
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative characterisation of non-monodisperse gold nanoparticle populations by X-ray scattering and electron microscopy†
Ye
Yang
 a,
Suiyang
Liao
b,
Zhi
Luo
b,
Runzhang
Qi
a,
Niamh
Mac Fhionnlaoich
a,
Francesco
Stellacci
bc and
Stefan
Guldin
*a
a,
Suiyang
Liao
b,
Zhi
Luo
b,
Runzhang
Qi
a,
Niamh
Mac Fhionnlaoich
a,
Francesco
Stellacci
bc and
Stefan
Guldin
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: s.guldin@ucl.ac.uk
bInstitute of Materials, École Polytechnique Fédérale de Lausanne (EPFL), Station 12, 1015 Lausanne, Switzerland
cInterfaculty Bioengineering Institute, École Polytechnique Fédérale de Lausanne (EPFL), Station 12, 1015 Lausanne, Switzerland
First published on 4th May 2020
Abstract
Accurate nanoparticle size determination is essential across various research domains, with many functionalities in nanoscience and biomedical research being size-dependent. Although electron microscopy is capable of resolving a single particle down to the sub-nm scale, the reliable representation of entire populations is plagued by challenges in providing statistical significance, suboptimal preparation procedures and operator bias. While alternative techniques exist that provide ensemble information in solution, their implementation is generally challenging for non-monodisperse populations. Herein, we explore the use of small-angle X-ray scattering in combination with form-free Monte Carlo fitting of scattering profiles as an alternative to conventional electron microscopy imaging in providing access to any type of core size distribution. We report on a cross-method comparison for quasi-monodisperse, polydisperse and bimodal gold nanoparticles of 2–7 nm in diameter and discuss advantages and limitations of both techniques.
1 Introduction
The preparation and accurate characterisation of sub-10 nm nanoparticles (NPs) plays a pivotal role in a multitude of chemical and biomedical applications, where size-dependent efficacy is often observed.1–3 Notably, the cutoff for efficient renal clearance is below 10 nm, and NPs with broad size distributions may impair biocompatibility.4 For gold nanoparticles (AuNPs), in particular, various important physicochemical properties are closely dependent on size. AuNP populations with mean diameters ranging from 2–10 nm manifest drastic differences not only concerning in vitro colloidal stability but also their intracellular and antimicrobial properties.5–8 Consequently, the quantitative assessment of the AuNP size distribution in a sample of interest is an indispensable routine that needs special attention.The most commonly implemented technique is transmission electron microscopy (TEM) imaging, combined with a plethora of software-based image analysis methods.9,10 As a direct imaging technique, TEM is a convenient tool to study both size and shape in ultra-high resolution. However, obtaining an accurate and representative size distribution of the AuNPs by TEM is challenged by limited sample counts, preparation procedures and operator bias in image acquisition and analysis.11–13 Considering the rather limited sample size, it is laborious to obtain results of statistical significance, especially for non-uniform or multimodal samples. Moreover, the drop casting for TEM sample preparation often results in drying artifacts, which vastly complicates image analysis.14,15 Even though fitting procedures with minimal human intervention meanwhile exist, image analysis in practice still commonly involves manual operations prone to user bias.10
To address these issues, a number of bulk-scale characterisation techniques for NPs have been developed as alternatives to TEM imaging, many of which manifest convincing advantages when assessing monodisperse NP populations. However, the analysis of non-uniform size distributions remains plagued by inherent weaknesses. For instance, dynamic light scattering (DLS) is able to probe the hydrodynamic size information of colloidal systems at both microscopic and nanoscopic scales, but non-monodisperse samples are typically not accurately described due to the size-dependent scattering cross-section.11,16 In addition, the interference of multiple scattering events may also impair the accuracy of DLS results, especially for NPs below 20 nm in diameter.17 X-ray diffraction (XRD) analysis enables to obtain the mean size of crystalline domains, either via the Scherrer formula or the more recent Fourier inversion method.18,19 However, this approach is hindered by issues such as the overweighting of larger crystallites as well as contributions from amorphous layers and lattice defects.14,20 Analytical ultracentrifugation (AUC) represent an alternative route to size and shape characterisation of NPs in solution.21 2D evaluation of sedimentation and diffusion coefficients offers pathways to representing non-uniform NP populations.22 However, in order to deduce accurate core size information from a hydrodynamic radius, additional information is required on the ligand shell and respective solvation layers, which is at times difficult to obtain. Two further techniques particularly suited for non-uniform NP populations are disc centrifugation analysis (DCA) and nanoparticle tracking analysis (NTA). Although DCA can track multimodal particle samples, it suffers from sizing underestimation as well as a strict requirement for well-matched calibrants.17,23 NTA allows to probe single NP in an ensemble through the mapping of individual Brownian trajectories.24 Nevertheless, it time-consuming to obtain large enough datasets for an accurate representation of NP populations is unable to resolve sub-10 nm NPs.17 Ultra-small nanoclusters may further be sized by 2D diffusion-ordered nuclear magnetic resonance spectroscopy (DOSY-NMR) via comparing the specific NMR signal peak of surface-bound ligands.25–27 In this case, size distribution analysis of non-uniform populations is particularly challenging due to ligand density fluctuation and peak line broadening, which results in altered resonance and chemical shift.28
An emerging technique to characterise the size distribution of colloidal ensembles is small-angle X-ray scattering (SAXS). Modern SAXS instruments are able to cover the NP diameter distribution ranging from 1–100 nm. The scattering length density (SLD) of gold is particularly pronounced in comparison to organic ligands and common solvents. This distinct feature of gold gives rise to significant contrast of the NP core and an excellent signal-to-noise ratio for resulting SAXS profiles. In principle, the size distribution of a NP sample may be calculated with an assumed functional form, i.e. lognormal, Gaussian, Boltzmann or Schultz–Zimm distribution.12 In a recent study on sub-5 nm hybrid NPs, SAXS enabled the simultaneous resolution of both core and shell, i.e. the size distribution of the silica core as well as the molecular mass dispersity of the polymer shell.29 In order to represent an arbitrary size distribution, a form-free regularisation approach is required, with the most widely adopted ones based on indirect Fourier transform, either involving form factor pattern matching and optimisation with least-squares methods.30–32 Nonetheless, the existing parametric regularisation methods are sensitive to externally derived boundary conditions such as the maximum diameter, which may incurr artificial oscillations of the pair distribution function.33 Consequently, a Monte Carlo (MC) method, based on model-free trial-and-error sampling, was proposed for unbiased NP size distribution analysis.34,35 Pioneered by Martelli and Di Nunzio and later refined by Pauw and co-workers, this MC method demonstrated comparable sizing capabilities to the established indirect Fourier transform, structure interference and maximum entropy methods.34–37 The recent development of the user-friendly software McSAS has further broadened the scope of this approach.38,39 In a cross-lab collaboration of unimodal AgNPs, the parametric fitting and the MC method led to consistent and similar results, highlighting the validity and reliability of employing the MC approach.40 The capabilities of the MC method in comparison to fitting to a presupposed model were further confirmed in a study of unimodal PbS nanocrystals.41 Despite these encouraging results, there remains a lack of comprehensive investigations utilising the MC-SAXS method, especially for characterising non-uniform NPs below 10 nm.
To this end, we present a systematic study of sizing quasi-monodisperse, polydisperse and bimodal thiol-capped AuNPs of 2–7 nm in diameter by conventional TEM and solution-based SAXS. The two characterisation techniques are based on fundamentally different working principles, i.e. single-particle direct imaging vs. ensemble scattering. We present feasible routes to obtain experimental data from both methods and provide a side-by-side comparison of the size distribution obtained for a variety of samples. By doing so, we aim to shed light on the general applicability of SAXS with MC fitting (MC-SAXS) for accurate NP size determination alongside a critical identification of its limitations.
2 Results and discussion
Homo-ligand AuNPs stabilised by 11-mercapto-1-undecanesulfonate (MUS) were prepared via thiol ligand exchange from four batches of differently-sized AuNPs that were initially protected by oleylamine (OAm).42 The core diameter of the four batches ranged from 2–7 nm and varying degrees of size dispersity (ESI, Table S1†). Two binary mixtures were prepared by mixing MUS-NP1 and MUS-NP4 at weight ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:5, labelled as MUS-B1 and MUS-B2, respectively (Table S2†).
:1 and 1:5, labelled as MUS-B1 and MUS-B2, respectively (Table S2†).
2.1 TEM imaging
TEM diameter histograms of MUS-NP1 and MUS-NP4 from three separate experimental sessions for both sample preparation and imaging are shown in Fig. 1 alongside a normalised overlay and representative micrographs. The count rate for each individual analysis was >2000. The micrographs for the rest of samples can be found in the ESI (Fig. S1†).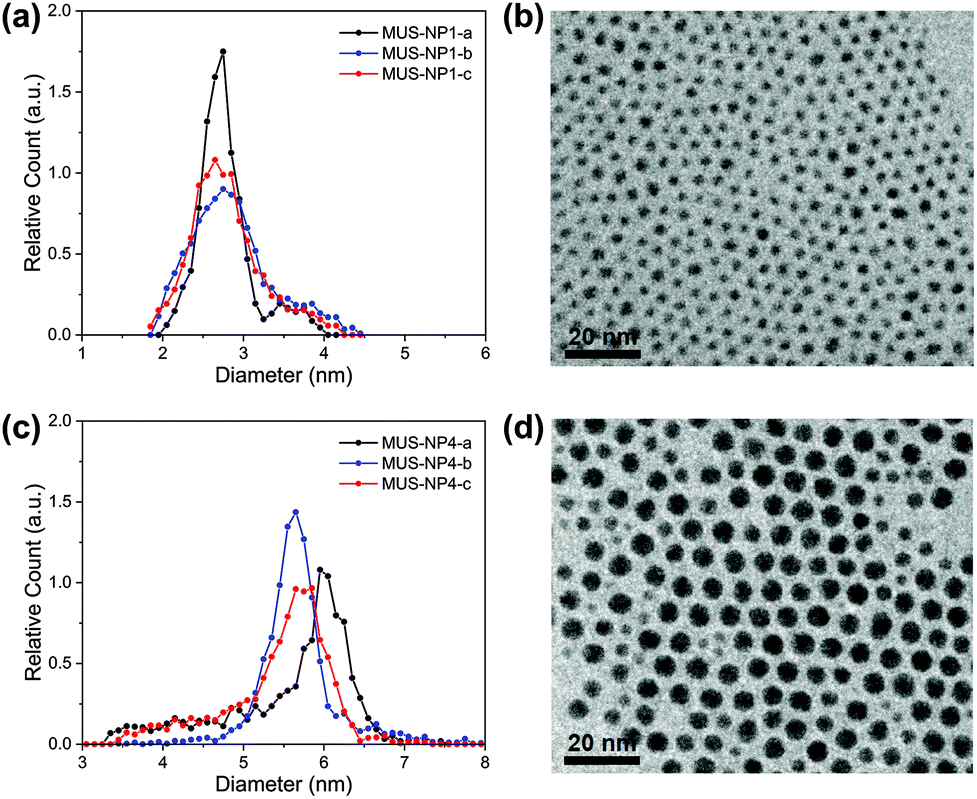 | ||
| Fig. 1 Diameter distribution histograms, normalised overlays and representative TEM image of (a) and (b) MUS-NP1 and (c) and (d) MUS-NP4. Three histograms were obtained per batch of AuNPs in three separate experiments (sample preparation and acquisition) from the identical sample solution. | ||
The mean size and standard deviation of MUS-NP1 were 2.8 ± 0.3, 2.9 ± 0.5 and 2.8 ± 0.4 for the three respective analyses with corresponding coefficient of variance of 12.4%, 17.7% and 15.9%, assuming a normal distribution (see ESI, Fig. S2a†). These significant differences are also depicted in the normalised overlays of the respective histograms, shown in Fig. 1b. A similar comparative analysis of the respective TEM experiments is presented for MUS-NP4 in Fig. 1c and d.
Further evidence for the disparity of the three sub-populations is provided by direct statistical comparison, namely the t-test, the Kolmogorov–Smirnov test and the information entropy test. The t-test assumes that both sub-populations follow a normal distribution with equal variance and determines whether the two groups have the same mean, i.e. originate from the same parental population. The corresponding p-values of the pairwise comparison are shown in Table S4 (ESI†), with the null hypothesis clearly rejected for all couples besides MUS-NP1-a and MUS-NP1-c, i.e. there were significant differences detected between the respective sub-populations. The more generic two-sample Kolmogorov–Smirnov test was also applied to determine whether the respective pairs of sub-populations follow the same distribution. The obtained p-values (Table S5†) were orders of magnitude below the significance level of 5%, suggesting that none of the sub-population would, from a statistical perspective, originate from the same AuNP population. We have recently introduced the concept of nanoparticle entropy, which provides a consistent and reliable comparative measure for the dispersity of particle populations.43 Herein, the normalised entropy value was 0.46, 0.69 and 0.63 for NP1-a, NP1-b, and NP1-c, respectively (Table S6†). This disparity is significant and could allow a population to be incorrectly identified as quasi-monodisperse (threshold En < 0.618). Importantly, the sample size tested for each population was sufficient to calculate a reliable estimate of the NP entropy; however, the variation between samples observed herein suggests that even count rates of more than 2000 AuNPs are insufficient to represent entire populations.
It is important to note that a sole reporting of mean values and standard deviation represents a simplification and is not suitable for size populations that deviate from normal distributions.44 The MUS-NP4 sub-populations displayed herein cannot be described by a normal distribution (ESI, Fig. S2b†). Nevertheless, significant differences are also apparent from qualitative comparison, most notably by the lack of a shoulder towards smaller AuNPs in MUS-NP4-b compared to MUS-NP4-a and MUS-NP4-c. Furthermore, the position of the main peak shifted from 5.6 nm to 6.0 nm between the three measurements. In contrast to other statistical measures, the normalised nanoparticle entropy can be calculated for any type of population regardless of its distribution. Herein, values of 0.49, 0.30 and 0.44 were obtained for NP4-a, NP4-b, and NP4-c, respectively (Table S6†), providing further evidence for the disparity between the sub-populations.
Overall these results indicate that the variation in sample preparation and analysis is statistically significant. These findings are in agreement with earlier studies, emphasising on the limitations of TEM size analysis when based on the conventional practice of sample preparation and image analysis applied herein.45,46
2.2 MC-SAXS analysis
The SAXS profiles of both single-type samples and binary mixtures were measured in aqueous solutions. For a better estimation of the form factor of individual AuNPs, 10 mM NaCl was added to increase the ionic strength of the medium and thus screen any medium- and long-range interaction between AuNPs. SAXS curves after background subtraction are presented as log–log plots in Fig. 2 for 5 mg ml−1 single-type solutions and 10 mg ml−1 binary mixtures, respectively. Qualitatively, both single-type and biomodal measured curves demonstrated pronounced form oscillations in the high q region and featureless flat profiles for q < 1 nm−1.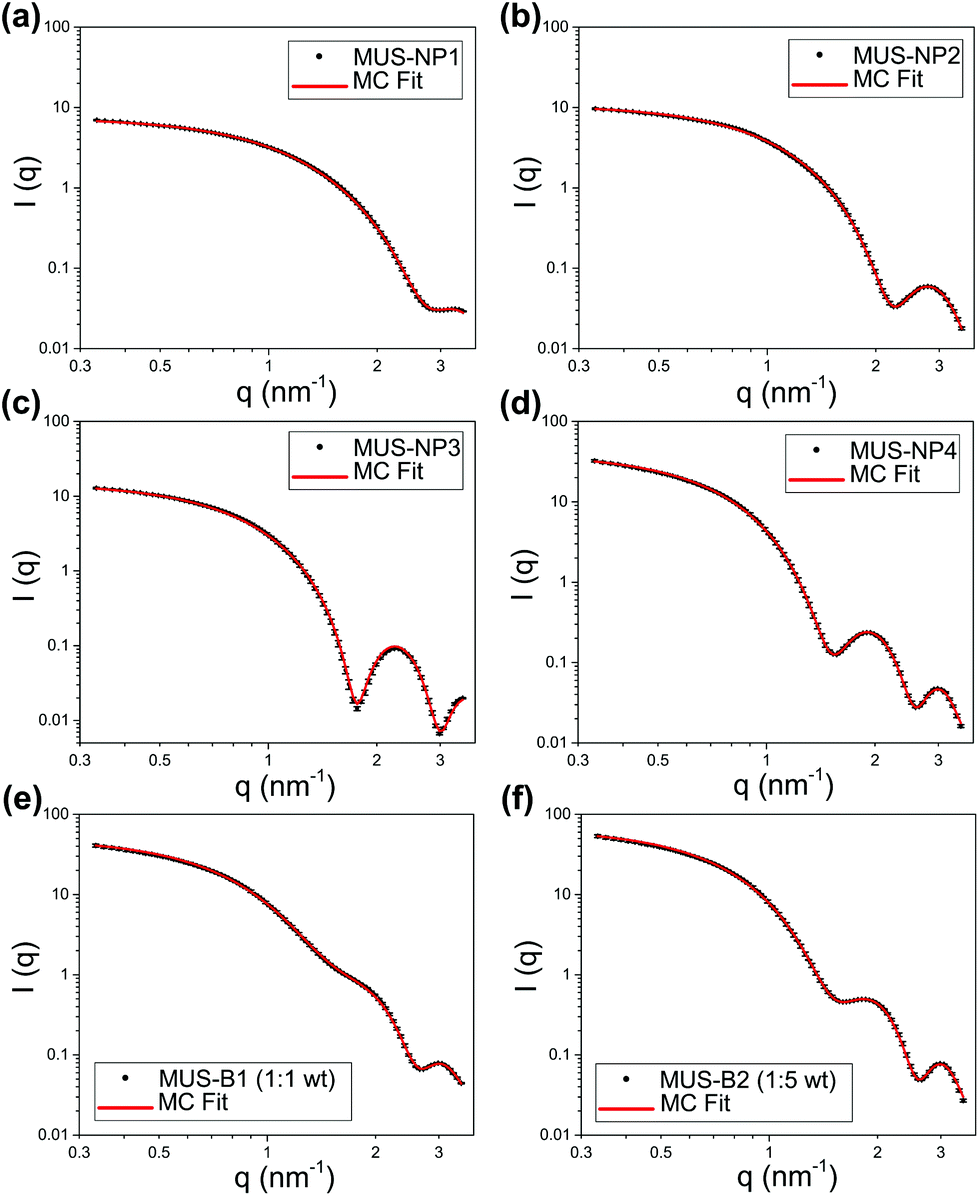 | ||
| Fig. 2 SAXS profiles and MC fitting (log–log plots) of (a) 5 mg ml−1 MUS-NP1, (b) 5 mg ml−1 MUS-NP2, (c) 5 mg ml−1 MUS-NP3, (d) 5 mg ml−1 MUS-NP4, (e) 10 mg ml−1 MUS-B1 (1:1 wt) and (f) 10 mg ml−1 MUS-B2 (1:5 wt). | ||
For single-type samples, the form oscillation in the high q region was at 3.2, 2.8, 2.2 and 1.9 nm−1 for MUS-NP1–NP4, respectively (Fig. 2a–d). This clear shift towards lower angles corresponds to a size increase of the scatterer, which was in line with the TEM results. Meanwhile, the curves of the binary mixtures followed closely the pattern overlapping of individual form oscillations observed in single-type samples (Fig. 2e and f).
Both, the date fitting of single-type and binary mixtures indicate that effective scattering occurred from individual AuNPs with non-interacting contributions, which we relate to three major factors: (1) the distinct SLD of gold gave rise to pronounced form factors in diluted solutions, (2) the ionic repulsion by MUS maintained excellent colloidal stability and prevented NP clustering or aggregation, (3) the addition of 10 mM NaCl resulted in charge screening, which disrupted medium- and long-range NP-NP ionic interaction. It is important to note that these features were key prerequisites for the implementation of MC fitting analysis. A summary of statistical information obtained from 10 number-weighted output distributions by independent MC fitting repetitions for each sample is shown in the ESI (Table S7†). Note the closely matched fitting curves and the negligible discrepancies between individual runs. These results indicate that the herein presented MC-SAXS method provides robust resultsfor non-monodisperse and bimodal AuNP populations with minimal external information.
2.3 Comparative studies
Fig. 3 summarizes the diameter distribution histograms obtained from TEM imaging and MC-SAXS for the single-type samples MUS-NP1 to MUS-NP4, containing AuNPs ranging from 2–7 nm. Some important characteristics emerge from a direct comparison.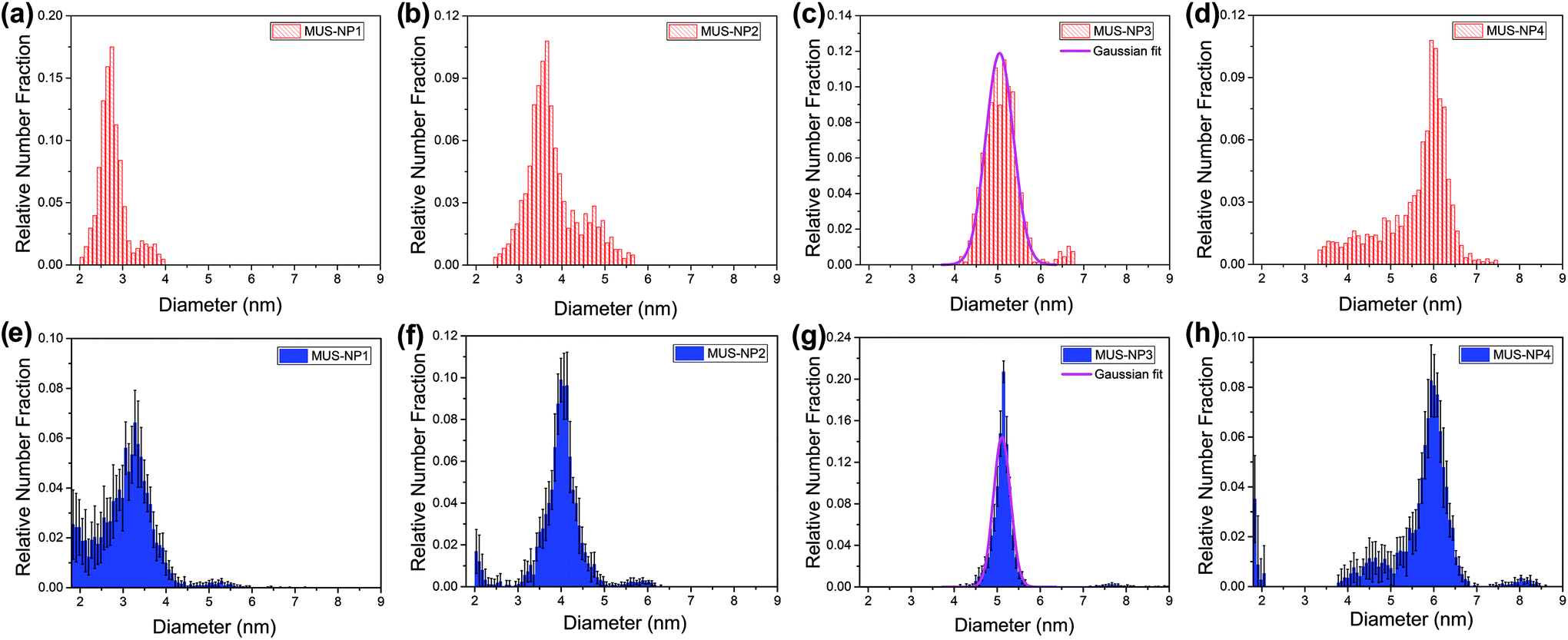 | ||
| Fig. 3 Diameter distribution analysis of single-type samples by TEM (red) and SAXS (blue): (a) and (e) MUS-NP1, (b) and (f) MUS-NP2, (c) and (g) MUS-NP3, (d) and (h) MUS-NP4. | ||
As evidenced by the Gaussian fit plotted alongside Fig. 3d and g, the results obtained for MUS-NP3 by both techniques follow a normal distribution, thus enabling to reliably compare mean diameter and standard deviation from statistical analysis. The obtained mean diameter was remarkably similar, with 5.0 nm and 5.1 nm calculated for TEM imaging and MC-SAXS, respectively. The spread of counts was broader in TEM analysis with a standard deviation of 0.3 nm in comparison to 0.2 nm obtained for MC-SAXS. The corresponding coefficient of variance was calculated alongside with 6.8% and 3.9%, respectively.
The shape and position of the main peak was found rather consistent for all AuNP populations across both techniques and within the observed fluctuations observed in TEM analysis of individual runs (see Fig. 1) The existence of shoulders or minority populations was found more pronounced in TEM than in MC-SAXS, but both techniques detected some degree of dispersity. For AuNPs below 3 nm, MC-SAXS provided a less defined spread and higher error values.
These trends were also observed in the binary mixtures MUS-B1 and MUS-B2, as shown in Fig. 4. A comparison of the data to expected simulated histograms via superposition of the single-type results can be found in the ESI (Fig. S3†).
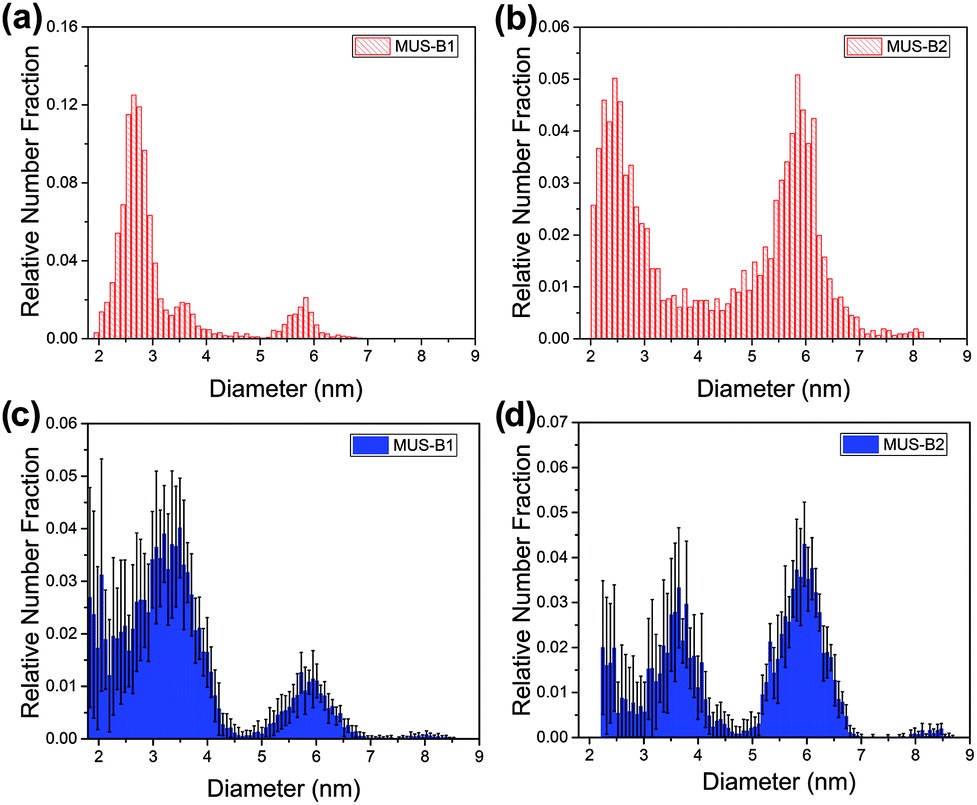 | ||
| Fig. 4 Diameter distribution analysis of binary mixtures by TEM (red) and SAXS (blue): (a) and (c) MUS-B1 (1:1 wt), (b) and (d) MUS-B2 (1:5 wt). | ||
The following discussion aims to systematically examine the advantages and limitations of each technique. In all samples, the variation between individual characterisation runs for the identical sample was observed to be significantly higher for TEM compared to SAXS. This may be related to the fact that (1) TEM is a local imaging technique, which carries limited statistical representation by examining only a small fraction of the whole sample population, and (2) the inspection of single particles and the experimenter-guided workflow of sample preparation, imaging and data analysis can entail errors from operator bias.12,13 Furthermore, the commonly used TEM procedures require the removal of the suspending liquid after drop-casting. This process is typically realised by vacuum or ambient drying, which often alters the dispersion state of sample materials and introduces artifacts obscuring accurate measurement.15 Furthermore, thresholding for image contrast may lead to an underestimation of the smaller-sized NPs as the outer part of smaller NPs with poor contrast is typically filtered when applying an overall threshold in common image analysis routines. All these factors generate discrepancies for representing the diameter distribution, which can impair comparable and reproducible data analysis.46 On the other hand, the diameter distribution observed in TEM results suggests a broader distribution, i.e. a higher dispersity, most notably in MUS-NP2–NP4. It is important to note that in contrast to SAXS, TEM measures individual NPs, and thus minority size fractions are equally counted and represented, offering an intrinsic advantage for arbitrary populations. In spite of several drawbacks, TEM still offers a number of advantages including accessibility and ease of data analysis, thus providing a rapid characterisation of AuNP core sizes and shapes, with a semi-quantitative estimation of the degree of homogeneity.14
By contrast, SAXS is an ensemble method which provides collective data in solution that can be used for detailed statistical analysis of in situ colloidal features. Due to the distinct X-ray SLD of the gold core and similar SLD values between alkanethiols and solvent molecules in a AuNP sample, SAXS permits the selective characterisation of the gold core while excluding the influence of the ligand shell and the solvation layer. Importantly, the histograms obtained by MC-SAXS as an ensemble technique display a similar level of detail to the ones obtained by TEM analysis, highlighting the potential of the form-free MC modelling, which extends the use of SAXS for non-monodisperse NP systems. It is important to note that the average diameter in SAXS histograms was slightly larger than those obtained by TEM for NP populations with significant fractions below 3 nm, notably MUS-NP1, MUS-B1 and MUS-B2. These findings are in agreement with the published results on unimodal PbS nanocrystals, where larger average diameter of 3.2 nm for the MC method in comparison to 2.8 nm when fitting the identical SAXS profile with a classical log-normal-based model.41 This limitation of the MC method for sub-3 nm NPs may be predominantly attributed to the diminishing scattering intensity in the high q region (as described by Porod's law), which entails significant data noise.12 Once more, this is in line with the PbS study, in which the scattering of unbound lead oleate molecules resulted in a secondary population at 2.3 nm in their MC fitting results.41 Consequently, the presented MC-SAXS is generally not suitable for characterising ultra-small AuNPs below 3 nm. Moreover, when considering the limitations in sample-to-detector distance of SAXS instrumentation as well as the reduced beam flux at the low q region, the MC-SAXS method may not provide accurate estimation of NPs above 100 nm in diameter without the use of ultra-small angle X-ray scattering (USAXS).
3 Conclusions
In summary, a comparative study of sub-10 nm AuNP diameter distribution characterised by TEM and SAXS is presented. Both methods provide consistent assessment of quasi-monodisperse AuNPs with an average core diameter of 5 nm, but distinct differences were observed for non-monodisperse and bimodal populations. The conventional TEM imaging method permitted facile characterisation of AuNPs with various size distributions, but it suffered from poor reproducibility and a lack of statistical significance. SAXS measurement, combined with subsequent model-free MC fitting, enabled reliable estimation of non-monodisperse size distributions with comprehensive statistical analysis. However, this method is not suitable for ultra-small NPs below 3 nm due to the reduced scattering intensity and the interference of small molecules. This study offers valuable insights into state-of-art AuNP characterisation methods and may guide the size distribution analysis on other material systems.4 Experimental section
4.1 Preparation of gold nanoparticles
The MUS ligand and MUS-AuNP syntheses via thiol-for-OAm ligand exchange were carried out following previously published procedures.42,47 Four batches of MUS-AuNPs weresynthesised with varying reaction temperatures at 40, 25, 15, 10 °C and labelled as MUS-NP1–NP4, respectively. We refer to the ESI† for experimental details.4.2 Transmission electron microscopy characterisation
The TEM samples were prepared by dipping a sample grid (Holey Carbon Film on Copper 400 mesh, EM Resolutions) into AuNPs solutions in dichloromethane (DCM). TEM images were acquired with a high-resolution JEM-2100 TEM system (200 keV, JEOL). The size distribution was determined by automated image analysis of the respective AuNP populations (count rates > 2000) using the software ImageJ. The size and circularity threshold were set as >2 nm2 and >0.6, respectively. To assess the reproducibility of TEM analysis, three individual sample grids were prepared from each of MUS-NP1 and MUS-NP4 sample solutions. Subsequent TEM imaging was performed in three separate experimental sessions.4.3 Small-angle X-ray scattering measurements and Monte Carlo fitting
Samples for solution-based SAXS were prepared in 10 mM NaCl aqueous solution at a concentration of 5 mg ml−1 for MUS-NP1–4 and 10 mg ml−1 for MUS-B1–B2. The SAXS measurements were carried out using a Ganesha 300XL (SAXSLAB) at 20 °C under vacuum with a high brilliance microfocus Cu-source (wavelength: 1.5418 Å). The SAXS data were recorded on a Pilatus 300 K solid-state photon-counting detector with a 2 mm beam stop for 1 h (q-range: 0.15 to 6.5 nm−1). After subtracting the scattering from the 10 mM NaCl buffer solution, the SAXS data were fed into the software McSAS (version 1.3) for size distribution analysis.38 In McSAS, the fitting of each SAXS dataset consisted of 10 individual repetitions with strict fitting criteria, namely a convergence criterion of χ2 < 1 and a minimum uncertainty estimate of 2%. Sphere model was chosen as the fit model and the ΔSLD (= SLCAu − SLDH2O) was input as 115.5 × 10–6 Å−2. Number-weighted histograms were generated in the post-fit analysis. The number of bins was set to be 100 in a size range of 1.8–9.0 nm in diameter.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 633635 (DIACHEMO). YY acknowledges the support by the UCL Overseas Research Scholarship and the Graduate Research Scholarship as well as Dr Han Wu and the EPSRC CNIE research facility service (EPSRC Award, EP/K038656/1) at UCL for the SAXS instrument access. NMF expresses gratitude for funding by the EPSRC under a Doctoral Training Partnership (EP/M507970/1). The authors are grateful to Dr Paulo Jacob Silva (EPFL) for providing MUS ligand.References
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- G. Bodelón, C. Costas, J. Pérez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzán, Nano Today, 2017, 13, 40–60 CrossRef.
- A. A. Burns, J. Vider, H. Ow, E. Herz, O. Penate-Medina, M. Baumgart, S. M. Larson, U. Wiesner and M. Bradbury, Nano Lett., 2009, 9, 442–448 CrossRef CAS PubMed.
- A. A. Sousa, S. A. Hassan, L. L. Knittel, A. Balbo, M. A. Aronova, P. H. Brown, P. Schuck and R. D. Leapman, Nanoscale, 2016, 8, 6577–6588 RSC.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS PubMed.
- Y. Jiang, S. Huo, T. Mizuhara, R. Das, Y.-W. Lee, S. Hou, D. F. Moyano, B. Duncan, X.-J. Liang and V. M. Rotello, ACS Nano, 2015, 9, 9986–9993 CrossRef CAS PubMed.
- T. J. Macdonald, K. Wu, S. K. Sehmi, S. Noimark, W. J. Peveler, H. du Toit, N. H. Voelcker, E. Allan, A. J. MacRobert, A. Gavriilidis and I. P. Parkin, Sci. Rep., 2016, 6, 39272 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- S. Mondini, A. M. Ferretti, A. Puglisi and A. Ponti, Nanoscale, 2012, 4, 5356–5372 RSC.
- E. J. Cho, H. Holback, K. C. Liu, S. A. Abouelmagd, J. Park and Y. Yeo, Mol. Pharm., 2013, 10, 2093–2110 CrossRef CAS PubMed.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128–11180 CrossRef CAS PubMed.
- M. M. Modena, B. Rühle, T. P. Burg and S. Wuttke, Adv. Mater., 2019, 1901556 CrossRef PubMed.
- H. Borchert, E. V. Shevchenko, A. Robert, I. Mekis, A. Kornowski, G. Grübel and H. Weller, Langmuir, 2005, 21, 1931–1936 CrossRef CAS PubMed.
- B. Michen, C. Geers, D. Vanhecke, C. Endes, B. Rothen-Rutishauser, S. Balog and A. Petri-Fink, Sci. Rep., 2015, 5, 9793 CrossRef CAS PubMed.
- Y. Dieckmann, H. Cölfen, H. Hofmann and A. Petri-Fink, Anal. Chem., 2009, 81, 3889–3895 CrossRef CAS PubMed.
- D. Mahl, J. Diendorf, W. Meyer-Zaika and M. Epple, Colloids Surf., A, 2011, 377, 386–392 CrossRef CAS.
- D. Zanchet, B. D. Hall and D. Ugarte, J. Phys. Chem. B, 2000, 104, 11013–11018 CrossRef CAS.
- B. D. Hall, D. Zanchet and D. Ugarte, J. Appl. Crystallogr., 2000, 33, 1335–1341 CrossRef CAS.
- S. Calvin, S. X. Luo, C. Caragianis-Broadbridge, J. K. McGuinness, E. Anderson, A. Lehman, K. H. Wee, S. A. Morrison and L. K. Kurihara, Appl. Phys. Lett., 2005, 87, 233102 CrossRef.
- K. L. Planken and H. Cölfen, Nanoscale, 2010, 2, 1849–1869 RSC.
- R. P. Carney, J. Y. Kim, H. Qian, R. Jin, H. Mehenni, F. Stellacci and O. M. Bakr, Nat. Commun., 2011, 2, 335 CrossRef PubMed.
- P. Bowen, J. Dispersion Sci. Technol., 2002, 23, 631–662 CrossRef CAS.
- I. Montes-Burgos, D. Walczyk, P. Hole, J. Smith, I. Lynch and K. Dawson, J. Nanopart. Res., 2010, 12, 47–53 CrossRef.
- R. H. Terrill, T. A. Postlethwaite, C.-h. Chen, C.-D. Poon, A. Terzis, A. Chen, J. E. Hutchison, M. R. Clark and G. Wignall, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- G. Canzi, A. A. Mrse and C. P. Kubiak, J. Phys. Chem. C, 2011, 115, 7972–7978 CrossRef CAS.
- K. Salorinne, T. Lahtinen, J. Koivisto, E. Kalenius, M. Nissinen, M. Pettersson and H. Häkkinen, Anal. Chem., 2013, 85, 3489–3492 CrossRef CAS PubMed.
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721–2739 CrossRef CAS.
- K. P. Barteau, K. Ma, F. F. E. Kohle, T. C. Gardinier, P. A. Beaucage, R. E. Gillilan and U. Wiesner, Chem. Mater., 2019, 31, 643–657 CrossRef CAS PubMed.
- O. Glatter, J. Appl. Crystallogr., 1977, 10, 415–421 CrossRef.
- D. I. Svergun, J. Appl. Crystallogr., 1991, 24, 485–492 CrossRef CAS.
- J. S. Pedersen, J. Appl. Crystallogr., 1994, 27, 595–608 CrossRef.
- S. Hansen and J. S. Pedersen, J. Appl. Crystallogr., 1991, 24, 541–548 CrossRef.
- S. Martelli and P. E. Di Nunzio, Part. Part. Syst. Charact., 2002, 19, 247–255 CrossRef CAS.
- B. R. Pauw, J. S. Pedersen, S. Tardif, M. Takata and B. B. Iversen, J. Appl. Crystallogr., 2013, 46, 365–371 CrossRef CAS PubMed.
- P. E. D. Nunzio, S. Martelli and R. R. Bitti, J. Dispersion Sci. Technol., 2005, 25, 491–501 CrossRef.
- J. M. Rosalie and B. R. Pauw, Acta Mater., 2014, 66, 150–162 CrossRef CAS.
- I. Bressler, B. R. Pauw and A. F. Thünemann, J. Appl. Crystallogr., 2015, 48, 962–969 CrossRef CAS PubMed.
- V. Geertsen, E. Barruet, F. Gobeaux, J.-L. Lacour and O. Taché, Anal. Chem., 2018, 90, 9742–9750 CrossRef CAS PubMed.
- B. R. Pauw, C. Kästner and A. F. Thünemann, J. Appl. Crystallogr., 2017, 50, 1280–1288 CrossRef CAS PubMed.
- J. Maes, N. Castro, K. De Nolf, W. Walravens, B. Abécassis and Z. Hens, Chem. Mater., 2018, 30, 3952–3962 CrossRef CAS.
- Y. Yang, L. A. Serrano and S. Guldin, Langmuir, 2018, 34, 6820–6826 CrossRef CAS PubMed.
- N. Mac Fhionnlaoich and S. Guldin, Chem. Mater. DOI:10.1021/acs.chemmater.0c00539.
- C. J. Murphy and J. M. Buriak, Chem. Mater., 2015, 27, 4911–4913 CrossRef CAS.
- R. F. Domingos, M. A. Baalousha, Y. Ju-Nam, M. M. Reid, N. Tufenkji, J. R. Lead, G. G. Leppard and K. J. Wilkinson, Environ. Sci. Technol., 2009, 43, 7277–7284 CrossRef CAS PubMed.
- Z. H. Chen, C. Kim, X.-b. Zeng, S. H. Hwang, J. Jang and G. Ungar, Langmuir, 2012, 28, 15350–15361 CrossRef CAS PubMed.
- R. P. Carney, Y. Astier, T. M. Carney, K. Voïtchovsky, P. H. J. Silva and F. Stellacci, ACS Nano, 2013, 7, 932–942 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C9NR09481D |
| This journal is © The Royal Society of Chemistry 2020 |