
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-peptide functionalized acetalated dextran-based nanoparticles for sequential targeting of macrophages during myocardial infarction†
Giulia
Torrieri
 a,
Flavia
Fontana
a,
Patrícia
Figueiredo
a,
Zehua
Liu
a,
Mónica P. A.
Ferreira
a,
Virpi
Talman
bc,
João P.
Martins
a,
Manlio
Fusciello
d,
Karina
Moslova
e,
Tambet
Teesalu
fg,
Vincenzo
Cerullo
dh,
Jouni
Hirvonen
a,
Heikki
Ruskoaho
b,
Vimalkumar
Balasubramanian
*a and
Hélder A.
Santos
*ah
a,
Flavia
Fontana
a,
Patrícia
Figueiredo
a,
Zehua
Liu
a,
Mónica P. A.
Ferreira
a,
Virpi
Talman
bc,
João P.
Martins
a,
Manlio
Fusciello
d,
Karina
Moslova
e,
Tambet
Teesalu
fg,
Vincenzo
Cerullo
dh,
Jouni
Hirvonen
a,
Heikki
Ruskoaho
b,
Vimalkumar
Balasubramanian
*a and
Hélder A.
Santos
*ah
aDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, FI-00014 Helsinki, Finland. E-mail: vimalkumar.balasubramanian@helsinki.fi; helder.santos@helsinki.fi
bDrug Research Program, Division of Pharmacology and Pharmacotherapy, Faculty of Pharmacy, University of Helsinki, FI-00140, Helsinki, Finland
cNational Heart and Lung Institute, Imperial College London, London W12 0NN, UK
dDrug Research Program, Division of Pharmaceutical Biosciences, Faculty of Pharmacy, University of Helsinki, FI-00140, Helsinki, Finland
eDepartment of Chemistry, University of Helsinki, FI-00014, Helsinki, Finland
fLaboratory of Cancer Biology, Institute of Biomedicine and Translational Medicine, Centre of Excellence for Translational Medicine, University of Tartu, Tartu, 50411, Estonia
gCancer Research Center, Sanford-Burnham Medical Research Institute, La Jolla, California 92037, USA
hHelsinki Institute of Life Science, HiLIFE, University of Helsinki, FI-00014 Helsinki, Finland
First published on 30th December 2019
Abstract
The advent of nanomedicine has recently started to innovate the treatment of cardiovascular diseases, in particular myocardial infarction. Although current approaches are very promising, there is still an urgent need for advanced targeting strategies. In this work, the exploitation of macrophage recruitment is proposed as a novel and synergistic approach to improve the addressability of the infarcted myocardium achieved by current peptide-based heart targeting strategies. For this purpose, an acetalated dextran-based nanosystem is designed and successfully functionalized with two different peptides, atrial natriuretic peptide (ANP) and linTT1, which target, respectively, cardiac cells and macrophages associated with atherosclerotic plaques. The biocompatibility of the nanocarrier is screened on both macrophage cell lines and primary macrophages, showing high safety, in particular after functionalization of the nanoparticles’ surface. Furthermore, the system shows higher association versus uptake ratio towards M2-like macrophages (approximately 2-fold and 6-fold increase in murine and human primary M2-like macrophages, respectively, compared to M1-like). Overall, the results demonstrate that the nanosystem has potential to exploit the “hitchhike” effect on M2-like macrophages and potentially improve, in a dual targeting strategy, the ability of the ANP peptide to target infarcted heart.
1. Introduction
Cardiovascular diseases (CVDs), and in particular, myocardial infarction (MI), are a global burden, causing 17.8 million deaths in 2017.1 The high mortality associated with CVDs is due to the inability of the human adult heart to replace the loss of cardiomyocytes (up to one billion may be lost in a MI)2 and restore the cardiac function.3 Fibrosis is taking place instead, leading to a chronic lethal syndrome, called heart failure (HF).4 Unfortunately, current therapies are still unsuccessful, because they are mainly aimed at managing symptoms of the disease, but unable to change the fate of cardiomyocytes.The advent of nanomedicine has brought new insights in innovative treatment strategies, currently at the pre-clinical stage, of CVD, in particular MI.5–10 Such approaches are very promising, but there is urgent need for smart targeting strategies. The infarcted tissue is challenging to address due to mechanical obstacles, like the constant pumping of the organ and the restless massive exchange of blood.11 Recently, Ferreira et al.,12 investigated the in vivo targeting abilities of different heart targeting peptides, demonstrating that atrial natriuretic peptide (ANP) is a peptide that allows nanoparticles to accumulate in the infarcted heart. However, ANP lacks the targeting exclusiveness towards cardiac tissue, because its receptors are expressed not only in the heart, but also in the kidneys, lungs, brain, testes, adipose tissue, adrenal gland tissues and vascular smooth muscle cells.13 Herein, we introduce an alternative strategy to improve the heart targeting properties of ANP. Considering the conspicuous involvement of inflammation during infarction, we explore the macrophage recruitment in order to “hitchhike” on macrophages and increase the ANP-functionalized nanoparticle accumulation in the infarcted heart. It is known that promptly after MI, neutrophils are recruited to the infarcted tissue, creating an oxidative stress milieu and producing a variety of inflammatory cytokines, which will further recruit on-site different populations of monocytes and macrophages.14,15 This monocyte/macrophage enrolment can be divided into two sequential phases. A first phase, which peaks at day 3 after the onset of MI, sees an accumulation of inflammatory type Ly-6Chigh monocytes/M1-type macrophages, which phagocytose dead cells and debris to make space for the evolving scar. Six days after the infarction onset, a second phase, characterized by the presence of healing Ly-6Cint/low monocytes/M2-type macrophages, supervenes and promotes the repair of the tissue with the release of the vascular endothelial growth factor (VEGF) and transforming growth factor beta (TGFβ), which support angiogenesis and collagen production, leading to the formation of un-functional scar tissue.14,15
Cellular hitchhiking is a very well-known approach used in the nanomedicine field.16 In the past, researchers exploited the long circulating properties of red blood cells,17,18 macrophages/monocytes19–22 and other cells23,24 to increase the circulation time17 and targeting abilities of specific nanocarriers,18 and avoid the rapid clearance of diagnostic and imaging agents.22 With this aim, we designed a putrescine-modified acetalated dextran (Putre-AcDEX)-based nanosystem, functionalized with ANP and TT1 peptides.
Acetalated dextran (AcDEX) was chosen because it is easy to modify and is highly biocompatible both as a starting material and in terms of degradation products,25 and due to its pH-responsiveness.26,27 The polymer was modified with putrescine to provide functional groups for further surface conjugations and to reduce the toxicity towards primary cardiomyocytes observed with spermine-modified nanoparticles.5 Spermine particles are highly positively charged even after the conjugations, due to free amine groups present in the structure. The system was loaded with two small hydrophobic compounds, CHIR99021 and SB203580, which have shown a synergistic effect in stimulating cardiomyocyte proliferation.28
To endow nanoparticles with heart targeting ability, the surface of the nanocarrier was modified with ANP and TT1 peptides, attached to the nanoparticles’ surface through a branched polyethylene glycol (PEG). Linear TT1 (Lin-TT1) (AKRGARSTA) is a well-established peptide for its tumor-homing abilities.29–31 The peptide addresses tumors by binding to the mitochondrial chaperone protein p32, normally expressed at the intracellular level, but translocated on the surface of tumor cells, tumor associated macrophages, tumor endothelial cells, as well as on macrophages associated with atherosclerotic plaques.30,32,33 The TT1 peptide, similar to integrin-binding arginine–glycine–aspartic acid (iRGD) and LyP-1 peptides, contains a cryptic basic sequence motif (C-end Rule or CendR motif), that is, following cell surface recruitment of the peptide by the p32-dependent mechanism, proteolytically processed to activate the CendR element and allow interaction of the peptide with the secondary receptor neutropilin-1 (NRP-1).34 This interaction initiates an uptake process similar to classical endocytosis.34
The aim of this work is to evaluate the successful conjugation of both peptide moieties on the nanoparticles’ surface, assessing the biocompatibility of the nanosystem and studying its interactions with both continuous macrophage cell lines and primary M1- and M2-like macrophages. For this purpose, we have tested the viability and uptake of both macrophage cell lines and primary macrophages treated with different concentrations of the nanosystem.
2. Results and discussion
2.1. Characterization of Putre-AcDEX nanoparticles
Putre-AcDEX nanoparticles were prepared by an oil-in-water (o/w) single emulsion method.35 The two hydrophobic compounds, CHIR99021 and SB203580 (abbreviated as C and S, respectively) were dissolved together with the polymer in the organic phase. Then, an aqueous solution of polyvinyl alcohol (PVA), which functions as a stabilizer, was added and the emulsion was created with the help of high energy sonication. Upon evaporation of the organic solvent for 3 h, the nanoparticles formed entrapped the compounds in a matrix-like structure. The surface of the nanoparticles was then functionalized with PEG and the two peptides, linTT1 and ANP, in order to achieve dual targeting. After preparation, the nanoparticles were characterized by dynamic light scattering (DLS) and electrophoretic light scattering (ELS) to obtain information about their size, polydispersity index (PDI), and zeta (ζ)-potential (Fig. 1A).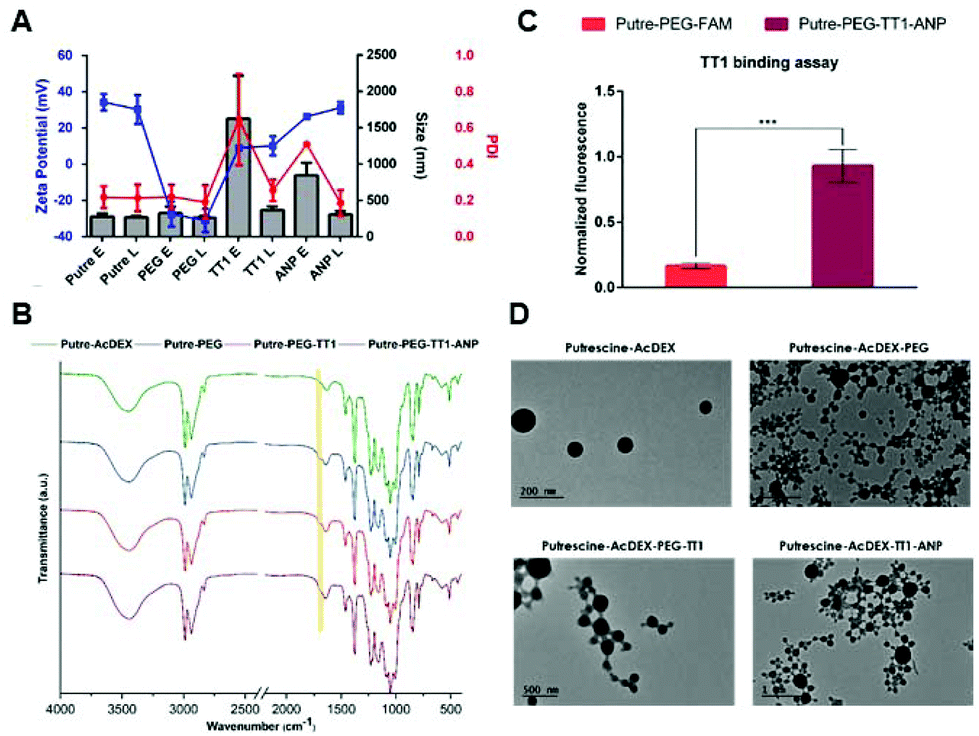 | ||
| Fig. 1 Physicochemical characterization of Putre-AcDEX nanoparticles. (A) Average size, PDI, and ζ-potential before and after the different conjugation steps. Letters E and L are used to differentiate between empty and loaded particles, respectively. The bare system is named Putre; PEGylated nanoparticles are abbreviated as PEG; particles conjugated with only TT1 peptide are named TT1, and the system after conjugation with all the peptides is called ANP. (B) KBr-FTIR spectra of the bare and functionalized nanoparticles. (C) TT1 binding assay. Fluorescent labelled nanoparticles conjugated with both ANP and TT1 peptides (Putre-PEG-TT1-ANP) were compared with PEGylated particles made fluorescent after conjugation with 6-carboxyfluorescein (Putre-PEG-FAM). (D) TEM images of the bare nanoparticles and after surface functionalization. Values are represented as the mean ± standard deviation (s.d.) (n ≥ 3 biological replicates in which each time 3 technical replicates have been used). The data were analysed by two-way ANOVA followed by a Tukey–Kramer post hoc test, using GraphPad Prism 7 software. Statistical significance was set at probabilities of *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
The nanoparticles were characterized by an increase of size after different conjugation steps. In particular, empty nanosystems showed a significant increase in hydrodynamic diameter after functionalization with the two peptides. Loaded particles did not show such a size increase, suggesting a stabilizing effect of the loaded compounds, C and S.
The PDI varied accordingly, showing higher values for empty particles after conjugation with linTT1 and ANP peptides, which are characteristic of heterogeneous nanoparticle suspensions,36 supporting the stabilizing theory of the loaded compounds. The charge of the nanoparticles was also measured, suggesting that the different components were effectively conjugated on the surface of the nanoparticles. The bare nanosystem had a high positive charge (ca. +40 mV), due to the presence of amine groups from putrescine. After PEGylation, the ζ-potential dropped to negative values (−30 mV), due to the presence of carboxyl-terminated groups in the PEG. The conjugation of the linTT1 peptide, reversed the charge to slightly positive values (ca. +10 mV), and this can be explained due to the presence of arginine residues in the peptide sequence. The charge had a further small increase of ca. 10 mV (reaching values of +20 mV) after conjugation of the ANP peptide, which is rich in positively charged arginine residues, and thus, responsible for the increase of the positive charge. The presence of PEG was furthermore confirmed by KBr-Fourier transform infrared (FTIR) spectroscopy (Fig. 1B).
The presence of the amide-indicative bands at 1565–1570 cm−1 (in-plane N–H bending and C–N stretching) and 1630–1640 cm−1 (amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) confirmed the formation of a covalent amide bond between the Putre-AcDEX and PEG, and the appearance of a shoulder at 1735 cm−1 (CO stretching from –COOH belonging to PEG and maleimide groups), underlined by the yellow band in Fig. 1B, denotes the presence of free carboxyl groups.
O stretching) confirmed the formation of a covalent amide bond between the Putre-AcDEX and PEG, and the appearance of a shoulder at 1735 cm−1 (CO stretching from –COOH belonging to PEG and maleimide groups), underlined by the yellow band in Fig. 1B, denotes the presence of free carboxyl groups.
The conjugation of linTT1 and ANP peptides did not change the FTIR spectrum, thus further methods were used to confirm the successful conjugation of the peptides onto the nanoparticle's surface. The presence of linTT1 was detectable due to its conjugation with carboxyfluorescein (FAM), which imparted fluorescence to the nanoparticles after conjugation with the peptide. Moreover, we performed a binding assay to ensure that the peptide was conjugated with the correct orientation, and thus, kept its functionality. The assay consisted of coating Ni-NTA magnetic agarose beads with the recombinant hexahistidine-tagged target of the linTT1 peptide, p32.37 Then, beads were incubated with FAM-labeled nanoparticles conjugated with the linTT1peptide or without it. Beads were finally washed and the nanoparticle-p32 complexes were released using imidazole elution buffer and the eluted fluorescence was measured. As shown in Fig. 1C, the eluted fractions collected from beads incubated with Putre-PEG-TT1-ANP nanoparticles, presented a significantly higher fluorescence compared to those collected from beads that interacted with particles without the peptide. This demonstrates that the functionally active linTT1 peptide was conjugated onto the nanoparticles’ surface. The effective conjugation of ANP was further confirmed by elemental analysis, which showed the presence of about 14.14 μg of ANP in 1 mg of Putre-PEG-TT1-ANP nanoparticles (Table S2†).
The nanoparticles’ morphology was obtained through transmission electron microscopy (TEM) (Fig. 1D), and the images showed round-shaped nanoparticles, with a diameter comprised between 100 and 200 nm, a bit smaller than what was observed with DLS. This phenomenon is in accordance with the fact that DLS measures the hydrodynamic diameter of the particles, whereas TEM refers to dry particles.38
2.2. Release studies and pH-dependent behavior
The release profile of the two encapsulated drug compounds, C and S, was evaluated in phosphate buffer saline (PBS) (pH 7.4) and acetate (pH 5.0) buffers, in order to mimic the physiological extracellular environment and the conditions found in acidic intracellular compartments after internalization in the cells, respectively.39 In addition, the determination of the drug loading degree (LD) and encapsulation efficiency (EE) values of bare and functionalized particles was also conducted as shown in Table S3.†The encapsulation of the drug molecules was done at a molar ratio C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :S of 1:2, which is the optimal ratio, according to the literature, in order to induce cardiomyocyte proliferation.40,41 Upon functionalization, there is a loss of encapsulated drugs, because some of the compounds might still be on the surface of the nanoparticles, even though they were washed thoroughly. Another reason could be that the system is saturated due to the high amount of compounds loaded at the beginning of the preparation process.
:S of 1:2, which is the optimal ratio, according to the literature, in order to induce cardiomyocyte proliferation.40,41 Upon functionalization, there is a loss of encapsulated drugs, because some of the compounds might still be on the surface of the nanoparticles, even though they were washed thoroughly. Another reason could be that the system is saturated due to the high amount of compounds loaded at the beginning of the preparation process.
However, the amount of encapsulated drugs was still enough to produce an effect on cells.42,43
As previously demonstrated,44–46 AcDEX-based nanoparticles should release drugs faster at acidic pH, because of the pH-dependent hydrolysis of acetal groups present in AcDEX. The results obtained are in accordance with what was previously reported, as shown in Fig. 2.
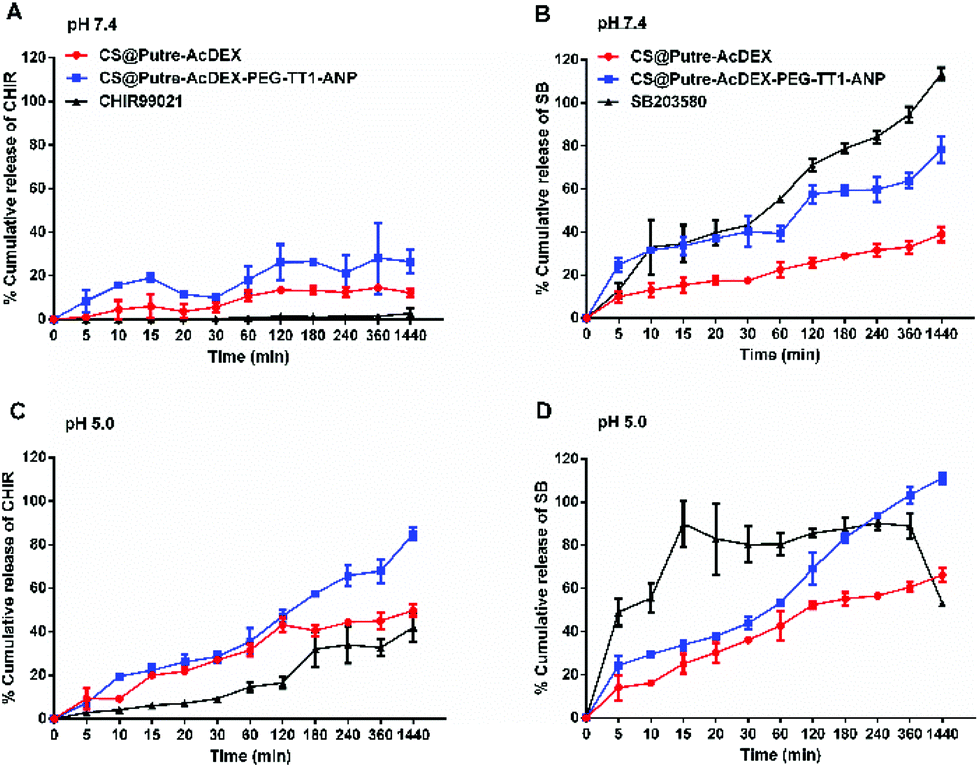 | ||
| Fig. 2 Release profiles of drug-loaded Putre-AcDEX nanoparticles. Release profiles of CHIR99021 (A and C) and SB203580 (B and D) from bare CS@Putre-AcDEX and functionalized CS@Putre-AcDEX-PEG-TT1-ANP at pH 7.4 and 5.0, at 37 °C. Data represented as mean ± s.d. (n ≥ 3 biological replicates in which each time 3 technical replicates have been used). | ||
At pH 5.0 both the drugs were released faster compared to pH 7.4 (Fig. 2C and D). Free drugs were used as controls and especially in the case of C, nanoparticles increased their dissolution rate in aqueous media. Also, the solubility of C was affected by pH, where the free drug was more soluble at pH 5.0 than at pH 7.4. For S at pH 5.0, it can be noted that the nanoparticles prevented drug degradation, because the free drug started degrading after 8 h, whereas the drug-loaded nanoparticles showed a release of the drug concentration up to 24 h. Moreover, the nanosystem conjugated with PEG and the peptides (named CS@Putre-AcDEX-PEG-TT1-ANP in the plot), presented a faster drug release of the drugs compared to the bare nanoparticles (named CS@Putre-AcDEX in the plot). This can be explained by the increased hydrophilicity of the nanosystem after surface modifications, which can promote the interaction with the solvent molecules and allow the release of the drugs entrapped in the polymer matrix close to the surface of the nanoparticles.47–49
Even though the drug release was faster at pH 5.0, there was a small amount of drugs released already at pH 7.4, which is justifiable by a slight degradation of AcDEX at this pH, as previously reported.5,50 This loss of drugs at pH 7.4 can also justify the decrease of the LD and EE values of the nanoparticles after the various conjugation steps. After PEGylation, washings were performed in a solution of 2% of sucrose at pH 7.4, in order to prevent the degradation of the maleimide groups of PEG needed to interact with the thiol groups of the TT1 peptide. Overall, these AcDEX-based nanoparticles were successfully designed to release payloads mostly in the acidic subcellular compartments of the cells, which can be considered optimal for intravenous administration. Furthermore, it is known that inflammation occurring in the ischemic myocardium lowers the interstitial pH to 6–6.5, which makes the AcDEX-based nanoparticles a valuable candidate for the sustained release of cargos in the infarcted heart.26,27
2.3. Cytocompatibility
The cytocompatibility of the developed nanosystem was assessed on macrophage cell lines, RAW 264.7 and KG-1, as well as on primary macrophages, of both human and murine origins. Cells were incubated with the nanoparticles for 24 and 48 h, since the drugs are released in 24 h and also because the desired effect on cardiomyocytes, produced by the compounds C and S (stimulation of cardiomyocyte proliferation by re-entry of cardiomyocytes in the cell cycle), takes time to manifest. At each time point, cell viability was assessed by CellTiter-Glo® luminescence assay.51 As shown in Fig. 3, both bare nanoparticles and Putre-AcDEX-PEG-TT1-ANP, empty and loaded, were safe towards RAW 264.7 and KG-1 cell lines.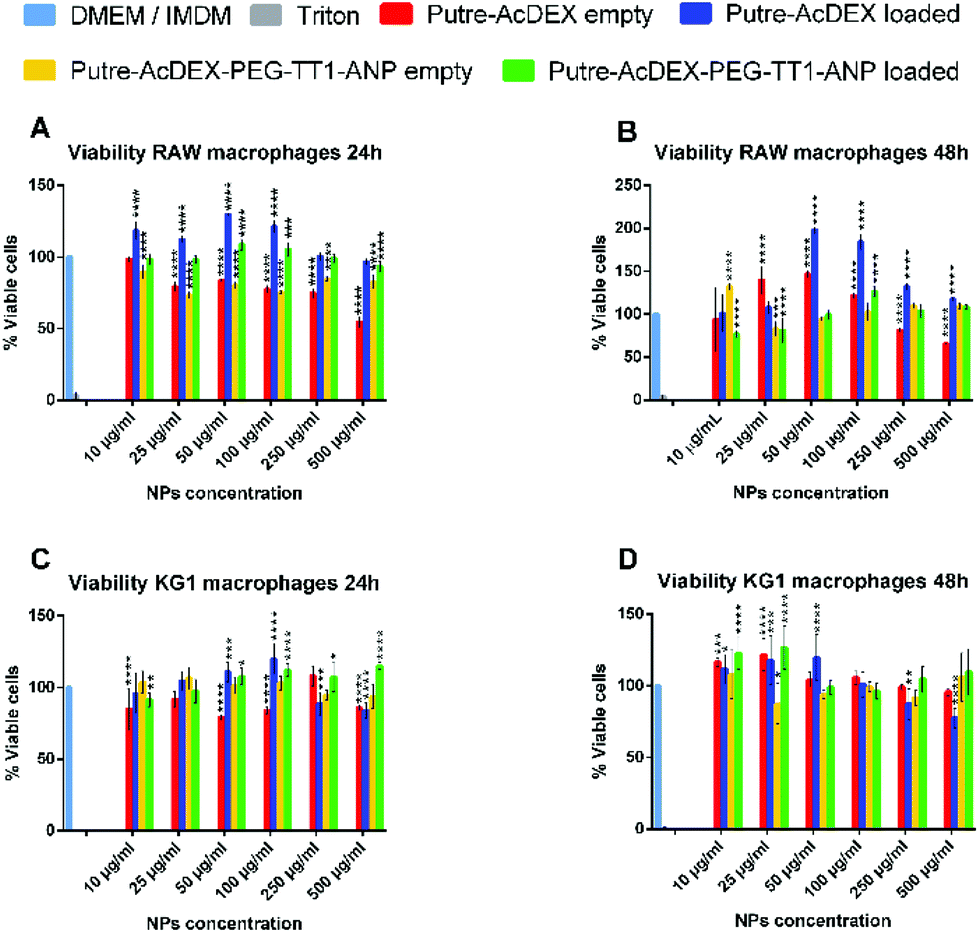 | ||
| Fig. 3 Cell viability of macrophage cell lines. Cytocompatibility studies were conducted to assess the safety of the produced nanoparticles on both RAW 264.7 (A–B) and KG-1 (C–D) cell lines. Values are represented as mean ± s.d. (n = 3 biological replicates in which each time 3 technical replicates have been used). A one-way ANOVA followed by a Tukey–Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at probabilities of *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 for comparison with the medium, which was used as the control in all the tests. | ||
Empty bare nanoparticles were slightly toxic at higher concentrations, probably due to the presence of putrescine, which endows the nanosystems with a high positive charge. Positively charged nanoparticles are toxic towards cells, because of the high interaction between the cationic surface groups of the nanoparticles and the negatively charged cell membranes, which cause disruption of the plasma-membrane integrity, production of a high number of autophagosomes and damage to cellular organelles, in particular mitochondria and lysosomes.52 After surface functionalization, the charge of the nanosystems is not highly positive anymore, thus the toxicity is reduced and the nanoparticles are more likely biocompatible. Moreover, the encapsulation of drugs corresponded to a significant increase in cell viability, suggesting a protective effect of the two compounds.53–55 Researchers demonstrated that the inhibition of p38 MAPK53 and activation of canonical wnt signaling55 pathways are implicated not only in the cytokine production of stimulated macrophages, but also in their proliferation. SB203580, a p38 MAPK inhibitor, promotes macrophage proliferation by increasing the stability of granulocyte-colony stimulating factor (G-CSF) mRNA, and thus, enhancing its expression at the post transcriptional level.53
The activation of the canonical wnt signaling instead, results in a proliferative effect through the upregulation of cyclin D1 protein expression.55
Next, the biocompatibility was also studied in primary macrophages, which showed a higher sensitivity to the positive nature of bare nanoparticles. As shown in Fig. 4, the nanoparticles without any surface modification drastically reduced the viability of both M1- and M2-like macrophages, with murine cells being the most sensitive. The cytotoxicity effect was both dose- and time-dependent. Also with primary macrophages, the surface functionalization and the loaded drugs improved the cell biocompatibility of the nanoparticles.
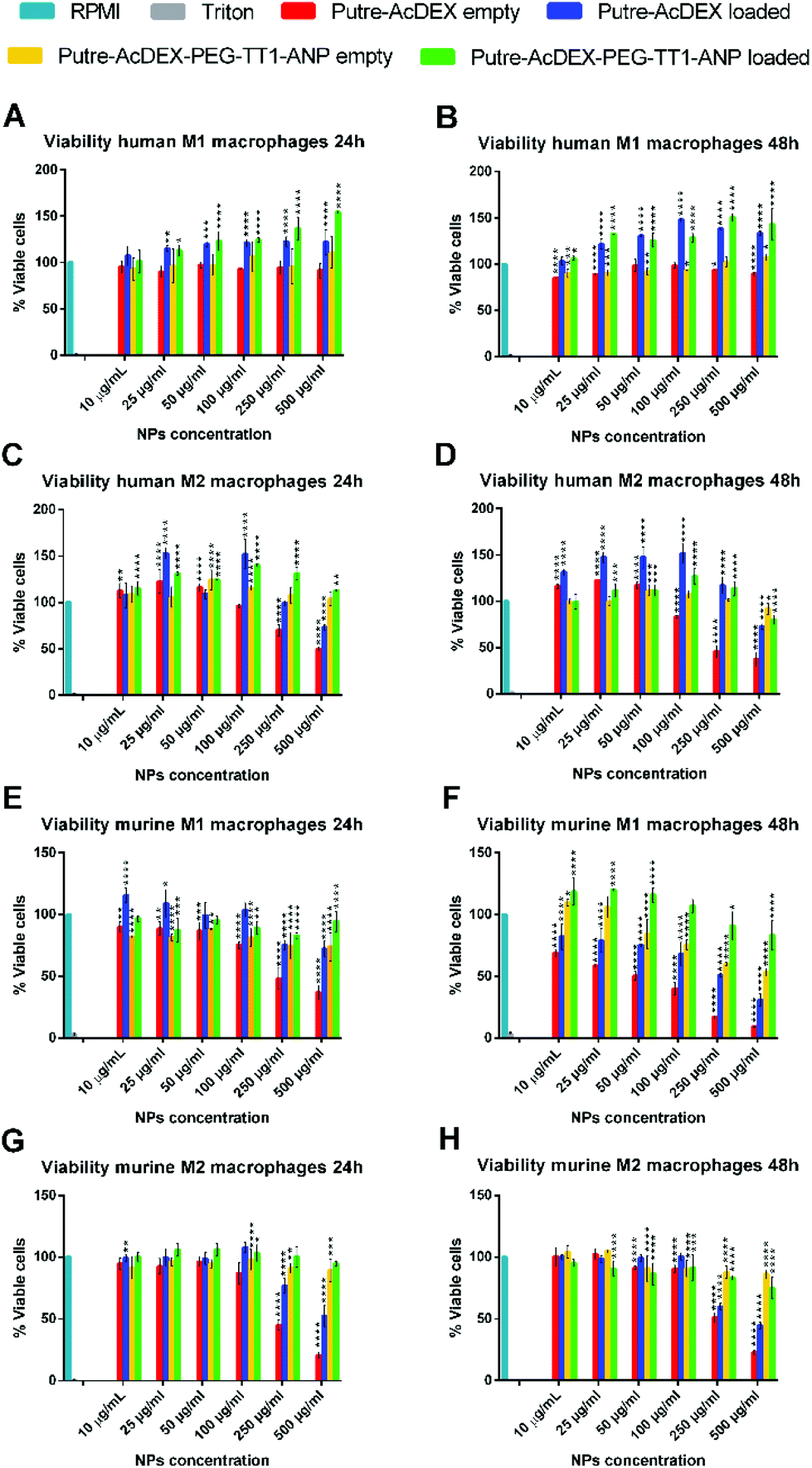 | ||
| Fig. 4 Cytocompatibility of primary macrophages. Cell viability studies on both human (A–D) and murine (E–H) M1-/M2-like macrophages. Biocompatibility was assessed at both 24 (A, C, E, G) and 48 h (B, D, F, H). Values are represented as mean ± s.d. (n = 3 biological replicates in which each time 3 technical replicates have been used). A one-way ANOVA followed by a Tukey–Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at probabilities of *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 for comparison with the medium, which was used as the control in all the tests. | ||
2.4 Cell–nanoparticle interactions with macrophage cell lines
Macrophage cell lines, RAW 264.7 and KG-1, were first screened to have preliminary indications on the behavior of the cells in the presence of nanocarriers. Based on the abovementioned cytocompatibility results, the nanoparticle concentration of 50 μg mL−1 was used in all uptake studies. The cell–nanoparticle interaction was studied by flow cytometry analysis, by incubating the cells for 1 h with fluorescently labelled nanoparticles, then washed and prepared for flow cytometry analysis, which was performed by the detection of fluorescence of AlexaFluor488®. Fig. 5 shows the median fluorescence intensity (MFI) values for the two cell lines treated with PEGylated and peptide-conjugated nanoparticles. MFI values are proportional to the extent of the cell–nanoparticle interaction.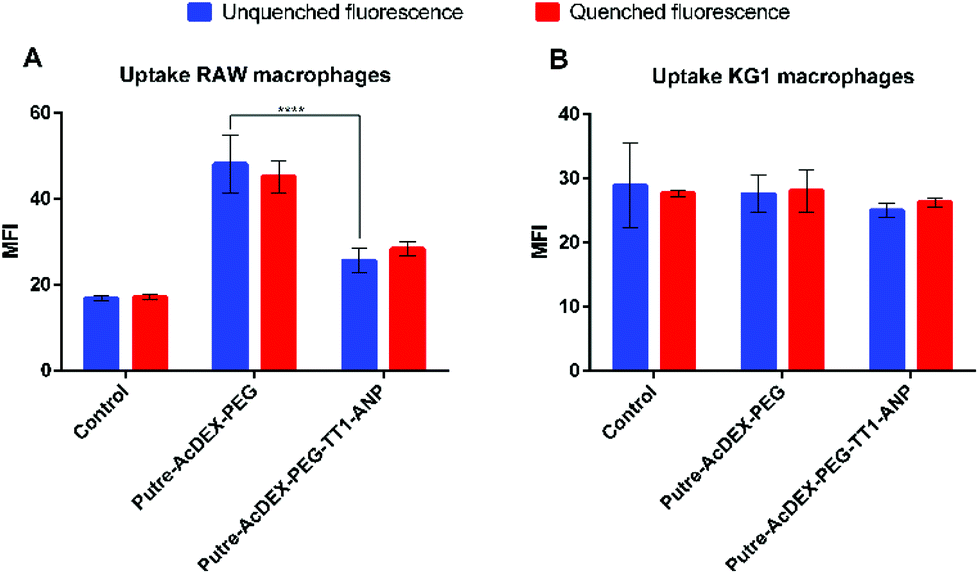 | ||
| Fig. 5 Quantitative uptake studies with macrophages cell lines. Uptake studies have been carried out with both RAW 264.7 (A) and KG-1 (B) cell lines incubated for 1 h with the nanoparticles. Values represent the MFI ± s.d. (n = 3 biological replicates in which each time 3 technical replicates have been used). A one-way ANOVA followed by a Tukey–Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at probabilities of ***p < 0.01 for comparison with Putre-AcDEX-PEG and Putre-AcDEX-PEG-TT1-ANP (both before fluorescence quenching with TB), and for Putre-AcDEX-PEG-TT1-ANP, before and after fluorescence quenching. | ||
It is known that macrophages express ANP receptors.56 However, ANP significantly influences the uptake properties of macrophages only after an appropriate incubation time (18 h).56 Thus, in this work we did not study the effect of ANP on different macrophages’ uptake profiles.
RAW 264.7 macrophages showed a preferential interaction with Putre-AcDEX-PEG as compared with Putre-AcDEX-PEG-TT1-ANP. It has been demonstrated that PEGylation improves the “stealth” properties of nanoparticles, and thus, it is expected that PEGylated nanoparticles interact less with cells.57 However, the internalization rate of PEGylated nanoparticles in this particular case was faster. This can be explained considering that the PEG attached to the particles had a low number of monomers per chain, and thus, it was expected to be internalized faster compared to a PEG with a longer chain.58 In contrast to flow cytometry results, confocal images in Fig. S1† showed that particles conjugated with both peptides interacted more with RAW macrophages as compared with the PEGylated nanosystems. Nonetheless, confocal images are only representative of part of the cell population and provide only a qualitative perspective on the interactions between cells and nanoparticles.
KG-1 cells, which are a human non-adherent macrophage cell line, did not show any difference in the interaction between the PEGylated nanosystems and the nanoparticles functionalized with both ANP and TT1 peptides. It is hypothesized that this could be due to the sedimentation of nanoparticles over time, excluding themselves from the interaction with the cells.59
2.5 Polarization of human and murine monocytes
Monocytes were isolated and collected from human blood and murine bone marrow. After collection, they were maturated and then polarized into M1- and M2-like macrophages by using different cocktails of cytokines (Scheme S1†). In order to confirm the impact of our polarization strategies, we assessed the surface expression of a panel of M1 and M2 phenotypic markers (Fig. S2 and S3†). For both MØ and LPS/IL-4 stimulated macrophages of human origin, CD86 and CD206 were used as M1 and M2 markers, respectively.60,61 Murine MØ macrophages were recognized by the expression of F4/80 and CD11b,62 whereas M1- and M2-like macrophages, were distinguished by the expression of CD206 and CD11c.63 Unstained cells were used as controls. Before polarization with lipopolysaccharide (LPS), the majority of the human monocytes matured with the granulocyte-macrophage colony-stimulating factor (GM-CSF) were CD206+CD86− (Fig. S2A†). Treatment with LPS shifted the expression of the markers towards an increase of CD86 positive cells (Fig. S2B†), thus suggesting that treatment with the cytokine was able to create a population of M1-like macrophages. Similarly, monocytes treated with the macrophage colony-stimulating factor (M-CSF), which were double positive for CD206 and CD86 (Fig. S2C†), showed a shift towards CD206+CD86− upon stimulation with interleukine-4 (IL-4) (Fig. S2D†). Murine cells exhibited major plasticity compared to human macrophages. Monocytes matured with both GM-CSF and M-CSF were, respectively, CD11b+F4/80− and double positive for F4/80 and CD11b (Fig. S3A†), typical monocyte markers. Then, LPS stimulated MØ macrophages became CD11c+CD206− macrophages (Fig. S3B†), whereas the cells treated with IL-4 were double positive (Fig. S3D†). The cellular morphology also changed depending on the cytokine treatment.2.6 Cell–nanoparticle interactions with primary macrophages
Finally, the interactions between the nanoparticles and primary macrophages were also assessed. Previous studies already evaluated the effect of macrophage polarization on nanoparticle uptake, but they obtained contrasting results, depending on the conditions used during the studies and, most importantly, on the type on nanoparticle used.64–66Ideally, for intravenous injectable nanosystems and the “hitchhike” effect to the infarcted heart by recruited macrophages to take place, the aim is to obtain a nanosystem that is able to associate with the macrophages’ surface, without being taken-up. To understand which type of macrophage was presenting a higher nanoparticle–cell association rate versus uptake, we evaluated the interactions between the hitchhiking nanoparticles and the differentiated M1- and M2-like macrophages derived from both human and murine precursors. Uptake studies were performed as previously done for continuous cell lines.
From the results, we observed that Putre-AcDEX-PEG-TT1-ANP nanoparticles generally interacted more with M2-like macrophages compared to M1-like ones (Fig. 6). The nanoparticle–cell association vs. uptake ratio was higher for M2-like macrophages in both human and murine cells, suggesting that M2 macrophages can act as more efficient carriers of nanoparticles into the infarcted heart.
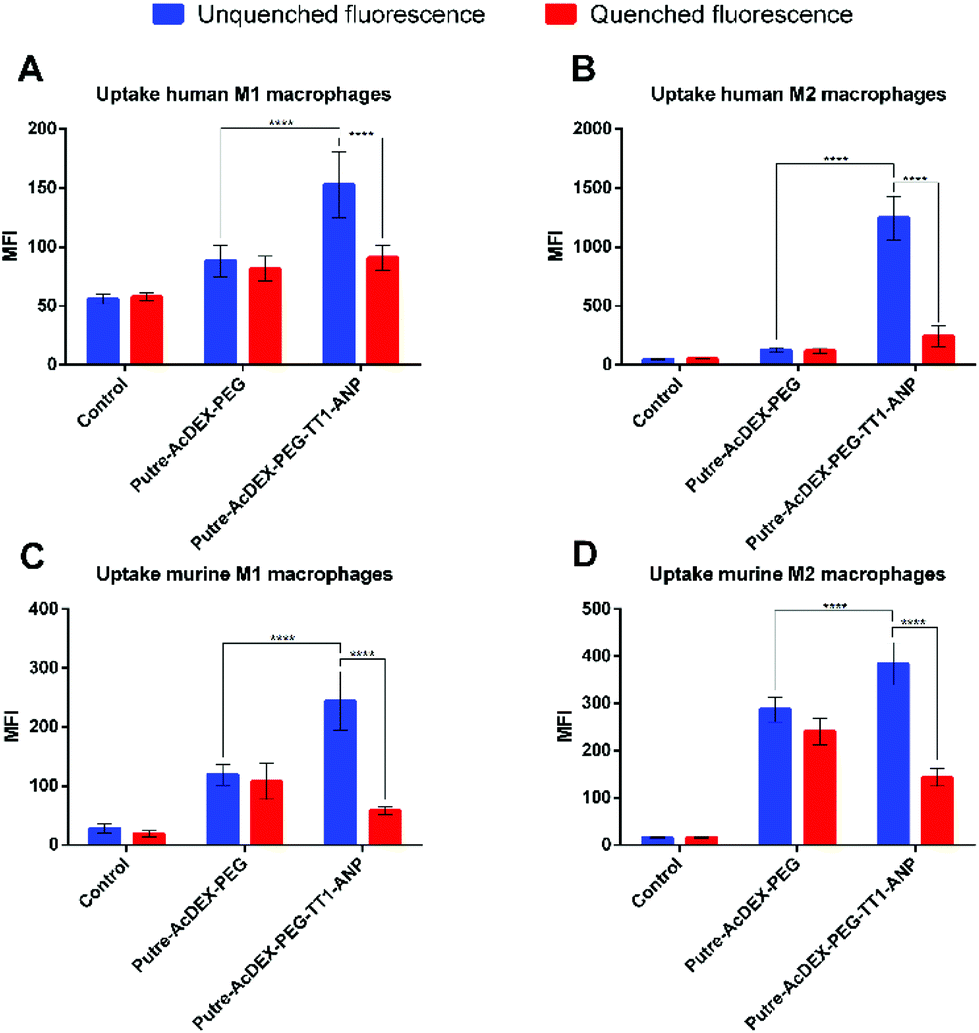 | ||
| Fig. 6 Quantitative cell uptake studies on primary M1- and M2-like macrophages. Interactions between Putre-AcDEX nanoparticles and cells were studied also on human (A, B) and murine (C, D) M1- and M2-like macrophages. The results are represented as MFI values ± s.d. (n = 3 biological replicates in which each time 3 technical replicates have been used). A one-way ANOVA followed by a Tukey–Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at probabilities of *p < 0.05 and ****p < 0.0001 for comparison between Putre-AcDEX-PEG and the Putre-AcDEX-PEG-TT1-ANP (both before fluorescence quenching with TB), and for Putre-AcDEX-PEG-TT1-ANP, before and after fluorescence quenching. | ||
Considering these findings and the bi-phasic accumulation of M1- and M2-like macrophages in the infarcted heart, the particles presented here can reach the infarcted heart in an optimal timeframe. The final aim of the designed nanosystem was to induce cardiomyocyte proliferation, restoring the loss of beating cells and cardiac function after infarction. The loaded drugs, C and S, were used for this aim, because it has been shown in the literature that they have a synergic effect in stimulating cardiomyocyte proliferation.28 In order to be successful in this strategy, we hypothesized that the first phase of macrophage recruitment is needed to phagocytose dead cells and debris, and to make space for the new regenerated tissue.14 Thus, the ability to exploit the recruitment of M2-like macrophages, suggests an increase of the chances for an optimal tissue remodelling, because the proliferation of cardiomyocytes would contrast the formation of scar tissue.
The results were confirmed also by qualitative cell uptake studies, using fluorescence confocal microscopy. Different macrophages were incubated with the nanoparticles for 1 h and then fixed and stained. Finally, the samples were imaged with an inverted confocal microscope. The images showed that the nanoparticles conjugated with both the peptides interacted more with the cells than the PEGylated ones. Moreover, in the images particles looked green and not yellow, suggesting that the nanoparticles were mainly associated with the surface of the macrophages, and not taken-up (Fig. S4†).
Finally, we also studied the mechanism of internalization of Putre-AcDEX-PEG-TT1-ANP nanoparticles. Since we observed higher cell–NP association than cell uptake, especially in M2-like macrophages, we also hypothesized whether the nanoparticles were inducing frustrated endocytosis. For this purpose, NP cell uptake mechanism studies were carried out and quantified as percentage of positive events. Primary macrophages were incubated with different compounds, in order to inhibit particular mechanisms of cell internalization. Cytochalasin D (CytoD) was used at two different concentrations to study the micropinocytosis and the role of actin in the endocytic process, because it is able to depolarize actin filaments.67,68 Nocodazole was used as a microtubule disruptor,69,70 in order to assess the influence of microtubules on the nanoparticle internalization. Genistein is an isoflavone used to inhibit caveoline-mediated endocytosis as a result of its suppressive effect on tyrosine kinases involved in this type of internalization.70–72 Clathrin-mediated uptake was inhibited by chlorpromazine, which interferes with clathrin disassembly and receptor recycling to the plasma membrane.73 Sodium azide was used to study whether the particles were taken-up by active transport or not, because it interferes with ATP production due to its ability to inhibit cytochrome c oxidase.74,75 Adsorptive-mediated uptake was inhibited by protamine sulphate.76 All primary macrophages internalized the nanoparticles preferentially by macropynocytosis, as shown in Fig. 7.
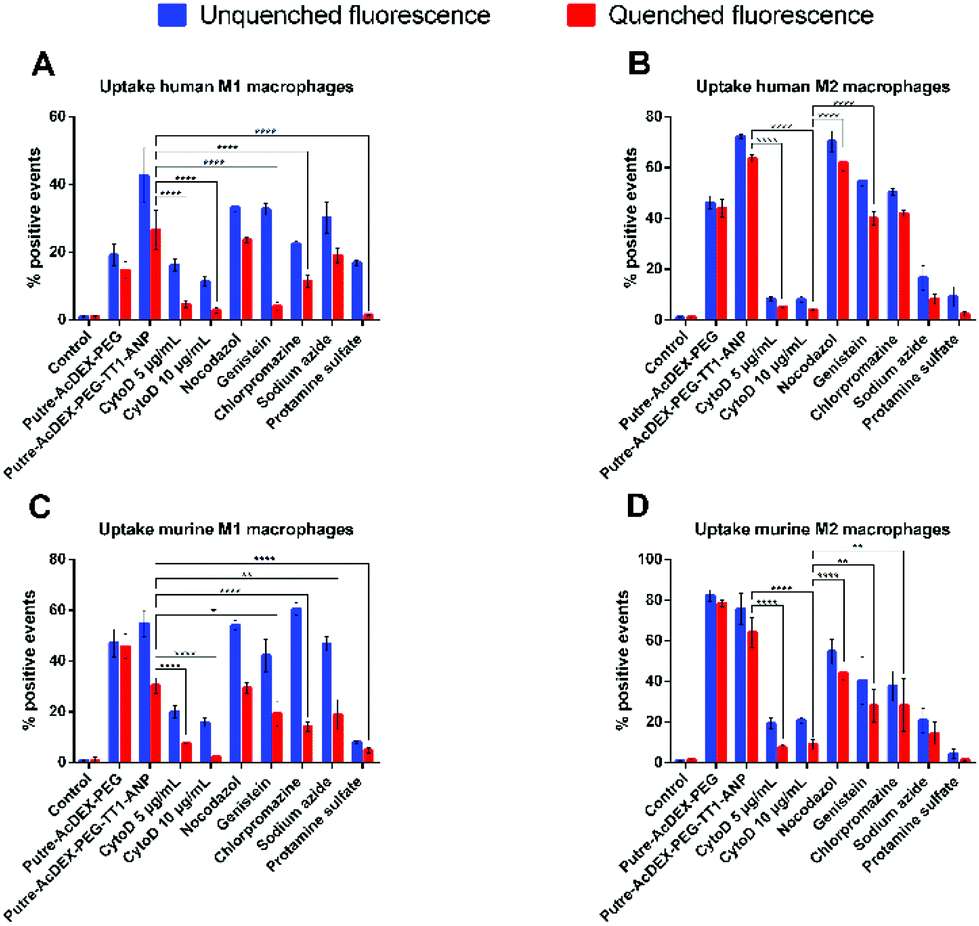 | ||
| Fig. 7 Study of the cell uptake mechanisms. Primary macrophages were incubated with different compounds, each one inhibiting a different mechanism of endocytosis, and then particles were added and the cell uptake evaluated by flow cytometry. Values represent the percentage of positive events ± s.d. (n = 3 biological replicates in which each time 3 technical replicates have been used). A one-way ANOVA followed by a Tukey–Kramer post hoc test was used for the statistical analysis. The significance levels of the differences were set at probabilities of *p < 0.1, ** p < 0.01, *** p < 0.001 and **** p < 0.0001. | ||
Macrophages treated with Cyto D presented lower values of percentage of positive events for nanoparticles taken-up as compared, for example, to genistein and chlorpromazine, which inhibit caveolin-mediated endocytosis and clathrin-mediated uptake, respectively. Human derived macrophages (Fig. 7A and B) had the lowest sensitivity to uptake inhibitors compared to murine cells (Fig. 7C and D). Also, it was notable that actin filaments, but not microtubules, were the cytoskeleton components involved in the uptake mechanism of the Putre-AcDEX-PEG-TT1-ANP nanoparticles. All these findings suggest the engagement of energy dependent pathways in the endocytosis of the produced nanoparticles, justifying the high association versus cell uptake observed in Fig. 6.
3. Conclusions
Herein, we report the design of a nanocarrier successfully decorated with two different peptides, TT1 and ANP, with promising targeting ability towards infarcted heart tissue, through the exploitation of macrophage recruitment. Putre-AcDEX nanoparticles after surface modifications were biocompatible towards both macrophage cell lines and primary macrophages of human and murine origin. Moreover, they showed preferential association versus uptake with M2-like macrophages (approximately 2-fold and 6-fold increase in murine and human primary macrophages, respectively, compared to M1-like), which makes them suitable candidates for achieving the “hitchhike” effect and target the infarcted heart in the later stage of the inflammatory response. Overall, the nanosystem developed here has the potential ability to exploit the post-infarction recruitment of M2-like macrophages, to provide new targeting strategies in the field of nanomedicines for heart disease treatment.Ethical statement
All experimental protocols with animals were approved by the Laboratory Animal Center of the University of Helsinki and the National Animal Experiment Board of Finland according to the EU's Guidelines for Accommodation and Care of Animals, following the Act (497/2013) and the Decree (564/2013) on Animal Experimentation approved by the Finnish Ministry of Agriculture and Forestry, and the EU Directive (2010/63/EU). For the primary cell cultures for in vitro experiments the animals were used after an internal license was authorized and approved by the Laboratory Animal Centre, University of Helsinki (Finland). All human blood samples were purchased from the Finnish Red Cross Blood Service (Veripalvelu) obtained from anonymous donors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Dr A. Rahikkala is acknowledged for providing protocols for PBMC isolation. Dr W. Li is acknowledged for helping with KBr-FTIR. Dr V. Balasubramanian acknowledges University of Helsinki Research Funds. Prof. Hélder A. Santos acknowledges financial support from the HiLIFE Research Funds, the Sigrid Jusélius Foundation and the Academy of Finland (grant no. 317042). T. Teesalu was supported by the European Union through the European Regional Development Fund (Project No. 2014-2020.4.01.15-0012) and by European Research Council grant GlioGuide from European Regional Development Fund. The authors also acknowledge the following core facilities funded by Biocenter Finland: Electron Microscopy Unity of the University for providing the facilities for TEM imaging, Flow cytometry, MST and Biacore core facilities of the University for the flow cytometer and the Light Microscopy Unit of the Institute of Biotechnology for the confocal microscope.References
- G. A. Roth, D. Abate and K. H. Abate, et al. , Lancet, 2018, 392, 1736 CrossRef.
- M. A. Laflamme and C. E. Murry, Nat. Biotechnol., 2005, 23, 845 CrossRef CAS PubMed.
- A. Uygur and R. T. Lee, Dev. Cell, 2016, 36, 362 CrossRef CAS PubMed.
- V. Talman and H. Ruskoaho, Cell Tissue Res., 2016, 365, 563 CrossRef CAS PubMed.
- M. P. A. Ferreira, V. Talman and G. Torrieri, et al. , Adv. Funct. Mater., 2018, 28, 1705134 CrossRef.
- R. C. Scott, J. M. Rosano and Z. Ivanov, et al. , FASEB J., 2009, 23, 3361 CrossRef CAS PubMed.
- J. Park, B. Kim and J. Han, et al. , ACS Nano, 2015, 9, 4987 CrossRef CAS PubMed.
- Y. Chang, E. Lee and J. Kim, et al. , Biomaterials, 2019, 192, 500 CrossRef CAS PubMed.
- Z. Fan, Z. Xu and H. Niu, et al. , J. Controlled Release, 2019, 311–312, 233 CrossRef CAS PubMed.
- M. P. A. Ferreira, S. Ranjan and S. Kinnunen, et al. , Small, 2017, 13, 1701276 CrossRef PubMed.
- M. Shin, H.-A. Lee and M. Lee, et al. , Nat. Biomed. Eng., 2018, 2, 304 CrossRef CAS PubMed.
- M. P. A. Ferreira, S. Ranjan and A. M. R. Correia, et al. , Biomaterials, 2016, 94, 93 CrossRef CAS PubMed.
- L. R. Potter, A. R. Yoder and D. R. Flora, et al. , Exp. Pharmacol., 2009, 191, 341 CAS.
- M. Nahrendorf, F. K. Swirski and E. Aikawa, et al. , J. Exp. Med., 2007, 204, 3037 CrossRef CAS PubMed.
- M. Nahrendorf and F. K. Swirski, Circ. Res., 2013, 112, 1624–1633 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, J. Controlled Release, 2014, 190, 531 CrossRef CAS PubMed.
- E. Chambers and S. Mitragotri, J. Controlled Release, 2004, 100, 111 CrossRef CAS PubMed.
- A. C. Anselmo, V. Gupta and B. J. Zern, et al. , ACS Nano, 2013, 7, 11129 CrossRef CAS PubMed.
- A. M. Brynskikh, Y. Zhao and R. L. Mosley, et al. , Nanomedicine, 2010, 5, 379 CrossRef CAS PubMed.
- H. Dou, C. B. Grotepas and J. M. McMillan, et al. , J. Immunol., 2009, 183, 661 CrossRef CAS PubMed.
- M.-R. Choi, R. Bardhan and K. J. Stanton-Maxey, et al. , Cancer Nanotechnol., 2012, 3, 47 CrossRef CAS PubMed.
- J. Choi, H.-Y. Kim and E. J. Ju, et al. , Biomaterials, 2012, 33, 4195 CrossRef CAS PubMed.
- M. T. Stephan, J. J. Moon and S. H. Um, et al. , Nat. Med., 2010, 16, 1035 CrossRef CAS PubMed.
- M. T. Stephan, S. B. Stephan and P. Bak, et al. , Biomaterials, 2012, 33, 5776 CrossRef CAS PubMed.
- T. Heinze, T. Liebert and B. Heublein, et al., Functional Polymers Based on Dextran BT - Polysaccharides II, ed. D. Klemm, Springer Berlin Heidelberg, Berlin, Germany, 2006, pp. 199–291 Search PubMed.
- S. Suarez, G. N. Grover and R. L. Braden, et al. , Biomacromolecules, 2013, 14, 3927 CrossRef CAS PubMed.
- S. L. Suarez, A. Muñoz, A. Mitchell and R. L. Braden, et al. , ACS Biomater. Sci. Eng., 2016, 2, 197 CrossRef CAS PubMed.
- H. Uosaki, A. Magadum and K. Seo, et al. , Circ.: Cardiovasc. Genet., 2013, 6, 624 CAS.
- S. Sharma, V. R. Kotamraju and T. Mölder, et al. , Nano Lett., 2017, 17, 1356 CrossRef CAS PubMed.
- L. Paasonen, S. Sharma and G. B. Braun, et al. , ChemBioChem, 2016, 17, 570 CrossRef CAS.
- E. Ruoslahti, Adv. Drug Delivery Rev., 2017, 110–111, 3 CrossRef CAS.
- J. Hamzah, V. R. Kotamraju and J. W. Seo, et al. , Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7154 CrossRef CAS PubMed.
- V. Fogal, L. Zhang and S. Krajewski, et al. , Cancer Res., 2008, 68, 7210 CrossRef CAS PubMed.
- H.-B. Pang, G. B. Braun and T. Friman, et al. , Nat. Commun., 2014, 5, 4904 CrossRef CAS PubMed.
- K. J. Kauffman, N. Kanthamneni and S. A. Meenach, et al. , Int. J. Pharm., 2012, 422, 356 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold and S. Ataei, et al. , Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- L. Simón-Gracia, P. Scodeller and S. S. Fuentes, et al. , Oncotarget, 2018, 9, 18682 CrossRef PubMed.
- S. Pabisch, B. Feichtenschlager and G. Kickelbick, et al. , Chem. Phys. Lett., 2012, 521, 91 CrossRef CAS PubMed.
- L. Kou, J. Sun and Y. Zhai, et al. , Asian J. Pharm. Sci., 2013, 8, 1 CrossRef CAS.
- F. B. Engel, M. Schebesta and M. T. Duong, et al. , Genes Dev., 2005, 19, 1175 CrossRef CAS PubMed.
- A.-S. Tseng, F. B. Engel and M. T. Keating, Chem. Biol., 2006, 13, 957 CrossRef CAS.
- J. F. Callahan, J. L. Burgess and J. A. Fornwald, et al. , J. Med. Chem., 2002, 45, 999 CrossRef CAS.
- D. B. Ring, K. W. Johnson and E. J. Henriksen, et al. , Diabetes, 2003, 52, 588 CrossRef CAS.
- E. M. Bachelder, T. T. Beaudette and K. E. Broaders, et al. , J. Am. Chem. Soc., 2008, 130, 10494 CrossRef CAS.
- E. R. Gillies, A. P. Goodwin and J. M. J. Fréchet, Bioconjugate Chem., 2004, 15, 1254 CrossRef CAS.
- J. L. Cohen, S. Schubert and P. R. Wich, et al. , Bioconjugate Chem., 2011, 22, 1056 CrossRef CAS PubMed.
- Y. Ahmad Nor, Y. Niu and S. Karmakar, et al. , ACS Cent. Sci., 2015, 1, 328 CrossRef CAS PubMed.
- M. U. Ghori and B. R. Conway, Am. J. Pharmacol. Sci., 2015, 3, 103 CAS.
- K. Frauke Pistel, A. Breitenbach and R. Zange-Volland, et al. , J. Controlled Release, 2001, 73, 7 CrossRef CAS PubMed.
- J. A. Cohen, T. T. Beaudette and J. L. Cohen, et al. , Adv. Mater., 2010, 22, 3593 CrossRef CAS PubMed.
- H. A. Santos, J. Riikonen and J. Salonen, et al. , Acta Biomater., 2010, 6, 2721 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577 CrossRef.
- S.-F. Chang, H.-C. Li and Y.-P. Huang, et al. , J. Biomed. Sci., 2016, 23, 3 CrossRef.
- A. Haghighat, D. Weiss and M. K. Whalin, et al. , Circulation, 2007, 115, 2049 CrossRef CAS PubMed.
- Y. Feng, Y. Liang and J. Ren, et al. , Kidney Dis., 2018, 4, 95 CrossRef PubMed.
- A. M. Vollmar, R. Förster and R. Schultz, Eur. J. Pharmacol., 1997, 319, 279 CrossRef CAS PubMed.
- D. E. Owens and N. A. Peppas, Int. J. Pharm., 2006, 307, 93 CrossRef CAS PubMed.
- Y. Li, M. Kröger and W. K. Liu, Biomaterials, 2014, 35, 8467 CrossRef CAS PubMed.
- C. B. Anders, J. J. Chess and D. G. Wingett, et al. , Nanoscale Res. Lett., 2015, 10, 448 CrossRef PubMed.
- J. C. Zarif, J. R. Hernandez and J. E. Verdone, et al. , BioTechniques, 2016, 61, 33 CrossRef CAS PubMed.
- S. Mia, A. Warnecke and X.-M. Zhang, et al. , Scand. J. Immunol., 2014, 79, 305 CrossRef CAS PubMed.
- K. Eske, K. Breitbach and J. Köhler, et al. , J. Immunol. Methods, 2009, 342, 13 CrossRef CAS PubMed.
- L. S. Bisgaard, C. K. Mogensen and A. Rosendahl, et al. , Sci. Rep., 2016, 6, 35234 CrossRef CAS PubMed.
- K. A. Binnemars-Postma, H. W. M. ten Hoopen and G. Storm, et al. , Nanomedicine, 2016, 11, 2889 CrossRef CAS PubMed.
- S. A. MacParland, K. M. Tsoi and B. Ouyang, et al. , ACS Nano, 2017, 11, 2428 CrossRef CAS PubMed.
- Y. Qie, H. Yuan and C. A. von Roemeling, et al. , Sci. Rep., 2016, 6, 26269 CrossRef CAS PubMed.
- J. A. Cooper, J. Cell Biol., 1987, 105, 1473 CrossRef CAS PubMed.
- D. A. Kuhn, D. Vanhecke and B. Michen, et al. , Beilstein J. Nanotechnol., 2014, 5, 1625 CrossRef PubMed.
- S. M. Liu, K.-E. Magnusson and T. Sundqvist, J. Cell. Physiol., 1993, 156, 311 CrossRef CAS PubMed.
- T. dos Santos, J. Varela and I. Lynch, et al. , PLoS One, 2011, 6, e24438 CrossRef CAS PubMed.
- K. H. Sit, B. H. Bay and K. P. Wong, In Vitro Cell. Dev. Biol.: Anim., 1993, 29, 395 CrossRef.
- L. Thors, J. Eriksson and C. J. Fowler, Br. J. Pharmacol., 2007, 152, 744 CrossRef CAS PubMed.
- Z. M. Qian, H. Li and H. Sun, et al. , Pharmacol. Rev., 2002, 54, 561 CrossRef CAS PubMed.
- M. C. Bennett, G. W. Mlady and Y.-H. Kwon, et al. , J. Neurochem., 1996, 66, 2606 CrossRef CAS PubMed.
- J. N. Stannard and B. L. Horecker, J. Biol. Chem., 1948, 172, 599 CAS.
- N. Shrestha, F. Araújo and M.-A. Shahbazi, et al. , Adv. Funct. Mater., 2016, 26, 3405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr09934d |
| This journal is © The Royal Society of Chemistry 2020 |