
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photon induced quantum yield regeneration of cap-exchanged CdSe/CdS quantum rods for ratiometric biosensing and cellular imaging†
Weili
Wang‡
 a,
Yifei
Kong‡
a,
Jun
Jiang
b,
Xin
Tian
b,
Shuang
Li
c,
Uchangi Satyaprasad
Akshath
a,
Christian
Tiede
d,
Nicole
Hondow
e,
Anchi
Yu
c,
Yuan
Guo
*f and
Dejian
Zhou
*a
a,
Yifei
Kong‡
a,
Jun
Jiang
b,
Xin
Tian
b,
Shuang
Li
c,
Uchangi Satyaprasad
Akshath
a,
Christian
Tiede
d,
Nicole
Hondow
e,
Anchi
Yu
c,
Yuan
Guo
*f and
Dejian
Zhou
*a
aSchool of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK. E-mail: d.zhou@leeds.ac.uk
bSchool of Biology & Basic Medical Sciences, Soochow University, Suzhou 215100, People's Republic of China
cDepartment of Chemistry, Renmin University of China, Beijing 100872, People's Republic of China
dSchool of Molecular and Cellular Biology and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK
eSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
fSchool of Food Science and Nutrition and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK. E-mail: y.guo@leeds.ac.uk
First published on 11th February 2020
Abstract
Full water-dispersion of commercial hydrophobic CdSe/CdS core/shell quantum rods (QRs) was achieved by cap-exchange using a dihydrolipoic acid zwitterion ligand at a low ligand:QR molar ratio (LQMR) of 1000. However, this process almost completely quenched the QR fluorescence, greatly limiting its potential in downstream fluorescence based applications. Fortunately, we found that the QR fluorescence could be recovered by exposure to near ultra-violet to blue light radiation (e.g. 300–450 nm). These “reborn” QRs were found to be compact, bright, and stable, and were resistant to non-specific adsorption, which make them powerful fluorescent probes in broad biomedical applications. We demonstrated their potential in two model applications: first, the QRs were conjugated with His8-tagged small antibody mimetic proteins (also known as Affimers) for the sensitive detection of target proteins via a Förster resonance energy transfer (FRET) readout strategy and second, the QR surface was functionalized with biotins for targeted imaging of cancer cells.
1. Introduction
Over the past three decades, fluorescent semiconductor nano-crystals (aka quantum dots, QDs) have been a major research focus owing to their unique size-dependent, bright, and stable fluorescence which makes them powerful fluorescent probes in broad biomedical applications.1–6 Compared to spherical QDs, elongated rod-shaped quantum rods (QRs) have a higher extinction coefficient and single particle brightness, making them potentially more powerful fluorescent probes.7–10 Moreover, their photophysical properties can be further tuned by altering the aspect (length/width) ratio, which can be extremely beneficial for certain biomedical applications. For example, the aspect ratio has been found to strongly affect the bioluminescence resonance energy transfer (BRET) between a CdSe/CdS core/shell QR and self-assembled firefly luciferase, and an aspect ratio of ∼3 gives the highest BRET ratio.9To date, most high-quality QRs have been synthesised using organometallic precursors in high boiling point coordinating solvents, through which their size and aspect ratio can be finely controlled.7,10 These QRs are naturally capped with hydrophobic ligands, making them only dispersible in non-polar organic solvents and hence are biologically incompatible. As a result, they must be made water-dispersible and biocompatible prior to any biomedical applications. In this regard, amphiphilic polymer encapsulation or coating with silica shells has been widely employed.1–3,7 Despite their success, these methods typically yield relatively bulky QRs (with hydrodynamic diameter, Dh > 20 nm) which can limit their applications in conditions that require compact sizes, e.g. imaging of crowded neuronal synapse, and particularly, in Förster resonance energy transfer (FRET) based applications.4–6,12–17 This is because of the inverse 6th power dependence of the FRET efficiency (E) with a donor–acceptor distance (r), E = 1/[1 + (r/R0)6], where R0 is the Förster radius of the FRET pair and r is the average QR–dye distance. To improve E, it is important to reduce r to a reasonable range (e.g. comparable to R0). Accordingly, cap-exchange is better suited here because it can produce compact sizes required to achieve a high FRET efficiency. In this regard, cap-exchange using poly(ethylene glycol) (PEG) or zwitterionic based ligands appending the dihydrolipoic acid (DHLA)-, poly-DHLA- or poly-histidine-based multi-dentate anchoring group has successfully transferred hydrophobic core/shell QDs into hydrophilic ones. The resulting QDs have been widely used in biosensing, bioimaging, cell tracking, and super-resolution imaging applications.14–21 Despite their success, these strongly chelating ligands have rarely been applied in CdSe/CdS QRs, the most widely studied QRs. In addition, a common drawback of cap-exchange using such strong chelating ligands has been a significant decrease in the fluorescence quantum yield (QY) of the cap-exchanged QDs, limiting their potential as fluorescent probes. Although this problem can be addressed by growing a thick inorganic shell to insulate the fluorescent core from the outer environment, making them more resilient to environmental changes caused by cap-exchange,22 it is not applicable for most commercial core/shell QDs which are known to have relatively thin shells. Moreover, growing a thick shell can significantly increase the overall QD size, leading to reduced FRET efficiency. Recently, we have found that shell etching during cap-exchange is a major factor responsible for the quenched fluorescence of commercial CdSe/ZnS QDs.12 By fully deprotonating the DHLA thiol groups to enhance the cap-exchange efficiency and reducing the ligand![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :QD molar ratio (LQMR) to the minimum, just sufficient to promote full QD cap-exchange and form isolated single QD dispersions, to minimise any possible shell etching, we have unveiled an ultra-efficient cap-exchange protocol which has successfully transferred the commercial hydrophobic CdSe/ZnS and CdSe/ZnSe/ZnS QDs into stable, compact, and water-dispersible ones while retaining >90% of their original fluorescence.12
:QD molar ratio (LQMR) to the minimum, just sufficient to promote full QD cap-exchange and form isolated single QD dispersions, to minimise any possible shell etching, we have unveiled an ultra-efficient cap-exchange protocol which has successfully transferred the commercial hydrophobic CdSe/ZnS and CdSe/ZnSe/ZnS QDs into stable, compact, and water-dispersible ones while retaining >90% of their original fluorescence.12
Here we extended this efficient cap-exchange method to commercial hydrophobic CdSe/CdS core/shell QRs (consisting of a CdSe dot in a CdS rod structure of approximately 3.6 nm width and 24 nm length as revealed by TEM imaging, see the ESI, Fig. S1†) with DHLA-based ligands. Similar to the CdSe/ZnS QDs reported previously, stable QR water-dispersions were readily prepared at a low LQMR of 1000. However, to our surprise, the cap-exchanged QRs were found to be almost fully quenched, making them unusable as fluorescent probes. Fortunately, we discovered that the fluorescence of the QR could be recovered slowly upon exposure to ambient room light and this photon activation process was accelerated by exposing to stronger lights in the near UV to visible region of the spectrum, with the light wavelength (λ) of 350–450 nm being optimal. Moreover, the fluorescence regenerated QRs were readily functionalised with Affimers (small antibody mimetic proteins)23 for FRET based ratiometric biosensing or biotins for targeted imaging of cancer cells.
2. Results and discussion
2.1. QR water-dispersion and characterisation
Scheme 1 shows the chemical structures of the lipoic acid (LA)-based ligands appending different terminal functional groups (e.g. zwitterion, PEG750, and PEG600-biotin) used in this study. These ligands were synthesized by following the literature procedures and purified by either column chromatography (for LA-PEG750 and LA-PEG600-biotin) or by high performance liquid chromatography (for LA-zwitterion).12,15 Details of the ligand synthesis procedures and their spectroscopic data were reported previously.12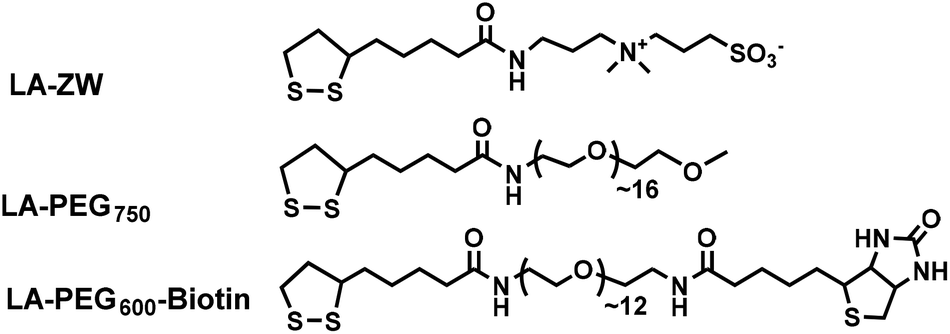 | ||
| Scheme 1 Chemical structures and their abbreviations of the ligands employed in this study. | ||
The procedures to transfer hydrophobic QRs into water-dispersed ones and the corresponding photographs following each step are shown in Fig. 1A/B. The cap-exchange conditions were optimised by performing cap-exchange under different LQMRs and monitoring the QR water-dispersion ability and hydrodynamic diameter (Dh), ensuring that a full water-dispersion and small Dh were obtained.12 The optimised procedures with 0.2 nmol QRs were as follows. First, commercial CdSe/CdS QR stock (20 μM stock, 10 μL in hexane) was precipitated by adding 500 μL of EtOH followed by centrifugation to remove any unbound free ligands. The QR pellet was dissolved in 100 μL CHCl3 and then 50 μL EtOH was added to make a uniform solution (Fig. 1B(i)). Meanwhile, the LA-ZW ligand (0.10 M, 2 μL in H2O) was reduced to DHLA-ZW by mixing with 1 molar equivalent of tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl, 0.10 M, 2 μL in H2O) for 15 min. The reduction of LA to DHLA by TECP was rapid and quantitative as reported previously.24 It could be readily visualised by the naked eye by the disappearance of the yellow colour (due to absorption of LA thiolane). After this, NaOH (0.10 M in EtOH, 12 μL, NaOH:DHLA-ZW ligand molar ratio = 6:1) was added to fully deprotonate DHLA di-thiol groups and to neutralise TCEP·HCl's four acid groups. The resulting solution was then added to the QR dispersion above and thoroughly mixed by shaking by hand for ∼1 min. The solution became turbid quickly, indicating the formation of DHLA-ZW capped QRs which had very low solubility in a mixed CHCl3/EtOH solvent (Fig. 1B(ii)). The suspension was centrifuged (3000 rpm for 10 s) and the QR was pelleted at the bottom of the tube, leaving the supernatant colourless (Fig. 1B(iii)). After careful removal of the supernatant, the QR pellet was dispersed in H2O (100 μL) to give a clear, stable solution (Fig. 1B(iva). The freshly prepared QRs were effectively non-emissive (Fig. 1B(ivb), but were readily transformed into brightly fluorescent ones after 4 h photon activation using a thin layer chromatography (TLC) UV lamp (λ = 365 nm, Fig. 1B(v)). The QR concentration was determined from its 1st exciton peak absorption at ∼545 nm using an extinction coefficient of 2.56 × 105 M−1 cm−1.17
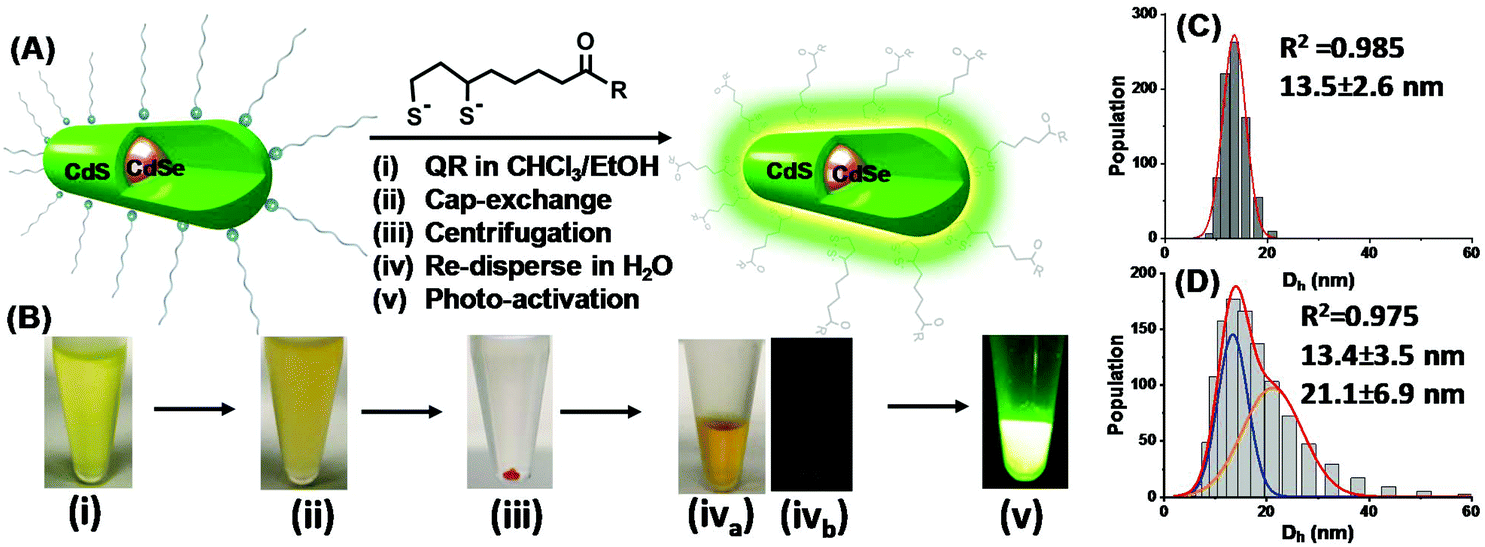 | ||
| Fig. 1 (A) Schematic showing the steps involved in transferring hydrophobic QRs into water-dispersed ones by cap-exchange using DHLA-ZW and photon activation. (B) Photographs showing (i) a CdSe/CdS QR (λEM ∼ 550 nm) dispersed in a mixed CHCl3/EtOH solvent, (ii) after mixing with the deprotonated DHLA-ZW ligand, (iii) after centrifugation; (iv) after removal of the supernatant and dispersion in H2O under ambient- (iva) or UV- (ivb) light illumination, (v) after exposure to 4 h UV light treatment of a TLC UV-lamp (λ = 365 nm) under UV light illumination. (C) Hydrodynamic diameter (Dh) histograms and Gaussian fitting of a freshly prepared DHLA-ZW capped QR showing a single species of 13.5 ± 2.6 nm (mean ± ½ FWHW, full width at half maximum). (D) The Dh histograms of the QR after 4 h UV light (λ = 365 nm) activation showing two Gaussian species of 13.4 ± 3.5 (∼43% area, blue line) and 21.1 ± 6.9 nm (∼57% area, orange line), respectively. | ||
The QR water-dispersion prepared with the DHLA-ZW ligand at a LQMR of 1000 (abbreviated as QR-ZW hereafter) was stable and showed no sign of precipitation or changes in the physical appearance after storage for at least 3 months in darkness (wrapped in aluminium foil). Its Dh sizes measured by dynamic light scattering showed a narrow size distribution which was fitted well by the Gaussian function (R2 = 0.985), giving a single Dh species of 13.5 ± 2.6 nm (mean ± ½ FWHM, FWHM = full width at half maximum obtained from Gaussian fitting, Fig. 1C).12 Despite not being spherical in shape, the QR's diffusion coefficient was comparable to that of a sphere of ∼13.5 nm, a value that lay in between the width and length of the QR, and was ∼1/2 that of the QR length after taking the DHLA-ZW ligand coating thickness into consideration. This indicated that the QR-ZW obtained here was made of isolated single QR particles, and not aggregated/clustered ones.
Theoretically, a cylinder shaped QR of 3.6 nm width and 24 nm length would have a total surface area of 291 nm2. Assuming that CdS adopted its stable wurtzite structure and terminated with a full Cd2+ outer layer, then each QR would have 1940 Cd2+ ions on its surface (see the ESI, Fig. S2†). Assuming that each thiolate coordinated to one Cd2+ ion, a LQMR of 970 with the DHLA-based ligand (each having 2 thiol groups) would completely cap all the QR surface Cd2+ ions and form a stable water-dispersion. This number matched well with the observation that a LQMR of 1000 was able to completely water-disperse the QR and form isolated single particles. Significantly, the LQMR used here to achieve complete QR water-dispersion was 1–2 orders of magnitude lower than that used in most literature methods,11,14–21 suggesting the ultra-efficiency of our cap-exchange method. Moreover, this calculation also indicated that there should be almost no free DHLA-ligands left in the system to produce any photo-chemical transformations.25 Nonetheless, this result was fully consistent with those obtained with the CdSe/ZnS and CdSe/ZnSe/ZnS QDs reported previously.12 Moreover, a TEM image of the cap-exchanged QRs revealed that they had almost identical particle shapes and sizes to the parent QRs prior to cap-exchange (ESI, Fig. S1†), suggesting no observable etching of the QR structure after cap-exchange. This result was also consistent with our above estimation that most of the added DHLA-ZW ligands should have capped onto the QR surface, leaving almost no free ligands that could etch the QR shell surface Cd2+ ions.12
2.2. Photon-induced QR fluorescence regeneration
Despite its success in promoting full QR water-dispersion, the resulting QR-ZW was found to be almost completely quenched, retaining just ∼2% of its original fluorescence (in CHCl3). In contrast, the CdSe/ZnS and CdSe/ZnSe/ZnS12 QDs were able to retain >90% of their original fluorescence under equivalent cap-exchange conditions (see the ESI, Fig. S3†).12 The difference here was likely due to the different band energy levels of the core and shell structures. Both the CdSe/ZnS and CdSe/CdS/ZnS QDs had a type-I core/shell band structure whose exciton carriers (electron and hole) were fully confined within the core,26 and thus a small change to the shell induced by cap-exchange would not significantly affect their fluorescence (see the ESI, Fig. S4†). However, the CdSe/CdS QR had a Quasi-type II core/shell band structure: the hole was confined in the core, but the electron was spread across both the core and shell structure.26 As a result, the QR is much more sensitive to even small changes to the shell: any new surface defects induced by cap-exchange could trap the excited electron and prevent its emissive recombination with the hole, giving rise to greatly quenched QR fluorescence.26 This problem greatly limited the potential use of the CdSe/CdS QRs as fluorescent probes in biomedical research. Moreover, this problem could not be solved by reducing the LQMR to the minimum (e.g. 1000 here) to minimize shell etching as we had achieved successfully with the type I QDs.12 Fortunately, we discovered that the fluorescence of the QRs gradually recovered over a few weeks upon exposure to ambient room light during storage, and the recovery rate was increased by exposing to UV light (λ = 365 nm, see the ESI, Fig. S5†).To investigate the driving force of this photon induced fluorescence regeneration, a freshly prepared QR-ZW was divided into 3 portions and exposed to different light conditions: UV light (a TLC UV lamp, λ = 365 nm), ambient room light, or in darkness (wrapped in aluminum foil). The QR exposed to UV light was found to recover rapidly, regaining ∼82% of its original fluorescence in 5 h (Fig. S5A†). The QR exposed to ambient room light recovered slowly, regaining ∼62% of its original fluorescence over 1 week (Fig. S5B†). The QR stored in darkness with occasional exposure to ambient room light (when taken out for inspection) showed very slow fluorescence recovery over 4 weeks (Fig. S5C†). This result revealed that both the intensity and wavelength of light radiation played an important role in the QR fluorescence recovery. Moreover, the photon activation behavior appeared to be general and applicable to other DHLA-based ligand capped QRs. For example, an initially “dark” DHLA-PEG750 capped QR showed greatly enhanced fluorescence after 4 h UV light activation (λ = 365 nm, via a TLC UV lamp), similar to that observed with QD-ZW. Moreover, it could retain the bright fluorescence for at least 1 month (see the ESI, Fig. S6†), suggesting that the photon activation effect was permanent.
To further investigate the dependence of QR fluorescence recovery on the wavelength of the exposing light, a freshly prepared QR-ZW water-dispersion (a different batch, λEM = ∼570 nm) was placed in a cuvette and its emission spectrum was repeatedly recorded using a fluorometer under a given excitation wavelength (e.g. λEX = 200, 300, 350, 400, 450 or 500 nm, respectively). The resulting scan number dependent fluorescence spectra revealed that QR fluorescence regeneration was only observed with λEX over a range of 300–450 nm, but not at a longer (e.g. 500 nm) or shorter (e.g. 200 nm) wavelength (see Fig. 2). In the longer λEX range, it appeared that the QR fluorescence could only be regenerated with photon energy higher than the 1P3/2 → 1Pe transition (λ = ∼460 nm as measured by transient absorption, see the next section). Interestingly, the photon activation process typically displayed three different stages. First, a lag phase where a minimal fluorescence increase was observed with an increase in the number of scans, followed by a photon activation phase where the QR fluorescence increased rapidly and roughly linearly with the number of scans, and finally a saturation phase where the QR fluorescence plateaued. λEX = 400 nm was found to be optimal for QR fluorescence regeneration: it gave the smallest number of scans in the lag phase (∼10 vs. ∼18 or ∼25 for λEX = 350 or 450 nm, respectively) and the highest increase rate per scan at the photon activation phase (ESI, Table S1†). Overall, >16 fold fluorescence enhancement was achieved with this batch of QR-ZW via photon-activation (Fig. 2D). This result thus established a facile route to control the QR fluorescence regeneration by simply applying a certain number of emission scans under a certain λEX.
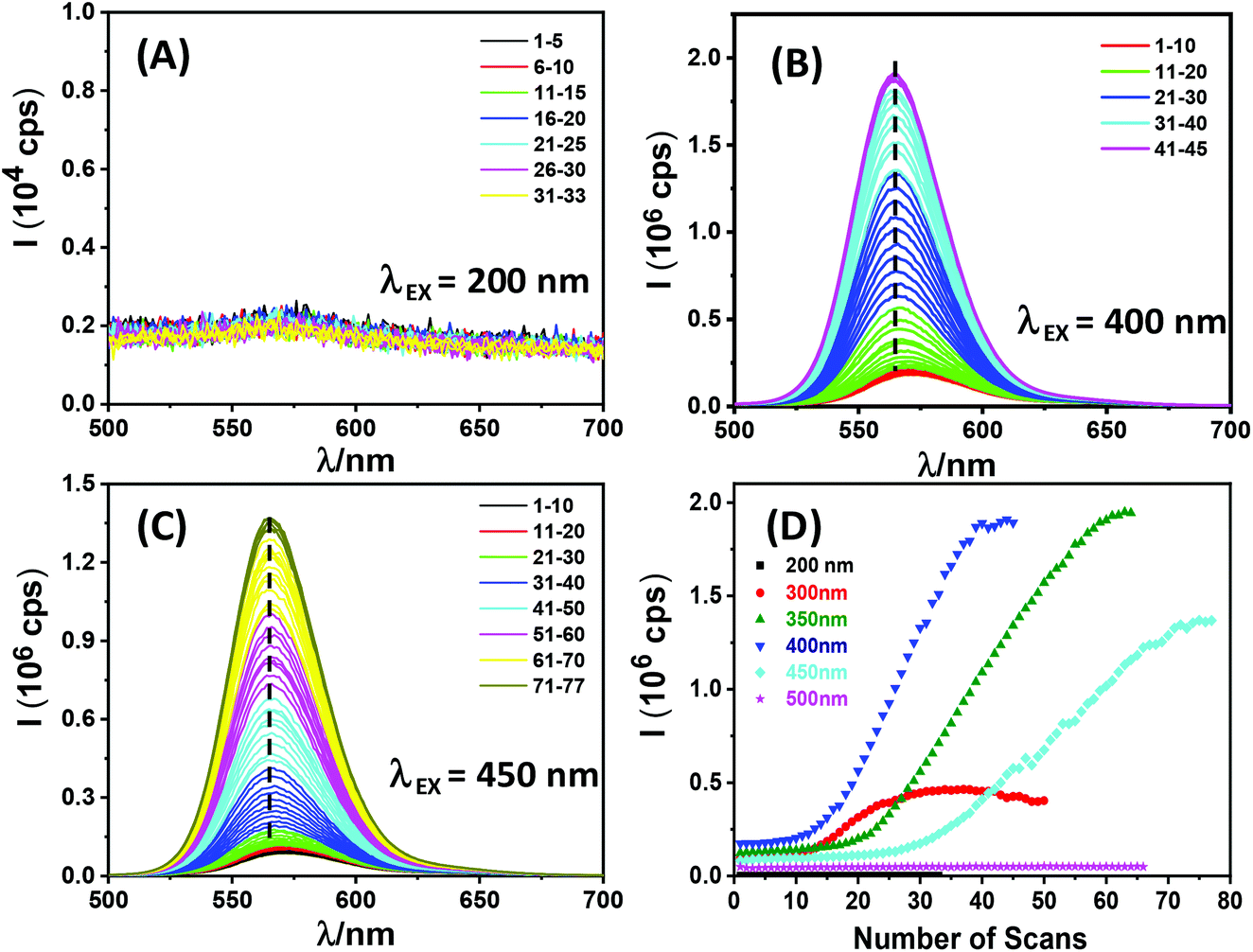 | ||
| Fig. 2 Fluorescence spectra of a freshly prepared DHLA-ZW ligand capped CdSe/CdS QR in H2O (5 nM) after repeated emission scans at a fixed excitation wavelength (λEX) of (A) 200 nm, (B) 400 nm and (C) 450 nm. (D) A plot of the QR fluorescence intensity versus the number of scans under different λEX. | ||
Interestingly, careful analysis of the fluorescence spectra during the repeated emission scans revealed a small but noticeable blue shift of the peak λEM at ∼6 nm (from ∼570 to ∼564 nm) with the photon regenerated QRs. Moreover, the onset of photon activation appeared to be coincident with the blue shift of the λEM. This result was consistent with the photo-oxidation induced fluorescence brightening mechanism previously observed with CdSe/CdS/ZnS QRs by the Alivisatos group10a and a CdSe/CdS QD by the Peng group.10b Moreover, the Dh distribution histograms of a UV-light regenerated QR (4 h exposure with a TLC UV lamp, λ = 365 nm) were best fitted by two Gaussian species, giving Dh values of 13.4 ± 0.3 (FWHM: 5.9 nm, 43% of area) and 21.1 ± 2.3 nm (FWHM: 11.8 nm, 57% of area), respectively (see Fig. 1D). The smaller species was almost identical to the freshly prepared QD-ZW particles, while the bigger ones could be assigned to clustered QRs after photon activation. Furthermore, prolonged exposure of the QRs to UV light radiation (e.g. >10 h, with a TLC UV lamp) produced extensive QR aggregation that eventually led to precipitation. This result was consistent with the reduced QR colloidal stability due to ligand depletion induced by photo-oxidation. All these results suggested that photo-oxidation induced fluorescence brightening10 was the most likely mechanism behind the observed QR fluorescence regeneration here.
Importantly, the photon regenerated QRs (4 h UV light exposure) still exhibited a Dh size useful for passive cancer tumour targeting via the enhanced permeation and retention (EPR) effect, a characteristic pathological condition of many cancer tumours.27a They were bigger than the gaps of healthy blood vessels (to reduce non-specific accumulation in healthy tissues) and the renal clearance threshold (∼8 nm, to allow for prolonged blood circulation time),27b but were smaller than the average gaps of the leaky blood vessels in tumours (e.g. up to 200 nm) and so could easily diffuse into and accumulate inside tumours. A combination of bright fluorescence and suitable Dh sizes for EPR based passive targeting made such QRs attractive for potential in vivo cancer tumour targeting and imaging applications.
To further investigate the mechanism of photon activation, we recorded the transient absorption spectra of a freshly prepared “dark” QR-ZW and two such QRs after photon activation via exposure to ambient room light for 15 and 30 days, respectively, on a femto-second transient absorption system.28 The transient absorption spectra showed two apparent negative bands, a weak band at 545 nm (corresponding to the first excitonic transition of the CdSe core, 1S3/2 → 1Se and 2S3/2 → 1Se) and a strong band at 460 nm (assigned to the 1P3/2 → 1Pe transition, see Fig. 3A).28,29 To better understand how photon activation affected the QR transient absorption, the time dependent decay curves at 470 nm were investigated. The decay curves of the freshly prepared “dark” QR could be fitted well by two exponential decays, while the photon regenerated QRs were best fitted by three exponential decays (Fig. 3B). The resulting fitting parameters are summarized in Table 1. The average lifetime (τave) of the QR was found to have increased considerably after photon activation, suggesting that photon activation reduced some rapid energy dispersion pathways (possibly due to the reduction of the QR surface electron traps via photon oxidation). Due to the short 1P to 1S relaxation lifetime, the lifetime from 1P3/2 to 1Pe approximated to the lifetime from 1S3/2 to 1Se.28,29 This result was consistent with the steady-state fluorescence spectra observed above.
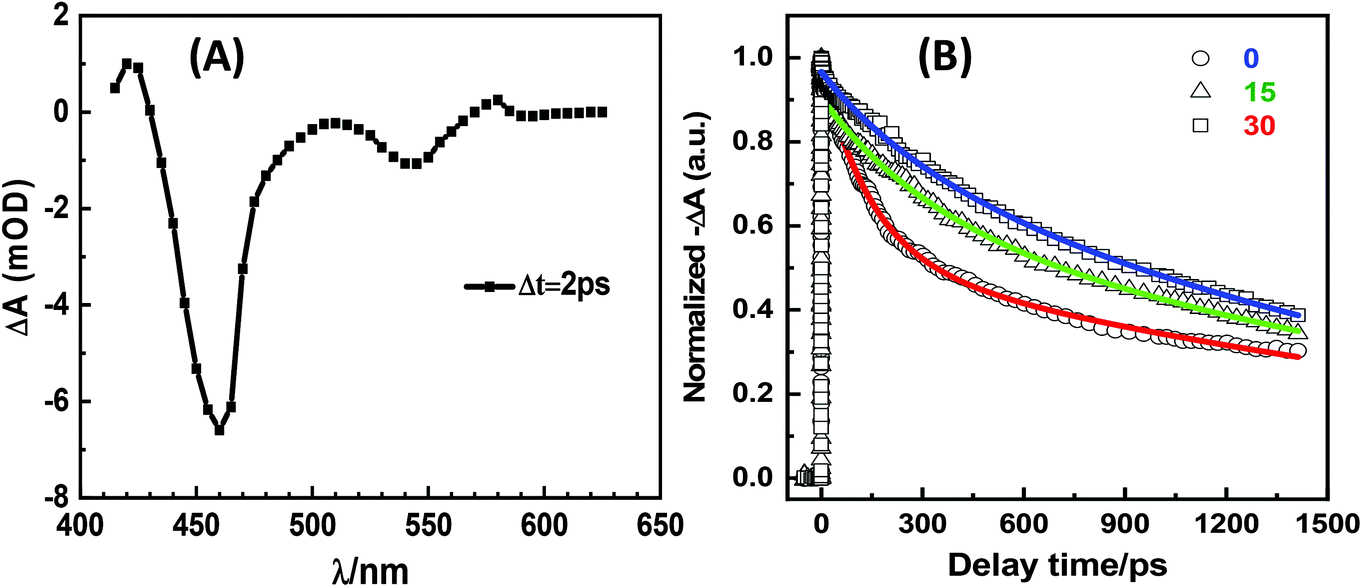 | ||
| Fig. 3 (A) Femtosecond transient absorption spectrum of the 30 day ambient room light activated QR in H2O recorded at 2 ps after 460 nm excitation. (B) Decay kinetics of the QRs in H2O after different light treatment conditions: freshly prepared without photon activation (○), after exposure to ambient room light for 15 (Δ) or 30 (□) days. Pump wavelength: 460 nm; probe wavelength: 470 nm. | ||
| Sample | a 1 | τ 1/ps | a 2 | τ 2/ps | a 3 | τ 3/ps | τ ave/ps |
|---|---|---|---|---|---|---|---|
| 0 | — | — | 0.46 | 145 ± 10 | 0.54 | 2330 ± 50 | 1320 ± 50 |
| 15 day | 0.09 | 3 ± 0.5 | 0.25 | 310 ± 10 | 0.66 | 2240 ± 50 | 1560 ± 50 |
| 30 day | 0.05 | 2 ± 0.5 | 0.57 | 770 ± 50 | 0.38 | 6400 ± 50 | 1650 ± 50 |
2.3. QR-Affimer for ratiometric biosensing
The high brightness of the photon regenerated QRs made them attractive for FRET-based sensing applications. To explore this potential, the QR-ZW was first photon regenerated by exposure to a TLC UV lamp (λ = 365 nm) for 4 h. Affimers were selected as the target protein binders due to their small molecular sizes (<1/10 of a full antibody)23 which could significantly reduce the FRET distance (r) and improve sensitivity.12 Here an anti-yeast SUMO (small ubiquitin-like modifier) Affimer was used to detect the target SUMO protein, acting as a model disease biomarker.23 The Affimer was modified by a C-terminal His8-tag for convenient QR conjugation via a facile metal-affinity self-assembly process as reported previously.4a,12,23First, we studied the QR-Affimer self-assembly by mixing an Atto-594 labeled Affimer with the QR-ZW (λEM ∼ 550 nm) under different molar ratios. The QR-Atto-594 FRET pair showed a good spectral overlap and an acceptable R0 of ∼5.5 nm (ESI, Fig. S7†), ensuring that efficient FRET could take place over a reasonable r. Meanwhile, their emission spectra showed minimal overlap, allowing for easy separation of the donor–acceptor emissions without the need for spectral deconvolution. All the fluorescence spectra were recorded at λEX = 450 nm, corresponding to the absorption minimum of Atto-594, to minimize the dye direct excitation background.6 A progressively quenched QR fluorescence together with a concurrently enhanced Atto-594 FRET signal (at ∼625 nm) were observed with an increase in the Affimer/QR ratio, which agreed well with the QR-sensitized Atto-594 dye FRET mechanism produced by QR-Affimer conjugation (ESI, Fig. S7C†). Fitting the FRET efficiency (E, determined from the QR donor quenching) dye/QR molar ratio relationship by a single QR donor in FRET interaction with an N identical acceptor model,14E = 1/[1 + (r/R0)6/N], gave an average FRET distance (r) of ∼5.9 nm (ESI, Fig. S7D†). Given the rod shape rather than the spherical structure of the QR, and the inverse 6th power dependence of E on r, the r value derived from the FRET studies here would predominantly represent Affimers assembled on the QR next to the fluorescent CdSe core at the centre of the rod as revealed from EDX mapping (see the ESI, Fig. S1†). Those assembled at the ends of the rod would be too far away to produce strong FRET with the QR, and hence would contribute very little to the FRET results. Nonetheless, this result indicated the formation of a compact QR-Affimer structure, at least in the central rod region, making it well-suited for FRET based sensing applications.12,13
We subsequently employed the QR-Affimer (unlabeled) conjugate for detecting the target SUMO protein. Here QR-ZW each linked with 40 copies of unlabeled Affimer was mixed with an increasing amount of the target SUMO protein (Atto-594 labelled). Fig. 4 shows that, with an increase in the SUMO concentration, the QR fluorescence was greatly quenched while the Atto-594 FRET signal was enhanced simultaneously, consistent with the QR-sensitized Atto-594 FRET mechanism induced by Affimer–SUMO binding. A plot of the peak intensity ratio of Atto-594 to the QR, I625/I549, versus the SUMO concentration revealed a two stage linear relationship: a slow phase at lower concentrations (from 0.2 to 20 pM, fitting equation: y = −1.95 + 0.12x, R2 = 0.983) and a rapid phase at higher concentrations (from 20 to 10000 pM, y = −2.37 + 0.41x, R2 = 0.925), respectively. The two stage FRET response described here appeared to resemble that of an earlier QD-FRET based DNA biosensor.17b The FRET ratio obtained with 5 pM protein was clearly above the limit of detection (indicated by the green dashed line, Fig. 4B) using the background + 3σ criteria (σ: standard deviation).30 This result suggested that the QR-Affimer sensor could ratiometrically detect the target protein down to the low pM level. This sensitivity was comparable to some of the most sensitive QD-FRET based sensors for protein detection.13
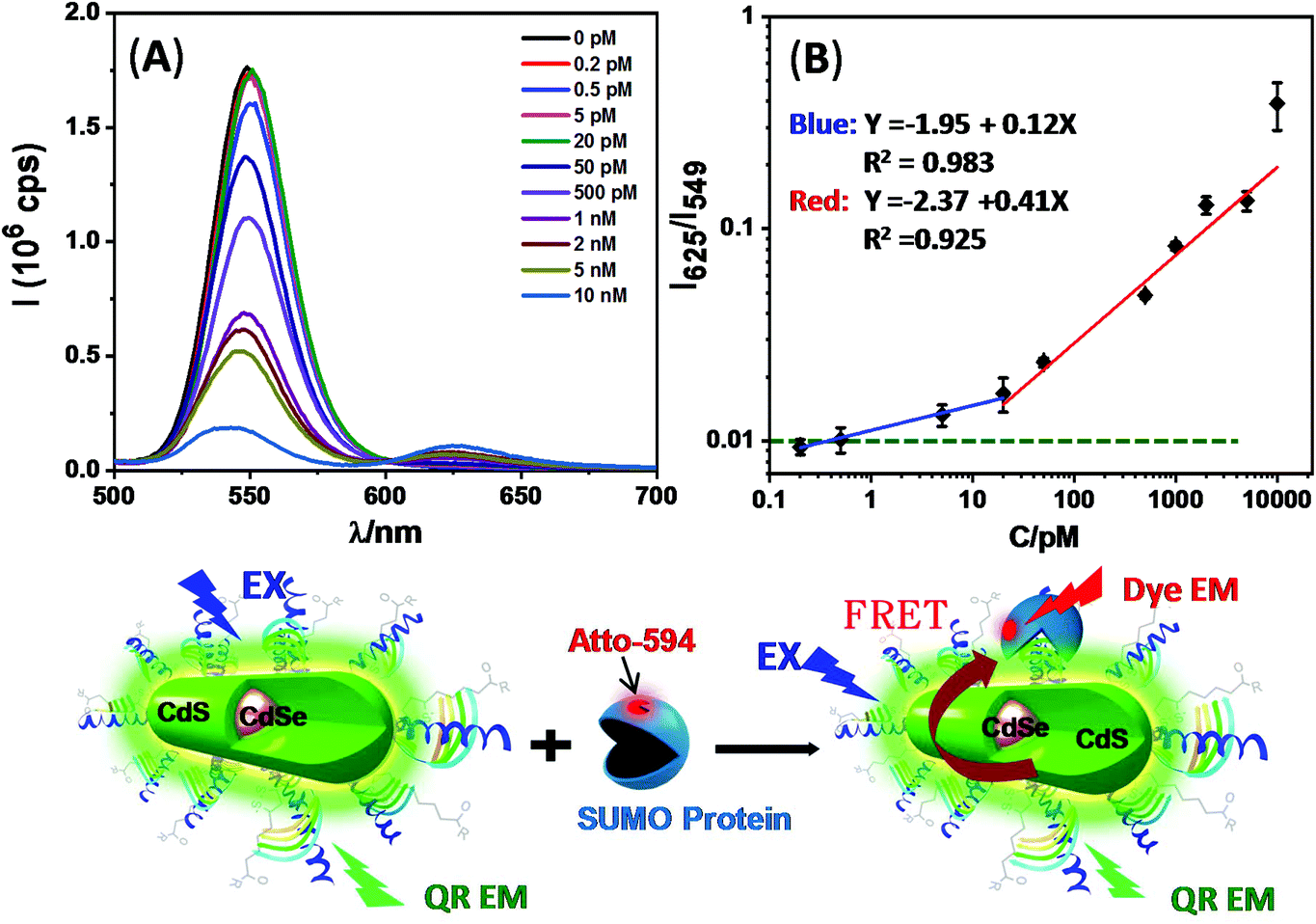 | ||
| Fig. 4 (A) Fluorescence spectra of the QR-Affimer conjugate (0.10 nM QR) after incubation with different amounts of Atto-594 labeled SUMO in PBS. (B) The relationship between the I625/I549 ratio versus the SUMO concentration (in the log–log plot). The data were best fitted in two linear functions, a slower increase between 0.2 and 20 pM (R2 = 0.983) and a sharper increase at 20–10000 pM (R2 = 0.925). The dashed green line indicates the limit of detection based on the background + 3σ criteria. The scheme beneath the figure showing the Affimer–SUMO protein binding induced FRET mechanism. Light symbols: blue (excitation light, EX); green (QR emission) and red (dye emission). | ||
We further investigated the potential of the QR-Affimer for detecting unlabeled proteins via a similar approach to that of the QD-Affimer reported previously.12 Here we first conjugated each QR with 1 copy of the anti-SUMO Affimer (via His8-tag self-assembly) and then blocked the QR surface with 40 copies of a control Affimer (also His8-tagged) showing no binding affinity to the target SUMO protein.23 An Atto-594 labeled SUMO protein (as the FRET reporter, 1 molar equivalent to the QR) was also added to develop the QR-Affimer sensing system.12 Addition of unlabeled SUMO proteins would compete with the labelled SUMO binding to the QR-Affimer, reducing the FRET efficiency and giving rise to the QR fluorescence recovery in a dose dependent manner. A plot of the I549/I625 ratio versus the SUMO concentration revealed that the signal of 1 nM SUMO was clearly above the level of background + 3σ, suggesting that it could detect unlabeled proteins down to the sub-nM level (see the ESI, Fig. S8†). This level of sensitivity was comparable to those of other sensitive QD-FRET sensors for the detection of unlabeled proteins.13
2.4. Preparation of QR-biotin for biosensing and cell imaging
Biotin-functionalized QRs were readily prepared by one-step cap-exchange using the mixed DHLA-PEG600-biotin and DHLA-ZW ligands. Here we found that cap-exchange using a total LQMR of 1200 (1100 for DHLA-ZW and 100 for DHLA-PEG600-biotin) could completely water-disperse the QR and form a stable dispersion (denoted as QR-biotin100 hereafter). Moreover, by varying the DHLA-PEG600-biotin to DHLA-ZW ligand ratio while keeping the total LQMR fixed at 1200 to perform cap-exchange, we were able to vary the QR surface biotin valency.12 Using this method, QR-biotin50 and QR-biotin500 were also prepared for cancer cell imaging (see the next section, the detailed procedures are given in the ESI†).QR-biotin100 was first employed for neutravidin detection via the strong biotin–neutravidin binding (Kd ∼ 10−15 M).31 As with the QR-ZW above, QR-biotin100 was photon activated and then incubated with an increasing amount of neutravidin (Atto-594 labelled). A good positive linear response between the FRET ratio and the neutravidin concentration over 0–15 nM with a detection limit of ∼0.2 nM was obtained (see the ESI, Fig. S9†). This sensor was highly specific: mixing Atto-594 labeled neutravidin (100 nM) with the control QR-ZW (1 nM, capped with the DHLA-ZW ligand only) gave no apparent quenching of the QR fluorescence. The FRET ratio of the control QR mixed with 100 nM neutravidin (corrected for dye direct excitation background) was 0.0042, <10% of that of 1 nM neutravidin binding to QR-biotin100. Given a positive linear relationship between the FRET ratio and the amount of QR bound proteins,4,6 this result indicated that the FRET signal obtained from the biotin–neutravidin binding was >1000 fold higher than that of non-specific adsorption, demonstrating an excellent sensing specificity.
Biotin is an important vitamin (known as vitamin H) and many cancer cells have shown to over-express biotin receptors, making it a useful ligand for cancer targeting.32–34 Here biotinylated QRs were used to target the model 4T1 breast cancer cells via their surface over-expressed biotin receptors. The 4T1 cancer cells (1.5 × 105 cells mL−1) were incubated with the QR-ZW control, QR-biotin50 or QR-biotin500 (all at 50 nM final QR concentration) for 4 h, washed with phosphate-buffered saline, and then treated with Hochest 33342 for nuclei staining. The cells were then imaged by laser scanning confocal microscopy using 488 nm laser excitation and the QR emission at >525 nm was collected and exhibited a green colour. The 4T1 cells treated with the control QR-ZW displayed almost no green fluorescence, suggesting very little cell uptake (top panel, Fig. 5). In contrast, cells treated with the QR-biotins showed strong green fluorescence, and the cellular QR fluorescence appeared to be positively correlated to the QR surface biotin valency (Fig. 5). These results are consistent with the biotin receptor mediated cell uptake mechanism,33,34 suggesting that the biotinylated QRs could be used for targeted fluorescence imaging of cancer cells.
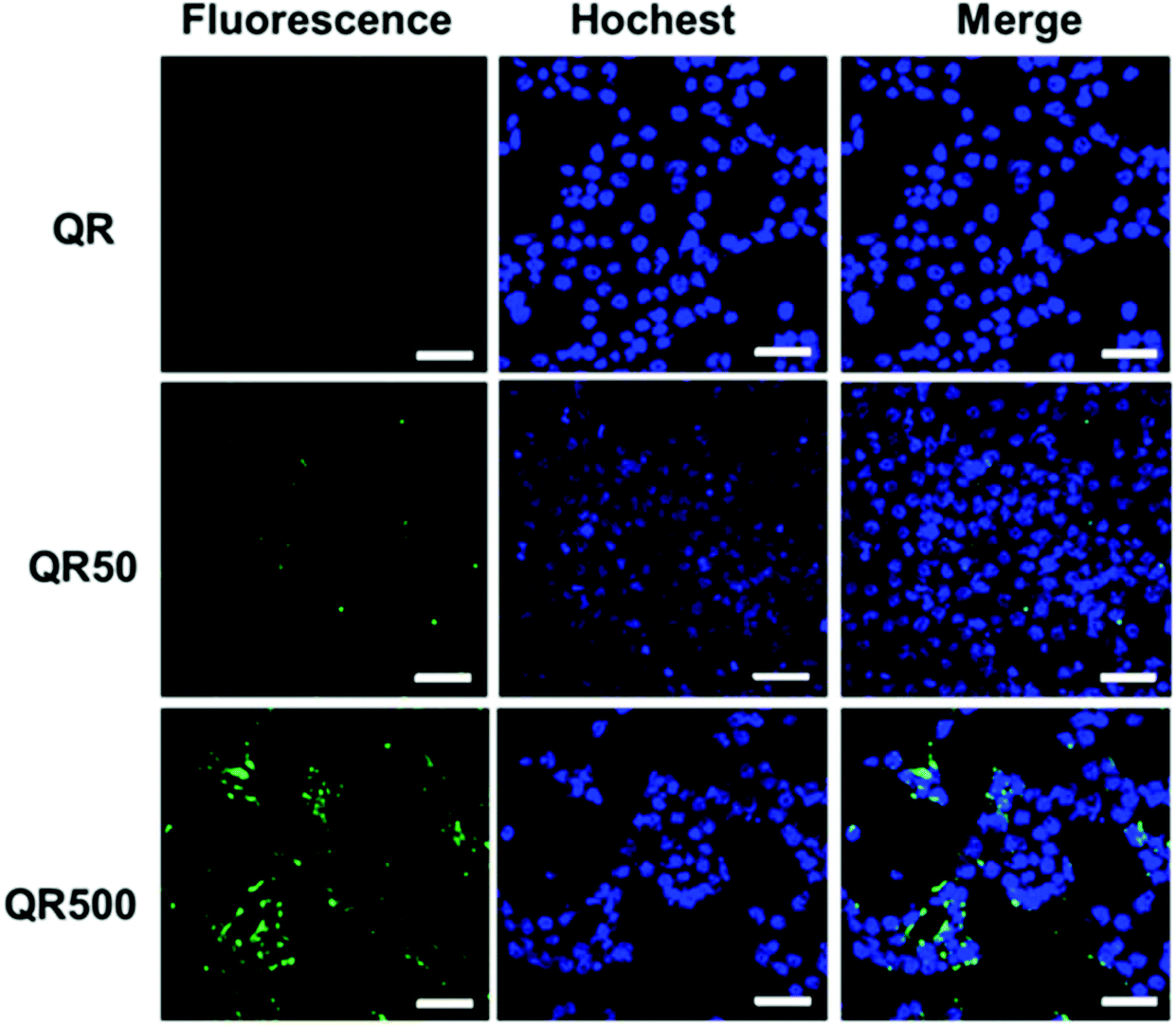 | ||
| Fig. 5 Confocal fluorescence images of 4T1 cells after 4 h treatment with 50 nM control QR-ZW (top, QR), QR-biotin50 (middle, QR50) or QR-biotin500 (bottom, QR500). From left to right, fluorescence images were recorded in the QR channel (green), Hochest channel (blue, staining nuclei) and the merged images of the QR/Hochest channels; scale bar = 50 μm. | ||
3. Conclusions
To sum up, a highly efficient cap-exchange method was employed to water-disperse hydrophobic CdSe/CdS QRs to produce compact, biocompatible, but almost completely quenched QRs. Fortunately, the resulting QR fluorescence could be effectively regenerated via a novel photon activation method. Such fluorescence recovered QRs were successfully conjugated with His8-tagged Affimers for the sensitive detection of 5 pM of a target protein. This cap-exchange method was successfully employed for the one-step preparation of biotin-functionalized QRs for the ratiometric detection of pM levels of neutravidin and also for specific imaging of cancer cells. Compared to the other methods reported in the literature, this method has the advantages of ease of operation (using air-stable ligands and rapid in situ reduction under ambient conditions), requirement of a minimal amount of ligands, and a simple light-activation process to regenerate QR fluorescence. The high brightness and compact structures of such photon regenerated QRs make them attractive fluorescent probes in broad biosensing and imaging applications, especially those relying on FRET-based readout strategies.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the University of Leeds, the Wellcome Trust (Grant No: 097354/Z/11/Z), the Biotechnology and Biological Sciences Research Council (grant no: BB/R007829/1) and the EU Horizon 2020 via a Marie Sklodowska-Curie Fellowship (grant no: 797597) for funding this research.References
- (a) M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013–2016 CrossRef CAS PubMed; (b) W. C. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- (a) X. Michalet, F. Pinaud, L. Bentolila, J. Tsay, S. Doose, J. Li, G. Sundaresan, A. Wu, S. Gambhir and S. Weiss, Science, 2005, 307, 538–544 CrossRef CAS PubMed; (b) K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed; (c) K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC; (d) I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17–45 RSC; (e) D. Zhou, Biochem. Soc. Trans., 2012, 40, 635–639 CrossRef CAS PubMed.
- (a) B. N. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed; (b) D. R. Larson, W. R. Zipfel, R. M. Williams, S. W. Clark, M. P. Bruchez, F. W. Wise and W. W. Webb, Science, 2003, 300, 1434–1436 CrossRef CAS PubMed.
- (a) I. L. Medintz, A. R. Clapp, F. M. Brunel, T. Tiefenbrunn, H. T. Uyeda, E. L. Chang, J. R. Deschamps, P. E. Dawson and H. Mattoussi, Nat. Mater., 2006, 5, 581–589 CrossRef CAS PubMed; (b) I. L. Medintz, M. H. Stewart, S. A. Trammell, K. Susumu, J. B. Delehanty, B. C. Mei, J. S. Melinger, J. B. Blanco-Canosa, P. E. Dawson and H. Mattoussi, Nat. Mater., 2010, 9, 676–684 CrossRef CAS PubMed; (c) C. Y. Zhang, H. C. Yeh, M. T. Kuroki and T. H. Wang, Nat. Mater., 2005, 4, 826–831 CrossRef CAS PubMed.
- (a) E. R. Smith, J. M. Luther and J. C. Johnson, Nano Lett., 2011, 11, 4923–4931 CrossRef CAS PubMed; (b) W. R. Algar, M. G. Ancona, A. P. Malanoski, K. Susumu and I. L. Medintz, ACS Nano, 2012, 6, 11044–11058 CrossRef CAS PubMed.
- (a) Y. Guo, C. Sakonsinsiri, I. Nehlmeier, M. A. Fascione, H. Y. Zhang, W. L. Wang, S. Pohlmann, W. B. Turnbull and D. J. Zhou, Angew. Chem., Int. Ed., 2016, 55, 4738–4742 CrossRef CAS PubMed; (b) Y. Guo, I. Nehlmeier, E. Poole, C. Sakonsinsiri, N. Hondow, A. Brown, Q. Li, S. Li, J. Whitworth, Z. Li, A. Yu, R. Brydson, W. B. Turnbull, S. Pohlmann and D. Zhou, J. Am. Chem. Soc., 2017, 139, 11833–11844 CrossRef CAS PubMed.
- (a) L. S. Li, J. Hu, W. Yang and A. P. Alivisatos, Nano Lett., 2001, 1, 349–351 CrossRef CAS; (b) A. Fu, W. Gu, B. Boussert, K. Koski, D. Gerion, L. Manna, M. Le Gros, C. A. Larabell and A. P. Alivisatos, Nano Lett., 2007, 7, 179–182 CrossRef CAS PubMed.
- (a) A. E. Albers, E. M. Chan, P. M. McBride, C. M. Ajo-Franklin, B. E. Cohen and B. A. Helms, J. Am. Chem. Soc., 2012, 134, 9565–9568 CrossRef CAS PubMed; (b) S. Halivni, A. Sitt, I. Hadar and U. Banin, ACS Nano, 2012, 3, 2758–2765 CrossRef PubMed; (c) K. Wu, L. J. Hill, J. Chen, J. R. McBridge, N. G. Pavlopolous, N. E. Richey, J. Pyun and T. Lian, ACS Nano, 2015, 9, 4591–4599 CrossRef CAS PubMed.
- (a) R. Alam, D. M. Fontaine, B. R. Branchini and M. M. Maye, Nano Lett., 2012, 12, 3251–3256 CrossRef CAS PubMed; (b) R. Alam, L. M. Karam, T. L. Doane, K. Coopersmith, D. M. Fontaine, B. R. Branchini and M. M. Maye, ACS Nano, 2016, 10, 1969–1977 CrossRef CAS PubMed.
- (a) L. Manna, E. C. Scher, L.-S. Li and A. P. Alivisatos, J. Am. Chem. Soc., 2002, 124, 7136–7145 CrossRef CAS PubMed; (b) J. J. Li, Y. A. Wang, W. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. Peng, J. Am. Chem. Soc., 2003, 125, 12567–12575 CrossRef CAS PubMed.
- D. Liu and P. T. Snee, ACS Nano, 2011, 5, 546–550 CrossRef CAS PubMed.
- W. Wang, Y. Guo, C. Tiede, S. Chen, M. Kopytynski, Y. Kong, A. Kulak, D. Tomlinson, R. Chen, M. McPherson and D. Zhou, ACS Appl. Mater. Interfaces, 2017, 9, 15232–15244 CrossRef CAS PubMed.
- (a) W. R. Algar, H. Kim, I. L. Medintz and N. Hildebrandt, Coord. Chem. Rev., 2014, 263, 65–85 CrossRef; (b) N. S. Hildebrandt, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- (a) A. R. Clapp, I. L. Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2004, 126, 301–310 CrossRef CAS PubMed; (b) E. R. Goldman, I. L. Medintz, J. L. Whitley, A. Hayhurst, A. R. Clapp, H. T. Uyeda, J. R. Deschamps, M. E. Lassman and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 6744–6751 CrossRef CAS PubMed; (c) I. L. Medintz, J. Konnert, A. Clapp, I. Stanish, M. Twigg, H. Mattoussi, J. M. Mauro and J. Deschamps, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9612–9617 CrossRef CAS PubMed.
- (a) K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987–13996 CrossRef CAS PubMed; (b) K. Susumu, B. C. Mei and H. Mattoussi, Nat. Protoc., 2009, 4, 424–436 CrossRef CAS PubMed; (c) N. Zhan, G. Palui, H. Grise, H. Tang, I. Alabugin and H. Mattoussi, ACS Appl. Mater. Interfaces, 2013, 5, 2861–2869 CrossRef CAS PubMed.
- M. Howarth, W. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397–399 CrossRef CAS PubMed.
- (a) D. Zhou, Y. Li, E. A. H. Hall, C. Abell and D. Klenerman, Nanoscale, 2011, 3, 201–211 RSC; (b) H. Zhang, G. Feng, Y. Guo and D. Zhou, Nanoscale, 2013, 5, 10307–10315 RSC; (c) D. Zhou, L. Ying, X. Hong, E. A. Hall, C. Abell and D. Klenerman, Langmuir, 2008, 24, 1659–1664 CrossRef CAS PubMed.
- (a) W. Liu, M. Howarth, A. B. Greytak, Y. Zheng, D. G. Nocera, A. Y. Ting and M. Bawendi, J. Am. Chem. Soc., 2008, 130, 1274–1284 CrossRef CAS PubMed; (b) E. Muro, T. Pons, N. Lequeux, A. Fragola, N. Sanson, Z. Lenkei and B. Dubertret, J. Am. Chem. Soc., 2010, 132, 4556–4557 CrossRef CAS PubMed.
- L. Ma, C. L. Tu, P. Le, S. Chitoor, S. J. Lim, M. U. Zahid, K. W. Teng, P. H. Ge, P. R. Selvin and A. M. Smith, J. Am. Chem. Soc., 2016, 138, 3382–3394 CrossRef CAS PubMed.
- W. H. Liu, A. B. Greytak, J. Lee, C. R. Wong, J. Park, L. F. Marshall, W. Jiang, P. N. Curtin, A. Y. Ting, D. G. Nocera, D. Fukumura, R. K. Jain and M. G. Bawendi, J. Am. Chem. Soc., 2010, 132, 472–483 CrossRef CAS PubMed.
- W. Wang, X. Ji, A. Kapur, C. Zhang and H. Mattoussi, J. Am. Chem. Soc., 2015, 137, 14158–14172 CrossRef CAS PubMed.
- (a) Y. Chen, J. Vela, H. Htoon, J. L. Casson, D. J. Werder, D. A. Bussian, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 5026–5027 CrossRef CAS PubMed; (b) J. Zhou, M. Zhu, R. Meng, H. Qin and X. Peng, J. Am. Chem. Soc., 2017, 139, 16556–16567 CrossRef CAS PubMed; (c) Y. Shi, R. Tan, M. Y. Gee and A. B. Greytak, ACS Nano, 2015, 9, 3345–3359 CrossRef PubMed.
- (a) C. Tiede, A. A. Tang, S. E. Deacon, U. Mandal, J. E. Nettleship, R. L. Owen, S. E. George, D. J. Harrison, R. J. Owens and D. C. Tomlinson, Protein Eng., Des. Sel., 2014, 27, 145–155 CrossRef CAS PubMed; (b) C. Tiede, R. Bedford, S. J. Heseltine, G. Smith, I. Wijetunga, R. Ross, D. AlQallaf, A. P. Roberts, A. Balls, A. Curd, R. E. Hughes, H. Martin, S. R. Needham, L. C. Zanetti-Domingues, Y. Sadigh, T. P. Peacock, A. A. Tang, N. Gibson, H. Kyle, G. W. Platt, N. Ingram, T. Taylor, L. P. Coletta, I. Manfield, M. Knowles, S. Bell, F. Esteves, A. Maqbool, R. K. Prasad, M. Drinkhill, R. S. Bon, V. Patel, S. A. Goodchild, M. Martin-Fernandez, R. J. Owens, J. E. Nettleship, M. E. Webb, M. Harrison, J. D. Lippiat, S. Ponnambalam, M. Peckham, A. Smith, P. K. Ferrigno, M. Johnson, M. J. McPherson and D. C. Tomlinson, eLife, 2017, 6, e24903 CrossRef PubMed.
- J. A. Burns, J. C. Butler, J. Moran and G. M. Whiteside, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- (a) J. A. Barltrop, P. M. Hayes and M. Calvin, J. Am. Chem. Soc., 1954, 76, 4348–4367 CrossRef CAS; (b) G. Palui, T. Avellini, N. Zhan, F. Pan, D. Gray, I. Alabugin and H. Mattoussi, J. Am. Chem. Soc., 2012, 134, 16370–16378 CrossRef CAS PubMed.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–252 CrossRef CAS PubMed.
- (a) D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed; (b) H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed.
- Y. Zhang, S. W. Yuan, R. Lu and A. C. Yu, J. Phys. Chem. B, 2013, 117, 7308–7316 CrossRef CAS PubMed.
- (a) V. I. Klimov, Annu. Rev. Phys. Chem., 2007, 58, 635–673 CrossRef CAS PubMed; (b) V. I. Klimov and D. W. McBranch, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 60, 13740–13749 CrossRef.
- (a) D. M. Rissin, C. W. Kan, T. G. Campbell, S. C. Howes, D. R. Fournier, L. Song, T. Piech, P. P. Patel, L. Chang and A. J. Rivnak, Nat. Biotechnol., 2010, 28, 595–599 CrossRef CAS PubMed; (b) L. D. S. Lapitan Jr., Y. Xu, Y. Guo and D. Zhou, Nanoscale, 2019, 11, 1195–1204 RSC.
- Thermo Scientific Pierce Protein Research Products 2009 Catalog. https://thermofisher.portica.de/system/includes/download.php?f=1033.
- N. Nateghian, N. Goodarzi, M. Amini, F. Atyabi, M. R. Khorramizadeh and R. Dinarvand, Chem. Biol. Drug Des., 2016, 87, 69–82 CrossRef CAS PubMed.
- (a) Y. Singh, R. Durga, K. K. Viswanadham, A. J. Kumar, J. G. Meher, K. Raval, S. Jaiswal, J. Dewangan, H. K. Bora, S. K. Rath, J. Lal and D. P. Mishra, Mol. Pharm., 2017, 14, 2749–2765 CrossRef CAS PubMed; (b) S. Park, E. Kim, W. Y. Kim, C. Kang and J. S. Kim, Chem. Commun., 2015, 51, 9343–9345 RSC.
- (a) L. Lv, Y. Guo, Y. Shen, J. Liu, W. Zhang, D. Zhou and S. Guo, Adv. Healthcare Mater., 2015, 4, 1496–1501 CrossRef CAS PubMed; (b) J. Wang, F. Wang, F. Li, W. Zhang, Y. Shen, D. Zhou and S. Guo, J. Mater. Chem. B, 2016, 4, 2954–2962 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr08060k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2020 |