
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtein-directed crystalline 2D fullerene assemblies†
Mantas
Liutkus‡
 a,
Alicia
López-Andarias‡
b,
Sara H.
Mejías
a,
Javier
López-Andarias
b,
David
Gil-Carton
c,
Ferran
Feixas
d,
Sílvia
Osuna
de,
Wakana
Matsuda
f,
Tsuneaki
Sakurai
f,
Shu
Seki
f,
Carmen
Atienza
*b,
Nazario
Martín
*bg and
Aitziber L.
Cortajarena
*ah
a,
Alicia
López-Andarias‡
b,
Sara H.
Mejías
a,
Javier
López-Andarias
b,
David
Gil-Carton
c,
Ferran
Feixas
d,
Sílvia
Osuna
de,
Wakana
Matsuda
f,
Tsuneaki
Sakurai
f,
Shu
Seki
f,
Carmen
Atienza
*b,
Nazario
Martín
*bg and
Aitziber L.
Cortajarena
*ah
aCIC biomaGUNE, Paseo de Miramón 182, E-20014 Donostia-San Sebastian, Spain. E-mail: alcortajarena@cicbiomagune.es
bDepartamento de Química Orgánica I, Facultad de Ciencias Químicas. Universidad Complutense de Madrid, E-28040 Madrid, Spain. E-mail: nazmar@quim.ucm.es; cmatienz@ucm.es
cCIC bioGUNE; Bizkaia Science and Technology Park, building 800, E-48160, Derio, Spain
dCompBioLab Group, Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Carrer Maria Aurèlia Capmany 69, 17003 Girona, Spain
eInstitució Catalana de Recerca i Estudis Avançats (ICREA), Pg. Lluís Companys 23, 08010 Barcelona, Spain
fDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Japan
gIMDEA-Nanoscience, Campus de Cantoblanco, E-28049 Madrid, Spain
hIkerbasque, Basque Foundation for Science, Mª Díaz de Haro 3, E-48013 Bilbao, Spain
First published on 8th January 2020
Abstract
Water soluble 2D crystalline monolayers of fullerenes grow on planar assemblies of engineered consensus tetratricopeptide repeat proteins. Designed fullerene-coordinating tyrosine clamps on the protein introduce specific fullerene binding sites, which facilitate fullerene nucleation. Through reciprocal interactions between the components, the hybrid material assembles into two-dimensional 2 nm thick structures with crystalline order, that conduct photo-generated charges. Thus, the protein–fullerene hybrid material is a demonstration of the developments toward functional materials with protein-based precision control of functional elements.
Introduction
In nature, proteins are the main components of cell structure, catalysts and cell regulators, due to the chemical stability and the structural diversity. These properties also make protein-based materials very appealing for different biotechnological and nanotechnological applications, such as optoelectronics,1 cell signalling,2,3 plasmonics,4 and catalysis,5,6 among others. As the performance of hybrid materials relies heavily on precise organisation of the functional elements within the materials and the emergent properties, many research efforts have been focussed on the design of encoded building blocks, which would allow reliable control over the arrangement of the active components as well as maintain long-range order. However, control of the coupling specificity between the active elements and engineered protein-based biomaterials remains a great challenge.Despite the advances in understanding of protein manipulation and creation of artificial constructions with enhanced properties, rational protein design is still far from straightforward due to the limited understanding of the sequence-structure–function relationship.7 The problem is somewhat mitigated when working with small repeat proteins, that are amenable to modular design and thus allow local modifications without affecting global structure. Consensus tetratricopeptide repeat (CTPR) protein has been particularly useful in the synthesis of hybrid materials. CTPR proteins are made of modules of 34 amino acids, folded into a helix-turn-helix motif; however, the fold is defined by only 8 conserved residues, thus allowing a lot of freedom for protein engineering. The modules generally appear repeated in tandem, from 2 to 20 repeats, generating a rigid right-handed superhelical structure8 that is able to function as stable scaffold in both biological9,10 and laboratory settings.11 Thus, CTPR proteins have been successfully used to build photoconductive and electroactive systems, combining covalent modification and non-covalent interactions, as well as stabilise fluorescent and electroactive clusters.12–15
In the current work, the CTPR proteins were engineered into scaffolds for C60 fullerene assembly. Carbon nanoforms combined with biomimetic nanomaterials have driven the advancement in nanoscience, biochemistry and materials chemistry in the recent years.16,17 C60 fullerene, since its discovery,18 has been of particular interest due to its unique chemical and physical properties.19–21 The hydrophobic nature of the carbon species allows the fullerene to form complexes with proteins, leading to biological activity.22,23 A systematic computational analysis suggested the fullerene cage is able to scan the protein surface and identify a suitable hydrophobic pocket to accommodate.24,25 Recently, fullerenes were co-crystallised with short helical peptides coordinating the C60 spheres through a single strategically placed aromatic residue,26 showing that the fullerene binding sites can be precisely defined. In the present work, tyrosine-based fullerene binding sites were engineered onto CTPR protein for fullerene coordination along the protein surface. The resulting protein–fullerene hybrid material assembles into two-dimension structures with long-range crystalline organisation in aqueous medium and improved photoconductivity through stabilisation and transmission of photo-generated charge carriers.
Results and discussion
CTPR proteins were identified as good candidates for hosting fullerenes due to high abundance of tyrosine (6 residues per repeat), that could form π–π stacking interactions with the fullerenes (Fig. 1a). Protein composed of eight repeats comprising a full superhelical turn, CTPR8, was chosen as the scaffold, given the stability and the well characterised properties of the eight-repeat protein. In addition to the shape compatibility to accommodate carbon nanoforms in the hydrophobic inner cavity,13 the CTPR protein also contains inward-pointing lysine residues that can further stabilise the protein-C60 conjugates.27–29 | ||
| Fig. 1 Schematic for CTPR8 and fullerene derivatives and characterisation of co-assemblies by FPLC chromatograms and UV-vis spectra. (a) The crystal structure of CTPR8 (PDB ID: 2HYZ)35,36 and the chemical structures of the fullerene derivatives; tyrosines (red) and lysines (blue) are highlighted. (b) FPLC chromatograms of CTPR8-C60 (red) and CTPR8-C60A (blue) hybrid materials and free CTPR8 (gray). (c) UV-visual spectra of CTPR8-C60 (red) and CTPR8-C60A (blue) hybrid materials and free CTPR8 (gray). | ||
Fullerene C60 was suspended in a solution of CTPR8 and the suspension was sonicated using a tip sonicator, after which the reaction was centrifuged to remove excess fullerene particles. Initial observations were promising, as the supernatant retained some of the dark colour, signifying that some of the otherwise completely water-insoluble fullerene was solubilised through interactions with the protein. The CTPR8-C60 hybrid material was purified from the supernatant by fast protein liquid chromatography (FPLC) (Fig. 1b). The absorption features of this new assembly confirmed the presence of the fullerene, with the appearance of the characteristic absorption bands at 266 and 343 nm and a shoulder around 450 nm (Fig. 1c). However, a substantial amount of free CTPR8 protein also eluted. The low extent of complex formation was attributed to poor dispersion of the fullerene in aqueous solutions even under sonication, thus coupling was attempted with a more water-soluble fullerene derivative C60 pyrrolidine tris-acid (C60A) (Fig. 1b). In this case, a higher degree of functionalisation was achieved, clearly observed with just the naked eye from the intense brown-solution (Fig. 1c). UV/vis spectra of the CTPR8-C60A co-assembly clearly showed a depletion of the characteristic C60 fullerene band at 330 nm and the loss of the feature band at 430 nm associated with the saturation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the C60 fullerene monoadduct, as well as considerable broadening of absorbance bands along the spectra (Fig. 1c). These absorption features of fullerene-based materials are typically attributed to interactions between fullerenes, e.g. in fullerene based colloids,30,31 C60 films and aggregates.32–34 In both CTPR8-C60 and CTPR8-C60A assemblies, the α-helical structure of the protein was unaffected by fullerene binding.
C double bond in the C60 fullerene monoadduct, as well as considerable broadening of absorbance bands along the spectra (Fig. 1c). These absorption features of fullerene-based materials are typically attributed to interactions between fullerenes, e.g. in fullerene based colloids,30,31 C60 films and aggregates.32–34 In both CTPR8-C60 and CTPR8-C60A assemblies, the α-helical structure of the protein was unaffected by fullerene binding.
The CTPR proteins were then reengineered to exert more specific interactions with the fullerene surface. Two additional tyrosine residues were introduced in positions 2 and 9 of helix A of every repeat, thus positioning two aromatic rings ca. 1 nm apart, optimal to clamp a fullerene molecule (Fig. 2a). The modified protein, CTPR8Y16, expressed well and showed the same helical structure as the original protein, based on CD spectroscopy (Fig. 2b). The mutant protein also showed improved fullerene binding, resulting in significantly greater amounts of hybrid material, with no free protein recovered in the case of the more water-soluble C60A during purification, as determined by gel filtration chromatography (Fig. 2c). The hybrid materials retained similar absorbance features to the CTPR8-based counterparts, with the CTPR8Y16-C60 showing discrete fullerene-characteristic bands and significant broadening of the absorption features with CTPR8Y16-C60A (Fig. 2d).
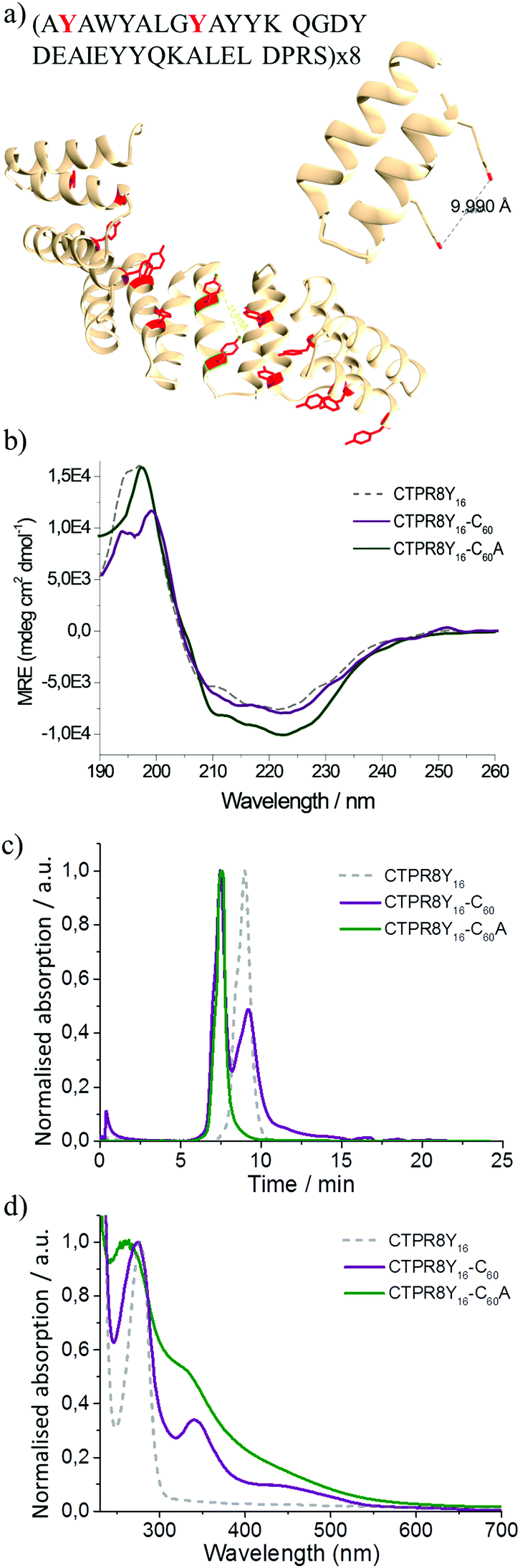 | ||
| Fig. 2 Schematic of modified protein and characterisation of co-assemblies by different techniques. (a) The model of CTPR8Y16 showing the newly introduced tyrosine clamps. (b) CD spectra of CTPR8Y16-C60 (purple) and CTPR8Y16-C60A (green) co-assemblies compared with CTPR8Y16 (gray). (c) FPLC gel filtration chromatograms of CTPR8Y16-C60 (purple) and CTPR8Y16-C60A (green) hybrid materials and free CTPR8Y16 (gray). (d) UV-visual spectra of CTPR8Y16-C60 (purple) and CTPR8Y16-C60A (green) hybrid materials and free CTPR8Y16 (gray). | ||
The CTPR8Y16-C60 and CTPR8Y16-C60A hybrids also retained the α-helical structure of the protein (Fig. 2b). More significantly, fullerene–bound protein became more resistant to thermal denaturation. With incrementally increased temperature, the free protein CTPR8Y16 reached midpoint denaturation at Tm = 70.5 °C. The pristine C60-based conjugate CTPR8Y16-C60 was marginally more stable, with Tm = 73.0 °C, while the midpoint denaturation temperature of CTPR8Y16-C60A could not be directly determined, as the protein was not completely unfolded even at 95 °C (Fig. S1†). Thermal denaturation of the complex was then monitored in the presence of increasing concentrations of urea, and Tm for CTPR8Y16-C60A was extrapolated to 90.0 °C (Fig. S2†). The observed enhancement of protein thermal stability can likely be explained by protein core stabilisation through C60 binding.
The CTPR8Y16-C60A assembly was then investigated by Raman scattering spectroscopy. For pristine C60 and C60A the Raman spectra showed the characteristic Ag(2) pentagonal pinch mode located at 1466 and 1462 cm−1, respectively. In the CTPR8Y16-C60A co-assembly, however, the Ag(2) mode was shifted towards lower frequencies at 1457 cm−1 (Fig. S3†).37 This significant downshift of the Raman Ag(2) peak for the CTPR8Y16-C60A hybrid material suggests possible electronic interactions between the protein and the electron-acceptor C60, which is in agreement with previous findings for the non-covalent modification of fullerene with electron donor units. In the current case, the presence of electron donor aromatic residues in the protein favours the interaction with the fullerene surface through π–π and OH⋯π interactions.38,39
Cryo-electron microscopy was employed to gain structural insights into the protein–fullerene assemblies. Well-defined structures with highly distinct features were visible in the micrographs of the CTPR8Y16-C60A assembly (Fig. 3a). 3D analysis of the structures revealed that the protein–fullerene hybrids assemble into thin 2D sheets of irregular shape, that are mostly flat, but may have a small twist (Fig. 3f and S4†). The sheets are ca. 2 nm thick (Fig. S5†), with dimensions of 20 nm × 20 nm on average, although reaching up to 50 nm (more precise measurements are not possible, as it is impossible to determine the precise orientation of the objects in vitrified drops). From a side view, the sheets appear made up of two parallel high contrast bands of ca. 0.7 nm, with a low contrast band of similar thickness between (Fig. S5†). The 2 nm thickness of the film is consistent with a monolayer of CTPR protein, matching the dimensions of CTPR fibres observed by TEM.40 The high contrast of the edge bands implies dense accretion of fullerenes, suggesting that the fullerenes are (predominantly) assembling on the surfaces of the protein monolayers. The thickness of the edge bands is also consistent with the fullerene frame size (Fig. 1a), thus strongly suggesting fullerene monolayer formation, although individual fullerenes deviating from this arrangement would not be detectable and cannot be ruled out.
 | ||
| Fig. 3 Cryo-electron microscopy analysis of the hybrid materials. Cryo-electron micrographs of (a) CTPR8Y16-C60A, (b) CTPR8-C60A, (c) CTPR8Y16-C60 and (d) CTPR8-C60 hybrid materials. (e) Close-up images of the areas marked with black squares in a–d. (f). A tilt series of a selected region of CTPR8Y16-C60A hybrid material at different degrees of sample tilt, showing the selected assembly from different perspectives. (g) Representative crystallites of CTPR8Y16-C60A hybrid material and the corresponding FFT images, showing the diffraction patterns of crystalline assemblies. The scale bar is 20 nm in all cases. | ||
From face view, the plaques have very low contrast due to their thickness, but they are identifiable due to very strong diffraction patterns. The assemblies are continuous throughout the formation, without gaps or fissures on the surface, which indicates that the assemblies are ordered on the molecular level, rather than maintained through random interactions. As the particles are dispersed in the vitrified fluid, diffraction patterns from individual aggregates can be studied (Fig. 3g). The observed interplanar spacing can be divided into two groups, localised in the 0.85–0.79 nm region and the region between 0.50 nm and 0.40 nm. The observed interplanar distances are consistent with the diffraction from ordered fullerenes, as the distances are far too big for salt crystals, but far too small to originate from crystallised protein. The observed variability of diffraction distances could be a consequence of differing orientations of the ordered assemblies within the vitrified fluid, exposing diffraction patterns from different planes, or could signify that slightly different arrangements are possible for the fullerenes.
The CTPR8-C60A assembly also formed similar 2D objects, but more sparsely and of smaller size, and without the two distinct bands in the side view, but rather of uniform contrast along the 2 nm thickness (Fig. 3b). The sample also produced similar diffraction patterns, although generally of lower intensity. This result strongly suggests that although the CTPR fold alone is able to facilitate fullerene nucleation, the “tyrosine clamps” direct fullerene assembly with much higher specificity, resulting in more extensive arrangements.
In the cases of CTPR8Y16-C60 and CTPR8-C60 where pristine fullerene was used, the thin sheets were not free-floating, but instead clumped into large clusters, with various independent and very strong diffraction patterns in different parts of the plaques (Fig. 3c, d and S6†). This behaviour is also explained by the proposed fullerene binding mode: when bound on the surface of the protein sheets, C60A self-orient to expose the hydrophilic tris-acid appendages to the solvent, thus contributing to the solubility of the superstructures, whereas pristine C60 fullerenes introduce hydrophobic patches of carbon, causing the sheets to coalesce. The diffraction patterns of the C60 assemblies showed on average smaller spacing (0.82–0.77 nm) than those of C60A (vide supra), which could be an indication that greater spacing is required to accommodate the pyrrolidine-tris acid groups (Fig. S6†).
To elucidate the molecular basis of the conjugation process, binding events were reconstructed with Molecular Dynamics (MD) simulations.41 Five replicas of 800 ns of MD were performed in the presence of eight [60]fullerene (either C60 or C60A) molecules and one CTPR8 unit (either CTPR8 or CTPR8Y16), simulating the CTPR8-C60, CTPR8-C60A, CTPR8Y16-C60 and CTPR8Y16-C60A co-assemblies. To characterise the spontaneous binding process, all [60]fullerene species were initially placed in the solvent at least 10 Å away from the protein surface and were allowed to freely diffuse from the solvent to the protein surface along the MD trajectory. From these spontaneous binding MD simulations, the interactions established between the fullerene species and CTPR8 units were monitored along time to identify the most visited binding sites (hot spots) on the protein surface. In all cases, [60]fullerene species rapidly collapsed over the protein surface, establishing transient interactions with the side chains of superficial protein residues. The initial recognition with CTPR8 took place in few nanoseconds of simulation time, and the fullerene species were then displaced over the protein surface until they identified a binding site (Fig. 4), where commonly they remained for the rest of the simulation. Both C60 and C60A prefer to interact with the CTPR modules located at both extremes of the CTPR8 unit, while permanent interactions with the inner hydrophobic cavity were observed less frequently. To identify the most visited binding sites, representative snapshots from the last 100 ns of each replica of the MD simulations were extracted and the position of each fullerene with respect to the protein surface was plotted (colour spheres in Fig. 4). CTPR8-C60, CTPR8-C60A and CTPR8Y16-C60 assemblies showed a wide dispersion of fullerene molecules all over the protein surface. These results indicate that although the fullerene species tend to be in contact with protein residues, these interactions are not stable along the MD simulation and among replicas. On the contrary, for the CTPR8Y16-C60A co-assembly, fullerene molecules accumulate in selected hot spots independently of which MD simulation is analysed. These results indicate that the new tyrosine residues favour the association and stabilisation of C60A at specific binding sites on the protein surface. These observations are in line with the highest fullerene/protein ratio experimentally observed for the CTPR8Y16-C60A co-assembly.
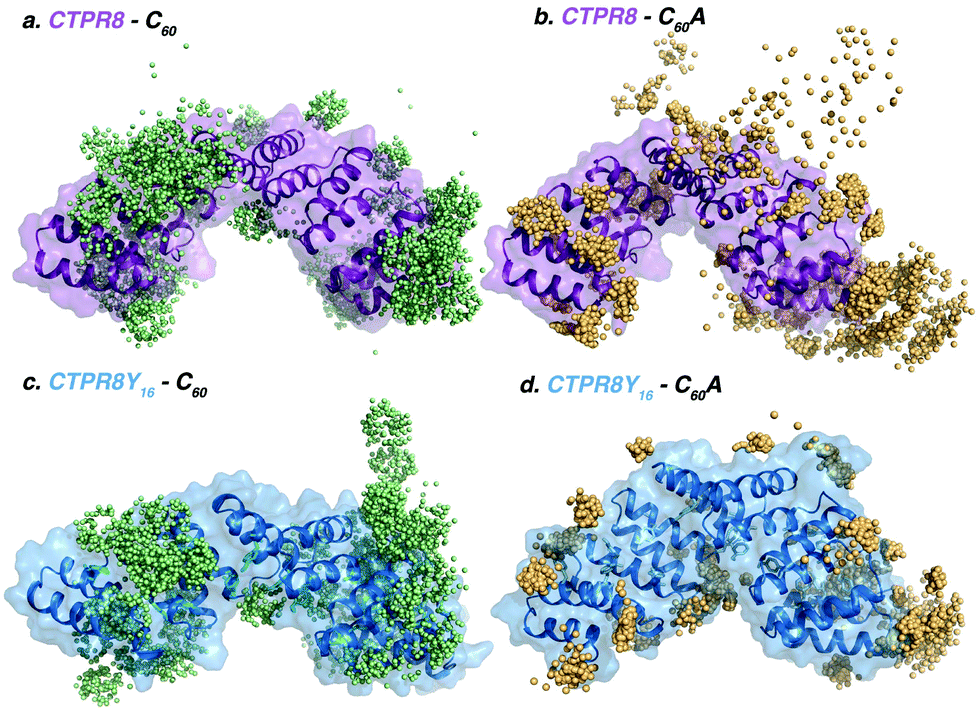 | ||
| Fig. 4 Representative snapshots extracted from the MD simulations of (a) CTPR8-C60, (b) CTPR8-C60A, (c) CTPR8Y16-C60 and (d) CTPR8Y16-C60A co-assemblies, where coloured spheres indicate the positions of pristine C60 (pale green) or C60A (orange) fullerene species on the protein surface during the last 100 ns of five replicas of MD simulations. The structure of the CTPR8 unit for each system corresponds to the last snapshot of the MD simulation of the first replica. | ||
To explore the binding mechanism of CTPR8Y16-C60A in more detail, the distance between the centre of mass of CTPR8Y16 and the centre of mass of each of the eight C60A molecules along the MD trajectory was monitored (Fig. 5a). All fullerenes were recognised by CTPR8Y16 in less than 100 ns of MD simulation time, reaching the final binding site after 300 ns, where C60A remained for the rest of the simulation. Six out of the eight fullerene molecules bound hot spots located more than 25 Å far away from the CTPR8Y16 centre of mass, indicating that C60A prefer to interact with the CTPR modules located at the N- and C-terminal domains of CTPR8Y16. Close examination of molecular interactions showed predominantly π–π interactions between C60A and some of the introduced tyrosine residues (Fig. 5c). Thus, the introduced tyrosine residues enhance supramolecular interactions with the aromatic surface of the fullerene, fixing the fullerene molecule in a particular binding site. In some hot spots, the fullerene is stabilised by π-stacking interactions with a total of six tyrosine residues, i.e. Tyr209, Tyr213, Tyr216, Tyr240, Tyr243, and Tyr247 (A in Fig. 5c), located at the extreme of the CTPR8 unit. These interactions are able to retain the C60A molecule in the hot spot for the rest of the simulation time. Additional binding sites are located at the inner hydrophobic cavity and display a broader range of interactions (B in Fig. 5c). The simulations reveal that Arg203 establishes cation⋯π interactions with C60A, the introduced Tyr138 on helix A displays OH⋯π interactions with the fullerene, while Lys81 and Lys115 stabilise the carboxylate groups of C60A by salt bridge interactions. Such strong interactions are not observed in the simulations where pristine C60 is used, with the fullerene more delocalised all over the protein surface. To gain a deeper insight into the recognition and binding processes, five replicas of 500 ns of MD simulations with 24 C60A molecules were performed for CTPR8Y16. Increasing the number of fullerenes led to their aggregation in groups of 6–8 molecules mediated by sodium cations. Interestingly, fullerene aggregates accumulate at the same binding sites observed previously with a smaller number of C60A (Fig. 5d).
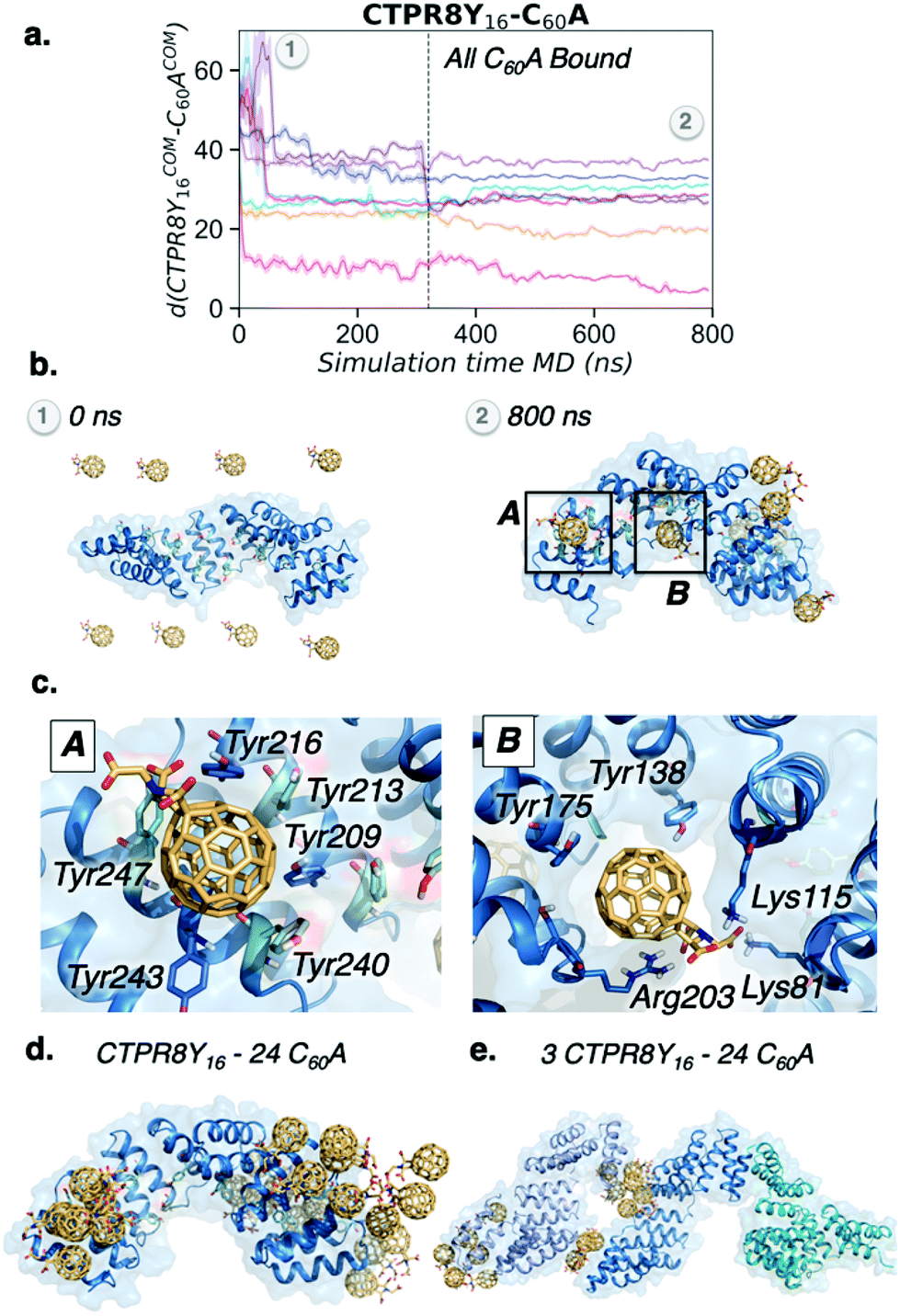 | ||
| Fig. 5 MD simulations of CTPR–fullerene binding. (a) Plot of the distance (in Å) between the centre of mass of CTPR8Y16 and the centre of mass of the eight C60A molecules along the 800 ns MD simulation trajectory. (b) Representative snapshot extracted from the MD simulations of the CTPR8Y16-C60A co-assembly at 0 ns and 800 ns simulation time. (c) Relevant residues of two representative binding sites of CTPR8Y16-C60A co-assembly. (d) Representative snapshot extracted from the MD simulations of the CTPR8Y16-24C60A co-assembly at 500 ns of MD simulation. (e) Representative snapshot extracted from the MD simulations of the 3CTPR8Y16-24C60A co-assembly at 200 ns of MD simulation. | ||
Finally, the potential role of C60A molecules on CTPR8Y16 aggregation was explored. To this end, five replicas of 200 ns of MD simulations were carried out with 24 C60A molecules and three CTPR8Y16 protein units. The simulations showed that fullerene molecules first aggregate and then contribute to the formation of CTPR8Y16 aggregates, acting as bridge molecules between CTPR8Y16 units (Fig. 5e). Despite the short simulation time of these simulations and the model system used, MD simulations revealed a direct contribution of C60A to the formation of CTPR8Y16 assemblies.
The simulated behaviour of fullerenes is consistent with experimental observations, as free C60A fullerenes aggregate in aqueous solution to form thin amorphous filaments (Fig. S7†). These filaments, however, have no uniform structure and produce no electron diffraction; the assembly is thus likely governed by non-specific interactions observed in the simulations. The contacts with the ordered surface of CTPR protein, however, seem to serve as nucleation points for crystalline-ordered fullerene assemblies. Reciprocally, the fullerenes appear to mediate and strengthen the interactions between the proteins that normally manifest in protein assemblies in solid state.4,42 The first 200 ns of MD simulation show that, from first contact with the fullerenes, CTPR proteins begin to assemble in a planar configuration (Fig. S8†). While during the simulations with 8 or 24 fullerene molecules preferential binding was observed to the more exposed binding hot spots at the termini of the protein, the large excess of fullerenes present during the conjugation process would result in the occupation of all or nearly all possible binding sites, effectively coating the protein surface. The sonication, that could not be easily simulated, would further increase the mobility of the fullerenes along the surface of the protein, facilitating the migration of the fullerenes into the more hindered binding sites.
Fullerene C60 crystallises in fcc lattice (a = 14.2 Å),43 transitioning into sc below 249 K (−24 °C);44,45 however, depending on the additives used during crystallisation, a range of different lattices and spatial arrangements has been achieved.46–50 As the protein and the fullerenes both actively contribute to the overall order of the hybrid assemblies, it is difficult to suggest the exact arrangement, and further research is needed to fully characterise the crystalline order. The analysis is greatly complicated by the low electron doses needed for cryo-electron microscopy, potentially resulting in loss of some of the finer diffraction peaks. The length of MD simulations is also limited by the complexity of the system, restricting the analysis to the initial moments of interaction. Thus, the transition of the fullerenes from the aggregate state (that forms prior to contact with protein) to ordered structures could not be observed. Nevertheless, the thickness of the assemblies and the continuous diffraction pattern throughout the assemblies suggest the fullerenes assemble in a monolayer, with the arrangement at least partially driven by fullerene interactions, rather than being entirely imposed by the protein. Furthermore, the crystalline arrangement of the hybrid material emerges and is stable in aqueous solution, with the fullerenes on the surface of the assemblies in contact with the aqueous phase. Under the right conditions, it is conceivable that the 2D lattice could be grown beyond the nanoscale, allowing to apply the potential of the carbonaceous assembly on a larger setting. Indeed, as the innate fullerene–fullerene contacts are maintained alongside with protein interactions, this new composite material is far more likely to retain some of the intrinsic electronic and photophysical properties of the constituent fullerenes.51,52
Hence, transient photoconductivity of the hybrid assemblies was assessed using the flash-photolysis time-resolved microwave conductivity (FP-TRMC) system.53–56 The electrode-less method, based on microwave dielectric spectroscopy, allows to measure conductivity due to local-scale motions of charge carriers, and is not limited by deep traps of charges at grain boundaries. Drop-cast films of CTPR–fullerene conjugates were photo-excited by 355 nm pulses, allowing to record time-dependent conductivity (Fig. 6a and S9†) from transiently photo-generated charge carriers (electrons and holes); the decay of the curve indicates the extinction of charges due to charge trapping and/or recombination processes. Photon density-normalised conductivity transients ϕ∑μ, represented as the product of the photocarrier generation yield (ϕ) and sum of electron and hole mobilities (∑μ), gave the maximum conductivities (∑μ)max of 3.0 × 10−8, 1.1 × 10−7, 3.5 × 10−8 and 1.2 × 10−7 m2 V−1 s−1 for CTPR8-C60, CTPR8-C60A, CTPR8Y16-C60 and CTPR8Y16-C60A, respectively.
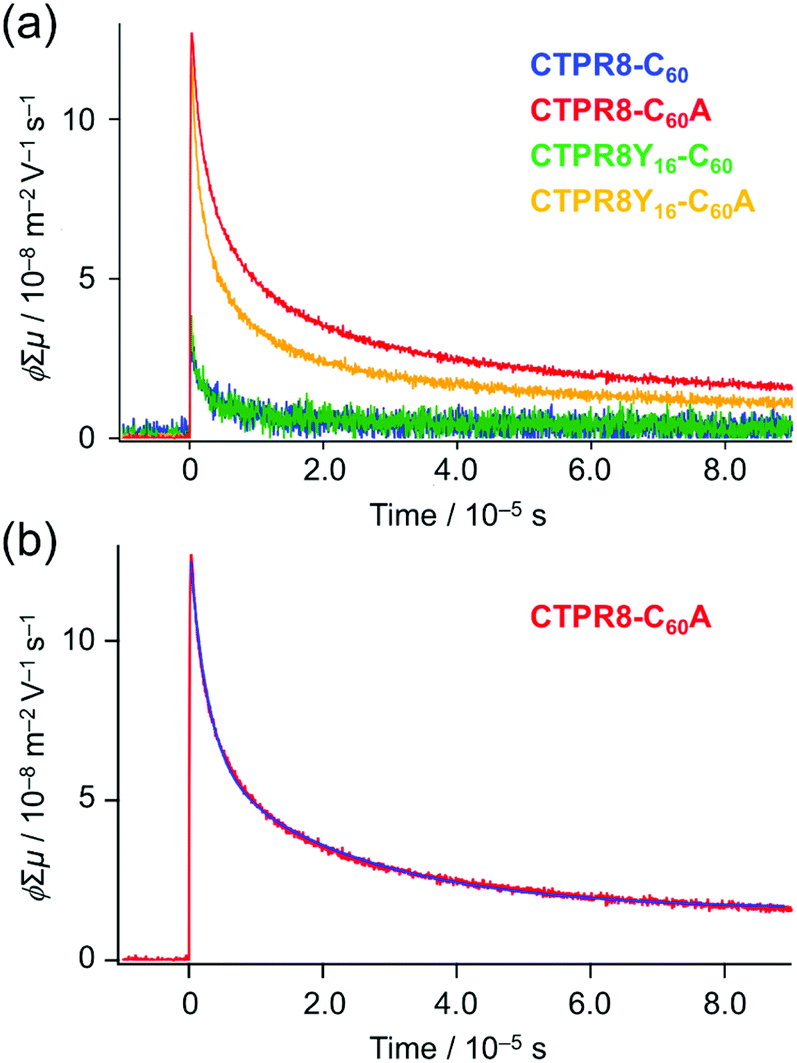 | ||
| Fig. 6 Photoconductivity of drop-cast films of CTPR8–fullerene hybrid materials. (a) Conductivity transients of CTPR8-C60 (blue), CTPR8-C60A (red), CTPR8Y16-C60 (green) and CTPR8Y16-C60A (yellow) co-assemblies upon photoexcitation with 355 nm pulses with photon density of 9.1 × 1015 cm−2. (b) Conductivity transient (red) with double-exponential fitted curve (blue) of CTPR8-C60A with photon densities of 9.1 × 1015 cm−2. | ||
The observed conductivity value is larger than what has been previously observed with fullerene derivatives51,52 that already have applications in material sciences. The film of CTPR8Y16 protein alone did not show significant photoconductivity. (Photoconductivity measurements of protein alone could not produce credible data using the current method, as the excitation light at 355 nm has very little interaction with the protein sample.) Thus, the observed photoconductivity for CTPR8Y16-C60A originates from charge transport through protein–immobilised fullerene contacts. The one-order magnitude larger conductivity of the co-assemblies with C60A, namely CTPR8-C60A and CTPR8Y16-C60A, as compared to the corresponding C60 assemblies (CTPR8-C60 and CTPR8Y16-C60) can likely be explained by stronger interactions among fullerene moieties, as is evidenced by Fig. 1c and 2d, allowing inter-fullerene charge transport pathways.
The decay curve, observed for CTPR8Y16-C60 at the excitation photon density of 9.1 × 1015 cm−2 (red in Fig. 6), was deconvoluted as an example by the fitting analysis and fitted with a sum of two exponential functions (blue in Fig. 6b)57
y(t) − y0 = y1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−k1t) + y2exp(−k2t), exp(−k1t) + y2exp(−k2t), | (1) |
Conclusions
The described system provides a rare example of protein-directed ordered assembly of fullerenes. The new-engineered CTPR proteins with tyrosine clamps allow to steer the assembly of fullerenes into crystalline arrangements, while promoting innate fullerene interactions. As a result, the hybrid material supports strong photocurrents, allowing photo-generated charge carriers to move through fullerene contacts. Thus, the presented system brings protein research one step closer to supramolecular assemblies governed by rationally designed proteins. In addition, to the best of our knowledge the CTPR–fullerene hybrid assemblies present a first instance of crystalline assemblies observed in liquid phase.Despite the advances in successful protein design, it is noteworthy that correct choice of a starting structure plays a crucial role. Based on the current and multiple other supramolecular assemblies, using repeat proteins as a scaffold for supramolecular ordering seems to be the winning strategy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Commission IRG-246688 Bionanotools (A. L. C.) and CIG project PCIG14-GA-2013-630978 (S. O.); the Spanish Ministry of Economy and Competitiveness (MINECO) BIO2016-77367-C2-1-R (A. L. C.), CTQ2014-520456-R (N. M.), PGC2018-102192-B-I00 (S. O.), and RTI2018-101032-J-I00 (F. F.); and the European Research Council ERC-320441-Chirallcarbon (N. M.), ERC-2014-CoG-648071-ProNANO (A. L. C.), and ERC-2015-StG-679001-NetMoDEzyme (S. O.). N. M. thanks the Community of Madrid Government (Photocarbon project. S2013/MIT-2841). C. A. thanks the Ramón y Cajal grant and J. L.-A. thanks the Spanish Ministry of Education for FPU grant. S. H. M. thanks the Basque Government for financial support through a PhD fellowship. F. F. thanks the European Community for MSCA-IF-2014-EF-661160-MetAccembly grant. S. O. and F. F. thank the Generalitat de Catalunya for group emergent CompBioLab (2017 SGR-1707). This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720 (CIC biomaGUNE).Notes and references
- A. Dimonte, S. Frache, V. Erokhin, G. Piccinini, D. Demarchi, F. Milano, G. D. Micheli and S. Carrara, Biomacromolecules, 2012, 13, 3503–3509 CrossRef CAS PubMed.
- D. L. Kaplan, Silk Polymers: Materials Science and Biotechnology : Developed from the Workshop on Silks: Biology, Structure, Properties, Genetics Sponsored by the Division of Polymer Chemistry: Charlottesville, Virginia, January 28–29, 1993, American chemical society, 1994 Search PubMed.
- X. Hu, P. Cebe, A. S. Weiss, F. Omenetto and D. L. Kaplan, Mater. Today, 2012, 15, 208–215 CrossRef CAS.
- S. H. Mejias, P. Couleaud, S. Casado, D. Granados, M. A. Garcia, J. M. Abad and A. L. Cortajarena, Colloids Surf., B, 2016, 141, 93–101 CrossRef CAS PubMed.
- J. H. Forstater, A. Kleinhammes and Y. Wu, Langmuir, 2013, 29, 15013–15021 CrossRef CAS PubMed.
- J. E. Gagner, W. Kim and E. L. Chaikof, Acta Biomater., 2014, 10, 1542–1557 CrossRef CAS PubMed.
- J. Clarke and L. Regan, Curr. Opin. Struct. Biol., 2010, 20, 480–481 CrossRef CAS PubMed.
- L. D. D'Andrea and L. Regan, Trends Biochem. Sci., 2003, 28, 655–662 CrossRef PubMed.
- A. L. Cortajarena, T. Y. Liu, M. Hochstrasser and L. Regan, ACS Chem. Biol., 2010, 5, 545–552 CrossRef CAS PubMed.
- M. E. Jackrel, A. L. Cortajarena, T. Y. Liu and L. Regan, ACS Chem. Biol., 2010, 5, 553–562 CrossRef CAS PubMed.
- A. L. Cortajarena, S. G. J. Mochrie and L. Regan, Protein Sci., 2011, 20, 1042–1047 CrossRef CAS PubMed.
- S. H. Mejías, J. López-Andarias, T. Sakurai, S. Yoneda, K. P. Erazo, S. Seki, C. Atienza, N. Martín and A. L. Cortajarena, Chem. Sci., 2016, 7, 4842–4847 RSC.
- J. López-Andarias, S. H. Mejías, T. Sakurai, W. Matsuda, S. Seki, F. Feixas, S. Osuna, C. Atienza, N. Martín and A. L. Cortajarena, Adv. Funct. Mater., 2018, 28, 1704031 CrossRef.
- S. H. Mejias, Z. Bahrami-Dizicheh, M. Liutkus, D. J. Sommer, A. Astashkin, G. Kodis, G. Ghirlanda and A. L. Cortajarena, Chem. Commun., 2019, 55, 3319–3322 RSC.
- A. Aires, I. Llarena, M. Moller, J. Castro-Smirnov, J. Cabanillas-Gonzalez and A. L. Cortajarena, Angew. Chem., Int. Ed., 2019, 58, 6214–6219 CrossRef CAS PubMed.
- M. Numata and S. Shinkai, Chem. Commun., 2011, 47, 1961–1975 RSC.
- N. Ousaka, F. Mamiya, Y. Iwata, K. Nishimura and E. Yashima, Angew. Chem., Int. Ed., 2017, 56, 791–795 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- N. Martín, L. Sánchez, B. Illescas and I. Pérez, Chem. Rev., 1998, 98, 2527–2548 CrossRef PubMed.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed.
- C. A. Reed and R. D. Bolskar, Chem. Rev., 2000, 100, 1075–1120 CrossRef CAS PubMed.
- S. H. Friedman, D. L. DeCamp, R. P. Sijbesma, G. Srdanov, F. Wudl and G. L. Kenyon, J. Am. Chem. Soc., 1993, 115, 6506–6509 CrossRef CAS.
- S. J. Vance, V. Desai, B. O. Smith, M. W. Kennedy and A. Cooper, Biophys. Chem., 2016, 214–215, 27–32 CrossRef CAS PubMed.
- M. Calvaresi and F. Zerbetto, ACS Nano, 2010, 4, 2283–2299 CrossRef CAS PubMed.
- M. Calvaresi, F. Arnesano, S. Bonacchi, A. Bottoni, V. Calò, S. Conte, G. Falini, S. Fermani, M. Losacco, M. Montalti, G. Natile, L. Prodi, F. Sparla and F. Zerbetto, ACS Nano, 2014, 8, 1871–1877 CrossRef CAS PubMed.
- K.-H. Kim, D.-K. Ko, Y.-T. Kim, N. H. Kim, J. Paul, S.-Q. Zhang, C. B. Murray, R. Acharya, W. F. DeGrado, Y. H. Kim and G. Grigoryan, Nat. Commun., 2016, 7, 11429 CrossRef CAS PubMed.
- R. Marega, F. De Leo, F. Pineux, J. Sgrignani, A. Magistrato, A. D. Naik, Y. Garcia, L. Flamant, C. Michiels and D. Bonifazi, Adv. Funct. Mater., 2013, 23, 3173–3184 CrossRef CAS.
- A. de Leon, A. F. Jalbout and V. A. Basiuk, Chem. Phys. Lett., 2008, 457, 185–190 CrossRef CAS.
- F. De Leo, J. Sgrignani, D. Bonifazi and A. Magistrato, Chem. – Eur. J., 2013, 19, 12281–12293 CrossRef CAS PubMed.
- D. M. Guldi, J. Phys. Chem. A, 1997, 101, 3895–3900 CrossRef CAS.
- H. Hungerbuehler, D. M. Guldi and K. D. Asmus, J. Am. Chem. Soc., 1993, 115, 3386–3387 CrossRef CAS.
- G. D. Pantoş, J.-L. Wietor and J. K. M. Sanders, Angew. Chem., Int. Ed., 2007, 46, 2238–2240 CrossRef PubMed.
- W. Kratschmer, L. D. Lamb, K. Fostiropoulos and D. R. Huffman, Nature, 1990, 347, 354–358 CrossRef.
- R. V. Bensasson, E. Bienvenue, M. Dellinger, S. Leach and P. Seta, J. Phys. Chem., 1994, 98, 3492–3500 CrossRef CAS.
- T. Kajander, A. L. Cortajarena, E. R. G. Main, S. G. J. Mochrie and L. Regan, J. Am. Chem. Soc., 2005, 127, 10188–10190 CrossRef CAS PubMed.
- T. Kajander, A. L. Cortajarena, S. Mochrie and L. Regan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 800–811 CrossRef CAS PubMed.
- A. Insuasty, C. Atienza, J. Luis López, J. Marco-Martínez, S. Casado, A. Saha, D. M. Guldi and N. Martín, Sci. Rep., 2015, 5, 14154 CrossRef CAS PubMed.
- B. C. Braden, F. A. Goldbaum, B.-X. Chen, A. N. Kirschner, S. R. Wilson and B. F. Erlanger, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12193 CrossRef CAS PubMed.
- W. H. Noon, Y. Kong and J. Ma, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6466 CrossRef CAS PubMed.
- S. H. Mejias, B. Sot, R. Guantes and A. L. Cortajarena, Nanoscale, 2014, 6, 10982–10988 RSC.
- D. A. Case, R. M. Betz, D. S. Cerutti, T. E. Cheatham, III, T. A. Darden, R. E. Duke, T. J. Giese, H. Gohlke, A. W. Goetz, N. Homeyer, S. Izadi, P. Janowski, J. Kaus, A. Kovalenko, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. Luchko, R. Luo, B. Madej, D. Mermelstein, K. M. Merz, G. Monard, H. Nguyen, H. T. Nguyen, I. Omelyan, A. Onufriev, D. R. Roe, A. Roitberg, C. Sagui, C. L. Simmerling, W. M. Botello-Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu, L. Xiao and P. A. Kollman, AMBER 2016, University of California, San Francisco, 2016 Search PubMed.
- T. Z. Grove, L. Regan and A. L. Cortajarena, J. R. Soc., Interface, 2013, 10, 20130051 CrossRef PubMed.
- D. R. Huffman, Phys. Today, 1991, 44, 22–29 CrossRef CAS.
- P. A. Heiney, J. E. Fischer, A. R. McGhie, W. J. Romanow, A. M. Denenstein, J. P. McCauley Jr., A. B. Smith and D. E. Cox, Phys. Rev. Lett., 1991, 66, 2911–2914 CrossRef CAS PubMed.
- G. Van Tendeloo, C. Van Heurck, J. Van Landuyt, S. Amelinckx, M. A. Verheijen, P. H. M. Van Loosdrecht and G. Meijer, J. Phys. Chem., 1992, 96, 7424–7430 CrossRef CAS.
- Y. Jin, R. J. Curry, J. Sloan, R. A. Hatton, L. C. Chong, N. Blanchard, V. Stolojan, H. W. Kroto and S. R. P. Silva, J. Mater. Chem., 2006, 16, 3715–3720 RSC.
- H. S. Shin, S. M. Yoon, Q. Tang, B. Chon, T. Joo and H. C. Choi, Angew. Chem., Int. Ed., 2008, 47, 693–696 CrossRef CAS PubMed.
- L. K. Shrestha, Y. Yamauchi, J. P. Hill, K. I. Miyazawa and K. Ariga, J. Am. Chem. Soc., 2013, 135, 586–589 CrossRef CAS PubMed.
- J. Kim, C. Park, I. Song, M. Lee, H. Kim and H. C. Choi, Sci. Rep., 2016, 6, 32205 CrossRef CAS PubMed.
- I. S. Neretin and Y. L. Slovokhotov, Russ. Chem. Rev., 2004, 73, 455–486 CrossRef CAS.
- S. Reboredo, R. M. Girón, S. Filippone, T. Mikie, T. Sakurai, S. Seki and N. Martín, Chem. – Eur. J., 2016, 22, 13627–13631 CrossRef CAS PubMed.
- T. Umeyama, S. Shibata, K. Igarashi, S. Takahara, T. Higashino, S. Seki and H. Imahori, Chem. Lett., 2017, 46, 1001–1003 CrossRef CAS.
- M. P. De Haas and J. M. Warman, Chem. Phys., 1982, 73, 35–53 CrossRef CAS.
- F. C. Grozema and L. D. A. Siebbeles, J. Phys. Chem. Lett., 2011, 2, 2951–2958 CrossRef CAS.
- S. Seki, A. Saeki, T. Sakurai and D. Sakamaki, Phys. Chem. Chem. Phys., 2014, 16, 11093–11113 RSC.
- O. G. Reid, D. T. Moore, Z. Li, D. Zhao, Y. Yan, K. Zhu and G. Rumbles, J. Phys. D: Appl. Phys., 2017, 50, 493002 CrossRef.
- C. E. A. F. Schafmeister, W. S. Ross and V. Romanovski, LEAP, University of California, San Francisco, 1995 Search PubMed.
- G. Rothenberger, J. Moser, M. Graetzel, N. Serpone and D. K. Sharma, J. Am. Chem. Soc., 1985, 107, 8054–8059 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, CD and thermal stability data, Raman spectra, additional cryo-electron micrographs and additional simulation data. See DOI: 10.1039/c9nr07083d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |