
Rapid and mass-producible synthesis of high-crystallinity MoSe2 nanosheets by ampoule-loaded chemical vapor deposition†

Abstract
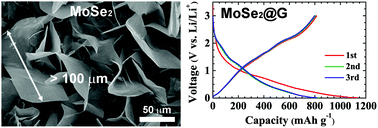
MoSe2 is an attractive transition-metal dichalcogenide with a two-dimensional layered structure and various attractive properties. Although MoSe2 is a promising negative electrode material for electrochemical applications, further investigation of MoSe2 has been limited, mainly by the lack of MoSe2 mass-production methods. Here, we report a rapid and ultra-high-yield synthesis method of obtaining MoSe2 nanosheets with high crystallinity and large grains by ampoule-loaded chemical vapor deposition. Application of high pressure to an ampoule-type quartz tube containing MoO3 and Se powders initiated rapid reactions that produced vertically oriented MoSe2 nanosheets with grain sizes of up to ∼100 μm and yields of ∼15 mg h−1. Spectroscopy and microscopy characterizations confirmed the high crystallinity of the obtained MoSe2 nanosheets. Transistors and lithium-ion battery cells fabricated with the synthesized MoSe2 nanosheets showed good performance, thereby further indicating their high quality. The proposed simple scalable synthesis method can pave the way for diverse electrical and electrochemical applications of MoSe2.
 Please wait while we load your content...
Please wait while we load your content...

