
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spin transition nanoparticles made electrochemically†
Guillermo
Pozo
a,
Patricia
de la Presa
bc,
Rafael
Prato
ad,
Irene
Morales
bc,
Pilar
Marin
bc,
Jan
Fransaer
d and
Xochitl
Dominguez-Benetton
 *ae
*ae
aSeparation and Conversion Technologies, VITO, Flemish Institute for Technological Research, Boeretang 200, 2400, Mol, Belgium. E-mail: xoch@vito.be
bInstituto de Magnetismo Aplicado, UCM-ADIF-CSIC, Universidad Complutense de Madrid, A6 22, 500 Km, 28230 Las Rozas, Spain
cDto. Física Materiales, UCM, Universidad Complutense de Madrid, 28040 Madrid, Spain
dDepartment of Materials Engineering, Surface and Interface Engineered Materials, Katholieke Universiteit Leuven, Kasteelpark Arenberg 44 – box 2450, 3001 Leuven, Belgium
eSIM vzw, Technologiepark 935, BE-9052 Zwijnaarde, Belgium
First published on 20th February 2020
Abstract
Materials displaying novel magnetic ground states signify the most exciting prospects for nanoscopic devices for nanoelectronics and spintronics. Spin transition materials, e.g., spin liquids and spin glasses, are at the forefront of this pursuit; but the few synthesis routes available do not produce them at the nanoscale. Thus, it remains an open question if and how their spin transition nature persists at such small dimensions. Here we demonstrate a new route to synthesize nanoparticles of spin transition materials, gas-diffusion electrocrystallization (GDEx), wherein the reactive precipitation of soluble metal ions with the products of the oxygen reduction reaction (ORR), i.e., in situ produced H2O2, OH−, drives their formation at the electrochemical interface. Using mixtures of Cu2+ and Zn2+ as the metal precursors, we form spin transition materials of the herbertsmithite family—heralded as the first experimental material known to exhibit the properties of a quantum spin liquid (QSL). Single-crystal nanoparticles of ∼10–16 nm were produced by GDEx, with variable Cu/Zn stoichiometry at the interlayer sites of ZnxCu4−x(OH)6Cl2. For x = 1 (herbertsmithite) the GDEx nanoparticles demonstrated a quasi-QSL behavior, whereas for x = 0.3 (0.3 < x < 1 for paratacamite) and x = 0 (clinoatacamite) a spin-glass behavior was evidenced. Finally, our discovery not only confirms redox reactions as the driving force to produce spin transition nanoparticles, but also proves a simple way to switch between these magnetic ground states within an electrochemical system, paving the way to further explore its reversibility and overarching implications.
Introduction
The synthesis of materials exhibiting spintronic properties is at the forefront of pursuit of novel magnetic ground states (i.e., quantum magnets). Spin transition materials refer to frustrated antiferromagnets, in which their spins are not aligned in a regular pattern (geometrical spin frustration), including spin glasses, quantum spin liquids (QSL), and high-temperature superconductivity materials.1,2 For instance, quantum spin liquids (QSL) were first realized by herbertsmithite,3 with chemical formula ZnCu3(OH)6Cl2. Due to its characteristics as a spin-liquid, herbertsmithite may have applications in quantum computing devices (e.g., for storage and memory purposes), and in quantum communications.1,4The copper ions in herbertsmithite are arranged in triangular grids known as kagomé lattices, wherein the spins are uncoupled.5 Each spin tends to be anti-parallel to its neighbors, but the geometric arrangement of the sites where the spins sit makes an ordered alignment impossible. So, the spins collapse into a fluctuating state.3,5,6 This non-trivial and disordered arrangement, results in magnetically-frustrated materials due to the low spin of the copper ions (S = 1/2). The entangled interactions continue to fluctuate, even at the lowest temperature of absolute zero.7,8 In such condition, the spins in other magnetic states would be already frozen, but in the quantum state, spins are never locked into a particular configuration.6 Compounds featuring such lattices are predicted to show no long-range magnetic order due to competing for antiferromagnetic interactions between nearest-neighbor spin centers,2,4–7,9–11 thus being also candidates for quantum spin transitions.
Before these materials can be effectively integrated into spintronic technologies, the competitive synthesis of herbertsmithite remains a significant challenge. A major synthesis problem is the production of sizeable amounts at relevant rates, with controllable physicochemical features. Moreover, envisioning small-scale applications, its synthesis at the nanoscale has not been reported. Thus, it is uncertain if its spin-liquid properties are preserved at such scale. This is also the case for other spin-liquid materials, as well as other spin-transition materials, such as spin-glasses.
The first method reported for synthesizing crystalline ZnxCu4−x(OH)6Cl2 (wherein x = 1 for herbertsmithite, and x = 0 and 0.3 < x < 1 for its polymorphs, clinoatacamite and paratacamite, respectively) was issued in 2012 (herbertsmithite was originally found in nature only in 2004).6 Herbertsmithite and its polymorphs are typically synthesized by hydrothermal, or solvothermal methods (e.g., 458 K–473 K). Related cuprate compounds of the form MCu3(OH)6Cl2 with divalent cations (i.e., M = Mg2+, Co2+, Fe2+, Mn2+, and Ni2+)2 and trivalent cations, MCu3(OH)6Cl3 (M = Y3+ or the lanthanides Nd3+ and Sm3+),12,13 have been achieved by unconventional solid-state reactions at higher temperature (e.g., 463 K). An ionothermal method, initially designed for the fabrication of new zeolitic solids, has also been described for the preparation of materials with kagomé lattices.14 However, as in the case of the methods previously described, the synthesis rates are rather slow (weeks- to months-scale). As an example, it took ten months to grow a 1 mm monocrystal of herbertsmithite, in a very complicated hydrothermal reactor.9,15
Although there is one work presenting the formation of nanoscale clinoatacamite by hydrothermal synthesis at 363 K–368 K, there are no equivalent methods available for producing nanoscale herbertsmithite. Furthermore, the study where nanoscale-clinoatacamite is reported does not evidence its magnetic (spin transition) properties, nor the control of other material features, such as crystallite size and stoichiometry, besides requiring the addition of a toxic organic buffer i.e. 2-(N-morpholino) ethanesulfonic acid.16
In general, all these methods are limited to meager production rates and have restrained the production of herbertsmithite (and its polymorphs) to limited samples of macroscale materials (i.e., mm-scale). Green synthesis routes are also preferred, but they are also not available thus far. The development of such routes will be a key step forward for innovations, growth, and competitiveness in the digital miniaturization arena—e.g., using quantum technologies in computing and electronics.
It has been experimentally proven for many materials that micro- and nano-scale dimensioned particles can differ in properties vs. their macro-scale counterparts. Especially, spin transition materials may display changes in their magnetic properties with size, and the effects at the nanoscale are mainly unknown.
In this work, we present the synthesis of spin transition nanoparticles. Specifically, using copper and zinc chlorides as precursors, our first goal was to achieve the synthesis of ZnxCu4−x(OH)6Cl2 compounds.
By using different concentrations and ratios of Cu and Zn, different values of x are targeted. The electrochemical method developed, gas-diffusion electrocrystallization (GDEx), results in these materials by the reactive precipitation of intermediaries issued from the oxygen reduction reaction (ORR) at a gas-diffusion cathode, with the metal precursors provided. GDEx has been formerly described for the synthesis of iron oxide nanoparticles with a controlled saturation magnetization.17 The materials resulting from the present research were investigated by X-ray powder diffraction (XRD), together with quantitative phase analysis (QPA, Rietveld method), infrared spectroscopy (IR) and inductively coupled plasma optical emission spectrometry (ICP-OES). The morphology and size of the nanoparticles were measured using transmission electron microscopy (TEM).
A second goal was to achieve well-controlled magnetic properties linked to the nano-sized ZnxCu4−x(OH)6Cl2 particles. Zero-field-cooled (ZFC) and field-cooled (FC) thermal dependence of magnetization (M) were used to distinguish magnetic phase transitions in these materials down to 2 K. Also, hysteresis loops at 2 K and 8 kA m−1 (100 Oe) were investigated for the different compositions.
We demonstrate that it is possible to use our novel electrochemical method for the production of spin transition nanoparticles (i.e., herbertsmithite, clinoatacamite, and paratacamite), in a fast, upscalable, atom- and electron-efficient, energy efficient, and environmentally-friendly way, setting a new precedent towards the industrial era of these materials.
Experimental
Gas-diffusion electrocrystallization reactor
The electrochemical reactor design described in Prato et al.,17 was used in this study. As a working electrode, 10 cm2 of VITO CoRE® multilayered carbon-based gas-diffusion cathodes were used to enable the GDEx process. The VITO CoRE® electrodes consisted of a current collector (stainless steel gauze), an active layer (i.e., porous electrically-conducting matrix), and a hydrophobic gas diffusion outer layer (porous PTFE). The composition of the active layer was 20% PTFE and 80% active carbon. Norit®SX1G (878 m2 g−1, Norit Americas Inc., USA) was employed as the active carbon source. The counter electrode (anode) consisted of a 10 cm2 of platinum sheet laser-welded to titanium (Ti) plate current collector. The anode and cathode compartments were separated by an ion-permeable separator (Zirfon® Perl UTP 500, Agfa, Belgium).18 A 3 M KCl saturated Ag/AgCl reference electrode (+212 mV vs. SHE) (REF 321, Radiometer Analytical, Hach, USA) was inserted in the proximity of the working electrode, via an external connector chamber, filled with 3 M KCl. A long cotton thread (e.g., 30 cm, packed within the external connector chamber) verged the reference and the working electrodes, through a small channel, with the purpose of establishing a microchannel enabling a continuous capillary-suction of the electrolyte. The thread was not treated, since due to its manufacturing process the wax layer that naturally covers cotton fibres is removed, making it more polar and hydrophilic due to exposure of the fibrous structure of cellulose, rich in hydroxyl (OH−) groups.19 All potentials here reported are referred versus the standard hydrogen electrode (SHE).Operation of the gas-diffusion electrocrystallization reactor
Experiments were conducted on a multichannel potentiostat (VMP-3, Bio-Logic SAS, France). The GDEx process was controlled chronoamperometrically at −0.15 VSHE and room temperature (18 °C) of synthesis. The pH in the catholyte was monitored using a sensor for continuous measurement of pH in liquid media (SE555X/1-NMSN pH sensor, Knick, Germany). A pH data logger was connected to the Bio-Logic potentiostat to simultaneously follow up the pH vs. charge (Q) evolution.The data logger consisted of a pH transmitter (A1491N-P1-10-0000, Knick, Germany) with a converter (P15000 H1, Knick, Germany) which transforms the 4–20 mA signal delivered by the transmitter into a 0–10 V signal.
All experiments were performed in batch mode with recirculation. The catholyte was recirculated at 15 L h−1 throughout the cathode compartment, where gas-diffusion electrocrystallization of Cu2+ and Zn2+ occurred. The total liquid volume of catholyte and anolyte was 500 mL, which was continuously stirred in the recirculation reservoir (i.e., a borosilicate glass bottle) using a polygonal, PTFE-coated rotating magnetic stirring bar (i.e., 200 rpm). The air was fed through the cathodic gas compartment at a flow rate of 200 ml min−1. A mass gas flow meter and controller (GF40 Bronkhorst hi-tech B.V, Netherlands) was set in place. An overpressure of 26 mbar over a water column (see Fig. 1 for a schematic layout) was applied at the gas exhaust.
 | ||
| Fig. 1 (A) Gas-diffusion electrocrystallization (GDEx), represented for a batch setup. The potentiostat employed is indicated as a power supply, due to its function. (B) Schematic representation of the parts of the multilayered gas-diffusion electrode employed as cathode. | ||
The GDEx-driven synthesis was carried out using a mixture of Cu2+ and Zn2+ ions as the metal precursors, and O2 (in air) as the oxidant gas through the gas-diffusion cathode. An acidified supporting electrolyte was employed as an anolyte and catholyte, composed by a NaCl aqueous solution (0.5 M), adjusted at pH 3 by adding HCl (1 M). In order to tune the formation of stable ZnxCu4−x(OH)6Cl2 products with the desired value for x, different [Cu2+]0/[Zn2+]0 ratios were evaluated. CuCl2·2H2O 99.999% (Sigma-Aldrich, Germany) and ZnCl2 99.999% (Sigma-Aldrich, Germany) were supplemented, respectively.
Oxygen (in air), provided through the gas chamber of the electrochemical cell (right side chamber, Fig. 1A), was electrochemically reduced at the active porous carbon layer of the gas-diffusion cathode (Fig. 1B). The products of the electrochemical ORR, profusely available at the electrochemical interface, reacted with the metals ions in solution (e.g., Cu2+, Zn2+), which are transported to the hydrophilic porous carbon on the cathode, via the aqueous electrolyte. When these metal ions met the ORR products (or highly reactive intermediaries) supersaturation was reached, resulting in the nucleation of hydroxides or oxides. Additive OH− concentration and supersaturation kept ongoing; thus secondary nucleation and crystal growth proceeded during the transient period of residence of the primary nuclei formed within the cathodic interface. The resulting precipitate progressively striped out, as a result of the flow and polarization conditions.20 The solid precipitate was collected in the electrolyte solution and left to sediment under stagnant conditions. Subsequently, the precipitate was washed with deionized water with a pH set at that of the final synthesis solution, re-centrifuged (Jouan CR422, France) at 3000 rpm, washed three times, and dried afterwards under a nitrogen atmosphere, resulting in a particulate powder.
Particle size, morphology and structure characterization
Magnetic characterization and analysis
The magnetic characterization was carried out on powder samples in a glycerin capsule and performed in a SQUID magnetometer Quantum Design MPMS-5S, with 5 T maximum applied field and temperature range from 2 K to 400 K. The ZFC–FC curves were measured at 8 kA m−1 (100 Oe) from 2 K to 300 K, with a step of 0.25 K from 2 K to 6 K, 0.5 K from 6 K to 12 K and more than 1 above 12 K. Before each ZFC measurement, a 3 T demagnetization field was applied in order to avoid any remanent magnetization in the superconductor coils. The hysteresis loops up to 5 T were measured at 2 K, 3 K, 4 K, 6 K, and 15 K.Results and discussion
Charge-driven stoichiometric and structural control
GDEx allows the selective preparation of ZnxCu4−x(OH)6Cl2 materials. By varying the [Cu2+]0/[Zn2+]0 ratio in the GDEx catholyte (wherein [Cu2+]0 and [Zn2+]0 correspond to the initial concentration of copper and zinc ionic precursors, respectively), together with a systematic control of the electric charge, the stoichiometric composition of the ZnxCu4−x(OH)6Cl2 products and their structural control, between Cu2(OH)3Cl and CuO, can be customized (Fig. 2). For instance, the formation of Cu2(OH)3Cl and CuO, for x = 0, and ZnxCu4−x(OH)6Cl2 within the range of 0.3 < x < 1 can be individually targeted, by controlling the operational conditions of GDEx. At x = 0, the initially-colorless solution of copper chloride (i.e., at pH 3) turns into an opalescent and greenish-colored dispersion upon reaching pH ∼5. The color change (see Fig. ESI 1†) can be attributed to the precipitation of Cu2(OH)3Cl (see the green line (a) in Fig. 2), by following reaction (1), in analogy to the mechanism proposed by Engelbrekt et al.16 | (1) |
 | ||
| Fig. 2 A suggested mechanism for the synthesis of spin transition nanoparticles. Effect of charge consumption on structural control of (a) Cu2(OH)3Cl vs. (b) CuO and stoichiometric composition control of spin transition compounds (ZnxCu4−x(OH)6Cl2) for (c) x = 0.3 (paratacamite) and (d) x = 1 (herbertsmithite). (e) The control experiment without metals in solution. | ||
A plateau of precipitation can be distinguished at pH 4.7–5.5, where 1630 C L−1 were consumed from a starting pH of 3. Upon reaching a pH of 5.5, the Cu2+ ions were fully removed from the solution (see Fig. ESI 1†), followed by a sharp increase of pH, up to 11—which was reached at a charge consumption of 1875 C L−1. Tenorite (CuO) formed rapidly under alkaline conditions (black line), according to reaction (2).
 | (2) |
The formation of ZnxCu4−x(OH)6Cl2 with x = 0.3 can be individually targeted with a combination of low charge consumption (778 C L−1), and a relatively low concentration of Cu2+ (7.3 mM) and Zn2+ (2.3 mM) as the metal precursors as shown in the red line (c) of Fig. 2.
The pH vs. charge evolution of sample x = 1, represented by the blue line (d) in Fig. 2, is distinct from that with x = 0, represented by the black line (b) in Fig. 2.
As zinc ions are substituted onto the interlayer site, a shorter plateau of precipitation is distinguished at pH 4.7–5.5, wherein 1000 C L−1 had been consumed from a starting pH of 3. This is attributed to the insertion of chloride ions as part of the chemical structure in the product form, which—concerning CuO implies a reduced consumption of hydroxide ions, as apparent from comparing reaction (2)vs. reaction (3).
 | (3) |
The extent of copper and zinc removal from the liquid phase is shown in Fig. ESI 2.† At a higher Cu2+ concentration of 574 mg L−1 and a Zn2+ concentration of 91 mg L−1, 100% of Cu2+ and 53% of Zn ions are removed from the liquid phase at a charge consumption of 2460 C L−1, while 300 mg L−1 of Cu2+ and 106 mg L−1 achieved 100% of Cu2+ and 44% removal of Zn ions at a charge consumption of 1810 C L−1.
The orange line in Fig. 2 was obtained, as a control experiment without metals in solution. The charge needed to raise the pH from 3 to 11 was 440 C L−1. The generation of peroxides and free radicals are transient intermediaries, formed together with OH− ions—as per the established mechanism of O2 reduction at non-catalyzed carbon electrodes.23,24 After oxygen diffuses to the electrocatalytic surface (i.e., activated carbon) of the gas diffusion electrode (GDE), O2 is reduced (see Fig. 2e). The imposed cathodic polarization conditions (e.g., −0.15 VSHE) drive this electrolysis mainly to OH− ions via a 4 electron (4e−) transfer (eqn (4)), as described in reactions proposed by Šljukić et al.23
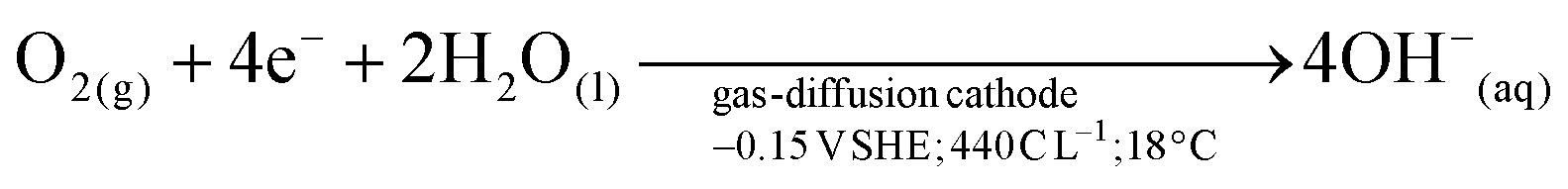 | (4) |
From the charge consumed, it is estimated that a profuse amount of OH− was produced (e.g., >1300 mol m−3) resulting from the oxygen reduction reaction, from early stages of the GDEx process.20 This would immediately result in a pH of 14 within the porosity of the gas-diffusion cathode,24 facilitating the onset for hydroxide supersaturation and hence for reactive precipitation (i.e., crystallization) at the electrochemical interface.
Spin transition nanoparticles characterization
The morphologies of the ZnxCu4−x(OH)6Cl2 particles made by GDEx, at different charge consumption extents, in which x = 0 for clinoatacamite and 0.3 < x < 1 for paratacamite and x = 1 for herbertsmithite, are shown in Fig. 3(a.2, c2, d2). The experimental evidence for x = 0, rendered (a2, a3) clinoatacamite (Cu2(OH)3Cl), which was produced as a mixture of faceted nanocrystals and large spiky nanowhiskers (16 ± 0.4 nm), when using a charge consumption of 987 C L−1, while only tenorite (CuO) microwhiskers of 1.5 μm (b2, b3), with needle-structures, were found at a high charge consumption of 1876 C L−1. This finding is consistent with that of Engelbrekt et al.,16 who demonstrated differences in the morphology of CuO microparticles concerning Cu2(OH)3Cl nanoparticles, which depended strongly on the reaction conditions by pH-controlled precipitation. Conversely, by adding the copper and zinc precursors in an adequate ratio, the formation of highly crystalline nanoparticles with sizes that seemingly correspond to single crystals of ZnxCu4−x(OH)6Cl2, is achieved. For samples with a stoichiometric coefficient of x = 0.3 (c2, c3), the nanocrystals size was (14 ± 0.2 nm), obtained with a charge consumption of 778 C L−1. When the stoichiometric coefficient was x = 1 (d2, d3), the nanocrystals size was 10 ± 0.1 nm, with a charge consumption of 2000 C L−1. | ||
| Fig. 3 Temperature dependence of mass magnetization (M) at 8 kA m−1 (100 Oe) for ZnxCu4−x(OH)6Cl2 and CuO as measured under ZFC–FC conditions; (a1–a2) for clinoatacamite; (b1–b2) for tenorite; (c1–c2) for paratacamite and (d1–d2) for herbertsmithite. TEM imaging for Cu4−xZnx (OH)6Cl2 products made by GDEx using 6 mM Cu2+ and 2 mM Zn2+ as precursors. | ||
The frequency of the spin transition nanoparticles sizes and the corresponding normal distribution fits are shown in Fig. ESI 3.† The addition of Zn2+ had a significant effect on the particle size and polydispersity degree. When the content of zinc was increased from x = 0.3 to x = 1, particle sizes reduced from 14 to 10 nm, even at higher alkaline pH (i.e., 11). The absence of Zn in the structure, under alkaline conditions, allows the rapid decomposition of Cu2(OH)3Cl to CuO microwhiskers of 1.5 μm with needle-structures.
The synthesis of particles at the nanoscale requires very specific supersaturation conditions, such as (1) short nucleation and growth periods, (2) at high rates, and (3) avoiding their immediate agglomeration.25 Correspondingly, the synthesis of spin transition nanoparticles by GDEx, is relies on these features, provided by the nature of the process and reagents employed, combined with the following aspects. The flowing conditions impose only a transient contact of metal ion precursors with the reactive species at the saturated electrochemical interface—rendering transient nucleation conditions.
The possibilities of growth are feeble, as encountering other metal ions is restricted by their high dilution. The rate of interfacial processes typically scales with available surface area (i.e., surface active sites) which is provided by the highly-porous GDE (i.e., >800 m2 g−1), facilitating a high rate of production of HO2−, OH− and their radicals, which quickly react with the metal ion precursors available at the interface.
Notably, the synthesis of ZnCu3(OH)6Cl2 is fast under the operational conditions here employed, with rates approaching 40 mg min−1. The process is highly reproducible, and it involves mild synthetic conditions (e.g., 291 K and atmospheric pressure), in contrast to all previous options reported thus far, such as hydrothermal or solvothermal methods (e.g., ∼458–473 K).6,15
The XRD patterns and IR spectra of 4 representative samples are shown in Fig. 4. The diffraction patterns of Fig. 5A are indexed with the following compounds; Zn0.25Cu3.75(OH)6Cl2 (ICSD-no. 192076), Zn0.85Cu3.15(OH)6Cl2 (ICSD-no. 424325), ZnO (ICSD-no. 26170), Cu2(OH)3Cl (ICSD-no. 64956) and CuO (ICSD-no. 67850).
 | ||
| Fig. 4 (A) XRD patterns Co-Kα (λ = 1.7928 Å) of solid products obtained using 7.3 mM Cu2+ and 2 mM Zn2+ as metal precursor. From top to bottom: Spin transition compounds with formula *ZnxCu4−x(OH)6Cl2 at x = 0, XRD patterns of (a) clinoatacamite (Cu2(OH)3Cl) x = 0 was produced using a charge consumption of 987 C L−1 (green line), while only (b) copper oxide (CuO) x = 0, at a charge consumption of 1876 C L−1 (black line). (c) Paratacamite x = 0.3 (red line); (d) herbertsmithite x = 1 + impurity of zincite (○ZnO) (blue line). The asterisk and circular markers show the most prominent peak positions of ZnxCu4−x(OH)6Cl2 and ZnO phases respectively. The square marker represents the polymorph Cu2(OH)3Cl. (B) FTIR absorption spectra of the synthesized ZnxCu4−x(OH)6Cl2 with x = 0 (a) and Cu2(OH)3Cl and (b) CuO; (c) x = 0.3 and (d) x = 1. The spectra graph is shown in two regions of the hydroxyl stretching (3500–3200 cm−1) and deformation (1000–8000 cm−1). Characteristic frequencies are indicated with vertical dashed lines). (C) M against field over temperature sweep at 2.0 K. Stoichiometric coefficient on the interlayer site ranges of x between 0 to 1. | ||
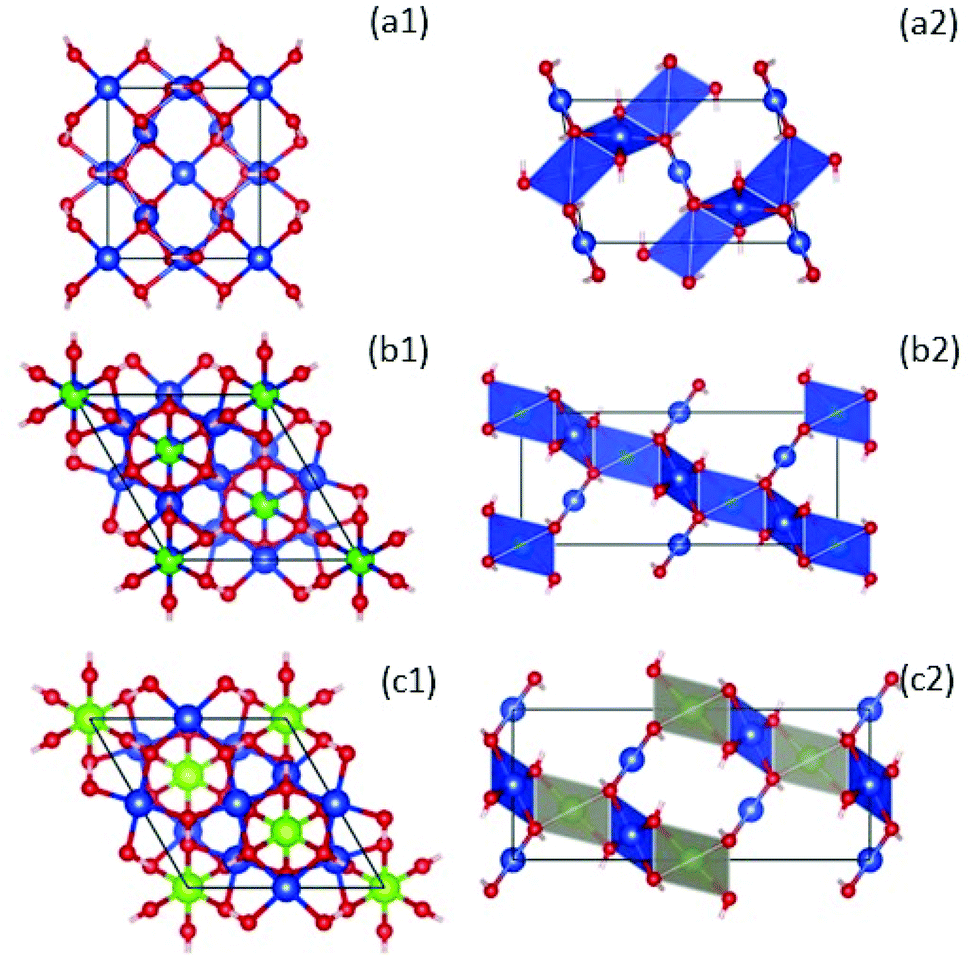 | ||
| Fig. 5 Left: Crystal structures of compounds with formula ZnxCu4−x(OH)6Cl2 at x = 0, clinoatacamite (a1),31 Zn-paratacamite (b1)10 and herbertsmithite (c1)9 looking down along the hexagonal c axis. Copper atoms are blue, zinc atoms are green, and oxygen (red). Chlorine and hydrogen atoms have been suppressed for clarity to emphasize better that the CuO4 units. Right: A side view of the crystal structure using a polyhedral representation, emphasizing the stacking of the kagome layers due to the coupling of CuO4 planar units by ZnO6 octahedra in herbertsmithite (c2). This figure was made using the Vesta – JP Minerals software, https://jp-minerals.org/vesta/en/, retrieving the XRD data indexed with the following compounds: Zn0.25Cu3.75(OH)6Cl2 (ICSD-no. 192076), Zn0.85Cu3.15(OH)6Cl2 (ICSD-no. 424325), Cu2(OH)3Cl (ICSD-no. 64956), which correspond to the theoretical charts closest to the stoichiometries we achieved experimentally (i.e., see Rietveld analysis in Table S1†). | ||
Table S1† compares the results obtained from quantitative phase analysis (QPA) by Rietveld refinement method and ICP-OES of powder samples. The synthesis of ZnxCu4−x(OH)6Cl2 compounds using less than 1000 C L−1, resulted in a product with stoichiometric coefficient x = 0.3 and the lowest amount of impurities, such as ZnO and Cu(OH)2 (Table S1†). At higher charge consumption (2000 C L−1), the synthesis of compounds with higher Zn content (x = 1) is differentiated, but a lower purity is obtained, due to the final pH of synthesis of 11 (see Fig. 2 and duplicate experiment data (d1, d2) in Table S1†).
The XRD traces of Cu2(OH)3Cl and ZnxCu4−x(OH)6Cl2 are indistinguishable by X-ray analysis (see Fig. 4A(a) vs. A (c)) due to the difficulty to differentiate Cu and Zn. The crystal structure of Cu2(OH)3Cl has been determined by several authors,10,16,26 which reported that pure Cu2(OH)3Cl occurs in the form of three different polymorphs: atacamite, clinoatacamite, and paratacamite. While the first two polymorphs constitute the thermodynamically-stable phase of Cu2(OH)3Cl at ambient temperatures (orthorhombic and monoclinic structure type), paratacamite (rhombohedral structure) has to be stabilized by partial substitution of Zn or Ni for Cu.26,27 According to the experimental work of Malcherek and Schlüter,26 the XRD diffraction pattern of ZnxCu4−x(OH)6Cl2 is weak and therefore easily mistaken by that of Cu2(OH)3Cl. Differences in the hydroxyl groups of clinoatacamite and herbertsmithite can be used for appropriate identification.
Thus, infrared spectroscopic (IR) studies of ZnxCu4−x(OH)6Cl2 and CuO were undertaken to overcome the barrier of crystal structure identification encountered with XRD (Fig. 4B). Our data indicate that Cu2(OH)3Cl and ZnCu3(OH)6Cl2 have close lying bands in the hydroxyl stretching region. However, a buried peak at 3400 cm−1 is unique for the Cu2(OH)3Cl structure and can be used as a fingerprint for its detection. Also, the slight shifts in the three major bands of the hydroxyl stretching region, allow us to confidently identify our synthesized Cu2(OH)3Cl as clinoatacamite. The IR spectrum of the synthesized CuO compounds supports the phase purity observed by XRD, with no indication of Cu2(OH)3Cl, and the stability of ZnCu3 (OH)6Cl2. Similar observations are reported using Raman spectroscopy and IR.16,28
Magnetic properties of spin transition nanoparticles
The thermal and field dependence of magnetization (M) over a temperature sweep down to 2.0 K are also reported in Fig. 3(a1, b1, c1 and d1) and 4C. The ZFC and FC thermal dependence of magnetization provides information about the magnetic properties of the spin transition nanoparticles at 8 kA m−1 (100 Oe) (see Fig. 3(a1, c1 and d1)). Tenorite (b1) is an antiferromagnetic semiconductor with Néel temperature (TN) of about 230 K (Fig. ESI 4†). The ZFC–FC curves and hysteresis loops (Fig. 4C) are typical for CuO samples.29On the contrary, on cooling and below T ∼8 K, the spin transition nanoparticles with formula ZnxCu4−x(OH)6Cl2 showed a transition, which also involves a strong antiferromagnetic (AFM) coupling down to 2 K, especially for clinoatacamite with x = 0. The strong AFM coupling of Cu atoms in the triangular lattice makes that the magnetic moments align against the applied field, giving as a result the negative magnetization reversal at low temperatures, especially in clinoatacamite (a.1). As the temperature increases, the small ferromagnetic (FM) contribution coming from the frustrated geometrical surface becomes to dominate. This effect has been observed in different systems (see “the phenomenon of negative magnetization and its implications”)30 and a similar behavior has been reported in Cu2(OH)3Cl.31,32
The negative magnetization shown for clinoatacamite allows us to differentiate between Zn-paratacamite compounds. The high magnetization value at FC in the clinoatacamite also confirms the coupling between a weak FM and a strong AFM interaction. These results are consistent with those of Colman et al.,2 who indicated the presence of a small amount of ferromagnetic impurities.
The introduction of non-magnetic Zn in the pyrochlores lattice produces changes in the crystalline structure (from monoclinic to rhombohedral) and the replacement of Cu2+ by non-magnetic Zn2+ weakens the AFM coupling and finally leads to a system of uncoupled spin, as can be seen from ZFC–FC curves for x = 0, 0.3 and 1.
Fig. 5 shows the crystal structure of ZnxCu4−x(OH)6Cl2 compounds, where clinoatacamite (x = 0) is a 3D distorted pyrochlore lattice antiferromagnet (a1; a2). The Zn2+ ions (green atoms) can be incorporated into the monoclinic clinoatacamite and converted to a rhombohedral structure with undistorted kagome layers forming a 3D framework structure.5,11
The presence of Zn2+ ions in paratacamite (b1; b2) and herbertsmithite (c1; c2) decrease the ordering temperature from 6.5 K to 4.5 K, which is an indication of spin frustration.
Herbertsmithite (x = 1) is proposed as an ideal 2D kagome with S = 1/2, and no long-range magnetic ordering is observed down to 50 mK.
Nanoparticles with a stoichiometric coefficient of x = 1 showed less long-range antiferromagnetic (AFM) ordering or spin freezing than paratacamite nanoparticles (x = 0.3). On the contrary, the higher TN = 6.5 K of Cu2(OH)3Cl with x = 0 compared to the TN = 4.5 K of compounds with x = 1 supports the stronger AFM coupling of the former; besides, the huge magnetization under FC of samples with x = 0 confirms the presence of spin glass due to spin frustration of the magnetic moments at the surface.
Magnetization against field over temperature sweep at 2.0 K was recorded to determine the nature of the magnetic component (Fig. 4C). The hysteresis loops at 2.0 K of Cu2(OH)3Cl shows a kick at low fields confirming the presence of two magnetic phases, one FM and the other AFM (the majority one). The high coercive field (Hc = 2500 Oe = 200 kA m−1) indicates also that the Cu2(OH)3Cl is a strong AFM.
Compounds with stoichiometry of ZnxCu4−x(OH)6Cl2 showed a paramagnetic-like behavior above the transition temperature and different magnetic couplings at low temperatures (see duplicate experiments in ESI 5†), where Cu2(OH)3Cl developed a stronger magnetic coupling than compounds with x = 1. The AFM behavior is weakened by the introduction of non-magnetic Zn2+, indicating the formation of spin–spin correlations; which is a common feature of quantum kagomé systems.2–4,6,13,33
The less ordering temperature in herbertsmithite is a clear indication of spin frustration, which inhibits the tendency of a spin to order under cooling conditions. However, the Néel temperature has not been suppressed as described in the synthesis of a single crystal of herbertsmithite.3
This result may be explained by the fact that below a critical size, magnetic particles become single domain in contrast with the usual multidomain structure of microcrystalline samples, which can exhibit unique phenomena such as unusual coercivities,34 quantum tunneling of the magnetization,35,36 and superparamagnetism.37,38 Another possible explanation for this is that the size reduction of herbertsmithite could affect the novel magnetic properties, due to the small volume or the high surface/volume ratio (spin canting effect),39,40 a phenomenon through which spins show a lack of full alignment at the surface. For example, Martínez et al.41 found a low-temperature surface spin-glass layer in Fe2O3 magnetic nanoparticles with a ferromagnetic core, indicating a magnetic disorder in a well-delimited surface layer of the nanoparticle. Aliyu et al.42 also found a low-temperature surface spin-glass layer in Pr0.5Ca0.5MnO3 very crystalline nanoparticles with an antiferromagnetic core and a ferromagnetic shell of a single unit cell thickness; the origin of the glassy state is the magnetic frustration of the ferromagnetic order of the shell.
The ferromagnetic component has been recently explained by muon spin rotation (μSR) studies, where a weak ferromagnetic moment appears in the paratacamite family, one around 18 K and the other one around 6 K, and where dynamics persist down to T = 0.8,31
However more in-depth investigation is needed to shed light on a spin canting effect in our spin transition nanoparticles. Although ZnxCu4−x(OH)6Cl2 compounds with x = 1 showed less long-range antiferromagnetic (AFM) ordering than nanocrystals with a stoichiometric coefficient of x = 0.3 down to 2 K, further research is required to elucidate if spin canting is responsible for the ferromagnetic component observed, which is in turn could increase the long-range antiferromagnetic (AFM) ordering of spin-transition nanoparticles.
Conclusions
The possibility of synthesizing spin transition nanoparticles using an electrochemical process with high and fast production rates is desirable for many reasons, notably the convenience of significantly reducing the temperature of synthesis and chemicals, providing a green route of synthesis.The magnetic properties, particle size and rate of the reaction can be precisely tuned (directly or indirectly) by manipulating the charge consumption, as well as the composition of the electrolyte. This process allows the careful preparation of Cu4−xZnx(OH)6Cl2 products. The synthesis is fast, reproducible and it involves mild synthetic conditions, running at 291 K and atmospheric pressure, in contrast previously reported methods.
We found faceted nanocrystals of around 10.4 ± 0.1 nm for ZnxCu4−x(OH)6Cl2 products with stoichiometric coefficient between x = 0.3 to 1. At a charge consumption of 987 C L−1, we produced a mixture of faceted nanocrystals and large spiky nanowhiskers of 16.1 ± 0.4 nm with a stoichiometric coefficient of x = 0. Only microwhiskers of CuO (1.5 μm) with needle-structures were found at higher charge consumption of 1876 C L−1.
Magnetic analyses demonstrate that nanoparticles with x = 1 in the formula ZnxCu4−x(OH)6Cl2 showed a small magnetic transition at TN = ∼4.5 K, with less long-range antiferromagnetic (AFM) ordering or spin freezing than nanoparticles with x = 0.3 down to 2 K.
ZFC–FC thermal dependence of M shows bifurcation during the transition at TN = ∼6 K in samples of stoichiometric coefficient between 0 and 0.3, which supports a long-range AFM ordering, spin freezing or spin-glass-behavior.
These results are in accord with recent studies of synthesis of spin transition compounds indicating that the presence of Zn allows the perfect kagomé distribution of the Cu2+ ions and consequently lead the absence of long-range ordering to the lowest measured temperature.
We finally suggest that the spin liquid behavior is sustained at the nanoscale in compounds of ZnCu3(OH)6Cl2. Our discovery not only confirms redox reactions as the driving force to produce spin transition nanoparticles, but also proves a simple way to switch between these magnetic ground states within an electrochemical system, paving the way to further explore its reversibility and overarching implications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Pozo acknowledges the funding from the European Union's Horizon 2020 research and innovation programme MSCA-IF-2017, under grant agreement no. 796320 (MAGDEx: Unmet MAGnetic properties in micro and nano-particles by synthesis through gas diffusion electrocrystallisation, (GDEx).X. Dominguez-Benetton thanks to VITO's strategic research funds and management for the possibility to conduct this pioneering research. GP, RP, JF, and XDB thank the support from the Flemish SIM MaRes programme, under grant agreement no. 150626 (Get-A-Met project). XDB and JF thanks the funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 654100 (CHPM2030 project). This work has been supported by the Ministerio Español de Economía y Competitividad (MINECO) RTI2018-095856-B-C21, and Comunidad de Madrid S2013/MIT-2850 NANOFRONTMAG projects. We would also like to acknowledge Myriam Mertens for fruitful discussions and her support on XRD analysis, as well as Kristof Tirez and Wilfried Brusten for assistance with analytical measurements.
References
- L. Balents, Nature, 2010, 464, 199–208 CrossRef CAS PubMed.
- R. H. Colman, A. Sinclair and A. S. Wills, Chem. Mater., 2011, 23, 1811–1817 CrossRef CAS.
- M. Shores, E. Nytko, B. Bartlett and D. Nocera, J. Am. Chem. Soc., 2005, 127, 13462–13463 CrossRef CAS PubMed.
- R. Colman, C. Ritter and A. Wills, Chem. Mater., 2008, 20, 2005–2007 CrossRef.
- M. R. Norman, Rev. Mod. Phys., 2016, 88, 1–14 CrossRef.
- T. Han, J. Helton, S. Chu, A. Prodi, D. Singh, C. Mazzoli, P. Müller, D. Nocera and Y. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 100402–100406 CrossRef.
- J. S. Helton, K. Matan, M. P. Shores, E. A. Nytko, B. M. Bartlett, Y. Yoshida, Y. Takano, A. Suslov, Y. Qiu, J. H. Chung, D. G. Nocera and Y. S. Lee, Phys. Rev. Lett., 2007, 98, 3–6 CrossRef PubMed.
- P. Mendels, F. Bert, M. A. De Vries, A. Olariu, A. Harrison, F. Duc, J. C. Trombe, J. S. Lord, A. Amato and C. Baines, Phys. Rev. Lett., 2007, 98, 1–4 CrossRef PubMed.
- M. P. Shores, E. A. Nytko, B. M. Bartlett and D. G. Nocera, J. Am. Chem. Soc., 2005, 127, 13462–13463 CrossRef CAS PubMed.
- M. D. Welch, M. J. Sciberras, P. A. Williams, P. Leverett, J. Schlüter and T. Malcherek, Phys. Chem. Miner., 2014, 41, 33–48 CrossRef CAS.
- P. Mendels and F. Bert, J. Phys. Soc. Jpn., 2010, 79, 011001 CrossRef.
- W. Sun, Y. X. Huang, Y. Pan and J. X. Mi, Dalton Trans., 2017, 46, 9535–9541 RSC.
- W. Sun, Y. X. Huang, S. Nokhrin, Y. Pan and J. X. Mi, J. Mater. Chem. C, 2016, 4, 8772–8777 RSC.
- F. Aidoudi, D. Aldous, R. Goff, A. Slawin, J. Attfield, R. Morris and P. Lightfoot, Nat. Chem., 2011, 3, 801–806 CrossRef CAS PubMed.
- T.-H. Han, J. S. Helton, S. Chu, D. G. Nocera, J. A. Rodriguez-Rivera, C. Broholm and Y. S. Lee, Nature, 2012, 492, 406–410 CrossRef CAS PubMed.
- C. Engelbrekt, P. Malcho, J. Andersen, L. Zhang, K. Ståhl, B. Li, J. Hu and J. Zhang, J. Nanopart. Res., 2014, 16(16), 2562 CrossRef.
- R. A. Prato, V. Van Vught, S. Eggermont, G. Pozo, P. Marin, J. Fransaer and X. Dominguez-Benetton, Sci. Rep., 2019, 9, 15370 CrossRef PubMed.
- P. Vermeiren, W. Adriansens and R. Leysen, Int. J. Hydrogen Energy, 1996, 21, 679–684 CrossRef CAS.
- D. Agustini, M. F. Bergamini and L. H. Marcolino-Junior, Lab Chip, 2016, 16, 345–352 RSC.
- X. Dominguez Benetton, Y. Alvarez, C. Porto-Carrero, K. Gijbels and S. Rajamani, US2018/0023201A1, 2018, 40.
- A. Belsly, M. Helderman, V. L. Karen and P. Ulkch, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 364–369 CrossRef PubMed.
- C. H. Görbitz, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 167–168 CrossRef PubMed.
- B. Šljukić, C. E. Banks and R. G. Compton, J. Iran. Chem. Soc., 2005, 2, 1–25 CrossRef.
- H.-H. Yang and R. L. McCreery, J. Electrochem. Soc., 2000, 147, 3420 CrossRef CAS.
- A. Mersmann and M. Löffelmann, Chem. Eng. Technol., 2000, 23, 11–15 CrossRef CAS.
- T. Malcherek and J. Schlüter, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 334–341 CrossRef CAS PubMed.
- R. S. W. Braithwaite, K. Mereiter, W. H. Paar and A. M. Clark, Mineral. Mag., 2004, 68, 13 CrossRef.
- G. Bertolotti, D. Bersani, P. P. Lottici, M. Alesiani, T. Malcherek and J. Schlüter, Anal. Bioanal. Chem., 2012, 402, 1451–1457 CrossRef CAS PubMed.
- D. Gao, Z. Yang, J. Zhang, G. Yang, Z. Zhu, J. Qi, M. Si and D. Xue, AIP Adv., 2011, 1, 042168 CrossRef.
- N. Kumar and A. Sundaresan, Solid State Commun., 2010, 150, 1162–1164 CrossRef CAS.
- X. G. Zheng, T. Kawae, Y. Kashitani, C. S. Li, N. Tateiwa, K. Takeda, H. Yamada, C. N. Xu and Y. Ren, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 4–7 Search PubMed.
- S. Chu, P. Müller, D. Nocera and Y. Lee, Appl. Phys. Lett., 2011, 98, 1–3 Search PubMed.
- M. Schmidt, F. M. Zimmer and S. G. Magalhaes, Phys. A, 2015, 438, 416–423 CrossRef.
- E. F. Kneller and F. E. Luborsky, J. Appl. Phys., 1963, 34, 656–658 CrossRef CAS.
- F. L. Pratt, P. J. Baker, S. J. Blundell, T. Lancaster, S. Ohira-Kawamura, C. Baines, Y. Shimizu, K. Kanoda, I. Watanabe and G. Saito, Nature, 2011, 471, 612–616 CrossRef CAS PubMed.
- D. Galetti, B. M. Pimentel, C. L. Lima and E. C. Silva, Phys. A, 2009, 388, 1105–1110 CrossRef.
- A. G. Kolhatkar, A. C. Jamison, D. Litvinov, R. C. Willson and T. R. Lee, Int. J. Mol. Sci., 2013, 14, 15977–16009 CrossRef PubMed.
- F. Arteaga-Cardona, K. Rojas-Rojas, R. Costo, M. A. Mendez-Rojas, A. Hernando and P. De La Presa, J. Alloys Compd., 2016, 663, 636–644 CrossRef CAS.
- R. Topkaya, O. Akman, S. Kazan, B. Aktaş, Z. Durmus and A. Baykal, J. Nanopart. Res., 2012, 14(1156), 14 Search PubMed.
- A. T. Ngo, P. Bonville and M. P. Pileni, J. Appl. Phys., 2001, 89, 3370–3376 CrossRef CAS.
- B. Martínez, X. Obradors, L. Balcells, a. Rouanet and C. Monty, Phys. Rev. Lett., 1998, 80, 181–184 CrossRef.
- H. D. Aliyu, J. M. Alonso, P. de la Presa, W. E. Pottker, B. Ita, M. Garcia-Hernández and A. Hernando, Chem. Mater., 2018, 30, 7138–7145 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr09884d |
| This journal is © The Royal Society of Chemistry 2020 |