
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrushing the surface: cascade reactions between immobilized nanoreactors†
Dalin
Wu
,
Serena
Rigo
,
Stefano
Di Leone
,
Andrea
Belluati
 ,
Edwin C.
Constable
,
Catherine E.
Housecroft
* and
Cornelia G.
Palivan
*
,
Edwin C.
Constable
,
Catherine E.
Housecroft
* and
Cornelia G.
Palivan
*
Department of Chemistry, University of Basel, BPR 1096, Mattenstrasse 24a, 4058 Basel, Switzerland. E-mail: catherine.housecroft@unibas.ch; cornelia.palivan@unibas.ch
First published on 26th November 2019
Abstract
Functionalization of hard or soft surfaces with, for example, ligands, enzymes or proteins, is an effective and practical methodology for the development of new applications. We report the assembly of two types of nanoreactors based upon poly(dimethylsiloxane)-block-poly(2-methyl-2-oxazoline) (PDMS-b-PMOXA) diblock copolymers as scaffold, uricase and lactoperoxidase as bio-catalysts located within the nanoreactors, and melittin as the biopores inserted into the hydrophobic shell. The nanoreactors were immobilized on poly(2-hydroxyethyl methacrylate)-co-poly(2-aminoethyl methacrylate hydrochloride) (PHEMA-co-P(2-AEMA·HCl) brushes-grafted wafer surfaces by utilizing the strong supramolecular interactions between biotin and streptavidin. The (PHEMA-co-P(2-AEMA·HCl) brushes on silicon surfaces were prepared by a surface initiating atom transfer radical polymerization (ATRP) “graft-from” technique. Cascade reactions between different surface-anchored nanoreactors were demonstrated by converting Amplex® Red to the fluorescent probe resorufin by using the H2O2 produced from uric acid and H2O. The detailed properties of the nanoreactors on the functionalized surface including the binding behaviours and cascade reactions were investigated using emission spectroscopy, transmission electron microscopy (TEM), light scattering (LS), atomic force microscopy (AFM) and a quartz crystal microbalance (QCM-D). The results are proof-of-principle for the preparation of catalytically functional engineered surface materials and lay the foundation for applying this advanced functional surface material in biosensing, implanting and antimicrobial materials preparation.
Introduction
Grafting of advanced functionalities onto hard (e.g. titania nanoparticles) or soft (e.g. polymer) surfaces is a critical and practical methodology achieving applications ranging from implanting,1 sensing,2 dye-sensitized solar cells3 to self-sterilizing systems.4 The development of surface functionalization is currently receiving significant research attention.5–8 Such functionalization typically originates from the immobilization of specific molecular species such as ligands and catalysts,9,10 biomolecules (peptides, sugars or proteins),8,11 polymers (thermo-responsive and pH responsive polymers)12,13 and various nano/micro-objects (inorganic nano/micro particles,14 liposomes15 or polymeric nano-objects16). In order to optimize the surface functionalities, several critical factors should be considered. Firstly, the stability of the functionalities is critical, especially when these are catalytic biomolecules because of fast degradation which may occur outside a biological environment.17 Secondly, the strength of surface–functionality interactions depends upon the choice of method by which functionalization is carried out, for example, through polymerization, covalent or hydrogen-bond formation, or ion–ion or host–guest interactions. However, whereas, for example, covalent bond formation leads to stronger surface–functionality binding than hydrogen-bond interactions, the former may require more demanding synthetic procedures.18,19 A third criterion to be considered where applications are in vivo is the biocompatibility of the surface itself.20Utilizing polymeric nano-objects, such as micelles and polymersomes to modify a surface is considered to be an advanced strategy for surface functionalization16 Compared with lipids, polymer-derived nano-objects exhibit greater thermodynamic stability leading to the functionalized surfaces having longer lifetimes and activities.21 The higher molecular weights of polymers compared to lipids militate against the uncontrollable leaking of small molecules from within the nano-comparts; this is a known problem with lipid nano-containers.22 Moreover, polymer-based nano-objects have the advantage of structural versatility of their components allowing variations in responsive properties, as well as permitting the incorporation of a wide variety of functional end groups.23–25
Polymer nano-compartments serve to create nanoreactors as ideal candidates for the development of reactions at the nanoscale by encapsulation of active molecules inside.21,26,27 The reactions take place inside the cavities of the compartments, where the active molecules are protected from the environment and perform their activity.21,26 The membrane can be intrinsically porous28 or can be generated by various approaches: use of responsive polymers,29 insertion of membrane proteins,21,27 treatment with membrane-permeabilizing peptides30 and chemical agents.31 Nanoreactors are essential for continuous production of active molecules within a spatially discrete aqueous environment by catalytic reactions. Products are able to diffuse out of the nanoreactor through the pores in the hydrophobic membrane.27 The ability of the polymer membrane to isolate encapsulated enzymes or catalysts is highly advantageous, preventing degradation or oxidation, and resulting in systems with extended periods of activity.23 Among others, Langowska et al. and Zhang et al. have reported penicillin acylase and ribitol dehydrogenase (RDH) encapsulated nanoreactors which were immobilized on a solid surface by amidation. The functional surfaces were applied as antibacterial materials and sugar-sensor chips.2,21 Although several systems comprising polymer-based nanoreactors immobilized on solid surfaces with various functionalities have been described,2,21,32 to the best of our knowledge, the immobilization of two different polymer-based nanoreactors, which can facilitate cascade reactions on the same surface, has not been demonstrated. The benefits of immobilizing two types of nanoreactors are: (1) to allow a tandem cascade reaction to happen on the surface and (2) to permit each reaction step to occur in a separate nanocompartment. In addition, direct immobilization of enzymes on polymer brushes as a complementary approach to perform reactions on surfaces and prepare catalytic functional surface materials may increase the amount of enzyme involved in the reaction. The use of immobilized nanoreactors has the advantage of protecting the biomolecules from detrimental environmental conditions, thus prolonging their activity.33
Immobilizing nano-objects on solid surfaces such as wafers has been widely reported.34–36 However, anchoring them on polymer brushes (a soft surface) is less well investigated. Polymer brushes are capable of providing a fluidic surface, thereby increasing the binding efficiency. Simultaneously, the biocompatibility of the surface can be greatly improved by using biocompatible polymers.35 Various methods have been applied to prepare polymer brushes on solid surfaces, for example, “graft-to” and “graft-from”.36 Of these two methodologies, the “graft-from” method has the advantage of favouring the formation of a high density of polymer brushes. The application of the reversible-deactivation radical polymerization, for example atom transfer radical polymerization (ATRP) on “graft-from” provides the polymer brushes with polydispersity values close to 1, and in comparison with traditional radical polymerization products, it gives relatively well defined molecular structures and abundant functional groups on the polymer chains.34–41
Here, we report tandem cascade reactions happening by product transfer between two types of nanoreactors which are immobilized on a polymeric soft surface. The advantage of nanoreactors working in tandem instead of co-encapsulation of different enzymes in one nanoreactor, is based on a simpler manner to control the ratio between the enzymes performing the cascade reaction and an increase in the encapsulation efficiency. We assembled two types of nanoreactors using poly(dimethylsiloxane)-block-poly(2-methyl-2-oxazoline) (PDMS-b-PMOXA) diblock copolymers as scaffold and melittin inserted into the membrane as a pore-former. The two distinct nanoreactors encapsulate uricase and lactoperoxidase (LPO), respectively. These two enzyme-encapsulated nanoreactors allow the tandem cascade reaction to occur in solution.42 Nanoreactors were immobilized on the poly(2-hydroxyethyl methacrylate)-co-poly(2-aminoethyl methacrylate hydrochloride) (PHEMA-co-P(2-AEMA·HCl) grafted wafer surface by supramolecular interactions between biotin and streptavidin. The choice of PHEMA as a hydrophilic and biocompatible polymer is based upon its applications in biomedical research.43 Grafting PHEMA onto the surface improves the biocompatibility of the surface, making it potentially applicable in vivo, for example in implanting materials and sensing chips. The interaction between biotin and streptavidin with real-time binding constants44 in the range 3.0 × 106 and 4.5 × 107 M−1 s−1 is regarded as one of the strongest intermolecular interactions in nature allowing it to serve as a highly efficient link between the nanoreactors and the PHEMA-co-P(2-AEMA·HCl) grafted surface. Critically, the protein streptavidin offers four receptor sites for biotin.45 Thus, once bound to the biotin-modified surface, streptavidin still retains additional free biotin-binding sites (Scheme 1). We now demonstrate a cascade reaction between the products of uricase encapsulated nanoreactors (uricase-nanoreactors) and LPO encapsulated nanoreactors (LPO-nanoreactors) (Scheme 2).
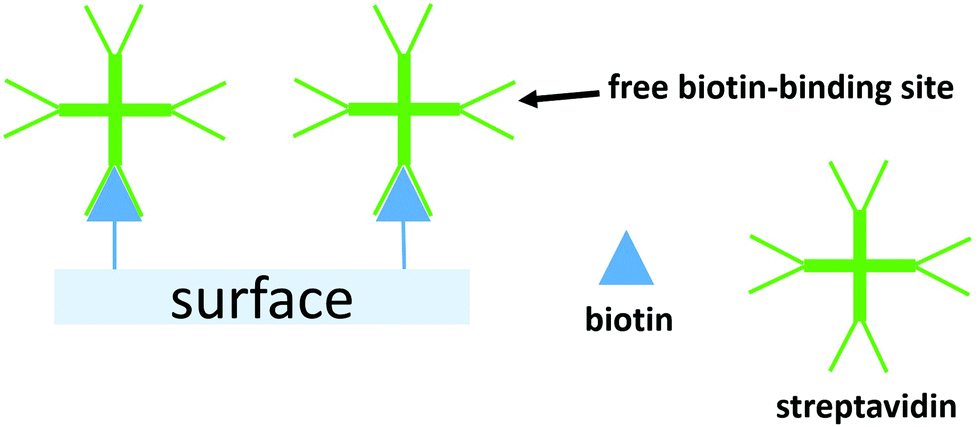 | ||
| Scheme 1 A schematic representation of a biotin-modified surface binding streptavidin and the free biotin-binding sites which remain. | ||
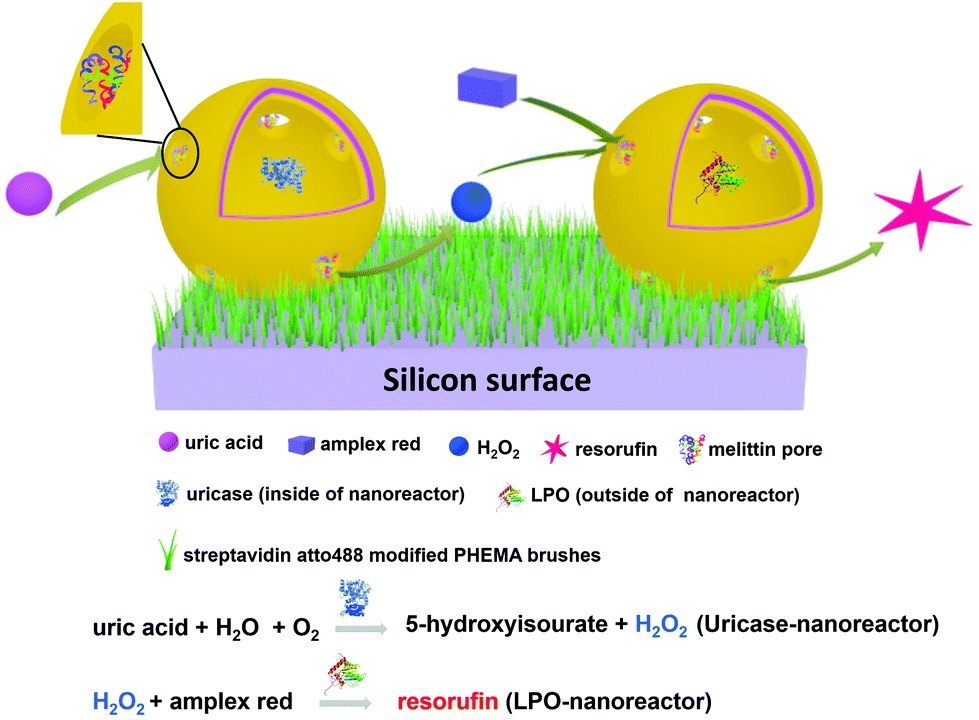 | ||
| Scheme 2 A schematic representation of the cascade reaction involving uricase encapsulated nanoreactors and LPO encapsulated nanoreactors. | ||
Experimental
Materials and general
Chemicals used are detailed in the ESI.† 2-Bromo-2-methyl-N-[3-(triethoxysilyl)propyl]propanamide37 and biotin N-hydroxysuccinimide ester38 were synthesized according to the published procedure. All experiments were performed at room temperature (r.t.) which was ca. 22 °C.Instrumental methods
1H NMR spectra were recorded at 298 K on a Bruker Avance III-500 NMR spectrometer. 1H NMR chemical shifts were referenced to residual solvent peaks with respect to δ(TMS) = 0 ppm. Details of the contact angle measurements, ellipsometry, atomic force microscopy (AFM), confocal scanning laser microscopy (CSLM), fluorescence correlation spectroscopy (FCS), transmission electron microscopy (TEM), dynamic and static light scattering (DLS and SLS), fluorescence spectroscopy, quartz crystal microbalance (QCM) with dissipation (QCM-D) are given in the ESI.†Preparation of atom transfer radical polymerization (ATRP) initiator modified surfaces
The surfaces of silicon wafers or quartz crystal microbalance chips (QCM chips) were first treated in an oxygen plasma (Plasma leaner, Harrick Plasma) for 10 min. After this, the active silicon wafer surface was functionalized by immersion in anhydrous toluene (40 mL) containing 2-bromo-2-methyl-N-[3-(triethoxysilyl)propyl]propanamide (100 μL) followed by shaking (50 rpm) overnight at r.t. Subsequently, the surfaces were washed with ethanol and dried under a gentle stream of air.Preparation of PHEMA-co-P(2-AEMA·HCl) modified surfaces
The ATRP initiator modified surfaces were first transferred into a Schlenk tube. Then, MeOH (10 mL), Millipore water (10 mL), 2-hydroxyethyl methacrylate (HEMA) (1 mL, 8.22 mmol), 2-aminoethyl methacrylate hydrochloride (100 mg, 0.60 mmol), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (87 μL, 0.54 mmol), CuBr2 (10 mg, 0.04 mmol) and CuBr (50 mg, 0.35 mmol) were added to the Schlenk tube in this order. The polymerization medium was degassed by bubbling argon through it for 30 min while cooling in an ice-bath before being sealed. Finally, the surface-initiating ATRP reaction was started by heating to 50 °C under an argon atmosphere. The polymerization time was 18 h. The PHEMA-co-P(2-AEMA·HCl) modified silicon wafers or chips were washed with toluene, acetone, H2O and ethanol before being dried under a gentle stream of air.Preparation of biotin modified surfaces
The PHEMA-co-P(2-AEMA·HCl) modified silicon wafers or QCM chips were first immersed in anhydrous DMSO (15 mL), then biotin N-hydroxysuccinimide ester (biotin-NHS, 15 mg, 0.04 mmol) and Et3N (10 μL, 0.07 mmol) were added to the DMSO solution. The solution was shaken (50 rpm) for two days at r.t. Finally, the biotin-modified silicon wafers or chips (biotin-surface) were washed with H2O and ethanol before being dried under a gentle stream of air.Preparation of streptavidin modified surfaces
The biotin-modified silicon wafers or QCM chips were immersed in streptavidin atto488 (40 μg mL−1) phosphate-buffered saline (PBS) solution (pH 7.3). The solution was then shaken (50 rpm) for two days at r.t. in the dark. Finally, the streptavidin atto488 modified surfaces (streptavidin-surface) were carefully washed with PBS and stored at 4 °C before use.Functional amphiphilic block copolymers: synthesis and characterization
PDMS21-PMOXA7-OH and PDMS28-PMOXA10-biotin diblock copolymers were synthesized according to a previously published procedure.46 The molecular weights, block ratio and chemical structures were characterized by 1H NMR spectroscopy. The averaged molecular weight and polydispersity index (PDI) were characterized by GPC (Polymer Standard Services, Germany) with PS beads as the solid phase and CHCl3 as the running phase (1.0 mL min−1 flow rate). The GPC traces were recorded using a refractive index detector.Biotin modified polymersomes (biotin-polymersomes): preparation
The biotin–polymersomes were prepared by the published film rehydration method.46 PDMS21-PMOXA7-OH (4 mg, 1.78 μmol) and PDMS28-PMOXA10-biotin (0.2 mg, 0.07 μmol) diblock copolymers were dissolved in EtOH (1 mL) in a 5 mL round bottomed flask. The solvent was then removed slowly under vacuum to form a polymer film on the bottom of flask. Afterwards, 1 mL of PBS buffer was added and the solution was further stirred by stirring bar at 300 rpm for 12 hours at r.t. The above biotin-modified polymersome solution was further homogenized by extrusion through a 200 nm cut-off membrane and purified by size extrusion chromatography with sepharose 4B (45–165 μM bead diameter) as solid phase and PBS as the running phase before use.Uricase-nanoreactors and LPO-nanoreactors preparation
First, stock solutions of enzyme (uricase or lactoperoxidase) (0.25 mg mL−1) and melittin (1 mM) in 1 mL of PBS were prepared. At the same time, the PDMS-b-PMOXA diblock copolymer film with the same constitution as the biotin-polymersomes (last section) were prepared under the same conditions as described above. Afterwards, 1 mL of enzyme (0.25 mg mL−1) and 60 μL of melittin (1 mM) PBS solution were added one after the other. The solution was further stirred (300 rpm) for 12 h at r.t. Finally, the nanoreactors were extruded through a 200 nm cut-off membrane and purified by size extrusion chromatography to remove un-encapsulated enzymes.For control experiments, polymersomes with encapsulated enzymes but without melittin were prepared using the same method as above.
Immobilization of nanoreactors on surfaces
First, a boron-dipyrromethene 630/650 (BodiPy 630/650) stock solution (100 μM in DMSO) was prepared. Then, 1 μL of this stock solution was added to 1 mL biotin-polymersomes PBS solution in order to dye the hydrophobic PDMS layer. Afterwards, streptavidin atto488-modified silicon wafers (1 cm × 1 cm) or QCM chips were immersed in PBS solutions of the BodiPy-dyed biotin-polymersomes or normal polymersomes (without biotin) in a well (diameter = 2 cm) of a tissue culture plate. At the same time, a PHEMA modified silicon surface (as control) was also immersed into the BodiPy dyed biotin-polymersomes PBS solution in a well (diameter = 2 cm). The solution was shaken gently for 48 h at r.t. in the dark. Finally, the surface was washed with PBS solution to remove non-surface-bound polymersomes.The surface with immobilized uricase-nanoreactors and LPO-nanoreactors for the cascade reaction was prepared by first mixing biotin-modified uricase-nanoreactors (0.5 mL, ∼2.5 mg mL−1) and biotin-modified LPO-nanoreactors (0.5 mL, ∼2.5 mg mL−1). Then the immobilization procedure on streptavidin atto488-modified silicon wafer surface was done as described above.
Characterization of nanoreactor catalytic ability in solution
For LPO-nanoreactors measurement, 5 μL LPO-nanoreactors or LPO-polymersomes (without melittin, as control) PBS solution (ca. 2.5 mg mL−1), 10 μL Amplex® Red (100 μM), 2 μL H2O2 (100 μM) and 183 μL PBS were mixed in the well first, then the kinetic fluorescence intensity was measured for 10 minutes at room temperature (ca. 22 °C). For uricase-nanoreactors, 80 μL uricase-nanoreactors or uricase-polymersomes (without melittin, as control) PBS solution (ca. 2.5 mg mL−1), 40 μL uric acid (200 μM), 60 μL Amplex® Red (100 μM), 20 μL horseradish peroxidase (0.1 mg mL−1) were mixed in the well first, then the kinetic fluorescence intensity was measured for 20 minutes at room temperature (ca. 22 °C).For the cascade reaction between uricase-nanoreactors and LPO-nanoreactors in PBS buffer, 80 μL uricase nanoreactors PBS solution (ca. 2.5 mg mL−1), 5 μL LPO nanoreactors PBS solution (ca. 2.5 mg mL−1), 40 μL uric acid (200 μM), and 60 μL Amplex Red (100 μM) were first mixed in the well first, then the kinetic fluorescence intensity was measured for 10 min at room temperature (ca. 22 °C).
Characterization of cascade reaction on surface
For the single nanoreactor reaction on the surface, the silicon wafer surface (1 cm × 1 cm) bearing anchored LPO-nanoreactors or normal LPO-polymersomes (without melittin) (1 cm × 1 cm) was first immersed in PBS (1 mL, pH 7.3) in the well (diameter 2 cm) of a tissue culture plate. Afterwards, H2O2 (40 μL, 100 μM) and Amplex® Red (200 μL, 100 μM) were added. The cell was covered in aluminium foil and was shaken (50 rpm) at r.t. Fluorescence measurements (λexc = 570 nm, λmaxem = 595 nm) of the upper PBS solution (200 μL) were recorded at different times, and each sample was measured three times. After each measurement, the solution was transferred back into the reaction cell. For the cascade reaction between uricase-nanoreactors and LPO-nanoreactors, the LPO- and uricase-nanoreactors immobilized silicon wafer (1 cm × 1 cm) or normal LPO-polymersomes and uricase-polymersomes (without melittin as biopores, as control) immobilized silicon wafer (1 cm × 1 cm) was first immersed in 1 mL PBS (pH 7.3) in the cell (diameter 2 cm). Afterwards, 200 μL uric acid PBS solution (200 μM) and 200 μL Amplex® Red PBS solution (100 μM) were added one after the other. The well was covered in aluminum foil and was shaken (50 rpm) at r.t. Fluorescence measurements (λexc = 570 nm, λmaxem = 595 nm) were taken as detailed above.Results and discussion
As discussed in the introduction, solid surfaces functionalized with biomolecules or catalysts have been reported,10,47 and we have also described the immobilization of functionally-active polymeric nanoreactors.2,16,21 One of the critical advantages of polymeric nanoreactor systems compared to a strategy of directly immobilizing enzymes or catalysts on solid surfaces is that encapsulation of the cargo (enzyme or catalyst) provides protection, thus minimizing degradation while at the same time retaining catalytic activity.23 Previously reported surfaces modified with enzymes, catalysts and nano-mimics utilize hard surfaces such as TiO2 and silicon.2,48 To the best of our knowledge, there are no previous examples of soft, polymeric surfaces functionalized with two different polymeric nanoreactors and the two types of nanoreactors can serve as the location for cascade reactions. PHEMA-based polymers are water soluble and are used widely in bio-related applications, such as drug delivery and hydrogels.49,50 The use of PHEMA can greatly improve biocompatibility of modified surfaces and increases their hydrophilicity.51 Moreover, the length and conformation of the PHEMA brushes on the surface can be controlled by adjusting the polymerization conditions. In addition, we selected biotin–streptavidin pairing to provide efficient binding of the polymeric nanoreactors to the PHEMA surface, as has been demonstrated for the attachment of dyes, proteins and liposomes to solid surfaces.52–54Streptavidin-functionalized polymeric surface (streptavidin-surface)
The strategy for preparing PHEMA-decorated surfaces is presented in Fig. 1. The ATRP initiators, 2-bromo-2-methyl-N-[3-(triethoxysilyl)propyl]propanamide, are first covalently linked to the activated silicon wafer surface (steps 1 and 2 in Fig. 1). After this first reaction, the contact angle (CA) values of the surface change from 0° to 65.0 ± 1.8° (digital images in Fig. 1, and see also Fig. S1†) demonstrating that the surface has become more hydrophobic and indirectly confirms the surface modification. In the next step, the well-established “graft-from” ATRP technique54 was applied to copolymerize the mixture of HEMA and 2-AEMA·HCl in the presence of CuBr, CuBr2 and PMDETA in a mixture of MeOH and H2O (step 3 in Fig. 1). It should be noted that the density of the primary amine groups (–NH2) and the thickness of the PHEMA-co-P(2-AEMA·HCl) polymer layer can be efficiently controlled by the ratio of the two monomers and the total amount of the monomers in the polymerization medium. In order to control the density of biotin molecules on the surface and support further streptavidin binding, the weight ratio of HEMA and 2-AEMA·HCl was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The ellipsometry measurements were performed in air and the results (shown in Fig. 1) show that the thickness of the polymeric layer is 11.4 ± 0.2 nm (averaged over 3 measurements). The calculated repeating unit of the PHEMA-co-P(2-AEMA·HCl) on the surface is 27 ± 1 assuming a typical carbon–carbon single bond length of 154 pm. Short brushes were synthesized to support the diffusion of the reactants necessary for the functionality of the nanoreactors which might have been hidden by long polymer brushes. Moreover, the CA values of the surface after polymerization decrease from 65.0 ± 1.8° to 46.3 ± 3.2° and this is consistent with a large number of hydrophilic groups (–OH and –NH2·HCl) from the PHEMA-co-P(2-AEMA·HCl). The biotin-modified surface (biotin-surface) was prepared by reacting biotin-NHS with the PHEMA-co-P(2-AEMA·HCl) on the surface through the NHS reaction in the presence of triethylamine in anhydrous DMF (step 4 in Fig. 1). Finally, the streptavidin-surface was obtained by immersing the biotin-surface into streptavidin atto488 (40 μg mL−1) PBS solution (pH 7.3) (step 5, Fig. 1).
:1. The ellipsometry measurements were performed in air and the results (shown in Fig. 1) show that the thickness of the polymeric layer is 11.4 ± 0.2 nm (averaged over 3 measurements). The calculated repeating unit of the PHEMA-co-P(2-AEMA·HCl) on the surface is 27 ± 1 assuming a typical carbon–carbon single bond length of 154 pm. Short brushes were synthesized to support the diffusion of the reactants necessary for the functionality of the nanoreactors which might have been hidden by long polymer brushes. Moreover, the CA values of the surface after polymerization decrease from 65.0 ± 1.8° to 46.3 ± 3.2° and this is consistent with a large number of hydrophilic groups (–OH and –NH2·HCl) from the PHEMA-co-P(2-AEMA·HCl). The biotin-modified surface (biotin-surface) was prepared by reacting biotin-NHS with the PHEMA-co-P(2-AEMA·HCl) on the surface through the NHS reaction in the presence of triethylamine in anhydrous DMF (step 4 in Fig. 1). Finally, the streptavidin-surface was obtained by immersing the biotin-surface into streptavidin atto488 (40 μg mL−1) PBS solution (pH 7.3) (step 5, Fig. 1).
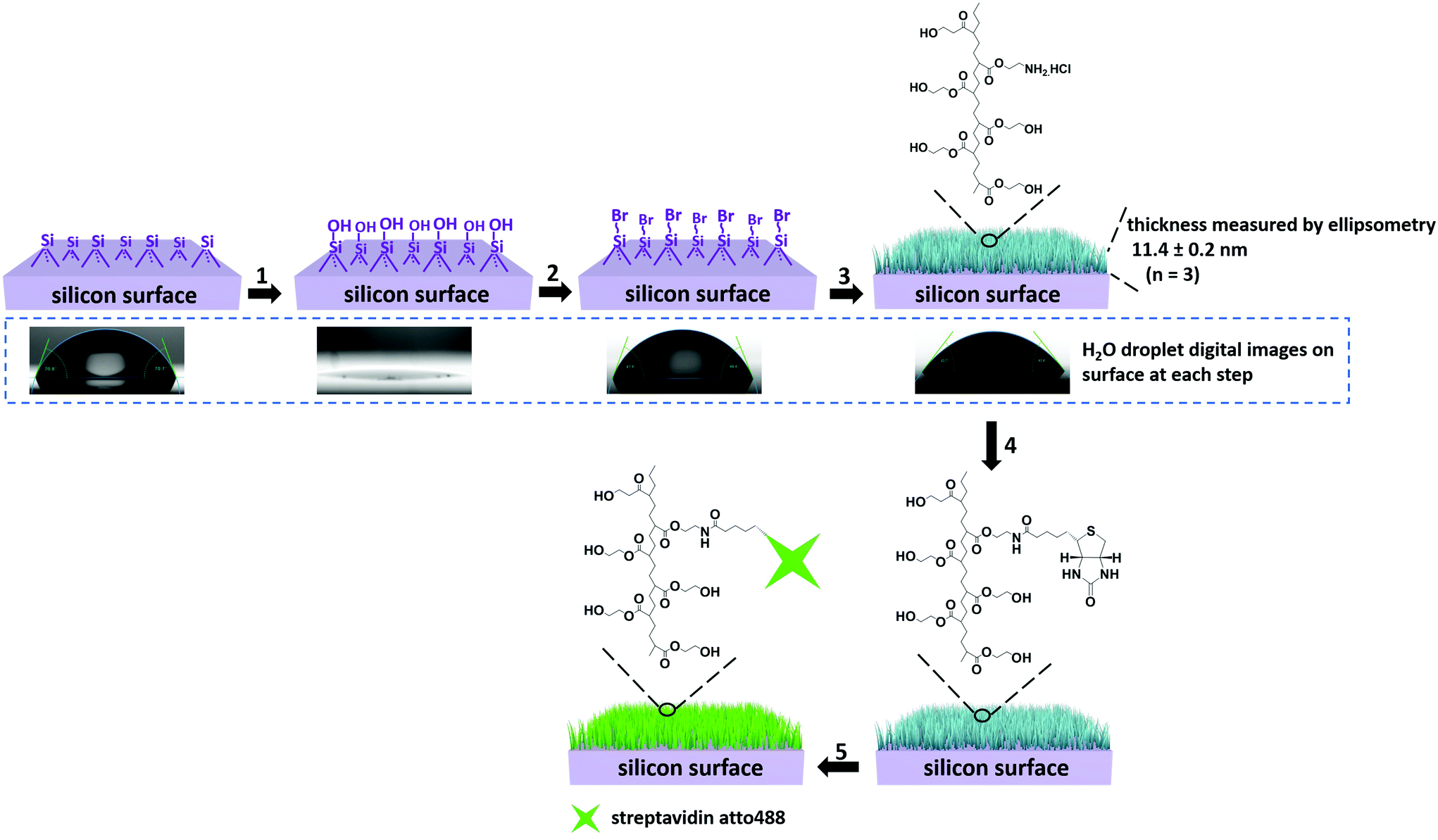 | ||
| Fig. 1 Schematic representation of the procedure of streptavidin-surface preparation. 1. Plasma treatment, 10 min, r.t., 2. 2-Bromo-2-methyl-N-(3-(trimethoxysilyl)propyl)propanamide, TEA, 15 h, r.t., 3. HEMA, 2-AEMA·HCl, PMDETA, CuBr, CuBr2, 18 h, 50 °C. The polymer-functionalized surface (‘brushes’) is represented in blue. 4. Biotin-NHS, TEA, 48 h, r.t., 5. Streptavidin atto488, PBS buffer (pH 7.3), 48 h, r.t. Ellipsometry data are shown at the top-right. The results of contact angle measurements are shown in the digital images. | ||
The surface topologies of the unfunctionalized wafers and the streptavidin-modified surfaces were determined by AFM in air (Fig. S2†). The roughness value (Rq) of the wafers increased from 0.36 nm to 2.15 nm after streptavidin molecules were attached onto the PHEMA grafting wafer surface.
The surfaces were also characterized by CLSM; streptavidin atto488 has an emission maximum at 523 nm. The CLSM image (Fig. 2A) shows strong fluorescence after the biotin-surface had been treated with streptavidin atto488. As a control, the PHEMA-co-P(2-AEMA·HCl) modified silicon wafer surface was also treated with streptavidin atto488 under the same conditions. The CLSM image (Fig. 2B) exhibits no fluorescence under similar conditions. The CLSM results demonstrate that the streptavidin-surface was successfully prepared in the four step-protocol shown in Fig. 1 with the streptavidin–biotin interaction being critical. We return to this later in the discussion of the QCM data.
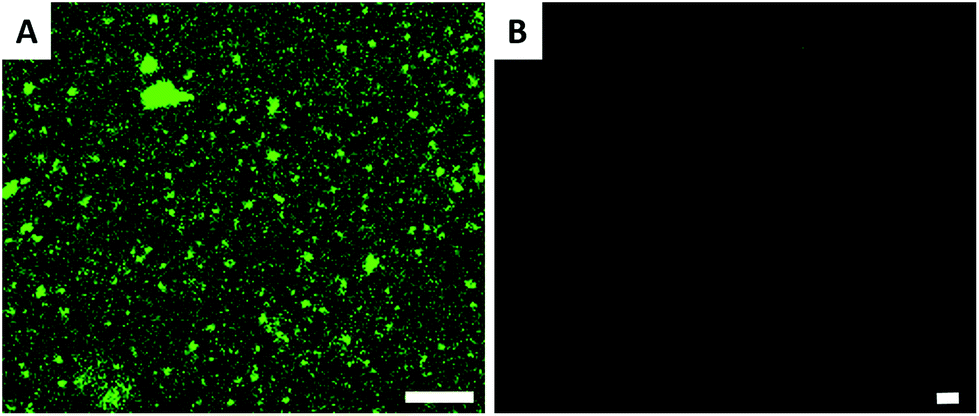 | ||
| Fig. 2 CLSM images of polymer modified surfaces after streptavidin atto488 treatment. (A) Biotin modified surface (biotin-surface) and (B) PHEMA-co-P(2-AEMA·HCl) modified surface. The length of the scale bar is 5 μm. | ||
Biotin modified polymersome preparation (biotin-polymersomes)
Polymersomes were prepared from PDMS21-PMOXA7-OH and PDMS28-PMOXA10-biotin diblock copolymers. These precursors were characterized by 1H NMR spectroscopy and GPC (see Fig. S3–S5†). The PDI values of the PDMS21-PMOXA7-OH (1.16) and PDMS28-PMOXA10-biotin (1.12) diblock copolymers are <1.2, demonstrating their narrow polydispersity. The biotin-polymersomes were prepared using the well-established film rehydration method;46 this method favours the formation of polymersomes with several hundred nanometers diameter for PDMS-PMOXA diblock copolymers.46 Because of the strong hydrophilicity of the biotin molecules, and longer PMOXA chain in PDMS28-PMOXA10-biotin compared with PDMS21-PMOXA7-OH, the biotin molecules can face towards the aqueous phase (not only external surface but also internal surface of polymersomes), rather than being buried in the hydrophobic PDMS shell. This makes the biotin units readily available for interaction with streptavidin. The density of the biotin units on the surface of the biotin-functionalized polymersomes can be finely controlled by adjusting the weight ratio of PDMS21-PMOXA7-OH to PDMS28-PMOXA10-biotin diblock copolymers during the self-assembly of the diblock copolymers in PBS buffer. A weight ratio of PDMS21-PMOXA7-OH to PDMS28-PMOXA10-biotin of 20 to 1 was used to achieve suitable density of biotin on surface of polymersomes. The density of biotin molecules on the surface should not perturb the polymersome integrity during the biotin–streptavidin interaction. The TEM micrograph (Fig. S6†) demonstrates the round polymersome structures formed by the mixture of PDMS21-PMOXA7-OH and PDMS28-PMOXA10-biotin. The ρ value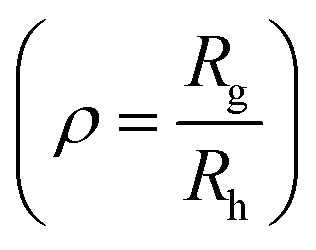 (Table S1†) of biotin-polymersomes characterized by LS is 0.97 and this also demonstrates a hollow structure of the polymersomes. The accessibility of the biotin molecules on the surface of the polymersomes with streptavidin atto488 was assessed by FCS. In Fig. 3, the fitted green curve (biotin-polymersomes treated with streptavidin atto488) exhibits a larger diffusion time (6970 ± 415 μs, averaged over 30 measurements) than the fitted blue curve (free streptavidin atto488) which has a diffusion time of 50.6 ± 1.3 μs (averaged over 30 measurements). For the biotin-polymersomes complexed with streptavidin atto488, there is only one fluorescence population, demonstrating that the streptavidin atto488 molecules added to the biotin-polymersome solution are all involved in streptavidin–biotin interactions. As a control, streptavidin atto488 was added to un-modified polymersomes (fitted purple curve in Fig. 3). The diffusion time was similar to that of free streptavidin atto488 (blue curve, Fig. 3) and there was no population with a longer diffusion time indicating that the streptavidin atto488 does not bind to the unmodified polymersomes through non-selective interactions. The normalized FCS results demonstrate that the biotin molecules on the surface of the polymersomes bind streptavidin atto488. The FCS measurements also show that the calculated hydrodynamic diameter of the supramolecular assembly formed between streptavidin atto488 and the biotin-polymersome according (Fig. 3, green curve) is 169 ± 17 nm. This is comparable with the value of 192 ± 10 nm derived from the DLS measurements (Table S1†).
(Table S1†) of biotin-polymersomes characterized by LS is 0.97 and this also demonstrates a hollow structure of the polymersomes. The accessibility of the biotin molecules on the surface of the polymersomes with streptavidin atto488 was assessed by FCS. In Fig. 3, the fitted green curve (biotin-polymersomes treated with streptavidin atto488) exhibits a larger diffusion time (6970 ± 415 μs, averaged over 30 measurements) than the fitted blue curve (free streptavidin atto488) which has a diffusion time of 50.6 ± 1.3 μs (averaged over 30 measurements). For the biotin-polymersomes complexed with streptavidin atto488, there is only one fluorescence population, demonstrating that the streptavidin atto488 molecules added to the biotin-polymersome solution are all involved in streptavidin–biotin interactions. As a control, streptavidin atto488 was added to un-modified polymersomes (fitted purple curve in Fig. 3). The diffusion time was similar to that of free streptavidin atto488 (blue curve, Fig. 3) and there was no population with a longer diffusion time indicating that the streptavidin atto488 does not bind to the unmodified polymersomes through non-selective interactions. The normalized FCS results demonstrate that the biotin molecules on the surface of the polymersomes bind streptavidin atto488. The FCS measurements also show that the calculated hydrodynamic diameter of the supramolecular assembly formed between streptavidin atto488 and the biotin-polymersome according (Fig. 3, green curve) is 169 ± 17 nm. This is comparable with the value of 192 ± 10 nm derived from the DLS measurements (Table S1†).
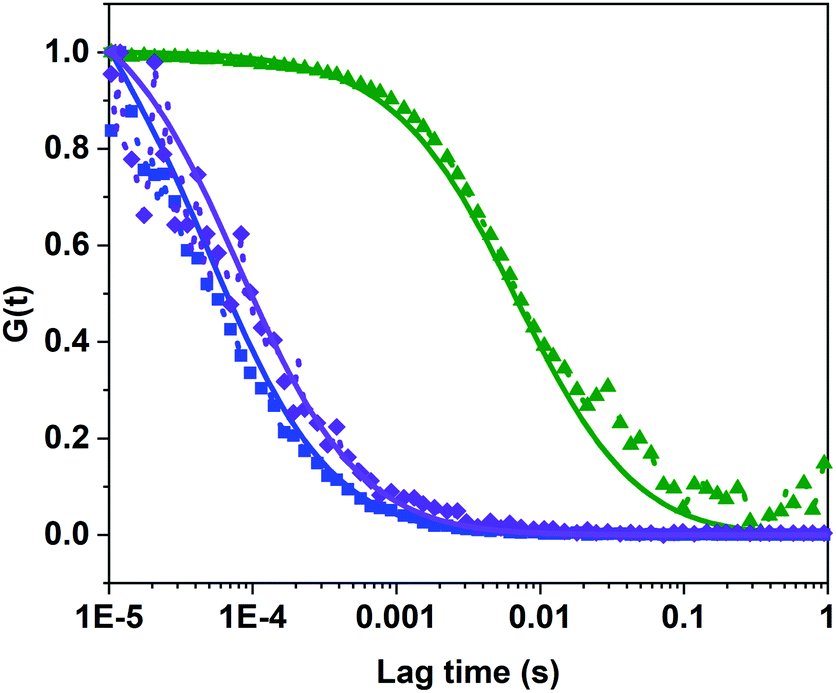 | ||
| Fig. 3 FCS results of binding behaviour of biotin-polymersomes with streptavidin atto488 in PBS buffer. Dots: normalized raw data; solid line: corresponding fitted data. Blue line: free streptavidin atto488, purple line: normal polymersomes treated with streptavidin atto488, green line: biotin-polymersomes treated with streptavidin atto488. | ||
Nanoreactor preparation
At this point, it is important to note the distinction between the terms polymersome (without melittin pores) and nanoreactor (functional polymersome, with melittin pores). For the preparation of the nanoreactors, two types of enzymes, lactoperoxidase (LPO) and uricase, were encapsulated into individual polymersomes during self-assembly of block copolymers to produce LPO-nanoreactors and uricase-nanoreactors. Uricase can efficiently catalyse the conversion of uric acid to 5-hydroxyisourate releasing H2O2 (Scheme 2). LPO is a natural antibacterial agent and peroxidase enzyme and can efficiently catalyse the reaction between Amplex® Red and H2O2 to form the strongly fluorescent resorufin (Scheme 2). As a result, the cascade reaction involving the two enzymes uricase and LPO and the conversion of Amplex® Red to resorufin can be utilized as a cascade reaction model with uric acid as the primary substrate. The membrane permeabilizing peptide, melittin, was added during the assembly of the nanoreactors. Because of the amphiphilicity of melittin, it is able to insert into the PDMS shell to serve as a biopore, thereby facilitating the passage of hydrophilic substances across the nanoreactor membrane.54 The TEM micrographs shown in Fig. 4 clearly reveal the round and uniform nanostructures of the uricase-nanoreactors (Fig. 4A) and LPO-nanoreactors (Fig. 4C) similar with uricase encapsulated polymersomes (Fig. 4B) and LPO encapsulated polymersomes (Fig. 4D). This indicates that the presence of LPO, uricase and melittin in the condition presented here does not influence the self-assembly of the PDMS-PMOXA diblock copolymers.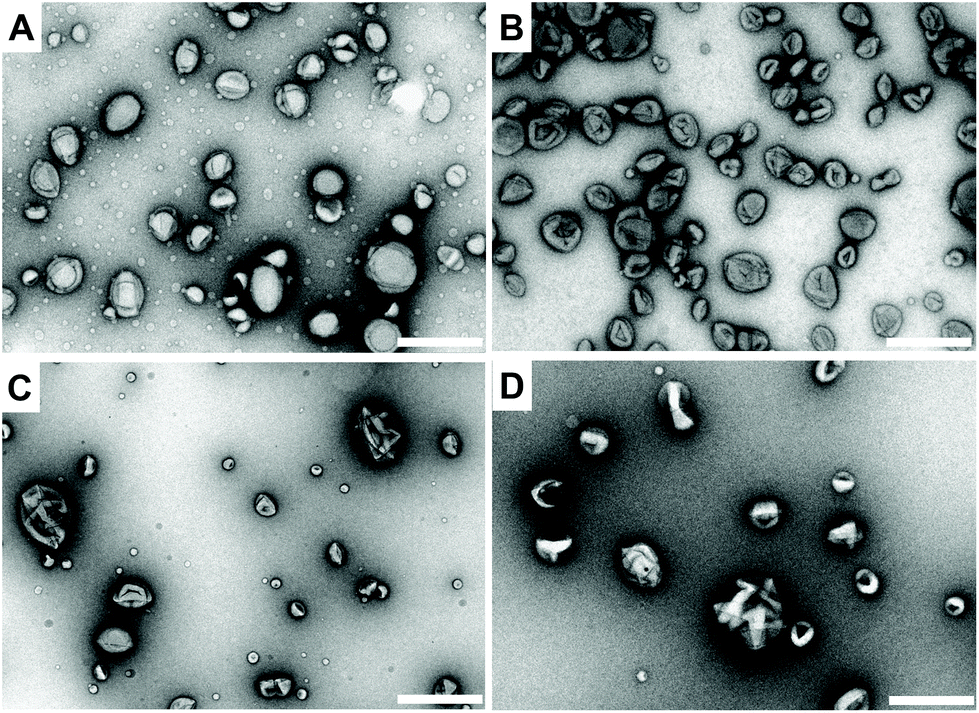 | ||
| Fig. 4 TEM images of the nanoreactors. (A) Uricase-nanoreactors, (B) uricase encapsulated polymersomes, (C) LPO-nanoreactors and (D) LPO encapsulated polymersomes. Scale bar is 500 nm. | ||
The hydrodynamic diameter of the uricase- and LPO-nanoreactors measured by DLS (measured angle 90°) are 228 ± 20 nm and 200 ± 20 nm, respectively. The ρ values 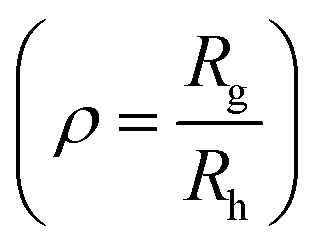 of the uricase- and LPO-nanoreactors are 0.99 and 0.93, respectively (Table S1†), demonstrating that the nanoreactors possess hollow aqueous cavities.56 Note that we obtained an enzyme encapsulation efficiency comparable to that reported by Belluati.42 The wrinkled nanostructures on the surface of nanoreactors in the TEM images (Fig. 4) are characteristic for TEM samples preparation, because of water evaporation inducing membrane collapse. Such collapse can further result in a decrease of the height values (measured by AFM) of the nanoreactors, as discussed in the next section.
of the uricase- and LPO-nanoreactors are 0.99 and 0.93, respectively (Table S1†), demonstrating that the nanoreactors possess hollow aqueous cavities.56 Note that we obtained an enzyme encapsulation efficiency comparable to that reported by Belluati.42 The wrinkled nanostructures on the surface of nanoreactors in the TEM images (Fig. 4) are characteristic for TEM samples preparation, because of water evaporation inducing membrane collapse. Such collapse can further result in a decrease of the height values (measured by AFM) of the nanoreactors, as discussed in the next section.
In order to quantify the average number of uricase and LPO molecules inside a polymersome, uricase (0.25 mg mL−1) and LPO (0.25 mg mL−1) were modified with atto655 and atto635, respectively, and then encapsulated inside the nanoreactors by using similar conditions as those reported by Beluatti.44 FCS was used to measure the brightness as counts per molecule of free uricase, LPO and the corresponding nanoreactors. The average value of the counts per molecule of uricase modified with atto655 was 8 ± 1.6 kHz and for a nanoreactor containing uricase was 79.8 ± 11.7 kHz. As the result, the average number of encapsulated uricase inside each nanoreactor was determined to be 10 ± 2. The average value of counts per molecule of LPO modified with atto635 was 0.41 ± 0.02 kHz and for a nanoreactor containing LPO was 3.50 ± 0.59 kHz. This leads to an average number of encapsulated LPO inside each nanoreactor of 9 ± 2. The number of encapsulated enzymes per nanoreactor is comparable with the encapsulation efficiency of these enzymes when loaded in PMOXA-PDMS-PMOXA copolymers nanoreactors by the film rehydration method.42,57
Catalytic activity of nanoreactors in solution
To test the activity of the uricase- and LPO-nanoreactors, fluorescence measurements were used to detect resorufin (λexc = 570 nm, λmaxem = 595 nm) formed by the reaction of Amplex® Red with H2O2 in the presence of LPO. Before the measurement, the PBS suspension containing the nanoreactors was subjected to size extrusion chromatography to remove non-encapsulated enzymes. Adding uric acid to the uricase-nanoreactors resulted in the production of H2O2, which was used by horseradish peroxidise, free in solution, to oxidize Amplex® Red (Fig. 5A and B). We emphasize that the H2O2 in the test experiment of LPO-nanoreactor (Fig. 5C) was directly added in the environment of LPO-nanoreactors to simulate the product of the first reaction and thus the “input signal” for the second reaction. The red curves in Fig. 5B and D show that polymersomes lacking melittin (i.e. unpermeabilized) were not active, with a very small fluorescence increase due to non-enzymatic autooxidation of the substrate.56–60 An apparent enzyme activity for LPO-nanoreactors of 2 × 10−6 μmol min−1 was obtained, in agreement with the previously reported activity of the nanoreactors.42 When the nanoreactors were combined in solution, the cascade reaction took place as indicated by the increase in the fluorescence intensity associated with the production of resorufin (Fig. 5E and F (black curve)). In contrast, when the nanoreactors were not permeabilized by insertion of melittin, the molecular flow was blocked, and thus the cascade reaction did not take place (Fig. 5F (red and blue curves). The slower kinetics of the cascade reaction between nanoreactors in solution has been already observed for similar catalytic compartments working in tandem.42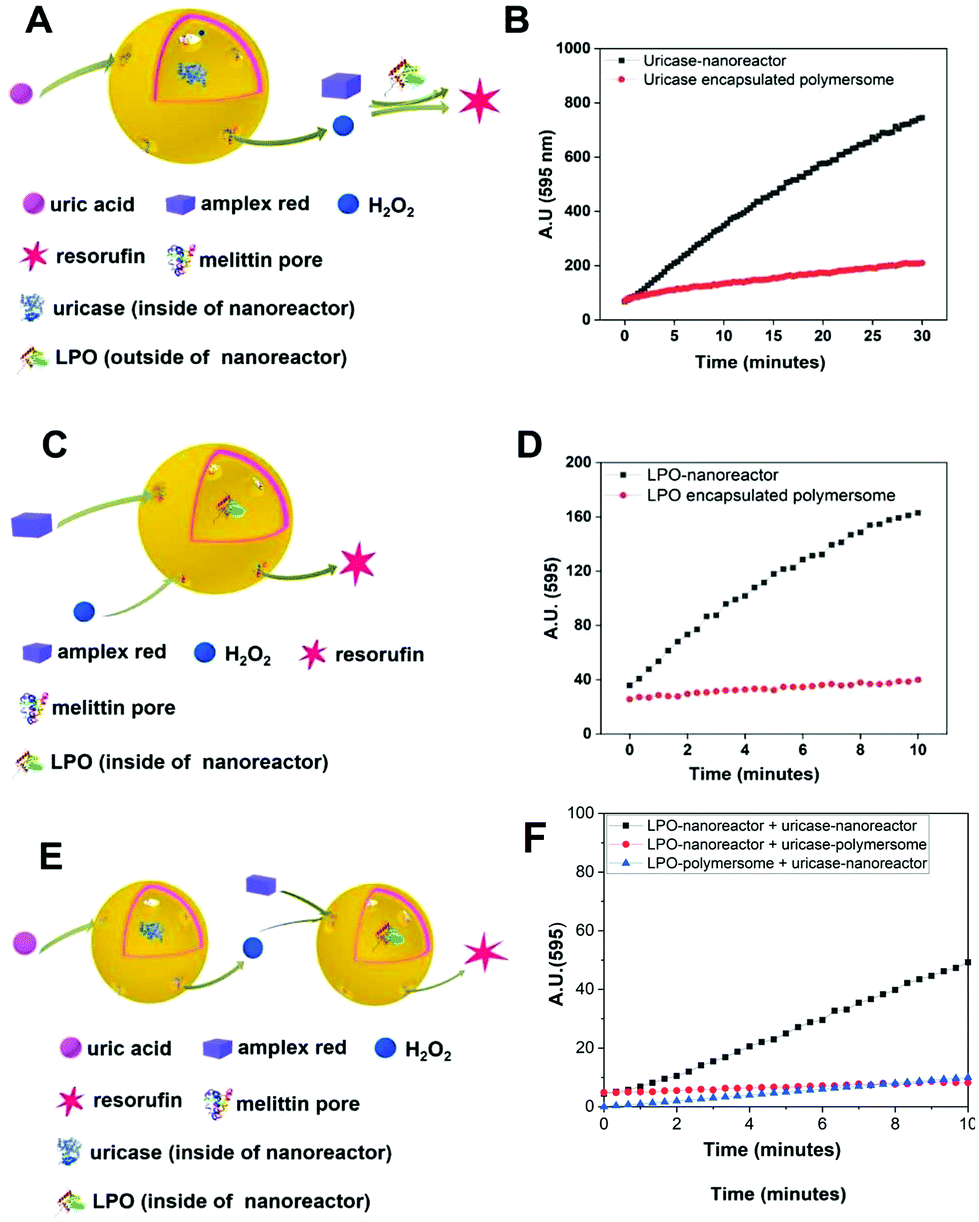 | ||
| Fig. 5 Schematic diagrams of the catalysis and fluorescence plots of resorufin produced inside of the nanoreactors in PBS solution. (A) Schematic diagram of catalysis in the uricase-nanoreactors, and (B) fluorescence plot of produced resorufin from solution by using H2O2 produced from uricase-nanoreactors (black) and uricase encapsulated polymersomes (as control) system (red). (C) Schematic diagram of catalysis in the LPO-nanoreactors, and (D) fluorescence plot of produced resorufin from LPO-nanoreactors (black) and LPO encapsulated polymersomes (as control) system (red). (E) Schematic diagram of cascade reaction between uricase-nanoreactors and LPO-nanoreactors in PBS solution and (F) fluorescence plot of produced resorufin from LPO-nanoreactors by using H2O2 produced from uricase-nanoreactors(black), uricase encapsulated polymersomes (as control) system (red) and LPO encapsulated polymersome (as control) (blue). | ||
Immobilization of biotin-polymersomes on streptavidin-surface
In order to characterize the binding of the biotin-polymersomes to the streptavidin-modified surface using CLSM, the hydrophobic PDMS layer of the biotin-polymersomes was first labelled with BodiPy630/650. CLSM was used to detect the fluorescence of the surfaces. Fig. 6 shows fluorescence at λmaxem = 523 nm arising from streptavidin atto488 (Fig. 6A) and at λmaxem = 650 nm from BodiPy (Fig. 6B) (i.e. from the BodiPy in the PDMS shell of biotin-polymersomes). Critically, the regions of the two emissions exactly overlap (Fig. 6C), and this is consistent with the presence of the biotin-polymersomes on the streptavidin-surface and of the coincidence of the positions of both entities. An inhomogeneous distribution of polymersomes on the surface, with the formation of clusters was obtained (Fig. 6).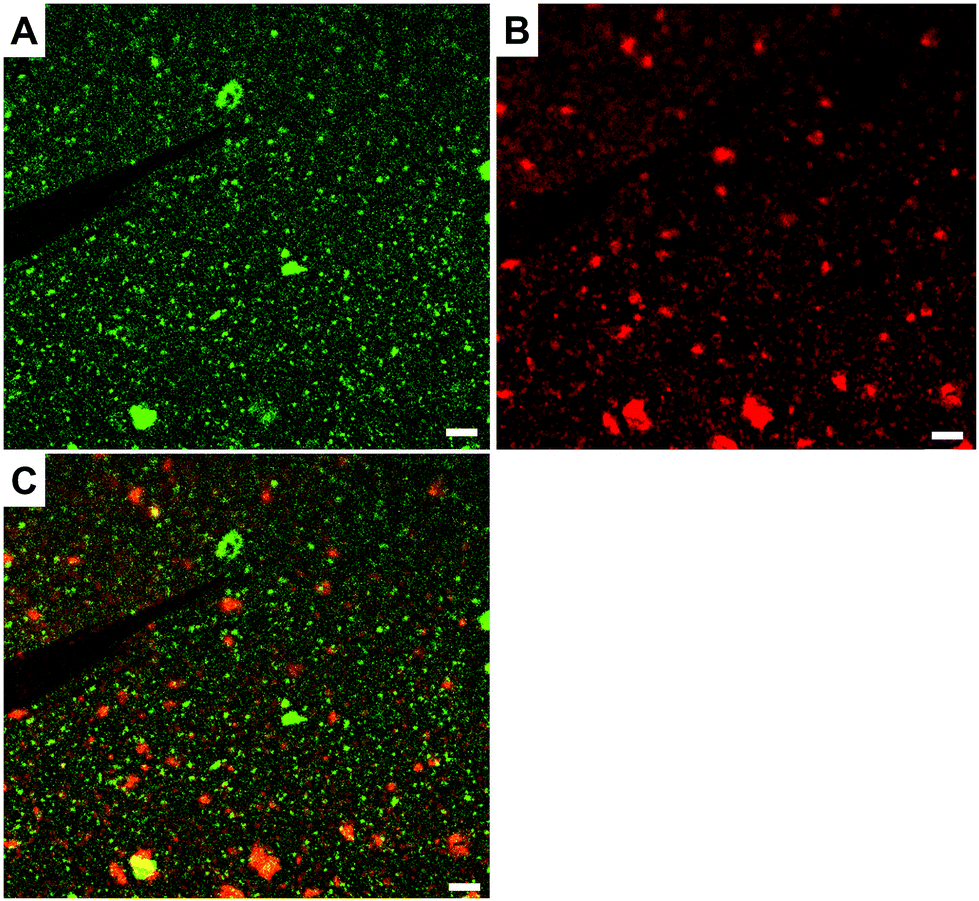 | ||
| Fig. 6 Double channels CLSM images of the biotin-polymersomes (dyed by BodiPy630/650) decorated on streptavidin-surface. (A) Channel with wavelength of laser 488 nm, (B) channel with wavelength of laser 633 nm and (C) merged image. Scale bar is 5 μm. | ||
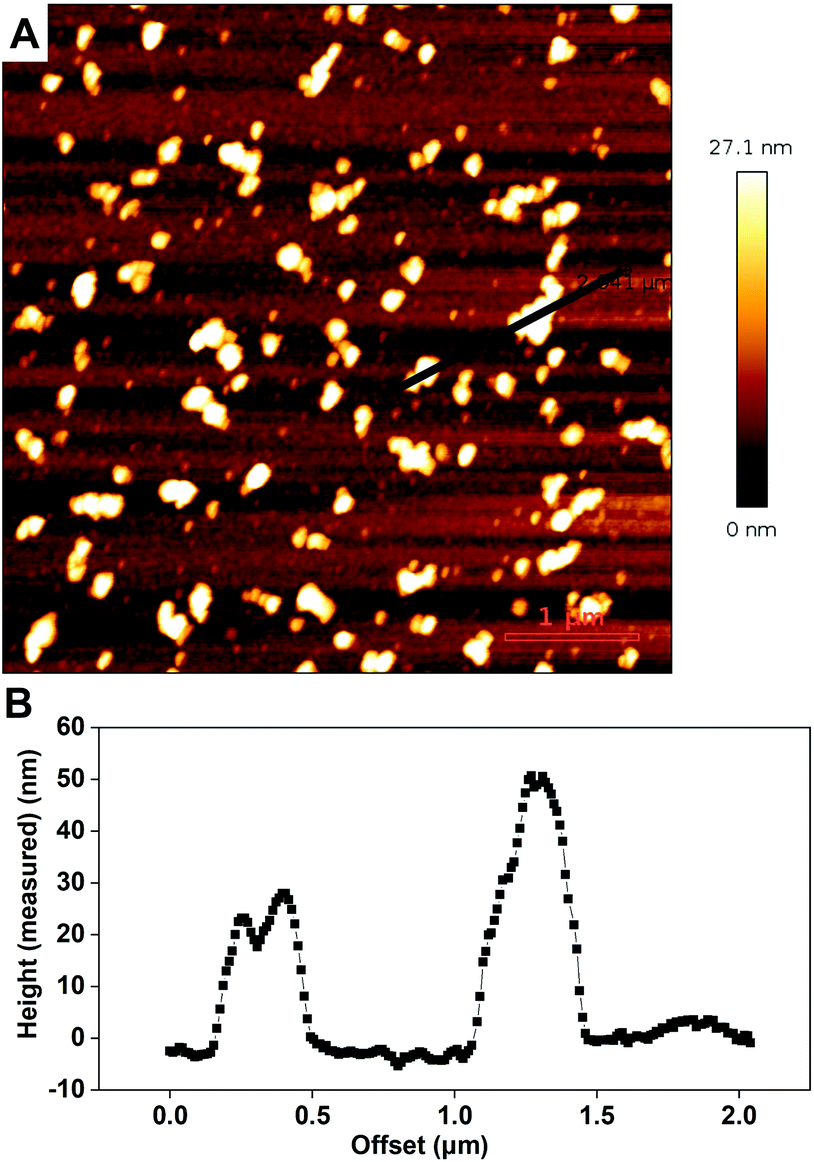
The presence and morphologies of the biotin-polymersomes on the streptavidin-surface were characterized using AFM in air (Fig. 7A). A comparison of AFM images of an unfunctionalized silicon wafer surface and a PHEMA-co-P(2-AEMA·HCl) grafted surface (Fig. S2†) confirms that only the streptavidin-surface exhibits nano-objects, mostly with diameters in the range 100 to 200 nm (Fig. 7B). However, Fig. 7B also shows diameters up to 500 nm, which is probably caused by the formation of polymersome clusters by weak interactions. The measured height values of the biotin-polymersomes on the surface are between 30 and 50 nm. This is less than the diameter of the biotin-polymersomes (200 nm) measured by DLS (90°) (Table S1†), and can be rationalized by the collapse of the biotin-polymersomers (Fig. S6†) in air. A similar phenomenon has been reported by others using PDMS-b-PMOXA diblock copolymers.2 Moreover, the AFM measurement in water (Fig. S7†) also demonstrates the presence of the biotin-polymersomes on surface of streptavidin modified surface. A dried specimen surface with immobilized biotin-polymersomes was rehydrated by addition of 2 mL of PBS and then observed by AFM (Fig. S7†). The height values of the biotin-polymersomes on the surface measured in H2O are lower than the size of biotin-polymersomes in solution obtained by DLS, probably caused by an incomplete rehydration of the dried biotin-polymersomes before the AFM measurement in aqueous solution. In addition, there are a few regions where the offset values of the nano-assemblies are larger than the diameter of a single polymersome, indicating a possible side-by-side arrangement of polymersomes during the immobilization on the surface (Fig. 7). In addition to CLSM and AFM, the detailed interaction behaviour between biotin-polymersomes and the streptavidin modified surface was investigated by QCM-D. The QCM-D method allows one to gain the information about the binding speed, binding strength and number of objects bound to the surface. The data were all obtained at the 7th overtone (see Experimental section in the ESI†). The value of frequency (7th overtone) decreases immediately after pumping a streptavidin-atto488 PBS solution (Fig. 8A, at time ∼6 minutes) into the QCM chamber. The value of frequency decreases until it becomes stable after 90 minutes with a frequency value −47.5 Hz. After that, the frequency value remains constant during the pumping of PBS buffer into the QCM-D chamber (from time ∼90 minutes). This can be rationalized in terms of the strong binding affinity of biotin for streptavidin (real-time binding constant 3.0 × 106–4.5 × 107 M−1 s−1).44 As there are four binding sites for biotin molecules on streptavidin molecules (Scheme 1), the free sites are still available for binding extra biotin molecules. The value of the frequency decreases significantly after pumping the PBS solution of the biotin-polymersomes into the QCM-D chamber (at time ∼120 minutes) as the biotin-polymersomes start to bind on the surface through the biotin–streptavidin interaction. The final frequency value (7th overtone) reaches a value of −185.2 Hz after 60 minutes. However, the frequency values in our system remain constant during the washing process with PBS buffer, which is consistent with the strong supramolecular interactions between biotin and streptavidin. This is despite the fact that the biotin molecules are covalently bound to the polymer chains and thereby experience relatively large steric hindrance. According to the decreasing value of the frequency (Δf), the weight of bonded streptavidin-atto488 and biotin-polymersomes can be calculated to be 0.71 ng cm−2 and 2.83 ng cm−2, respectively, using the Sauerbrey equation (Δm = −CΔf),55 where C is a proportionality constant, depending on the quartz properties (C = 18 ng cm−2 Hz−1).47
 | ||
| Fig. 7 The AFM characterization result of biotin-polymersome decorated streptavidin-surface (measured in air). (A) AFM image of the streptavidin-surface immobilized by biotin-polymersomes and (B) a height profile along the white line shown in A on the surface from the AFM image. | ||
 | ||
| Fig. 8 (A) QCM-D plot of the biotin modified QCM-D chip treated by streptavidin atto488 and biotin-polymersomes PBS solution, (B) AFM image of the above QCM chip after being washed by PBS and Millipore water and (C) height curve of the two selected biotin-polymersomes from above AFM image. | ||
In addition, the corresponding dissipation plot (Fig. 8A, red) shows that during the streptavidin-atto488 binding on the biotin-surface, the dissipation curve remains straight (time = 0–90 minutes). However, binding of the biotin-polymersomes results in a dramatic increase in the dissipation value from 0 to 50 × 10−6 (time = 120–150 minutes) and this can be interpreted in terms of the increasing flexibility of the surface after nanosized polymersomes have been anchored. While, for PHEMA-co-P(2-AEMA·HCl) modified QCM-D chips (without biotin on surface), after pumping streptavidin atto488 PBS solution and biotin-polymersomes, the frequency curve remained constant (Fig S8†) demonstrating no streptavidin atto488 and biotin-polymersomes were immobilized on surface.61
An AFM image of the QCM-D chip surface after treatment with streptavidin-atto488 and then biotin-polymersomes, and finally being washed by PBS and Millipore water, confirms the presence of the polymersomes on top of the chip surface (Fig. 8B). The measured height value range of the polymersomes (Fig. 8C) is 60 to 90 nm and this is smaller than their hydrodynamic diameter (192 ± 10 nm) in PBS. This is explained by their collapse in air as previously discussed.
Proof-of-principle reactions involving nanoreactors immobilized on soft surfaces
Fig. 5 illustrates H2O2 can be produced by uric acid, H2O and O2 inside of the uricase-nanoreactors and resorufin can be generated by Amplex® Red and H2O2 inside of LPO-nanoreactors in PBS. In contrast to the conditions for the reaction in PBS solution described earlier, there are a number of points to consider when we are dealing with nanoreactors which are anchored to a surface through the biotin–streptavidin interaction. Firstly, the PHEMA-co-P(2-AEMA·HCl) chains (Fig. 1) may partially block the diffusion of the hydrophilic reactants into and out of the nanoreactors. This will be especially true for parts of the nanoreactors that face the surface. Secondly, immobilization of the nanoreactors on a surface restricts their 3D Brownian motion. It might be expected that these two factors may restrict the observed catalytic activity of the surface-bound nanoreactors with respect to the activity of those tested in solution.First, we tried a simple case – a reaction catalysed only by LPO-nanoreactors anchored to a surface. Fig. 9A shows the schematic presentation of the LPO-catalysed reaction in the LPO-nanoreactors attached on surface in which Amplex® Red reacts with added H2O2 to give fluorescent resorufin. Encouragingly, the intensity of fluorescence (λmaxem = 595 nm) originating from resorufin increases with time (0–28 h) (Fig. 9B (black curve)). A control experiment in which only LPO encapsulated polymersomes were immobilized on the surface was also carried out and in this case there was no increase in fluorescence at λmaxem = 595 nm indicating that no resorufin was being produced (Fig. 9B, red curve). The fluorescence intensity at λmaxem = 595 for pure PBS buffer was zero (Fig. 9B, blue curve). The changes in the gradient of the black curve in Fig. 9B indicate that the rate of production of resorufin in the first 5 h is faster than after 5 h, presumably because the rate of reaction is dependent upon the concentration of the reactants. The colour of the PBS solution with LPO-nanoreactors immobilized on the surface changed from colourless to red after being stored for one week, while, LPO-encapsulated polymersome immobolized soft surface without melittin is still colourless (Fig. S9†), demonstrating the catalytic reaction of transferring Amplex® Red into resorufin in presence of H2O2 happened inside of the LPO-nanoreactors successfully on surface.
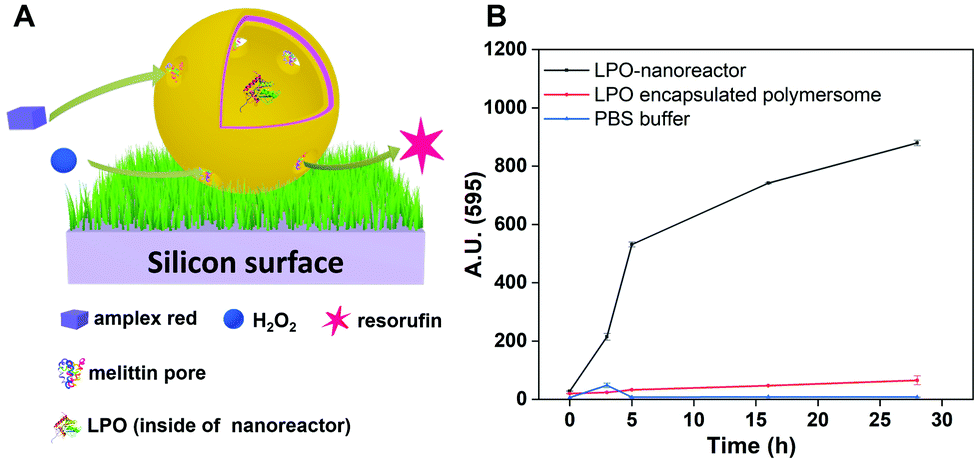 | ||
| Fig. 9 (A) Schematic presentation of the reaction in the LPO-nanoreactor anchored on a soft surface and (B) measured fluorescence intensity of resorufin in solution produced in and released from the immobilized LPO-nanoreactors (black curve), immobilized LPO encapsulated polymersomes (red curve) and PBS solution (blue curve). | ||
Cascade reactions involving nanoreactors immobilized on soft surfaces
After confirming the catalytic ability of surface-anchored LPO-nanoreactors, we next combined uricase- and LPO-nanoreactors immobilized on a single streptavidin-surface. We emphasize two points. Firstly, by using the same weight ratio (20:1) of the PDMS21-PMOXA7-OH and PDMS28-PMOXA10-biotin diblock copolymers during the preparation of the nanoreactors, we ensured that the density of the biotin domains on the surface of the two different nanoreactors was the same. Secondly, the concentration of two types of the nanoreactors was the same (∼2.5 mg mL−1). As a result, we may assume that there were equal numbers of uricase- and LPO-nanoreactors on the streptavidin-surface. Amplex® Red and uric acid were added to the uricase- and LPO-nanoreactors immobilized on the streptavidin-surface (see Experimental section). The fluorescence intensity of the resorufin produced in the solution (see schematic diagram in Fig. 10A) increased linearly with the reaction time from 0 to 40 h (Fig. 10B, black curve, 3 measurements per data point). In a control experiment, the nanoreactors were replaced by polymersomes containing uricase or LPO but without melittin biopores. The observed fluorescence over time and the very low and near-constant value is consistent with a low level of resorufin production (red curve in Fig. 10A), as expected from non-enzymatic substrate oxidation.55,56 In this case, the reaction rate decreased significantly. The reaction kinetics of the nanoreactors in tandem in solution was completely blocked when the distance between them was above 1 μm.42
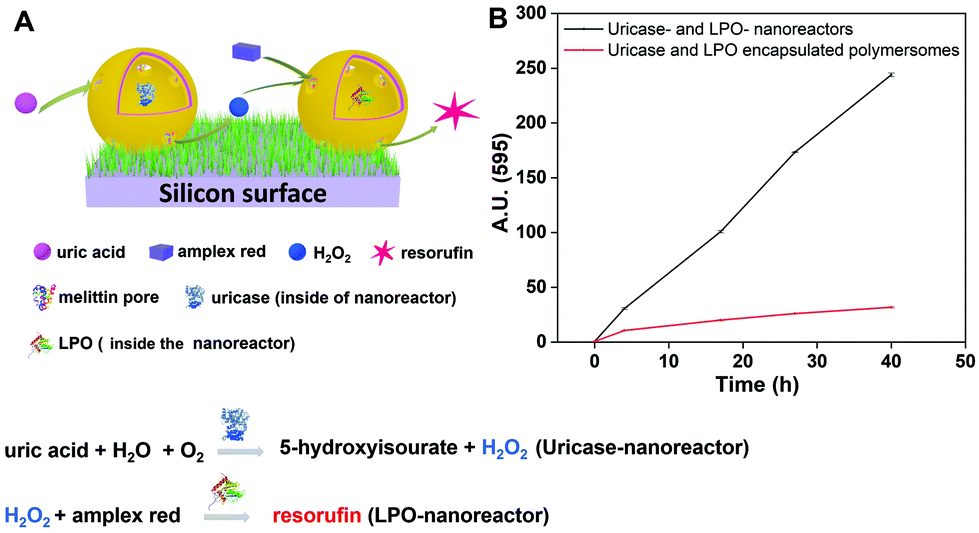 | ||
| Fig. 10 (A) Schematic diagram of the cascade reaction on uricase- and LPO-nanoreactors immobilized surface, (B) fluorescence intensity of resorufin in PBS solution produced by uricase- and LPO-nanoreactors immobilized soft surface (black curve) and uricase and LPO encapsulated polymersomes (without melittin as control) immobilized soft surface (red curve). | ||
These results demonstrate that the cascade reaction involving the encapsulated enzymes LPO and uricase and the conversion of Amplex® Red to resorufin occurs when the enzymes are encapsulated in separate nanoreactors which are anchored to a soft, polymer surface. In addition, the colour of the PBS solution with uricase- and LPO-nanoreactors immobilized on the surface changed from colourless to light red after being stored for nine days, and the control sample (immobilized uricase and LPO encapsulated polymersomes without melittin) is still colourless (Fig. S10†), demonstrating the production of resorufin through the cascade reaction between uricase- and LPO-nanoreactors on soft surface.
As expected, the reaction kinetics of the nanoreactors in tandem when surface-immobilized (Fig. 10) are significantly slower than in PBS solution due to the intrinsic specificity of each experiment (Fig. 5F). There are various factors inducing this decrease in the kinetics of the cascade reaction when the nanoreactors are immobilized: (i) the number of attached nanoreactors decreases upon the immobilization and washing procedures, (ii) attachment of nanoreactors on polymer brushes limits the accessibility of substrates and products into/from the polymersomes compared to bulk conditions where nanoreactors are moving freely in solution, (iii) the fraction of melittin pores inserted into the section of polymersomes facing the brushes does not contribute to the permeability of the polymersome, and (iv) the distance between the immobilized nanoreactors varies and when higher than 1 μm, it decouples the nanoreactors. Importantly, even in these conditions, the cascade reaction takes place on the surface and further optimization is expected to improve its efficiency. We emphasize that the conditions for the nanoreactors in tandem in solution and when immobilized are different due to the specificity of each set-up, which prevents a direct comparison being made.
While direct immobilization of enzymes on polymer brushes as a complementary approach to perform reactions on surfaces might increase the amount of enzymes involved in the reaction, the use of immobilized nanoreactors has the advantage to protect the biomolecules from harmful environmental conditions, thus prolonging their activity.
Conclusions
We have demonstrated that functional polymeric nanoreactors encapsulating uricase or LPO and containing melittin as biopores can be successfully produced by using PDMS-b-PMOXA diblock copolymers. The catalytic activity of the encapsulated enzymes was first confirmed in solution using the cascade reaction involving the enzymes LPO and uricase and the conversion of Amplex® Red to resorufin, the latter being detected by its fluorescence. Following this proof-of-principle reaction, PHEMA-co-P(2-AEMA·HCl)-modified silicon surfaces were prepared by co-immobilizing uricase- and LPO-nanoreactors making use of the strong supramolecular interaction between biotin and streptavidin. The presence and nanostructured architectures of the nanoreactors on surface were characterized by AFM, CLSM and QCM-D. Moreover, the catalytic activity of the encapsulated enzymes LPO and uricase were retained after surface-immobilization of the nanoreactors. Cascade reactions using uric acid and Amplex® Red as the model reactants with the production of fluorescent resorufin was successfully performed.This successful investigation of a cascade reaction a on polymeric soft surface will allow us to take the concept further in the development of functional surface materials for application in various domains, such as advanced biosensing, implanting and the preparation of antimicrobial materials. Future efforts will reveal how such nanoreactors in tandem on surfaces can be optimized by adjusting the encapsulation efficiency, the distance between the nanoreactors, and their accessibility for molecular exchange. Our concept of different immobilized-nanoreactors working in tandem faciliates a straightforward extension to other target reactions by a change of the enzymes inside the nanoreactors. In addition, immobilizing two types of nanoreactors between which the cascade reaction is able to happen in a controllable way, for example, selectively anchoring on a patterned surface, is a challenging research direction aimed at further optimization of multifunctional surfaces.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Swiss National Science Foundation for support through the NCCR Molecular Systems Engineering (MSE, grant number 51NF40-182895), and we also thank the University of Basel for financial support. D. W. would like to thank Prof. Wolfgang Meier (University of Basel) for his comments and support during accomplishing the project.References
- T. Wei, Q. Yu and H. Chen, Adv. Healthcare Mater., 2019, 8, 1801381–1801404 CrossRef CAS PubMed.
- X. Zhang, M. Lomora, T. Einfalt, W. Meier, N. Klein, D. Schneider and C. G. Palivan, Biomaterials, 2016, 89, 79–88 CrossRef CAS PubMed.
- F. J. Malzner, C. E. Housecroft and E. C. Constable, Inorganics, 2018, 6, 57–73 CrossRef.
- H. C. Pappas, S. Phan, S. Yoon, L. E. Edens, X. Meng, K. S. Schanze, D. G. Whitten and D. J. Keller, ACS Appl. Mater. Interfaces, 2015, 7, 27632–27638 CrossRef CAS PubMed.
- J. R. Wayment and J. M. Harris, Anal. Chem., 2009, 81, 336–342 CrossRef CAS PubMed.
- K. Jaskiewicz, M. Makowski, M. Kappl, K. Landfester and A. Kroeger, Langmuir, 2012, 28, 12629–12636 CrossRef CAS PubMed.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed.
- S. Cavallaro, J. Horak, P. Hååg, D. Gupta, C. Stiller, S. S. Sahu, A. Görgens, H. K. Gatty, K. Viktorsson, S. El Andaloussi, R. Lewensohn, A. E. Karlström, J. Linnros and A. Dev, ACS Sens., 2019, 4, 1399–1408 CrossRef CAS PubMed.
- O. Wahlsten, A. Gunnarsson, L. Simonsson Nyström, H. Pace, S. Geschwindner and F. Höök, Langmuir, 2015, 31, 10774–10780 CrossRef CAS PubMed.
- K. Motokura, K. Saitoh, H. Noda, W.-J. Chun, A. Miyaji, S. Yamaguchi and T. Baba, Catal. Sci. Technol., 2016, 6, 5380–5388 RSC.
- X. Han, J. Zheng, F. Lin, K. Kuroda and Z. Chen, Langmuir, 2018, 34, 512–520 CrossRef CAS PubMed.
- K. Hu, J. Tang, S. Cao, Q. Zhang, J. Wang and Z. Ye, J. Phys. Chem. C, 2019, 123, 9066–9073 CrossRef CAS.
- X. Wang and M. G. McCord, J. Appl. Polym. Sci., 2007, 104, 3614–3621 CrossRef CAS.
- V. Hornebecq, M. Antonietti, T. Cardinal and M. Treguer-Delapierre, Chem. Mater., 2003, 15, 1993–1999 CrossRef CAS.
- J. Voskuhl, C. Wendeln, F. Versluis, E.-C. Fritz, O. Roling, H. Zope, C. Schulz, S. Rinnen, H. F. Arlinghaus, B. J. Ravoo and A. Kros, Angew. Chem., Int. Ed., 2012, 51, 12616–12620 CrossRef CAS PubMed.
- S. Rigo, G. Gunkel-Grabole, W. Meier and C. G. Palivan, Langmuir, 2019, 35, 4557–4565 CrossRef CAS PubMed.
- A. Patel, M. Patel, X. Yang and A. K. Mitra, Protein Pept. Lett., 2014, 21, 1102–1120 CrossRef CAS PubMed.
- L. S. Penn and H. Wang, Polym. Adv. Technol., 1994, 5, 809–817 CrossRef CAS.
- K. S. Mali, N. Pearce, S. D. Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC.
- B. D. Ratner, in Host Response to Biomaterials, ed. S. F. Badylak, Academic Press, Oxford, 2015, pp. 37–51 Search PubMed.
- K. Langowska, C. G. Palivan and W. Meier, Chem. Commun., 2012, 49, 128–130 RSC.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- M. E. Idrissi, C. E. Meyer, L. Zartner and W. Meier, J. Nanobiotechnol., 2018, 16, 63–76 CrossRef PubMed.
- Z. Deng, Y. Qian, Y. Yu, G. Liu, J. Hu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2016, 138, 10452–10466 CrossRef CAS PubMed.
- C. Nardin, S. Thoeni, J. Widmer, M. Winterhalter and W. Meier, Chem. Commun., 2000, 1433–1434 RSC.
- T. Nishimura, Y. Sasaki and K. Akiyoshi, Adv. Mater., 2017, 29, 1702406 CrossRef PubMed.
- O. Rifaie-Graham, S. Ulrich, N. F. B. Galensowske, S. Balog, M. Chami, D. Rentsch, J. R. Hemmer, J. Read de Alaniz, L. F. Boesel and N. Bruns, J. Am. Chem. Soc., 2018, 140, 8027–8036 CrossRef CAS PubMed.
- M. Lomora, F. Itel, I. A. Dinu and C. G. Palivan, Phys. Chem. Chem. Phys., 2015, 17, 15538–15546 RSC.
- M. Spulber, A. Najer, K. Winkelbach, O. Glaied, M. Waser, U. Pieles, W. Meier and N. Bruns, J. Am. Chem. Soc., 2013, 135, 9204–9212 CrossRef CAS PubMed.
- A. Belluati, V. Mikhalevich, S. Yorulmaz Avsar, D. Daubian, I. Craciun, M. Chami, W. Meier and C. Palivan, Biomacromolecules, 2019 Search PubMed , Under Revision.
- K. Langowska, J. Kowal, C. G. Palivan and W. Meier, J. Mater. Chem. B, 2014, 2, 4684–4693 RSC.
- M. Ben Haddada, J. Blanchard, S. Casale, J.-M. Krafft, A. Vallée, C. Méthivier and S. Boujday, Gold Bull., 2013, 46, 335–341 CrossRef.
- J. Eklöf, T. Gschneidtner, S. Lara-Avila, K. Nygård and K. Moth-Poulsen, RSC Adv., 2016, 6, 104246–104253 RSC.
- M. Gosecka and T. Basinska, Polym. Adv. Technol., 2015, 26, 696–706 CrossRef CAS.
- N. Ayres, Polym. Chem., 2010, 1, 769–777 RSC.
- M. Fantin, A. A. Isse, A. Venzo, A. Gennaro and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 7216–7219 CrossRef CAS PubMed.
- P. Chmielarz, Polym. Adv. Technol., 2018, 29, 470–480 CrossRef CAS.
- S. Park, P. Chmielarz, A. Gennaro and K. Matyjaszewski, Angew. Chem., Int. Ed., 2015, 54, 2388–2392 CrossRef CAS PubMed.
- Y. Sun, S. Lathwal, Y. Wang, L. Fu, M. Olszewski, M. Fantin, A. E. Enciso, G. Szczepaniak, S. Das and K. Matyjaszewski, ACS Macro Lett., 2019, 8, 603–609 CrossRef CAS.
- A. Belluati, I. Craciun, J. Liu and C. G. Palivan, Biomacromolecules, 2018, 19, 4023–4033 CrossRef CAS PubMed.
- J.-P. Montheard, M. Chatzopoulos and D. Chappard, J. Macromol. Sci. C, 1992, 32, 1–34 CrossRef.
- M. Srisa-Art, E. C. Dyson, A. J. deMello and J. B. Edel, Anal. Chem., 2008, 80, 7063–7067 CrossRef CAS PubMed.
- A. Jain and K. Cheng, J. Controlled Release, 2017, 245, 27–40 CrossRef CAS PubMed.
- D. Wu, M. Spulber, F. Itel, M. Chami, T. Pfohl, C. G. Palivan and W. Meier, Macromolecules, 2014, 47, 5060–5069 CrossRef CAS.
- C. Draghici, J. Kowal, A. Darjan, W. Meier and C. G. Palivan, Langmuir, 2014, 30, 11660–11669 CrossRef CAS PubMed.
- L. de la Garza, Z. V. Saponjic, N. M. Dimitrijevic, M. C. Thurnauer and T. Rajh, J. Phys. Chem. B, 2006, 110, 680–686 CrossRef CAS PubMed.
- A. J. Cadotte and T. B. Demarse, J. Neural Eng., 2005, 2, 114–122 CrossRef PubMed.
- J. K. Rao, D. V. Ramesh and K. P. Rao, Biomaterials, 1994, 15, 383–389 CrossRef CAS PubMed.
- J. Gu, X. Li, H. Ma, Y. Guan and Y. Zhang, Polymer, 2017, 110, 114–123 CrossRef CAS.
- E. Kang, J.-W. Park, S. J. McClellan, J.-M. Kim, D. P. Holland, G. U. Lee, E. I. Franses, K. Park and D. H. Thompson, Langmuir, 2007, 23, 6281–6288 CrossRef CAS PubMed.
- H. Orelma, L. Johansson, I. Filpponen, O. J. Rojas and J. Laine, Biomacromolecules, 2012, 13, 2802–2810 CrossRef CAS PubMed.
- Y. Ishizuka-Katsura, T. Wazawa, T. Ban, K. Morigaki and S. Aoyama, J. Biosci. Bioeng., 2008, 105, 527–535 CrossRef CAS PubMed.
- L. Wu, U. Glebe and A. Böker, Polym. Chem., 2015, 6, 5143–5184 RSC.
- D. L. Elbert and J. A. Hubbell, Annu. Rev. Mater. Sci., 1996, 26, 365–394 CrossRef CAS.
- O. Stauch, R. Schubert, G. Savin and W. Burchard, Biomacromolecules, 2002, 3, 565–578 CrossRef CAS PubMed.
- P. Tanner, V. Balasubramanian and C. G. Palivan, Nano Lett., 2013, 13, 2875–2883 CrossRef CAS PubMed.
- M. Afzal, S. Matsugo, M. Sasai, B. Xu, K. Aoyama and T. Takeuchi, Biochem. Biophys. Res. Commun., 2003, 304, 619–624 CrossRef CAS PubMed.
- B. Zhao, K. Ranguelova, J. Jiang and R. P. Mason, Free Radicals Biol. Med., 2011, 51, 153–159 CrossRef CAS.
- B. D. Vogt, E. K. Lin, W. Wu and C. C. White, J. Phys. Chem. B, 2004, 108, 12685–12690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: List Fig. S1 is values of contact angles of surfaces, Fig. S2 is AMF image of surface, Fig. S3 and S4 are 1H NMR spectrum of polymers, Fig. S5 is GPC plot of polymers, Table S1 is summary of LS data, Fig. S6 is TEM image of polymersomes, Fig. S7 is AFM image of polymersome on surface, Fig. S8 is QCM-D plot, Fig. S9 and S10 are digital images of catalytic reaction solution. See DOI: 10.1039/c9nr08502e |
| This journal is © The Royal Society of Chemistry 2020 |