PBMA82 and PMMA100 (results on the smaller NP filled composites can be found in the ESI†). With the following nanoscale analysis, we investigate how the structural evolution of the NPs, PMMA100 and PBMA82, influences the mechanism that gives rise to enhanced strainability and seemingly prolonged plastic deformation – properties necessary for enhancement in otherwise potentially viable, renewable materials.
Ex situ TEM of CNF–NPs double networks
Nanocomposite samples were microtomed to provide high-resolution cross-sectional TEM images of the films (Fig. 4). Each of the samples show dark ribbons oriented largely orthogonal to the casting direction of the film. The CNF-PMMA100 has obvious spherical NPs distributed within the network of CNFs with no obvious change to their shape (Fig. 4B). The CNF-PBMA82 has instead semi-continuous domains of clustered NPs (Fig. 4C), formed from the production of the nanocomposites, and therefore show very little resemblance to the original spherical NP shape seen in water dispersion. The low Tg core of the PBMA NP allows the filler to completely lose its shape and spread throughout the CNFs, like a blended polymer. It is important to note that the TEM image shown of the PBMA composite has undergone heat treatment above the Tg of PBMA in order to secure it in epoxy for microtoming. SEM imaging of the sample in Fig. S2,† performed without heating, supports the lack of coherent NP structure being present, though the degree to which this blending occurs may be exacerbated in the TEM. Despite this, high resolution transmission imaging of the composites support the structures observed in previously published SEM images.15 | ||
| Fig. 4 Cross-sectional TEM of (A) CNF, (B) CNF-PMMA100 and (C) CNF-PBMA82. Scale bars are 4 μm. | ||
In situ SAXS analysis of CNF–NPs double networks during tensile testing
The following part of this study aims at investigating the nanostructures of CNFs and the CNF-latex double networks using SAXS, first focusing on systems measured at 50% RH and 1% strain rate. The results in Fig. 5 show the reduced SAXS patterns of the materials, focusing on the difference between scattering in the horizontal (red line) and vertical (blue line) directions. By integrating the pattern perpendicular versus parallel to the strain we see the divergence of the resulting intensities. As seen in Fig. 5A, the CNF reference demonstrates no obvious divergence in the scattering, as there are no contrasting scatters in the system that would appear to be reorganizing (only one inset image of the 2D image is included in this case). The CNF-PMMA100 composite scattering, seen in Fig. 5B, shows a scattering curve clearly resembling that of spherical particles within the network of CNFs. Scattering on the NPs in dilute solution indicates that the well-dispersed NP form factor has a radius of 51 nm with a polydispersity of 0.085 (see ESI Fig. S3† for fitting). For high concentrations of NPs within the CNF network, a Percus–Yevick structure factor is incorporated to fit the composite scattering. With the increase in strain, the scattering intensity sharply diverges, more notably in the direction parallel to strain. The very subtle change in horizontal scattering is dwarfed by the stark decrease in vertical scattering intensity. At some point during the strain, the vertically integrated intensity begins to increase again, though some hysteresis in the effective structure factor is apparent (notice the reduced depth of the oscillating intensity). A similar trend is observed for the CNF-PBMA82 composites. These samples, unlike the rigid PMMA NPs, provide a scattering pattern only vaguely reminiscent of a spherical form factor with the −4 slope in the Porod region, and a slight turning over at low q, around where we could expect this sized NP form factor to be. A spherical form, even of very high polydispersity, could not be used to fit the curves due to the high degree of coalescence, seen in the TEM image in Fig. 4C. Despite this loss of their initially spherical form, the composite shows a similar trend in scattering anisotropy during strain, where the vertically integrated intensity drops during strain, but then increases again slightly before fracture. The latexes of smaller size (32–37 nm) show similar SAXS pattern transitions, but the seemingly high level of aggregation for these composites, as indicated from the scattering, make it more difficult to discern these morphological transitions (see ESI Fig. S4†). | ||
| Fig. 5 SAXS curves measured during in situ tensile strain measurements – increasing arbitrarily shifted intensities indicate measurements before strain, at maximum change in anisotropy during strain, at maximum anisotropy, and at sample failure. Samples measured are (A) CNF reference, (B) CNF-PMMA100, and (C) CNF-PBMA82. SAXS curves were integrated horizontally (red) and vertically (blue) to demonstrate the appearance of anisotropy with increasing strain time. Black dashed curve is the isotropically integrated data from pre-strain scattering, shifted up with each measurement to more clearly show the relative changes in intensity. Insets: 2D SAXS patterns before strain (left) and at maximum anisotropy (right), again demonstrating the visible anisotropy in the scattering pattern. | ||
A primary aim of this study was to analyze how the nanocompositeś anisotropy changes with the type of latex and the correlating mechanical properties, thereby leading to evidence of the mechanism inside the double-network. To help monitor this divergence in scattering, an anisotropy factor (AF) was calculated from the scattering invariants where AF = (Qhor − Qvert)/Qvert, and  . This ad hoc method of monitoring changes in scattering helps to easily quantify changes in the structural alignment of NPs in directions perpendicular and parallel to strain, though we will note later how this method is further challenged at high strain. Fig. 6A shows the change in AF tracked alongside the simultaneous force measured during continuous sample strain. Without any scatterer present in the CNF reference film on the SAXS length-scale there appears to be no change in AF with increasing strain, though some small amount of inherent anisotropy in the scattering exists from the CNF fibrils. The PMMA- and PBMA-filled nanocomposites, however, both show a gradual increase in AF for increasing strain, with a notably similar trend prominently appearing at the transition from the elastic to plastic deformation strain. The CNF-PMMA100 nanocomposite is able to reach further strains, upon which the AF begins to plateau, and even drop back down. As noted in the 1D scattering, this is not a return of the NPs to their initial state, but an apparent relaxation to another structure. To further probe this structural change, we perform the same strain experiment out to 12.5% strain (sufficiently into the plastic deformation region), and then hold the sample to relax. The AF of the CNF reference film again remains unaffected. The CNF-PMMA100 and CNF-PBMA82 samples again sharply increase in anisotropy up until the cessation of strain, at which point the PMMA sample AF value drops slightly at very short times, then very slowly settles down to lower and lower values (note the time scale spanning into hours). This tracks closely with the relaxation of the sample stress measurement. The PBMA sample, upon cessation, similarly halts the sharply increases trend, however the AF in this case has a gradual increase over long times.
. This ad hoc method of monitoring changes in scattering helps to easily quantify changes in the structural alignment of NPs in directions perpendicular and parallel to strain, though we will note later how this method is further challenged at high strain. Fig. 6A shows the change in AF tracked alongside the simultaneous force measured during continuous sample strain. Without any scatterer present in the CNF reference film on the SAXS length-scale there appears to be no change in AF with increasing strain, though some small amount of inherent anisotropy in the scattering exists from the CNF fibrils. The PMMA- and PBMA-filled nanocomposites, however, both show a gradual increase in AF for increasing strain, with a notably similar trend prominently appearing at the transition from the elastic to plastic deformation strain. The CNF-PMMA100 nanocomposite is able to reach further strains, upon which the AF begins to plateau, and even drop back down. As noted in the 1D scattering, this is not a return of the NPs to their initial state, but an apparent relaxation to another structure. To further probe this structural change, we perform the same strain experiment out to 12.5% strain (sufficiently into the plastic deformation region), and then hold the sample to relax. The AF of the CNF reference film again remains unaffected. The CNF-PMMA100 and CNF-PBMA82 samples again sharply increase in anisotropy up until the cessation of strain, at which point the PMMA sample AF value drops slightly at very short times, then very slowly settles down to lower and lower values (note the time scale spanning into hours). This tracks closely with the relaxation of the sample stress measurement. The PBMA sample, upon cessation, similarly halts the sharply increases trend, however the AF in this case has a gradual increase over long times.
 | ||
| Fig. 6 Measured AF values plotted on the left axis (dots) with the simultaneously measured force of stress on the right axis (dashed line) over time at 50% RH and 1% strain for (brown) CNF reference film, (blue) CNF-PMMA100, and (green) CNF-PBMA82. Tensile measurements performed (A) to failure, and (B) to 12.5% strain and held. | ||
The increase in adhesive interactions between the nanoscale components within the network (CNFs and latex) likely leads to either increases in E-modulus, strength or strainability, depending on the mechanism at nanoscale, followed by the structural evolution and organization of the latex NPs. During tensile testing, the vertical SAXS integration follows the tensile direction and the horizontal along the width. Therefore, depending on the initial homogeneity of NPs in either direction, this relative shift in scattering will correspond to the relative change in organization and structural evolution of the latex inside. Fig. 1 provides a visual image for the hypothesis of how this re-organization might look at the nanoscale and in the double network, indicating the layered structures of CNFs (4A) and how the latex facilitates a disruption of the layered structure (4B). Such CNF networks and their layered structures (Fig. 1A) created by film formation have been visualized in literature recently by Walther et al. when they attempted to correlate structure–property relationships using counterions and the effect of humidity.31 It is important to remember that the structural inferences from SAXS provide length scales only out to ∼200 nm, roughly twice the diameter of the NPs. This means that the scattering provides very local changes in the NP reorganization, rather than CNF length scales, which span micron. (Ex situ USAXS scans probing out to the micron scale did not provide any obvious indication of observable structures at that length scale, though these measurements are notably performed with a 1D detector. See ESI Fig. S5†). Having said that, we notice from the structural change a horizontal clumping of NPs as the sample compresses in the lateral direction while they become stretched and spread vertically. The clumping of NPs is further supported by a growing structure peak at ∼85 nm. This suggests that the NPs, ∼100 nm, are somehow clumping together with center-to-center distances smaller than that of their radii, likely due to a softening of the corona, allowing the PMMA centres to sink closer. The cationic polyelectrolyte PDMAEMA chains in the corona are not typically “soft” in a dry state, but they show softening with the increased humidity in the system, along with their fibril interactions.
The temporal resolution allowed by the synchrotron beamline made it possible to detect simultaneous wide-angle X-ray scattering (WAXS) of the samples during tensile testing, to determine if the crystalline scattering from the CNFs changes during strain. However, no clear difference was observed due to strain in the CNF reference nor the nanocomposites (see ESI Fig. S6†). From these results, it appears that the CNFs can neither organize nor align enough to increase the crystallinity during strain.
Effect of humidity on the CNF–NPs double networks
The previous section focused on samples analyzed in situ at 50% RH, consistent with that of the ex situ laboratory environment where Instron measurements were performed. The mechanical section provides data at 20, 50, and 80% RH, demonstrating the intuitive result that increases in humidity allow for higher strains, with relatively minimal changes in strength. SAXS patterns of the CNF-PMMA100 samples show increasing AF values with increasing humidity (Fig. 7). This suggests that the higher humidity allows for more NP rearrangement prior to fracture, correlated to the increased strain. The increased amount of water inside the double network makes the cationic corona of the PISA-latex more solvated and flexible, possibly allowing re-organization to occur, while still maintaining some adhesion to the surfaces of CNFs. Interestingly, at the highest humidity (80%) the structure within the network changes drastically well before the time of fracture. This allows for a more detailed investigation of the change in horizontal versus vertical scattering that causes the decrease in AF that occurs at high strain, despite the lack of recovery to the initial structure. This is emphasized in the scattering of the 80% RH tests in Fig. 7B where the scattering along both directions has significantly diverged from that of the initial, isotropic sample. It is not clear how much of this final change is due to structural changes between the NPs or coalescing of the particle interfaces through a more flexible corona, discussed further below. | ||
| Fig. 7 In situ measurements on CNF-PMMA100 performed at (purple) 20%, (green) 50%, and (yellow) 80% RH. SAXS on the sample (A) at the time of maximum AF and (B) just before failure. (Circles and triangles are integrated horizontally and vertically, respectively. Black dashed lines are isotropically integrated SAXS prior to strain.) (C) Simultaneous (dots) AF and (dashed line) force measurements over time. | ||
Structural evolution and organization of high Tg NPs within double networks
From the high resolution SAXS, structural information can be determined for the PMMA100 latex inside the network of CNFs throughout the applied strain by tensile testing. The structural evolution of the NPs provides a deeper understanding of the mechanism for increased ductility of the double network. The scattering of the well-defined spherical NPs distributed within the CNF network, as visualized in TEM (Fig. 4B) and SEM (Fig. 1), can then be fitted, corresponding to the strain-induced transitions. It is hypothesized that the toughening mechanism during in situ tensile testing should manifest in either a change in the particle shape (coalescence or ellipsoidal stretching) or inter-particle structural changes, either of which would appear in the scattering patterns. SAXS data for CNF PMMA100 was fit, accounting for the structural changes seen during strain testing (see ESI Fig. S7†), in order to quantitatively describe the directional anisotropy seen from the straining of the material in situ. Form factor and Percus–Yevick structure factor fits do not support a clear change in NP shape (i.e. spherical to ellipsoidal). The ex situ TEM also provides no evidence for drastic changes in the NP (PMMA100) shape. It is important to note that the more smeared scattering, with higher intensity and less oscillations, seen at high strain in the high humidity tests is in fact similar to what would be expected of more ellipsoidal particles. However, the similar contrast of the core and corona could allow for changes in the form factor through changes in interpenetration of the PDMAEMA coronas while still maintaining a rigid PMMA core.Provided that the NPs (PMMA100) do not change their core shape, we break down the evolution of the NP structure into three regimes following the force and AF of the samples. The ability to reach each regime is notably dependent on the RH of the experiment. The first regime sees a slight increase in anisotropy as NPs move from their initially random, isotropic dispersion into a more anisotropic arrangement. This occurs in the elastic regime as NPs are flexibly squeezed together horizontally and spread out vertically, which continues into the plastic deformation of the sample. The second regime follows the stable, constant increase anisotropy during plastic deformation into a plateau of AF where there is little change in the organization of the NPs, possibly indicating the strong adhesion of the NPs to the CNFs due to no movement. Around the occurrence of this plateau in AF, there is a reproducible inflection in the measured force, transitioning the plastic response of the material at high strain. Finally, at higher humidity (80% RH), the plateau is quickly followed by a third regime, where a turnover in the AF correlates with an impending upturn in the force, transitioning the plastic response of the material at high strain, reminiscent of a strain hardening effect. The decrease in AF is, again, not a recovery of the initial NP organization, but a shift in NP structure postulated to be caused by NPs, adhered to CNF surfaces and moving with the bundles as they slip like layers in CNF reference, thus colliding vertically. The shearing of particles into vertical stacks and subsequent CNF alignment supports the upturn seen in the force measurement. The vertical scattering in this regime could no longer be fit simply with the Percus–Yevick structure model and would likely need a combination of forms and structures to represent its scattering pattern. By breaking up the analysis this way we can propose some mechanism for the mechanical response of the composite that correlates with the reorganization of NPs.
Conclusions
The nanocomposites of 75 wt% CNFs and 25 wt% PISA-latexes, with PDMAEMA-stabilizing corona, have been successfully analyzed by SAXS with both ex situ (lab-scale) and in situ (synchrotron beamline). The SAXS data provide structural information about the fillers before and during strain measurements. All materials’ nanostructures as visualized by SAXS analysis correlated well with cross-section imaging using TEM and SEM. The 200% increase in strain-at-break observed only for the CNF-PMMA100 composites compared to CNF reference and other types of NPs (altering core stiffness and size), but shown at all humidity's investigated (20, 50 and 80% RH). The nanoscale mechanism, hypothesized previously as a stick-and-slip between the NP and the CNFs, was indirectly supported and expanded by showing a distinct reorganization of NPs inside the networks through in situ SAXS analysis. This reorganization was quantified by an increase in anisotropy in the nanocomposites, showing large differences between CNF references and CNF–NP nanocomposites (specifically CNF-PMMA100). Furthermore, the effect of humidity on the re-organization of NPs in these materials was highlighted by the appearance of a third regime in the CNF-PMMA100 samples, where the material shows a drop in anisotropy before failure. The tailoring of the assembly of these specific NPs (with PDMAEMA-corona) together with CNFs in water dispersions, enable them to disburse uniformly and disrupt the otherwise more layered 2D network formation. Upon strain, the reorganization of the 3D double network and the NP–CNF interactions lead to a stick-and-slip mechanism during plastic deformation, that results in a more strainable material. The applied PISA-latex at 100 nm diameter, with its PMMA-core and PDMAEMA-corona, is perfectly designed to facilitate the stick-and-slip mechanism, relating back to the actual film formation of the nanocomposites whereby balancing electrostatic interaction and controlled flocculation during drying we create a well-distributed 3D double-network.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Knut and Alice Wallenberg foundation through the Wallenberg Wood Science Centre (WWSC) for financial support. Thanks to Professor Sanat Kumar for helpful discussions and research support. This research used the 11-BM CMS beamline of National Synchrotron Light Source-II (NSLS-II), Brookhaven National Laboratory (BNL), a U.S. Department of Energy User Facility operated for the Office of Science by BNL under contract No. DE-SC0012704. Thanks to Masafumi Fukuto and Ruipeng Li for their assistance at the CMS beamline. Thanks also to Alexandros Alexakis for his help running the SAXS experiments. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Proposal No. GUP-62379. Thanks to Columbia University for funding AMJ.References
- I. Sakurada, Y. Nukushina and T. Ito, J. Polym. Sci., 1962, 57, 651–660 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- A. J. Benítez and A. Walther, J. Mater. Chem. A, 2017, 5, 16003–16024 RSC.
- T. Saito, Y. Nishiyama, J.-L. Putaux, M. Vignon and A. Isogai, Biomacromolecules, 2006, 7, 1687–1691 CrossRef CAS PubMed.
- L. Wågberg, G. Decher, M. Norgren, T. Lindström, M. Ankerfors and K. Axnäs, Langmuir, 2008, 24, 784–795 CrossRef PubMed.
- T. Benselfelt, J. Engström and L. Wågberg, Green Chem., 2018, 20, 2558–2570 RSC.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- I. Siró, D. Plackett, M. Hedenqvist, M. Ankerfors and T. Lindström, J. Appl. Polym. Sci., 2011, 119, 2652–2660 CrossRef.
- T. Saito, R. Kuramae, J. Wohlert, L. A. Berglund and A. Isogai, Biomacromolecules, 2012, 14, 248–253 CrossRef PubMed.
- M. Shimizu, T. Saito, H. Fukuzumi and A. Isogai, Biomacromolecules, 2014, 15, 4320–4325 CrossRef CAS.
- P. Larsson, L. Berglund and L. Wågberg, Cellulose, 2014, 21, 323–333 CrossRef CAS.
- M. Wang, A. Olszewska, A. Walther, J.-M. Malho, F. H. Schacher, J. Ruokolainen, M. Ankerfors, J. Laine, L. A. Berglund, M. Österberg and O. Ikkala, Biomacromolecules, 2011, 12, 2074–2081 CrossRef CAS PubMed.
- E. L. Vinay Kumar, E. Lazarus, P. Salminen, D. Bousfield and M. Toivakka, Nord. Pulp Pap. Res. J., 2016, 31, 7 Search PubMed.
- V. Favier, G. R. Canova, J. Y. Cavaillé, H. Chanzy, A. Dufresne and C. Gauthier, Polym. Adv. Technol., 1995, 6, 351–355 CrossRef CAS.
- J. Engström, F. L. Hatton, L. Wågberg, F. D'Agosto, M. Lansalot, E. Malmström and A. Carlmark, Polym. Chem., 2017, 8, 1061–1073 RSC.
- P. Akcora, H. Liu, S. K. Kumar, J. Moll, Y. Li, B. C. Benicewicz, L. S. Schadler, D. Acehan, A. Z. Panagiotopoulos, V. Pryamitsyn, V. Ganesan, J. Ilavsky, P. Thiyagarajan, R. H. Colby and J. F. Douglas, Nat. Mater., 2009, 8, 354 CrossRef CAS PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- S. Mourdikoudis, R. M. Pallares and N. T. K. Thanh, Nanoscale, 2018, 10, 12871–12934 RSC.
- J. Zhang, S. Hu, J. Rieger, S. V. Roth, R. Gehrke and Y. Men, Macromolecules, 2009, 42, 4795–4800 CrossRef CAS.
- U. M. Garusinghe, V. S. Raghuwanshi, C. J. Garvey, S. Varanasi, C. R. Hutchinson, W. Batchelor and G. Garnier, Colloids Surf., A, 2017, 513, 373–379 CrossRef CAS.
- T. Boursier, I. Chaduc, J. Rieger, F. D'Agosto, M. Lansalot and B. Charleux, Polym. Chem., 2011, 2, 355–362 RSC.
- G. Bouhadir, N. Legrand, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1999, 40, 277–280 CrossRef CAS.
- S. H. Thang, Y. K. Chong, R. T. A. Mayadunne, G. Moad and E. Rizzardo, Tetrahedron Lett., 1999, 40, 2435–2438 CrossRef CAS.
- T. Saito, M. Hirota, N. Tamura, S. Kimura, H. Fukuzumi, L. Heux and A. Isogai, Biomacromolecules, 2009, 10, 1992–1996 CrossRef CAS PubMed.
- S. Katz, R. P. Beatson and A. M. Scallan, Sven. Papperstidn., 1984, 87, 48–53 Search PubMed.
- N. T. Cervin, E. Johansson, J.-W. Benjamins and L. Wågberg, Biomacromolecules, 2015, 16, 822–831 CrossRef CAS PubMed.
- D. Horn, in Progress in Colloid & Polymer Science, ed. G. Lagaly, F. H. Müller and A. Weiss, Steinkopff, 1978, vol. 65, ch. 28, pp. 251–264 Search PubMed.
- L. Carlsson, A. Fall, I. Chaduc, L. Wågberg, B. Charleux, E. Malmström, F. D'Agosto, M. Lansalot and A. Carlmark, Polym. Chem., 2014, 5, 6076–6086 RSC.
- H. Fukuzumi, T. Saito, T. Iwata, Y. Kumamoto and A. Isogai, Biomacromolecules, 2008, 10, 162–165 CrossRef PubMed.
- F. L. Hatton, J. Engström, J. Forsling, E. Malmström and A. Carlmark, RSC Adv., 2017, 7, 14947–14958 RSC.
- A. J. Benítez, J. Torres-Rendon, M. Poutanen and A. Walther, Biomacromolecules, 2013, 14, 4497–4506 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr10964a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2020 |