
Enhanced sodium storage kinetics by volume regulation and surface engineering via rationally designed hierarchical porous FeP@C/rGO†

Abstract
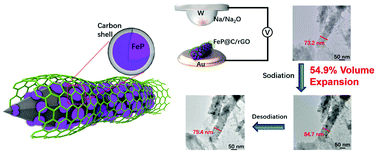
Transition metal phosphides, such as iron phosphide (FeP), have been considered as promising anode candidates for high-performance sodium ion batteries (SIBs) owing to their high theoretical capacity. However, the development of FeP is limited by large volume change, low electrical conductivity and sluggish kinetics with sodium ions. Moreover, the sodium storage kinetics and dynamics behavior in FeP are still unclear. Herein, improved sodium storage ability of FeP is achieved by volume regulation and surface engineering via a rationally designed hierarchical porous FeP@C/rGO nanocomposite. This FeP@C/rGO nanocomposite exhibits excellent rate capability and long cycle life as the anode of SIBs. Specifically, the FeP@C/rGO nanocomposite delivers high specific capacities of 635.7 and 343.1 mA h g−1 at 20 and 2000 mA g−1, respectively, and stable cycling with 88.2% capacity retention after 1000 cycles. The kinetics and dynamics studies demonstrate that the superior performance is attributed to the rationally designed hierarchical porous FeP@C/rGO with a high capacitive contribution of 93.9% (at 2 mV s−1) and a small volume expansion of only 54.9% by in situ transmission electron microscopy (TEM) measurement. This work provides valuable insights into understanding the phase evolution of FeP during the sodiation/desodiation process for designing high-performance SIBs.
- This article is part of the themed collection: Nanoscale 2021 Lunar New Year Collection
 Please wait while we load your content...
Please wait while we load your content...

