
Revisiting Pt/TiO2 photocatalysts for thermally assisted photocatalytic reduction of CO2†
 b
Xintong
Zhang
*a
and
b
Xintong
Zhang
*a
and
Abstract
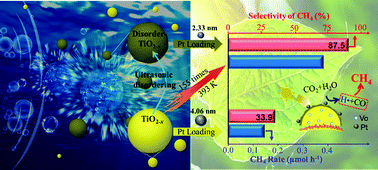
Artificial photosynthesis by a semiconductor–oxide-based photocatalysis is presently challenging due to low CO2 conversion rates and poor product selectivity. To promote CO2 reduction, Pt/TiO2 has been deemed as a classic photocatalyst. In this study, we restudy Pt/TiO2 for the thermally assisted photocatalytic reduction of CO2 and reveal a different story between photocatalysis and photothermal catalysis. For example, when using disordered Pt/TiO2−x, the CO2 conversion via photocatalysis at 298 K is not impressive. However, when the system temperature is increased to 393 K, the CO2 conversion rate is significantly enhanced by a factor of 155 as compared to that obtainable from pristine TiO2; further, surprisingly high selectivity of CH4 (87.5%) could be achieved. Thermally coupled photocatalysis yields the enhanced evolution of H2 side products over Pt (4.06 nm)/TiO2 and promoted H2 splitting over Pt (2.33 nm)/TiO2, which is seldom observed in conventional Pt/TiO2 photocatalysis. The synergy of improved charge separation at the Pt/TiO2−x interface induced by surface disordering and accelerated H2 consumption near smaller Pt nanoparticles by thermal assistance are believed to be critically important for the simultaneous enhancement of CO2 conversion rates and CH4 product selectivity. This study inspires revisiting not only Pt/TiO2 but also reactivating other semiconductor–oxide-based photocatalysts for use in thermally assisted photocatalysis.
 Please wait while we load your content...
Please wait while we load your content...

