DOI:
10.1039/C9NR09875E
(Paper)
Nanoscale, 2020,
12, 5038-5047
Reversible cation exchange on macroscopic CdSe/CdS and CdS nanorod based gel networks†
Received
20th November 2019
, Accepted 23rd January 2020
First published on 28th January 2020
Abstract
Over the past decades, cation exchange reactions applied to nanoparticles have opened up synthetic pathways to nanocrystals, which were not accessible by other means before. The limitation of cation exchange on the macroscopic scale of bulk materials is given by the limited ion diffusion within the crystal structure. Lyogels or aerogels are macroscopic, highly voluminous, porous materials composed of interconnected nanoscopic building blocks and hence represent a type of bridge between the macroscopic and the nanoscopic world. To demonstrate the feasibility of cation exchange on such macroscopic nanomaterials, the cation exchange on CdSe/CdS core/shell and CdS nanorod based lyogels to Cu2−xSe/Cu2−xS and Cu2−xS and the reversible exchange back to CdSe/CdS and CdS lyogels is presented. These copper-based lyogels can also be used as an intermediate state on the way to other metal chalcogenide-based macroscopic structures. By reversed cation exchange back to cadmium an additional proof is given, that the crystal structures remain unchanged. It is shown that cation exchange reactions can also be transferred to macroscopic objects like aerogels or lyogels. This procedure additionally allows the access of aerogels which cannot be synthesized via direct destabilization of the respective colloidal solutions.
Introduction
The assembly of colloidal semiconductor nanoparticles into highly porous, voluminous macroscopic 3D structures like lyogels or aerogels can combine the physicochemical properties of nanoparticles with the size of bulk materials. The assembly of semiconductor nanocrystals with defined shapes like quantum dots (QDs), nanorods (NRs), nanoplatelets (NPLs) or of colloidal metal nanoparticles for different applications (e.g. photocatalysis,1–3 sensors for chemical detection4–6) has received vast attention over the past decades. Nowadays, there is a large variety of II–VI semiconductor composed aerogels available, such as CdS,7,8 CdSe,8,9 CdTe,10 ZnS7 or metal nanoparticle based aerogels, such as Ag,11–13 Au,14 Pt,11,15 and Pd.16 Furthermore, reports can be found regarding the synthesis of self-supported aerogels, where the building blocks are semiconductor heterostructure nanoparticles, which are composed of two or more components, such as CdSe/ZnS QDs,17 CdSe/CdS NRs,18 and CdSe/CdS NPLs,19 taking advantage of those elaborate band structures due to their beneficial physical (optical, electrical, spectroelectrical) properties. The key challenge of synthesizing macroscopic aerogels from nanoscopic building blocks is the choice of a right destabilization agent for inducing a controlled assembly to form a porous and voluminous self-supporting network. Based on the solvent and surface chemistry of the nanoparticles, certain specific destabilization reagents are already known to form self-supported lyogels, which can e.g. be hydrogen peroxide,18–23 hydrazine,15 tetranitromethane,22 or salts.13 The as-synthesized lyogels can be transformed to aerogels by supercritical drying, according to the well-established Kistler's route.24 These macroscopic gel networks have the advantages of a high surface-to-volume ratio, good accessibility of the surface, and are rare examples of materials with nanoscopic properties within a macroscopic structure. One drawback of monolithic cadmium chalcogenide nanoparticle based aerogel structures is their poor mechanical stability, when immersed in liquid again as well as in a strong air stream, which can be increased, for example, by reinforcement with an additional silica shell.25 Nevertheless, due to the advantages, macroscopic gel networks were used in different fields, e.g. electrocatalysis,26,27 photoelectrochemical applications,6,28 and photocatalysis.1,3,29 In the recent past, different working groups investigated cation exchange reactions on nanocrystals with different shapes and material composition.30–35 Due to the short diffusion path of the cations within the nanoparticle crystal, even cation exchange reactions which are unknown (or extremely slow) on the bulk scale can occur. In nanoparticle systems, the cation exchange process can take place in the millisecond regime.36 Brock and co-workers published the first and so far only work on cation exchange experiments on macroscopic cadmium selenide quantum dot based gel structures.37 Their study focused on spherical CdSe quantum dots with a size of 4 nm which were used as building blocks for the gelation. The achieved CdSe lyogels were treated with silver ions to obtain Ag2Se aerogels by the cation exchange reaction.
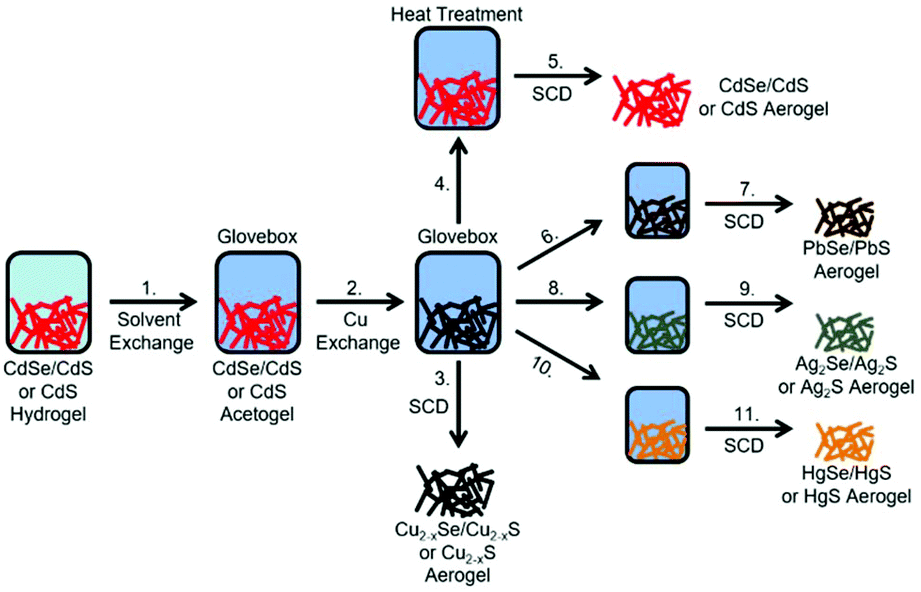
In the present work, we want to expand the cation exchange reaction on macroscopic lyogel networks by using nanorod shaped cadmium sulfide based building blocks. In addition to pristine CdS NRs, dot-in-rod shaped CdSe/CdS NR heterostructures were also investigated as building blocks for macroscopic lyogel network formation to establish aerogels with new material composition via cation exchange reactions. Initially, CdSe/CdS NRs and CdS NRs were synthesized in organic media, phase transferred by ligand exchange to aqueous media and gelated with H2O2 to form macroscopic CdSe/CdS NR and CdS NR lyogels according to our previously described method.18 Subsequently, these gels are subjected to various cation exchange procedures. In the first cation exchange reaction, we investigated the formation of the copper based lyogels composed of Cu2−xSe/Cu2−xS and Cu2−xS. Since copper chalcogenides are known to readily undergo cation exchange reactions due to the high copper ion mobility in these systems, the copper chalcogenide based lyogels were treated with different metal ions (Cd2+, Pb2+, Ag+, Hg2+). By cation back exchange from copper chalcogenide based lyogels to cadmium chalcogenide based aerogels the reversibility of the cation exchange reaction preserving morphology, crystal structure and optical properties was demonstrated (Fig. 1). Copper chalcogenide based lyogels were also used as the intermediate state to achieve NIR emitting lead chalcogenide based gels (PbSe/PbS), as well as silver chalcogenide and mercury chalcogenide based gels (Ag2Se/Ag2S, HgSe/HgS, Ag2S, HgS). Finally, all dot-in-rod shaped or rod shaped nanocrystal-based lyogels obtained by cation exchange were transformed to aerogel networks by supercritical drying. To confirm the composition and structure of the synthesized aerogels, powder X-ray diffraction (PXRD) patterns and energy dispersed X-ray spectra were recorded. The influence of the cation exchange on the morphology of the gel network was investigated via transmission electron microscopy (TEM) and scanning electron microscopy (SEM). Specific surface area measurements were also carried out to investigate changes in porosity due to the cation exchange. The synthesis strategy of cation exchange reactions on well-established nanoparticle-based gel structures developed in this work allows the synthesis of novel classes of aerogels, while at the same time the search for the right destabilization method for each individual colloidal nanoparticle solution is prevented. Hence, cation exchange reactions at macrostructures significantly widen the amount of accessible aerogel materials.
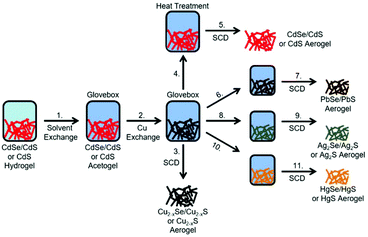 |
| | Fig. 1 Cation exchange mechanism in several steps. 1. Solvent exchange from water to dry acetone and transfer to a glovebox for an inert environment. 2. and 3. Copper cation exchange and supercritical drying (SCD) to obtain Cu2−xSe/Cu2−xS and Cu2−xS aerogels. 4. and 5. Cadmium cation back-exchange, heat treatment and supercritical drying to obtain CdSe/CdS and CdS aerogels. 6. and 7. Lead cation exchange and SCD to obtain PbSe/PbS aerogels. 8. and 9. Silver cation exchange and SCD to obtain Ag2Se/Ag2S and Ag2S aerogels. 10. and 11. Mercury cation exchange and SCD to obtain HgSe/HgS and HgS aerogels. | |
Results and discussion
Initial structures – CdSe/CdS and CdS NRs and their aerogel structures
The gel building blocks used in this work were CdSe/CdS nanorods (NRs) and CdS NRs synthesized according to the procedure from Carbone et al. in a so-called seeded growth synthesis.38 In a two-step synthesis, rod-type CdS shells were grown on previously synthesized CdSe and CdS seeds. From the TEM micrographs (Fig. 2A and B) it can be observed that the gel building blocks were homogeneously grown rod-shaped nanoparticles (NPs) with lengths of 36.4 ± 3.1 nm for CdSe/CdS NRs and 16.6 ± 1.6 nm for CdS NRs and thicknesses of 6.3 ± 1.3 nm for CdSe/CdS NRs and 5.0 ± 0.9 nm for CdS NRs. Additionally, the nanoparticle solutions were analysed by absorbance and emission measurements which are shown in Fig. 2 (A, B bottom). The absorbance spectra of CdSe/CdS NRs (Fig. 2, A bottom) in organic (black line) and aqueous solution (blue line) exhibit nearly the same curve progression for the CdS absorption onset of 477 nm. The emission maximum at 616 nm of CdSe/CdS NRs is the same for the organic (black dashed line) and the aqueous solution (blue dashed line) indicating an unchanged charge carrier recombination in the CdSe core of the core/shell nanorods. The photoluminescence quantum yield (PLQY) was also measured and is 45.9% ± 3.2% for the organic CdSe/CdS NRs and 9.5% ± 1.6% for the aqueous CdSe/CdS NRs. The absorbance spectra of CdS NRs (Fig. 2, B bottom) shows a slightly hypsochromic shift of the CdS absorption onset from 465 nm in organic (black line) to 455 nm in aqueous media (green line). The emission spectra (Fig. 2, B bottom) of the organic (black dashed line) and aqueous (green dashed line) solution have the same emission maxima at 479 nm.
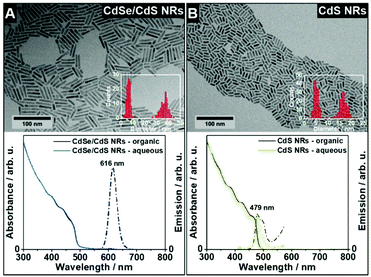 |
| | Fig. 2 TEM micrographs, absorbance spectra and emission spectra of CdSe/CdS NRs (A) and CdS NRs (B). Absorbance spectra (solid line) and emission spectra (dashed line) of CdSe/CdS NRs and CdS NRs in organic solution (black line) and aqueous solution (blue or green line). | |
From SEM micrographs (Fig. 3A and B top) both materials exhibit a porous structure, which is built from interconnected NRs. Additionally, TEM micrographs show the tip-to-tip connection of the single NRs within the gel network, whose thicknesses are 5.6 nm ± 0.9 nm for the CdSe/CdS NRs and 4.7 nm ± 0.5 nm for the CdS NRs. Compared to the initial NRs the thicknesses are slightly decreased, which can be caused by increasing the aging time in the presence of H2O2 resulting in a partial surface oxidation of the NRs.18 The absorption spectra of the CdSe/CdS NR aerogel (Fig. 3A, bottom) and CdS NR aerogel (Fig. 3B, bottom) show a slight hypsochromic shift of the CdS absorption band to 470 nm (CdSe/CdS NRs) and 465 nm (CdS NRs), compared to the NR solution, which can be explained by the change of the dielectric constant of the surrounding media. For both model systems, the particle–particle contact in the aerogel structures results in a bathochromic shift of the emission maxima of around 20 nm. For the CdSe/CdS NR based aerogel a PLQY of 28.2% ± 0.3% was measured. The emission spectrum of the CdS NR aerogel (Fig. 3B, bottom, dashed line) shows an additional trap-state emission in the higher wavelength regime. The PXRD patterns of the CdSe/CdS and CdS NR aerogels (Fig. 3C) show the hexagonal CdS crystal structure. Due to the much lower volume fraction of CdSe seeds within the CdSe/CdS NRs, CdSe reflections are not visible. All cation exchange experiments described in this article were performed on acetogels consisting of CdSe/CdS NRs and CdS NRs.
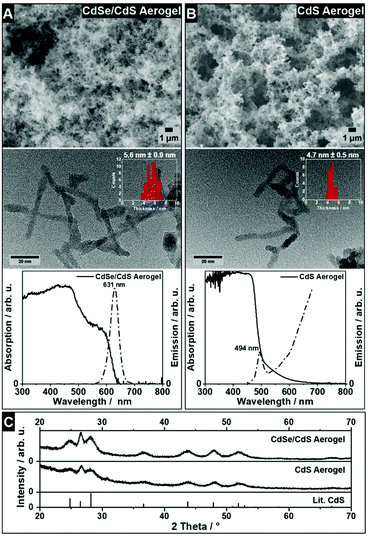 |
| | Fig. 3 SEM micrographs, TEM micrographs and absorption spectra and emission spectra of CdSe/CdS NR aerogel (A) and CdS NR aerogel (B). Absorption spectra and emission spectra are shown by solid lines and dashed lines, respectively. PXRD pattern (C) of CdSe/CdS NRs, CdS NRs and reference XRD pattern of hexagonal CdS (greenockite, ICDD Card No. 01-080-0006). | |
Reversible cation exchange on CdSe/CdS NR and CdS NR based gel networks
The above-described CdSe/CdS NR and CdS NR hydrogels were used as the starting material for cation exchange reactions. Established by recent studies published in the literature for cation exchange reactions at single nanocrystals, the copper chalcogenide phase was used as an intermediate for further cation exchange reactions.31,39–41 To show the reversibility of the cation exchange, the copper chalcogenide acetogels were also exchanged back to CdSe/CdS NR and CdS NR acetogels and heated for several weeks to recover their original fluorescence properties.39 Microscopic characterization techniques (SEM and TEM) were performed in order to analyse the morphological changes in the aerogel structures due to the cation exchange reactions. From the TEM and SEM micrographs (Fig. 4A–D) of the copper chalcogenide aerogels (Fig. 4A and B) and the back-exchanged cadmium chalcogenide aerogels (Fig. 4C and D) it can be seen that the porous gel structure is not affected by the cation exchange. The TEM images of all cation exchanged aerogels show a retained tip-to-tip connection of the single NRs within the porous structures. Furthermore, the shape of the nanorod building blocks within the porous structure is unaltered. The elemental compositions were analysed via energy-dispersive X-ray spectroscopy (EDXS) using scanning electron microscopy and the obtained EDXS spectra are shown in the ESI (Fig. S1†).
 |
| | Fig. 4 SEM (top), TEM micrographs (bottom), and photographs (inset) of the Cu2−xSe/Cu2−xS NR aerogel (A), Cu2−xS NR aerogel (B), back-exchanged CdSe/CdS NR aerogel (C), and back-exchanged CdS NR aerogel (D). | |
To clarify the impact of the cation exchange reaction on the porosity of the aerogel structure, krypton physisorption experiments (Fig. 5) of the copper chalcogenide based aerogels (Fig. 5B Cu2−xSe/Cu2−xS and Fig. 5D Cu2−xS) were conducted and the obtained specific surface areas are compared to those of the initial cadmium-based (Fig. 5A CdSe/CdS and Fig. 5C CdS) aerogel structures. The surface areas derived from the Brunauer–Emmett–Teller (BET) method42 are 98 m2 g−1 (CdSe/CdS) and 75 m2 g−1 (CdS) for the initial aerogel structures. After the cation exchange reaction the copper chalcogenide based aerogels exhibit a surface area of 103 m2 g−1 (Cu2−xSe/Cu2−xS) and 98 m2 g−1 (Cu2−xS). It can be concluded that the cation exchange has no significant impact on the porosity of the aerogel structure according to the BET evaluation. Furthermore, all values are comparable to previously reported surface areas of metal chalcogenide aerogels, which are in the range of 90–240 m2 g−1.5,17,25
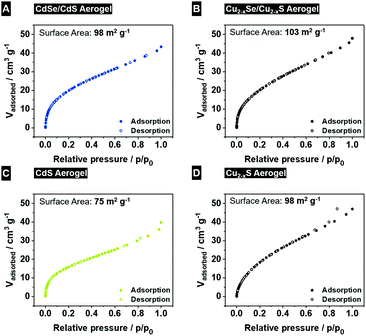 |
| | Fig. 5 Krypton adsorption and desorption curves of initial CdSe/CdS (A) and CdS (C) aerogels, and copper cation exchanged Cu2−xSe/Cu2−xS (B) and Cu2−xS (D) aerogels. | |
Powder X-ray diffraction (PXRD) patterns of the copper chalcogenide NR based aerogels (Cu2−xSe/Cu2−xS and Cu2−xS) and the back-exchanged cadmium chalcogenide NR based aerogels (CdSe/CdS and CdS) are illustrated in Fig. 6. Both copper exchanged aerogels (upper part) show the diffraction pattern of the orthorhombic Cu2S structure (chalcocite), without any reflections of the CdS starting material. The PXRD patterns of the back-exchanged CdSe/CdS and CdS aerogels show reflections of the hexagonal CdS phase (greenockite). Due to the low amount of the CdSe core material compared to the surrounding CdS media, no CdSe reflections can be observed. Besides the hexagonal CdS phase no additional reflections are visible, so that a total back-exchange from 2 Cu+ to Cd2+ can be assumed. Moreover, the results prove that the NR networks are perfectly penetrable and the accessibility of the NC sites is ensured in all systems.
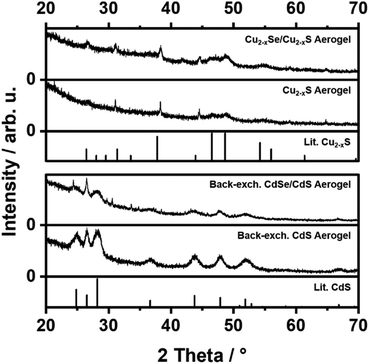 |
| | Fig. 6 PXRD patterns of exchanged Cu2−xSe/Cu2−xS, Cu2−xS, back-exchanged CdSe/CdS and back-exchanged CdS aerogels. Reference PXRD patterns of orthorhombic Cu2−xS (chalcocite, ICDD Card No. 00002-1294) and hexagonal CdS (greenockite, ICDD Card No. 01-080-0006) crystal structures. | |
All cation exchanged aerogels were also characterized by optical spectroscopy (Fig. 7A–C). The absorption spectra of the copper chalcogenide based aerogels (Fig. 7A) show a broad absorption band in the range of 800–2000 nm which can be attributed to the localized surface plasmon resonance (LSPR) of doped copper chalcogenides. The LSPR position depends on the stoichiometric composition of Cu2−xS and Cu2−xSe NPs which is well known from the literature.41,43,44 The absorption spectra of the back-exchanged CdSe/CdS aerogel (Fig. 7B) show the CdSe core absorption at 605 nm and the CdS absorption band at 480 nm. The absorption spectra of the back-exchanged CdS aerogel show the CdS absorption at 477 nm. Due to the heat treatment and the related healing of defects induced by cation exchange the emission was regained. The emission maxima of the back-exchanged CdSe/CdS NR aerogel (λmax = 617 nm, Fig. 7B) and the CdS NR aerogel (λmax = 477 nm, Fig. 7C) are shifted hypsochromically compared to the initial CdSe/CdS NR and CdS NR aerogel (Fig. 3A and B). The shift can be explained by an etching process during the cation exchange experiments, since chloride ion containing precursors were used. The reoccurrence of the photoemission (PLQY of 1.3% ± 0.5% for the back-exchanged CdSe/CdS NR aerogel) is a very good indicator that the back-exchange is a quantitative process since already the smallest amounts of Cu ions incorporated into CdSe/CdS NRs are known to completely quench their PL.
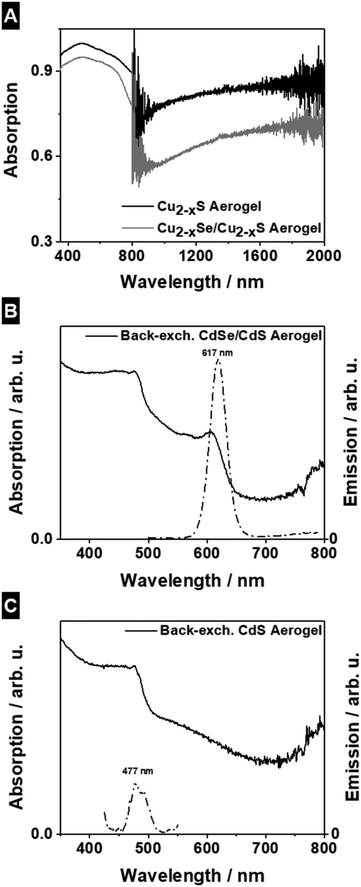 |
| | Fig. 7 Absorption spectra of Cu2−xSe/Cu2−xS and Cu2−xS aerogels (A), absorption- and emission spectra of back-exchanged CdSe/CdS aerogel (B) and back-exchanged CdS aerogel (C). | |
Cation exchange reactions with Pb2+, Ag+ and Hg2+ ions
Copper chalcogenide based lyogels were also used as intermediate states to synthesize other metal (lead, silver, mercury) chalcogenide based aerogels with interesting (opto)electronic properties, like photoluminescence in the NIR range, by cation exchange reactions. Based on recent studies published in the literature for cation exchange reactions at single nanocrystals, Cu2−xSe/Cu2−xS NR based solvogels were used for lead cation exchange reactions to achieve a NIR emitting PbSe/PbS NR based aerogel.39,45 From TEM and SEM micrographs (Fig. 8A and B), it can be seen that the porous macroscopic gel network is not affected by the cation exchange reaction. Furthermore, the TEM image shows a retained nanorod shape and a tip-to-tip connection of the single nanocrystals within the porous structure. The absorbance spectra (Fig. 8C) show a broad band in the NIR range between 1000 nm–1750 nm, which can be attributed to the excitonic absorption of the PbSe core, combined with contributions of the PbS shell.46,47 The emission maximum at around 1540 nm is in good agreement with the band-edge excitonic transitions for PbSe in the range of 0.5–1.1 eV, which can be found in the literature.48–50 The elemental composition of the PbSe/PbS aerogel was analyzed by EDX-SEM and PXRD. The EDX spectra (Fig. 8D) show only elemental lines of Pb, Se, and S, without any contamination of Cu or Cd. Furthermore, the PXRD pattern (Fig. 8E) shows the diffraction pattern of the cubic PbS structure. These findings all strongly indicate a quantitative cation exchange reaction and demonstrate the versatility of our approach and the possibility of accessing new and functional materials like NIR emitting aerogels.
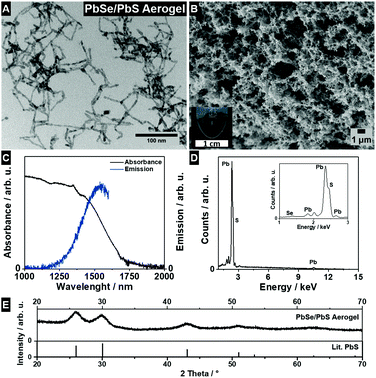 |
| | Fig. 8 TEM micrograph (A), SEM micrograph and photograph (B), absorbance- and emission spectra (C) recorded from PbSe/PbS acetogel, SEM-EDX spectra (D), and PXRD pattern of the PbSe/PbS aerogel (E). The reference PXRD pattern of cubic PbS (galena, ICDD Card No. C5-592). | |
As already mentioned, exchange reactions with silver and mercury cations were also tested. From SEM micrographs (Fig. 9A–D) it can be seen that in all cases the porous structure is retained. The silver chalcogenide aerogels seem to contain Ag nanoparticles as the side product (Fig. 9A and B). On the SEM images of the mercury chalcogenide based aerogels (Fig. 9C and D) large crystals are visible. The diffraction patterns (see ESI Fig. S3†) of the silver chalcogenide aerogels show the reflections of the monoclinic acanthite Ag2S phase combined with reflections from the cubic Ag phase, and the diffraction pattern of mercury chalcogenide aerogels shows the hexagonal HgS phase with additional reflections of Hg2Cl2. TEM images of silver and mercury chalcogenide aerogels show differently shaped nanocrystals compared to the initial nanorod shape within the macroscopic structure and additional nanocrystals with a high contrast, which can be attributed to be pristine Ag nanoparticles, as well as Hg2Cl2 crystals (ESI Fig. S2†). It can be assumed that during the cation exchange from copper to silver or mercury, the replaced Cu+ ions are able to reduce the excess of silver or mercury ions, which are present in solution, resulting in the growth of the side products (Ag NPs and Hg2Cl2).
 |
| | Fig. 9 SEM micrographs and photographs of Ag2Se/Ag2S (A), Ag2S (B), HgSe/HgS (C), and HgS (D) aerogels. | |
In order to understand the silver and mercury exchanges via the copper phase on the macroscale, all cation exchange reactions were also carried out on the nanoscale. Therefore, the pristine NRs were treated with the same synthesis procedure, expect that the reaction time was shortened. The performed cation exchange reactions at pristine nanorod samples (Ag2Se/Ag2S, HgSe/HgS) show nearly the same results (change of the nanocrystal shape or growth of side products) compared to the macroscopic counterparts. The results are shown and discussed in detail in the ESI (Fig. S5†). Here it should be mentioned that the cation exchange reactions were performed using a ten times higher concentration of the starting material and exchange precursors compared to the literature for colloidal dispersed NPs, which can be a reason for the different results. These high concentrations are however necessary to achieve complete ion exchange on the macroscopic gel structures, since these cannot be diluted to the concentration of typical colloidal solutions.
Conclusions
In the present work the limitation of cation diffusion within a macroscopic material was circumvented by using a highly porous and voluminous macroscopic gel network, which was built from nanoscopic CdSe/CdS and CdS NR building blocks. It was shown that via copper ion treatment the initial cadmium-based hydrogel structures can be transferred to copper-based Cu2−xSe/Cu2−xS and Cu2−xS aerogels. In addition, the back-exchange from copper to cadmium ions was investigated to demonstrate the reversibility of the cation exchange process. By heat treatment of the back-exchanged CdSe/CdS and CdS NR aerogels the photoluminescence properties were successfully reclaimed. Regaining the photoluminescence is a good indication for quantitative reversibility of the ion exchange process. SEM, TEM and BET measurements showed that the treatment with different precursor solutions did not affect the porous morphology of the gel networks. First experiments and results based on copper chalcogenide lyogels as the intermediate state and the successful exchange with lead, as well as tested exchanges with silver and mercury ions show that there is a wide range of possible cation exchange reactions on macroscopic gel networks which broadens the accessibility of aerogel materials from these components in future. To conclude, the cation exchange process on macroscopic gel networks opens up new perspectives towards novel classes of aerogels by using a well-known and controllable starting nanoparticle-based gel network.
Experimental section
Chemicals
Cadmium oxide (CdO, 99.998%), methanol (anhydrous, 99.9%), and selenium (Se, 99.999%) were purchased from Alfa Aesar. Bis(trimethylsilyl)sulfide (97%), silver nitrate (99.85%), tri-n-butylphosphine (97%), tri-n-octylphosphine oxide (TOPO, 99%) and tri-n-octylphosphine (TOP, 97%) were purchased from ABCR. Toluene (99.9%) and methanol (≥99.5%) were purchased from Carl Roth. Octadecylphosphonic acid (ODPA, ≥99%), hexylphosphonic acid (HPA, ≥99%), and tributyl phosphine (97%) were purchased from PCI Synthesis. Cadmium nitrate tetrahydrate (99%), hydrogen peroxide (H2O2, 35%), lead acetate trihydrate (>99%), mercaptopropionic acid (MPA, 99%), mercury chloride (99.5%), octadecene (ODE, 90%), potassium hydroxide (KOH, ≥85%), sulfur (S, 99.98%), tetrakis(acetonitrile)copper hexafluorophosphate ([Cu(CH3CN)4]PF6, 97%) were purchased from Sigma Aldrich. Acetone (dried, 99.9%) was purchased from Merck.
Synthesis of CdSe seeds, CdS seeds and growth of the CdS shell
CdSe seeds and CdS seeds syntheses and CdS shell growth followed the method of Carbone et al. with certain modifications.38 For a detailed description of the syntheses see the ESI.†
Phase transfer of CdSe/CdS NRs and CdS NRs
The as-synthesized CdSe/CdS NRs and CdS NRs were transferred to aqueous solution by the method of Kodanek et al.51 For a detailed description of the phase transfer see the ESI.†
Synthesis of acetogels and cation exchange reactions
In the following Experimental section all information before the slash character addresses the synthesis with CdSe/CdS NRs and all information behind the slash character belongs to the synthesis with CdS NRs. All cation exchange experiments take place in the glovebox under an argon or nitrogen atmosphere.
Synthesis of CdSe/CdS NR and CdS NR acetogels
The CdSe/CdS NR and CdS NR acetogels were prepared following the method of Sánchez-Paradinas et al. with certain modifications.18 For a detailed description of the acetogel preparation see the ESI.†
Copper cation exchange
The copper cation exchange was adapted from the method of Miszta et al. for core/shell nanocrystals and applied on CdSe/CdS NR and CdS NR based gel structures with certain modifications.31 A copper exchange solution with a molar ratio of Cd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu 1:10 was used and 65 mg/141.5 mg (0.34 mmol/0.76 mmol) [Cu(CH3CN)4]PF6 was dissolved in 1.5 mL dry methanol. The solution was slowly injected at the edges of the NR based acetogel. After 12 h, the acetogels were washed 5 times with 1.5 mL dry acetone to purify the supernatant solution. Then, again 1.5 mL methanolic copper exchange solution (65 mg/141.5 mg [Cu(CH3CN)4]PF6) was slowly injected to the acetogels and left there for additional 12 h. Subsequently, the acetogels were washed 5 times with 1.5 mL dry methanol to remove excess copper and cadmium cations. Afterwards, the gels were washed 8 times over several hours with 1.5 mL dry acetone and stored overnight. The following day, before the transfer to the critical point dryer, the gels were washed 2 additional times with 1.5 mL dry acetone.
:Cu 1:10 was used and 65 mg/141.5 mg (0.34 mmol/0.76 mmol) [Cu(CH3CN)4]PF6 was dissolved in 1.5 mL dry methanol. The solution was slowly injected at the edges of the NR based acetogel. After 12 h, the acetogels were washed 5 times with 1.5 mL dry acetone to purify the supernatant solution. Then, again 1.5 mL methanolic copper exchange solution (65 mg/141.5 mg [Cu(CH3CN)4]PF6) was slowly injected to the acetogels and left there for additional 12 h. Subsequently, the acetogels were washed 5 times with 1.5 mL dry methanol to remove excess copper and cadmium cations. Afterwards, the gels were washed 8 times over several hours with 1.5 mL dry acetone and stored overnight. The following day, before the transfer to the critical point dryer, the gels were washed 2 additional times with 1.5 mL dry acetone.
Cd cation back exchange
The cadmium ion back-exchange from copper based acetogels was adapted from the method of Jain et al. for cadmium chalcogenide nanocrystals and applied on the CdSe/CdS NR and CdS NR based gel structures with certain modifications.39 For the reversible cation exchange an exchange cation ratio of Cu:Cd 1:100 was used. For the exchange solution 536 mg/1.17 g Cd(NO3)2·4H2O (1.74 mmol/3.79 mmol) and 100 μL TBP were dissolved in 1.5 mL dry methanol. The solution was slowly injected at the edges of the gel and left for 24 h. After that, the gels were washed 5 times with 1.5 mL dry methanol and freshly prepared cadmium exchange solution was injected and left once more for 24 h. The methanol washing step and the replacement of the cadmium cation exchange solution was repeated. After a total reaction time of 96 h the gels were washed 5 times with 1.5 mL dry methanol and over the time of several hours 8 times with 1.5 mL dry acetone and stored overnight. On the subsequent day, the gels were washed again 2 times with 1.5 mL dry acetone. The gels were placed in an oven at 50 °C for 3 weeks in order to complete the cation exchange and to obtain the photoluminescence back.
Lead cation exchange
The cation exchange from Cu to Pb was performed at CdSe/CdS aerogels by following the method of Bouet et al. for core/shell nanocrystals and applied to our acetogel system.45 The atomic ratio from copper to lead was set to 1:47. For the lead cation exchange solution 89.6 mg Pb(Ac)2·3H2O (0.236 mmol) and 67 μL tributyl phosphine were dissolved in 1.5 mL dry methanol. The exchange mixture was replaced every 24 h and after a total time of 168 h the exchanged solvogels were washed 5 times with methanol and 10 times with dry acetone.
Silver and mercury cation exchange
The cation exchanges from Cu to Ag, and Cu to Hg were also realized following the method of Miszta et al. for core/shell nanocrystals and applied to our acetogel system.31 The atomic ratio from copper to silver was set to 1:1.8 and for copper to mercury was set as 1:3. For the silver cation exchange solution 10.5 mg/96.5 mg AgNO3 (0.06 mmol/0.56 mmol), and for mercury cation exchange 35.4 mg/206.2 mg HgCl2 (0.13 mmol/0.76 mmol) were dissolved in 1.5 mL dry methanol. The exchange mixture was replaced every 24 h and after 96 h the exchanged solvogels were washed 5 times with methanol and 10 times with dry acetone.
Preparation of aerogels
After the exchange procedures the washed acetogels were placed in a critical point dryer (E3100, Quorum Technologies) and the acetone phase was substituted with a liquid carbon dioxide phase. Therefore, the critical point dryer was flushed for 10 min with liquid CO2, closed completely and left overnight. After 24 h the critical point dryer was flushed again for 10 min. At least, the temperature of the critical point dryer was increased, so the aerogels were dried at 31.5 °C and 75.8 bar.
Atomic absorption spectroscopy
The Cd concentration of the aqueous CdSe/CdS NR/CdS NR solution was determined by atomic absorption spectroscopy (AAS) using a Varian AA140 instrument supplied with an air/acetylene (1.5:3.5) flame atomizer. The samples were prepared by dissolving defined amounts of CdSe/CdS NRs and CdS NRs in 500 μL fresh prepared aqua regia and diluting them in 50 mL volumetric flasks. Additionally, five Cd2+ solutions with known concentrations were measured to obtain a calibration curve.
Transmission electron microscopy
The morphology of the CdSe/CdS NR/CdS NR in solution and all aerogels were investigated by transmission electron microscopy (TEM) using a FEI Tecnai G2 F20 TMP (CS = 2 mm, CC = 2 mm), equipped with a 200 kV field emission gun. Micrographs were obtained in bright field mode. For grid preparation out of solution, 10 μL of the purified and diluted sample was dropcast on a 300 mesh carbon coated copper grid, purchased from Quantifoil. For grid preparation of the aerogels the grid was pressed slightly to the top of the aerogel.
Scanning electron microscopy
The morphology of the aerogel samples was investigated by scanning electron microscopy (SEM) using a JEOL JSM 6700F instrument equipped with a cold field emission gun electron source. The elementary components of the aerogels were analysed by energy-dispersive X-ray spectroscopy (EDXS) and the software Oxford Instruments INCA 300.
Spectroscopic characterization
UV/Vis absorbance spectra of the solutions were recorded in transmission mode using an Agilent Cary 5000 absorption spectrophotometer. Therefore, 10 μL of the organic NR solution was diluted to 3 mL and measured inside fluorescence quartz cuvettes purchased from Hellma Analytics. The absorption spectra of the copper-based aerogels were recorded in reflection mode using a Cary 5000 spectrophotometer from Agilent as well equipped with an Agilent DRA-2500 integrating sphere. The absorption spectra of back-exchanged CdSe/CdS and CdS aerogels were acquired with a Fluoromax-4 equipped with a Quanta-ϕ integrating sphere (HORIBA). Photoluminescence (PL) emission spectra were obtained using a Horiba Fluoromax-4. Photoluminescence quantum yields (PLQYs, measured in absolute mode by dividing the number of emitted photons by the number of absorbed photons) of the samples were recorded using a Horiba Dual-FL spectrophotometer equipped with a Quanta-ϕ integrating sphere (Horiba). PL emission spectra in the NIR range were obtained using an Edinburgh FLS 1000 spectrometer with an NIR InGaAs PMT detector with a spectral range of 1000–1600 nm.
Krypton physisorption
Krypton physisorption was measured with an Autosorb-1 instrument from Quantachrome operating at 87 K. Prior to physisorption measurements, the samples were degassed under vacuum at room temperature for 24 h. Surface areas were estimated by applying the Brunauer–Emmett–Teller (BET) equation.
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) patterns of the aerogel samples were measured in transmission mode using an STEO StadiP diffractometer with CuKα1 radiation with a wavelength of λ = 1.54060 Å and a Ge(111) monochromator. The powders were ground before the measurements and placed between X-ray amorphous polymer foils. Powder X-ray diffraction patterns of the nanorod solutions were measured in reflection using a Bruker D8 Advance with a CuKα1 source operating at 40 kV and 30 mA. The samples have been prepared by drop-casting a concentrated NR solution on a single crystalline silicon sample holder.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors (N. C. B., F. L.) are grateful for financial support from the German Federal Ministry of Education and Research (BMBF) within the framework of the program NanoMatFutur, support code 03X5525. Furthermore, the project leading to these results has in part received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant agreement no 714429). The author N. C. B. thanks the DFG (research grant BI 1708/4-1). The project has in parts been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the Cluster of Excellence PhoenixD (EXC2122). The author D. D. thanks the DFG (research grant 1580/5-1). The authors thank Prof. Caro and Prof. Feldhoff for access to a scanning electron microscope and powder X-ray diffraction measurements, as well as the Laboratorium of Nano- and Quantum Engineering for support. M. S. and M. S. thank the Hannover School for Nanotechnology for financial support.
Notes and references
- J. Puskelova, L. Baia, A. Vulpoi, M. Baia, M. Antoniadou, V. Dracopoulos, E. Stathatos, K. Gabor, Z. Pap, V. Danciu and P. Lianos, Chem. Eng. J., 2014, 242, 96–101 CrossRef CAS.
- F. Rechberger and M. Niederberger, Mater. Horiz., 2017, 4, 1115–1121 RSC.
- L. Korala, J. R. Germain, E. Chen, I. R. Pala, D. Li and S. L. Brock, Inorg. Chem. Front., 2017, 4, 1451–1457 RSC.
- B. J. Riley, J. Chun, W. Um, W. C. Lepry, J. Matyas, M. J. Olszta, X. Li, K. Polychronopoulou and M. G. Kanatzidis, Environ. Sci. Technol., 2013, 47, 7540–7547 CrossRef CAS.
- Q. Yao and S. L. Brock, Nanotechnology, 2010, 21, 115502 CrossRef.
- A. Schlosser, L. C. Meyer, F. Lübkemann, J. F. Miethe and N. C. Bigall, Phys. Chem. Chem. Phys., 2019, 21, 9002–9012 RSC.
- J. L. Mohanan, I. U. Arachchige and S. L. Brock, Science, 2005, 307, 393–397 CrossRef.
- S. L. Brock, I. U. Arachchige and K. K. Kalebaila, Comments Inorg. Chem., 2006, 27, 103–126 CrossRef CAS.
- I. R. Pala, I. U. Arachchige, D. G. Georgiev and S. L. Brock, Angew. Chem., Int. Ed., 2010, 49, 3661–3665 CrossRef CAS PubMed.
- N. Gaponik, A. Wolf, R. Marx, V. Lesnyak, K. Schilling and A. Eychmüller, Adv. Mater., 2008, 20, 4257–4262 CrossRef CAS.
- N. C. Bigall, A.-K. Herrmann, M. Vogel, M. Rose, P. Simon, W. Carrillo-Cabrera, D. Dorfs, S. Kaskel, N. Gaponik and A. Eychmüller, Angew. Chem., Int. Ed., 2009, 48, 9731–9734 CrossRef CAS PubMed.
- X. Gao, R. J. Esteves, T. T. H. Luong, R. Jaini and I. U. Arachchige, J. Am. Chem. Soc., 2014, 136, 7993–8002 CrossRef CAS.
- K. G. S. Ranmohotti, X. Gao and I. U. Arachchige, Chem. Mater., 2013, 25, 3528–3534 CrossRef CAS.
- D. Wen, W. Liu, D. Haubold, C. Zhu, M. Oschatz, M. Holzschuh, A. Wolf, F. Simon, S. Kaskel and A. Eychmüller, ACS Nano, 2016, 10, 2559–2567 CrossRef CAS PubMed.
- S. Naskar, A. Freytag, J. Deutsch, N. Wendt, P. Behrens, A. Köckritz and N. C. Bigall, Chem. Mater., 2017, 29, 9208–9217 CrossRef CAS.
- M. Zareie Yazdan-Abad, M. Noroozifar, A. R. Modaresi Alam and H. Saravani, J. Mater. Chem. A, 2017, 5, 10244–10249 RSC.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2006, 128, 7964–7971 CrossRef CAS.
- S. Sánchez-Paradinas, D. Dorfs, S. Friebe, A. Freytag, A. Wolf and N. C. Bigall, Adv. Mater., 2015, 27, 6152–6156 CrossRef PubMed.
- S. Naskar, J. F. Miethe, S. Sánchez-Paradinas, N. Schmidt, K. Kanthasamy, P. Behrens, H. Pfnür and N. C. Bigall, Chem. Mater., 2016, 28, 2089–2099 CrossRef CAS.
- T. Gacoin, L. Malier, J. Boilot and P. Cedex, J. Mater. Chem., 1997, 7, 859–860 RSC.
- Q. Yao and S. L. Brock, Inorg. Chem., 2011, 50, 9985–9992 CrossRef CAS.
- I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2007, 129, 1840–1841 CrossRef CAS PubMed.
- A.-K. Herrmann, P. Formanek, L. Borchardt, M. Klose, L. Giebeler, J. Eckert, S. Kaskel, N. Gaponik and A. Eychmüller, Chem. Mater., 2014, 26, 1074–1083 CrossRef CAS.
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- P. Rusch, F. Niemeyer, D. Pluta, B. Schremmer, F. Lübkemann, M. Rosebrock, M. Schäfer, M. Jahns, P. Behrens and N. C. Bigall, Nanoscale, 2019, 11, 15270–15278 RSC.
- W. Liu, A. K. Herrmann, N. C. Bigall, P. Rodriguez, D. Wen, M. Oezaslan, T. J. Schmidt, N. Gaponik and A. Eychmüller, Acc. Chem. Res., 2015, 48, 154–162 CrossRef CAS PubMed.
- W. Liu, P. Rodriguez, L. Borchardt, A. Foelske, J. Yuan, A. K. Herrmann, D. Geiger, Z. Zheng, S. Kaskel, N. Gaponik, R. Kötz, T. J. Schmidt and A. Eychmüller, Angew. Chem., Int. Ed., 2013, 52, 9849–9852 CrossRef CAS PubMed.
- F. Lübkemann, J. F. Miethe, F. Steinbach, P. Rusch, A. Schlosser, D. Zámbó, T. Heinemeyer, D. Natke, D. Zok, D. Dorfs and N. C. Bigall, Small, 2019, 15, 1970212 CrossRef.
- A. Freytag, C. Günnemann, S. Naskar, S. Hamid, F. Lübkemann, D. Bahnemann and N. C. Bigall, ACS Appl. Nano Mater., 2018, 1, 6123–6130 CrossRef CAS PubMed.
- G. D. Moon, S. Ko, Y. Min, J. Zeng, Y. Xia and U. Jeong, Nano Today, 2011, 6, 186–203 CrossRef CAS.
- K. Miszta, G. Gariano, R. Brescia, S. Marras, F. De Donato, S. Ghosh, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2015, 137, 12195–12198 CrossRef CAS PubMed.
- S. Gupta, S. V. Kershaw and A. L. Rogach, Adv. Mater., 2013, 25, 6923–6944 CrossRef CAS PubMed.
- B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759–19770 CrossRef CAS.
- P. Adel, A. Wolf, T. Kodanek and D. Dorfs, Chem. Mater., 2014, 26, 3121–3127 CrossRef CAS.
- A. Wolf, L. Diestel, F. Lübkemann, T. Kodanek, T. Mohamed, J. Caro and D. Dorfs, Chem. Mater., 2016, 28, 7511–7518 CrossRef CAS.
- E. M. Chan, M. A. Marcus, S. Fakra, M. ElNaggar, R. A. Mathies and A. P. Alivisatos, J. Phys. Chem. A, 2007, 111, 12210–12215 CrossRef CAS PubMed.
- Q. Yao, I. U. Arachchige and S. L. Brock, J. Am. Chem. Soc., 2009, 131, 2800–2801 CrossRef CAS PubMed.
- L. Carbone, C. Nobile, M. De Giorgi, F. Della Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- P. K. Jain, L. Amirav, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 9997–9999 CrossRef CAS PubMed.
- B. Sadtler, D. O. Demchenko, H. Zheng, S. M. Hughes, M. G. Merkle, U. Dahmen, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 5285–5293 CrossRef CAS PubMed.
- D. Dorfs, T. Hartling, K. Miszta, N. C. Bigall, M. R. Kim, A. Genovese, A. Falqui, M. Povia and L. Manna, J. Am. Chem. Soc., 2011, 133, 11175–11180 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- Y. Xie, A. Riedinger, M. Prato, A. Casu, A. Genovese, P. Guardia, S. Sottini, C. Sangregorio, K. Miszta, S. Ghosh, T. Pellegrino and L. Manna, J. Am. Chem. Soc., 2013, 135, 17630–17637 CrossRef CAS PubMed.
- J. M. Luther, P. K. Jain, T. Ewers and A. P. Alivisatos, Nat. Mater., 2011, 10, 361–366 CrossRef CAS PubMed.
- C. Bouet, D. Laufer, B. Mahler, B. Nadal, H. Heuclin, S. Pedetti, G. Patriarche and B. Dubertret, Chem. Mater., 2014, 26, 3002–3008 CrossRef CAS.
- A. Sashchiuk, L. Amirav, M. Bashouti, M. Krueger, U. Sivan and E. Lifshitz, Nano Lett., 2004, 4, 159–165 CrossRef CAS.
- P. K. Jain, L. Amirav, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2010, 132, 9997–9999 CrossRef CAS PubMed.
- A. D. Andreev and A. A. Lipovskii, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15402–15404 CrossRef CAS.
- B. L. Wehrenberg, C. Wang and P. Guyot-Sionnest, J. Phys. Chem. B, 2002, 106, 10634–10640 CrossRef CAS.
- H. Du, C. Chen, R. Krishnan, T. D. Krauss, J. M. Harbold, F. W. Wise, M. G. Thomas and J. Silcox, Nano Lett., 2002, 2, 1321–1324 CrossRef CAS.
- T. Kodanek, H. M. Banbela, S. Naskar, P. Adel, N. C. Bigall and D. Dorfs, Nanoscale, 2015, 7, 19300–19309 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: EDX spectra and further XRD diffractograms. See DOI: 10.1039/c9nr09875e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Peter
Behrens
c,
Peter
Behrens