
Penetration of the blood–brain barrier and the anti-tumour effect of a novel PLGA-lysoGM1/DOX micelle drug delivery system†
 *ad
*ad
Abstract
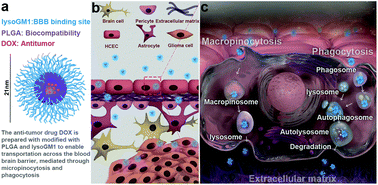
Effective treatment of glioma and other central nervous system (CNS) diseases is hindered by the presence of the blood–brain barrier (BBB). A novel nano-delivery vehicle system composed of PLGA-lysoGM1/DOX micelles was developed to cross the BBB for CNS treatment. We have shown that doxorubicin (DOX) as a model drug encapsulated in PLGA-lysoGM1 micelles can achieve up to 3.8% loading efficiency and 61.6% encapsulation efficiency by the orthogonal test design. Our in vitro experiments demonstrated that PLGA-lysoGM1/DOX micelles had a slow and sustainable drug release under physiological conditions and exhibited a high cellular uptake through the macropinocytosis and the autophagy/lysosomal pathways. In vivo experimental studies in zebrafish and mice confirmed that PLGA-lysoGM1/DOX micelles could cross the BBB and be specifically accumulated in the brain. Moreover, an excellent anti-glioma effect was observed in intracranial glioma-bearing rats. Therefore, PLGA-lysoGM1/DOX micelles not only effectively can cross the BBB, but our results also suggest that they have great potential for anti-glioma therapy and other central nervous system diseases.
- This article is part of the themed collections: Editor’s Choice: Recent breakthroughs in nanobiotechnology and 2020 Nanoscale HOT Article Collection
 Please wait while we load your content...
Please wait while we load your content...

