
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePLGA protein nanocarriers with tailor-made fluorescence/MRI/PET imaging modalities†
Yajie
Zhang
 a,
Miguel
García-Gabilondo
b,
Alba
Grayston
b,
Irene V. J.
Feiner
c,
Irene
Anton-Sales
a,
Rodrigo A.
Loiola
d,
Jordi
Llop
ce,
Pedro
Ramos-Cabrer
fg,
Ignasi
Barba
h,
David
Garcia-Dorado‡
h,
Fabien
Gosselet
d,
Anna
Rosell
*b and
Anna
Roig
*a
a,
Miguel
García-Gabilondo
b,
Alba
Grayston
b,
Irene V. J.
Feiner
c,
Irene
Anton-Sales
a,
Rodrigo A.
Loiola
d,
Jordi
Llop
ce,
Pedro
Ramos-Cabrer
fg,
Ignasi
Barba
h,
David
Garcia-Dorado‡
h,
Fabien
Gosselet
d,
Anna
Rosell
*b and
Anna
Roig
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), 08193 Bellaterra, Catalonia, Spain. E-mail: anna.roig@csic.es
bNeurovascular Research Laboratory, Vall d'Hebron Institut de Recerca, Universitat Autònoma de Barcelona, 08035 Barcelona, Catalonia, Spain. E-mail: anna.rosell@vhir.org
cRadiochemistry and Nuclear Imaging Group, CIC biomaGUNE, Basque Research and Technology Alliance (BRTA), 20014 San Sebastian, Guipúzcoa, Spain
dUniversity of Artois, Blood-Brain Barrier Laboratory (BBB Lab), UR2465, F-62300 Lens, France
eCIBERES, Centro de Investigación Biomédica en Red, 28029 Madrid, Spain
fMagnetic Resonance Imaging Laboratory, CIC biomaGUNE, Basque Research and Technology Alliance (BRTA), 20014 San Sebastian, Guipúzcoa, Spain
gIkerbasque, Basque Foundation for Science, 48013 Bilbao, Spain
hCardiovascular Diseases Research Group, Vall d'Hebron University Hospital and Research Institute, Universitat Autònoma de Barcelona, 08035 Barcelona, Spain
First published on 23rd January 2020
Abstract
Designing theranostic nanocarriers with high protein payload and multimodality tracking without cross interferences between the different imaging probes and the delicate protein cargo is challenging. Here, chemical modifications of poly(lactic-co-glycolic acid) (PLGA) to produce nanocapsules (NCs) that incorporate several imaging moieties are reported. The biocompatible and biodegradable PLGA-NCs can be endowed with a magnetic resonance imaging (MRI) reporter, two fluorescence imaging probes (blue/NIR) and a positron emission tomography (PET) reporter. The modular integration of these imaging moieties into the shell of the NCs is successfully achieved without affecting the morphochemical properties of the nanocarrier or the protein loading capacity. In vivo biodistribution of the NCs is monitored by MRI, PET and NIRF and the results from different techniques are analyzed comparatively. The viabilities of two different human endothelial cells in vitro show no toxicity for NC concentration up to 100 μg mL−1. The morbidity of mice for 2 weeks after systemic administration and the hepatic/pancreatic enzymes at the plasma level indicate their in vivo biosafety. In summary, the new theranostic PLGA nanoplatform presented here shows versatile in vitro/in vivo multimodal imaging capabilities, excellent biosafety and over 1 wt% protein loading.
Introduction
Drug delivery nanocarriers can be extremely advantageous to administer insoluble, sensitive or multi-component drugs by protecting the encapsulated therapeutic molecules from clearance, inactivation or degradation, as well as by reducing potential toxicity, increasing blood circulation time or tissue specificity.1–5 Poly(lactic-co-glycolic acid) (PLGA) has demonstrated optimal properties for the encapsulation of a large variety of therapeutic agents and it is extensively employed in nanomedicine.6,7 Moreover, PLGA-based nanocarriers have been used as drug delivery systems to administer proteins,8,9 DNA10 and anticancer drugs among others.1,11,12 Nonetheless, despite their compositional similarities, any novel PLGA-based drug delivery system will require a thorough evaluation to address potential toxicity issues and study the pharmacokinetics and the pharmacodynamics of each specific carrier. Such preclinical in vitro and in vivo studies, comprising multiple biological characterization phases, could be expedited by endowing the carrier with complementary imaging capacities. For instance, in vitro cellular uptake can be easily imaged with fluorescence13,14 while radioimaging15,16 or near infrared (NIR) imaging1,17 are well suited for in vivo biodistribution studies. However, the simultaneous incorporation of all these imaging probes into a single nanocarrier unnecessarily increases the complexity of the system, potentially limiting translation to the clinics. Moreover, any additional component of the theranostic carrier will have an enormous impact on the manufacturing process, production cost and in complying with the medical regulations. In this sense, a drug delivery nanocarrier easily adaptable to incorporate one or several imaging moieties in a modular approach can be a smart strategy to guide the carrier preclinical development while guaranteeing that a simpler formula is used for the final product. However, this strategy is suitable only when the inclusion or removal of the imaging probes do not affect the size, shape and main compositional traits of the final carrier.Furthermore, conventional approaches to physically blend imaging probes within the carrier can lead to incomplete conclusions or misinterpretations on the carrier's biodistribution.18,19 For example, loading two different molecules as probes into the same carrier led to opposite conclusions regarding the kinetics of brain-specific delivery.19 A growing body of evidence suggests that new methods will be required to monitor nanocarriers’ biodistribution avoiding interference of the imaging moieties with the encapsulated drug or leaching of the imaging probes.19,20
Here, we report on PLGA nanocapsules (NCs) as a multimodal theranostic platform for in vivo drug delivery. We describe the chemical synthetic routes to covalently label the PLGA with biocompatible small molecule fluorophores or radioligands. It involves the fabrication of PLGA NCs integrating several moieties for their in vitro and in vivo imaging. Specifically, superparamagnetic iron oxide nanoparticles (SPIONs) enabled magnetic resonance imaging (MRI), fluorescence imaging at different emitting wavelengths (blue and NIR) and 89Zr-labeling enabled positron emission tomography (PET) imaging. We show that the morphology and size of PLGA NCs are not altered by either the incorporation or the removal of one of the several imaging probes. In all cases, the imaging moieties are chemically attached to the PLGA shell matrix as opposed to being entrapped in the core of the nanocapsule.13–15,17 This approach is advantageous to avoid interferences of the imaging moieties with the cargo in the core.21 This is especially important for delicate payloads such as proteins, enzymes or microRNAs. For this, we have used bovine serum albumin (BSA) as a model protein to evaluate the protein loading capability.
Experimental section
All reagents were purchased from Sigma Aldrich unless otherwise specified. The Ethics Committee of Animal Experimentation of the Vall d'Hebron Research Institute approved all experimental animal procedures (protocol number 70.18), which were performed in accordance with the Spanish legislation and the Directives of the European Union. Experiments performed at CIC biomaGUNE were approved by the Ethical Committee of the Institution and by the corresponding authorities (Departamento de Promoción Económica, Turismo y Medio Rural, Diputación Foral de Guipúzcoa; project codes PRO-AE-SS-059 and PRO-AE-SS-067).Modification and functionalization of the commercial PLGA
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolic ratio 50:50, RG502H, Mw 12000) was activated and reacted with ethylenediamine. The carboxylic groups were firstly activated with N,N′-dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS). 0.025 mmol of PLGA was dissolved in 8 ml of dichloromethane (DCM), and then 0.1 mmol of DCC in 1 mL of DCM and 0.1 mmol of NHS in 0.5 mL of acetone were added into the polymer solution under magnetic stirring. PLGA-NHS activation was achieved after 4 h at room temperature (RT). The coupling reaction was accomplished by adding an excess amount of ethylenediamine (0.15 mmol) in 1 mL of DCM to the solution and further stirring for 2 h. The majority of the insoluble by-product, dicyclohexylurea, was removed by centrifuging the solution after reaction at 9391 rcf for 15 min at 4 °C and discarding the pellet. The supernatant was filtered (0.2 μm PTFE syringe filter) to remove any trace of the insoluble by-product. PLGA-NH2 was precipitated by slowly pouring the supernatant into an excess amount of ethanol (10-fold), where the un-reacted NHS/DCC/ethylenediamine was separated from PLGA-NH2 due to their solubility in ethanol. Precipitated PLGA-NH2 was redissolved in DCM and reprecipitated in ethanol twice for purification. The purified PLGA-NH2 was dried under vacuum.
171 rcf for 60 min to remove the insoluble p-NCS-Bz-DFO. The supernatant was further filtered (0.2 μm PTFE syringe filter) to remove the traces of p-NCS-Bz-DFO. Finally PLGA-DFO was precipitated by slowly dropping the supernatant into 10-fold diethyl ether, collected by centrifugation and dried under vacuum.
:glycolic ratio 50:50, RG502H, Mw 12000) was activated and reacted with ethylenediamine. The carboxylic groups were firstly activated with N,N′-dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS). 0.025 mmol of PLGA was dissolved in 8 ml of dichloromethane (DCM), and then 0.1 mmol of DCC in 1 mL of DCM and 0.1 mmol of NHS in 0.5 mL of acetone were added into the polymer solution under magnetic stirring. PLGA-NHS activation was achieved after 4 h at room temperature (RT). The coupling reaction was accomplished by adding an excess amount of ethylenediamine (0.15 mmol) in 1 mL of DCM to the solution and further stirring for 2 h. The majority of the insoluble by-product, dicyclohexylurea, was removed by centrifuging the solution after reaction at 9391 rcf for 15 min at 4 °C and discarding the pellet. The supernatant was filtered (0.2 μm PTFE syringe filter) to remove any trace of the insoluble by-product. PLGA-NH2 was precipitated by slowly pouring the supernatant into an excess amount of ethanol (10-fold), where the un-reacted NHS/DCC/ethylenediamine was separated from PLGA-NH2 due to their solubility in ethanol. Precipitated PLGA-NH2 was redissolved in DCM and reprecipitated in ethanol twice for purification. The purified PLGA-NH2 was dried under vacuum.
171 rcf for 60 min to remove the insoluble p-NCS-Bz-DFO. The supernatant was further filtered (0.2 μm PTFE syringe filter) to remove the traces of p-NCS-Bz-DFO. Finally PLGA-DFO was precipitated by slowly dropping the supernatant into 10-fold diethyl ether, collected by centrifugation and dried under vacuum.
Synthesis of SPIONs coated with oleic acid (OA-SPIONs)
SPIONs were synthesized through a microwave assisted thermal decomposition in a microwave oven (Discover SP, CEM Corporation) at a frequency of 2.45 GHz and 300 W of power and then coated with oleic acid (OA).23 Briefly, the process to obtain homogeneous OA-SPIONs with an average diameter of 9 nm was as follows. In a microwave reaction glass tube 3.5 mmol of iron precursor Fe(acac)3 was dissolved in 4.5 mL of benzyl alcohol. The tube was closed and microwave irradiation occurred with the power set at 300 W. The solution was kept at 60 °C for 5 min to fully dissolve the precursor and subsequently heated to 210 °C and kept at this temperature for 30 min and then cooled down to room temperature. After the reaction, a black colored dispersion formed which suggested the formation of a magnetic material. Then 3.17 mmol of oleic acid in 4 mL of toluene was added to the product dispersion and sonicated for 1 h. Then 5-fold of acetone was added followed by centrifugation to sediment the product pellet. The pellet was redispersed in 4 mL of toluene and transferred into a glass vial, a magnet was attached on the wall and after 5 s the suspension containing SPIONs with smaller size and lower magnetization was discarded. Then the pellet was redispersed in 6 mL of DCM followed by centrifugation at 1075 rcf for 5 min to sediment the unstable big particles. The OA-SPIONs of 9 nm stable in DCM were precipitated by adding 5-fold acetone and centrifugation. The final black product was dried under vacuum and redispersed in DCM at the concentration required for use.Fabrication of PLGA-based NCs and encapsulation of BSA
BSA encapsulated PLGA-based NCs were prepared by the double emulsion solvent evaporation method. Briefly, 50 μL of the inner aqueous phase (W1) containing BSA (30 mg mL−1) was emulsified in 500 μL of DCM organic phase (O) composed of 50 mg of different proportions of PLGA/functional PLGA and a certain amount of OA-SPIONs by sonication at 200 W for 28 s (VC505, Sonics & Materials, Inc., USA) to form the first emulsion (W1/O). Then 2 ml of external aqueous phase (W2) with PVA (20 mg mL−1) was added and the second emulsion (W1/O/W2) was formed by sonication for an additional 28 s. The temperature during the whole emulsion process was kept at 4 °C by using an ice bath. The resulting double emulsion was poured into 50 mL of MilliQ water and mechanically stirred at RT for 2 h to allow complete evaporation of the organic solvent and formation of NCs. Finally, the NC pellet was obtained by centrifugation at 9391 rcf for 15 min and washed three times with 50 mL of MilliQ water and then lyophilized in 6 mL of trehalose (2 mg mL−1) aqueous solution. The as-obtained powder was stored at 4 °C with desiccant silica gel.Physicochemical characterization of the polymers, SPIONs and PLGA-NC
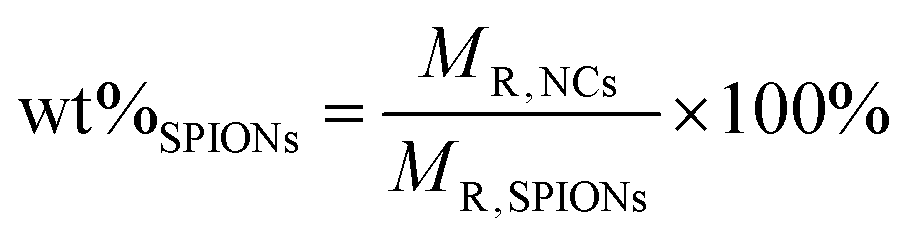
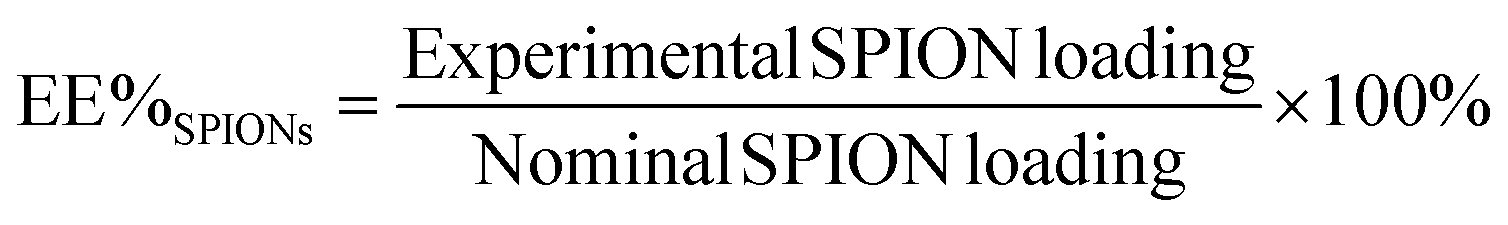
In vitro and in vivo MRI of the NC·SPIONs
In vitro agarose phantoms of NCs were prepared by filling a series of glass microtubes with solutions of 0.63% agarose in water (Conda, Madrid), into which different amounts of NCs were admixed. The corresponding iron doses (mmol L−1) were calculated according to the wt% of SPIONs in each sample. T2 weighted images (T2WI) of the phantoms were acquired at 7 T in a 70/30 Bruker USR Biospec system (Bruker GmbH, Ettlingen) as follows: multi-slice multi-echo (MSME) sequence with echo time (TE) = 8 ms, repetition time (TR) = 2600 ms, average N = 2, matrix size = 160 × 160, field of view (FOV) = 24 × 24 mm, 14 slices of 1 mm thickness, and spectrometer bandwidth (BW) of 474 Hz per pixel. Quantitative T2 values were obtained from hand-drawn regions of interest (ROIs) by using curve fitting in the Image Sequence Analysis (ISA) Tool (ParaVision v.5.1).Balb/c female mice (16–17 weeks of age, n = 2) were anesthetized with isoflurane (IsoFlo; Abbott Laboratories). T2WI were acquired pre- and 15–20 min post-administration of NCs (1.6 mg in 150 μL of saline through tail vein) using the following imaging parameters: MSME pulse sequence, 20 echo times with ΔTE = 8 ms, TR = 2.6 s, N = 2 averages, a matrix of 256 × 256 points covering a FOV of 25.6 × 25.6 mm2, giving a resolution of 100 μm in plane, 14 continuous slices of 1 mm thickness, and BW = 75 kHz. The images were acquired with a respiratory gating by using a SAII Model 1030 monitoring & gating system (Smal Animals Instruments, Stony Brook, NY, USA). Fat suppression was achieved by a saturation pulse of 1050 Hz. T2 maps were constructed off-line using Image-J 1.50b (National Institutes of Health), and data were fitted to a three parameter exponential decay equation (S = A + S0exp(−TE/T2), where S represents signal intensity, A, S0 and T2 are the fitting parameters), achieved by the “exponential decay with offset” equation of the fitting routine of ImageJ. Signal intensities in T2WI and T2 maps were quantified manually in selected ROIs using ImageJ. Afterwards mice were euthanized and organs were preserved for histological analyses.
In vitro and in vivo NIRF imaging of NC·PLGA-Cy7.5
A series of concentrations of NCs in 100 μL of saline (n = 3) were prepared in a 96-well plate and imaged using a Xenogen IVIS® spectrum (λex/λem 710/820 nm). BALB/cAnNRj male mice (Janvier; 7–8 weeks of age, n = 2) were deeply anesthetized with isoflurane and were injected via the tail vein with NCs at a dose of 1.6 mg in 200 μL of saline. The NIRF images were acquired on a Xenogen IVIS® spectrum imaging system at 0.5 h, 1 h and 3 h post injection. At the end of the 3 h scan, the mice were euthanized to image the signal intensity of the principal organs ex vivo together with blood and urine.In vivo PET imaging of 89Zr labelled NC·PLGA-DFO
NC·PLGA-DFO were labelled with 89Zr as follows: [89Zr]ZrC2O4 was produced in house following a standard protocol. The as-obtained 1500 μL of 1 M oxalic acid containing 89Zr (8.2 mCi, 303.4 MBq) were neutralized with 2 M sodium carbonate (pH = 7.2) and the volume was adjusted to 3.5 mL with 0.5 M HEPES buffer. Six aliquots (1.6 mg each) of the lyophilized particles were dispersed in 50 μL MQ-water, and 450 μL of the freshly prepared 89Zr-solution was added to each and mixed well by sonication. After incubating overnight at room temperature the particles were purified by centrifugation and washed two times with MQ-water followed by one wash with saline to remove all non-bound 89Zr. The pellet of the last wash was dispersed in 120 μL of saline with an average calculated radiochemical yield of 70.1% (decay-corrected). As the achieved samples were too high in activity, a one to one dilution with non-labelled particles was performed. The final injections (150 μL) were performed intravenously via the tail vein of healthy female mice (Crl:CD1(ICR)) with an average injected activity of 161 μCi (6.0 MBq).
Imaging studies were conducted using positron emission tomography (PET) in combination with computerized tomography (CT), using the β- and X-cube microsystem of Molecubes. Static whole body images (1 bed) were acquired in a 511 keV ± 30% energetic window at 1 h, 4 h, 23 h and 47 h post injection (acquisition time 5 min for 1 h and 4 h, 20 min for 23 h and 47 h). PET images were analyzed using PMOD image analysis software (PMOD Technologies Ltd, Zurich, Switzerland).
Prussian blue stain
Liver samples from the mice subjected to PET were embedded in an optimal cutting temperature (OCT) compound and kept at −80 °C until sliced in 8 μm sections with a cryostat. After rehydration, the sections were stained with a Prussian blue iron stain kit (Polysciences Inc, USA) following the manufacturer's protocol except that potassium ferrocyanide:hydrochloric acid solution was kept for a total of 40 minutes and nuclear Fast Red for 1 minute. Images were acquired after dehydration with a transmitted light microscope (Leica, Germany).
In vitro and in vivo toxicity evaluation of the NCs
BSA loading content
The albumin content was determined directly using the CBQCA protein assay kit (Invitrogen™ ref. C6667), which determines the protein concentration based on the production of fluorescent products measurable at λex/λem = 450 nm/550 nm via non-covalent interaction between CBQCA and primary aliphatic amines of proteins. This highly sensitive fluorescence-based method showed compatibility with DMSO, SPIONs, detergents and other substances which interfere with many commonly used protein determination methods. Lyophilized NCs encapsulating albumin as well as empty NCs as control were fully dissolved in DMSO at 100 mg mL−1. The protein contents in the NC lysates were measured and calculated based on the difference in fluorescence with control and a calibration curve drawn with standard albumin solutions, and the protein contents in the BSA solutions used for encapsulation were also measured. All determinations were performed in duplicate for each NC batch. The experimental BSA loading content was expressed as μg of BSA per mg of NCs (μg mg−1). The encapsulation efficiency (EE%) was calculated as follows: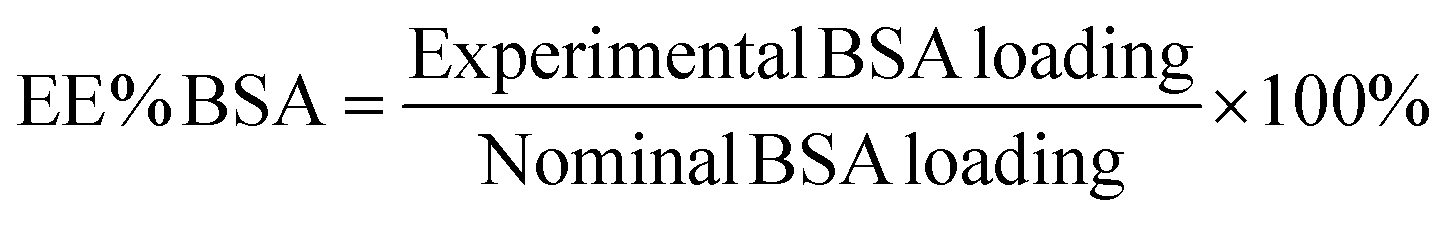
Results and discussion
PLGA NCs reported here incorporate several biocompatible multimodal imaging modalities (Fig. 1): fluram and cyanine 7.5 (Cy7.5) as fluorescent probes in the blue and the near-infrared (NIR) wavelengths, 89Zr chelated with DFO as a radio imaging probe and SPIONs as a MRI contrast agent. The fluorescent probes and the DFO chelator were anchored to an aminated PLGA, PLGA-NH2, resulting from the modification of the commercial carboxylic acid-terminated PLGA. BSA encapsulated PLGA NCs could then be prepared by a double emulsion solvent evaporation method where selected imaging moieties were incorporated into the PLGA shell by adding the desired proportions of a given functional PLGA and oleic acid coated SPIONs to the organic phase while the BSA was added to the aqueous phase. | ||
| Fig. 1 Schematic illustration of a PLGA NC as a tailor-made multimodal theranostic platform. | ||
Chemical modification of the PLGA with functional moieties
Fig. 2 shows the schematic representation for terminal modifications and functionalization of the PLGA. Firstly, the carboxylic acid end group of PLGA was activated with DCC and NHS to conjugate ethylenediamine and form PLGA-NH2, bearing a free primary amine group in the PLGA end (Fig. 2a). An excess amount of ethylenediamine (6:1 ethylenediamine:PLGA mole ratio) was used to suppress the coupling reaction of PLGA–PLGA that could occur due to the homo-functional crosslinker ethylenediamine. Subsequently, the three functional moieties with amine-reactive groups (fluram, Cy7.5, DFO) could be respectively integrated at the end of the PLGA molecule through coupling reactions (Fig. 2b). Further details are reported in the Experimental section.
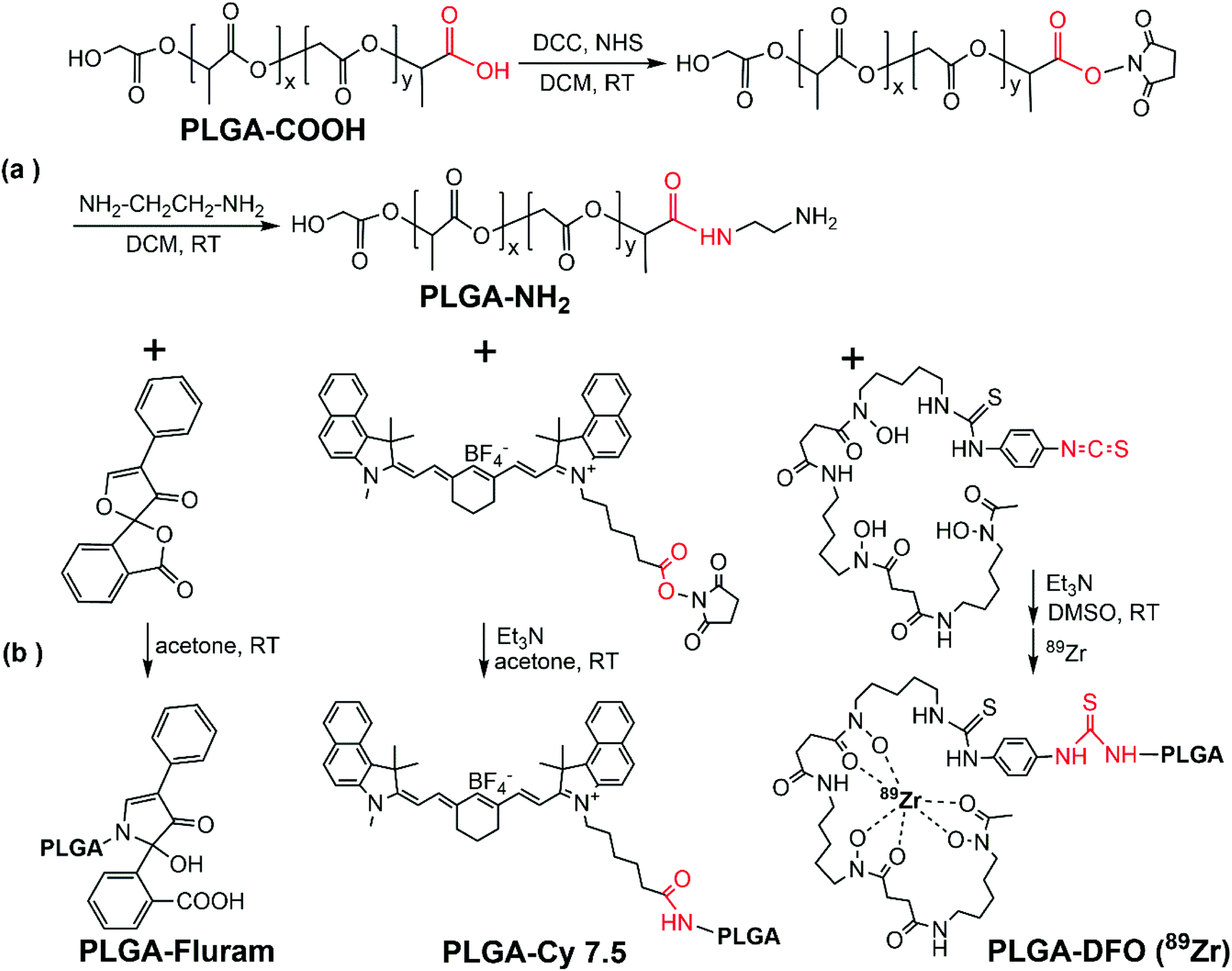 | ||
| Fig. 2 PLGA chemical modifications. Synthetic schemes of terminal modifications and functionalization of the PLGA. | ||
ATR-FTIR spectra of the commercial PLGA-COOH and the PLGA-NH2 product confirmed their similar chemical structure and successful modification (Fig. S1 in the ESI†). The reaction efficiency of PLGA-COOH to PLGA-NH2 was quantified by measuring the conjugated amount of fluram to the –NH2 groups. The UV absorbance of the PLGA-NH2 product (Mw 12000 g mol−1) in chloroform at 390 nm due to the conjugation of fluram was compared with a standard calibration curve constructed from different concentrations of ethylenediamine prepared under the same conditions (Fig. S2†). A reaction efficiency of 86% of PLGA-COOH to PLGA-NH2 was estimated, which means that the molar fraction of pure PLGA-NH2 in the product was 0.86.
Fluram is a non-fluorescent compound that reacts quickly and almost quantitatively with primary amines (R-NH2) to form a fluorescent derivative, which emits strong fluorescence at around 480 nm when excited at 390 nm.22 The conjugation of this compound to the –NH2 end of PLGA for fluorescence application is very advantageous considering that neither free fluram itself nor its hydrolysis product show fluorescence. As shown in Fig. 3a, the characteristic absorption peak of the fluorophore at 390 nm occurred in the UV-Vis spectrum of the conjugation product PLGA-fluram but this was not observed either in PLGA-NH2 or in fluram. The disappearance of the 309 nm absorption band of fluram in the PLGA-fluram product indicates that the purification process removes the vast majority of free fluram. The fluorescence of the obtained PLGA-fluram was evaluated and the excitation and emission spectra are depicted in Fig. 3b. Importantly, the fluorescence intensity of PLGA-fluram decreased only slightly (<5%) after continuous UV excitation at the maximum excitation wavelength for 3 h, thus exhibiting a photostability just slightly lower (2%) than rhodamine-B, a widely used commercial fluorescent dye (Fig. 3c). The photostability of PLGA-fluram using a confocal microscope configuration is also included in Fig. S3b.† The fluorescence intensity of NCs fabricated by PLGA-fluram decreased by only 13% after 10 min of continuous illumination at 10% of laser power (401 nW). Considering that the laser power applied to observe stained cells is generally less than 10%, our fluorescent NCs would exhibit good photostability under in vitro conditions. Note that the calculated quantum yield of PLGA-fluram (18%) (Fig. S3a†) is much higher than the values reported for fluorescent proteins such as green fluorescent protein (GFP) (7.3%) and its blue variants (7.9%),27 and similar to other polymeric fluorescent materials such as poly(amido amine)s.28,29 Finally, the calculated fluorescent brightness of PLGA-fluram (25056) is also much higher than that of other reported biodegradable photo-luminescent poly(lactide-co-glycolide) (BPLP-co-PLGA) (6550–19230)30 and other frequently used small organic dyes such as Alexa Fluor 430 (8800) or DAPI (15660). The fluorescence intensities of the fabricated NCs show linear dependency on the NC concentration, and NCs can be clearly imaged with a fluorescence microscope (Fig. 3d). Although in this work we have focused on in vivo imaging modalities, the remarkable fluorescence properties shown here endow the PLGA-fluram system with very interesting features for in vitro fluorescence tracking studies.
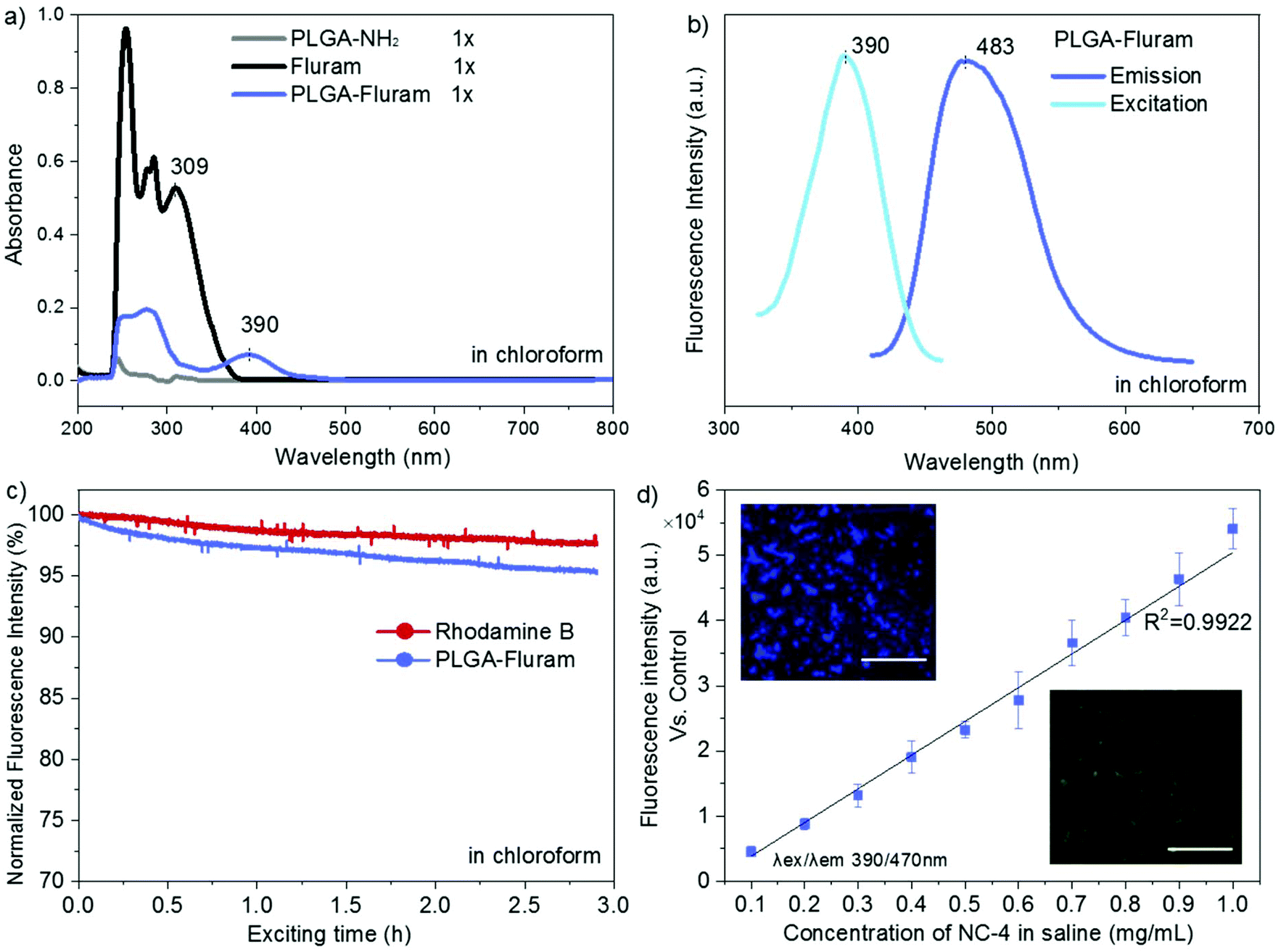 | ||
| Fig. 3 PLGA-fluram system spectroscopic and fluorescence characterization. (a) UV-Vis absorption spectra of PLGA and PLGA/fluram samples; (b) excitation and emission spectra of PLGA-fluram; (c) photostability evaluation of PLGA-fluram and control dye rhodamine B, fluorescence intensity expressed as the percentage vs. time 0; (d) fluorescence intensity of different concentrations of NC·PLGA-fluram (NC-4) vs. NC-control (NC-3) measured by using a microplate reader (n = 3, values represent mean ± SEM); insert: lyophilized NC-4 observed under a fluorescence microscope with DAPI/GFP filters, scale bar 60 μm. | ||
Next, the NIR fluorophore Cy7.5 was introduced into the terminal amine of PLGA-NH2via an amide bond between the PLGA terminal amine group and amine-reactive Cy7.5 NHS ester. The absorption band of Cy7.5 appeared in the UV-Vis spectrum of the PLGA-Cy7.5 product, which confirmed the successful conjugation of Cy7.5 (Fig. S4a†). The amount of conjugated Cy7.5 in the PLGA-Cy7.5 product was quantified by comparing the fluorescence intensity of the PLGA-Cy7.5 product with freeCy7.5 (Fig. S4b†) in DMSO. Pure PLGA-Cy7.5 with a molar fraction of 0.65 was estimated in the product and a conjugation efficiency of 76% of the Cy7.5 NHS ester to PLGA-NH2 was calculated considering that the fraction of pure PLGA-NH2 is 0.86.
For radiolabelling functionalization the 89Zr chelator DFO was introduced at the end of PLGA-NH2 by forming a thiocarbamide bond between the PLGA terminal amine group and the amine reactive –NCS group in the DFO derivative p-NCS-Bz-DFO. Compared to the IR spectrum of PLGA-NH2, some new bands corresponding to the DFO conjugate appear in the spectrum of PLGA-DFO, indicating the successful conjugation of DFO (Fig. S1†). The amount of conjugated DFO in the PLGA-DFO product was quantified by measuring the amount of unreacted PLGA-NH2 through the fluram method described above (Fig. S2c†). Pure PLGA-DFO with a molar fraction of 0.75 was estimated in the product, and a conjugation efficiency of 88% of p-NCS-Bz-DFO to PLGA-NH2 was calculated considering that the fraction of pure PLGA-NH2 is 0.86.
Fabrication of PLGA NCs with selected combinations of imaging moieties
PLGA NCs with encapsulated BSA were prepared by a double emulsion solvent evaporation method.31 Imaging moieties were incorporated into the PLGA shell by adding to the organic phase various proportions of functionalized PLGA as well as the oleic acid (OA) coated SPIONs (Table 1). Compositional changes of the organic phase did not affect the encapsulation process, and all types of NCs prepared showed mean hydrodynamic sizes within a diameter range of 250–300 nm and a polydispersity index (PdI) <0.2. As seen in Table 1 from NC-1 to NC-3, neither increasing the SPION size nor increasing two-fold its loading affected the mean size of the NCs compared to plain PLGA NCs (NC-0). Note that for the same loading of SPIONs, the magnetization of NC-2 is much higher than that of NC-1 given the larger size of SPIONs for NC-2. Therefore optimal formulation regarding SPIONs (6 wt%, with 9 nm nanoparticles) yields an average saturation magnetization value of 4.06 emu g−1 NCs which is more than four-fold larger than our previously reported value (0.90 emu g−1).31 As will be shown later, the increase of the magnetic moment of NCs resulting from using larger size SPIONs and increasing their load will positively impact on the MRI signal intensity.32| Sample type | PLGA information (wt%) | SPIONs | M R (emu g−1) | M S (emu g−1) | Size (DLS) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLGA | PLGA-Fluram | PLGA-DFO | PLGA-Cy7.5 | PLGA-PEG | Sizea (nm) | Loadingb (wt%) | EE%b | d.nm | PdI | |||
| a The size of SPIONs was determined by statistics from TEM images (Fig. S5†). b SPION loading and entrapment efficiency (EE%) were determined by SQUID measurements at 5 K. c The fraction of SPIONs in OA-SPIONs was determined by thermogravimetric analysis (TGA) and the magnetization is expressed as emu g−1 SPIONs (Fig. S6†). | ||||||||||||
| NC-0 | 100 | — | — | — | — | — | — | — | — | — | 255 | 0.10 |
| NC-1 | 100 | — | — | — | — | 6 | 2.9 | 92% | 0.4 | 1.0 | 279 | 0.17 |
| NC-2 | 100 | — | — | — | — | 9 | 3.0 | 90% | 0.8 | 1.9 | 264 | 0.19 |
| NC-3 | 100 | — | — | — | — | 9 | 6.1 | 95% | 1.6 | 3.9 | 276 | 0.18 |
| NC-4 | 50 | 50 | — | — | — | 9 | 6.2 | 95% | 1.6 | 4.7 | 260 | 0.15 |
| NC-5 | 40 | — | 60 | — | — | 9 | 6.3 | 97% | 1.6 | 4.3 | 271 | 0.19 |
| NC-6 | 40 | — | — | 60 | — | 9 | 5.9 | 91% | 1.5 | 3.6 | 245 | 0.11 |
| NC-7 | — | 50 | 50 | — | — | 9 | 5.9 | 91% | 1.5 | 3.7 | 268 | 0.18 |
| NC-8 | 93 | — | — | — | 7 | 9 | 6.0 | 92% | 1.6 | 3.8 | 265 | 0.13 |
| OA-SPIONsc | 6 | 14.0 | 50.8 | |||||||||
| 9 | 26.1 | 75.1 | ||||||||||
Furthermore, the size of NCs as well as the encapsulation efficiency (EE%) of SPIONs were not affected when various proportions of PLGA-fluram (NC-4), PLGA-DFO (NC-5) or PLGA-Cy7.5 (NC-6) were incorporated into the PLGA matrix, even when incorporating various moieties simultaneously (NC-7). Importantly, we had found that the PLGA-based NCs can be fabricated using either up to 100 wt% of any modified PLGA (PLGA-DFO, PLGA-Cy7.5 or PLGA-fluram) or with any mixed proportion of them without affecting the morphology of NCs and EE% of SPIONs. This can be explained by the forming process of the NCs: the different functional moieties at the end of the comparatively extra-long PLGA molecules do not affect the film-forming of PLGA and the entrapment of SPIONs in the polymeric matrix. Finally, PEGylation of the PLGA NCs was achieved by adding a given amount of the commercial amphiphilic block copolymer PLGA-PEG (PEG Mn 5000 and PLGA Mn 7000) to the organic phase. The morphology and size of NCs (NC-8) are maintained up to a maximal fraction of 7 wt% amphiphilic PLGA-PEG (3 wt% PEG, Table 1) and are not affected when co-incorporating other hydrophobic modified PLGA (i.e., PLGA-Fluram/PLGA-DFO/PLGA-Cy7.5) (data not shown).
Fig. 4 shows a representative scanning electron microscopy (SEM) image of the NCs (NC-3) presenting homogeneous spherical morphologies. The inset is a dynamic light scattering (DLS) size distribution histogram of lyophilized NCs redispersed in water. In this case, the mean hydrodynamic diameter of the NCs is 276 nm with a polydispersity index (PdI) of 0.18 reflecting good redispersibility of the NCs in water after the lyophilization process. As mentioned, all types of NCs listed in Table 1 had similar morphologies and sizes (Fig. S7†), which were considered to be suitable for intravenous administration.33 The upper inset in Fig. 4 shows a representative broken NC, exposing the hollow core, ideal for the loading of protein drugs. The transmission electron microscopy (TEM) image in the lower inset revealed the embedding and finely homogeneous distribution of the SPIONs within the polymeric matrix.
 | ||
| Fig. 4 Morphological data of a representative PLGA NC system (NC-3). SEM image of lyophilized PLGA based NCs with the upper inset showing the hollow core of the NC; lower inset TEM image shows a spherical NC with SPIONs visible as black spots, uniformly distributed in the polymer matrix; DLS curve of NC water suspension after lyophilization with a mean diameter of 276 nm and 0.18 polydispersity. | ||
NC magnetic properties, accumulation and biodistribution by MRI
The magnetic hysteresis loop at room temperature of lyophilized NC-3 is displayed in Fig. 5a showing the superparamagnetic character of the NCs (lack of coercivity). The superparamagnetic behavior of the NCs after SPION encapsulation is important since in the absence of an external magnetic field, it ensures lack of interactions among NCs thus minimizing the risk of embolization for i.v. administration. The magnetic retention of NCs is illustrated with the use of an external magnet. As seen in Fig. 5b, the colloidal water suspension of 2 mg mL−1 of NC-3 forms a round pellet next to the magnet after 7 h. The solution becomes completely clear indicating that a great majority of the NCs are retained by the magnet. Previous experiments using magnetic PLGA nanoparticles34 or microparticles loaded with only 1 wt% iron oxide35 already demonstrated magnetic retention in rodent brain or knees, indicating that our protein loaded magnetic NCs with a higher magnetic load (6 wt%, MS = 4 emu g−1) may still have better characteristics to address magnetic targeting.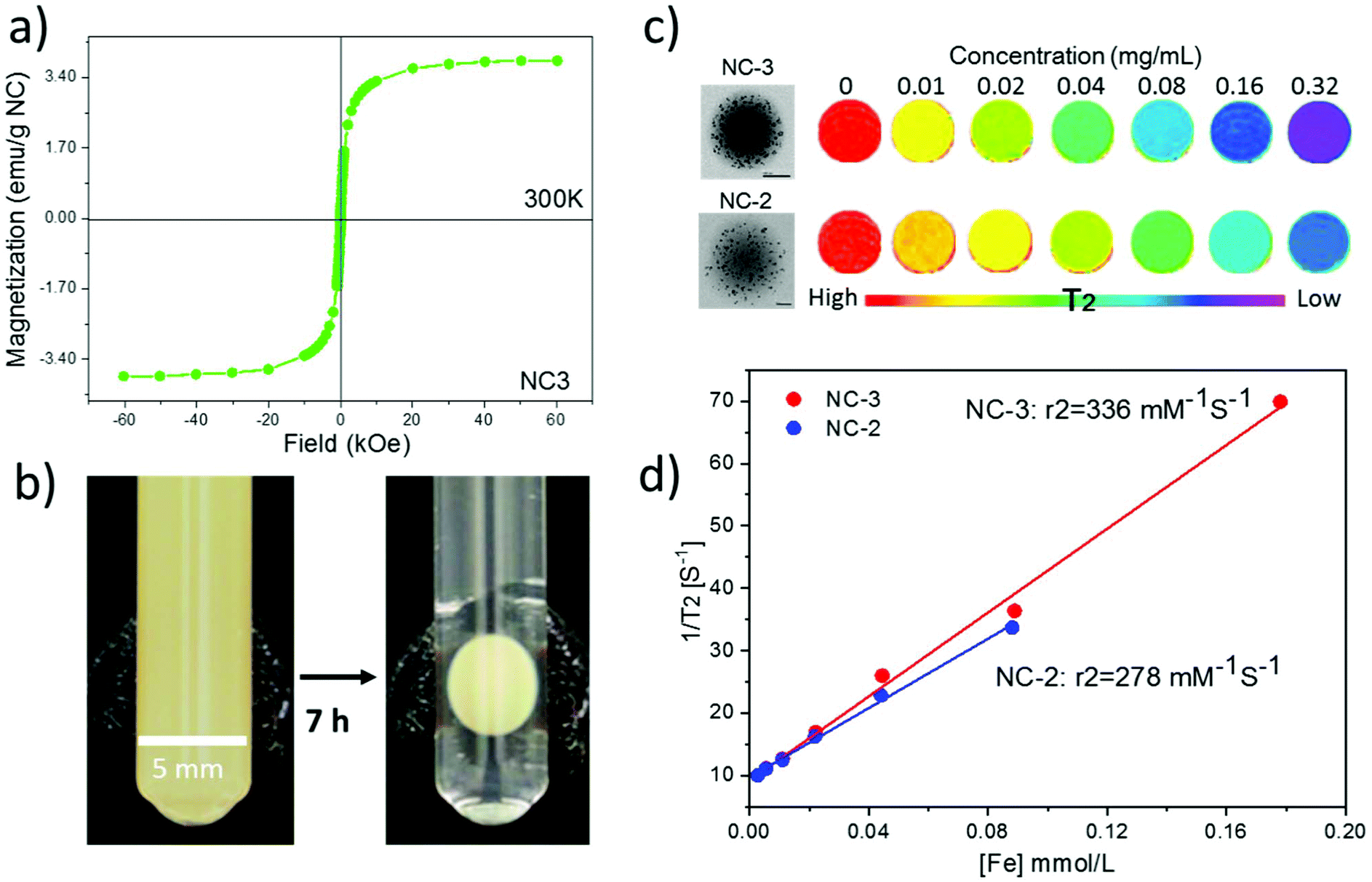 | ||
| Fig. 5 Magnetic characterization, retention and MRI phantoms of PLGA–SPIONs NCs. (a) Hysteresis loop of the lyophilized batch NC-3 at 300 K, showing superparamagnetism at room temperature; (b) glass tube with water suspension of NC-3 at a concentration of 2 mg mL−1. A permanent magnet (diameter 8 mm, surface field ∼0.4 T) was placed on the wall. After 7 h, the NCs adsorbed to the tube wall on the magnet side; (c) T2-weighted phantoms obtained by mixing lyophilized NCs with agarose at increasing NC concentrations, on the left are TEM images of NCs; (d) evaluation of r2 relaxivities for NC-3 and NC-2 (3 wt% and 6 wt%; 9 nm diameter). | ||
Phantom studies were conducted to confirm the MRI performance of the NCs. Phantoms from two batches (NC-2 and NC-3) with different SPION loadings (3 wt% and 6 wt%; 9 nm diameter) at a series of concentrations were prepared (Fig. 5c). Spin–spin relaxation time (T2) weighted color-coded MRI images clearly exhibit signal decay in a concentration dependent manner. Importantly, at the same concentration NC-3 with 6 wt% SPIONs showed a higher T2 signal decay rate than NC-2 with 3 wt% SPIONs indicating that NC-3 will be easier to track in vivo by MRI. Furthermore, the transverse relaxivity (r2) value at 7 Tesla of NC-3 (336 mM−1 s−1) was higher, as expected, than that of NC-2 (278 mM−1 s−1) as seen in Fig. 5d, indicating that the denser packing of SPIONs in a single NC of NC-3 endows the NCs with a larger magnetic moment and increases its efficiency as a spin–spin relaxation agent.36 Compared with other clinically used SPION systems such as Feridex (98 mM−1 s−1) and Resovist (151 mM−1 s−1),37 the much higher r2 value in our final NC formula (6 wt% SPIONs of 9 nm in diameter) is expected to be useful for in vivo MRI tracking of the NCs.
As the representative abdominal images show in Fig. 6b, a much clearer increase of the contrast of the liver was observed in both T2WI and T2 maps after NC administration, illustrating the fast accumulation of NCs in the liver. Quantitative ROI analysis in Fig. 6c shows a 55% drop of signal intensity (SI) on T2W images and a 26% drop of T2 relaxation time in the liver, while a negligible signal drop was observed in the kidney, which further demonstrate the large accumulation of NCs in this organ. To further confirm the presence of NCs in the tissue, Prussian blue staining on liver sections confirmed the presence of ferric iron (Fig. 6d) after the in vivo MRI.
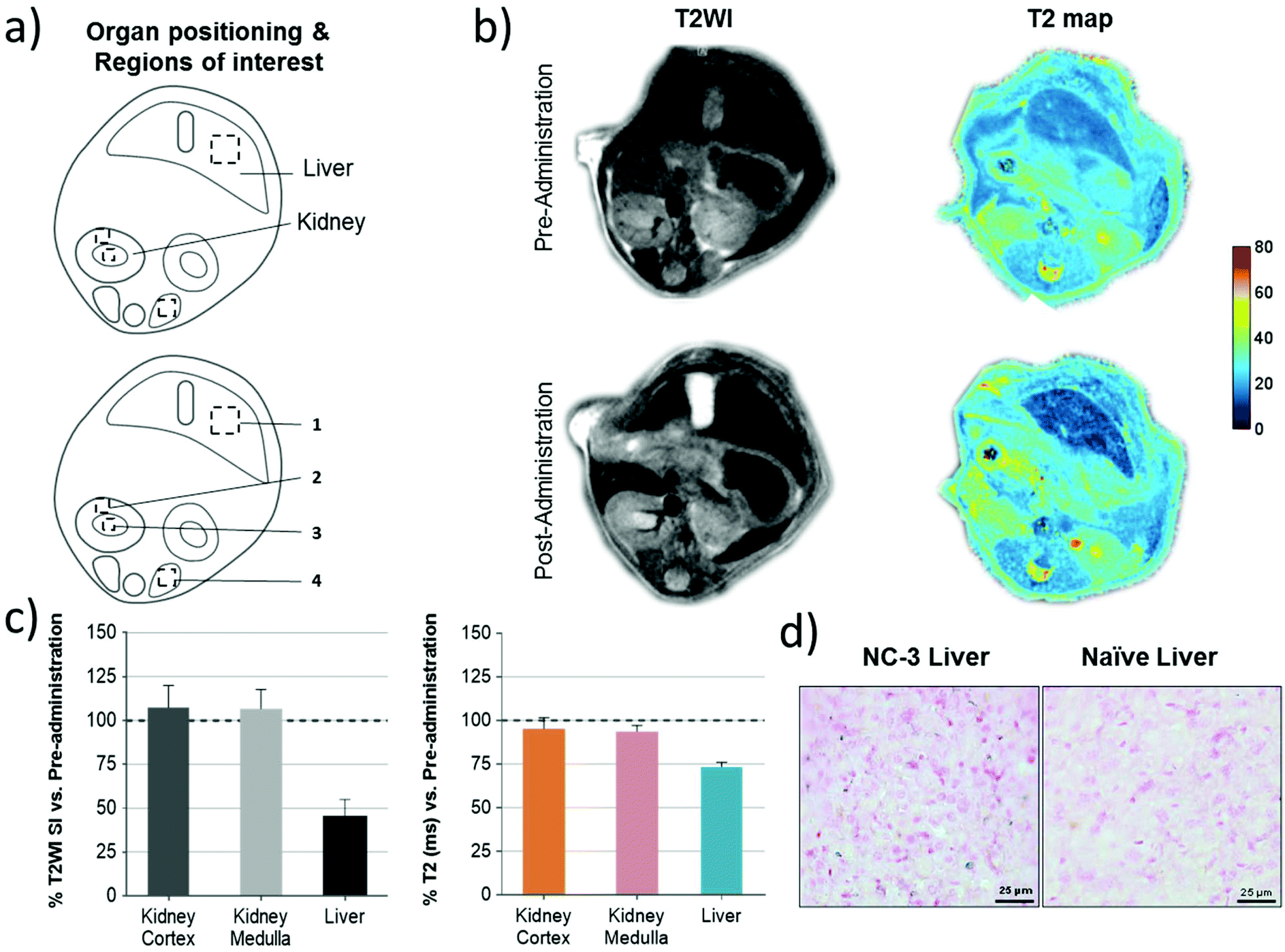 | ||
| Fig. 6 In vivo mouse MRI of the PLGA–SPIONs NCs and liver Prussian blue iron-stain. (a) Representation of the abdominal region with organ positioning and ROI quantification in the kidney and liver (1: liver, 2: kidney cortex, 3: kidney medulla, 4: paravertebral muscle used for normalization). (b) Representative images of T2W and T2 maps before and after NC-3 administration. (c) Signal intensity (SI) and relaxation time (ms) were calculated for each ROI corresponding to the analyzed anatomical structures which were then corrected by muscle ROI SI. Final data are represented as post-administration SI or T2 relaxation as the percentage of the pre-administration values (mean ± SEM; n = 2). (d) Representative images of Prussian blue stain for ferric iron in liver sections of naïve and treated animals administered with NC-3 in the MRI study (blue spots). | ||
Biodistribution of the NCs monitored by PET
With a ∼3 day half-life, 89Zr-based PET imaging holds great potential as a useful tool for long-term monitoring of the dynamic biodistribution, biodegradation and clearance pathway of nanoparticles in vivo.38 Our results showed that 89Zr-labelling has a yield of about 70% for NC·PLGA-DFO (NC-5) after overnight incubation at room temperature at a concentration of 3 mg mL−1. A DFO challenge study has been reported to demonstrate the radiostability of 89Zr-labelled nanoparticles.39 In our study, the as-obtained NC·PLGA-DFO-89Zr was suspended in 1 mM DFO aqueous solution and incubated at 37 °C for 23 h. Then the NCs were separated by centrifugation and 100% were still labelled, with no presence of 89Zr-DFO in the supernatant, demonstrating a strong binding affinity of 89Zr to the NC·PLGA-DFO.89Zr labelled NC·PLGA-SPIONs-DFO-89Zr (NC-5) were then injected via the tail vein with the same iron dosage as for the MRI study (1.68 mg Fe per kg−) to anesthetized mice. Static whole body PET-CT images were acquired at 1 h, 4 h, 23 h and 47 h p.i. Dominant liver, spleen and lung uptake was observed (Fig. 7a) and confirmed by the quantitative ROI analysis (Fig. 7b) within two days, which can be related to phagocytic cell uptake by the mononuclear phagocyte system,1 as expected for i.v. injected PLGA nanoparticles of this size.17 Interestingly, noticeable bladder accumulation (∼22%ID cm−3) was observed at 1 h p.i., with a sharp decrease at 4 h p.i. (∼6%ID cm−3) likely due to uncontrolled animals’ urination between scans. The early stage activity in the bladder could also be confirmed by continuous PET-CT acquisition (Fig. S8†), which showed a linear increase of activity within 3 h p.i. It is hard to assign the activity in the urine to the detachment of free 89Zr from the NCs, since detached free 89Zr is a well-known osteophilic cation with a fast and retained uptake in bones,38,40 which is not detectable in our case. In the study of Chen et al.,38 for both free 89Zr-oxalate or 89Zr labelled SiO2 nanoparticles, fast and sustained uptake of free/detached 89Zr in bones and joints was observed from the first few hours up to three weeks, while the accumulation in the bladder was negligible. We thus hypothesized that the erosion of the soft polymeric NCs had happened at an early stage in the complex blood circulation environment. The detached 89Zr-molecular fragments from the NCs, either by degradation of ester bonds in PLGA molecular chains or thiocarbamide bonds between PLGA and DFO or by mechanical force, with a size smaller than 15 nm (ref. 41) underwent renal clearance and were observed in the bladder. Additional experiments showed erosion of NCs clearly observed after in vitro incubation in mouse plasma at 37 °C for 24 h (Fig. 7c). Small holes appeared on the NCs while under the same conditions NCs incubated in saline did not show the same eroded surface (Fig. 7d). Considering the in vivo blood circulation setting, the erosion of PLGA NCs would be much faster as reported by Swider.42 Not surprisingly, at late time points the concentration of radioactivity in the bladder decreases likely due to the fast accumulation of the NCs in the liver/spleen.
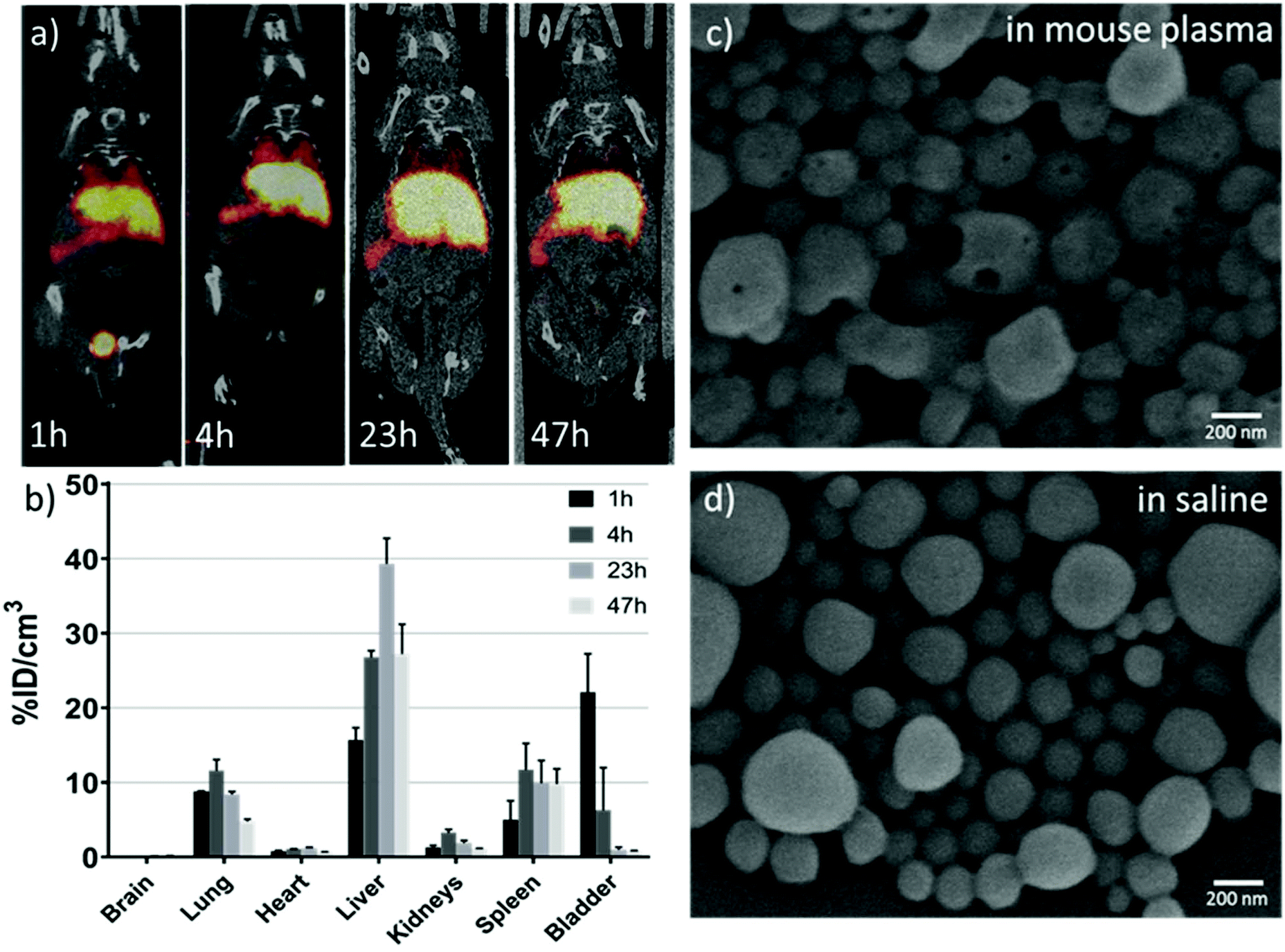 | ||
| Fig. 7 PET biodistribution study of PLGA-SPIONs-DFO-89Zr NCs. (a) Representative PET-CT coronal images of mice obtained at different time points after intravenous (i.v.) administration of NC·PLGA-SPIONs-DFO-89Zr (NC-5). PET images (maximum intensity projections) have been co-registered with CT images (slices) of the same animals for localization of the radioactive signal; (b) accumulation of radioactivity, expressed as a percentage of injected dose per cm3 of tissue (%ID cm−3) at 1 h, 4 h, 23 h and 47 h p.i. (mean ± SEM of n = 2); (c) and (d) SEM images of the NCs after incubation for 24 h at 37 °C in mouse plasma or saline. | ||
NC biodistribution monitored by near infrared fluorescence (NIRF) imaging
To investigate the feasibility of the formulation of NC·PLGA-Cy7.5 (NC-6) for in vivo NIRF imaging, in vitro fluorescence performance was firstly checked. The in vitro fluorescence intensity of NC-6 was linearly dependent on the concentrations of NCs, showing strong fluorescence and no quenching up to a high level of 1 mg mL−1 (Fig. 8a). Real-time whole-body imaging revealed a dominant abdominal distribution of the NCs with a total radiant efficiency (TRE) of ∼42.40 vs. control at 2 h p.i. At the end of the observation, main organs including brain, heart, lungs, liver, spleen, kidneys, and bladder were collected. Fig. 8c shows a representative ex vivo fluorescence picture of those organs and Fig. 8d shows the quantified result of the biodistribution of the NCs. Dominant liver, spleen and lung accumulation was observed which was consistent with the data acquired with PET. Note that we also did see the bladder signal during the real-time imaging, which further demonstrated that the erosion of NCs really occurred at the early stage p.i., considering that the NIRF imaging is less sensitive than PET. The unobservable ex vivo bladder signal may result from the uncontrolled urination and urine sample collection.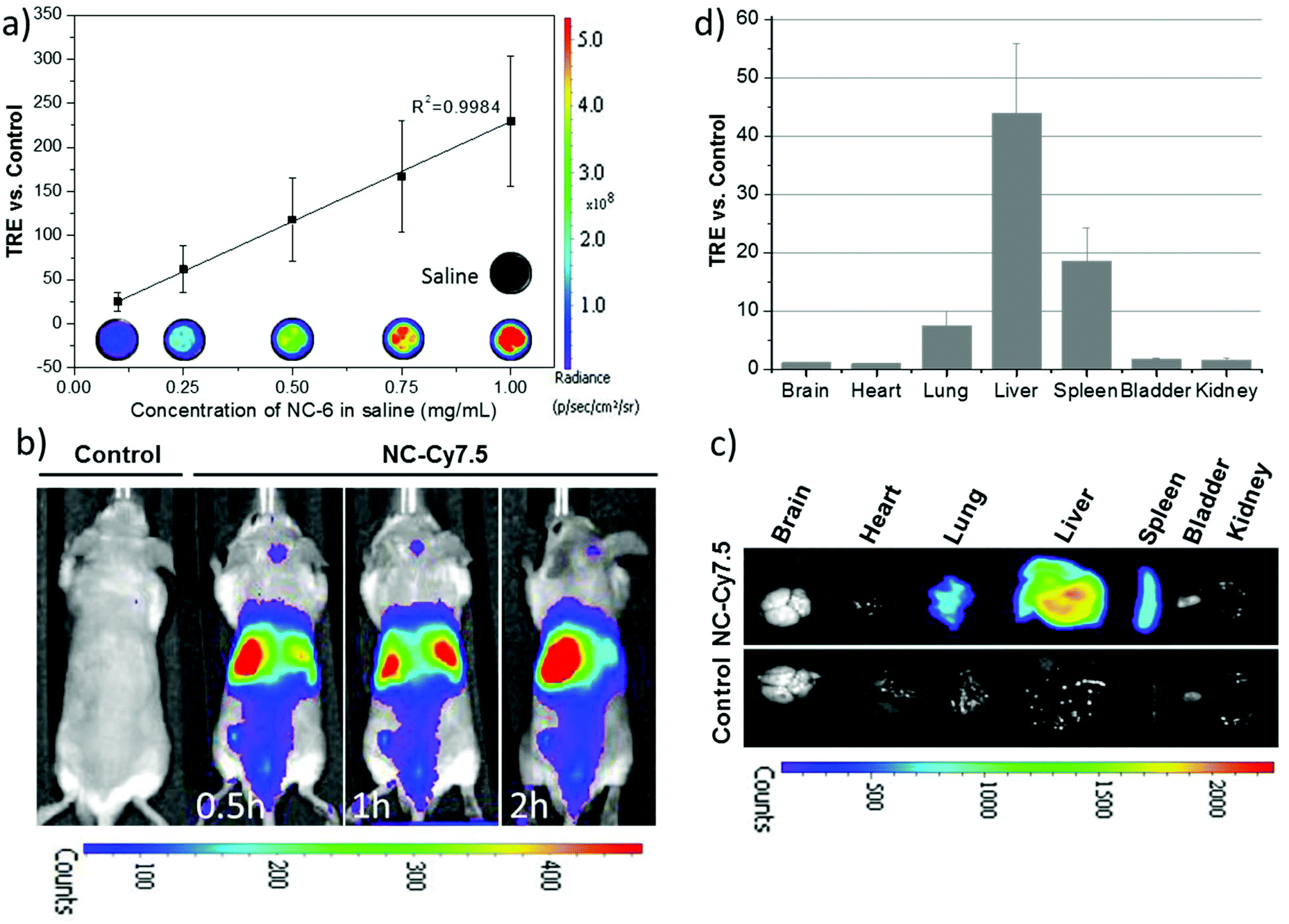 | ||
| Fig. 8 Fluorescence molecular imaging of PLGA-SPIONs-Cy7.5 NCs. A Xenogen IVIS® imaging system (λex/λem 710/820 nm) was used to monitor the NIRF signal of NC-6, and the fluorescence intensity was quantified as total radiant efficiency (TRE) vs. the corresponding control. (a) Quantitative in vitro NIRF intensity of different concentrations of NCs vs. control saline (n = 3, mean ± SEM) and representative images of NCs in the well; (b) representative whole body in vivo images obtained at different time points after i.v. injection; (c) representative ex vivo images of excised organs at the end of the in vivo experiment; (d) ex vivo quantification of biodistribution of NCs determined by the averaged organ TRE vs. the control mouse (n = 2, mean ± SEM). | ||
From the above discussion, similar biodistribution of the NCs with different imaging probes for MRI/PET/NIRF was evidenced. That was expected since the only difference among the NCs for these three imaging modalities is the existence or not of small molecule 89Zr-DFO or Cy7.5 at the end of the PLGA molecular chain. As we have demonstrated, the imaging probes in the matrix of the PLGA shell do not affect the size of the NCs, which is a key attribute influencing the biodistribution and pharmacokinetics of the nanocarriers.17,41,43 Thus, the incorporation or removal of these small molecules in the shell does not affect the NCs’ in vivo biodistribution and tailor-made capsules can be fabricated with the appropriate imaging probe needed for each specific in vivo and in vitro study. Moreover, simultaneous multimodal imaging is an option through custom-tuning the functional moieties in the capsules. Despite that bare (non-PEGylated) NCs were used here, the PLGA NCs can also be easily pegylated as shown for the NC-8 system (3 wt% PEG Mn 5000) and modify the blood circulation time of NCs. Moreover, the magnetic functionality could be exploited for magnetic targeting in a selected organ or tissue.
In vitro and in vivo safety studies of NCs
In vitro and in vivo safety studies of NC-3 (PLGA-SPIONs NCs) were conducted considering that this is the simplest therapeutic formulation for magnetically targeted delivery. We tested the effect of NC-3 on the proliferation/viability of human endothelial cells since endothelial cells lining the blood vessels are the first NC target following their intravenous administration. Thus, human brain endothelial cells (hCMEC/D3), a cell line derived from brain microcirculation, and primary cultures of human CD34+-derived endothelial cells were exposed to NC-3 and its cytotoxicity was evaluated by performing a MTT assay. As shown in Fig. 9a, the cells did not exhibit a decrease in viability after being exposed for 48 h to the NCs from 25 to 100 μg mL−1, and the differences in cell viability between groups were not significant (ANOVA >0.05), thus our results suggest that the NCs are biocompatible for the studied range of concentrations and time. Thereafter, the systemic effect of NCs via i.v. administration was evaluated. Fig. 9b shows that after injection of NCs, all animals maintained the body weight within normal values for two weeks (no differences between groups at the end of the experimental period), with no significant morbidity and no potential adverse effects. At the same time, blood sample tests to monitor potential liver and pancreatic toxicity showed that none of the biomarker enzymes were significantly different between groups and similar to those measured in non-treated mice (Fig. 9c; ANOVA >0.05), suggesting a normal activity of these organs. All the above results reinforce the biosafety of this formulation of nanocarriers for future clinical translational practice.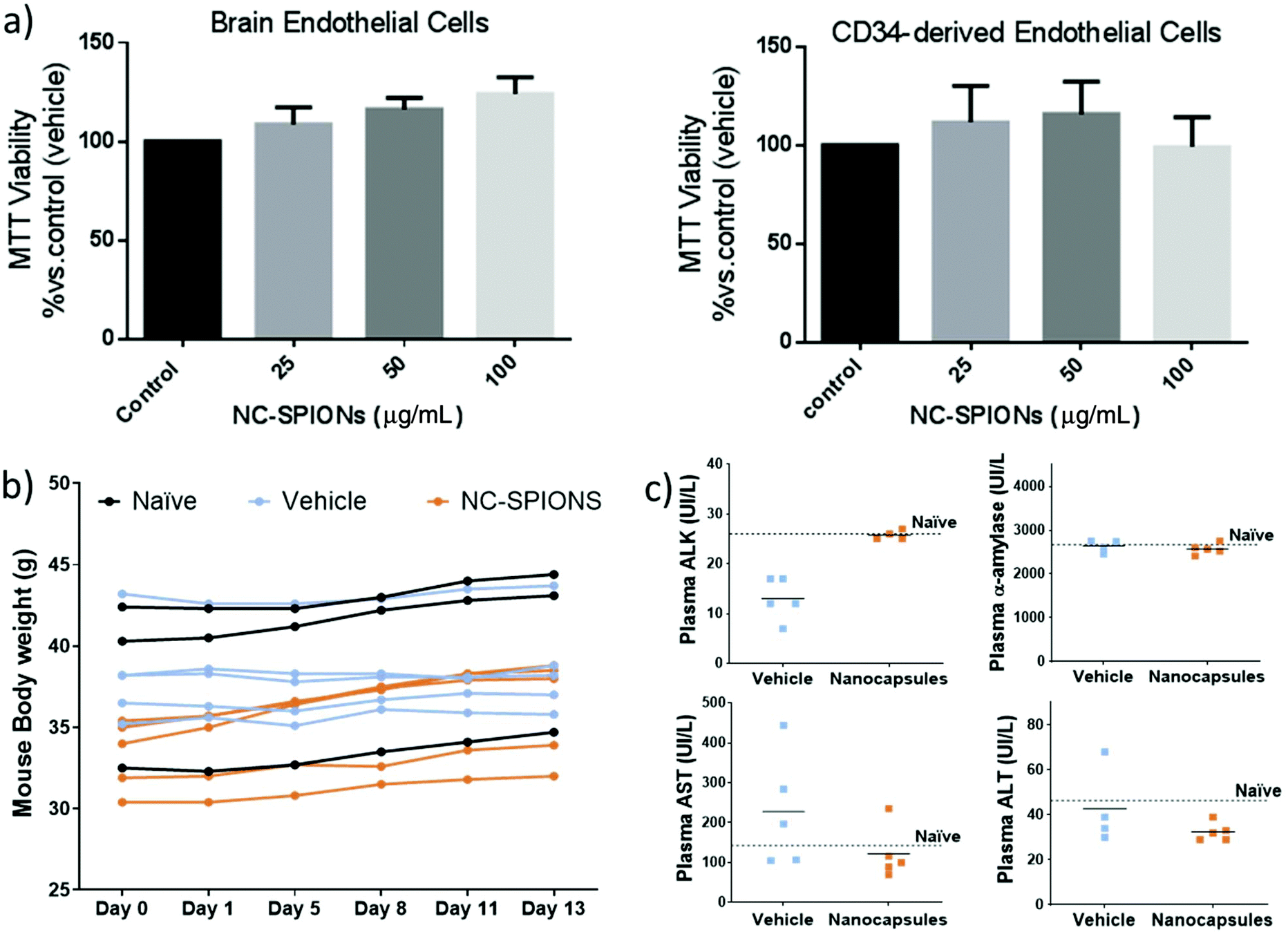 | ||
| Fig. 9 In vitro and in vivo safety of PLGA–SPION NCs (NC-3). (a) Cell viability after exposing to an increasing dose of NCs for 48 h, expressed as a percentage of untreated cells (control media), determined by MTT assay (n = 3, mean ± SEM); (b) the curve of body weight of the observed mice throughout the experiment, the naïve group was not treated serving as control (n = 3), the vehicle group received 150 μL of saline (n = 5) and the NC group received 0.84 mg NCs in 150 μL of saline (n = 5); (c) liver and pancreatic toxicity after 13 days of NC injection based on specific enzyme levels in plasma: alanine aminotransferase (ALT), alkaline phosphatase (ALK), aspartate aminotransferase (AST) and α-amylase. | ||
Protein loading
The protein loading content was determined as 11.3 ± 1.0 μg BSA per mg NCs (1.13 wt%) with an encapsulation efficiency of 46.0 ± 1.4%, the average of six different batches measured. The protein loading of 1.13 wt% is much higher than the reported value (0.03 wt%) of vessel endothelial growth factor loaded PLGA NCs31 and the EE% is comparable to the PLGA NCs loaded with neurotrophin-3 (47 ± 2%) or brain-derived neurotrophic factor (47 ± 7%).8Conclusions
By covalently bonding PLGA with small molecules of fluorophores and radioligands as well as by incorporating iron oxide nanoparticles we propose here chemical synthetic approaches to transform PLGA NCs into a sensitive, multimodal theranostic platform for drug delivery. We have shown that the size of PLGA NCs remained unaltered after incorporation or removal of one or several imaging moieties. We have proposed a modular approach for capsules with multimodal capacities to be used at several development phases and investigated by different imaging modalities including MRI, fluorescence at different emitting wavelengths (blue and NIR) or 89Zr-labeling enabled positron emission tomography (PET) imaging. In all cases, the imaging moieties are chemically attached to the PLGA and with minimal interaction with the encapsulated drug thus avoiding leaching and interferences with the therapeutic agent.The theranostic PLGA NCs show no toxicity in vitro or in vivo. In this regard, the NCs did not affect the viability of two different human endothelial cells (brain endothelial cells and CD34+ derived endothelial cells) for concentration up to 100 μg mL−1. In vivo biosafety was shown based on the lack of morbidity of mice for 2 weeks after the systemic NC administration and the fact that none of the biomarker enzymes monitored were significantly different between groups and similar to those measured in non-treated mice.
Finally, we have shown that the NCs can contain over 1 wt% of protein in their core achieved at the fabrication level. Interestingly, PLGA NCs decorated with iron oxide nanoparticles can be exploited for magnetic retention or magnetic guiding and biodistribution could be modified by using PEG-PLGA NCs.
Author contributions
Y. Zhang contributed to the experimental design, data acquisition and analysis, and manuscript writing. M. García contributed to protein loading and in vitro safety study. A. Grayston contributed to the in vivo safety and IVIS study. I. Feiner and J. Llop contributed to the radiolabelling and PET study. R. Loiola and F. Gosselet participated in the in vitro safety study. P. Cabrer, I. Barba and D. Dorado contributed to the MRI study. I. Anton participated in the NC preparation. A. Rosell and A. Roig contributed to the design of experiments, coordination of the study, and manuscript writing. All authors revised the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The Acciones Complementarias program from the Instituto de Salud Carlos III, Spain, co-financed by the European Regional Development Fund (AC17/00004 grant) as part of the MAGBBRIS project (Euronanomed III 8th joint call), and the RETICS INVICTUS RD16/0019/0021 are acknowledged. The Miguel Servet program (CPII15/00003) and PFIS program (FI17/00073) from Fondo de Investigaciones Sanitarias-Instituto de Salud Carlos III and ERDF supported A. Rosell and A. G. research contracts. This work was also partially supported by the Spanish Ministry of Science, Innovation and Universities through the grants PCIN-2017-090, RTI2018-096273-B-I00, CTQ2017-87637-R, SAF2017-87670-R, SEV-2015-0496 and MDM-2017-0720 and by the Generalitat de Catalunya grants (2017SGR765 & 2017SGR1427). Yajie Zhang is supported by the China Scholarship Council (CSC). FG and RAL are supported by the French national agency (ANR) through the grant ANR-17-ENM3-0005-01. The authors thank Dr Vanessa Gómez-Vallejo, Dr Unai Cossío, Dr Xabier Rios and Zuriñe Baz for support in labelling and PET in vivo experimentation. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- R. Klippstein, J. T. W. Wang, R. I. El-Gogary, J. Bai, F. Mustafa, N. Rubio, S. Bansal, W. T. Al-Jamal and K. T. Al-Jamal, Small, 2015, 11, 4704–4722 CrossRef CAS PubMed.
- T. Sudha, D. J. Bharali, M. Yalcin, N. H. Darwish, M. Debreli Coskun, K. A. Keating, H. Y. Lin, P. J. Davis and S. A. Mousa, Int. J. Nanomed., 2017, 12, 1305–1315 CrossRef CAS PubMed.
- R. Yang, J. Xu, L. Xu, X. Sun, Q. Chen, Y. Zhao, R. Peng and Z. Liu, ACS Nano, 2018, 12, 5121–5129 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- Z. Ouyang, T. Tan, C. Liu, J. Duan, W. Wang, X. Guo, Q. Zhang, Z. Li, Q. Huang and P. Dou, Biomaterials, 2019, 205, 50–63 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- M. M. Pakulska, I. E. Donaghue, J. M. Obermeyer, A. Tuladhar, C. K. McLaughlin, T. N. Shendruk and M. S. Shoichet, Sci. Adv., 2016, 2, e1600519 CrossRef PubMed.
- X. Qin, C. Yu, J. Wei, L. Li, C. Zhang, Q. Wu, J. Liu, S. Q. Yao and W. Huang, Adv. Mater., 2019, 31, 1902791 CrossRef CAS PubMed.
- A. Harguindey, D. W. Domaille, B. D. Fairbanks, J. Wagner, C. N. Bowman and J. N. Cha, Adv. Mater., 2017, 29, 1700743 CrossRef PubMed.
- C. J. Bowerman, J. D. Byrne, K. S. Chu, A. N. Schorzman, A. W. Keeler, C. A. Sherwood, J. L. Perry, J. C. Luft, D. B. Darr, A. M. Deal, M. E. Napier, W. C. Zamboni, N. E. Sharpless, C. M. Perou and J. M. DeSimone, Nano Lett., 2017, 17, 242–248 CrossRef CAS PubMed.
- B. Colzani, L. Pandolfi, A. Hoti, P. A. Iovene, A. Natalello, S. Avvakumova, M. Colombo and D. Prosperi, Int. J. Nanomed., 2018, 13, 957–973 CrossRef CAS PubMed.
- W. Tao, X. Zeng, J. Wu, X. Zhu, X. Yu, X. Zhang, J. Zhang, G. Liu and L. Mei, Theranostics, 2016, 6, 470–484 CrossRef CAS PubMed.
- P. Kumar, T. V. Treuren, A. P. Ranjan, P. Chaudhary and J. K. Vishwanatha, Nanotechnology, 2019, 30, 265101 CrossRef CAS PubMed.
- A. C. Souza, A. L. Nascimento, N. M. de Vasconcelos, M. S. Jeronimo, I. M. Siqueira, L. R-Santos, D. O. Cintra, L. L. Fuscaldi, O. R. Pires Junior, R. Titze-de-Almeida, M. F. Borin, S. N. Bao, O. P. Martins, V. N. Cardoso, S. O. Fernandes, M. R. Mortari, A. C. Tedesco, A. C. Amaral, M. S. Felipe and A. L. Bocca, Eur. J. Med. Chem., 2015, 95, 267–276 CrossRef CAS PubMed.
- J. Llop, P. Jiang, M. Marradi, V. Gomez-Vallejo, M. Echeverria, S. Yu, M. Puigivila, Z. Baz, B. Szczupak and C. Perez-Campana, J. Mater. Chem. B, 2015, 3, 6293–6300 RSC.
- L. J. Cruz, M. A. Stammes, I. Que, E. R. van Beek, V. T. Knol-Blankevoort, T. J. A. Snoeks, A. Chan, E. L. Kaijzel and C. Lowik, J. Controlled Release, 2016, 223, 31–41 CrossRef CAS PubMed.
- M. M. Abdel-Mottaleb, A. Beduneau, Y. Pellequer and A. Lamprecht, Int. J. Pharm., 2015, 494, 471–478 CrossRef CAS PubMed.
- R. L. Cook, K. T. Householder, E. P. Chung, A. V. Prakapenka, D. M. DiPerna and R. W. Sirianni, J. Controlled Release, 2015, 220, 89–97 CrossRef CAS PubMed.
- S. Florinas, M. Liu, R. Fleming, L. Van Vlerken-Ysla, J. Ayriss, R. Gilbreth, N. Dimasi, C. Gao, H. Wu and Z.-Q. Xu, Biomacromolecules, 2016, 17, 1818–1833 CrossRef CAS PubMed.
- K. Qian, J. Wu, E. Zhang, Y. Zhang and A. Fu, Int. J. Nanomed., 2015, 10, 4149 CAS.
- S. Udenfriend, S. Stein, P. Bohlen, W. Dairman, W. Leimgruber and M. Weigele, Science, 1972, 178, 871–872 CrossRef CAS PubMed.
- E. Carenza, V. Barcelo, A. Morancho, J. Montaner, A. Rosell and A. Roig, Acta Biomater., 2014, 10, 3775–3785 CrossRef CAS PubMed.
- J. Yang, Y. Zhang, S. Gautam, L. Liu, J. Dey, W. Chen, R. P. Mason, C. A. Serrano, K. A. Schug and L. Tang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 10086–10091 CrossRef CAS PubMed.
- R. Cecchelli, S. Aday, E. Sevin, C. Almeida, M. Culot, L. Dehouck, C. Coisne, B. Engelhardt, M. P. Dehouck and L. Ferreira, PLoS One, 2014, 9, e99733 CrossRef PubMed.
- F. Shimizu, Y. Sano, M. a. Abe, T. Maeda, S. Ohtsuki, T. Terasaki and T. Kanda, J. Cell. Physiol., 2011, 226, 255–266 CrossRef CAS PubMed.
- K. Mauring, V. Krasnenko and S. Miller, J. Lumin., 2007, 122, 291–293 CrossRef.
- D. Wang, T. Imae and M. Miki, J. Colloid Interface Sci., 2007, 312, 8–13 CrossRef CAS PubMed.
- W. Yang and C. Y. Pan, Macromol. Rapid Commun., 2009, 30, 2096–2101 CrossRef CAS PubMed.
- J. Hu, J. Guo, Z. Xie, D. Shan, E. Gerhard, G. Qian and J. Yang, Acta Biomater., 2016, 29, 307–319 CrossRef CAS PubMed.
- E. Carenza, O. Jordan, P. Martinez-San Segundo, R. Jiřík, Z. Starčuk Jr., G. Borchard, A. Rosell and A. Roig, J. Mater. Chem. B, 2015, 3, 2538–2544 RSC.
- Y.-w. Jun, Y.-M. Huh, J.-s. Choi, J.-H. Lee, H.-T. Song, S. Kim, S. Kim, S. Yoon, K.-S. Kim and J.-S. Shin, J. Am. Chem. Soc., 2005, 127, 5732–5733 CrossRef CAS PubMed.
- R. Gref, M. Lück, P. Quellec, M. Marchand, E. Dellacherie, S. Harnisch, T. Blunk and R. Müller, Colloids Surf., B, 2000, 18, 301–313 CrossRef CAS PubMed.
- Y. Cui, M. Zhang, F. Zeng, H. Jin, Q. Xu and Y. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 32159–32169 CrossRef CAS PubMed.
- N. Butoescu, C. A. Seemayer, G. Palmer, P. A. Guerne, C. Gabay, E. Doelker and O. Jordan, Arthritis Res. Ther., 2009, 11, R72 CrossRef PubMed.
- E. Taboada, R. Solanas, E. Rodríguez, R. Weissleder and A. Roig, Adv. Funct. Mater., 2009, 19, 2319–2324 CrossRef CAS.
- Y. X. Wang, Quant. Imaging Med. Surg., 2011, 1, 35–40 Search PubMed.
- F. Chen, S. Goel, H. F. Valdovinos, H. Luo, R. Hernandez, T. E. Barnhart and W. Cai, ACS Nano, 2015, 9, 7950–7959 CrossRef CAS PubMed.
- E. Boros, A. M. Bowen, L. Josephson, N. Vasdev and J. P. Holland, Chem. Sci., 2015, 6, 225–236 RSC.
- D. S. Abou, T. Ku and P. M. Smith-Jones, Nucl. Med. Biol., 2011, 38, 675–681 CrossRef CAS PubMed.
- M. Cataldi, C. Vigliotti, T. Mosca, M. Cammarota and D. Capone, Int. J. Mol. Sci., 2017, 18, 1249 CrossRef PubMed.
- E. Swider, S. Maharjan, K. Houkes, N. K. van Riessen, C. G. Figdor, M. Srinivas and O. Tagit, ACS Appl. Bio Mater., 2019, 2(3), 1131–1140 CrossRef CAS PubMed.
- J. M. Caster, S. K. Yu, A. N. Patel, N. J. Newman, Z. J. Lee, S. B. Warner, K. T. Wagner, K. C. Roche, X. Tian, Y. Min and A. Z. Wang, Nanomedicine, 2017, 13, 1673–1683 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr10620k |
| ‡ In memory of our colleague Dr Garcia-Dorado who passed away on August 16th 2019. |
| This journal is © The Royal Society of Chemistry 2020 |