
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adsorbate-induced structural evolution changes the mechanism of CO oxidation on a Rh/Fe3O4(001) model catalyst†
Zdenek
Jakub
 a,
Jan
Hulva
a,
Paul T. P.
Ryan
bc,
David A.
Duncan
b,
David J.
Payne
c,
Roland
Bliem
a,
Manuel
Ulreich
a,
Patrick
Hofegger
a,
Florian
Kraushofer
a,
Matthias
Meier
ad,
Michael
Schmid
a,
Ulrike
Diebold
a and
Gareth S.
Parkinson
*a
a,
Jan
Hulva
a,
Paul T. P.
Ryan
bc,
David A.
Duncan
b,
David J.
Payne
c,
Roland
Bliem
a,
Manuel
Ulreich
a,
Patrick
Hofegger
a,
Florian
Kraushofer
a,
Matthias
Meier
ad,
Michael
Schmid
a,
Ulrike
Diebold
a and
Gareth S.
Parkinson
*a
aInstitute of Applied Physics, TU Wien, 1040 Vienna, Austria. E-mail: parkinson@iap.tuwien.ac.at
bDiamond Light Source, Harwell Science and Innovation Campus, Didcot, OX11 0DE, UK
cDepartment of Materials, Imperial College London, South Kensington, London, SW7 2AZ, UK
dUniversity of Vienna, Faculty of Physics and Center for Computational Materials Science, 1090 Vienna, Austria
First published on 26th February 2020
Abstract
The structure of a catalyst often changes in reactive environments, and following the structural evolution is crucial for the identification of the catalyst's active phase and reaction mechanism. Here we present an atomic-scale study of CO oxidation on a model Rh/Fe3O4(001) “single-atom” catalyst, which has a very different evolution depending on which of the two reactants, O2 or CO, is adsorbed first. Using temperature-programmed desorption (TPD) combined with scanning tunneling microscopy (STM) and X-ray photoelectron spectroscopy (XPS), we show that O2 destabilizes Rh atoms, leading to the formation of RhxOy clusters; these catalyze CO oxidation via a Langmuir–Hinshelwood mechanism at temperatures as low as 200 K. If CO adsorbs first, the system is poisoned for direct interaction with O2, and CO oxidation is dominated by a Mars-van-Krevelen pathway at 480 K.
Introduction
Supported rhodium catalysts show high activity for a number of reactions, including CO oxidation.1–4 It is generally accepted that Rh nanoparticles supported on rigid, irreducible substrates catalyze CO oxidation via the Langmuir–Hinshelwood (L–H) or Eley–Rideal (E–R) mechanisms, where the CO and O2 molecules either coadsorb on the surface (L–H), or one of the molecules reacts directly from the gas phase with the other molecule adsorbed on the surface (E–R).6–12 However, the reactivity of the system is often affected by the choice of support, and reducible supports enable an alternative pathway, the so-called Mars-van-Krevelen (MvK) mechanism. Here, oxygen is extracted from the metal oxide lattice to oxidise CO, and the lattice is repaired by O2 from the gas phase.14,16–19 Metal–support interactions are not always beneficial, however, and encapsulation by the support can completely deactivate the catalyst.13,14,20,21 Improving a catalyst's activity, selectivity, and stability requires the optimization of atomic-scale processes that are very difficult to ascertain on working catalysts. This motivates the use of well-defined model systems studied by the surface science approach, which can provide atomic-scale insights.In this paper we present how CO oxidation proceeds on a Rh/Fe3O4(001) model “single-atom” catalyst. Using a combination of temperature-programmed desorption (TPD), scanning tunneling microscopy (STM), and X-ray photoelectron spectroscopy (XPS), we show the system is active for CO oxidation by both L–H and MvK mechanisms, depending on which of the reactants the surface is exposed to first. Oxygen adsorption leads to formation of superoxo (O2)− species bound to Rh1 adatoms, which rapidly agglomerate to small RhxOy clusters. The clusters contain weakly bound oxygen, which reacts with CO below room temperature. In contrast, CO adsorption on the bare Rh1 species poisons the system for O2 adsorption, but CO oxidation nevertheless takes place via a MvK mechanism at ca. 480 K. Two different deactivation mechanisms are observed: the oxidised clusters become inactive after the first desorption of the weakly bound oxygen, while the Rh1 adatoms incorporate into the support lattice as the CO desorbs.
Methods
The experiments were carried out using two independent ultra-high vacuum (UHV) setups. The STM data were acquired in a two-vessel UHV chamber consisting of a preparation chamber (p < 10−10 mbar) and an analysis chamber (p < 7 × 10−11 mbar) equipped with an Omicron μ-STM operated at room temperature in constant-current mode with electrochemically etched tungsten tips. The chamber further includes commercial XPS, Auger electron spectroscopy and low-energy electron diffraction instruments. The STM images were corrected for distortion and creep of the piezo scanner as described in ref. 22.The TPD and XPS data were acquired in a second chamber equipped with a liquid-He cryostat, a home-built molecular beam source, a quadrupole mass spectrometer (Hiden HAL 3F PIC), a monochromatized Al/Ag twin anode X-ray source (Specs XR50 M, FOCUS 500), a hemispherical analyzer (Specs Phoibos 150), a low-energy electron diffraction setup (Specs ErLEED), an ion source (Specs IQE 12/38) and a UV source (Specs UVS 10/35). This chamber was specifically designed for surface chemistry studies of single-crystal oxide samples; full details are given in ref. 23. The samples were mounted on a Ta sample plate using Ta clips, with a thin Au foil placed between the sample plate and the sample to ensure good thermal contact. The sample plate was attached to the liquid-He cryostat via Ta rods, and the temperature was measured by a K-type thermocouple spot-welded on the sample plate. A molecular beam is formed by expansion of 0.53 mbar 18O2 or 0.27 mbar 13CO through two differentially pumped stages. This results in a well-defined beam spot on the sample surface with a diameter of 3.35 ± 0.17 mm and a top-hat intensity profile.23 The TPD spectra were acquired with a 1 K s−1 heating ramp.
All the experiments were conducted on natural Fe3O4(001) single crystals (SurfaceNet GmbH or Surface Preparation Laboratory) prepared by sputtering (1 keV Ar+ or Ne+, 10 min) and annealing in UHV (930 K, 10 min). Every other annealing step was done in a partial pressure of O2 (pO2 = 5 × 10−7 mbar, 20 min) to avoid reduction of the surface. Rhodium was deposited using Focus e-beam evaporators, and the deposition rate was calibrated by temperature-stabilized quartz crystal microbalances (QCM). One monolayer (ML) is defined as one atom per (√2 × √2)R45° unit cell of the Fe3O4(001), which corresponds to 1.42 × 1014 atoms per cm2.
Results
Rh1 species on Fe3O4(001)
Fig. 1a and b shows STM images of the Fe3O4(001) surface before and after deposition of Rh. The undulating rows running in the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] direction correspond to 5-fold coordinated Fe atoms in surface octahedral sites (Feoct, see model in the inset of Fig. 1a). In each pair of neighbouring terraces, the surface Feoct row direction is rotated by 90°, as expected for the spinel structure (not shown here). Surface oxygen atoms are not visible in the STM images because they possess no density of states near the Fermi level, but their positions are precisely known from diffraction studies and density functional theory calculations.24,25 This Fe3O4(001) surface reconstruction is based on subsurface cation vacancies (therefore it's referred to as the SCV reconstruction), and it has been shown previously to support dense arrays of single metal atoms, preventing the adatom mobility and cluster formation even at elevated temperatures.15,24,26–28 This is due to the existence of highly stable adsorption sites with a nearest-neighbour distance of 0.84 nm, in which the adatoms are 2-fold coordinated to the surface oxygen atoms, essentially occupying a bulk continuation tetrahedral site (schematic representation shown in the uppermost inset of Fig. 2a). After deposition of Rh at room temperature, isolated Rh1 adatoms appear as bright protrusions (red circles in Fig. 1b) sitting between the Feoct rows. In addition to the single Rh1 adatoms, we also observe larger elongated species (cyan circle in Fig. 1b), which in subsequent STM scans exhibit a distinctive hopping motion over the adsorption site (Fig. 1c and d). We identify these species as metastable Rh2 dimers that can be split into two Rh1 adatoms by bias pulsing or high-bias scanning. Fig. 1e and f shows this process on two STM images acquired on the same spot: in Fig. 1e two Rh2 dimers are observed (cyan arrows), Fig. 1f shows pairs of isolated Rh1 adatoms (smaller paired arrows) appearing instead of the Rh2 after the surface was scanned with a +3 V bias. The observed hopping of the Rh2 is restricted to the given adsorption site, thus it might be also considered a restructuring of the dimer and does not lead to destabilization of the system by diffusion or agglomeration. Apart from the dimer hopping, no mobility of Rh species is observed in subsequently acquired STM images at room temperature under UHV conditions. The Fe3O4(001) surface supports the Rh1 adatoms even at significantly higher coverages; Fig. S1† shows an STM image of 0.5 ML Rh/Fe3O4(001) featuring a high density of Rh1 adatoms, and XPS spectra acquired on an Fe3O4(001) surface with varying Rh coverage between 0.1 and 0.8 ML Rh showing virtually no difference in Rh 3d peak shape or position.
10] direction correspond to 5-fold coordinated Fe atoms in surface octahedral sites (Feoct, see model in the inset of Fig. 1a). In each pair of neighbouring terraces, the surface Feoct row direction is rotated by 90°, as expected for the spinel structure (not shown here). Surface oxygen atoms are not visible in the STM images because they possess no density of states near the Fermi level, but their positions are precisely known from diffraction studies and density functional theory calculations.24,25 This Fe3O4(001) surface reconstruction is based on subsurface cation vacancies (therefore it's referred to as the SCV reconstruction), and it has been shown previously to support dense arrays of single metal atoms, preventing the adatom mobility and cluster formation even at elevated temperatures.15,24,26–28 This is due to the existence of highly stable adsorption sites with a nearest-neighbour distance of 0.84 nm, in which the adatoms are 2-fold coordinated to the surface oxygen atoms, essentially occupying a bulk continuation tetrahedral site (schematic representation shown in the uppermost inset of Fig. 2a). After deposition of Rh at room temperature, isolated Rh1 adatoms appear as bright protrusions (red circles in Fig. 1b) sitting between the Feoct rows. In addition to the single Rh1 adatoms, we also observe larger elongated species (cyan circle in Fig. 1b), which in subsequent STM scans exhibit a distinctive hopping motion over the adsorption site (Fig. 1c and d). We identify these species as metastable Rh2 dimers that can be split into two Rh1 adatoms by bias pulsing or high-bias scanning. Fig. 1e and f shows this process on two STM images acquired on the same spot: in Fig. 1e two Rh2 dimers are observed (cyan arrows), Fig. 1f shows pairs of isolated Rh1 adatoms (smaller paired arrows) appearing instead of the Rh2 after the surface was scanned with a +3 V bias. The observed hopping of the Rh2 is restricted to the given adsorption site, thus it might be also considered a restructuring of the dimer and does not lead to destabilization of the system by diffusion or agglomeration. Apart from the dimer hopping, no mobility of Rh species is observed in subsequently acquired STM images at room temperature under UHV conditions. The Fe3O4(001) surface supports the Rh1 adatoms even at significantly higher coverages; Fig. S1† shows an STM image of 0.5 ML Rh/Fe3O4(001) featuring a high density of Rh1 adatoms, and XPS spectra acquired on an Fe3O4(001) surface with varying Rh coverage between 0.1 and 0.8 ML Rh showing virtually no difference in Rh 3d peak shape or position.
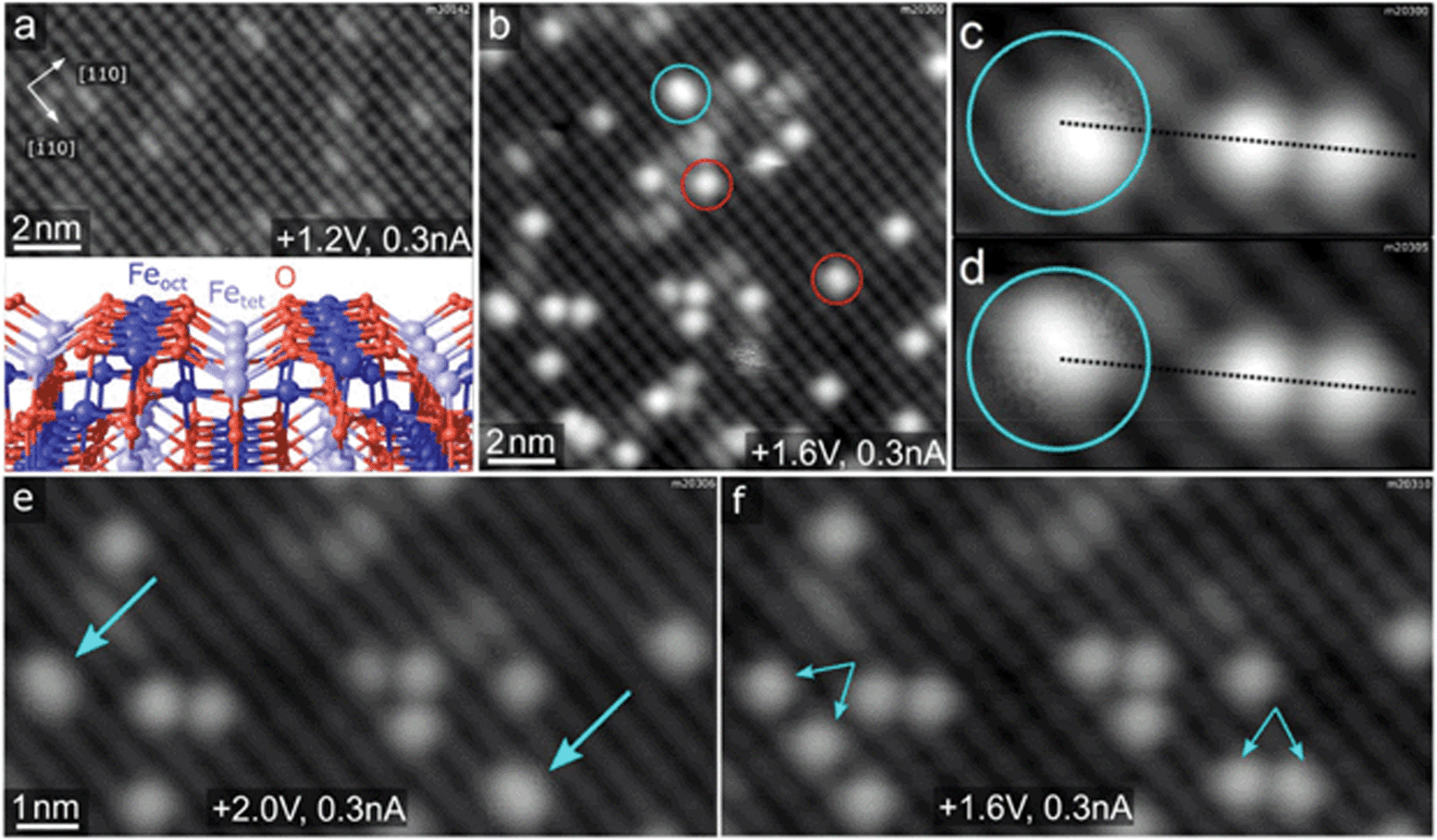 | ||
| Fig. 1 Characterization of the Rh/Fe3O4(001) system by STM. (a) STM images of pristine Fe3O4(001) surface show undulating rows of Feoct running in the [10] direction. The small bright species on the Feoct rows are surface hydroxyls.5 The inset shows a model of the Fe3O4(001) surface with the octahedral Feoct, tetrahedral Fetet and O atoms being represented by dark blue, light blue and red spheres, respectively. (b) After deposition of 0.2 ML Rh at room temperature, the majority species are Rh1 adatoms (red circles), but we also observe slightly elongated species which we identify as Rh2 dimers (cyan circles). (c and d) The Rh2 dimers exhibit characteristic hopping motion in subsequent STM images (broken lines for guidance). (e and f) Scanning with a +3 V bias results in dissociation of the Rh2 dimers (cyan arrows in panel e) into pairs of single Rh1 adatoms (paired cyan arrows in panel f). The +3 V scan was done between acquisition of images shown in (e) and (f). | ||
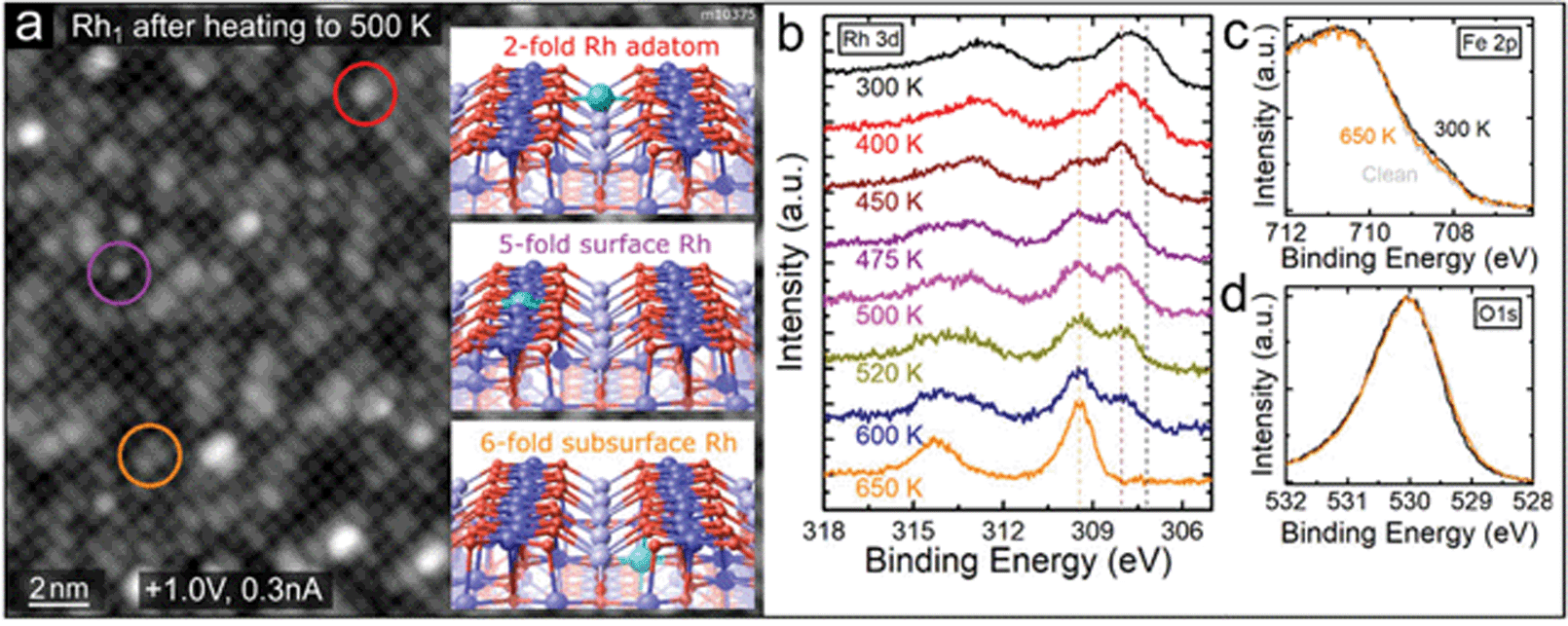 | ||
| Fig. 2 Rh1 adatoms incorporate into the Fe3O4(001) support upon annealing above 450 K. (a) STM images after deposition of 0.4 ML Rh and heating to 500 K show predominantly circular protrusions (highlighted by purple colour) and elongated protrusions (orange colour) on the Feoct rows. Only a very small number of Rh1 remain in the 2-fold coordinated site between the Feoct rows (red colour). We identify the new species as Rh1 incorporated in the octahedral cation sites near the surface, consistent with previously reported studies of various metal adatoms on Fe3O4(001).14,15 Schematic representations of these sites are shown in the insets and labelled with the color of the corresponding circle. (b) Rh 3d region of the XPS spectra (monochromatized Al Kα, grazing emission) taken after heating to different temperatures (measured after cooling back to 300 K). Upon heating to 400 K the Rh 3d peak maximum shifts to 308.0 eV, as the low binding-energy shoulder at 307.2 eV decreases. Heating above 450 K leads to an increase of the 309.5 eV component, which becomes dominant above 600 K. We attribute this component to Rh incorporated into the octahedral sites of the Fe3O4(001) surface. (c and d) The Fe 2p and O 1s peaks corresponding to the first and last spectra shown in (b). Notably, in the Fe 2p region the shoulder at ≈708.5 eV slightly increases upon Rh deposition, indicating an increased amount of Fe2+ near the surface. This shoulder decreases again upon heating as the Rh cations diffuse into deeper layers. | ||
At elevated temperatures, the Rh1 adatoms are unstable against incorporation into the Fe3O4(001) support. Such a phenomenon has been observed for several different metal adatoms on Fe3O4(001) before and the foreign adatom can either substitute a 5-fold coordinated surface Feoct or incorporate in the vacant octahedral site of the SCV reconstruction.13–15,29 Schematic representations of the three possible sites of Rh1 near the surface are shown in insets in Fig. 2a. The incorporation of Rh takes place at temperatures above ≈450 K and can be followed both in STM and XPS, as shown in Fig. 2a and b. STM images taken after annealing to 500 K show predominantly elongated (orange circle) and circular (purple circle) features on the Feoct rows, which is consistent with Rh1 incorporated into the subsurface 6-fold site or surface 5-fold site, respectively (STM simulations of different metal adatoms in the same sites are provided in ref. 14, 15 and Fig. S2†). Additionally, we observe an increased number of bigger brighter features, presumably Rh clusters, and a very small number of remaining 2-fold Rh1 adatoms (red circle). Fig. 2b shows the Rh 3d region of XPS spectra taken after depositing 0.6 ML Rh and heating to different temperatures. After deposition at 300 K, the Rh 3d peak maximum is located at ≈307.7 eV, but its broad shape suggests convolution of several components. Apart from the main signal corresponding to 2-fold Rh1, the additional XPS components might come from the Rh2 dimers or Rh1 adatoms residing in the vicinity of surface defects or step edges. On different samples, small variations of the peak maximum between 307.7 and 308.1 eV were observed, most likely depending on the relative number of these species. After heating to 400 K we observe a decrease on the low-binding-energy side of the peak (around ≈307.2 eV) and the peak maximum shifts to 308.0 eV. Heating above 450 K leads to an increase of a higher-binding-energy component at 309.5 eV and decrease of the signal at 308.0 eV. After heating to 650 K, the 308.0 eV peak is no longer observed, and the 309.5 eV component is dominant. Prolonged heating above this temperature leads to a decrease and eventual disappearance of the Rh signal in XPS, consistent with diffusion of Rh into the Fe3O4 bulk (Fig. S3†). Thus, we attribute the 309.5 eV component to Rh incorporated into the Fe3O4(001) substrate.
The Fe 2p and O 1s regions only show small differences with Rh deposition and heating (Fig. 2c and d). Upon Rh deposition, an Fe 2p shoulder at ≈708.5 eV slightly increases in comparison to the clean surface, indicating an increased amount of Fe2+ near the surface.30 This shoulder decreases again upon heating, presumably because the Rh incorporates and thus the number of near-surface cations decreases.
Exposing Rh1/Fe3O4(001) to O2
When the Rh1/Fe3O4(001) system is exposed to O2 at room temperature, the Rh1 adatoms become mobile and begin to agglomerate. Fig. 3a–c shows three frames of an STM movie acquired while keeping a constant background pressure of 2 × 10−10 mbar O2 in the analysis chamber. Single adatoms (highlighted by red circles) form well-resolved oxidised Rh2Ox dimers (yellow rectangles) and subsequently bigger RhxOy clusters (purple rectangle). Over the course of the STM movie, no O2 adsorption was observed on the Rh2 dimers (cyan circle). After dosing a higher amount of O2 in the preparation chamber (50 Langmuir, where 1 L = 1.33 × 10−6 mbar s), the majority species observed in STM are small RhxOy clusters, although some adatoms remain (Fig. 3d). Scanning of the oxidised RhxOy species is difficult because the tip is very unstable, with frequent tip changes suggesting the presence of weakly bound species that interact with the STM tip. A comparison of the cluster density following saturation exposure with the initial adatom coverage suggests that the majority contain at most 2–3 Rh atoms. The fact that similar Rh agglomeration was observed after O2 exposure in the preparation chamber suggests that the mobility observed in STM movies is not solely tip-induced. After heating the surface with oxidised RhxOy clusters to 400 K, no structural differences are observed, but the imaging becomes much more stable, suggesting desorption or trapping of the weakly bound species. | ||
| Fig. 3 STM of Rh1/Fe3O4(001) and its interaction with oxygen. (a–c) Three frames from an STM movie acquired while keeping a constant O2 background pressure of 2 × 10−10 mbar. Panel (a) shows a majority of single Rh1 adatoms (red circles). During the O2 exposure these species first form well-resolved Rh2Ox clusters (yellow rectangles in panels (b) and (c)) and subsequently bigger nanoparticles (purple rectangle in panel (c)). (d) After a high dose (50 L) of O2 at room temperature the imaging becomes unstable, and the majority species are larger bright clusters of varying size and shape. A few Rh1 adatoms remain; one of them is highlighted by the red circle. | ||
Fig. 4a shows O2 TPD measurements from the clean Fe3O4(001) surface (grey curve) and from the surface with 0.6 ML Rh (orange and brown curves). On the pristine Fe3O4(001) surface, O2 physisorbs below 70 K (desorption peak labelled α in Fig. 4a, full spectrum shown in Fig. S4†), but in our work this phase could not be saturated due to the O2 dosing temperature (60 K) being within this desorption peak. Thus, desorption already takes place before the start of the heating ramp and the magnitude and shape of the α peak varies in repeated measurements due to slightly varying time between dosing and heating. Above 100 K, the O2 TPD acquired on the pristine surface exhibits two distinct peaks, labelled β and γ in Fig. 4a and Fig. S4,† which presumably correspond to desorption from defects, domain boundaries, and/or step edges. The presence of Rh1 adatoms on the surface prior to O2 dosing leads to an increase of these two desorption peaks and induces a sharp new desorption peak, δ, at ≈330 K. When the TPD measurement was repeated on the same sample (following termination of the previous heating ramp at 570 K), the δ peak was no longer present (brown curve in Fig. 4a), and a similar result was observed in a separate experiment when the first heating ramp ended already at 380 K, before any Rh incorporation took place (evidenced by XPS spectra).
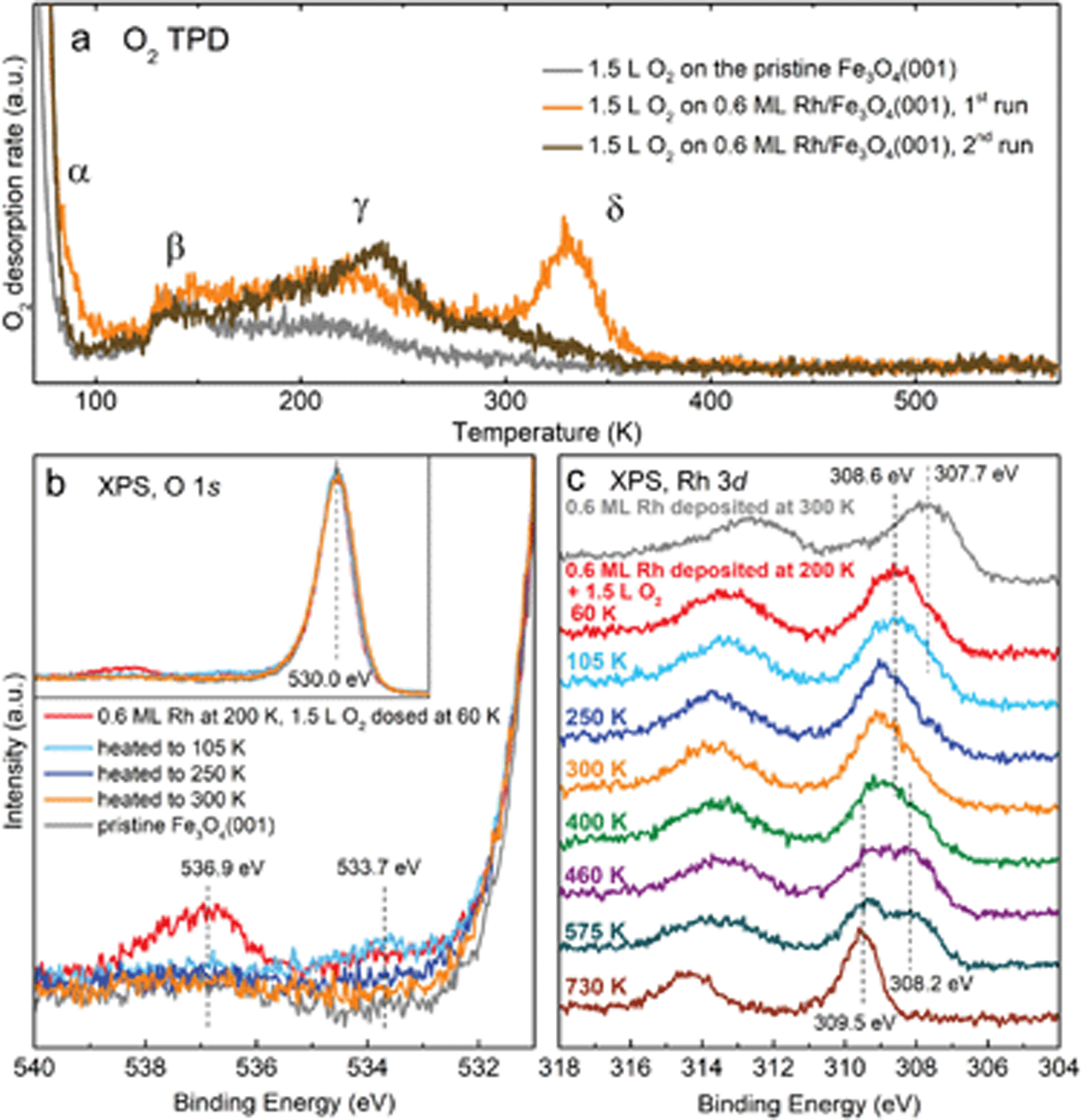 | ||
| Fig. 4 Spectroscopic characterization of O2 adsorption on Rh/Fe3O4(001) by TPD and XPS. In all spectra, 1.5 L O2 was dosed at 60 K. (a) O2 TPD spectra acquired in the first (orange curve) and second (brown curve) dosing on 0.6 ML Rh/Fe3O4(001). Notably, the δ peak at ≈330 K appears only in the first dosing experiment. Reference spectra of the clean surface are plotted in grey. (b and c) Details of the O 1s and Rh 3d XPS regions (monochromatized Al Kα, grazing emission) of 0.6 ML Rh/Fe3O4(001) after O2 dosing and heating to different temperatures. Compared to the pristine Fe3O4(001), we identify two new components in the O 1s region at 533.7 and 536.9 eV. The spectra taken after heating above room temperature look identical to the orange curve taken at 300 K. The whole O 1s region is shown in the inset in (b). (c) Adsorption of O2 at 60 K causes a ≈0.9 eV shift of the Rh 3d peak, and a further shift is observed upon heating to 300 K. Heating above ≈500 K leads to growth of the 309.5 eV component corresponding to Rh incorporated into the substrate. Up to 300 K, the spectra were taken at the temperatures given; for higher temperatures, the spectra were taken after cooling the sample back to 300 K. | ||
XPS characterization of the O2 adsorption on Rh/Fe3O4(001) is shown in Fig. 4b and c. Spectra taken directly after Rh deposition at 200 K and dosing 1.5 L O2 at 60 K reveal two new components in the O 1s region at 533.7 and 536.9 eV (red curve in Fig. 4b). The 536.9 eV signal quickly decreases over time and completely disappears within several minutes, which is consistent with the O2 physisorbed on Fe3O4(001) and readily desorbing at 60 K. The 533.7 eV component remains unchanged upon heating to 105 K, but disappears upon heating above 250 K (blue and orange curves in Fig. 4b). After heating to 300 K (orange curve), the spectrum in the O 1s region closely resembles the spectrum acquired on a pristine Fe3O4(001) surface (grey curve), save for a very small and broad shoulder between 530–535 eV, which can be attributed to a small amount of water adsorbed on surface defects.31 Upon heating to higher temperatures up to 730 K, no significant change is observed in the O 1s region.
The adsorption of O2 can also be followed in the Rh 3d region. Fig. 4c shows spectra taken after Rh deposition (0.6 ML), dosing O2 at 60 K, and heating to different temperatures. Directly after Rh deposition at room temperature the shape of the Rh 3d5/2 peak has a maximum at ≈307.7 eV, but as mentioned previously, its shape suggests convolution of several components. A small shoulder at ≈309.5 eV corresponds to Rh incorporated in the octahedral sites of the support (see Fig. 2); this component can be minimized by depositing Rh at lower temperature (Fig. S5†). After dosing O2 at 60 K, the maximum of the Rh 3d5/2 peak shifts by ≈0.9 eV to 308.6 eV. Heating to 300 K induces a further shift to ≈309.0 eV, and this position remains the same with heating to 400 K. At this temperature, a small shoulder at 308.2 eV appears. Heating above 460 K causes growth of the 309.5 eV component corresponding to incorporated Rh and slight increase of the signal at 308.2 eV. Above 700 K only the signal from the incorporated Rh remains. The Fe 2p region does not show any significant changes with O2 adsorption and heating (spectra shown in Fig. S6†). The question of how the changes in XPS spectra correlate to the O2-induced Rh1 agglomeration observed by STM is covered in the discussion section.
Exposing Rh1/Fe3O4(001) to CO
The same experimental approach as utilized above for O2 was used to study CO adsorption on the Rh1 adatoms. Fig. 5 shows five frames of an STM movie recorded while keeping a background pressure of 5 × 10−9 mbar CO in the analysis chamber. In panel (a), the vast majority of the Rh1 species have an apparent height of ≈180 pm (red circle). Over the course of the movie, the apparent height of the individual Rh1 species abruptly decreases to 95–125 pm (panels b–d) and eventually almost all of the Rh1 appear darker (panel (e)). We attribute this change to the formation of monocarbonyl RhCO, and the reduction of the apparent height to the modification of empty Rh states near the Fermi level through the interaction with CO. With higher doses of CO, we also observe double-lobed species oriented perpendicular to the surface Fe rows, which we assign as Rh(CO)2 dicarbonyls (orange arrow in Fig. 5e and Fig. S7†). Noncontact atomic force microscopy (ncAFM) measurements of the Rh carbonyls acquired with a CO-terminated tip (Fig. S7†) appear strikingly similar to those observed in the previously published study of Ir1/Fe3O4(001),14 and one would indeed expect similar behavior for these two metals. As in the Ir case, the concentration of the dicarbonyl species never exceeded 25% of the total, which we attribute to the low pressures used in our experiments. Neither the monocarbonyls nor dicarbonyls appear to interact with molecular O2, as no changes were observed upon subsequent O2 exposure in room-temperature STM measurements. | ||
| Fig. 5 Adsorption of CO on the Rh1/Fe3O4(001) followed by STM. In panel (a), acquired in UHV, the vast majority of the Rh1 adatoms (red circle) have an apparent height of ≈180 pm. (b–d) During exposure in 5 × 10−9 mbar CO, the individual Rh1 change their apparent height one by one to ≈95–125 pm. We identify these as Rh1(CO) species. (e) After a CO dose of ≈10 L, the majority of Rh1 are darker. The images also show a few double-lobed features (orange arrow), which we attribute to Rh1(CO)2 dicarbonyls. Detailed images of these species are shown in Fig. S5.† | ||
In 13CO TPD experiments on 0.5 ML Rh/Fe3O4(001), we observe a 13CO desorption peak with a maximum at ≈530 K, which has an additional shoulder at ≈450 K (Fig. 6a). Comparison to the spectrum acquired on the pristine Fe3O4(001) reveals that all the desorption signal above 250 K is related to Rh, and that the presence of Rh leads to disappearance of a defect-related 13CO desorption peak at ≈190 K. On Rh/Fe3O4(001), a small 13CO2 signal is observed at ≈480 K, which we attribute to CO oxidation via a Mars-van-Krevelen mechanism. This phenomenon has been observed for Ir single atoms and Pt clusters on Fe3O4(001).14,18 No CO desorption is observed above 580 K and in XPS spectra the C 1s region is featureless after heating to these temperatures (Fig. 6c). The CO desorption features below 200 K in the TPD spectra correspond to CO adsorption on the clean Fe3O4(001) surface.32 Focusing on the Rh 3d and C 1s regions of the XPS spectra, CO adsorption at 60 K induced a shift of the Rh 3d peak maximum to 308.5 eV accompanied by the emergence of C 1s peak components at 290.6 eV and 287.3 eV. The 290.6 eV peak corresponds to adsorption of CO on the bare Fe3O4(001) surface.32 Heating to 300 K leads to desorption of CO from Fe3O4(001), so that only CO bound to the Rh species remains (287.3 eV component in the C 1s region). The Rh 3d peak maximum shifts to lower binding energy (308.2 eV), and this position stays constant upon heating to higher temperatures. After the CO desorption from the Rh species above ≈500 K, the majority of the Rh incorporates into the surface, clearly evidenced by the increase in intensity of the component at 309.5 eV. Repeated 13CO TPD measurements feature a small desorption peak at ≈410 K (Fig. S8†), which we attribute to CO desorption from Rh1 substituting an Fe atom in the 5-fold surface site. The Fe 2p region of the XPS spectra taken after CO adsorption and heating to different temperatures shows the same shape as the spectra taken without the CO (see Fig. 2c and Fig. S6†).
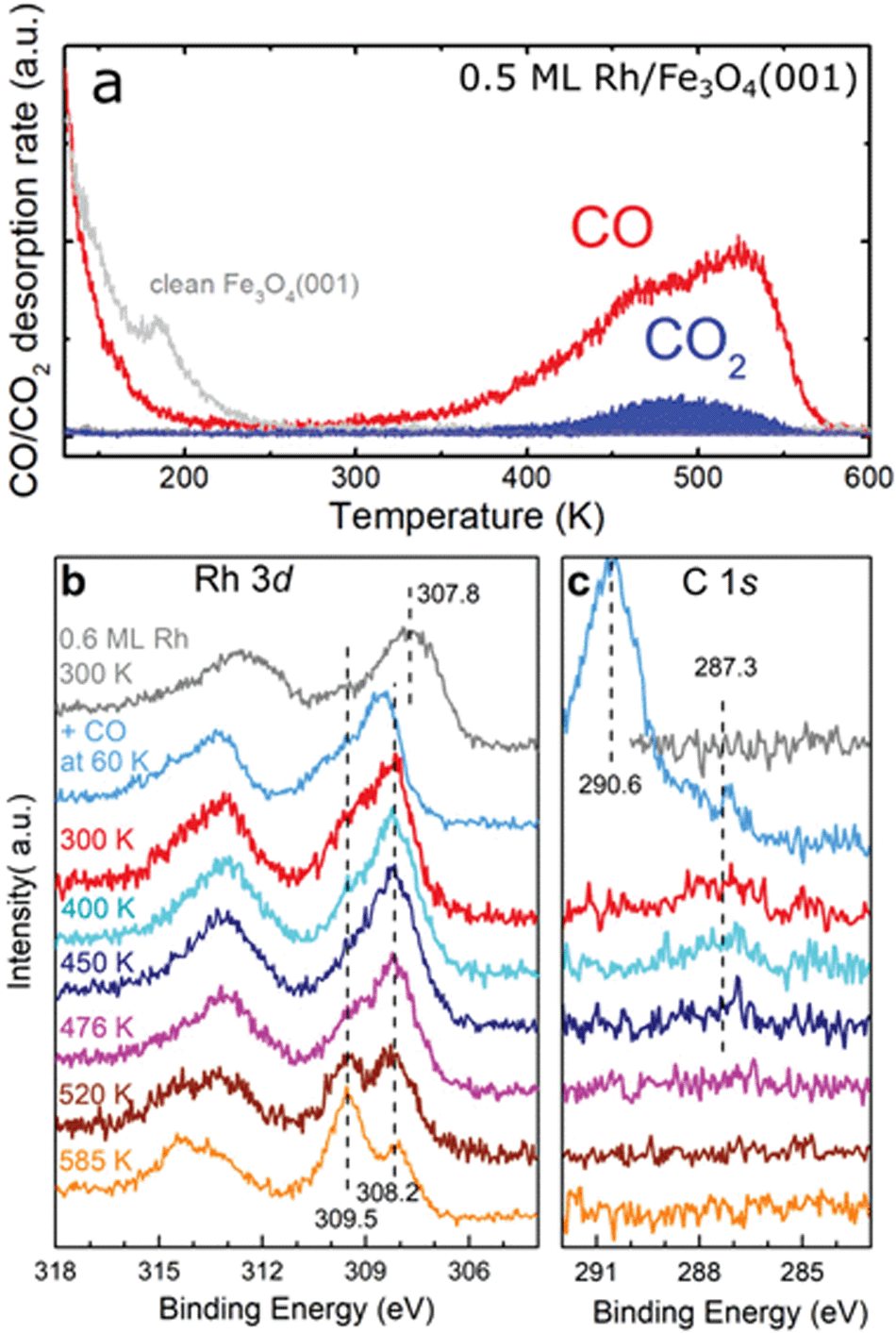 | ||
| Fig. 6 TPD and XPS characterization of CO adsorbed on Rh/Fe3O4(001). (a) CO desorbs from 0.5 ML Rh/Fe3O4(001) in a peak with a maximum around 530 K. A smaller signal of CO2 is observed peaking around 480 K. (b) Adsorption of CO at 60 K on 0.6 ML Rh/Fe3O4(001) shifts the Rh 3d photoemission peak maximum to 308.5 eV, but heating to 300 K shifts the maximum to 308.2 eV. With annealing to temperatures above 500 K, a new component develops at 309.5 eV, due to Rh incorporated into the Fe3O4(001) support. (c) In the C 1s region of the XPS spectra, adsorption of CO at 60 K results in a dominant feature at 290.6 eV, corresponding to CO adsorbed at the Fe3O4(001) surface,32 and a smaller peak at 287.3 eV, corresponding to CO adsorbed on the Rh species. The 287.3 eV component is still observed after heating to 450 K. Above this temperature, no C 1s signal is observed in XPS. | ||
Sequential dosing experiments
Sequential 13CO and 18O2 dosing experiments were performed to find out whether the Rh adatoms or RhxOy clusters can co-adsorb CO and O2. Fig. 7a–d shows the CO (red) and CO2 (blue and orange) signals desorbing from a 0.5 ML Rh/Fe3O4(001) sample exposed to CO and O2 at different temperatures and in a different order. The blue area corresponds to CO2 formed via a MvK mechanism (13C16O2, mass 45), while the orange area indicates CO2 formed by a L–H mechanism (13C16O18O, mass 47). The corresponding 18O2 signal (mass 36) acquired simultaneously is shown in Fig. S9.†Fig. 7a provides a reference in which the sample was saturated by 13CO at 300 K. Dosing O2 on the sample predosed with 13CO at 130 K (panel b) leads to a lower CO desorption signal, and the overall amount of CO2 produced is increased. The CO2 signal produced via a MvK mechanism is comparable to the previous case, but additionally we observe a low, broad 13C16O18O signal between 200–500 K corresponding to CO2 formed by a L–H mechanism. Overall, roughly 35% of the CO2 produced is formed by the L–H channel.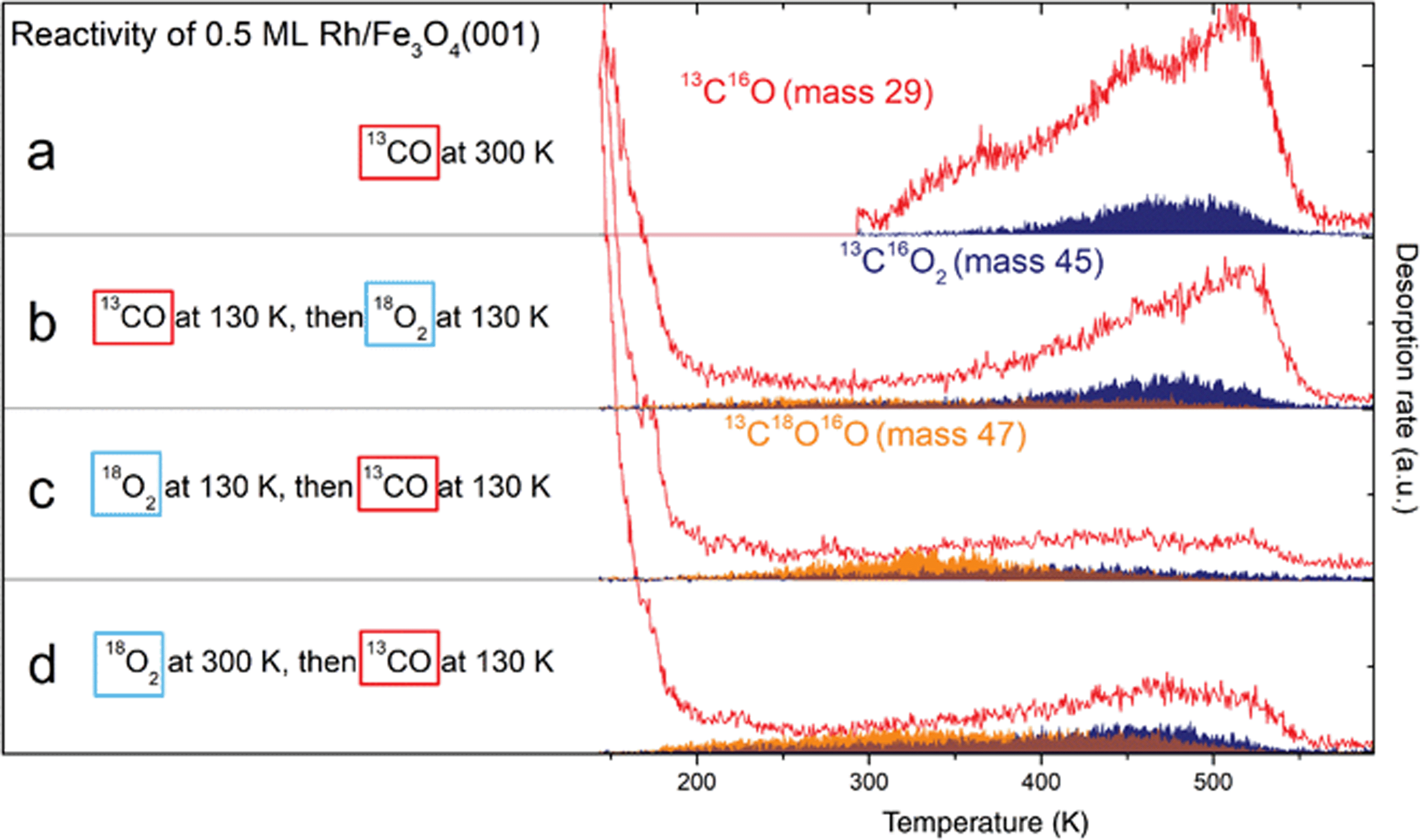 | ||
| Fig. 7 Probing the reactivity of 0.5 ML Rh/Fe3O4(001) by TPD after sequential dosing of 13CO and 18O2. Red traces correspond to 13CO, blue areas to 13C16O2 produced via a MvK mechanism and orange areas correspond to 13C16O18O produced via a L–H mechanism. (a) Dosing 13CO at 300 K results in spectra comparable to Fig. 6a. (b) Dosing 13CO at 130 K, then 18O2 at 130 K leads to a lower 13CO desorption signal, but overall higher 13CO2 production below 400 K. (c) Dosing 18O2 first at 130 K, then 13CO at 130 K leads to significant CO2 signal produced by the L–H mechanism, the MvK channel is suppressed. (d) Dosing 18O2 at 300 K, then 13CO at 130 K leads to similar results as (c), but with more CO2 produced above 400 K in both reaction channels. | ||
The situation changes dramatically when the same gases are dosed with the order reversed (panel c). Dosing the 18O2 before the 13CO at 130 K leads to a significantly enhanced mass 47 signal, which now exhibits a clear maximum at ≈340 K. The sum of masses 29, 45 and 47 is however significantly reduced compared to the previous CO-first experiments, which suggests that, overall, less CO can adsorb on the system once it is saturated with O2 at 130 K, but a significantly higher proportion of adsorbed CO is converted to CO2. The overall amount of CO2 produced is comparable to the case shown in Fig. 7b, but now the majority (≈60%) forms via the L–H channel. If the system is predosed with O2 at 300 K prior to CO adsorption, the observed CO2 TPD signal shown in Fig. 7d is significantly enhanced between 400 and 500 K in both reaction channels. In this experiment the amount of formed CO2 is the highest (≈35% higher than in the previous two cases), and approximately 50% is formed by the L–H channel. These experiments, with O2 predosing at 130 and 300 K, likely differ in that the oxygen-induced Rh1 diffusion should be suppressed at low temperature, and additional oxygen is bound at sites linked to Rh species (see Fig. 4a). No CO2 formation by any mechanism is observed in the absence of Rh species (see Fig. S10†).
Discussion
The experimental data acquired on the Rh/Fe3O4(001) model catalyst clearly show that Rh1 adatoms sinter rapidly when exposed to O2 at room temperature, forming small RhxOy clusters that are active for CO oxidation. Given the extremely low O2 pressure used in the STM movies, it seems likely that just one O2 molecule is sufficient to create a mobile oxidised Rh species, probably Rh1O2. This suggests that oxygen adsorption significantly weakens the interaction of the Rh adatom with the support. DFT calculations for oxygen adsorption on the Rh1/Al2O3 system predict significant electron transfer from Rh to O2, and the formation of superoxo (O2)− species.3 In the O 1s XPS data taken after O2 adsorption below 100 K (Fig. 4b) we do observe a peak at 533.7 eV, which is close to the position expected for superoxo species.33 However, this peak disappears after heating above 250 K, suggesting a further reaction with Rh takes place. We also observe a strong positive binding energy shift in the Rh 3d XPS peak maximum (Fig. 4c) after O2 dosing at 60 K (308.6 eV), which shifts further to ≈309.0 eV upon heating above 250 K. Even though the position observed at 60 K is already close to that expected for RhO2 (308.6 eV)34 or Rh2O3 (308.3 eV),34,35 and thus could be already interpreted as a signal corresponding to the oxidised Rh clusters, the further shift above 250 K accompanied with the disappearance of the superoxo component in the O 1s region suggests that the agglomeration takes place between 100 K and 250 K, and the O 1s signal of the resulting RhxOy nanoparticles is indistinguishable from that of the O2− in the Fe3O4 support.Our room-temperature STM movies of O2 exposure (Fig. 3a–c) suggest that the mobile Rh1O2 species interact strongly with bare Rh adatoms to form a well-defined double protrusion (presumably with Rh2O2 stoichiometry). The addition of yet another mobile Rh1O2 seems common, and would in principle yield clusters of nominal Rh3O4 stoichiometry. These clusters appear larger, and have a scratchy appearance due to the interaction with the STM tip. The cluster density observed following 50 L O2 exposure suggests that the clusters rarely grow larger than 2–3 Rh atoms, but it cannot be concluded how many O2 molecules can ultimately be adsorbed on each cluster, nor whether the formation of these clusters results in the abstraction of one or more O atoms. Nevertheless, the O2 TPD measurements clearly show that some of the O atoms are weakly bound, and desorb as molecular O2 just above room temperature in the absence of CO (Fig. 4a). The Rh 3d spectrum is barely changed by this desorption; only a very small shoulder appears at ≈308.2 eV (green curve in Fig. 4c), which suggests the clusters remain oxidised. This shoulder gets significantly more pronounced after heating to 460 and 575 K (purple and cyan curves in Fig. 4c), and at the same time the component corresponding to incorporated Rh appears (309.5 eV). In the TPD spectra there is no O2 desorption signal observed above 380 K (up to 660 K, where the acquisition was stopped), therefore it seems likely that the oxygen from RhxOy clusters gets accommodated in the Fe3O4 lattice as the Rh incorporates, and the 308.2 eV component in this case indicates an intermediate step between RhxOy on the surface and Rh incorporated in the lattice. Repeating the oxygen exposure (after its desorption at ≈350 K, but before the Rh incorporation at higher temperatures) does not replenish the weakly bound O2, suggesting that the clusters restructure to a stable configuration, and that the weakly bound oxygen was a consequence of the agglomeration process.
When the O2-sintered sample is exposed to CO at low temperature and TPD is performed, CO2 is found to evolve from the sample in a broad signal between 200–500 K (Fig. 7d). Isotopic labelling of the reactants shows that the majority of the CO2 formed below ≈400 K is produced by the L–H channel, with the molecular O2 being the oxidising agent. The observation of a similar result when the initial O2 exposure was performed at 130 K instead of 300 K hints that O2-induced sintering happens already at low temperatures. Interestingly, the CO2 signal at higher temperature is reduced in intensity when the O2 exposure is performed at 130 K. The comparison of these two datasets suggests that the oxygen bound above 300 K (δ peak in Fig. 4a) is facilitating the CO2 formation by the L–H channel, and the oxygen adsorbed below 300 K likely blocks a small number of sites for CO adsorption. An alternative explanation is that at 300 K the 18O2 spills over to the Fe3O4 surface, which leads to the increased CO2 production above 400 K. In this case the process would be MvK, but due to the previous spillover from Rh it would be measured in both 13C16O2 and 13C16O18O signals. Reexposing the surface to O2 following CO2 desorption is not expected to facilitate further CO2 production via a L–H mechanism, because the remaining RhxOy clusters seem to be inert for further interaction with O2.
When the Rh1/Fe3O4(001) sample is exposed to CO first, the Rh adatoms do not become mobile at room temperature. We have recently shown that CO adsorption allows Ir1 adatoms to achieve highly-stable square-planar environments,14 and our STM/ncAFM images for Rh1 (Fig. S7†) appear very similar. In ref. 14, the enhanced stability was rationalized by analogy to Ir(I) complexes, and it seems reasonable to expect similar behavior for Rh(I), which can also adopt the preferred square planar d8 configuration. The XPS spectra acquired on the CO-saturated Rh/Fe3O4(001) system show almost no change in the Rh 3d region when heated to temperatures between 300 K and 450 K, suggesting most of the Rh carbonyls still exist at 450 K, and are still present before the CO2 production takes place via a MvK mechanism at ≈480 K. Exposing the CO-saturated sample to O2 does not lead to visible changes in STM or XPS, nor does it lead to significant low-temperature CO2 production. We conclude that CO poisons the Rh adatoms for O2 adsorption, at least under our low-pressure conditions. We thus do not form OCOO species, which have been proposed as possible intermediates in prior studies of CO oxidation by Rh-based single-atom catalysts.36,37 This could be because the Rh atom is inaccessible to the O2 molecule once the CO is present, or because adsorbing both molecules on the same Rh adatom is energetically unfavourable. The situation could be different at higher pressures where the kinetic limitations are more easily overcome. A more likely scenario, however, is that stable Rh(CO)2 dicarbonyls would be formed and the catalyst would remain poisoned for CO oxidation via a L–H mechanism. Our TPD experiments show that a small amount of CO is oxidised to CO2 at high temperature via a MvK mechanism, but this is followed by immediate incorporation of Rh into the support, which ultimately deactivates the catalyst.
Conclusions
This study presents an atomic-scale view on a range of phenomena relevant for heterogeneous catalysis by single atoms and sub-nano clusters. We show that O2 adsorption initially leads to formation of superoxo species bound to Rh1 adatoms, which already at room temperature rapidly agglomerate to small RhxOy clusters. CO adsorption, on the other hand, completes the preferred square-planar coordination environment of the Rh1 adatoms, stabilizing the Rh1 in place while poisoning them for O2 adsorption. Both the oxidised RhxOy clusters and Rh1CO carbonyls show activity for CO oxidation, but via very different pathways: oxidised clusters catalyse the reaction via a Langmuir–Hinshelwood mechanism at temperatures as low as 200 K, while the presence of Rh carbonyls at 300 K allows CO2 formation via a Mars-van-Krevelen mechanism at ≈480 K. These results highlight the importance of a careful characterization of the catalyst during the exposure to the reactants and show that the gas composition can play a crucial role in the structural evolution of the catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GSP, JH and MM acknowledge funding from the Austrian Science Foundation (FWF) Start Prize Y847-N20. ZJ and RB acknowledge support from the TU Wien Doctoral Colleges TU-D and Solids4fun, respectively.References
- H. Guan, J. Lin, B. Qiao, X. Yang, L. Li and S. Miao, et al., Catalytically Active Rh Sub-Nanoclusters on TiO2 for CO Oxidation at Cryogenic Temperatures, Angew. Chem., Int. Ed., 2016, 55(8), 2820–2824 CrossRef CAS PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen and G. W. Graham, et al., Adsorbate-mediated strong metal–support interactions in oxide-supported Rh catalysts, Nat. Chem., 2016, 9, 120 CrossRef PubMed.
- T. K. Ghosh and N. N. Nair, Rh1/γ-Al2O3 Single-Atom Catalysis of O2 Activation and CO Oxidation: Mechanism, Effects of Hydration, Oxidation State, and Cluster Size, ChemCatChem, 2013, 5(7), 1811–1821 CrossRef CAS.
- J. Gustafson, O. Balmes, C. Zhang, M. Shipilin, A. Schaefer and B. Hagman, et al., The Role of Oxides in Catalytic CO Oxidation over Rhodium and Palladium, ACS Catal., 2018, 8(5), 4438–4445 CrossRef CAS.
- G. S. Parkinson, Iron oxide surfaces, Surf. Sci. Rep., 2016, 71(1), 272–365 CrossRef CAS.
- H.-J. Freund, G. Meijer, M. Scheffler, R. Schlögl and M. Wolf, CO Oxidation as a Prototypical Reaction for Heterogeneous Processes, Angew. Chem., Int. Ed., 2011, 50(43), 10064–10094 CrossRef CAS PubMed.
- A. Beniya and S. Higashi, Towards dense single-atom catalysts for future automotive applications, Nat. Catal., 2019, 2(7), 590–602 CrossRef.
- S. Sinthika, S. T. Vala, Y. Kawazoe and R. Thapa, CO Oxidation Prefers the Eley–Rideal or Langmuir–Hinshelwood Pathway: Monolayer vs Thin Film of SiC, ACS Appl. Mater. Interfaces, 2016, 8(8), 5290–5299 CrossRef CAS PubMed.
- M. A. Newton, D. Ferri, G. Smolentsev, V. Marchionni and M. Nachtegaal, Room-temperature carbon monoxide oxidation by oxygen over Pt/Al2O3 mediated by reactive platinum carbonates, Nat. Commun., 2015, 6, 8675 CrossRef CAS PubMed.
- Q. Pan, X. Weng, M. Chen, L. Giordano, G. Pacchioni and C. Noguera, et al., Enhanced CO Oxidation on the Oxide/Metal Interface: From Ultra-High Vacuum to Near-Atmospheric Pressures, ChemCatChem, 2015, 7(17), 2620–2627 CrossRef CAS.
- Y. Lu, J. Wang, L. Yu, L. Kovarik, X. Zhang and A. S. Hoffman, et al., Identification of the active complex for CO oxidation over single-atom Ir-on-MgAl2O4 catalysts, Nat. Catal., 2019, 2(2), 149–156 CrossRef CAS.
- A. Halder, L. A. Curtiss, A. Fortunelli and S. Vajda, Perspective: Size selected clusters for catalysis and electrochemistry, J. Chem. Phys., 2018, 148(11), 110901 CrossRef PubMed.
- P. T. P. Ryan, Z. Jakub, J. Balajka, J. Hulva, M. Meier and J. T. Küchle, et al., Direct measurement of Ni incorporation into Fe3O4(001), Phys. Chem. Chem. Phys., 2018, 20(24), 16469–16476 RSC.
- Z. Jakub, J. Hulva, M. Meier, R. Bliem, F. Kraushofer and M. Setvin, et al., Local Structure and Coordination Define Adsorption in a Model Ir1/Fe3O4 Single-Atom Catalyst, Angew. Chem., Int. Ed., 2019, 58(39), 13961–13968 CrossRef CAS PubMed.
- R. Bliem, J. Pavelec, O. Gamba, E. McDermott, Z. M. Wang and S. Gerhold, et al., Adsorption and incorporation of transition metals at the magnetite Fe3O4(001) surface, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92(7), 075440 CrossRef.
- L. DeRita, J. Resasco, S. Dai, A. Boubnov, H. V. Thang and A. S. Hoffman, et al., Structural evolution of atomically dispersed Pt catalysts dictates reactivity, Nat. Mater., 2019, 18(7), 746–751 CrossRef CAS PubMed.
- K. Zhao, H. Tang, B. Qiao, L. Li and J. Wang, High Activity of Au/γ-Fe2O3 for CO Oxidation: Effect of Support Crystal Phase in Catalyst Design, ACS Catal., 2015, 5(6), 3528–3539 CrossRef CAS.
- R. Bliem, J. van der Hoeven, A. Zavodny, O. Gamba, J. Pavelec and P. E. de Jongh, et al., An Atomic-Scale View of CO and H2 Oxidation on a Pt/Fe3O4 Model Catalyst, Angew. Chem., Int. Ed., 2015, 54(47), 13999–14002 CrossRef CAS PubMed.
- D. Widmann and R. J. Behm, Active Oxygen on a Au/TiO2 Catalyst: Formation, Stability, and CO Oxidation Activity, Angew. Chem., Int. Ed., 2011, 50(43), 10241–10245 CrossRef CAS PubMed.
- K. Zhang, S. Shaikhutdinov and H.-J. Freund, Does the Surface Structure of Oxide Affect the Strong Metal–Support Interaction with Platinum? Platinum on Fe3O4(001) versus Fe3O4(111), ChemCatChem, 2015, 7(22), 3725–3730 CrossRef CAS.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec and R. Kosak, et al., Carbon monoxide-induced adatom sintering in a Pd–Fe3O4 model catalyst, Nat. Mater., 2013, 12(8), 724–728 CrossRef CAS.
- J. I. J. Choi, W. Mayr-Schmölzer, F. Mittendorfer, J. Redinger, U. Diebold and M. Schmid, The growth of ultra-thin zirconia films on Pd3Zr(0001), J. Phys.: Condens. Matter, 2014, 26(22), 225003 CrossRef CAS PubMed.
- J. Pavelec, J. Hulva, D. Halwidl, R. Bliem, O. Gamba and Z. Jakub, et al., A multi-technique study of CO2 adsorption on Fe3O4 magnetite, J. Chem. Phys., 2017, 146(1), 014701 CrossRef PubMed.
- R. Bliem, E. McDermott, P. Ferstl, M. Setvin, O. Gamba and J. Pavelec, et al., Subsurface cation vacancy stabilization of the magnetite (001) surface, Science, 2014, 346(6214), 1215–1218 CrossRef CAS PubMed.
- B. Arndt, R. Bliem, O. Gamba, J. E. S. van der Hoeven, H. Noei and U. Diebold, et al., Atomic structure and stability of magnetite Fe3O4(001): An X-ray view, Surf. Sci., 2016, 653, 76–81 CrossRef CAS.
- Z. Novotný, G. Argentero, Z. Wang, M. Schmid, U. Diebold and G. S. Parkinson, Ordered Array of Single Adatoms with Remarkable Thermal Stability: Au/Fe3O4(001), Phys. Rev. Lett., 2012, 108(21), 216103 CrossRef PubMed.
- R. Bliem, R. Kosak, L. Perneczky, Z. Novotny, O. Gamba and D. Fobes, et al., Cluster Nucleation and Growth from a Highly Supersaturated Adatom Phase: Silver on Magnetite, ACS Nano, 2014, 8(7), 7531–7537 CrossRef CAS.
- M. Meier, Z. Jakub, J. Balajka, J. Hulva, R. Bliem and P. K. Thakur, et al., Probing the geometry of copper and silver adatoms on magnetite: quantitative experiment versus theory, Nanoscale, 2018, 10(5), 2226–2230 RSC.
- R. Gargallo-Caballero, L. Martin-Garcia, A. Quesada, C. Granados-Miralles, M. Foerster and L. Aballe, et al., Co on Fe3O4(001): Towards precise control of surface properties, J. Chem. Phys., 2016, 144(9), 094704 CrossRef PubMed.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Investigation of multiplet splitting of Fe 2p XPS spectra and bonding in iron compounds, Surf. Interface Anal., 2004, 36(12), 1564–1574 CrossRef CAS.
- M. Meier, J. Hulva, Z. Jakub, J. Pavelec, M. Setvin and R. Bliem, et al., Water agglomerates on Fe3O4(001), Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5642–E5E50 CrossRef CAS PubMed.
- J. Hulva, Z. Jakub, Z. Novotny, N. Johansson, J. Knudsen and J. Schnadt, et al., Adsorption of CO on the Fe3O4(001) surface, J. Phys. Chem. B, 2018, 122(2), 721–729 CrossRef PubMed.
- D. Halwidl, W. Mayr-Schmölzer, M. Setvin, D. Fobes, J. Peng and Z. Mao, et al., A full monolayer of superoxide: oxygen activation on the unmodified Ca3Ru2O7(001) surface, J. Mater. Chem. A, 2018, 6(14), 5703–5713 RSC.
- Y. Abe, K. Kato, M. Kawamura and K. Sasaki, Rhodium and Rhodium Oxide Thin Films Characterized by XPS, Surf. Sci. Spectra, 2001, 8(2), 117–125 CrossRef CAS.
- S. Blomberg, E. Lundgren, R. Westerström, E. Erdogan, N. M. Martin and A. Mikkelsen, et al., Structure of the Rh2O3(0001) surface, Surf. Sci., 2012, 606(17), 1416–1421 CrossRef CAS.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, Exploration of High-Performance Single-Atom Catalysts on Support M1/FeOx for CO Oxidation via Computational Study, ACS Catal., 2015, 5(2), 544–552 CrossRef CAS.
- E. A. Wovchko and J. T. Yates, Activation of O2 on a Photochemically Generated RhI Site on an Al2O3 Surface: Low-Temperature O2 Dissociation and CO Oxidation, J. Am. Chem. Soc., 1998, 120(40), 10523–10527 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr10087c |
| This journal is © The Royal Society of Chemistry 2020 |