
SPION decorated exosome delivery of TNF-α to cancer cell membranes through magnetism†
Manjiao
Zhuang
a,
Xuelian
Chen
b,
Dan
Du
b,
Jiamei
Shi
b,
Mian
Deng
b,
Qian
Long
b,
Xiaofei
Yin
b,
Yayu
Wang
c and
Lei
Rao
 *b
*b
aWest China School of Pharmacy, Sichuan University, Chengdu 610041, China
bDepartment of Biomedicine, Collaborative Innovation Center of Sichuan for Elderly Care and Health, Chengdu Medical College, Chengdu 610500, China. E-mail: YZYCDYX@cmc.edu.cn
cDepartment of Cell Biology, Institute of Biological Medicine, Jinan University, Guangzhou 510632, China
First published on 5th December 2019
Abstract
Tumor necrosis factor (TNF-α) is capable of inducing apoptosis and is a promising candidate for genetic engineering drugs in cancer therapy; however, the serious side-effects of TNF-α hinder their clinical application. In the present study, a method for preparing fusion proteins of cell-penetrating peptides (CPP) and TNF-α (CTNF-α)-anchored exosomes coupled with superparamagnetic iron oxide nanoparticles (CTNF-α-exosome-SPIONs) with membrane targeting anticancer activity has been demonstrated. To acquire exosomes with TNF-α anchored in its membrane, a CTNF-α expression vector was constructed and a stable mesenchymal stem cell cell line that expressed CTNF-α was established. Conjugating transferrin-modified SPIONs (Tf-SPIONs) onto CTNF-α-exosomes through transferrin–transferrin receptor (Tf-TfR) interaction yields CTNF-α-exosome-SPIONs with good water dispersibility. The incorporation of TNF-α into exosomes and the conjugation of SPIONs significantly enhanced the binding capacity of TNF-α to its membrane-bound receptor TNFR I, thus increasing the therapeutic effects. CTNF-α-exosome-SPIONs significantly enhanced tumor cell growth inhibition via induction of the TNFR I-mediated apoptotic pathway. In vivo studies using murine melanoma subcutaneous cancer models showed that TNF-α-loaded exosome-based vehicle delivery enhanced cancer targeting under an external magnetic field and suppressed tumor growth with mitigating toxicity. Taken together, our results suggest that CTNF-α-exosome-SPIONs showed great potential in membrane targeting therapy.
Introduction
Cancer is a serious threat to human health with high mortality.1 Melanoma is one of the most lethal forms of cancer with high morbidity and mortality among human cancers worldwide.2 Fortunately, advances in surgery and multi-agent chemotherapy have improved the surgical outcome and the quality of life of patients in the past few years. However, the therapeutic activity of most clinical anticancer drugs is hindered by their toxicity to normal cells and lack of targeting.3 The emergence of nano-biomaterials has enabled new prospects for drug therapies in cancer. Cooperative, synergistic therapies using a nanomaterial system can significantly reduce the required dose of anticancer drugs, mitigate toxic side effects, and increase target drug delivery.4The application of proteins and peptides as human therapeutics has rapidly increased in recent years, and genetic engineering drugs are one of the top-selling drugs in the global market with products that target TNFR alone generating $30.5 billion in 2013.5 TNF-α was the first cytokine to be employed for cancer treatment. Additionally, TNF-α significantly inhibited VEGF-induced HUVEC proliferation, thus restraining tumor angiogenesis.6 Despite the promising anti-cancer potential of TNF-α, its clinical application is limited by its severe toxicity after systemic administration.7 When administered directly, TNF-α is unable to target tumors specifically, thus leading to poor distribution and therapeutic effects, as well as serious undesirable adverse effects.8 Moreover, the potential therapeutic benefit of using TNF-α is also limited by its poor stability in vivo.9 Recent attempts have been made to reduce its systemic toxicity, for example, via passive targeting, cell-based therapy, gene therapy, and fusion proteins.10–12 Motivated by the development of biotechnology and nanotechnology, the applications of nanomedicine in cancer therapy, diagnosis and cell imaging have made tremendous achievements.13 However, the utilization of nanotechnology in TNF-α-based therapies in cancer has rarely been reported.
The development or identification of a promising delivery system for effectively conveying therapy is indispensable for combatting disease.14 The ideal delivery system should be safe and efficient with optimal bioavailability and should mitigate toxicity and immunogenicity. Recent studies have demonstrated that exosomes are ideal drug and gene delivery vehicles with various advantages over the existing synthetic delivery systems, given their low immunogenicity and high tolerance.
Exosomes are nano-sized membrane vesicles existing in eukaryotic fluids, such as blood, urine, and the medium of cultured cells.15 Exosomes not only possess critical nanoparticle (NP) characteristics, for example, an enhanced permeability and retention (EPR) effect and passive targeting, but also exhibit unique properties, such as targeting specificity, as well as their intrinsic biological effects on the targeted cells due to the exosome cellular origin.16,17 Delimited by the lipid bi-layer, exosomes contain various molecular constituents of the producing cell and expose the extracellular domain of various transmembrane proteins at their surface.18 Their specific origin enables exosomes to participate in intercellular communication through the transfer of cargo between cells. Therefore, various reports have shown that exosomes can be used as effective delivery nanovesicles to deliver small interfering RNA and microRNAs, RNA, or chemotherapeutics to improve treatment in cancer.19–23
Moreover, recent studies have also reported that exosomes may be used as naturally derived nanovesicles to deliver protein therapeutics.23–25 In general, cells transfected with a recombinant vector plasmid can be used to generate exosomes with specific proteins anchoring onto its membrane. It is well known that mesenchymal stem cells (MSCs) are promising tools in the fields of constructing stable expression cell lines. Moreover, exosomes derived from MSCs have been demonstrated to exhibit hyperproductivity and hypoimmunogenicity; therefore, MSCs are an ideal cell line to generate exosomes carrying certain proteins.26
Although impressive progress has been made in the progression of exosome isolation techniques, it is challenging to rapidly and efficiently isolate exosomes. For example, differential ultracentrifugation for exosome isolation is laborious; in addition to its high workloads, exosomes isolated using differential ultracentrifugation are typically contaminated with proteins.27 A recent study has demonstrated that exosomes endowed with magnetic properties can be magnetically separated and efficiently modulated by magnetic force (MF) and thus provide exosomes with favorable targeting properties.28
Targeted drug delivery can significantly improve drug distribution and reduce the dosage of drugs and adverse effects on the body.29 Recently, SPIONs have received substantial attention due to their applications as magnetic nanoplatforms for drug targeting carriers.30,31 SPIONs can be targeted to the required area through external magnets. The stability of the iron oxide particles is integrant for SPIONs to prevent agglomeration in a biological medium. Chitosan (CS) is an effective and common material used to modify NPs with therapeutically desirable characteristics, such as biocompatibility.32 CS has been widely used for the dispersion of SPIONs in aqueous medium for the purpose of drug delivery.33 Notably, CS NPs have been demonstrated as carriers for protein or gene therapeutics in recent years.34 In this study, carboxylated CS is used for better solubility.
Therefore, our aim was to design a synergistic system via conjugation of SPIONs to the surface of exosomes with TNF-α anchored in the membrane. Briefly, recombinant plasmids that contained the sequence of CPP and TNF-α were constructed to establish the stable MSC cell line expressing TNF-α. Exosomes anchored with TNF-α were subsequently derived from MSCs and conjugated with SPIONs to construct CTNF-α-exosome-SPIONs. The anticancer activity and the underlying mechanisms of CTNF-α-exosome-SPIONs against cancer cells were investigated. Our results showed that the exosome delivery system significantly enhanced the binding capacity of TNF-α to its membrane-bound receptor, which enhanced TNF-α-mediated cell apoptosis. The in vivo study demonstrated that SPIONs endowed CTNF-α-exosome-SPIONs with an excellent targeting ability, which enhanced the cancer inhibition effect and decreased the toxicity.
Scheme 1 summarizes the synthesis of CTNF-α-exosome-SPIONs, with details provided in the subsequent sections. Instead of intracellular delivery, the ligand of TNF-α located on the cell membrane, therefore, gene engineering rather than electroporation was used to construct CTNF-α-exosome-SPIONs with membrane targeting ability. To construct the recombinant expression vector encoding a fusion peptide (CTNF-α) comprising TNF-α and cell-penetrating peptides (CPP), overlapping polymerase chain reaction (PCR) was used (Scheme 1A).35 CPP are small peptides with lipotropic activity that can spontaneously incorporate into cell membranes and are successfully used as delivery vectors for therapeutic molecules.36 Fusion with CPP enables TNF-α to anchor in the cell membrane, thus producing exosomes with TNF-α anchoring in its membrane (CTNF-α-exosomes). Moreover, a short peptide was introduced between TNF-α and CPP to optimize their flexibility.37 The MSC cell line stably expressing CTNF-α was selected to produce CTNF-α-exosomes (Scheme 1B).
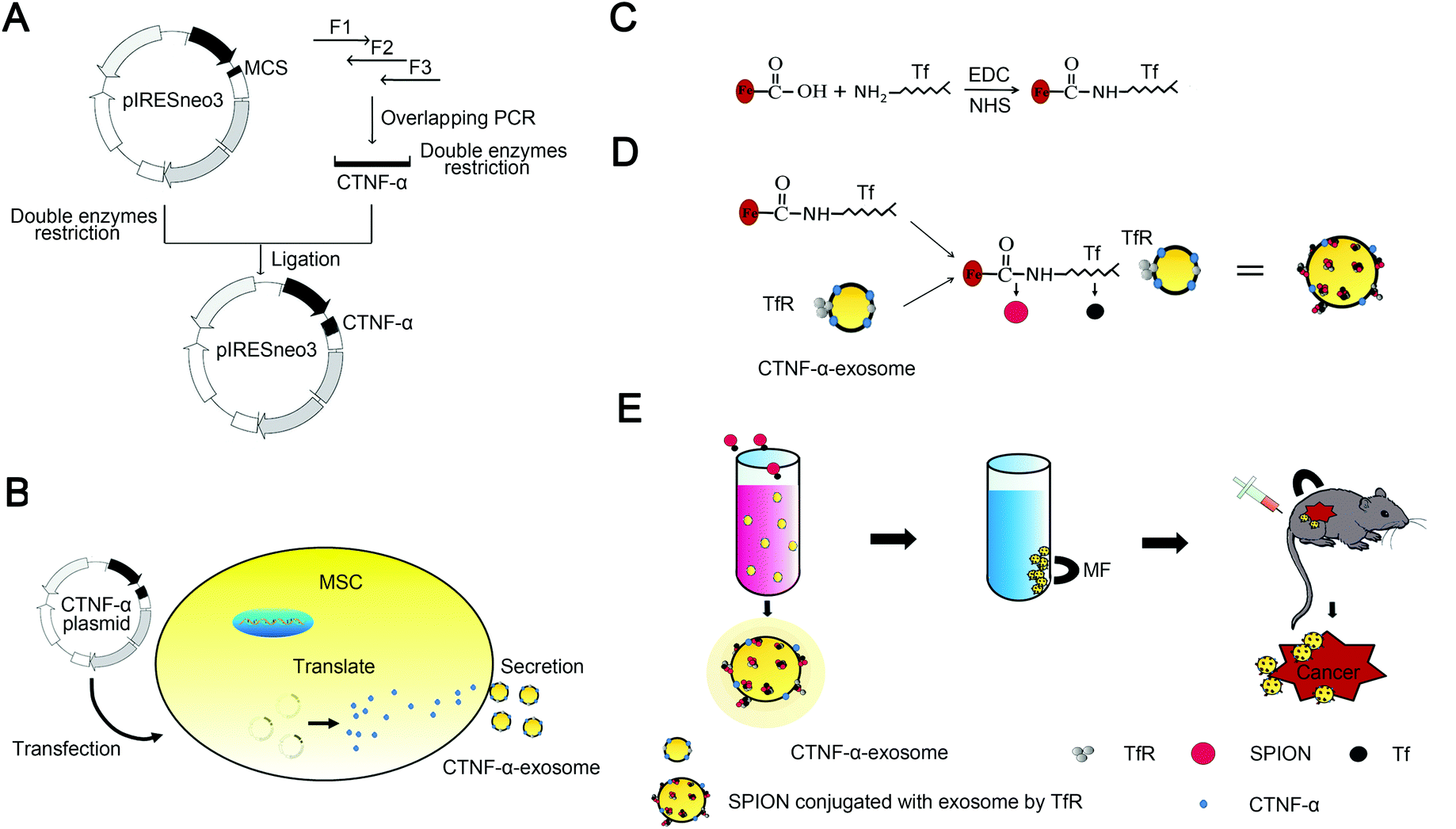 | ||
| Scheme 1 Schematic illustration for the preparation of the CTNF-α-exosome-SPION. (A) Construction of the recombinant plasmid encoding CTNF-α; (B) plasmids encoding CTNF-α were transfected to MSCs to produce CTNF-α-exosomes. (C) Synthesis of a Tf modified SPION (Tf-SPION); (D) preparation of the CTNF-α-exosome-SPION; (E) separation of the CTNF-α-exosome-SPION and magnetic targeting of the CTNF-α-exosome-SPION in vivo. | ||
Scheme 1C and D depict the synthetic process of SPION conjugated exosomes. In the present study, size-controlled SPIONs were prepared by the addition of carboxylated CS. The COO− groups in carboxylated CS on the surface of SPIONs could interact with the NH3+ of transferrin (Tf) in the presence of carbodiimide (EDC) and N-hydroxysulfosuccinimide sodium salt (NHS), thereby obtaining Tf-modified SPIONs (Tf-SPIONs) as described in Scheme 1C. It well known that exosomes contain various membrane proteins, including abundant transferrin receptors (TfR).15 As a result of the high affinity of Tf and TfR, Tf-SPIONs and CTNF-α-exosomes could self-assemble to form CTNF-α-exosome-SPIONs in aqueous solution (Scheme 1D). Under an external MF, CTNF-α-exosome-SPIONs could be easily collected, which may be beneficial for their biomedical applications. SPIONs as drug carriers could enhance the drug concentration in the target location with the aid of MF and improve the tumor inhibition capacity of TNF-α both in vitro and in vivo (Scheme 1E).
Materials and methods
Materials
MSC NutriStem® XF medium was purchased from Biological Industries (Kibbutz Beit-Haemek, Israel). GIBCO® Mouse (C57BL/6) MSC was purchased from Thermo Fisher Scientific (Waltham, MA, USA). RPMI-1640 medium or DMEM was purchased from Sigma (St Louis, MO, USA). MTT was purchased from Merck (Darmstadt, German). Primers were synthesized by BGI (Shenzhen, Guangdong, China). Restriction enzymes, FeCl2·4H2O, FeCl3·6H2O, NH3·H2O, CS, EDC, NHS, Tf and phosphotungstic acid were obtained from Sigma (St Louis, MO, USA). The dialysis tube, co-immunoprecipitation kit, BCA kit, Sephadex G-25, and mitochondrial membrane potential kit (JC-1) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). The hematoxylin and eosin staining kit and immunohistochemical kit were obtained from Boster Company (Wuhan, Hubei, China). Cleaved caspase-3 (Asp175) (5A1E) rabbit mAb, cleaved caspase-8 (Asp391) (18C8) rabbit mAb, TNF-α (D1G2) rabbit mAb and TfR I (D317 K) rabbit mAb were obtained from Cell Signaling Technology (Danvers, MA, USA). Cleaved-caspase-2 antibody [EPR16796] (ab179520) was obtained from Abcam (Cambridge Science Park, England). The anti-CD9 antibody and anti-CD63 antibody were obtained from Sigma (St Louis, MO, USA).Cell lines and cell culture
Several tumor cell lines and normal cell lines including A375 human melanoma cell lines, MCF-7 human breast adenocarcinoma cell lines, A549 human lung carcinoma cell lines, Colo201 human colon adenocarcinoma cell lines, HCM human myocardial primary cells, HUVECs human umbilical vein endothelial primary cells, HKC human embryonic kidney epithelial primary cells and L929 mouse fibroblast primary cells were obtained from the China Academia Sinica Cell Repository (Shanghai, China). All cell lines were maintained in either RPMI-1640 medium or DMEM supplemented with penicillin (100 units per mL), fetal bovine serum (10%), and streptomycin (50 units per mL) at 37 °C in a humidified incubator under a 5% CO2 atmosphere. Cells were plated in 6-well plates (1 × 105 cells per well) for western blotting studies and in 96-well plates (0.5 × 104 cells per well) for viability assays. After 2 days of culture, cells were used for various experiments.Construction of recombinant plasmids and their transfection
The pIREsneo3 plasmid was obtained from FengHui Biotechnology Company (Hunan province, China). The vector plasmid contains a multiple cloning site (MCS) flanked by left and right inverted/direct repeat elements.38,39 A gene fragment could insert into the plasmid MCS via double enzyme restriction. Overlapping extension PCR was employed to generate recombinant fusion genes, including genes of TNF-α, CPP and a flexible peptide. Based on the sequence of the extracellular fragment of TNF-α, the sequence of CPP (KETWWETWWTEWSQPKKKRKV) and the sequence of flexible peptide (GGGGSGSG),40,41 two pairs of primers were designed for amplifying the target gene fragments. The extracellular fragment of the TNF-α gene was amplified from cDNA by PCR with the forward primer F1 and reverse primer R1. Similarly, the fragment of the CPP gene was amplified using the primers F2 and R2, and the full-length recombinant fusion protein gene was amplified using the primers F1 and R2.42 The extracellular fragment of the TNF-α gene was fused directly to the N-terminus of the CPP by overlap-PCR from the PCR products (extracellular fragment of TNF-α and CPP genes) with the forward primer F2 and reverse primer R1 as described in Fig. S1.† The sites for the restriction digest Nhe I and BamH I were introduced into F1 and R2, respectively. The primer sequences are designed by using NCBI and shown in Table S1.† The full-length recombinant fusion protein gene of the PCR products was cloned into the pIREsneo3 plasmid by DNA ligase, after being digested by Nhe I and BamH I, thereby generating the construct pIREsneo3-CTNF-α. The resulting constructs were confirmed via DNA sequencing by BGI (Chengdu, China). MSCs were transfected with recombinant plasmids that expressed pIREsneo3-CTNF-α using Lipofectamine 2000 (Invitrogen, Carlsbad, CA).43Preparation and modification of SPIONs
SPIONs can be synthesized by a typical chemical co-precipitation of Fe2+ and Fe3+ in ammonium hydroxide solution according to the following reaction: FeCl2 + 2FeCl3 + 8NH3·H2O = Fe3O4 + 8NH4Cl + 4H2O. Briefly, 5 mL FeCl3·6H2O (2.5 mol L−1) was mixed with 8.75 mL FeCl2·4H2O (2.5 mol L−1) at pH 5.5. Different concentrations of carboxylated CS were added and mixed well (Fig. S2A†). The pH value was adjusted close to 10. The mixture was subsequently placed in a water bath at 80 °C for 1 h–8 h to select the optimum process time, with dialysis for 48 h to desalt against distilled water to generate CS-SPIONs.CS-SPIONs (200 μL, 1 mg mL−1) were mixed with EDC and NHS at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:3 (pH 5.5). This reaction mixture was incubated at room temperature for 1 h. Then, 5 μL of 2-mercaptoethanol was added to terminate the reaction. The activated CS-SPIONs were purified by magnetic separation and were resuspended in 200 μL PBS (pH 7.4). Then, 10 μg Tf was added, and the mixture was incubated for 12 h at 4 °C in a closed environment. Finally, Tf-SPIONs were purified by magnetic separation and washed with PBS three times. The resulting solution was stored at 4 °C until it was used for magnetic separation.
:2:3 (pH 5.5). This reaction mixture was incubated at room temperature for 1 h. Then, 5 μL of 2-mercaptoethanol was added to terminate the reaction. The activated CS-SPIONs were purified by magnetic separation and were resuspended in 200 μL PBS (pH 7.4). Then, 10 μg Tf was added, and the mixture was incubated for 12 h at 4 °C in a closed environment. Finally, Tf-SPIONs were purified by magnetic separation and washed with PBS three times. The resulting solution was stored at 4 °C until it was used for magnetic separation.
The synthesis of CTNF-α-exosome-SPIONs
MSCs that are transfected with recombinant plasmids that expressed pIREsneo3-CTNF-α were seeded in a cell culture dish (10 cm) and cultured for 3 days. Once the confluence reached 80%, 30 mL of the cell culture supernatant that contained exosomes was harvested and dialyzed against PBS for 24 h. Thereafter, the cell culture supernatant was centrifuged at 200g for 5 min to eliminate cells. The supernatant was then centrifuged at 12000g for 45 min and filtered through a 0.22 μm filter membrane to remove dead cells and cell debris. The resulting supernatant that contained CTNF-α-exosomes was mixed with 200 μL Tf-SPION solution (0.5 mg mL−1) and incubated for 4 h at 4 °C. Following this step, CTNF-α-exosome-SPIONs were obtained by magnetic separation and resuspended in PBS. CTNF-α-exosome-SPIONs were stored at 4 °C prior to use. The concentration of SPIONs was found to be 2.31 μg and the concentration of Tf was found to be 0.104 μg in every 1 μg of CTNF-exosome-SPIONs (protein concentration). The exosome markers CD 9, CD 63 and TNF-α of the magnetic separation mixture were assayed by western blot to verify whether the exosome was well conjugated to Tf-SPIONs. The protein concentration of CTNF-α-exosome-SPIONs was detected by the BCA assay and an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES, OPTIMA 2000DV, PerkinElmer, USA) was used for detecting the iron element. The Tf content in CTNF-α-exosome-SPIONs was measured by using the ELISA kit. Before the detection, CTNF-α-exosome-SPIONs were concentrated using a Termovap Sample Concentrator (Organomation, South Berlin, MA, USA).
Characterization of CTNF-α-exosome-SPIONs
To analyze the size distribution, a Zetasizer Nano (Malvern Instruments, Malvern, UK) was used. The structures of SPION, CS-SPION and TF-SPION were analyzed and characterized by Fourier-transform infrared spectroscopy (FT-IR). FT-IR spectra of the samples were recorded on an Equinox 55 IR spectrometer in the range 4000–400 cm−1 using the KBr-disk method. The morphology of the CTNF-α-exosome-SPIONs was characterized by TEM.Cellular binding of CTNF-α-exosome-SPIONs
To detect the cellular binding of CTNF-α-exosome-SPIONs to TNFR I on tumor cells, the pIREsneo3 plasmid that contained the green fluorescent protein (GFP) gene was applied to produce the fusion protein CTNF-α with green fluorescent dyes.44,45 Moreover, exosomes were labeled with the red fluorescent dye 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil) in advance according to the instructions.The cellular binding of CTNF-α-exosome-SPION/MF was monitored and qualitatively assessed by fluorescence microscopy (Carl Zeiss Meditec AG, Jena, Germany). Briefly, cells cultured in 6-well plates until 70% confluence were incubated with CTNF-α-exosome-SPION/MF for 1 h. The cells were then washed three times with PBS and measured at 470 nm.
Simultaneously, flow cytometry assay was also conducted to elevate the cellular binding efficiency of CTNF-α-exosome-SPION/MF. After being incubated with CTNF-α-exosome-SPION/MF for 1 h, the cells were harvested and washed with PBS, and dispersed in 1 mL of PBS. The cellular binding was measured by detecting the fluorescence intensity of TNF-α via flow cytometry (Becton Dickinson, USA).
Loading rate
Samples for the calibration curve were prepared by using various amounts of TNF-α (25, 50, 75, 100, 125, 150, 175, 200, 225, and 250 pg mL−1) or BSA (100, 200, 300, 400, 500, 600, 700, 800, 900, and 1000 μg mL−1). The concentration of TNF-α or BSA versus the OD value was calculated by least squares linear regression using a weighting factor of 1/x2 (the reciprocal of the squared concentration), thus the calibration curve of BSA or TNF-α was constructed. The correlation coefficient was used to evaluate the linearity. The loading rate of exosome-SPIONs was calculated using the following equation: loading rate = (CTNF-α quantification/BSA quantification) × 100% (3).Pharmacokinetic studies
Kunming mice (10 weeks old, 18–22 g) were randomly divided into two groups of six mice each: group-I was administered TNF-α at a dose of 5 mg kg−1; group-II was administered CTNF-α-exosome-SPIONs in accordance with the concentration of TNF-α in group-I. Compounds were delivered via a tail vein injection. At different time intervals post injection, 10 μL blood samples were collected from the tail vein in heparinized micropipettes and rapidly mixed with 1 mL of PBS. The collected samples were centrifuged at 10000 rpm for 10 min, and the supernatants were then detected by using the human TNF-α Elisa Kit.34
Co-immunoprecipitation assay
A375 cells were seeded in 6-well plates at 1 × 105 cells per well for 48 h. The cells were then incubated with TNF-α, CTNF-α-exosome-SPION and CTNF-α-exosome-SPION/MF at a concentration of 2.5 nmol L−1 for 1 h prior to lysis and extraction. After protein extraction, 500 μL supernatant was incubated with a rabbit polyclonal antibody for TNF-α at 4 °C for 1 h and mixed with 20 μL protein A- or protein G-Sepharose beads (GE Healthcare) by rotating at 4 °C for 1 h. The complexes were washed with RIPA buffer and were subjected to western blotting using a polyclonal antibody for TNFR I.46MTT assay
Cells were seeded in 96-well plates at 0.5 × 104 cells per well for 24 h. The cells were then incubated with TNF-α, CTNF-α-exosome-SPION or CTNF-α-exosome-SPION/MF at different concentrations for 24 h. After 24 h, 100 μL MTT solution (0.5 mg mL−1) was added and incubated for another 4 h at 37 °C, and then the plate was shaken every 30 minutes. 100 μL of methyl alcohol is added to each well and resuspended until all crystals have dissolved. Thereafter, the absorbance at 490 nm was measured by using a SpectraMax M5 microplate reader (Molecular Devices, LLC, Sunnyvale, CA, USA).47Colony-forming assay
The colony formation ability assay was performed as previously described.48 A375 cells were seeded in 6-well plates at 1000 cells per mL and allowed to attach for 24 h. The cells were subsequently treated with 2.5 nmol L−1 of TNF-α, CTNF-α-exosome-SPION or CTNF-α-exosome-SPION/MF separately. The cells were incubated with fresh medium of TNF-α, CTNF-α-exosome-SPION and CTNF-α-exosome-SPION/MF once every 3 days for an additional 12 days and stained with 0.5% crystal violet after fixation in 4% paraformaldehyde. The morphology of the cells was subsequently observed using a microscope (uX71, Olympus Corp, Tokyo, Japan), and the colony formation efficiency was used to evaluate the effects of the different treatments.Flow cytometric analysis
The cell cycle distribution was analyzed following TNF-α, CTNF-α-exosome-SPION and CTNF-α-exosome-SPION/MF treatment using flow cytometry. Briefly, A375 cells were treated for 24 h, harvested, washed once with PBS and fixed using 75% ethanol at 4 °C overnight. Following centrifugation at 800g for 5 min, the cells were then resuspended and stained using a cell cycle analysis kit, according to the manufacturer's protocol. The cell cycles were subsequently analyzed using a flow cytometer (Becton Dickinson, USA).Evaluation of mitochondrial membrane potential
The mitochondrial membrane potential was determined using the JC-1 probe. A375 cells were treated with 2.5 nmol L−1 of TNF-α, CTNF-α-exosome-SPION or CTNF-α-exosome-SPION/MF for 24 h, and then the cells were loaded with 1 mg L−1 of JC-1 at 37 °C for 10 min. Then a fluorescence microscope (Carl Zeiss Meditec AG, Jena, Germany) was used to analyze the fluorescence intensity.49Biodistribution and magnetic targeting for cancer in vivo
TNF-α and CTNF-α-exosome-SPIONs were labeled with a fluorescent dye to show their traces. TNF-α and CTNF-α-exosome-SPIONs were labeled with NHS-CY5.5 (mass ratio of 100:1) in pH 8.5 buffer solution. After incubation for 4 h, CY5.5-labeled TNF-α was separated by HPLC, and CY5.5-labeled CTNF-α-exosome-SPIONs were separated magnetically.28,50 The separated CY5.5-labeled CTNF-α-exosome-SPIONs were washed three times with PBS and were resuspended in PBS. The concentration of CTNF-α was measured using a TNF-α Elisa Kit.
The B16F10 melanoma tumor model was generated by subcutaneous injection of 2 × 106 B16F10 cells that were suspended in serum free 1640 medium using BALB/c female mice. The cells were allowed to grow for 2 weeks until the tumors were approximately 300 × mm3 in volume. The mice were randomly divided into four groups. One group was the control group, and two groups were intravenously injected with Cy5.5-labeled TNF-α and a CTNF-α-exosome-SPION solution (5 mg of TNF-α-equiv per kg of body weight). The fourth group was intravenously injected with the same dose of Cy5.5-labeled CTNF-α-exosome-SPION solution, while a magnet (MF density: 1 T) was placed over the surface of the cancerous tumor using Steri-Strip tape for 24 h. In vivo fluorescence imaging was performed using the IVIS Imaging System (Caliper Life Sciences, Boston, USA). The mice were subsequently euthanized, and the tumors, tissues, and organs were dissected and subjected to ex vivo fluorescence imaging. The fluorescence imaging results and the average ratio intensities were recorded using the same IVIS spectrum imaging system.51
Additionally, ICP-OES was used to determine the biodistribution and the targeting effect of the CTNF-α-exosome-SPIONs. The heart, liver, spleen, lungs, kidneys and tumors in each experimental group were harvested. 4 mL of concentrated nitric acid and 1 mL of concentrated hydrochloric acid were added to the sample (0.1 g) in a tube and incubated overnight for dissolving tissue. Metal analysis was performed using ICP-OES.55
Immunohistochemical staining
Immunohistochemical staining for activated caspases 2, 3, and 8 was conducted according to standard protocols. Paraffin sections were reacted with an activated caspase-specific antibody at a dilution of 1:500, which recognizes large fragments of activated caspase. Five-micrometer-thick tissue sections were deparaffinized in xylene and hydrated in ethanol, and the endogenous peroxidase activity was blocked by soaking in 3% H2O2 for 10 min. Antigen retrieval was conducted by incubating tissue sections in a microwave in 10 mmol L−1 Tris-HCl buffer (pH 8.0) that contained 1 mmol L−1 EDTA. Sections were blocked with 1% normal bovine calf serum in 50 mmol L−1 Tris buffered saline (TBS, pH 7.6) and incubated with the diluted primary antibodies at 4 °C overnight. After washing with TBS, the slides were incubated with peroxidase–biotin–avidin for 60 min. Diaminobenzidine was used as the chromogen. The sections were then counterstained with hematoxylin.53
Statistical analysis
All experiments were carried out at least in triplicate of independent experiments and the results were expressed as mean + standard deviation. The statistical difference was assessed by one-way analysis of variance. Unpaired two-tailed t-tests and paired two-tailed t-tests were used to determine the statistical significance between the test groups as appropriate. P < 0.05 was considered statistically significant (*) and P < 0.01 was considered statistically highly significant (**).Results and discussion
CTNF-α-exosome-SPION morphology and characterization
Previous studies have demonstrated that iron oxide NPs exhibit superparamagnetic behavior at sizes <25 nm.56 SPIONs with a smaller diameter have a larger specific surface area, which favorably enhances their adsorption. In the present study, size-controlled SPIONs were prepared by carboxylated CS. The synthesis conditions have a significant influence on the chemical, structural and physical properties of iron oxides.54 The optimal concentration and reaction time of carboxylated CS were investigated to obtain SPIONs with an ideal diameter. SPIONs with a diameter of 10 nm were obtained using CS at the concentration of 0.08 mg mL−1 for 1 h (Fig. S2B†). The size distribution of carboxylated CS modified SPIONs (CS-SPION) used in the following experiment is shown in Fig. 1A.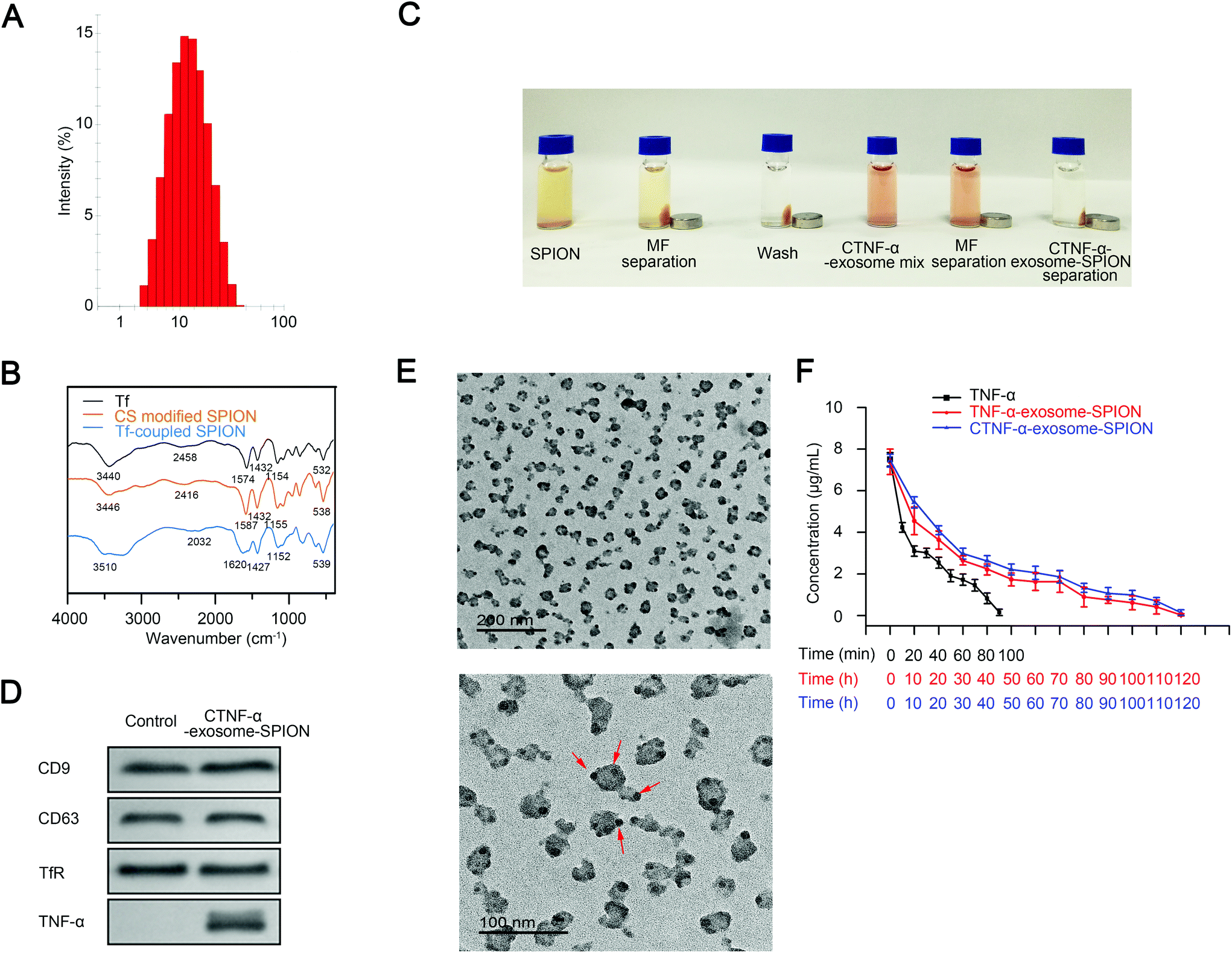 | ||
| Fig. 1 CTNF-α-exosome-SPION morphology and characterization (A) size distribution of the SPION; (B) FT-IR of Tf, CS modified SPION (CS-SPION) and Tf-coupled SPION (Tf-SPION); (C) images of the CTNF-α-exosome-SPION construction process; (D) western blot analysis of specific exosome marker proteins (CD9 and CD63), TfR and TNF-α in the redispersed CTNF-α-exosome-SPION or exosome-SPION (control); (E) TEM images of the CTNF-α-exosome-SPION under different magnifications; (F) concentration–time profile of CTNF-α in Kunming mice following the tail-vein injection of 5 mg kg−1 TNF-α or CTNF-α-exosome-SPION. Six independent experiments, significance levels are shown as *p < 0.05 and **p < 0.01. | ||
To construct CTNF-α-exosome-SPIONs, a TF-SPION was first synthesized by conjugating Tf to the CS-decorated SPION surface. Fig. 1B shows the Fourier-transform infrared spectroscopy (FT-IR) analysis of Tf, CS-SPION and Tf-SPION. The peak of the CS-SPION at 538 cm−1 was attributed to the Fe–O group. The peaks at 1155 cm−1 corresponded to the stretching vibration of C–O, which is the characteristic peak of CS. The peaks of Tf that appeared at 1574 cm−1 and 1432 cm−1 were assigned to the first and secondary –CO–NH– groups of Tf. The peaks of Tf at 3440 cm−1 and 2458 cm−1 corresponded to –OH and C–H, respectively. The characteristic O–H group peak in the Tf-SPION at 3510 cm−1 was relatively higher than that of Tf. The slight shift of the O–H group was likely due to the conjugation of Tf to the SPION surface. Specifically, the amide group from Tf was different in the Tf-SPION, which indicates linking of Tf to the nanoparticles via the amide groups in carboxylated CS. In conclusion, chemical bond changes occurred during the process of conjugation.
A coupling reaction of the CS-SPION and Tf was conducted at 4 °C for 12 h in 200 μL borate buffer (20 mM, pH 8.5) (Fig. 1C, SPION), and free Tf was removed by magnetic separation (Fig. 1C MF separation). The Tf-SPION was subsequently washed with PBS, and magnetic separation was performed again (Fig. 1C, wash). Next, the Tf-SPION was mixed with the medium of the MSC cell line that contained CTNF-α-exosomes at 4 °C for 4 h (Fig. 1C CTNF-α-exosome mixture). CTNF-α-exosomes coupled with the Tf-SPION through the self-combination of Tf and TfR, thus obtaining the final magnetic nanocomposite CTNF-α-exosome-SPION. CTNF-α-exosome-SPIONs were harvested from the medium by magnetic separation (Fig. 1C MF separation). Finally, CTNF-α-exosome-SPIONs were washed with PBS (Fig. 1C CTNF-α-exosome-SPION separation).
Western blot analysis showed that MSCs transfected with the recombinant plasmid successfully expressed CTNF-α. Moreover, CTNF-α could not be detected in exosomes from MSCs transfected with an empty vector. Two typical marker proteins of exosomes (CD9 and CD63)57 and TfR were also detected in CTNF-α-exosome-SPIONs harvested by magnetic separation, which confirmed the successful conjunction of CTNF-α-exosomes to Tf-SPION (Fig. 1D). CTNF-α-exosome-SPIONs were further characterized using a transmission electron microscope (TEM). Our results showed that CTNF-α-exosome-SPIONs were well dispersed. Nanoparticles with good water dispersibility are essential to medical applications. As shown in Fig. 1E, the exosome surface was uniformly surrounded with SPIONs.
Taken together, it can be concluded that CTNF-α-exosome-SPIONs were successfully constructed with good water dispersibility.
Drug loading and half-life
To determine the drug loading profile, the calibration curves of BSA or TNF-α were first constructed. The representative linear equations were protein concentration (μg mL−1) = 0.0009 × OD + 0.0717 (1) for BSA and TNF-α concentration (pg mL−1) = 0.0028 × OD + 0.0842 (2) for TNF-α, with the coefficient of correlation invariably greater than 0.99 (Fig. S3†). The CTNF-α-loading rate of CTNF-α-exosome-SPIONs was found to be 3.21%, which indicates that CTNF-α-exosome-SPIONs possessed a high loading capacity. The high loading rate of the nano-delivery system could decrease the dose of CTNF-α, thus minimizing potential side effects.Fig. 1F demonstrates the plasma concentration–time profiles of TNF-α, TNF-α-exosome-SPIONs and CTNF-α-exosome-SPIONs. TNF-α displayed a short life span, which is consistent with the previous study. However, when loaded into exosomes, TNF-α was detectable for 120 hours. Moreover, the blood circulation of TNF-α, TNF-α-exosome-SPIONs and CTNF-α-exosome-SPIONs in vivo was further investigated by using Pksolver. TNF-α was characterized with a rapid distribution and elimination phase (t1/2α: 0.15 min, t1/2β: 4.19 min), while TNF-α exhibited a relatively slow distribution phase and elimination phase (t1/2α: 0.49 h and t1/2β: 3.48 h for TNF-α-exosome-SPIONs; t1/2α: 0.75 h and t1/2β: 3.89 h for CTNF-α-exosome-SPIONs) following the application of exosomes and the conjugation of the Tf-SPION. The prominent improvement in pharmacokinetics was most likely attributed to the protection of exosomes. Therefore, it could be concluded that the prolonged in vivo half-life of CTNF-α-exosome-SPIONs increased the acting time of TNF-α, which would most likely improve its antitumor properties and reduce the dose of TNF-α. To investigate whether Tfs in the serum can compete and dissociate the Tf-SPION, the particle stability of CTNF-α-exosome-SPIONs in serum was examined for 7 days by measuring particle sizes. We magnetically separated and redispersed CTNF-α-exosome-SPIONs, the magnetic separation performance of CTNF-α-exosome-SPIONs maintained stability and no obvious aggregation was observed in serum for 7 days (Fig. S4A†). The high stability of CTNF-α-exosome-SPIONs provides support for translating these findings into future medical applications. Moreover, the concentration of TNF-α was also elevated for 7 days. As shown in Fig. S4B,† when loaded into exosome-SPIONs, a relatively high concentration of TNF-α was detectable for up to 7 days. This phenomenon can be explained by the fact that exosomes provide a protective barrier against elimination and degradation.
CTNF-α-exosome-SPION delivery and distribution
TNFR I, a transmembrane protein, can be completely activated by both the membrane-bound and soluble trimeric forms of TNF-α. The cell apoptosis function of TNF-α depends on the binding of TNF-α to its receptor TNFR I.58 As shown in the schematic, with the help of moderate MF and SPIONs, exosomes delivered CTNF-α to cancer cell membranes, which increased the binding capacity of CTNF-α to TNFR I, thus facilitating TNFR I-mediated cell apoptosis (Fig. 2A). To evaluate the binding capacity between CTNF-α and TNFR I, co-immunoprecipitation (CO-IP) was performed. The results indicated that CTNF-α-exosome-SPIONs significantly enhanced the binding of TNF-α to its receptor, which likely occurred because cell derived-exosomes could fuse with cell membranes, thus increasing the receptor-binding capacity. Moreover, the binding activity of CTNF-α and TNFR I was further facilitated by additional MF (Fig. 2B). These results indicate that the combination of the exosome-based delivery system and active magnetic targeting can clearly enhance the TNF-α–TNFR I combination, which consequently enhances TNFR I-mediated cell apoptosis.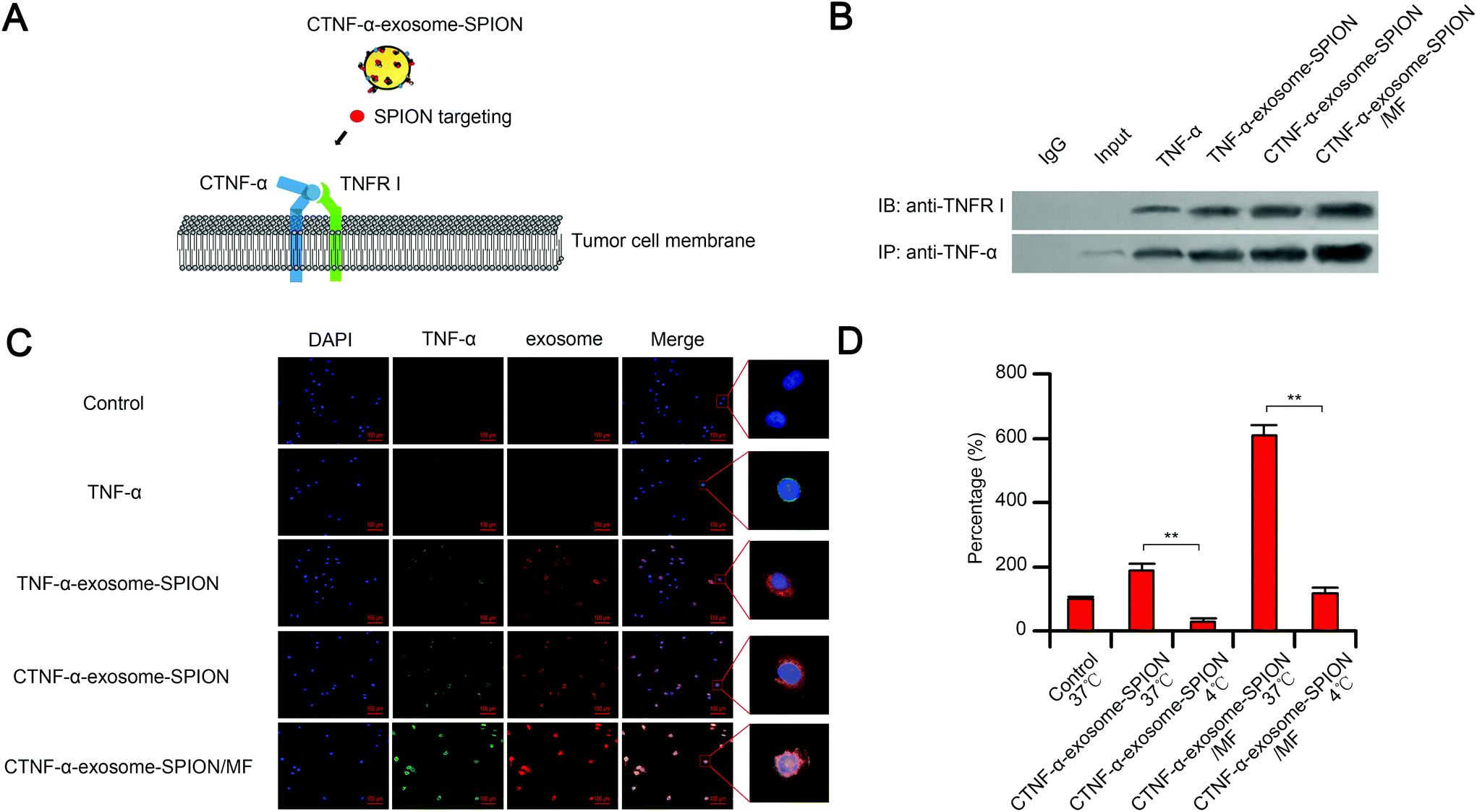 | ||
| Fig. 2 CTNF-α-exosome-SPION delivery and distribution. (A) Schematic overview of CTNF-α-exosome-SPION binding to TNFR I; (B) binding abilities of TNF-α and TNFR I were analyzed by CO-IP; (C) membrane fusion of the CTNF-α-exosome-SPION was monitored by fluorescence microscopy. Exosomes were labeled with Dil, and TNF-α was fused with GFP; (D) membrane fusion of the CTNF-α-exosome-SPION at 4 °C. Prior to treatment with CTNF-α-exosomes or CTNF-α-exosome-SPION, cells were incubated at 37 °C or 4 °C. The membrane fusion of the CTNF-α-exosome or CTNF-α-exosome-SPION was monitored by fluorescence microscopy. Cells treated with TNF-α at 37 °C were considered a control. Six independent experiments, significance levels are shown as *p < 0.05 and **p < 0.01. | ||
Selective cellular binding of therapeutic agents remains a substantial obstacle for cancer therapy. The nano-delivery system can increase the cellular binding of various peptides, which protects them against degradation and enhances their targeting and selectivity. In this study, the cellular binding of CTNF-α-exosome-SPIONs in cancer cells was investigated by measuring the fluorescence intensity. Fig. 2C demonstrates that in comparison with TNF-α or TNF-α-exosome-SPIONs, fusing CPP with TNF-α significantly increased the cellular binding of CTNF-α. Moreover, in the presence of MF, most exosome-based nanoparticles were further internalized into the cell membrane, thus facilitating the cellular binding of CTNF-α. This phenomenon can be rationalized by the fact that exosomes are composed of natural lipid bilayers, which can readily interact with cellular membranes. Additionally, SPION-decorated exosomes tend to accumulate on the surface of cells with the help of MF, thereby elevating the cellular binding of CTNF-α. Furthermore, flow cytometry assays were also conducted to evaluate the cellular binding efficiency. As shown in Fig. S5,† The average cellular binding efficiencies of the TNF-α and TNF-α-exosome-SPIONs were 2.86% and 3.73% respectively. CTNF-α-exosome-SPIONs displayed a significantly increased cellular binding efficiency of 12.08% which was approximately 4.22 times that with TNF-α. In the presence of MF, the cellular binding efficiency of CTNF-α-exosome-SPIONs was increased to 25.92%. These results further demonstrate higher cellular binding efficiency for the CTNF-α-exosome-SPION/MF. Taken together, our results showed that with the help of MF, exosomes enhance the cellular binding of CTNF-α through membrane fusion, which likely enhances the activation of TNFR I-mediated cell apoptosis.
The mobility of cell membranes is decreased at low temperatures because of a weaker physical resistance of the membrane or a potentially higher level of reactive oxygen radicals at low temperatures.59 The membrane mobility has an important impact on the cytokine-receptor binding capacity. Our results demonstrated that a low temperature of 4 °C remarkably reduced the fusion ability of CTNF-α-exosome-SPIONs to melanoma cell membranes in the presence or absence of MF, which may result in a lower binding capacity of TNF-α-TNFR I (Fig. 2D). This effect can be explained by the finding that the mobility of cell membranes was decreased at low temperatures (4 °C). Thus, it can be concluded that the cellular binding of CTNF-α-exosome-SPIONs substantially depends on the membrane mobility of target cells.
Moreover, the mechanism of CPP mediated TNF-α anchoring to the cell membrane was investigated. Firstly, TNF-α was loaded into exosome-SPIONs by electroporation to construct TNF-α-exosome-SPIONs. The membrane proteins of TNF-α-exosome-SPIONs or CTNF-α-exosome-SPIONs were efficiently extracted and separated, thereafter, TNF-α was detected by western blotting. As shown in Fig. S6A,† only a small amount of TNF-α could be detected in the membrane of exosome-SPIONs that are loaded with TNF-α by electroporation, whereas a large amount of TNF-α was found in the membrane of CTNF-α-exosome-SPIONs constructed by genetic engineering. Thereafter, co-immunoprecipitation was carried out to characterize the cellular binding capacity of TNF-α to its membrane receptor TNFR-I. Compared with TNF-α-exosome-SPIONs, CTNF-α-exosome-SPIONs significantly enhanced the binding of TNF-α to its receptor, which likely occurred because the lipotropic activity enables CPP to mediate TNF-α anchoring to the exosome membrane, thus increasing TNF-α's membrane distribution (Fig. S6B and S6C†). Therefore, it can be concluded that fusing CPP with TNF-α by genetic engineering endowed CTNF-α-exosome-SPIONs with membrane targeting ability.
Induction of apoptotic cell death by CTNF-α-exosome-SPION/MF
The in vitro cytotoxic effects or apoptosis-promoting effects of CTNF-α-exosome-SPION/MF were monitored via the MTT assay, including normal and cancer cell lines. As shown in Fig. S7,† CTNF-α-exosome-SPION/MF exhibited the maximum inhibition of the cell viability of cancer cell lines (A375, MCF-7, A549 and Colo201), followed by the CTNF-α-exosome-SPION, with a minimum found for the TNF-α group, similarly, CTNF-α-exosome-SPION/MF displayed a higher cytotoxicity toward normal cells compared with TNF-α or CTNF-α-exosomes in vitro. Although CTNF-α-exosome-SPION/MF displays higher toxicity toward normal cells, an external MF is generally placed over the surface of the tumor site rather than the normal tissue in practical use, therefore, a smaller amount of CTNF-α-exosome-SPIONs is accumulated in normal tissue. Thereafter, IC50 was calculated according to the results of MTT to further characterize the cytotoxicity. As shown in Fig. 3A, CTNF-α-exosome-SPION/MF displayed a broad-spectrum inhibition against A375, MCF-7, A549 and Colo201 cancer cells with IC50 values of approximately 5 nmol L−1. Compared with TNF-α or CTNF-α-exosome-SPIONs without MF, CTNF-α-exosome-SPION/MF displayed a higher cytotoxicity with a lower IC50 value toward tumor cells, which indicates that the combination of the exosome-based nano-delivery system and magnetic targeting can enhance the cytotoxicity of an antitumor drug. Exosomes isolated from cells contain a wide range of components, which may affect the anti-cancer activity of CTNF-α-exosome-SPIONs, therefore, the anti-cancer activity of SPIONs or exosomes was detected by MTT. As shown in Fig. S8,† SPIONs or exosomes have no effects on cell viability.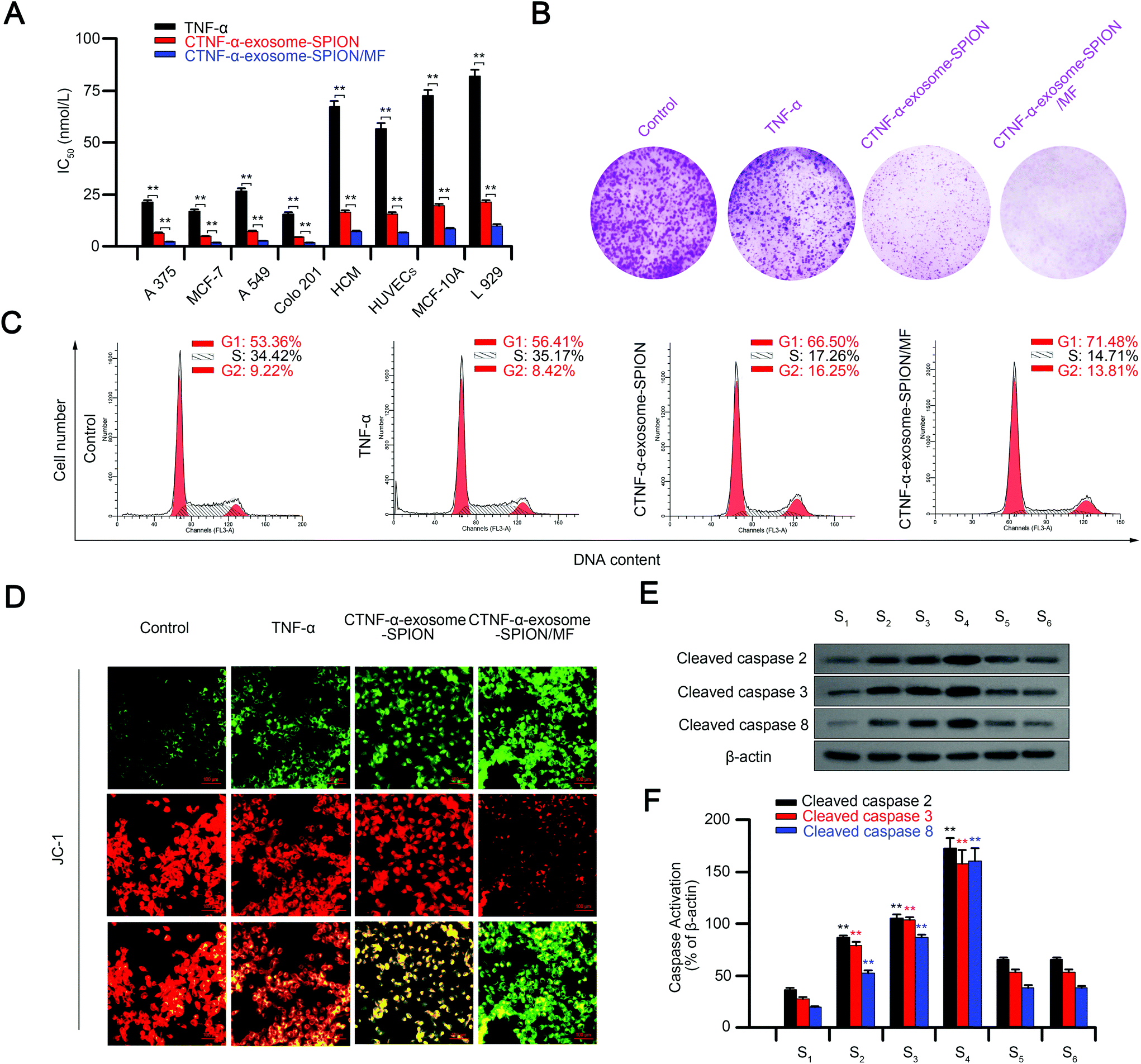 | ||
| Fig. 3 Induction of apoptotic cell death by CTNF-α-exosome-SPION/MF. (A) Cytotoxic effects of TNF-α, CTNF-α-exosome-SPION and CTNF-α-exosome-SPION/MF on various human cancer cells and normal cells after 48 h incubation; (B) the colony formation and survival fraction of A375 were observed after treatment with TNF-α, CTNF-α-exosome-SPION or CTNF-α-exosome-SPION/MF; (C) DNA cell cycle rates in A375 cells were analyzed using flow cytometry; (D) effects of TNF-α, CTNF-α-exosome-SPION and CTNF-α-exosome-SPION/MF on mitochondrial membrane potential using probe JC-1; (E, F) western blot analysis and quantification of apoptosis-related proteins were investigated after different treatments. S1: control, S2: TNF-α, S3: CTNF-α-exosome-SPION, S4: CTNF-α-exosome-SPION/MF, S5: CTNF-α-exosome-SPION/MF + etanercept, S6: CTNF-α-exosome-SPION/MF + z-VAD-fmk. Six independent experiments, significance levels are shown as *p < 0.05 and **p < 0.01. | ||
The results of the colony formation assay indicated that cells treated with TNF-α displayed substantially smaller and fewer colonies. CTNF-α-exosome-SPIONs enhanced the inhibitory effects of CTNF-α on the colony formation. Notably, after co-treatment with external MF and CTNF-α-exosome-SPIONs, the colony formation and survival fraction of A375 cells were at the minimum level (Fig. 3B).
The distribution of the cell cycle and apoptosis were analyzed using flow cytometry. Regulation of the cell cycle is essential to cell survival, including the detection and repair of genetic damage, as well as the inhibition of uncontrolled cell division.60Fig. 3C shows representative DNA histograms from PI staining cells, which indicated that the exposure of A375 cells to different treatments for 24 h led to a clear increase in the proportion of apoptotic cells, as reflected by the increased G1 populations. Treatment with TNF-α resulted in an increase of the G1 population from 53.36% to 56.41%. Notably, the G1 population increased to 66.5% using the exosome delivery system, while it increased to 71.48% by the combination of CTNF-α-exosome-SPIONs and MF. Apoptotic cell death was further confirmed by the mitochondrial membrane potential as detected by using JC-1 (Fig. 3D). The decline of the mitochondrial membrane potential is an early landmark event of apoptosis.61 It may be observed from microscopic images that compared with TNF-α or CTNF-α-exosome-SPIONs, the introduction of CTNF-α-exosome-SPION/MF exhibited the maximum decrease in the mitochondrial membrane potential. These results indicated that CTNF-α-exosome-SPION/MF achieved an excellent tumor inhibitory efficacy.
Apoptosis is a mechanism of programmed cell death. The caspases involved in apoptosis can be divided into two groups, the initiator caspases, such as caspases 2, 8, and 9, and the effector caspases, such as caspase 3.62 TNF-α stimulation can specifically cause the activation of caspases 2, 3, and 8 and lead to cell apoptosis.63 The presence of elevated cleaved-caspases 2, 3, and 8 confirmed that CTNF-α-exosome-SPION/MF could induce cell apoptosis in a TNF-α dependent manner. Moreover, the application of etanercept (20 μmol L−1) (Fig. S9†), a compound that blocks the toxic effects of TNF-α, or Z-VAD-FMK (20 μmol L−1), a cell-permeant pan caspase inhibitor, specifically and competitively inhibited CTNF-α-exosome-SPION-induced caspase 2, 3, and 8 activation or cell apoptosis (Fig. 3E and F and Fig. S10†).
In vivo distribution of CTNF-α-exosome-SPIONs
To determine the tumor targeting capacity of CTNF-α-exosome-SPIONs, the biodistribution of Cy5.5-labeled TNF-α or CTNF-α-exosome-SPIONs was investigated on A375 tumor-bearing model mice utilizing noninvasive near-infrared fluorescence (NIRF). As shown in Fig. 4A, the fluorescence signals suffered from attenuation and disappeared 24 h after injection with TNF-α because of the poor half-life of TNF-α in vivo. A weak fluorescence signal was observed in the tumors of the CTNF-α-exosome-SPION or TNF-α-exosome-SPION without the application of MF at 24 h due to the enhanced EPR effect and the prolonged half-life endowed by the protection of exosomes. The Cy5.5 signal of the CTNF-α-exosome-SPION was clearly observed at the tumor site in the presence of MF for 24 h, which indicates the excellent magnetic-targeting ability of the CTNF-α-exosome-SPION. After in vivo imaging, the mice were euthanized, and tumor tissues and organs were excised for ex vivo imaging. As shown in Fig. 4B, CTNF-α was predominantly accumulated in the tumor and liver with the application of the exosome-based nano-delivery system in the presence of MF, which was consistent with the in vivo findings. Quantification of the fluorescence intensity in normal tissues and tumor tissues was also evaluated. As shown in Fig. 4C, compared with the CTNF-α-exosome-SPION group, the fluorescence intensity of the tumor tissues increased approximately 10-fold by the application of MF.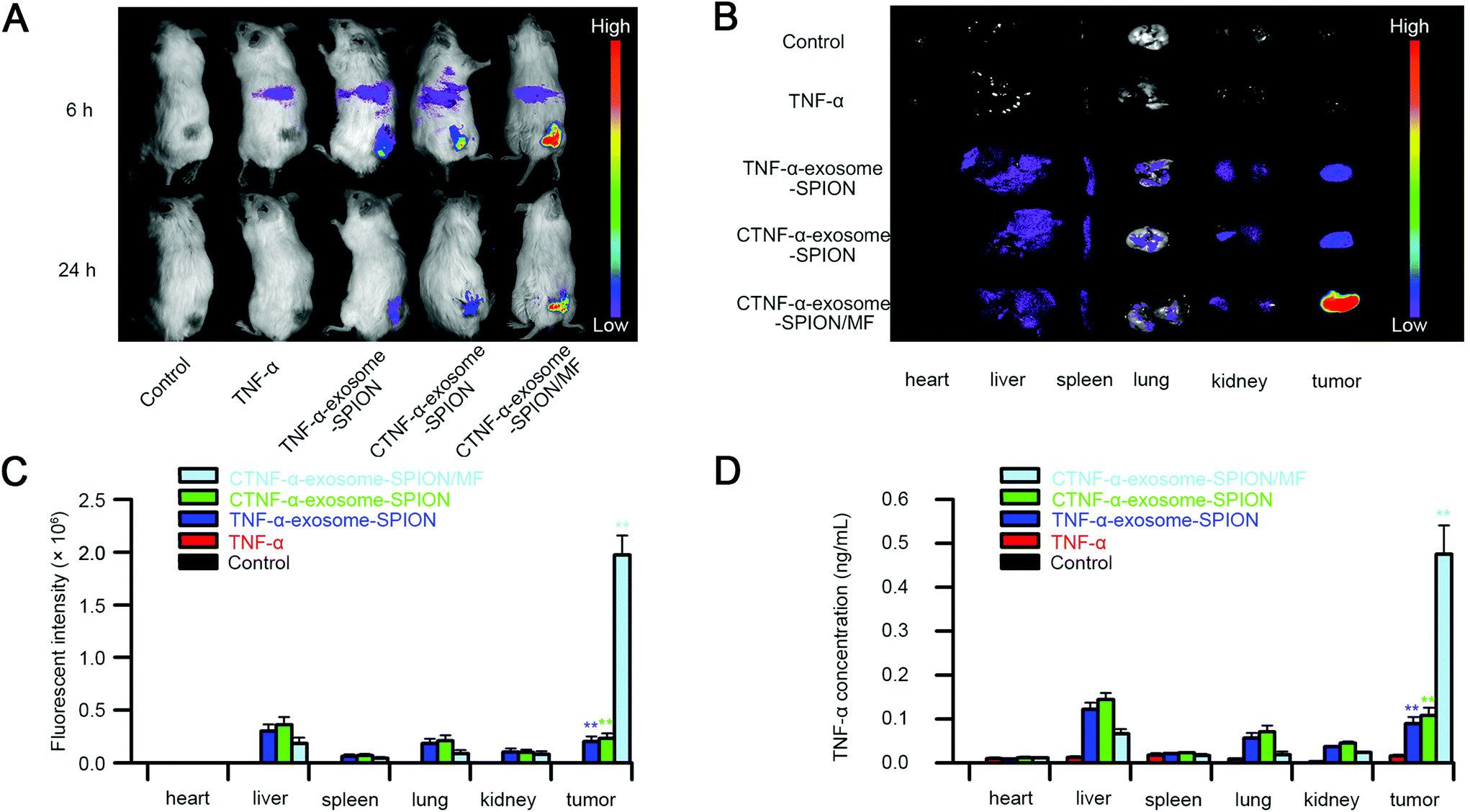 | ||
| Fig. 4 In vivo tumor targeting ability of the CTNF-α-exosome-SPION. (A) NIRF imaging of the cy5.5-labeled CTNF-α-exosome-SPION in model mice after 6 h or 24 h intravenous injection in the absence or presence of external MF; (B) representative ex vivo NIRF optical images of tumors and major organs; (C) fluorescence intensity of the cy5.5-labeled CTNF-α-exosome-SPION in tumors and major organs; (D) CTNF-α concentration in cancers and major organs. Six independent experiments, significance levels are shown as *p < 0.05 and **p < 0.01. | ||
To further investigate the biodistribution of the CTNF-α-exosome-SPION and evaluate its tumor targeting capacity, the TNF-α concentration of tumors and other organs was further investigated. The determined results of TNF-α's organized distribution indicate that the combined utilization of the CTNF-α-exosome-SPION and external MF had the maximum TNF-α concentration in the tumor, followed by the CTNF-α-exosome-SPION and TNF-α-exosome-SPION, with a minimum in the TNF-α group (Fig. 4D); these findings indicate that CTNF-α tends to accumulate and maintain an effective concentration in tumors for a longer period with the help of exosomes and external MF. Additionally, ICP-OES results (Fig. S11†) showed that remarkably more iron content was present in the tumor tissue after administration with the CTNF-α-exosome-SPION in the presence of MF than in the absence of MF which further confirms that with the help of MF, the SPION-based delivery system can be efficiently guided to their targeting location. These results provided solid evidence that CTNF-α-exosome-SPION/MF possessed an outstanding tumor targeting ability with remarkable potential for antitumor efficacy through a combination of EPR and active magnetic targeting. Targeted drug delivery using CTNF-α-exosome-SPION/MF can markedly increase the drug concentration in the tumor site and decrease the drug dosage, as well as the drug's side effects on the body.
Tumor suppression by the CTNF-α-exosome-SPION
We further investigated the in vivo activity of the CTNF-α-exosome-SPION using a subcutaneously grafted murine melanoma model. TNF-α, identified as cachectin, can induce cachexia with symptoms of weight loss, muscle atrophy or fatigue;64 thus, the body weight was evaluated before the end of the experiment. As shown in Fig. 5A, the mice in the TNF-α group or those in the CTNF-α-exosome-SPION group in the absence of MF exhibited a notable decrease in body weight. In contrast, no change in body weight was detected in the CTNF-α-exosome-SPION/MF group because of its excellent magnetic targeting ability.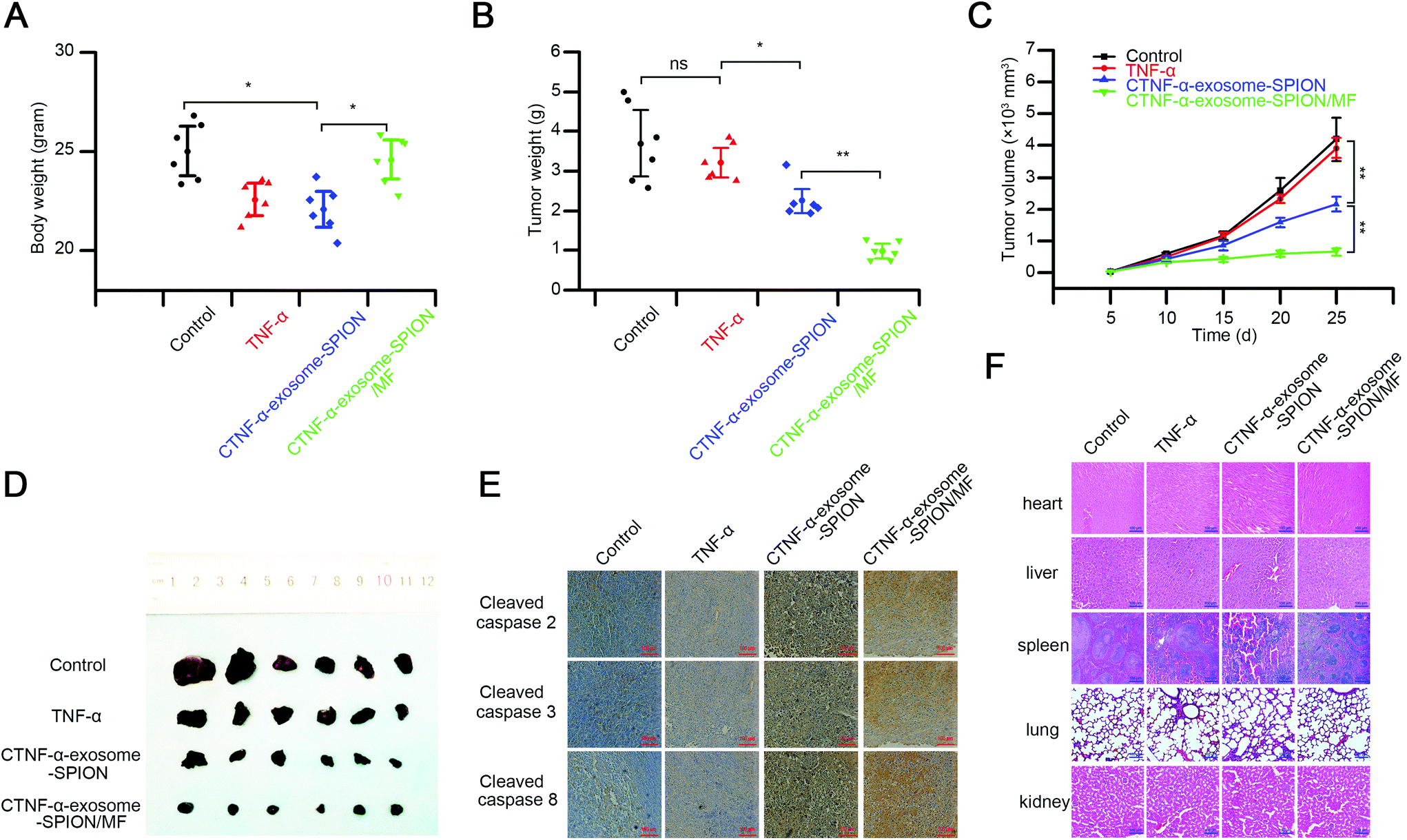 | ||
| Fig. 5 In vivo antitumor activity and toxicity of the CTNF-α-exosome-SPION. (A) Average body weight of mice; (B) average mass of excised tumors; (C) tumor growth monitored at different time-points and volume calculation; (D) tumor tissues obtained from euthanized mice; (E) immunohistochemical staining of cleaved caspases 2, 3 and 8 in tumor sections from different groups; (F) HE-stained major organ tissues (heart, liver, spleen, lungs and kidneys) obtained from laboratory mice. Six independent experiments, significance levels are shown as *p < 0.05 and **p < 0.01. | ||
The tumor volume was detected to examine the kinetics of cancer growth, which was calculated as (length × width2)/2.65 Previous studies have demonstrated that exosomes as a drug delivery system had no effect on tumor growth.27,66 The results indicated that the treatments with TNF-α showed a weak therapeutic effect in tumor inhibition (Fig. 5B). Compared with the control or TNF-α, the EPR effect of exosomes contributed to the better tumor growth inhibition capacity of CTNF-α-exosome-SPIONs. Moreover, treatment of CTNF-α-exosome-SPIONs in the presence of MF decreased the tumor volumes more effectively than the single treatments with CTNF-α-exosome-SPIONs, which validates that the application of MF significantly increased the efficacy of CTNF-α-exosome-SPIONs. The tumor weights of the CTNF-α-exosome-SPION/MF treated group as shown in Fig. 5C were also clearly reduced compared with the model group. The morphology of the isolated tumor in Fig. 5D further confirmed that the combination of CTNF-α-exosome-SPIONs and MF exhibited the best tumor growth suppression efficacy in vivo.
Moreover, the histological assays of the tumors and other tissues after treatment were performed using hematoxylin and eosin (HE) staining or immunohistochemistry. The HE staining in Fig. S12† and immunohistochemistry study in Fig. 5E showed that CTNF-α-exosome-SPION/MF possessed the best apoptosis induction capacity with its cleaved-caspase-2, 3, and 8 activation at a maximum level, closely followed by the CTNF-α-exosome-SPION group, which was consistent with the in vitro results. These findings indicated that with the aid of MF, CTNF-α-exosome-SPIONs could induce tumor-targeted melanoma death via TNF-α-mediated apoptosis.
Despite the promising anti-cancer potential of TNF-α, its clinical application remains hindered by the severe toxicity after systemic administration. Therefore, the toxicity of CTNF-α-exosome-SPION/MF was further investigated by examining the pathological changes in the major organs of the mice (Fig. 5F). Pathological changes in the liver, lungs and spleen were observed in the TNF-α and CTNF-α-exosome-SPION groups and compared with the control group that received saline. For example, extensive necrosis, accompanied by strong infiltration of inflammatory cells in the liver were observed in both the TNF-α and CTNF-α-exosome-SPION groups. Congestion in the pulmonary capillary and an irregular thickened wall were also recorded in the lung. In comparison with TNF-α or CTNF-α-exosome-SPION/MF, the prolonged in vivo plasma half-life and weak targeting capacity enabled TNF-α-exosome-SPIONs to accumulate in the normal tissue, thereby increasing TNF-α concentration which was harmful to normal tissue. Thus, the H&E staining of the normal tissue in the mice treated with CTNF-α-exosome-SPIONs exhibited significant changes compared with the control group or TNF-α group. Under the effective anticancer dose, no clear tissue damage was observed in the CTNF-α-exosome-SPION/MF treated mice. Overall, this study demonstrates an effective and safe strategy for the targeted therapy of human cancer.
Conclusion
In the present study, we demonstrated a method for preparing CTNF-α-anchored exosomes that are coupled with SPIONs with enhanced membrane anticancer activity. CTNF-α-exosomes with membrane targeting ability were successfully acquired from MSCs by fusing CPP with TNF-α via genetic engineering. Conjugating a Tf-SPION onto CTNF-α-exosomes through Tf-TfR interaction yields CTNF-α-exosome-SPIONs, thus endowing CTNF-α-exosomes with superparamagnetic properties. The EPR effect of exosomes and active targeting properties of SIPONs enable TNF-α to gain favorable targeting properties, better antitumor activity and lower toxicity; thus, CTNF-α-exosome-SPIONs were demonstrated as a promising approach for cancer therapy. The most promising aspect of this study is that CTNF-α-exosome-SPIONs can deliver peptide drugs to the cytomembrane rather than the cytoplasm. Most peptide drug receptors are distributed in the cell membrane. Therefore, our research provides a useful strategy for cytomembrane targeting of peptide drugs. Taken together, our study indicated that the incorporation of genetic engineering with nanotechnology shows a promising prospect in tumor therapy and the application of peptide drugs.Abbreviations
| SPION | Superparamagnetic iron oxide nanoparticle |
| TNF-α | Tumor necrosis factor-alpha |
| Tf | Transferrin |
| TfR | Transferrin receptor |
| TNFR I | Tumor necrosis factor receptor I |
| NP | Nanoparticle |
| EPR | Permeability and retention |
| MSCs | Mesenchymal stem cells |
| MF | Magnetic force |
| CS | Chitosan |
| CPP | Cell-penetrating peptides |
| EDC | Carbodiimide |
| NHS | N-Hydroxysulfosuccinimide sodium salt |
| TfR | Transferrin receptors |
| FT-IR | Fourier-transform infrared spectroscopy |
| GFP | Green fluorescent protein |
| HE | Hematoxylin and eosin |
| Dil | 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate |
| BSA | Bovine serum albumin |
| CY5.5 | Cyanine5.5 NHS ester |
| TEM | Transmission electron microscope |
| CO-IP | Co-immunoprecipitation |
| MTT | Tetramethylimidazolium salt |
| JC-1 | 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine |
| Z-VAD-FMK | Carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone |
| NIRF | Near infrared fluorescent |
| HRP | Horseradish peroxidase |
| TMB | 3,3′,5,5′-Tetramethyl benzidine |
| CTNF-α | Fusion protein of cell-penetrating peptides and TNF-α. |
Conflicts of interest
The authors declare no potential conflict of interest.Acknowledgements
This work was supported by the Collaborative Innovation Center of Sichuan for Elderly Care and Health (number YLZBZ1517), the Education Department of Sichuan Province (number 16ZA0292) and the Chengdu Medical College Foundation (number CYZ15-07).References
- C. Fitzmaurice, D. Dicker, A. Pain, H. Hamavid, M. Moradilakeh, M. F. Macintyre, C. Allen, G. Hansen, R. Woodbrook and C. Wolfe, The Global Burden of Cancer 2013, JAMA Oncol., 2015, 1(4), 505 CrossRef PubMed.
- A. Jemal, S. S. Devesa, P. Hartge and M. A. Tucker, Recent trends in cutaneous melanoma incidence among whites in the United States, J. Natl. Cancer Inst., 2001, 93(9), 678–683 CrossRef CAS PubMed.
- S. Parveen and S. K. Sahoo, Polymeric nanoparticles for cancer therapy, J. Drug Targeting, 2018, 16(2), 108–123 CrossRef PubMed.
- O. C. Farokhzad and R. Langer, Impact of Nanotechnology on Drug Delivery, ACS Nano, 2009, 3(1), 16–20 CrossRef CAS PubMed.
- G. Walsh, Biopharmaceutical benchmarks 2014, Nat. Biotechnol., 2014, 32(10), 992–1000 CrossRef CAS PubMed.
- H. Misato, H. Naohiko, S. Miho and M. Masahiko, IL-6/sIL-6R trans-signalling, but not TNF-alpha induced angiogenesis in a HUVEC and synovial cell co-culture system, Rheumatol. Int., 2009, 29(12), 1449–1454 CrossRef PubMed.
- J. Shen, Z. Xiao, Q. Zhao, M. Li, X. Wu, L. Zhang, W. Hu and C. H. Cho, Anti-cancer therapy with TNFα and IFNγ: A comprehensive review, Cell Proliferation, 2018, e12441 CrossRef PubMed.
- P. D. Lyons and E. N. Benveniste, Cleavage of membrane-associated ICAM-1 from astrocytes: involvement of a metalloprotease, Glia, 2015, 22(2), 103–112 CrossRef.
- E. T. Creagan, J. S. Kovach, C. G. Moertel, S. Frytak and L. K. Kvols, A phase I clinical trial of recombinant human tumor necrosis factor, Cancer, 1988, 62(12), 2467 CrossRef CAS PubMed.
- L. Borsi, E. Balza, B. Carnemolla, F. Sassi, P. Castellani, A. Berndt, H. Kosmehl, A. Biro, A. Siri and P. Orecchia, Selective targeted delivery of TNFalpha to tumor blood vessels, Blood, 2003, 102(13), 4384–4392 CrossRef CAS PubMed.
- J. Treisman, P. Hwu, J. R. Yannelli, G. E. Shafer, R. Cowherd, D. Samid and S. A. Rosenberg, Upregulation of Tumor Necrosis Factor-α Production by Retrovirally Transduced Human Tumor-Infiltrating Lymphocytes Using trans -Retinoic Acid, Cell. Immunol., 1994, 156(2), 448–457 CrossRef CAS PubMed.
- S. R. Murugesan, M. Akiyama, D. A. Einfeld, T. J. Wickham and C. R. King, Experimental treatment of ovarian cancers by adenovirus vectors combining receptor targeting and selective expression of tumor necrosis factor, Int. J. Oncol., 2007, 31(4), 813 CAS.
- C. R. Kagan, L. E. Fernandez, Y. Gogotsi, P. T. Hammond, M. C. Hersam, A. E. Nel, R. M. Penner, C. G. Willson and P. S. Weiss, Nano Day: Celebrating the Next Decade of Nanoscience and Nanotechnology, ACS Nano, 2016, 10(10), 9093–9103 CrossRef CAS PubMed.
- Z. Yang, J. Xie, J. Zhu, C. Kang, C. Chiang, X. Wang, X. Wang, T. Kuang, F. Chen and Z. Chen, Lt, Functional exosome-mimic for delivery of siRNA to cancer: in vitro and in vivo evaluation, J. Controlled Release, 2016, 243(10), 160–171 CrossRef CAS PubMed.
- C. Thery, L. Zitvogel and S. Amigorena, Exosomes: composition, biogenesis and function, Nat. Rev. Immunol., 2002, 2(8), 569–579 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, S. Zhang, Z. B. Deng, W. Grizzle, D. Miller and H. G. Zhang, Exosomes are endogenous nanoparticles that can deliver biological information between cells, Adv. Drug Delivery Rev., 2013, 65(3), 342–347 CrossRef CAS PubMed.
- C. Liu and C. Su, Design strategies and application progress of therapeutic exosomes, Theranostics, 2019, 9(4), 1015–1028 CrossRef CAS PubMed.
- A. Parodi, R. Molinaro, M. Sushnitha, M. Evangelopoulos, J. O. Martinez, N. Arrighetti, C. Corbo and E. Tasciotti, Bio-inspired engineering of cell- and virus-like nanoparticles for drug delivery, Biomaterials, 2017, 147(5), 155 CrossRef CAS PubMed.
- H. Valadi, K. Ekström, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lötvall, Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells, Nat. Cell Biol., 2007, 9(6), 654 CrossRef CAS PubMed.
- Y. Tian, S. Li, J. Song, T. Ji, M. Zhu, G. J. Anderson, J. Wei and G. Nie, A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy, Biomaterials, 2014, 35(7), 2383 CrossRef CAS PubMed.
- C. J. Su, O. Y. Kim, M. Y. Chang, D. S. Choi, T. Y. Roh, J. Park, J. Nilsson, J. Lötvall, Y. K. Kim and S. G. Yong, Bioinspired Exosome-Mimetic Nanovesicles for Targeted Delivery of Chemotherapeutics to Malignant Tumors, ACS Nano, 2014, 8(1), 7698–7710 Search PubMed.
- T. Tian, H. X. Zhang, C. P. He, S. Fan, Y. L. Zhu, C. Qi, N. P. Huang, Z. D. Xiao, Z. H. Lu and B. A. Tannous, Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy, Biomaterials, 2018, 150(1), 137–149 CrossRef CAS PubMed.
- Y. Hong, G. Nam, E. Koh, S. Jeon, G. B. Kim, C. Jeong, D. Kim, Y. Yang and I. A. Kim, Exosome as a Vehicle for Delivery of Membrane Protein Therapeutics, PH20, for Enhanced Tumor Penetration and Antitumor Efficacy, Adv. Funct. Mater., 2017, 28(5), 1801301 Search PubMed.
- S. F. Ahmed, N. Das, M. Sarkar, U. Chatterjee, S. Chatterjee and M. K. Ghosh, Exosome-mediated delivery of the intrinsic C-terminus domain of PTEN protects it from proteasomal degradation and ablates tumorigenesis, Mol. Ther., 2015, 23(2), 255–269 CrossRef CAS PubMed.
- A. Longatti, C. Schindler, A. Collinson, L. Jenkinson and N. Tigue, High affinity single-chain variable fragments are specific and versatile targeting motifs for extracellular vesicles, Nanoscale, 2018, 10(29), 14230–14244 RSC.
- S. C. Tao, S. C. Guo, M. Li, Q. F. Ke, Y. P. Guo and C. Q. Zhang, Chitosan Wound Dressings Incorporating Exosomes Derived from MicroRNA-126-Overexpressing Synovium Mesenchymal Stem Cells Provide Sustained Release of Exosomes and Heal Full-Thickness Skin Defects in a Diabetic Rat Model, Stem Cells Transl. Med., 2016, 6(3), 2016–0275 Search PubMed.
- C. MP, L. D, V. C, R. G and B. C, Exosomal-like vesicles are present in human blood plasma, Int. Immunol., 2005, 17(7), 879–887 CrossRef PubMed.
- H. Qi, C. Liu, L. Long, Y. Ren, S. Zhang, X. Chang, X. Qian, H. Jia, J. Zhao and J. Sun, Blood Exosomes Endowed with Magnetic and Targeting Properties for Cancer Therapy, ACS Nano, 2016, 10(3), 3323 CrossRef CAS PubMed.
- H. B. You and K. Park, Targeted drug delivery to tumors: Myths, reality and possibility, J. Controlled Release, 2011, 153(3), 198–205 CrossRef PubMed.
- M. Wahajuddin and S. Arora, Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers, Int. J. Nanomed., 2012, 7(7), 3445–3471 CrossRef PubMed.
- Y. Wang, C. Xu, Y. N. Chang, L. Zhao, K. Zhang, Y. Zhao, F. Gao and X. Gao, Ultrasmall Super-paramagnetic Iron Oxide Nanoparticle for T2-weighted Magnetic Resonance Imaging, ACS Appl. Mater. Interfaces, 2017, 9(34), 1003 Search PubMed.
- A. M. D. Campos, Y. Diebold, E. L. S. Carvalho, A. Sanchez and M. J. Alonso, Chitosan nanoparticles as new ocular drug delivery systems: in vitro stability, in vivo fate, and cellular toxicity, Pharm. Res., 2004, 21(5), 803 CrossRef PubMed.
- J. Varshosaz, The promise of chitosan microspheres in drug delivery systems, Drug Delivery, 2007, 4(3), 263 CAS.
- R. Lei, M. Yi, M. Zhuang, T. Luo, Y. Wang and H. An, Chitosan-decorated selenium nanoparticles as protein carriers to improve the in vivo half-life of the peptide therapeutic BAY 55–9837 for type 2 diabetes mellitus, Int. J. Nanomed., 2014, 9(1), 4819–4828 Search PubMed.
- M. Terai and R. D. Burk, Identification and characterization of 3 novel genital human papillomaviruses by overlapping polymerase chain reaction: HPV89, HPV90, and HPV91, Br. J. Dermatol., 2010, 162(6), 1186–1197 CrossRef PubMed.
- B. Gupta, T. S. Levchenko and V. P. Torchilin, Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides, Adv. Drug Delivery Rev., 2005, 57(4), 637–651 CrossRef CAS PubMed.
- P. Zhou, S. Hou, Z. Bai, Z. Li, H. Wang, Z. Chen and Y. Meng, Disrupting the intramolecular interaction between proto-oncogene c-Src SH3 domain and its self-binding peptide PPII with rationally designed peptide ligands, Artif. Cells, Nanomed., Biotechnol., 2018, 46(6), 1122–1131 CrossRef CAS.
- A. M. Geurts, Y. Yang, K. J. Clark, G. Liu, Z. Cui, A. J. Dupuy, J. B. Bell, D. A. Largaespada and P. B. Hackett, Gene transfer into genomes of human cells by the sleeping beauty transposon system, Mol. Ther., 2003, 8(1), 108 CrossRef CAS.
- P. B. Hackett, S. C. Ekker, D. A. Largaespada and R. S. Mcivor, Sleeping beauty transposon-mediated gene therapy for prolonged expression, Adv. Genet., 2005, 54(1), 189 CAS.
- S. Deshayes, T. Plénat, P. Charnet, G. Divita, G. Molle and F. Heitz, Formation of transmembrane ionic channels of primary amphipathic cell-penetrating peptides. Consequences on the mechanism of cell penetration, Biochim. Biophys. Acta, Biomembr., 2006, 1758(11), 1846–1851 CrossRef CAS.
- E. G. Stanzl, B. M. Trantow, J. R. Vargas and P. A. Wender, Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications, Acc. Chem. Res., 2013, 46(12), 2944–2954 CrossRef CAS.
- Z. Lei, Z. Pan, X. Kang, Y. Yun, H. Kang, Z. Na, J. M. Rosati and X. Jiao, Amino acids 89–96 of Salmonella typhimurium flagellin represent the major domain responsible for TLR5-independent adjuvanticity in the humoral immune response, Cell. Mol. Immunol., 2014, 12(5), 625–632 Search PubMed.
- R. Miyagawa, K. Tano, R. Mizuno, Y. Nakamura, K. Ijiri, R. Rakwal, J. Shibato, Y. Masuo, A. Mayeda and T. Hirose, Identification of cis- and trans-acting factors involved in the localization of MALAT-1 noncoding RNA to nuclear speckles, RNA, 2012, 18(4), 738 CrossRef CAS PubMed.
- R. E. Campbell, O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias and R. Y. Tsien, A monomeric red fluorescent protein, Proc. Natl. Acad. Sci. U. S. A., 2012, 7877–7882 Search PubMed.
- M. Chalfie, Y. Tu, G. Euskirchen, W. W. Ward and D. C. Prasher, Green fluorescent protein as a marker for gene expression, Science, 1994, 263(5148), 802–805 CrossRef CAS PubMed.
- F. Cuello, R. A. Schulze, F. Heemeyer, H. E. Meyer, S. Lutz, K. H. Jakobs, F. Niroomand and T. Wieland, Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gbeta subunits. Complex formation of NDPK B with Gbeta gamma dimers and phosphorylation of His-266 IN Gbeta, J. Biol. Chem., 2003, 278(9), 7220–7226 CrossRef CAS.
- L. Ma, X. Yao, Z. Wei, G. Xin, Z. Xing, N. Hai and H. Wen, Deoxyarbutin displays antitumour activity against melanoma in vitro and in vivo through a p38-mediated mitochondria associated apoptotic pathway, Sci. Rep., 2017, 7(1), 7197 CrossRef.
- K. Hak-Su, K. Myung-Jin, K. Eun Ju, Y. Yang, M. S. Lee and J. S. Lim, Berberine-induced AMPK activation inhibits the metastatic potential of melanoma cells via reduction of ERK activity and COX-2 protein expression, Biochem. Pharmacol., 2012, 83(3), 385–394 CrossRef.
- L. Perico, M. Morigi, C. Rota, M. Breno, C. Mele, M. Noris, M. Introna, C. Capelli, L. Longaretti and D. Rottoli, Human mesenchymal stromal cells transplanted into mice stimulate renal tubular cells and enhance mitochondrial function, Nat. Commun., 2017, 8(1), 1–16 CrossRef CAS PubMed.
- G. Li, Y. Xing, J. Wang, P. S. Conti and K. Chen, Near-infrared fluorescence imaging of CD13 receptor expression using a novel Cy5.5-labeled dimeric NGR peptide, Amino Acids, 2014, 46(6), 1547–1556 CrossRef CAS PubMed.
- V. Mumprecht, M. Honer, B. Vigl, S. T. Proulx, E. Trachsel, M. Kaspar, N. E. Banziger-Tobler, R. Schibli, D. Neri and M. Detmar, In vivo Imaging of Inflammation- and Tumor-Induced Lymph Node Lymphangiogenesis by Immuno-Positron Emission Tomography, Cancer Res., 2010, 70(21), 8842 CrossRef CAS PubMed.
- I. Tusa, S. Gagliardi, A. Tubita, S. Pandolfi, C. Urso, L. Borgognoni, J. Wang, X. Deng, N. S. Gray and B. Stecca, ERK5 is activated by oncogenic BRAF and promotes melanoma growth, Oncogene, 2018, 37(77), 2601–2614 CrossRef CAS PubMed.
- Z. J. Messenger, J. R. Hall, D. D. Jima, J. S. House, H. W. Tam, D. A. Tokarz and R. C. Smart, C/EBPbeta deletion in oncogenic Ras skin tumors is a synthetic lethal event, Cell Death Dis., 2018, 9(11), 1054–1054 CrossRef PubMed.
- C. Kilkenny, W. J. Browne, I. C. Cuthill, M. Emerson and D. G. Altman, Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research, Animal, 2014, 4(1), e1000412 Search PubMed.
- L. Su, B. Zhang, Y. Huang, Z. Fan and Y. Zhao, Enhanced cellular uptake of iron oxide nanoparticles modified with 1,2-dimyristoyl-sn-glycero-3-phosphocholine, RSC Adv., 2017, 7(60), 38001–38007 RSC.
- H. Maleki, A. Simchi, M. Imani and B. F. O. Costa, Size-controlled synthesis of superparamagnetic iron oxide nanoparticles and their surface coating by gold for biomedical applications, J. Magn. Magn. Mater., 2012, 324(23), 3997–4005 CrossRef CAS.
- A. Tan, J. Rajadas and A. M. Seifalian, Exosomes as nano-theranostic delivery platforms for gene therapy, Adv. Drug Delivery Rev., 2013, 65(3), 357–367 CrossRef CAS PubMed.
- E. Chalecka-Franaszek, H. B. Weems, A. T. Crowder, B. M. Cox and T. E. Côté, Immunoprecipitation of high-affinity, guanine nucleotide-sensitive, solubilized mu-opioid receptors from rat brain: coimmunoprecipitation of the G proteins G(alpha o), G(alpha i1), and G(alpha i3), J. Neurochem., 2010, 74(3), 1068–1078 CrossRef PubMed.
- L. Camus, B. E. Grøsvik, J. F. Børseth, M. B. Jones and M. H. Depledge, Stability of lysosomal and cell membranes in haemocytes of the common mussel (Mytilus edulis): effect of low temperatures, Mar. Environ. Res., 2000, 50(1), 325–329 CrossRef CAS PubMed.
- Z. Darzynkiewicz, S. Bruno, G. D. Bino, W. Gorczyca, M. A. Hotz, P. Lassota and F. Traganos, Features of apoptotic cells measured by flow cytometry, Cytometry, Part A, 2010, 13(8), 795–808 CrossRef PubMed.
- J. D. Ly, D. R. Grubb and A. Lawen, The mitochondrial membrane potential (Δψm) in apoptosis; an update, Apoptosis, 2003, 8(2), 115 CrossRef CAS PubMed.
- N. A. Thornberry and Y. Lazebnik, Caspases: enemies within, Science, 1998, 281(5381), 1312 CrossRef CAS PubMed.
- R. Menon, S. J. Lombardi and S. J. Fortunato, TNF-α Promotes Caspase Activation and Apoptosis in Human Fetal Membranes, J. Assist. Reprod. Genet., 2002, 19(4), 201–204 CrossRef PubMed.
- B. Beutler, D. Greenwald, J. D. Hulmes, M. Chang, Y. C. E. Pan, J. Mathison, R. Ulevitch and A. Cerami, Identity of tumour necrosis factor and the macrophage-secreted factor cachectin, Nature, 1985, 316(6028), 552–554 CrossRef CAS PubMed.
- C. Liu, J. Wen, Y. Meng, K. Zhang, J. Zhu, Y. Ren, X. Qian, X. Yuan, Y. Lu and C. Kang, Efficient delivery of therapeutic miRNA nanocapsules for tumor suppression, Adv. Mater., 2015, 27(2), 292–297 CrossRef CAS PubMed.
- Y. Wang, X. Chen, B. Tian, J. Liu, L. Yang, L. Zeng, T. F. Chen, A. Hong and X. G. Wang, Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer, Theranostics, 2017, 7(5), 1360–1372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr05865f |
| This journal is © The Royal Society of Chemistry 2020 |