
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The scaffold-forming steps of plant alkaloid biosynthesis
Benjamin R.
Lichman
 *
*
Centre for Novel Agricultural Products, Department of Biology, University of York, York YO10 5DD, UK. E-mail: benjamin.lichman@york.ac.uk
First published on 3rd August 2020
Abstract
Alkaloids from plants are characterised by structural diversity and bioactivity, and maintain a privileged position in both modern and traditional medicines. In recent years, there have been significant advances in elucidating the biosynthetic origins of plant alkaloids. In this review, I will describe the progress made in determining the metabolic origins of the so-called true alkaloids, specialised metabolites derived from amino acids containing a nitrogen heterocycle. By identifying key biosynthetic steps that feature in the majority of pathways, I highlight the key roles played by modifications to primary metabolism, iminium reactivity and spontaneous reactions in the molecular and evolutionary origins of these pathways.
 Benjamin Lichman | Dr Benjamin Lichman received his MSc in Natural Sciences from the University of Cambridge and his PhD in biochemistry from University College London. His doctoral research, under the guidance of Professors John Ward and Helen Hailes, focussed on the mechanism and applications of norcoclaurine synthase. He then joined the group of Prof Sarah O'Connor at the John Innes Centre to investigate iridoid biosynthesis in plants. In 2018 he was appointed a lecturer at the Centre for Novel Agricultural Products at the University of York. Benjamin is currently a UKRI Future Leaders Fellow and manages a research group investigating the origins and applications of plant natural product biosynthesis. |
1. Introduction
1.1. Background
Alkaloids from plants have remarkable powers to harm, heal and reveal; this has led to their use in herbal medicine and related practises across continents and cultures for millennia. Alkaloids were foundational in the development of organic chemistry and were among the first pharmaceuticals developed. They retain a privileged position in modern medicine, used widely to treat pain, cancer, dementia and countless other ailments (Fig. 1).1,2 Despite losing pace to synthetic drug discovery approaches in recent decades, plant alkaloids remain a viable source of bioactive compounds with considerable therapeutic potential.3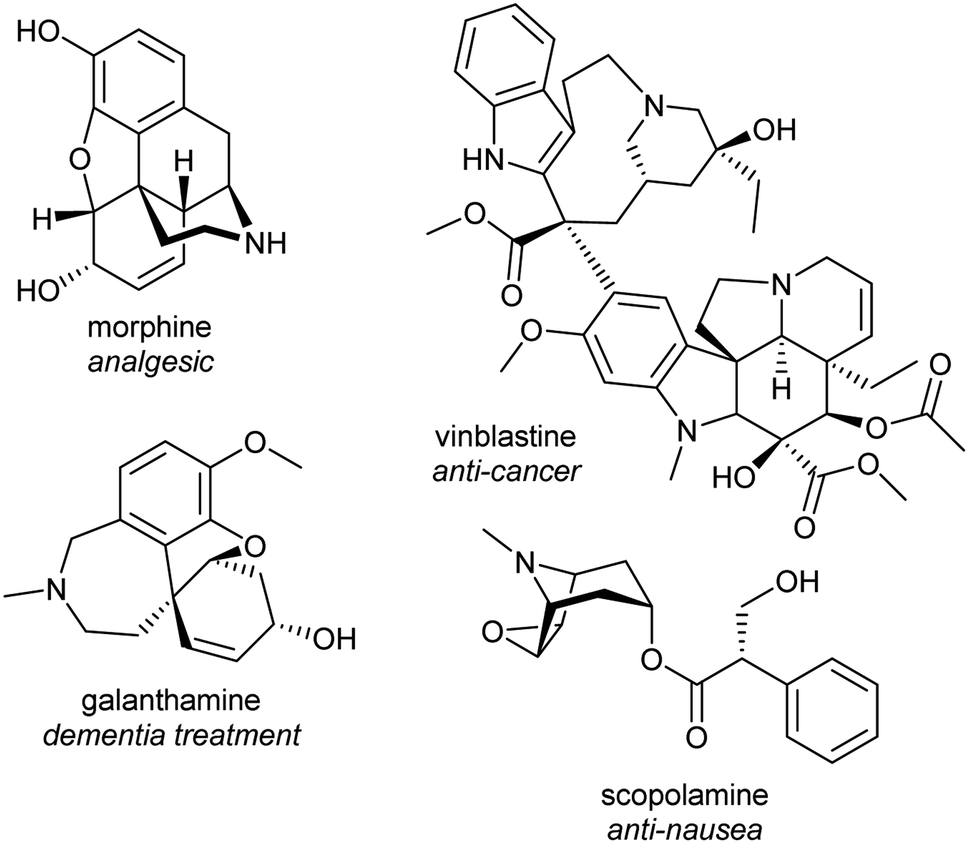 | ||
| Fig. 1 Examples of pharmaceutical alkaloids. | ||
It is only through the elucidation of plant alkaloid biosynthesis—determining chemical intermediates, characterising enzymes and sequencing genomes—that we can truly appreciate and understand the synthetic ingenuity of nature in constructing these complex compounds. There has been remarkable progress in the discovery and characterisation of enzymes over the last decade, triggered by advances and expanded use of sequencing technologies.4,5 Genome assemblies of alkaloid producing plants have revealed certain biosynthetic genes co-localise in clusters, aiding gene discovery and providing insight into evolution and pathway regulation.6–8
The investigation of plant alkaloid biosynthesis can lead to new tools and technologies. For example, enzymes can be employed as biocatalysts for the in vitro formation of new-to-nature compounds.9,10 Genes and pathways can be heterologously expressed in genetically tractable and rapidly growing organisms in an attempt to increase alkaloid yield, purity or access new-to-nature compounds through pathway modifications.11–13 Understanding the genomic basis for alkaloid biosynthesis can also inform efforts to breed plants with higher compound yields or lower toxicity.14
1.2. Definition of alkaloids
Alkaloids were originally defined as alkaline substances extracted from a plant with a biological activity.15 This definition has been refined multiple times to include compounds from outside the plant kingdom, as well as those that share a biosynthetic origin with alkaloids but do not have a basic nitrogen.16,17 In the broadest sense, alkaloids are nitrogen-containing compounds derived from secondary, or specialised, metabolism. However, it is useful to categorise alkaloids further, not with the intention of excluding compounds, but to help comprehend similarities and differences in the compounds' origins (Fig. 2). The true alkaloids, referred to as complex alkaloids in this review, are compounds in which the nitrogen atom is derived from an amino acid and is part of a heterocycle. Proto-alkaloids, or simple alkaloids, are amines derived from amino acids but do not have a nitrogen heterocycle. Pseudo-alkaloids are nitrogen-containing metabolites in which the nitrogen is introduced at a late stage through an enzymatic process such as transamination.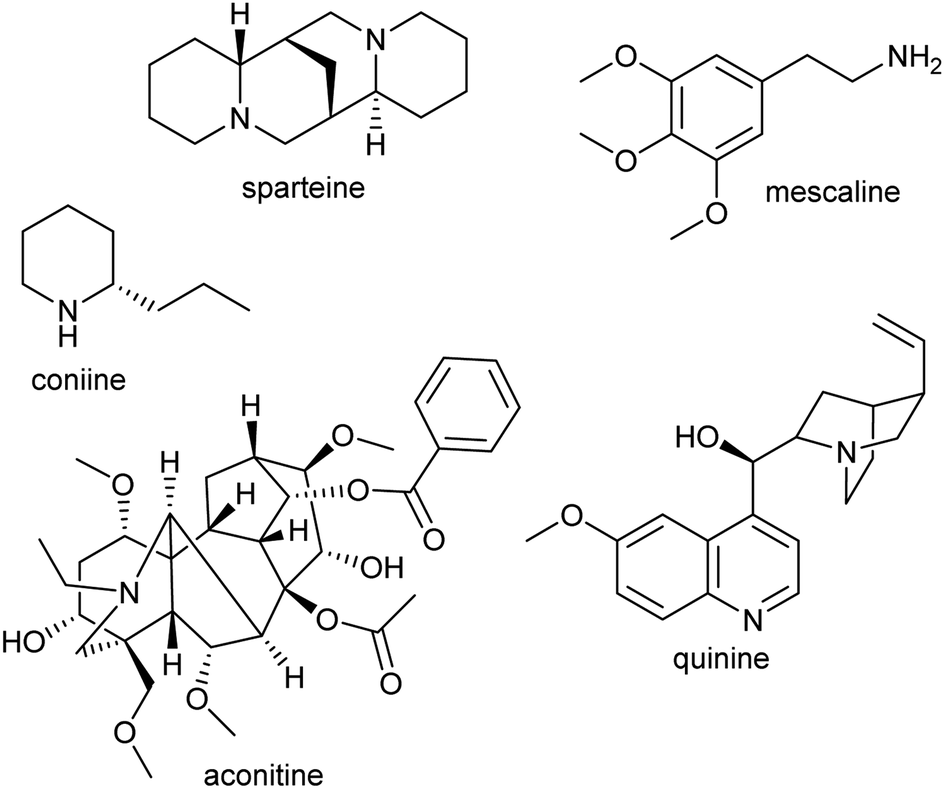 | ||
| Fig. 2 Types of alkaloids. Sparteine and quinine are true alkaloids, derived from lysine and tryptophan respectively. Mescaline is a proto-alkaloid. Coniine and aconitine are pseudo-alkaloids, with the nitrogen inserted through transamination. | ||
1.3. Major alkaloid classes
This review focusses on complex ‘true’ alkaloids from plants: compounds derived from an amino acid containing a nitrogen heterocycle. Despite their wide structural diversity and taxonomic distribution, the biosynthetic origins of these compounds follows a unifying chemical logic centred around the formation and reactivity of the iminium cation.18 I will examine the first steps of alkaloid biosynthetic pathways: the transition from primary to specialised metabolism and formation of the defining scaffold. The metabolic, enzymatic and evolutionary origins of these steps will be reviewed, including the latest developments in the field.This review will cover all major complex alkaloid families in plants. Compounds derived from tyrosine include benzylisoquinolines, tetrahydroisoquinolines and the Amaryllidaceae alkaloids. Although not classically described as alkaloids, the tyrosine-derived betalains share many biosynthetic features with alkaloids and will be discussed. The tryptophan-derived monoterpene indole alkaloids are included. The anthranilate derived acridone and quinoline alkaloids are only briefly mentioned, as they do not subscribe to the biosynthetic patterns described in this review. Polyamine derived alkaloids including piperideine, tropane, quinolizidine, pyrrolizidine and Nicotiana alkaloids are all included. It is hoped that the principles of biosynthesis discussed in this review can be applied to many alkaloid families not directly mentioned.
1.4. Patterns in biosynthesis
The first steps in an alkaloid pathway are the most crucial, acting as the gateway to a new chemical space. They are important metabolically, as they direct flux away from primary and into specialised metabolism. They are significant chemically and enzymatically as they involve the formation of a new molecular structure. They also play a foundational role in the evolution of the pathways.There are four steps that are typically present in the first steps of complex alkaloid biosynthesis: (i) accumulation of an amine precursor, (ii) accumulation of an aldehyde precursor, (iii) formation of an iminium cation and (iv) a Mannich-like reaction (Scheme 1). This final step is often considered the “scaffold-forming”, signature, or first-committed step into a pathway.
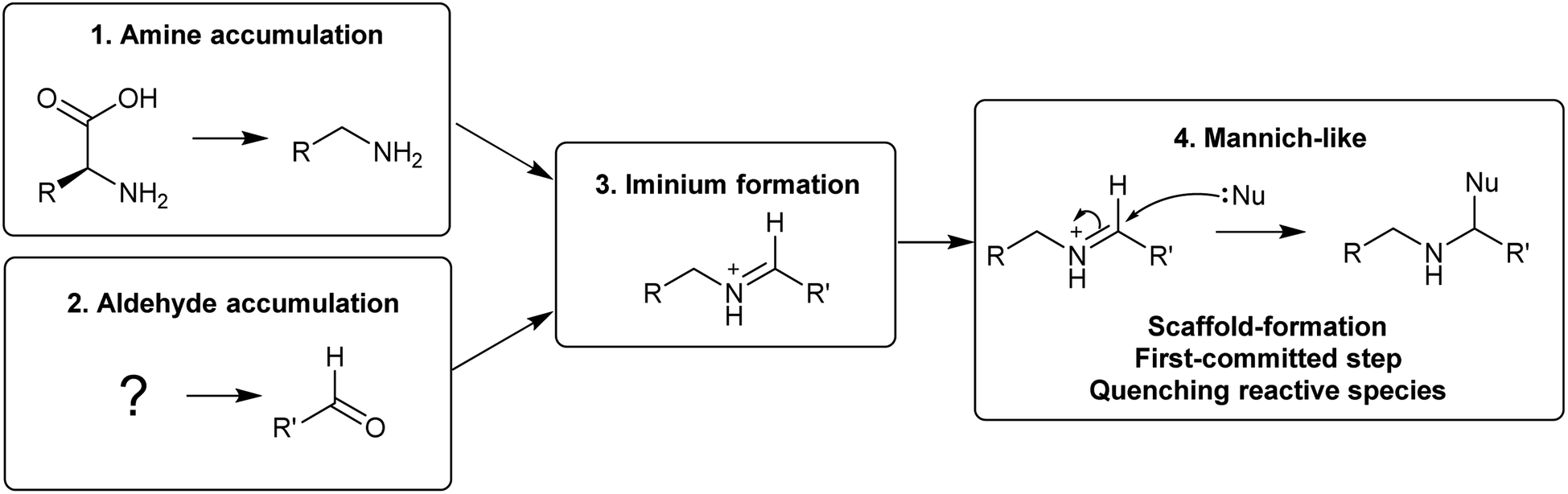 | ||
| Scheme 1 Themes in complex alkaloid biosynthesis explored in this review. | ||
A major origin of variation in these generalised steps is whether they are intermolecular or intramolecular (Table 1). For example, polyamine-derived alkaloids generally form a cyclic iminium through intramolecular condensation, followed by an intermolecular Mannich-like reaction. In contrast, the Pictet–Spengler step that contributes to major alkaloid families, such as the benzylisoquinolines, is an intermolecular condensation, involving formation of an iminium intermediate, followed immediately by an intramolecular Mannich-like reaction.
| Alkaloid type | Amine origin | Aldehyde origin | Iminium | Mannich-like reaction |
|---|---|---|---|---|
| Nicotine | Polyamine, putrescine | Intramolecular | Cyclic iminium, pyrollinium | Intermolecular, nicotinic acid |
| Tropane | Polyamine, putrescine | Intramolecular | Cyclic iminium, pyrollinium | Intermolecular, polyketide |
| Lycopodium | Polyamine, cadaverine | Intramolecular | Cyclic iminium, piperideine | Intermolecular, polyketide |
| Quinolizidine | Polyamine, cadaverine | Intramolecular | Cyclic iminium, piperideine | Intermolecular, dimerization |
| Pyrrolizidine | Polyamine, homospermidine | Intramolecular | Cyclic iminium, pyrollinium | Intramolecular |
| Betalains | Tyrosine, L-DOPA | Tyrosine, betalamic acid | Intermolecular, spontaneous | None |
| Benzylisoquinolines | Tyrosine, dopamine | Tyrosine, 4-HPAA | Intermolecular, enzyme catalysed | Intramolecular, Pictet–Spengler |
| Monoterpene indoles | Tryptophan, tryptamine | Terpene, secologanin | Intermolecular, enzyme catalysed | Intramolecular, Pictet–Spengler |
| Amaryllidaceae | Tyrosine, tyramine | Phenylpropanoid, benzaldehyde | Intermolecular | Intramolecular, reduction |
| Ipecac | Tyrosine, dopamine | Terpene, secologanin | Intermolecular | Intramolecular, Pictet–Spengler |
| Colchicine | Tyrosine, dopamine | Phenylpropanoid, dihydrocinnamaldehyde | Intermolecular | Intramolecular, Pictet–Spengler |
There have been many excellent general reviews of alkaloids,16,17,19 and many detailed reviews of individual alkaloid families.14,20–34 In order to differentiate this review, and to highlight its focus on the patterns in the early steps of biosynthesis, it will not be structured by alkaloid class but instead by the particular step in the aforementioned biosynthetic model (Scheme 1). By viewing alkaloid biosynthesis in this holistic manner, it may be possible to gain insights from one pathway that can be applied to aid elucidation of another.
2. Amine accumulation
The nitrogen in complex alkaloids originates from amino acid metabolism. The majority of alkaloids do not directly incorporate an amino acid, but instead use a primary amine derivative. Simple primary amines are ubiquitous in green plants, and have a variety of both essential and specialised roles in development, reproduction and stress responses.35–38 In the general pattern of alkaloid biosynthesis proposed in this review, amines accumulate beyond typical concentrations to enable flux to be directed into alkaloid metabolism without disruption to existing pathways. This usually requires changes to metabolism bought about by gene duplication. The contribution of primary metabolism to plant chemical diversity has been recently reviewed.39For essential or abundant amines, such as putrescine or tyramine, accumulation may result from higher expression or duplication of a pre-existing gene without modification to the enzyme's substrate scope. Accumulation of amines that are typically at low concentrations or absent, such as cadaverine or homospermidine, may require the evolution of new enzyme activity through modification of substrate scope.40 Other possible routes to the accumulation of amines include boosting the concentration of amino acid precursors, or reducing degradative catabolic pathways. The amines contributing to alkaloid biosynthesis can be split into two categories: polyamines, derived from lysine, arginine and ornithine; and aromatic amines, derived from tryptophan and tyrosine.
2.1. Polyamines
The polyamines putrescine, spermine and spermidine play essential roles in plants, for example in seed development41 and protein translation35,42,43 (Fig. 3). Cadaverine is found only in selected plant taxa (e.g. legumes) and contributes to stress responses.44 The simple polyamines putrescine and cadaverine are precursors to major alkaloid classes containing pyrrolinium or piperideine moieties respectively. The triamine dimer of putrescine, homospermidine, is a precursor to the pyrrolizidine alkaloids.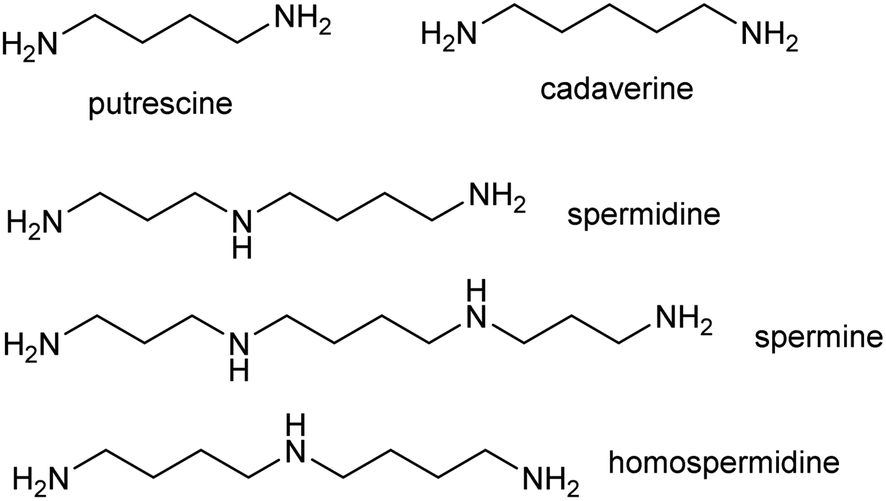 | ||
| Fig. 3 Polyamines in plants. Putrescine, spermidine and spermine are ubiquitous in green plants whereas cadaverine and homospermidine only accumulate in selected taxa. | ||
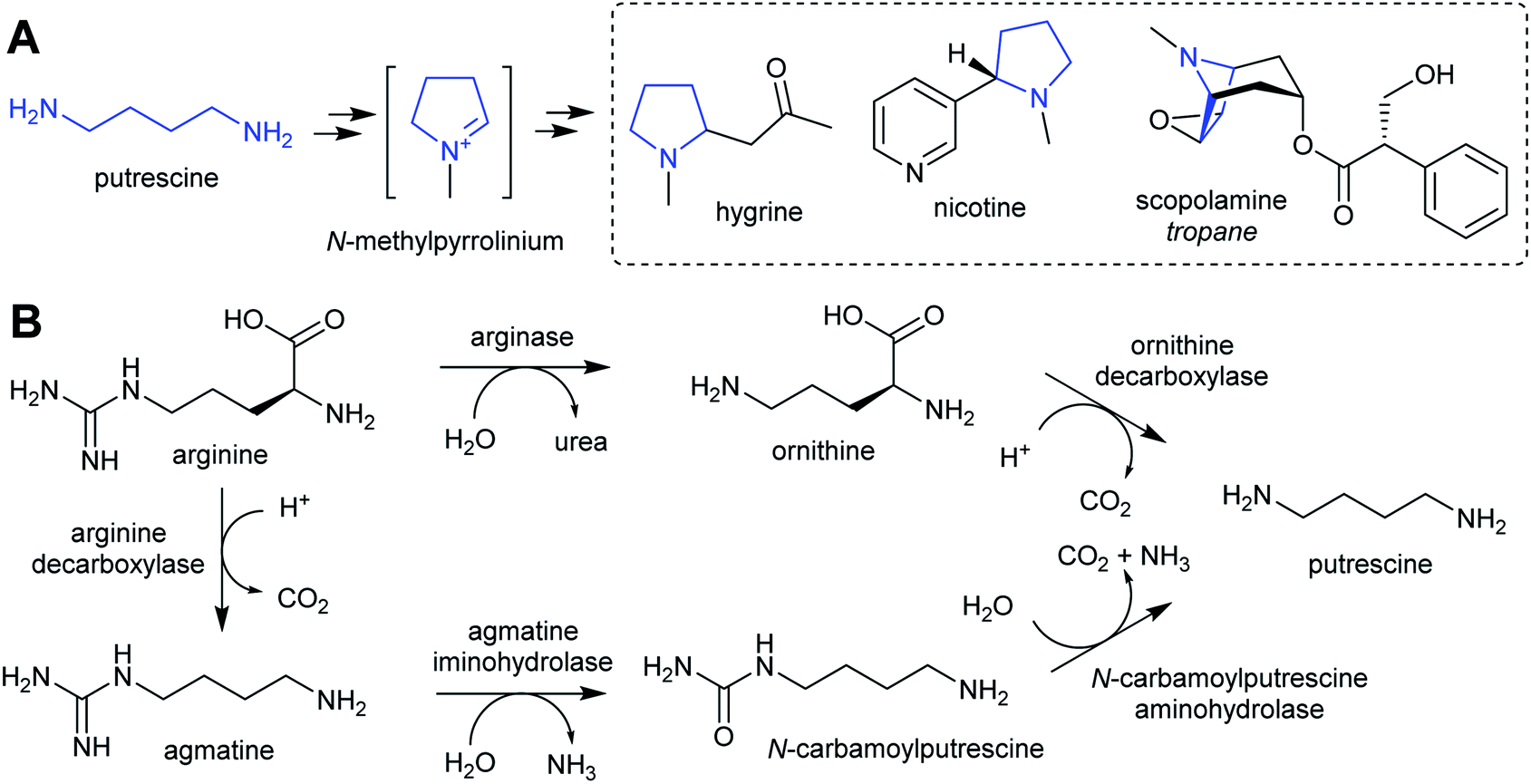 | ||
| Scheme 2 Putrescine derived alkaloids. (A) Putrescine derived alkaloids (moiety originating from putrescine highlighted in blue). (B) Biosynthesis of putrescine from ornithine or arginine. | ||
The nightshade family (Solanaceae) includes species rich in putrescine-derived alkaloids, such as Nicotiana tobaccum (nicotine) and tropane alkaloid producers Atropa belladonna and Solanum sp. In this family, the ornithine pathway appears to be the major source of putrescine for alkaloid biosynthesis.51,52 A number of Solanaceae species contain two copies of OrnDC, originating from a putative duplication event prior to the emergence of the family.53 In Nicotiana, one OrnDC co-expresses with alkaloid biosynthesis genes in the roots.53 The duplication of OrnDC may have led to tissue specific accumulation of putrescine, ultimately enabling the formation of diverse putrescine-derived alkaloids in Solanaceous plants.
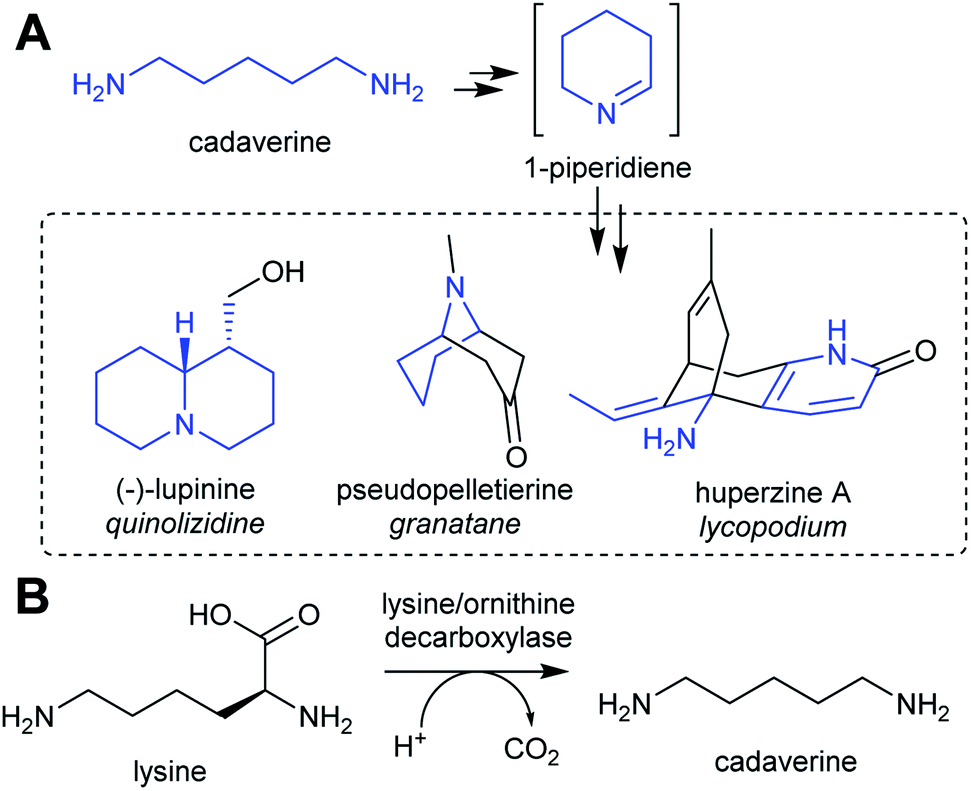 | ||
| Scheme 3 Cadaverine derived alkaloids. (A) Cadaverine derived alkaloids (portion originating from cadaverine highlighted in blue). Huperzine A labelling based on predicted biosynthetic pathway.57 (B) Biosynthesis of cadaverine from lysine. | ||
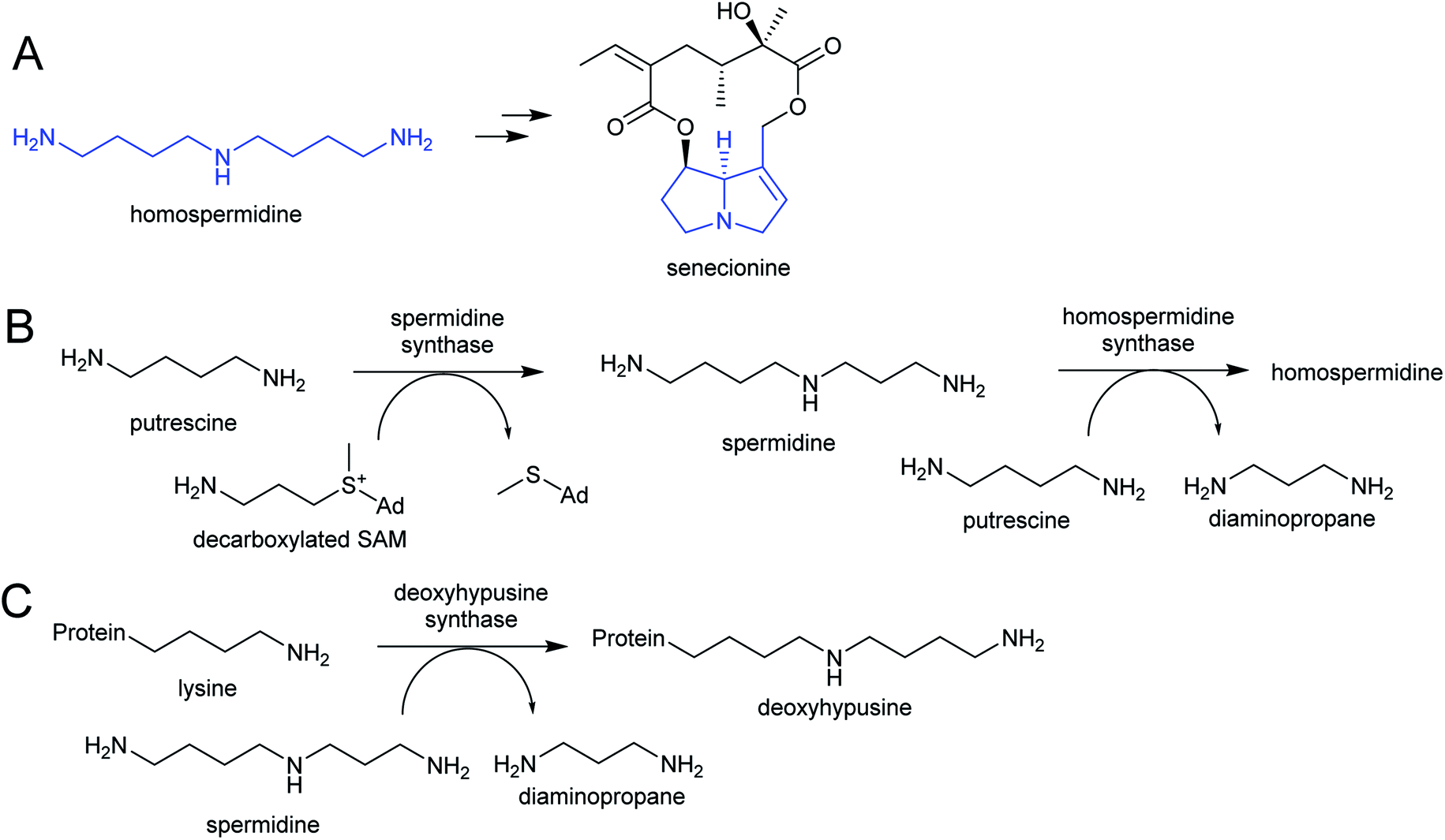 | ||
| Scheme 4 Homospermidine derived alkaloids. (A) Homospermidine and a derived pyrrolizidine alkaloid (moiety derived from homospermidine, the necine base, highlighted in blue). (B) Biosynthetic route to homospermidine from putrescine (Ad = adenosyl). (C) Activity of deoxyhypusine synthase. Protein represents eIF5A precursor. | ||
Homospermidine synthase (HSS) has evolved from deoxyhypusine synthase (DHS) on at least six independent occasions.42,58,60,61 Deoxyhypusine synthase has a key role in eukaryotic translation, modifying the initiation factor eIF5A with an aminobutyl group derived from spermidine (Scheme 4C). It is also able to accept putrescine as a substrate in place of eIF5A. Duplications and neofunctionalisations have led to enhancement of putrescine activity and reduction of eIF5A activity to yield HSS.61 Convergence is also evident on the residue level, with identical active site substitutions occurring in each independent HSS origins.62
2.2. Aromatic amines
The aromatic amino acids tyrosine and tryptophan are precursors to major alkaloid families, typically via their primary amine derivatives, tyramine and tryptamine. Simple tyramine and tryptamine-derived products are ubiquitous in green plants (Fig. 4). For example, tyramine contributes to the structure of suberin, a cell wall biopolymer,37 and hordenine, found in barley, is a derivative of tyramine with anti-fungal properties.38 Tryptophan contributes to a number of simple proto-alkaloids in plants via indole including indole-3-acetic acid and the indigo precursor indican. Tryptamine is a precursor to serotonin and psychoactive proto-alkaloids such as dimethyltryptamine.36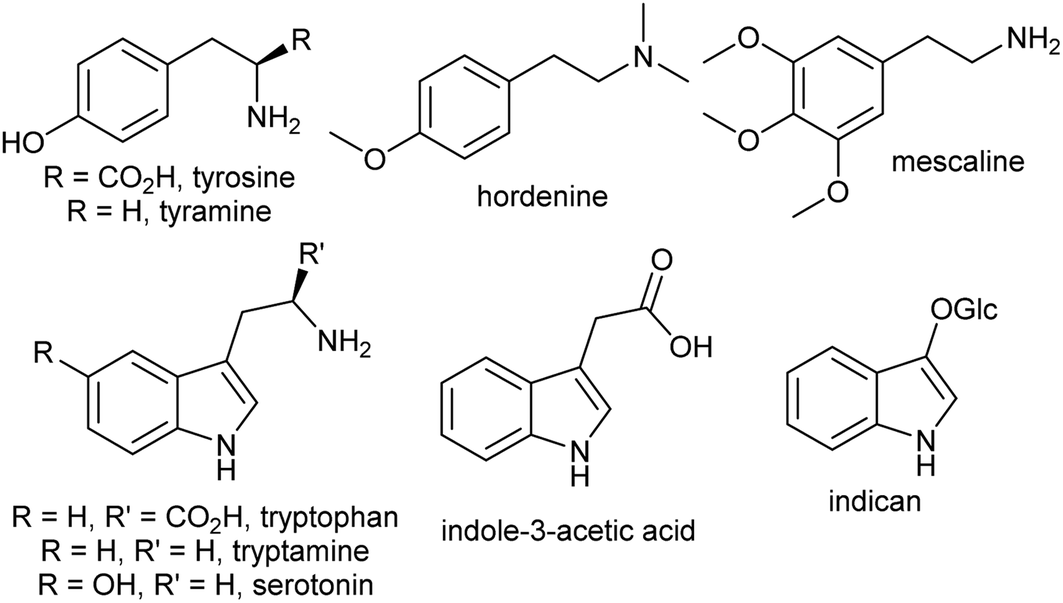 | ||
| Fig. 4 Examples of simple aromatic amines in plants derived from tyrosine and tryptophan. | ||
Tyramine and tryptamine are derived from tyrosine and tryptophan by decarboxylation, catalysed respectively by tyrosine decarboxylase (TyrDC)63 and tryptophan decarboxylase (TrpDC).64,65 These decarboxylases are ubiquitous in green plants: TyrDCs and TrpDCs form distinct clades in the plant aromatic amino acid decarboxylase (AAAD) protein family; both appear to have originated in the angiosperm ancestor.66 Despite their evolutionary distance, they share active site and substrate recognition sites: substitution of a single residue in Papaver somniferum TyrDC with the equivalent amino acid from Catharanthus roseus TrpDC (S372G) enables TyrDC to accept indolic substrates, and the complementary mutation in CrTrpDC has the equivalent effect, allowing it to accept phenolic substrates.67
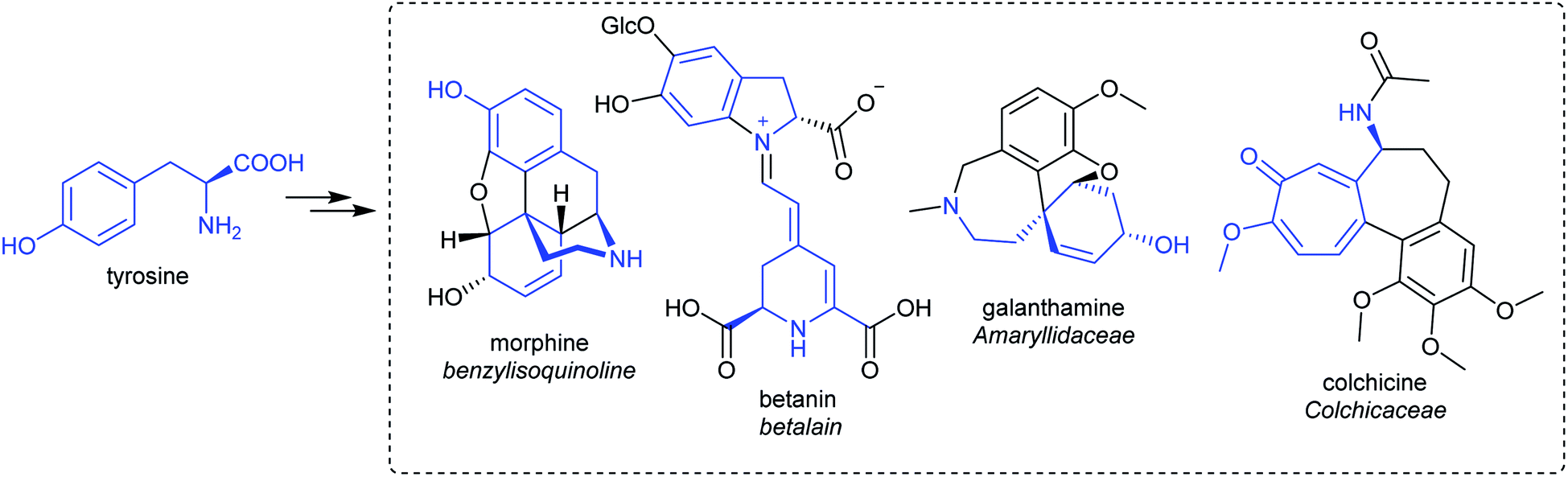 | ||
| Scheme 5 Tyrosine derived alkaloids. Portion from tyrosine highlighted in blue. | ||
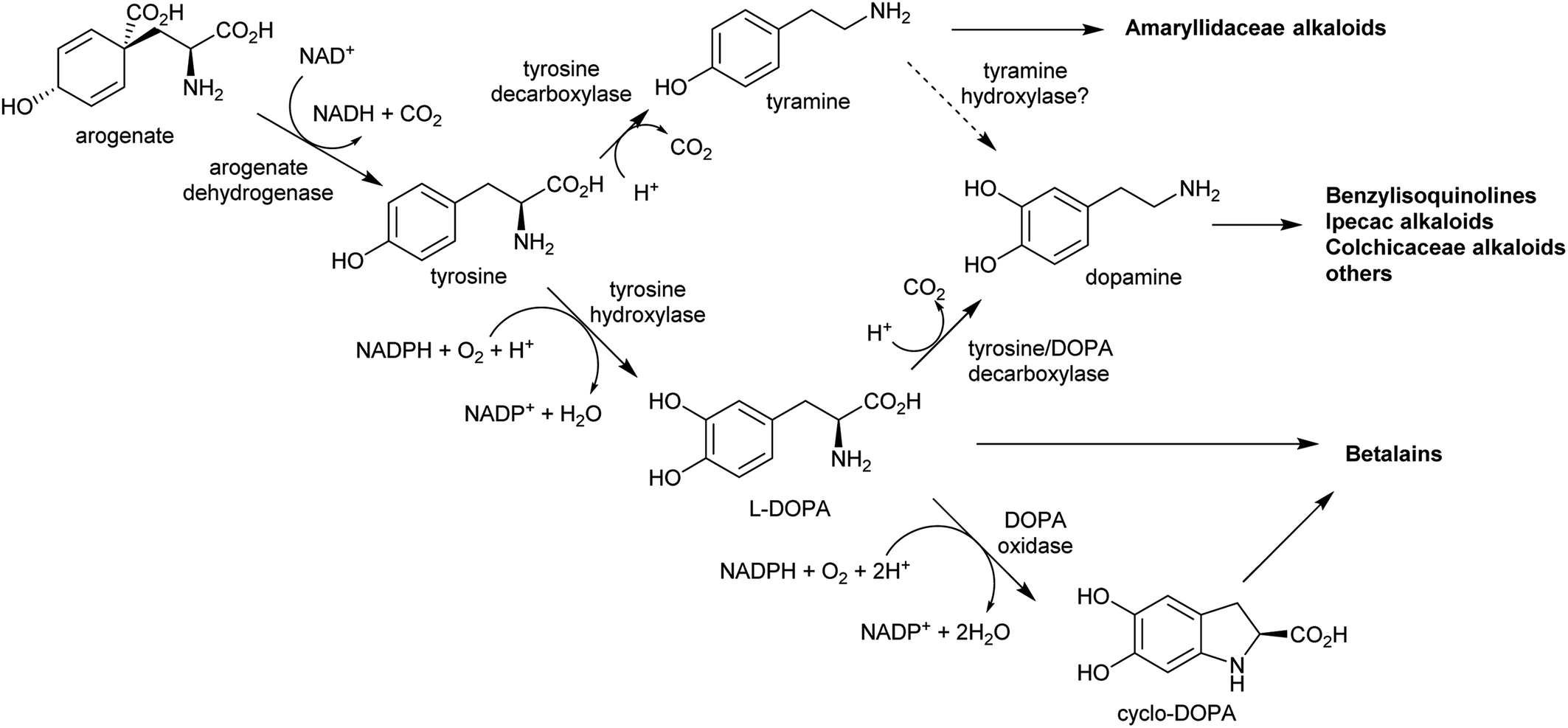 | ||
| Scheme 6 Origins of tyrosine derived alkaloids. Biosynthesis of dopamine has not been elucidated outside of betalain producing plants. Tyrosine hydroxylase and DOPA oxidase have only been described in betalain biosynthesis. Arogenate dehydrogenase is typically inhibited by tyrosine, but in betalain producing plants a second arogenate dehydrogenase is present lacking sensitivity to tyrosine. | ||
In the BIA producing Papaver species, dopamine accumulates to a high concentration (∼16% cellular dry weight of P. bracteatum) and is stored in the vacuole.74,75 Tyramine accumulates up to 5 mM in the latex of a low-alkaloid variety of P. somniferum.76 These observations indicate that tyrosine derived amines accumulate in BIA producing plants; however, the metabolic and evolutionary origins of this accumulation remain unclear.
By contrast, the metabolic and evolutionary origins of tyrosine accumulation in betalain producing plants is well characterised. Betalain pigments are derived from tyrosine, but unlike typical alkaline alkaloids, they incorporate amino acids directly without decarboxylation. The non-proteinogenic amino acids L-DOPA and cyclo-DOPA are precursors to betalains, and are formed through hydroxylation and oxidative cyclisation of tyrosine catalysed by cytochrome P450s in the CYP76AD1-subfamily.77 The hydroxylation is equivalent to the missing step in BIA dopamine biosynthesis. The CYP76AD1-subfamily in the betalain producing Caryophyllales form two clades,78 with enzymes in the β-clade (CYP76AD5/6/15) catalysing the hydroxylation of tyrosine,79,80 and enzymes in the α-clade (CYP76AD1-4) capable of catalysing both tyrosine hydroxylation and L-DOPA oxidative cyclisation to form cyclo-DOPA.79–82 As betacyanins are derived from cyclo-DOPA whereas betaxanthins are not, the ratio of α- and β-clade enzymes can determine the ratio of betaxanthins and betacyanins (see Section 4.2.2).83
The metabolic origins of the betalain pathway lie in primary metabolism and tyrosine biosynthesis.84 Arogenate dehydrogenases (AroDH) catalyse the formation of tyrosine and typically have strong product feedback inhibition.85 In the Caryophyllales, there are two clades of AroDH paralogs: AroDHβs, which have the typical tyrosine sensitivity, and AroDHαs, which show relaxed sensitivity to tyrosine. These two clades emerged in the ancestor of the core Caryophyllales, prior to the emergence of betalain biosynthesis: AroDHαs may cause accumulation of tyrosine required for betalain biosynthesis. This shows how changes in primary metabolism resulting in the accumulation of metabolites may have led to the evolution of new pathways.
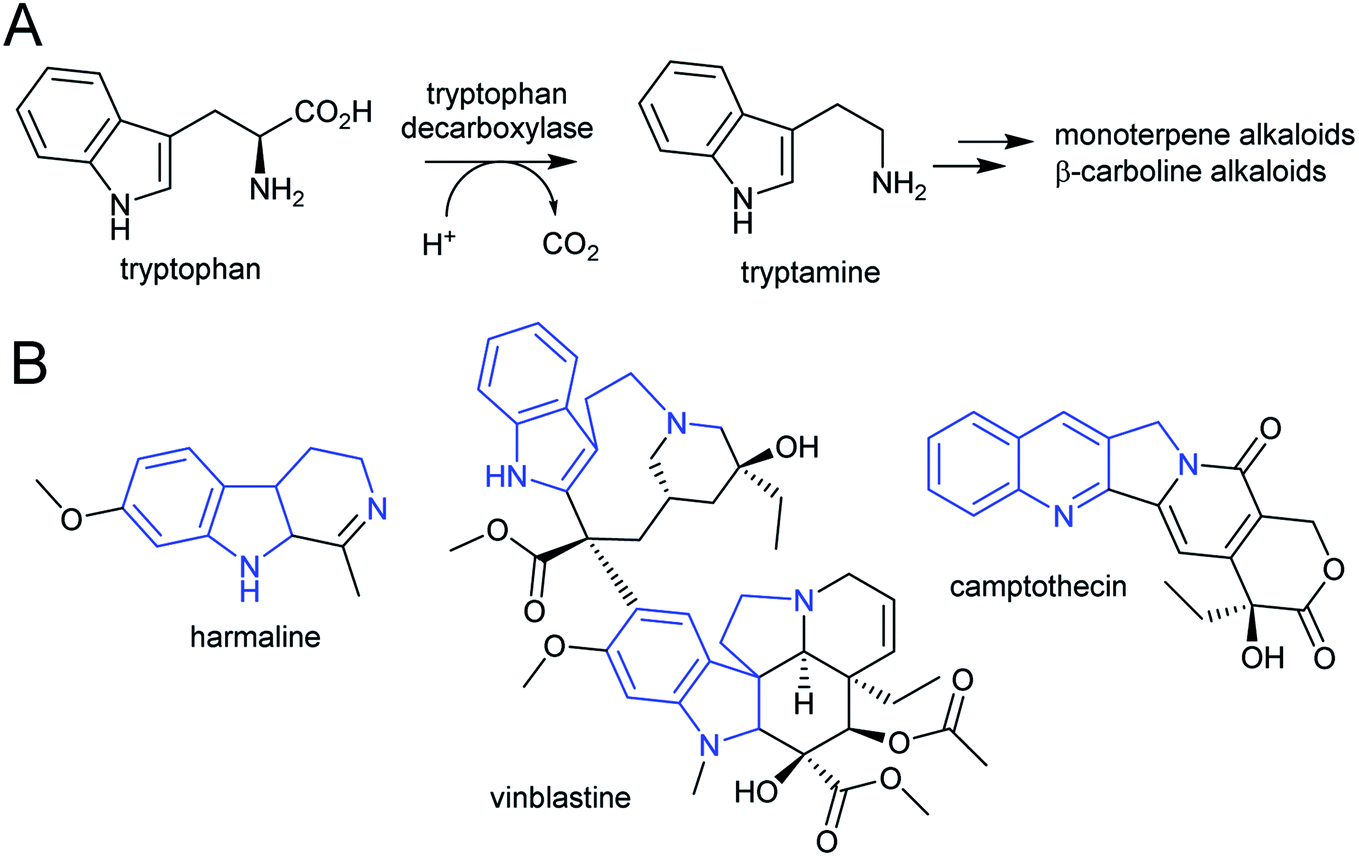 | ||
| Scheme 7 Tryptophan derived alkaloids. (A) Origin of tryptamine from tryptophan. (B) Alkaloids derived from tryptamine. Portion from tryptophan highlighted in blue. | ||
The acridone and quinoline alkaloids, derived from the tryptophan precursor anthranilate, do not follow the typical alkaloid biosynthetic pattern described in this review.90 However, their origins are informative regarding how precursors can accumulate (Scheme 8). Anthranilate is formed from chorismate through a reaction catalysed by anthranilate synthase (AS), which is typically feedback inhibited by tryptophan, enabling regulation of the tryptophan accumulation.91 The acridone alkaloid producing species Ruta graveolens has two copies of the ASα subunit: ASα2 is constituently expressed and inhibited by tryptophan, whereas ASα1 is upregulated upon elicitation and has reduced sensitivity to tryptophan.92,93 ASα1 enables the accumulation of anthranilate that ultimately leads to increased alkaloid biosynthesis.
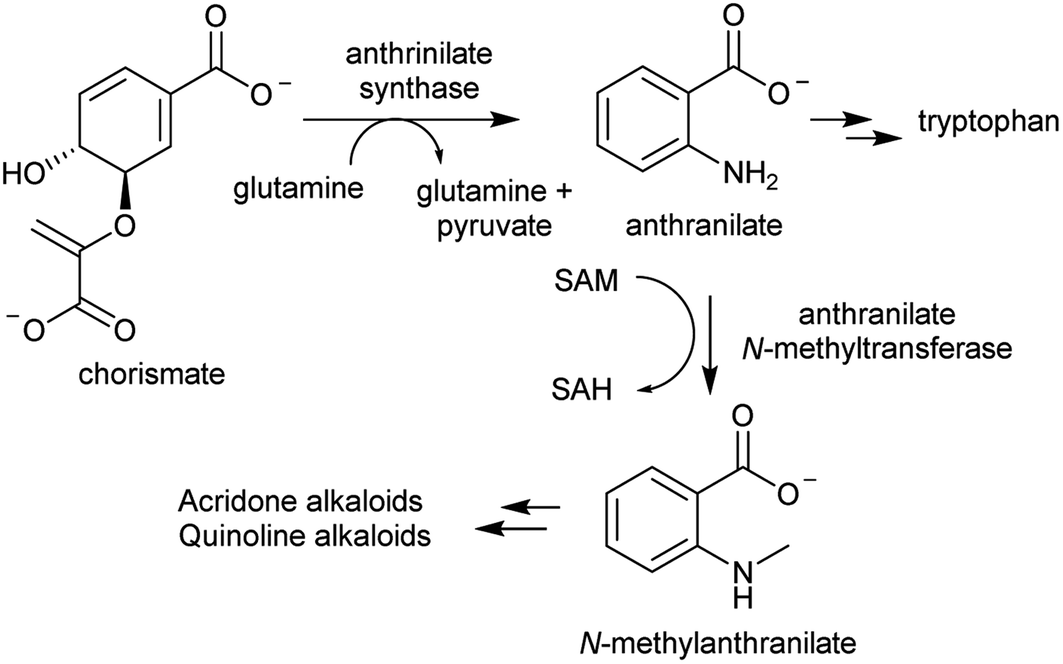 | ||
| Scheme 8 Biosynthetic origins of acridone and quinoline alkaloids. Anthrinilate synthase is typically inhibited by tryptophan, but in acridone/quinoline alkaloid producing plants a second anthrinilate synthase is present lacking sensitivity to tryptophan. | ||
2.3. Summary
The nitrogen atom defines alkaloids. Therefore understanding how nitrogen is channelled into alkaloid biosynthesis is vital for our comprehension of their metabolic and evolutionary origins. Many of the amine precursors of complex alkaloids are present in the majority of plant taxa. Modifications to the metabolism of these precursors can result in their accumulation and channelling into alkaloid biosynthesis. In both betalain and acridone biosynthesis core metabolic enzymes have been duplicated, and their feedback inhibition reduced, enabling accumulation of precursors.84,93 In the Solanaceae, duplication of OrnDC may have led to the accumulation of putrescine.53In pathways where the amine precursor is not part of primary metabolism, enzyme evolution has occurred. This includes the repeated evolution of HSS from DHS in pyrrolizidine alkaloid biosynthesis,58 and of Lys/OrnDC from OrnDC for piperideine alkaloid formation.55 These events have happened multiple times independently due to the inherent promiscuity of the ancestral enzymes.
Does accumulation of amine precursors presage alkaloid evolution? Or does modification to primary metabolism serve to push flux through a pre-existing pathway? Phylogenetic analyses of genes involved in the formation of amine precursors in betalain (AroDH), piperideine (Lys/OrnDC) and pyrrolinium biosynthesis (OrnDC) place enzyme duplication/evolution events prior to the emergence of alkaloid producing taxa.53,55,84 Whilst further verification is needed of this chronology, it suggests that amine accumulation prefigures the emergence of alkaloid biosynthesis.
3. Aldehyde accumulation
Complex alkaloid biosynthesis typically requires an aldehyde precursor. The aldehyde must accumulate to a high concentration and in close proximity to the amine precursor to enable the next stage of general alkaloid biosynthesis, formation of a reactive iminium intermediate. For polyamine-derived alkaloids, this aldehyde is formed through oxidative deamination of a terminal amine on the polyamine precursor, forming an amino aldehyde. In aromatic amine derived alkaloids, the aldehyde moiety is present on a distinct molecule, typically a secondary metabolite.3.1. Amino aldehydes
The oxidative deamination of a primary amine on a polyamine produces an amino aldehyde. As described below (Section 4.1), this leads to the spontaneous intramolecular formation of a cyclic iminium, which is a key electrophilic intermediate in alkaloid biosynthesis. The catabolism of polyamines in primary metabolism involves oxidative deamination catalysed by copper-containing amine oxidases (CuAOs) or FAD-dependent polyamine oxidases.94 The enzymes responsible for aldehyde formation in polyamine derived alkaloid biosynthesis appears to have been derived from these catabolic enzymes.95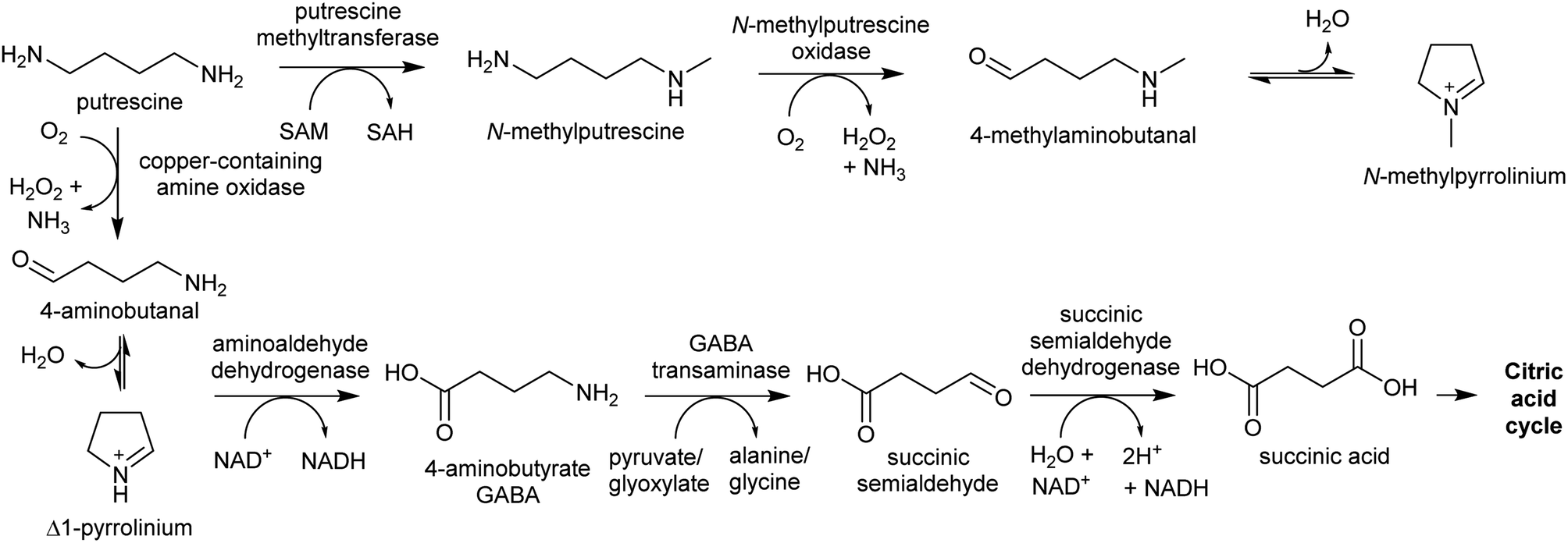 | ||
| Scheme 9 Metabolite fate of putrescine. Typically, putrescine is converted to GABA and shuttled into the citric acid cycle. In plants producing putrescine-derived alkaloids, a portion of putrescine is shuttled into alkaloid biosynthesis through methylation and oxidation. | ||
PMTs are SAM-dependent methyltransferases that evolved from spermidine synthases (SPDSs).100,101 Wild-type SPDSs demonstrate PMT activity, indicating that neofunctionalisation of SPDS to PMT can occur with ease.101 The PMT involved in tropane and nicotine biosynthesis emerged from a SPDS duplication in the Solanales lineage, prior to the origin of the Solanaceae.53,101
MPO is a copper-containing amine oxidase localised to the peroxisome.95 The Solanaceae MPO originated from a CuAO homolog generated by a whole genome triplication event.53,95 As part of polyamine catabolism, peroxisomal CuAOs oxidise putrescine to 4-aminobutanal, which is then directed into the TCA cycle via 4-aminobutyrate and succinate (Scheme 9).102,103 It is possible that the methyl group introduced by PMT prevents recognition by catabolic enzymes, allowing N-methylaminobutanal to accumulate.
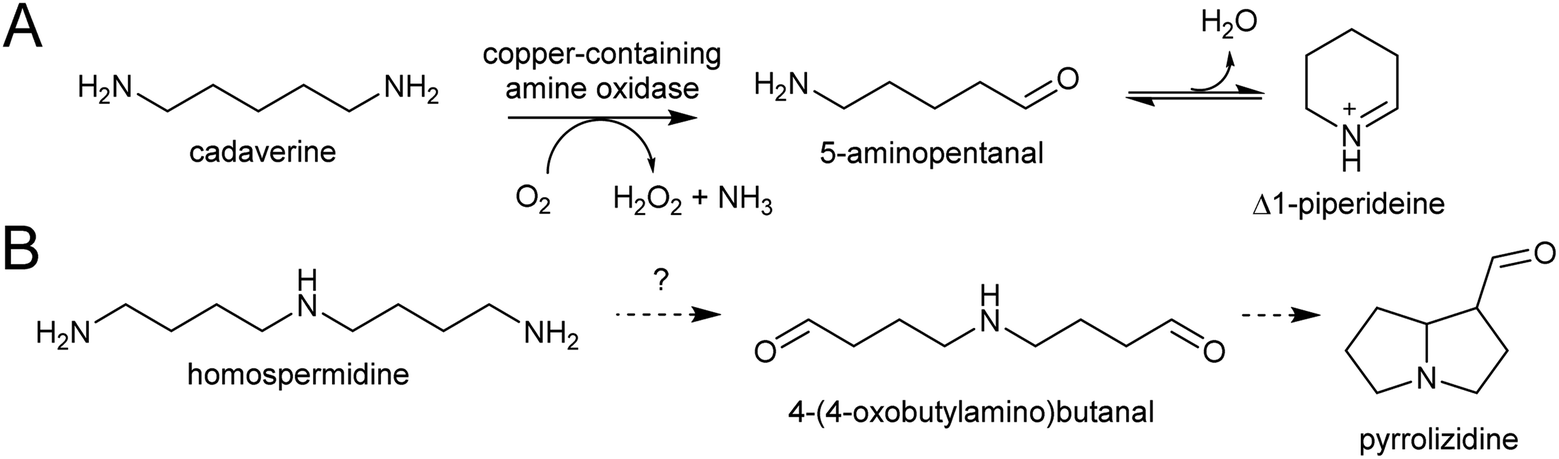 | ||
| Scheme 10 Polyamine oxidation steps. (A) Oxidative deamination of cadaverine to 1-piperidiene. (B) Putative double deamination of homospermidine in pyrrolizidine alkaloid formation. | ||
3.2. Amino acid origins
Aldehydes that are incorporated into amino-acid derived alkaloids accumulate independently of the amine precursor. Often, the aldehyde is a specialised metabolite, already present at high concentration. Alternatively, aldehyde accumulation could be triggered by gene duplications or other changes to metabolism.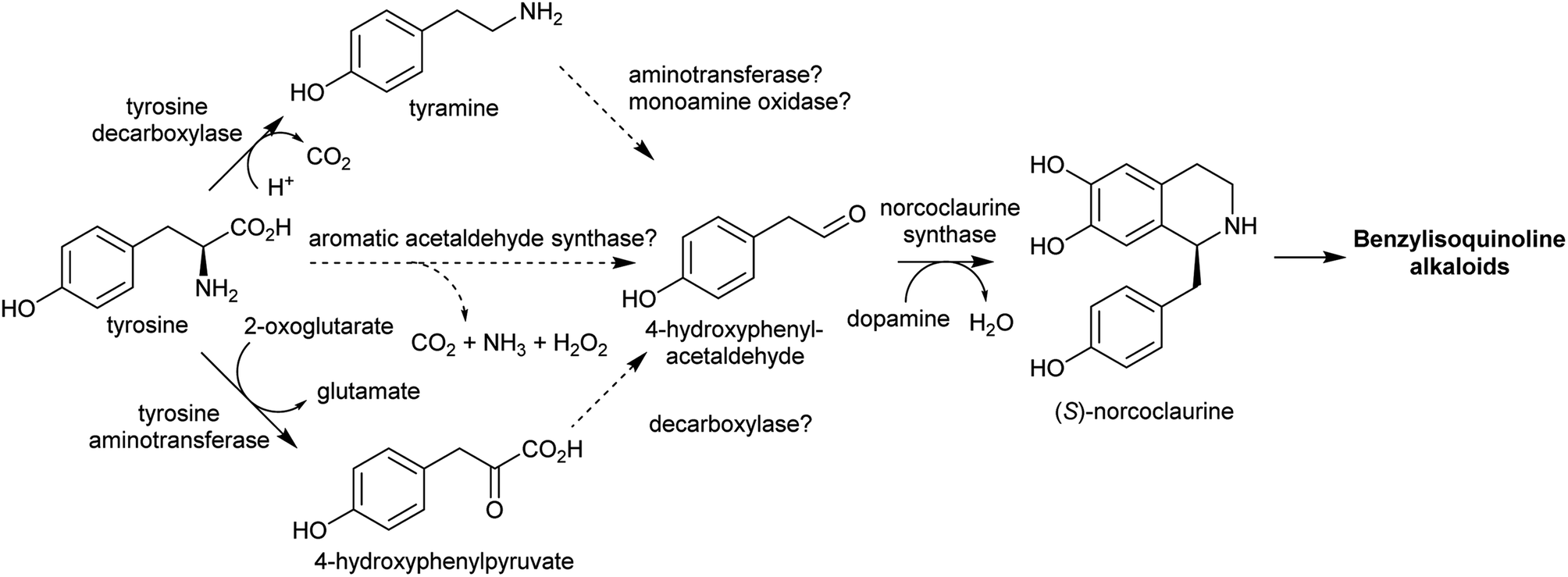 | ||
| Scheme 11 Biosynthesis of 4-hydroxyphenylacetaldehyde and its incorporation into benzylisoquinoline alkaloids. | ||
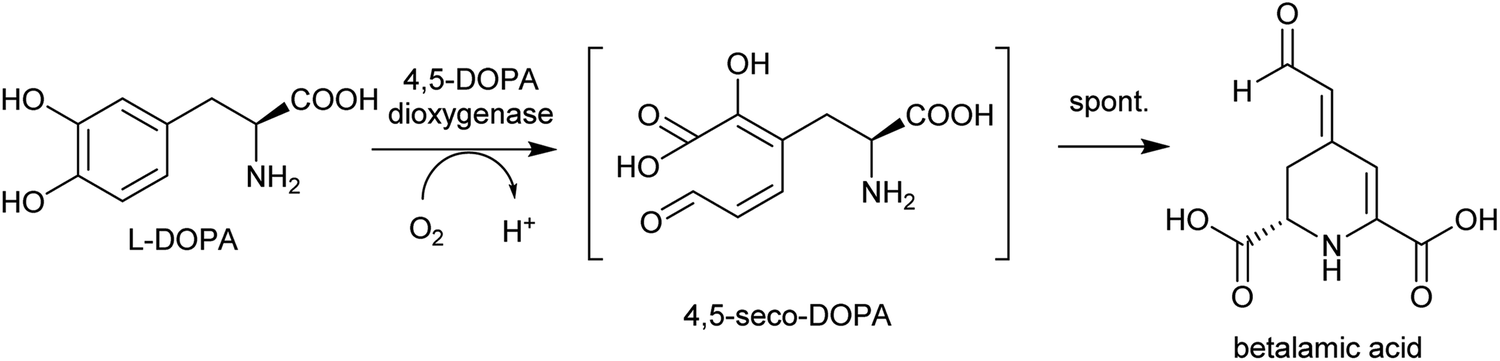 | ||
| Scheme 12 Biosynthesis of betalamic acid. | ||
3.3. Phenylpropanoids
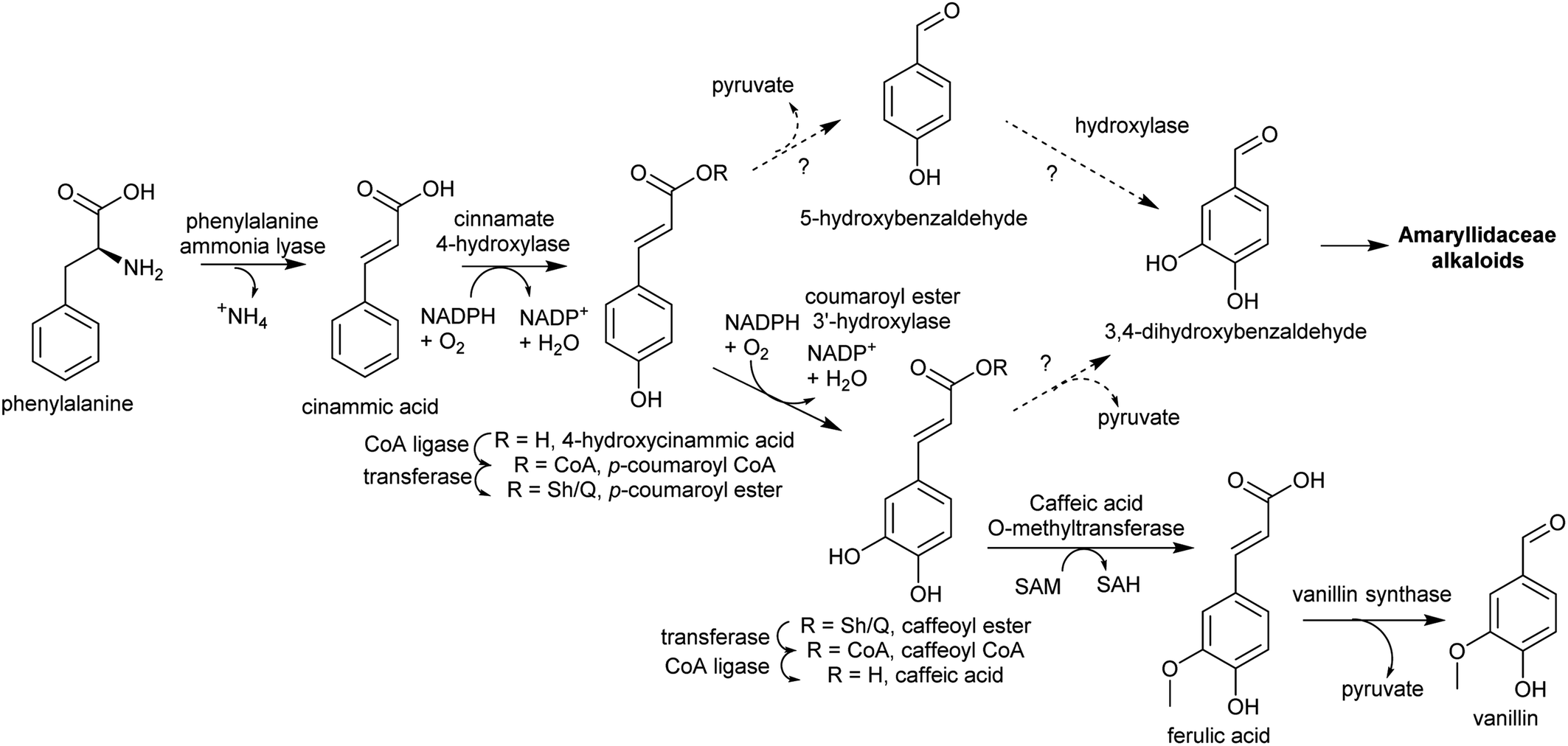 | ||
| Scheme 13 Substituted benzaldehyde biosynthesis. Pathway to vanillin from Vanilla planifolia.116 Coumaroyl ester hydroxylation may occur on CoA or Sh/Q ester. Sh = shikimate, Q = quinate. | ||
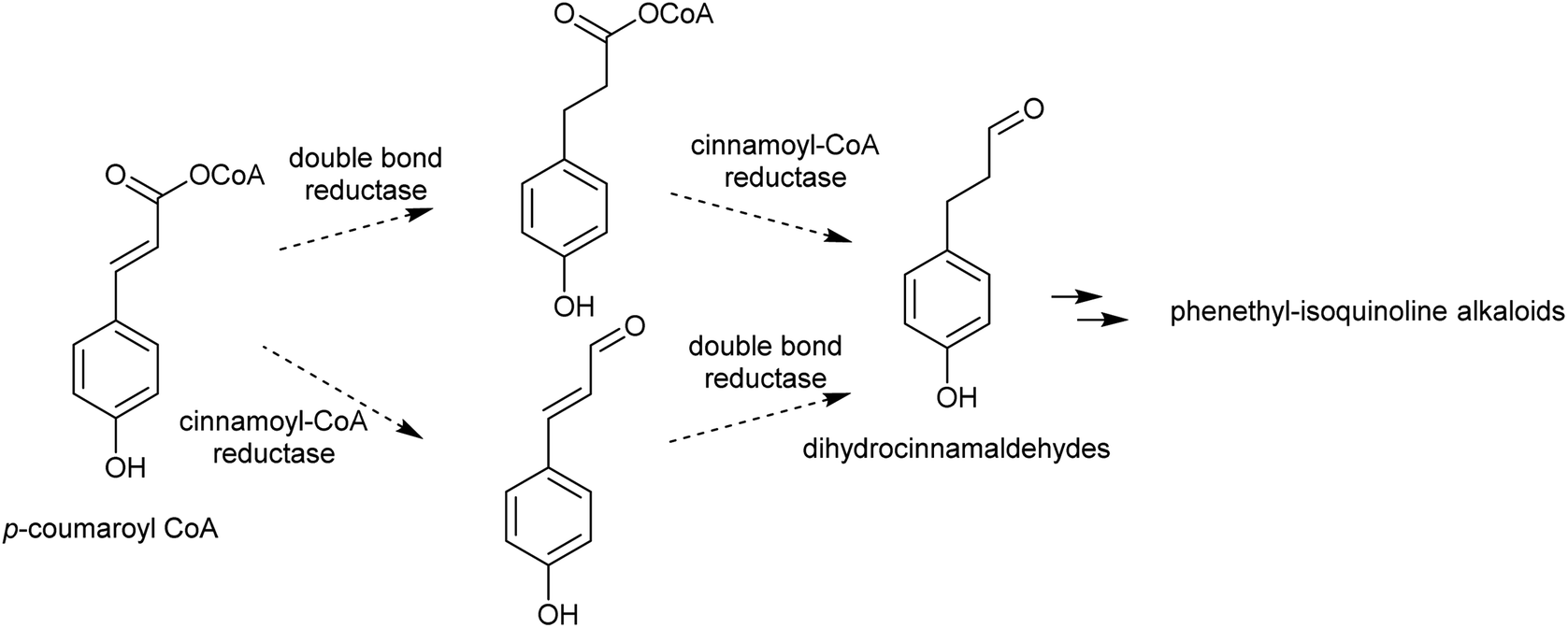 | ||
| Scheme 14 Biosynthesis of dihydrocinnamaldehydes. | ||
3.4. Secologanin
Iridoids are a widespread class of secondary metabolites derived from the monoterpene pathway.122 Within the plant kingdom, iridoids are largely restricted to the Asterids,123 where they typically occur as glycosides providing chemical defence against biting herbivores.124 Seco-iridoids, such as secologanin, are a subset of iridoids where the 5-membered carbon ring has been oxidatively cleaved. These seco-iridoids are found in multiple plants where alkaloids are absent.125,126 In C. roseus (Gentianales), secologanin is the precursor to all monoterpene indole alkaloids (MIAs). It is also a precursor to the ipecac alkaloids (Carapichea ipecacuanha [Gentianales] and Alangium salviifolium [Cornales]) (Scheme 15A).127–129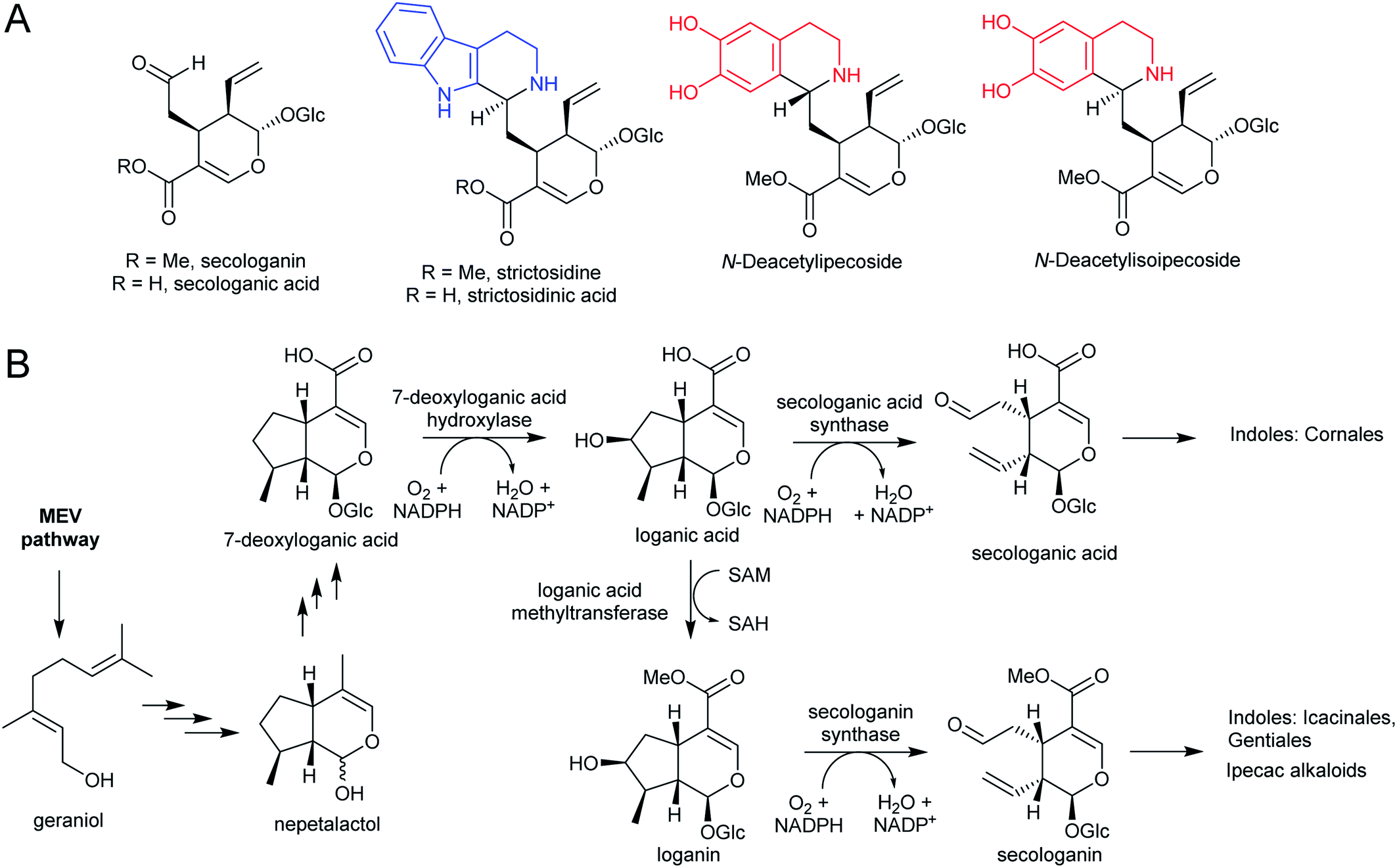 | ||
| Scheme 15 Iridoid monoterpene derived alkaloids. (A) Iridoid secologanin/secologanic acid and derived alkaloids. Strictosidine/strictosidinic acid derived from trypamine (blue). Ipecac alkaloids derived from dopamine (red). (B) Biosynthesis of secologanin/secologanic acid. | ||
The iridoid biosynthetic pathway has been elucidated in C. roseus, and derives from geraniol (Scheme 15B).122 Secologanin is formed from loganin through the action of secologanin synthase (SLS, CYP72A1).130 SLS is closely related to another enzyme involved in iridoid biosynthesis, 7-deoxyloganic acid hydroxylase (CYP72A224), indicating that substrate recognition is key to CYP evolution.131C. roseus contains at least two copies of SLS.132
The MIA pathway in Camptotheca acuminata (Cornales) proceeds via secologanic acid and not secologanin.133 Consequently, strictosidinic acid and not strictosidine is the first MIA. Cytochrome P450s (CYP72A565 and CYP72A610) from C. acuminata are able to catalyse both the hydroxylation 7-deoxyloganic acid to form loganic acid and the oxidative cleavage of loganic acid to form secologanic acid.134 These are closely related to secologanin synthase (CYP72A1) and 7-deoxyloganic acid hydroxylase (CYP72A224).
Nothapodytes nimmoniana (Icacinales) also produces camptothecin, though is more closely related to Gentianales than Cornales, and the pathway appears to proceed using the typical Gentianales route via secologanin.135 Icaninales and Gentianales may share an MIA origin, and this is supported by characterisation of an active secologanin synthase from N. nimmoniana (NnCYP72A1).136
4. Iminium formation
A primary amine can condense reversibly with an aldehyde to form an electrophilic iminium cation, also known as a Schiff base (Scheme 16). In water, the equilibrium is typically on the side of the reactants, and the concentration of iminium ions is low.137–139 The position of the equilibrium can shift towards the products if the iminium is cyclic or conjugated.140,141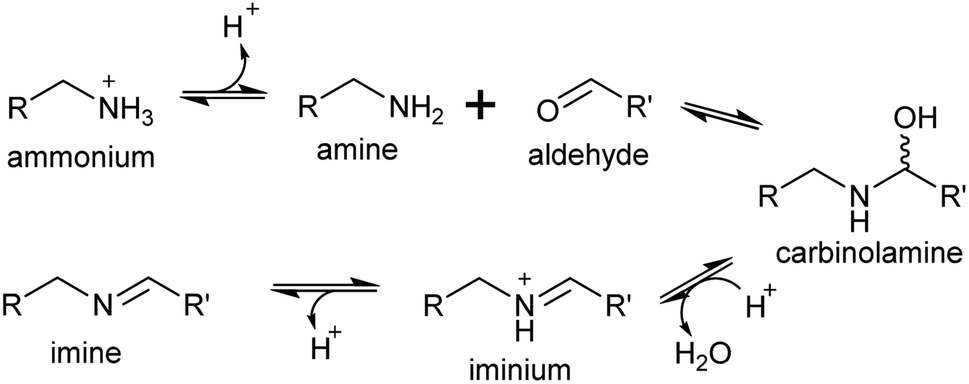 | ||
| Scheme 16 Mechanism of iminium formation. | ||
Surprisingly, the ratio of neutral imine to charged iminium in solution is largely dependent on the pKaH of the amine precursor (∼9–10), rather than the pKaH of the iminium itself (∼7), meaning that at physiologically relevant pHs the reactive iminium will be more abundant than the imine.137 Iminiums are highly electrophilic and form rapidly, so even when they not at high concentrations, the product of their reaction with a nucleophile can accumulate, provided the reaction is irreversible. This reaction of an iminium with a nucleophile is described here as a Mannich-like reaction, which quenches the reactivity of both the iminium and nucleophile.
Enzyme catalysed formation of iminiums is often futile due to their high reactivity in water: no enzymes catalysing only iminium formation have been described. However, iminiums can be enzyme substrates, such as in imine reductases, which typically take preformed iminiums (typically conjugated or cyclic) from solution as substrates.140 Yet, iminium formation can be enzyme catalysed when coupled directly to a quenching reaction, such as reduction.142
4.1. Intramolecular
The electrophilic intermediates in polyamine derived alkaloid biosynthesis are cyclic iminiums such as N-methylpyrollinium and Δ1-piperideine, formed from linear amino aldehydes through intramolecular condensation (Scheme 17). These rings form spontaneously, without the requirement for catalysis, as demonstrated by chemical syntheses.143,144 Even in water, the cyclic iminium will be abundant. The CuAO enzymes forming the amino aldehyde are localised to the peroxisome, and the cyclic iminiums will form upon ejection into the solvent or possibly in the enzyme active site.95,105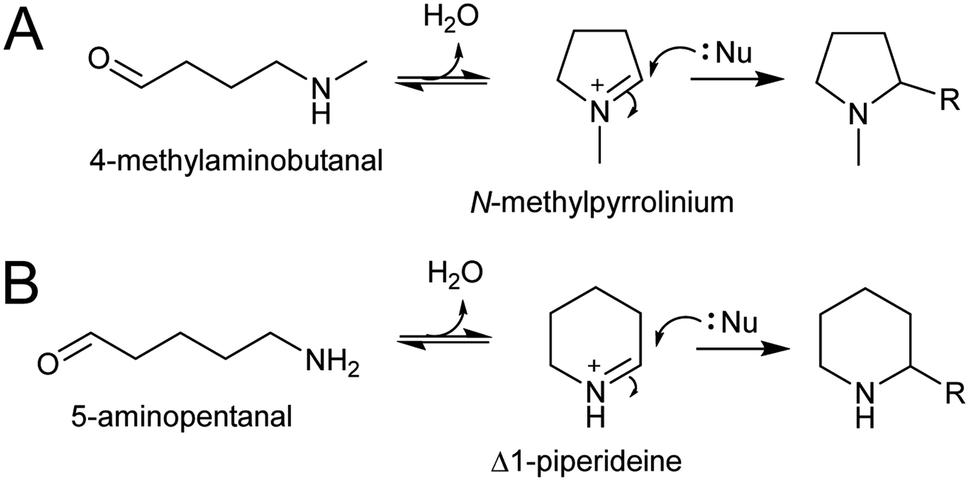 | ||
| Scheme 17 Polyamine-derived alkaloid formation. Cyclic iminium formation is followed by a Mannich-like reaction. | ||
4.2. Intermolecular
In the biosynthesis of alkaloids derived from aromatic amines, the iminium intermediate is formed through an intermolecular condensation and is non-cyclic. Typically, these iminiums are at very low concentration in water. This potentially unfavourable equilibrium can be overcome through rapid quenching of the transient iminium through an intramolecular nucleophile, as seen in Pictet–Spengler reactions (see Section 5.2); or through formation of a highly stable conjugated iminium as demonstrated in betalain biosynthesis (Section 4.2.2).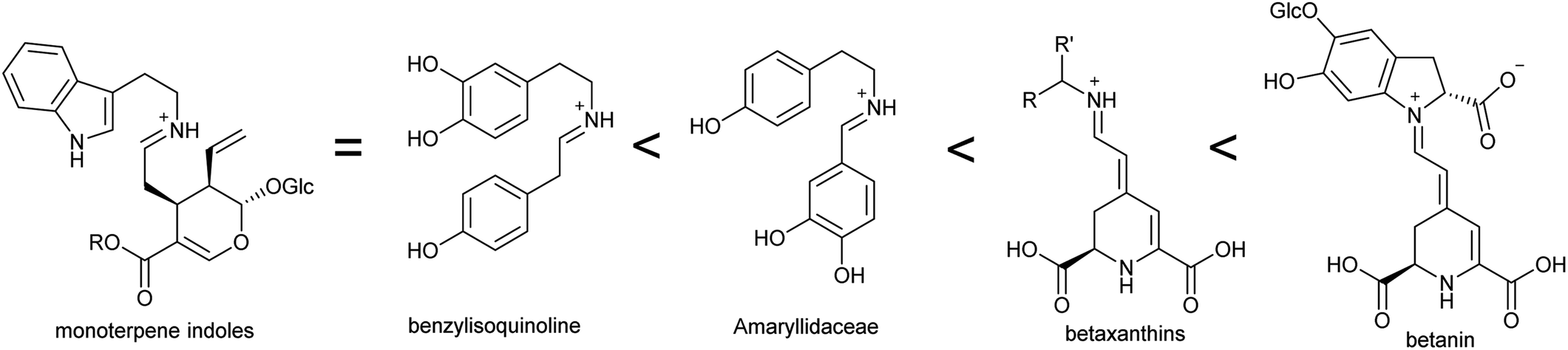 | ||
| Fig. 5 Stability of iminium intermediates. Predicted trend in iminium intermediates in alkaloid biosynthesis based on conjugation. | ||
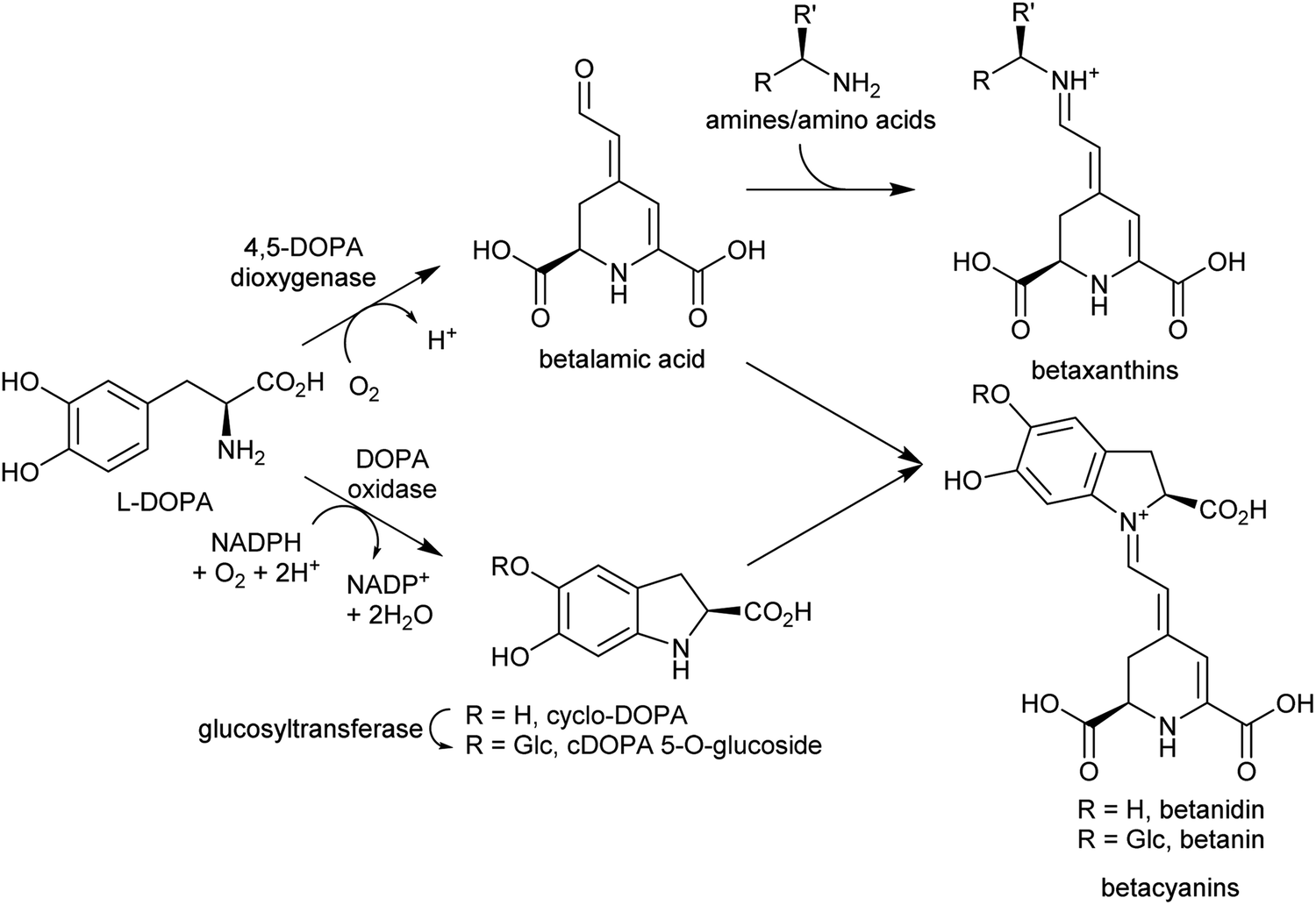 | ||
| Scheme 18 Betalain biosynthesis. Iminium formation steps are spontaneous without requiring enzyme catalysis. | ||
Betalains are all derived from the aldehyde containing betalamic acid, but can be divided into two categories depending on the amine donor: the red betacyanins are derived from cyclo-DOPA, and the yellow betaxanthins are derived from amino acids or other amines (e.g. dopamine, tyramine) (Scheme 18). The iminium formation step appears to occur spontaneously and does not require enzyme catalysis.150 The specific betalain formed therefore appears to depend on the concentrations of amines present. For example, in plants expressing betalain biosynthesis genes heterologously, the ratio of betacyanins and betaxanthins can be controlled solely by varying the amount of the L-DOPA oxidase activity present (see Section 2.2.1). This enzyme forms cyclo-DOPA, and its presence enhances betacyanins at the expense of betaxanthins.83
Like many other pigments, betalains are stored in the vacuole. However, the key enzymes required for cyclo-DOPA and betalamic acid biosynthesis are cytoplasmic.151 Neither the subcellular location nor control mechanisms of iminium condensation have been determined. The vacuole represents one possible location for iminium formation: the reaction may be promoted in acidic conditions, and betalain selectivity could be controlled by the accumulation of specific amines by tonoplast (vacuolar) transporters. Identification and characterisation of tonoplast transporters in betalain producers would help resolve this question.
5. Mannich-like reaction
The key step in many alkaloid biosynthetic pathways is a Mannich-like reaction, in which the iminium reacts irreversibly with a nucleophile. This reaction quenches reactive species and causes the formation of a new carbon–carbon bond; it is therefore very energetically favourable, to the extent that in many cases the reaction can proceed without enzyme catalysis.This reaction can be considered the first committed step into an alkaloid pathway, as the specific combination of electrophile and nucleophile defines alkaloid subtype and is irreversible. Furthermore, it is typically the scaffold-forming step and establishes a heterocyclic structure that is central to alkaloid identity, with impacts on downstream reactions and compound bioactivities.
5.1. Polyamine-derived
In polyamine-derived alkaloids, the Mannich-like reaction is typically intermolecular. The nucleophiles are of diverse origin and give rise to specific alkaloid sub-types. These nucleophiles must accumulate in the same subcellular location as the iminium. As the accumulating non-amine component of the alkaloid, they are equivalent to aldehydes required in aromatic amino acid derived alkaloids.To date, no enzymes have been described that catalyse the intermolecular Mannich-like reactions of alkaloid biosynthesis, and it is possible that the in planta reactions occur spontaneously with no enzyme catalysis. The intermolecular nature of the reaction has led to a modular organisation of in polyamine alkaloid biosynthesis, where a range of different nucleophiles may combine with either N-methylpyrollinium or Δ1-piperideine to generate chemical diversity.
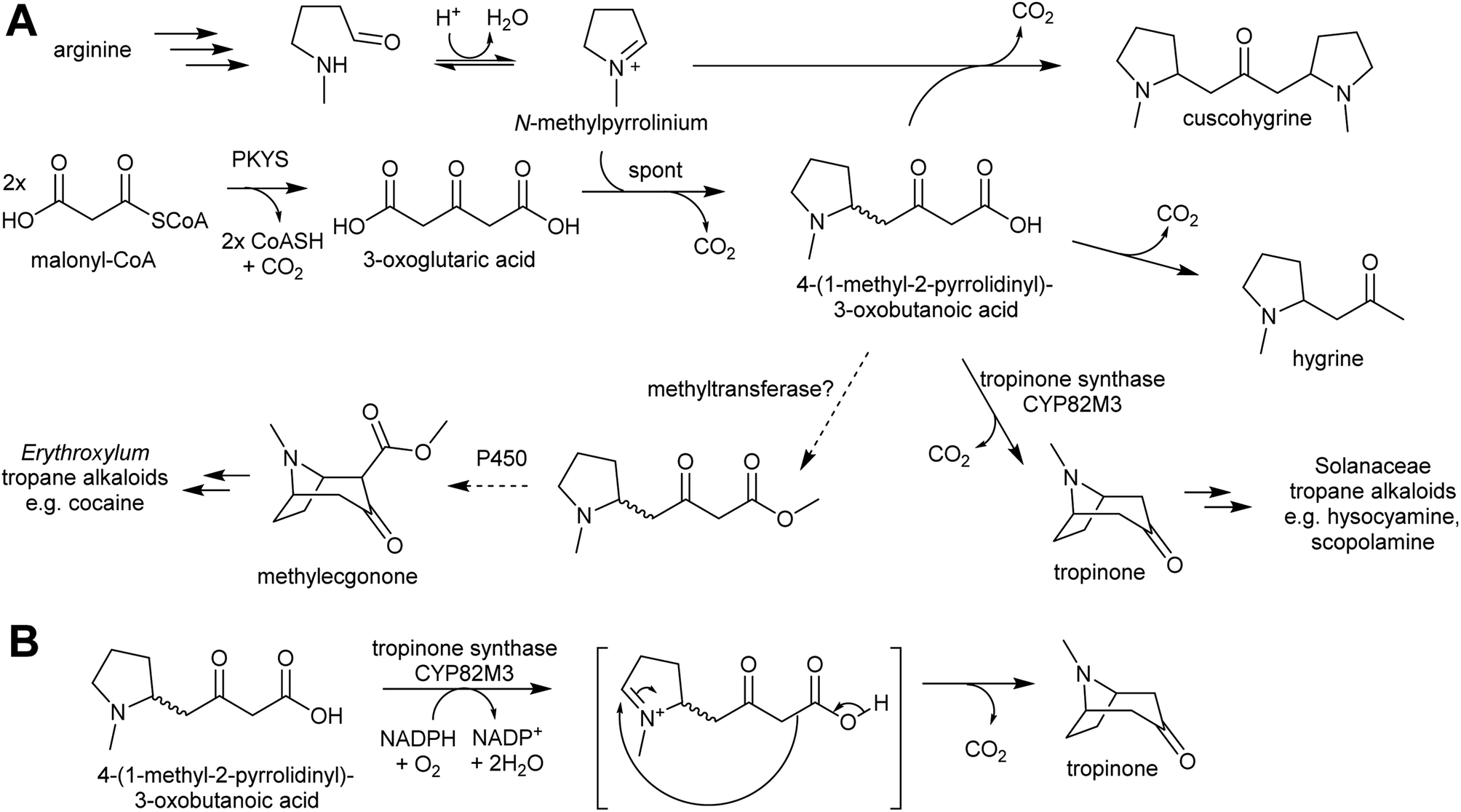 | ||
| Scheme 19 Tropane alkaloid biosynthesis. (A) Outline of tropane alkaloid biosynthesis as elucidated in Solanaceae. Methyltransferase step in Erythroxylum is hypothetical. (B) Hypothetical mechanism of tropinone synthase. Note that a methylester would not decarboxylate, giving rise to methylecgonone. | ||
Pyrrolidine ketide synthase (PYKS), a type III polyketide synthase, catalyses the formation of 3-oxoglutaric acid from two malonyl-CoA units. PYKS has an active site with the typical type-III PKS catalytic triad for substrate loading and chain extension (Cys166, His305 and Asn338), but has additional residues (Arg-134 and Ser-340) which limit the enzyme to a single extension step by interacting with the substrate carboxylate moiety.153
When incubated with PYKS, N-methylpyrollinium and malonyl-CoA form 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid, a key intermediate.152,153 PYKS forms 3-oxoglutaric acid from malonyl-CoA and releases it into the solvent where it spontaneously reacts with N-methylpyrollinium through a decarboxylative Mannich condensation.153 PYKS does not utilise N-methylpyrollinium as a starter unit, nor does the enzyme catalyse the condensation reaction. This observation is supported by the racemic nature of 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid, and by the presence of the side products hygrine and cuscohygrine, which may be formed through decarboxylation or a second decarboxylative Mannich condensation respectively.153
The 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid intermediate can be converted to tropinone through the action of tropinone synthase (CYP82M3).152 The mechanism of this is unknown, but a possible route is formation of a pyrollinium cation through hydroxylation and dehydration (Scheme 19B). This intermediate then undergoes a further, possibly spontaneous, intramolecular decarboxylative Mannich condensation to yield tropinone.
Tropane alkaloids emerged independently Erythroxylaceae and Solanaceae.99 However, in Erythroxylum coca (Erythroxylaceae) alkaloids, the tropane ring has an additional carboxymethyl group. This could arise if the carboxylic acid moiety of 4-(1-methyl-2-pyrrolidinyl)-3-oxobutanoic acid is methylated prior to cyclisation (Scheme 19).152 The enzymes catalysing the formation of tropane alkaloids in the Erythroxylaceae are yet to be fully described.155
The solution of the key step in tropane biosynthesis has implications for other pathways. The 3-oxoglutaric acid nucleophile can also react with Δ1-piperideine, leading to a set of homologous compounds and pathways to those derived from N-methylpyrollinium. For example, quenching of Δ1-piperideine by 3-oxoglutaric acid through a decarboxylative Mannich reaction yields oxo-2-piperidine-butanoic acid. This intermediate could decarboxylate to yield pelletierine, a homolog of hygrine, or undergo oxidative cyclisation to yield norpseudopelletierine, a homolog of tropinone (Scheme 20A).22,153 Pseudopelletierine is a precursor to the granatane alkaloids. The biosynthesis of lycopodium alkaloids such as huperzine is likely to proceed via aldol addition of oxo-2-piperidine-butanoic acid to pelletierine (Scheme 20B).57,156
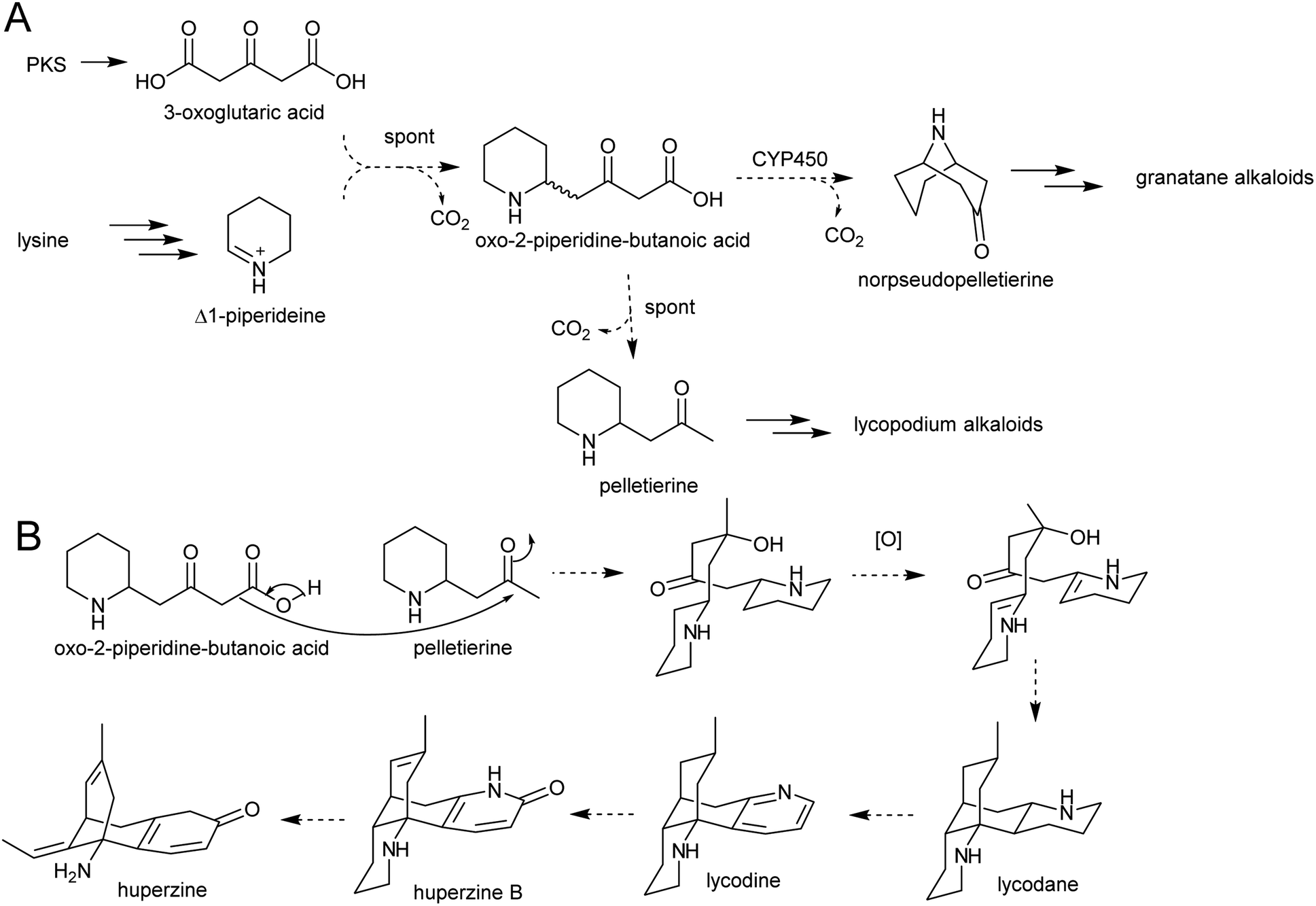 | ||
| Scheme 20 Hypothesised formation of piperideine alkaloids. (A) Proposed formation of piperideine derived alkaloids based on known formation of tropane alkaloids. (B) Possible route to lycopodium alkaloids such as huperzine.57 | ||
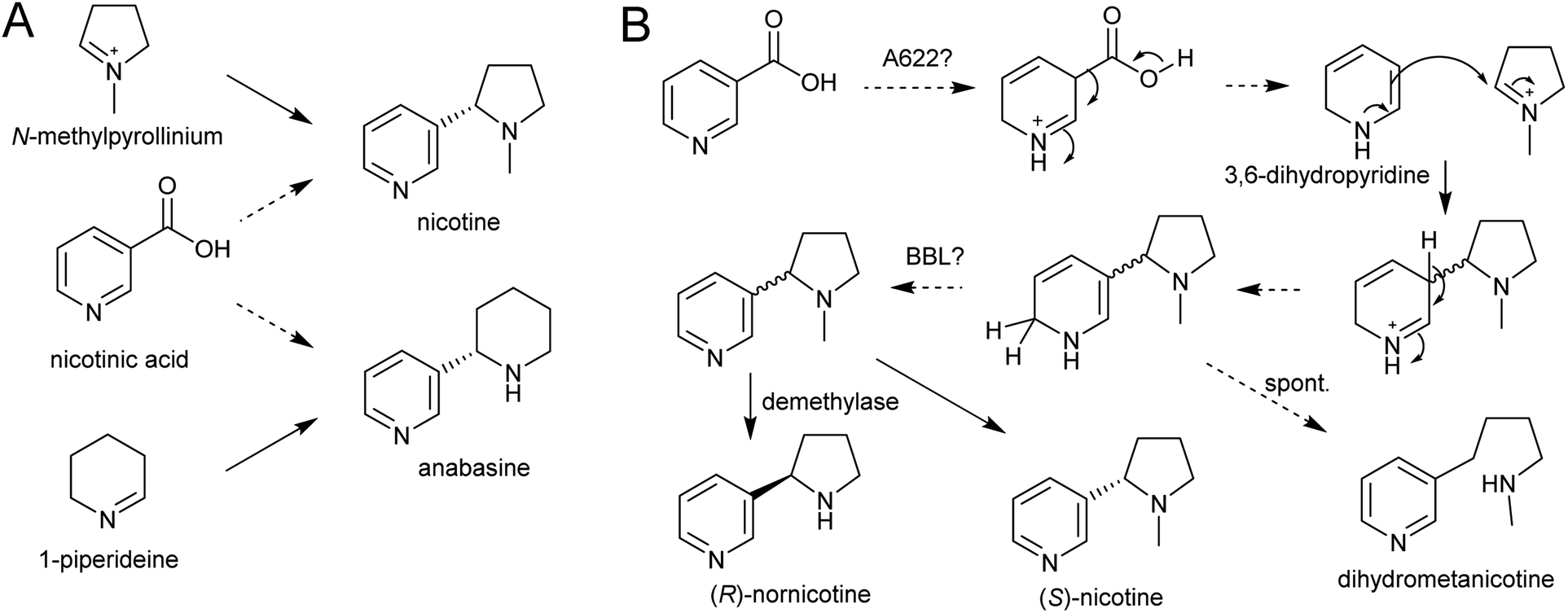 | ||
| Scheme 21 Nicotine biosynthesis. (A) Origins of nicotine and anabasine. (B) Putative biosynthetic pathway for nicotine. | ||
A gene encoding an NADPH-dependent reductase, A622, has a major role in activating nicotinic acid, however neither the substrate nor product structure is known.157,158 It is hypothesised that the nucleophilic compound attacking N-methylpyrollinium or Δ1-piperideine may be 3,6-dihydronicotinic acid or 3,6-dihydropyridine (Scheme 21B).159
The final step in nicotine biosynthesis is catalysed by vacuolar located flavin-containing oxidases with similarity to berberine-bridge enzymes (BBLs).160 Knocking out all six BBL paralogs in tobacco results in a nicotine-free plant.161 Knocking-down BBL expression results in the accumulation of a reduced nicotine metabolite (dihydrometanicotine), indicating that the enzyme acts after the condensation step, oxidising the pyridine ring (Scheme 21B).19,160 A similar oxidation is likely to be involved in anabasine production.
It is possible that, like the quenching step in tropane alkaloid biosynthesis, the reaction between N-methylpyrollinium or Δ1-piperideine and the nicotinic acid derivative is spontaneous. This is supported by the observation that the enantiomeric excess of the (S)-enantiomer in natural nicotine is caused by enantioselective enzymatic demethylation of (R)-nicotine to (R)-nornicotine and not selective cyclisation.162 The increase in anabasine to nicotine ratio through increased concentration of precursors could also be indication of this.163 Based on the location of BBL it is possible the Mannich-like reaction occurs in the vacuole.
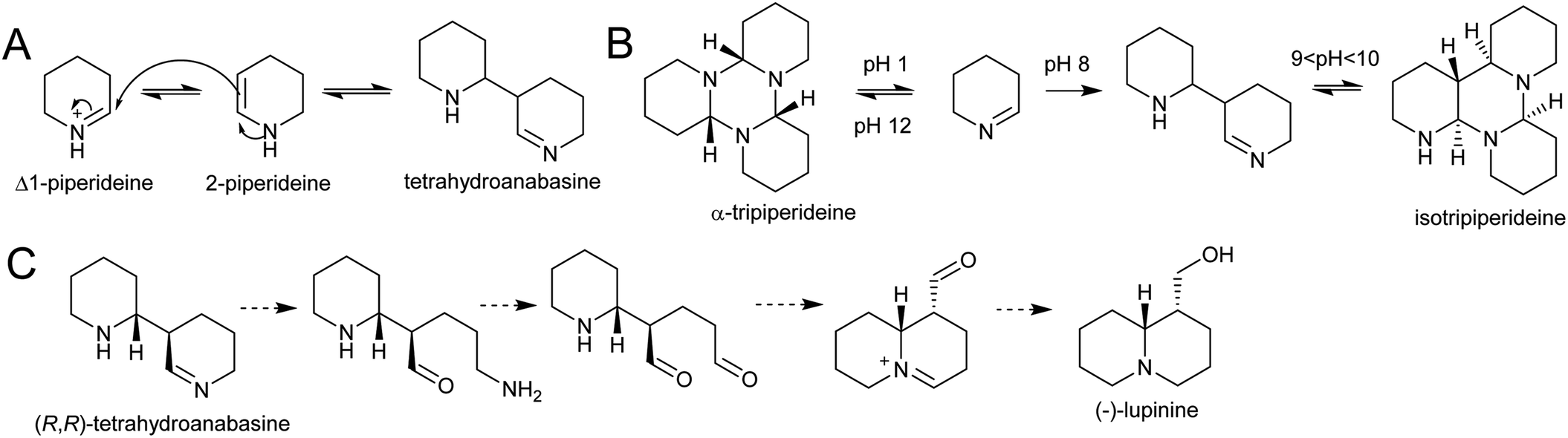 | ||
| Scheme 22 Quinolizidine alkaloid biosynthesis. (A) Dimerisation of Δ1-piperideine. (B) Behaviour of Δ1-piperideine in solution.143 (C) Hypothetical origin of (−)-lupinine from energetically viable piperideine dimer precursor. | ||
Density functional theory calculations indicate that the dimerization reaction between Δ1-piperideine and 2-piperideine occurs spontaneously to form the (R,R) or (S,S)-tetrahydroanabasine, but formation of the (R,S)- or (S,R)-tetrahydroanabasine dimers proceeds more slowly.165 The diastereoselectivity of the spontaneous reaction matches the biosynthetic origins of compounds such as (−)-lupinine, a major alkaloid found in Lupinus (Scheme 22C).32 However, quinolizidines are often found in high enantiomeric excess, suggesting that a stereoselective enzyme catalyses the dimerization step, or downstream enzymes exert enantioselectivity.
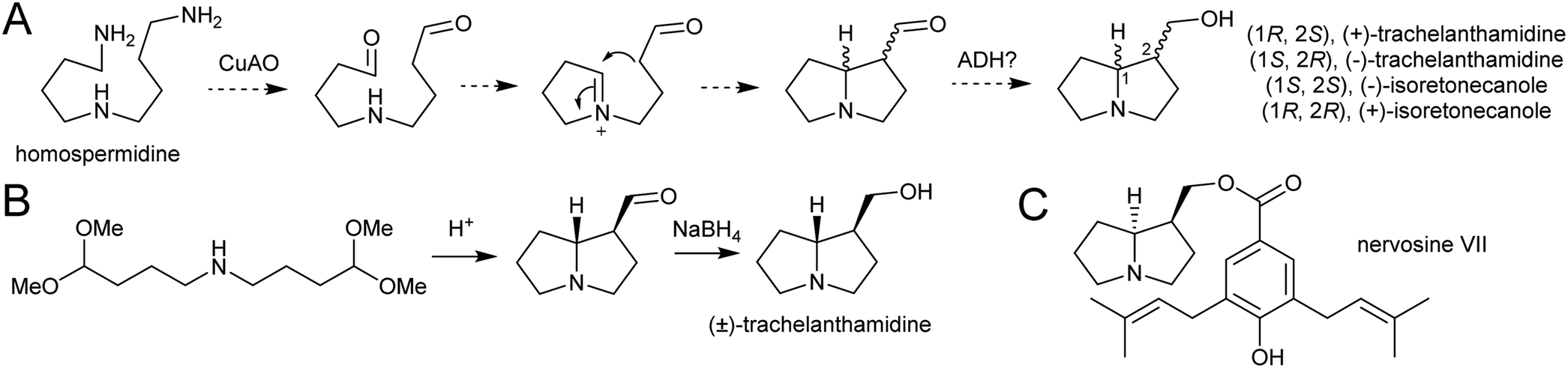 | ||
| Scheme 23 Pyrrolizidine alkaloid biosynthesis. (A) Proposed route from homospermidine to pyrrolizidine base precursor. (B) Biomimetic synthesis of trachelanthamidine from Takano et al. 1981.144 C. Structure of nervosine VII, a cis-pyrrolizidine. | ||
5.2. Aromatic amino-acid derived
Major classes of alkaloids generated from aromatic amines proceed via a Pictet–Spengler reaction. In this reaction, the intermolecular condensation of an amine and aldehyde forms an iminium, which is then quenched through intramolecular electrophilic aromatic substitution.166,167 The tethered nucleophilic aromatic system bypasses the requirement for accumulation of a distinct nucleophile. Mechanistic and biocatalytic aspects of Pictet–Spenglerases have been reviewed recently.10 The entry to the tyramine derived Amaryllidaceae alkaloids does not involve a Pictet–Spengler reaction; instead, the Mannich-like step is a reduction, in which the hydride can be considered an intermolecular nucleophile.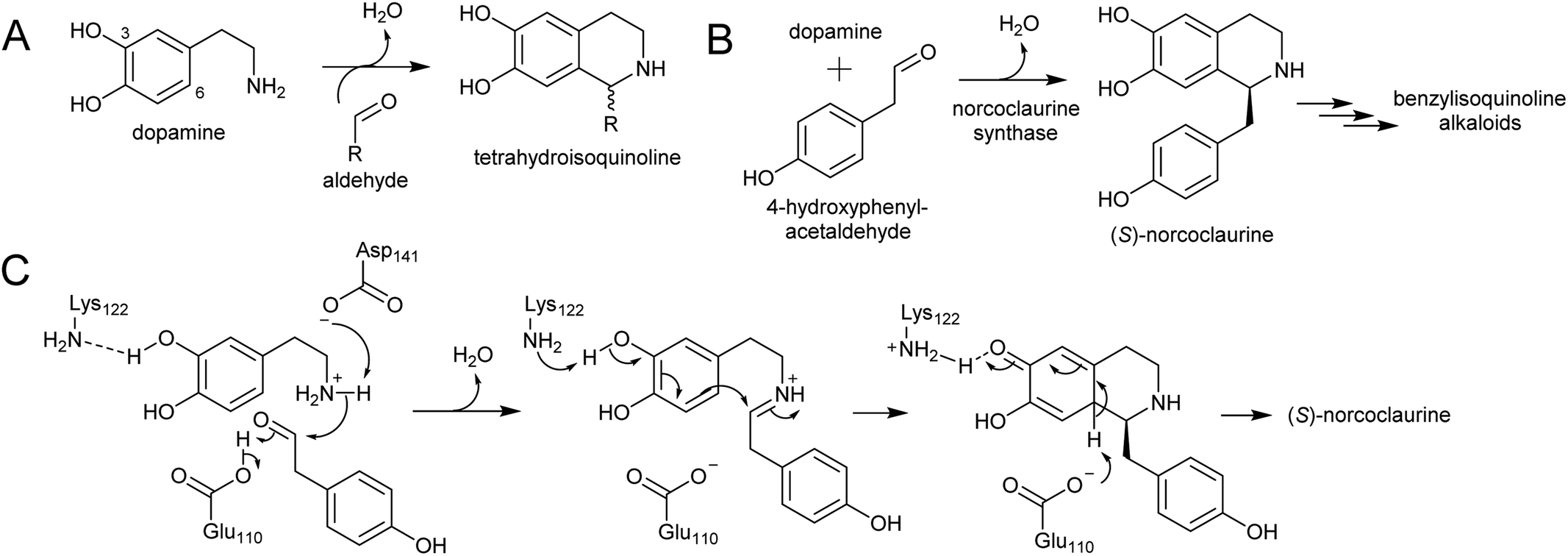 | ||
| Scheme 24 Formation of tetrahydroisoquinolines. (A) General formation of tetrahydroisoquinolines from dopamine and an aldehyde. (B) Reaction catalysed by norcoclaurine synthase. (C) Mechanism of norcoclaurine synathase. | ||
In benzylisoquinoline alkaloid biosynthesis, norcoclaurine synthase (NCS) catalyses the Pictet–Spengler condensation between dopamine and 4-HPAA, yielding (S)-norcoclaurine (Scheme 24B).170–173 NCS is a member of the pathogenesis-related 10/Bet-v1 (PR10) protein family, a diverse set of small (15–25 kDa), soluble proteins notable for their ability to bind diverse ligands.172,174 NCSs from Ranunculaceae (e.g. Thalictrum flavum) are single domain proteins, but NCS from the Papaveraceae, which includes opium poppy (P. somniferum), are present as fused repeats, with up to four consecutive domains on the same polypeptide chain.175 Recently, two more PR10s involved in BIA biosynthesis have been identified: thebaine synthase176 and neopinone isomerase.177 Outside BIA metabolism, PR10s have roles in defence responses178 and binding intermediates in secondary metabolism pathways.179,180
NCS contains an N-terminal signal peptide, which targets the enzyme to the vacuole via the endoplasmic reticulum.172 As previously described, the vacuole may be a site with high dopamine concentrations.75 However, it has been proposed that the primary site of NCS activity is en route to the vacuole, in the lumen of the endoplasmic reticulum, possibly in alkaloid specific vesicles.23,181 The details of subcellular trafficking in BIA biosynthesis including the primary location of NCS activity has not yet been resolved.
The mechanism of NCS (Scheme 24C) has been elucidated through structural182,183 and computational analyses.147 Dopamine binds in the enzyme active site prior to 4-HPAA, with the catechol 3-OH H-bonding to the Lys-122 residue deep in the active site and the residues Glu-110 and Asp-141 interact with the dopamine amine group.148 Iminium formation probably involves acid/base catalysis from Glu-110 and Asp-141, and may also involve changes to the enzyme structure.183 The key steps of C–C bond formation and subsequent loss of proton are catalysed by Lys-122 and Glu-110 respectively.
To date, all characterised NCS enzymes are from Ranunculales. The control of the Pictet–Spengler reaction in BIA biosynthesis outside this order, such as in sacred lotus (Nelumbo nucifera, Proteales) or the magnoliids, is unknown. NCS activity was found to be widely distributed in plant extracts from across angiosperms, including those not producing BIAs, but the proteins responsible for this were not identified.171 A number of NCS homologs are present in the sacred lotus genome and some feature some key NCS catalytic residues; however, these have not been characterised.184 Phylogenetic analysis of NCSs across published genomes place these lotus sequences away from the NCS clade indicating an independent origin of BIAs in lotus.185 Sacred lotus produces BIAs with both (S)- and (R)-stereoisomers,186 suggesting that: (i) both (S)- and (R)-selective NCSs are present, (ii) there is an epimerisation step in the pathway or (iii) the Pictet–Spengler reaction is not enzyme catalysed.
The species Alangium salviifolium (syn. A. lanmarckii, Cornaceae) and Carapichea ipecacuanha (syn. Psychotria ipecacuanha, Cephaelis ipecacuanha, Rubiaceae) produce alkaloids derived from the Pictet–Spengler condensation of dopamine and secologanin (Scheme 25A). Despite similarities in the biosynthetic pathways, the species' evolutionary distance suggests they were acquired independently. A. salviifolium cell-free extract catalyses the condensation of dopamine and secologanin into both epimer products: (1R)-deacetylipecoside and (1S)-deacetylisoipecoside.187 An enzyme with stereoselective (1R)-deacetylipecoside synthase activity was partially purified from A. salviifolium, though no sequence information was determined.129 In alkaloid biosynthesis in ipecac (C. ipecacuanha), both (1R)-deacetylipecoside and (1S)-deacetylisoipecoside are intermediates with separate metabolic fates, though enzymes catalysing their formation are not known.127,188 The ipecac alkaloid emetine is formed through a second Pictet–Spengler condensation.
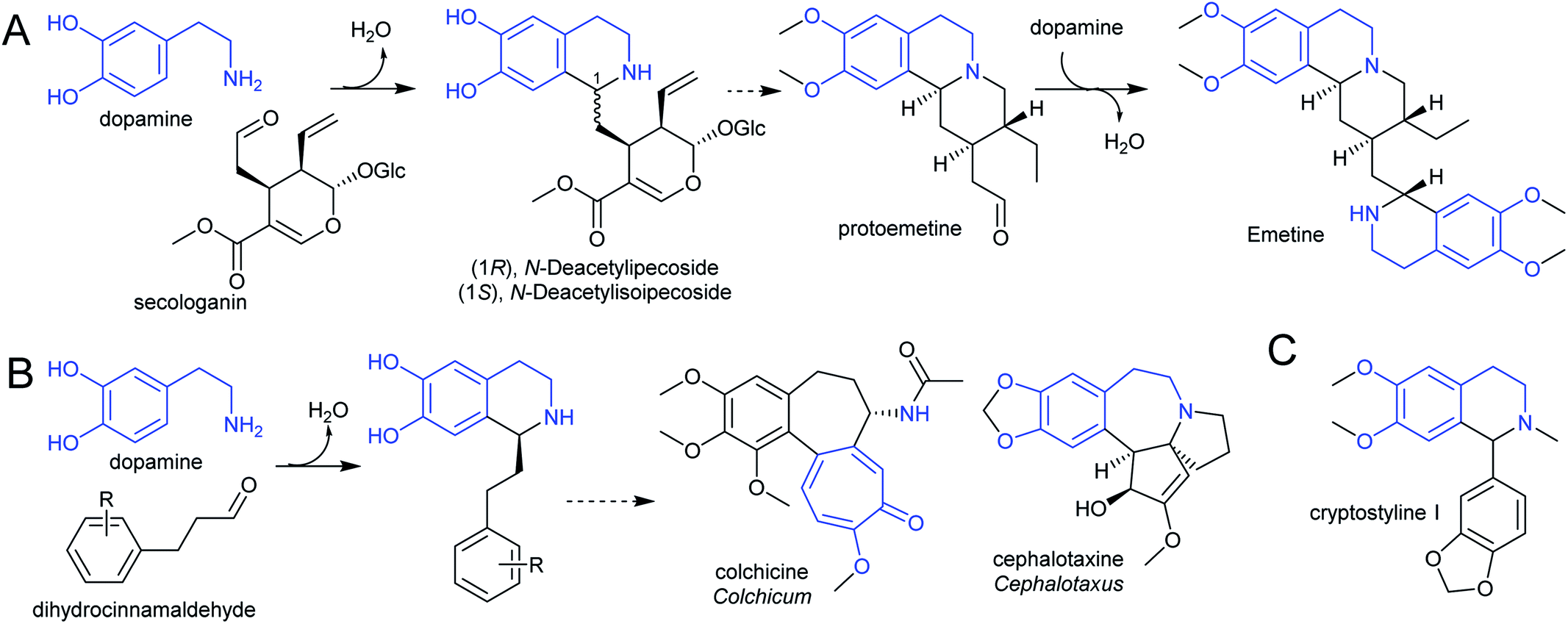 | ||
| Scheme 25 Alkaloids derived from dopamine. (A) Alkaloids derived from dopamine and secologanin. Emetine is found in C. ipecacuanha and is derived from a second Pictet–Spengler reaction. (B) Alkaloids derived from dopamine and dihydrocinnamaldehyde. (C) Alkaloids derived from dopamine and substituted benzaldehyde. | ||
There are multiple examples of alkaloids derived from dopamine and dihydrocinnamaldehyde derivatives, such as those found in the Colchicaceae (e.g. Colchicum, Schelhammera) and Cephalotaxus (Scheme 25B).20,119 Cryptostylines (Cryptostylis) are products of Pictet–Spengler condensation of dopamine and benzaldehydes (Scheme 25C). To date, no enzymes have been reported catalysing the Pictet–Spengler step in these pathways.
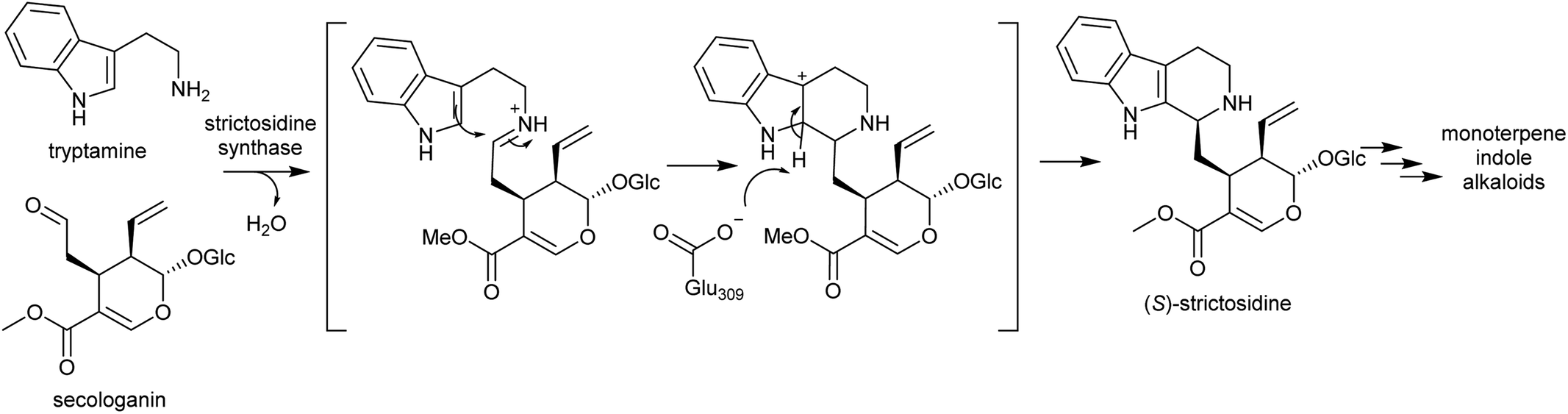 | ||
| Scheme 26 Strictosidine synthase reaction. | ||
Strictosidine synthases are part of the nucleophilic attack six-bladed β-propeller (N6P) superfamily, which includes enzymes such as paraoxonases and lactonohydrolases, and likely evolved from metal dependent enzymes.190,191 Strictosidine synthase-like (SSL) proteins are ubiquitous in plants, and although their activities are largely unknown, they appear to have diverse functions including in anther development192 and defence response.193
In plants, SS is localised to the vacuole of laticifer cells.194,195 However, the enzymes involved in the formation of its substrates, tryptophan decarboxylase and secologanin synthase, are in the cytoplasm and endoplasmic reticulum respectively. Strictosidine glucosidase, the enzyme that converts strictosidine into reactive aldehyde intermediates, is localised to the nucleus.196 Therefore, transportation of substrates and products are necessary. The efflux of strictosidine across the vacuolar membrane (tonoplast) is controlled by CrNPF2.9, a member of the nitrate/peptide transporter family (NPF).197 The transporters involved in the vacuolar import of tryptamine and secologanin are unknown.
Strictosidine synthase (SS) has so far only been characterised from Gentianales. In MIA biosynthesis in Camptotheca acuminata (Cornales), the precursor to MIAs is 3-(S)-strictosidinic acid,133 but a putative strictosidinic acid synthase has not been identified, and there is no clear ortholog to SS in the transcriptome or genome.198 Based on the substrates employed, it is possible that the MIA pathways in Camptotheca and the Gentianales are convergent, and if so, the Pictet–Spenglerase is unlikely to be orthologous to SS. In contrast, MIA biosynthesis in Nothapodytes nimmoniana (Icaninales) and Gentianales are likely to share a common origin,136 and accordingly a putative strictosidine synthase homologous to those in Gentianales has been identified in its transcriptome.199
The Harmala alkaloids (Peganum harmala) may be derived from a Pictet–Spengler condensation of a tryptophan-derived amine with pyruvate. However, few details of the biosynthesis have been elucidated.200
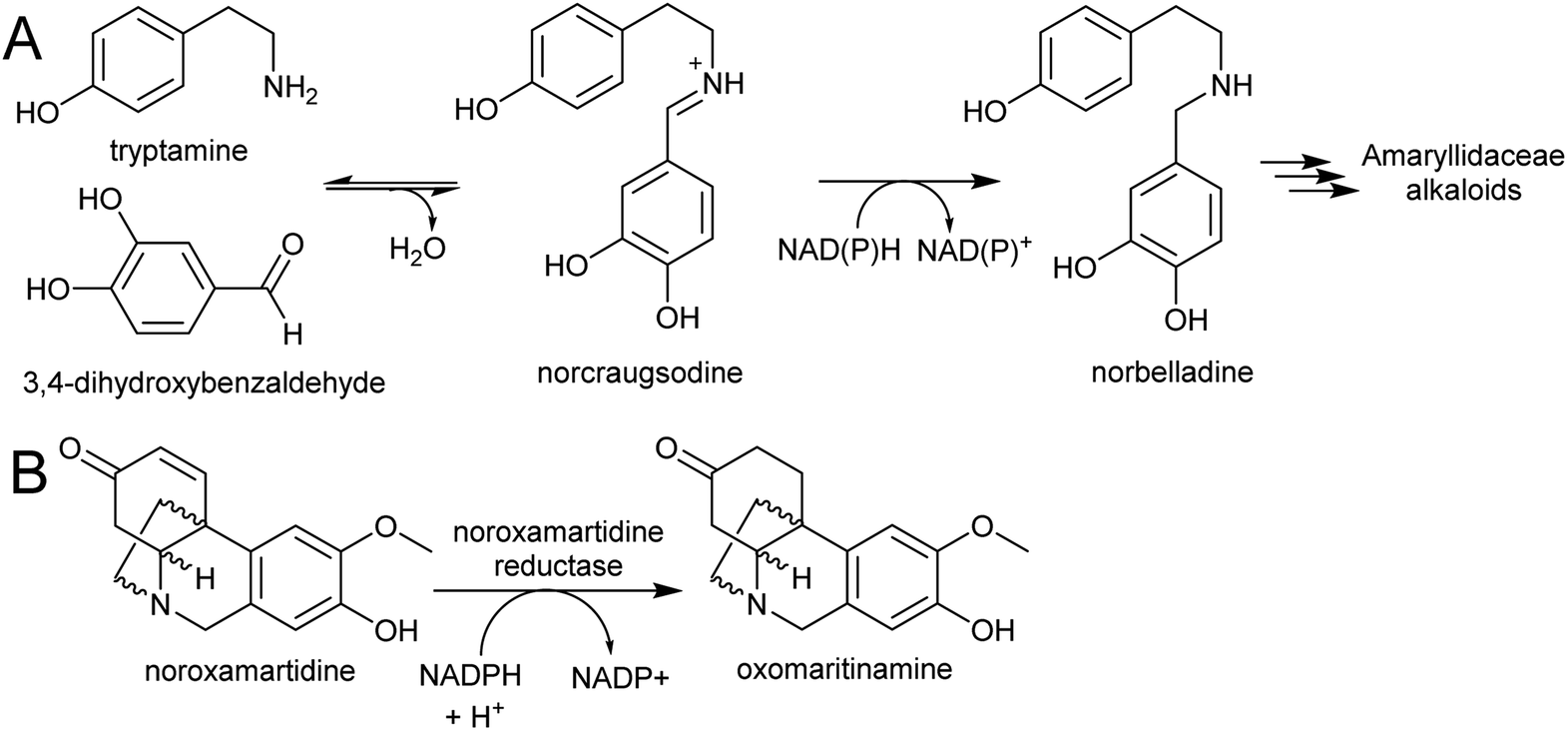 | ||
| Scheme 27 Amaryllidaceae alkaloid biosynthesis. (A) Entry point into the Amaryllidaceae alkaloid biosynthesis by condensation and reduction. (B) Reaction catalysed by noroxamartidine reductase. | ||
An NADPH-dependent short chain reductase from N. pseudonarcissus capable of catalysing the formation of norbelladine from trypamine and 3,4-dihydroxbenzaldehyde has been reported.201 However, this enzyme was characterised as noroxomaritidine reductase, as this activity was 400× faster than the nobelladine synthase activity (Scheme 27B). The low norbelladine synthase activity indicates this is a side reaction and unlikely to be relevant in planta. However, the result shows in principle that norbelladine can be formed by a reductase incubated with tyramine and 3,4-dihydroxybenzaldehyde.
Surprisingly, a homolog of norcoclaurine synthase (NCS), has been proposed to catalyse the formation of norbelladine from tyramine and 3,4-dihydrobenzaldehyde.202 Whilst it is conceivable that an NCS-type enzyme could aid in iminium formation, it is unlikely to catalyse the subsequent reduction step. Therefore, the key step in AA biosynthesis, the formation of norbelladine, remains unresolved. There are now plenty of transcriptomic resources for AA biosynthesis across multiple species, and these will surely lead to the discovery of new enzyme activities in the near future.203–205
6. Discussion
Numerous alkaloid biosynthetic pathways exist in plants, and many of these follow the pattern described in this review: amine and aldehyde accumulate and condense into an iminium, which is quenched by a nucleophile in a Mannich-like scaffold-forming reaction. This model of alkaloid biosynthesis highlights how plants exploits simple chemical logic to construct complex molecules.18 It also enables us to gain insight into how these pathways may have evolved.6.1. Precursor accumulation
The key requirement for alkaloid biosynthesis is the accumulation, or increased production, of precursors in a location specific manner. Amine accumulation occurs due to changes in amino acid or polyamine metabolism. This can be triggered by gene duplication leading to neofunctionalisation through loss of feedback inhibition,84 shifts in substrate specificity61 or changes to regulation.206 Complex alkaloids require a second molecule to accumulate. In the case of aromatic amino acid derived alkaloids, this is an aldehyde, whereas for polyamine-derived alkaloids, the accumulating compound is a nucleophile. Aldehydes may be generated directly from accumulating amino acid precursors (e.g. betalamic acid, 4-HPAA) or may be co-opted from existing secondary metabolism (e.g. secologanin). Nucleophiles in polyamine-derived alkaloid biosynthesis have a diverse origin, in secondary metabolism (e.g. polyketides) or from duplicated pathways (e.g. nicotinic acid).6.2. Subcellular localisation
To form a specific alkaloid scaffold, the two reacting species must accumulate in the same subcellular compartment. Whist this compartment may differ between alkaloid pathways, possible locations include the peroxisome or vacuole. Accumulating polyamines are oxidised into amino aldehydes by promiscuous peroxisomal CuAOs and form cyclic iminiums. If a nucleophile is transported to or synthesised in the peroxisome they could react with the cyclic iminiums prior to export to the cytoplasm.The vacuole seems a likely location for alkaloid biosynthesis: accumulating compounds may be sequestered in the vacuole to prevent cellular damage. Aldehydes for example, are cytotoxic due to their ability to cross-link DNA or proteins. Alkaloid precursors and products have been measured at high concentration in the vacuole.75,207 Furthermore, the Pictet–Spengler reaction in MIA biosynthesis occurs in the vacuole197 and other enzymes involved in key scaffold forming steps in alkaloid biosynthesis have vacuolar targeting sequences (e.g. NCS, BBL).160,172 Reactive specialised metabolites are often sequestered in the vacuole and its acidic conditions may promote spontaneous reactions.207,208 As the pathway matures and comes under greater regulation,206 the reaction may migrate to a different subcellular location.
6.3. Spontaneous reactions
In a cellular environment, two molecules at high concentration with complementary reactivity may combine spontaneously, without enzyme catalysis. Spontaneous reactions can lead to a leap in chemical complexity, as they are not reliant on existing enzyme mechanisms or substrate constraints. Therefore, new chemical scaffolds can be formed without the evolution of new enzyme activities.Recently the Mannich-like reaction from tropane alkaloid biosynthesis has been shown to occur without enzyme catalysis.153 In other polyamine-derived alkaloids, evidence for spontaneous quenching can be found in the presence of multiple stereoisomers in planta162 or in biomimetic reactions.143,144,164 Furthermore, this non-catalysed reaction can account for the modularity of nucleophile and electrophile. An exemplar of the modularity is tropane and nicotine alkaloid biosynthesis in the Solanaceae: both require N-methylpyrollinium but utilise nucleophiles of different origins. Notably, the gene duplications required to cause accumulation of N-methylpyrollinium appeared prior to the ancestor of the Solanaceae, whilst the genes required for the formation of nicotinic acid-like nucleophile only arose in the Nicotiana genus. Therefore, specific taxa have evolved different ‘solutions’ to an abundance of N-methylpyrollinium by accumulating different nucleophiles that react with the iminium and form bioactive compounds. Similarly, different electrophiles can also react with the same nucleophile, such as the homologous tropane and granatane alkaloids derived from 3-oxoglutaric acid plus N-methylpyrollinium or Δ1-piperideine respectively.
Iminium formation in betalain biosynthesis is spontaneous and does not involve enzyme catalysis.150 The Pictet–Spengler reactions of MIA and BIA are enzyme catalysed, but they have a low activation barrier and can occur in relatively mild aqueous conditions.72,209 With sufficient concentration of precursors, the Pictet–Spengler reaction can occur within a cell without a specific catalyst.169 Therefore, it is conceivable that the Pictet–Spengler step emerged prior to the origins of a corresponding Pictet–Spenglerase.
6.4. Enzyme evolution
If the new compound formed from a spontaneous reaction confers an advantage—through the development of new bioactivities or reduction in cellular toxicity—an enzyme may be recruited to enhance its reaction rate or provide stereoselectivity. Therefore, a reaction that occurs spontaneously and rapidly in vitro may still have an associated enzyme in vivo. This has been highlighted recently with the identification of enzymes for steps in BIA biosynthesis previously thought to be spontaneous.176,177Enzymes that have evolved to catalyse a “spontaneous” reaction are somewhat unusual in that they often evolve from protein families with no obvious connection to the reaction or pathway (e.g. NCS, SS). In structurally related alkaloid pathways with independent origins, enzymes catalysing similar “spontaneous” steps would not be orthologous and could have evolved from a variety of protein starting points: they may not be easy to identify through homology. In this sense these enzymes have similarities to the [4 + 2]-cyclases, which are from diverse protein families.210
It is possible that proteins have evolved to influence the stereochemical course of the reaction without necessarily catalysing it. Such scaffolding proteins are sometimes referred to as dirigent proteins, and have been identified in lignin211 and iridoid biosynthesis.212 Although none has been identified in alkaloid biosynthesis to date, they could account for the enantiomeric enrichment observed in polyamine-derived alkaloids.
6.5. Pathway evolution
This model of alkaloid biosynthesis suggests that the key factor in the evolution of new alkaloids are modifications to existing primary and secondary metabolism, and not the emergence of a new enzyme activity. Scaffold-forming enzymes have been considered crucial for catalysing the first-committed step into alkaloid pathways. However, at least in the early stages of alkaloid pathway evolution, such steps may occur without enzyme catalysis.This fits with a general model of metabolite-enzyme coevolution where enzymes catalysing rate-limiting steps are the first to be recruited into a new pathway.213 When the scaffold-forming step has a low energy barrier, it will not be rate-limiting. Instead, the upstream steps that determine the concentration of precursors will limit flux, and enzymes boosting these will emerge first. Even some downstream steps with high energy barriers—such as those catalysed by methyltransferases, cytochrome P450 and dehydrogenases—may be established before an enzyme catalysing the scaffold-forming step emerges. A combination of phylogenetics and biochemistry may be able to interrogate the relative timings of enzyme evolution in a pathway.
7. Conclusion
Methods for the discovery and characterisation of enzymes from plant specialised metabolism have reached maturity,4 and we will continue to see rapid progress in the elucidation of alkaloid biosynthesis.214 These discoveries will feed into synthetic biology and metabolic engineering, and consequently, the yields and variety of alkaloids available from heterologous production methods will increase.12,215 Phylogenomic approaches are beginning to reveal the evolutionary origins of specialised metabolism,216 and with ever increasing sequence data, these can be applied to alkaloid producing plants. Furthermore, investigations into aspects of alkaloid biosynthesis such as how flux is channelled from primary metabolism,39 how promoters have evolved206 and how pathways are compartmentalised197,207 will help reveal how alkaloid biosynthesis is integrated into a complete cellular context.Alkaloids have fascinated scientists for centuries due to their structural intricacies and profound bioactivities. Despite these complexities, alkaloid formation is largely governed by a simple and modular chemical logic, centred around iminium reactivity (e.g.Scheme 28).18,164,217 The ability of nature to generate functional and diverse molecules from simple physical principles will provide inspiration for chemists and biologists for many decades to come.
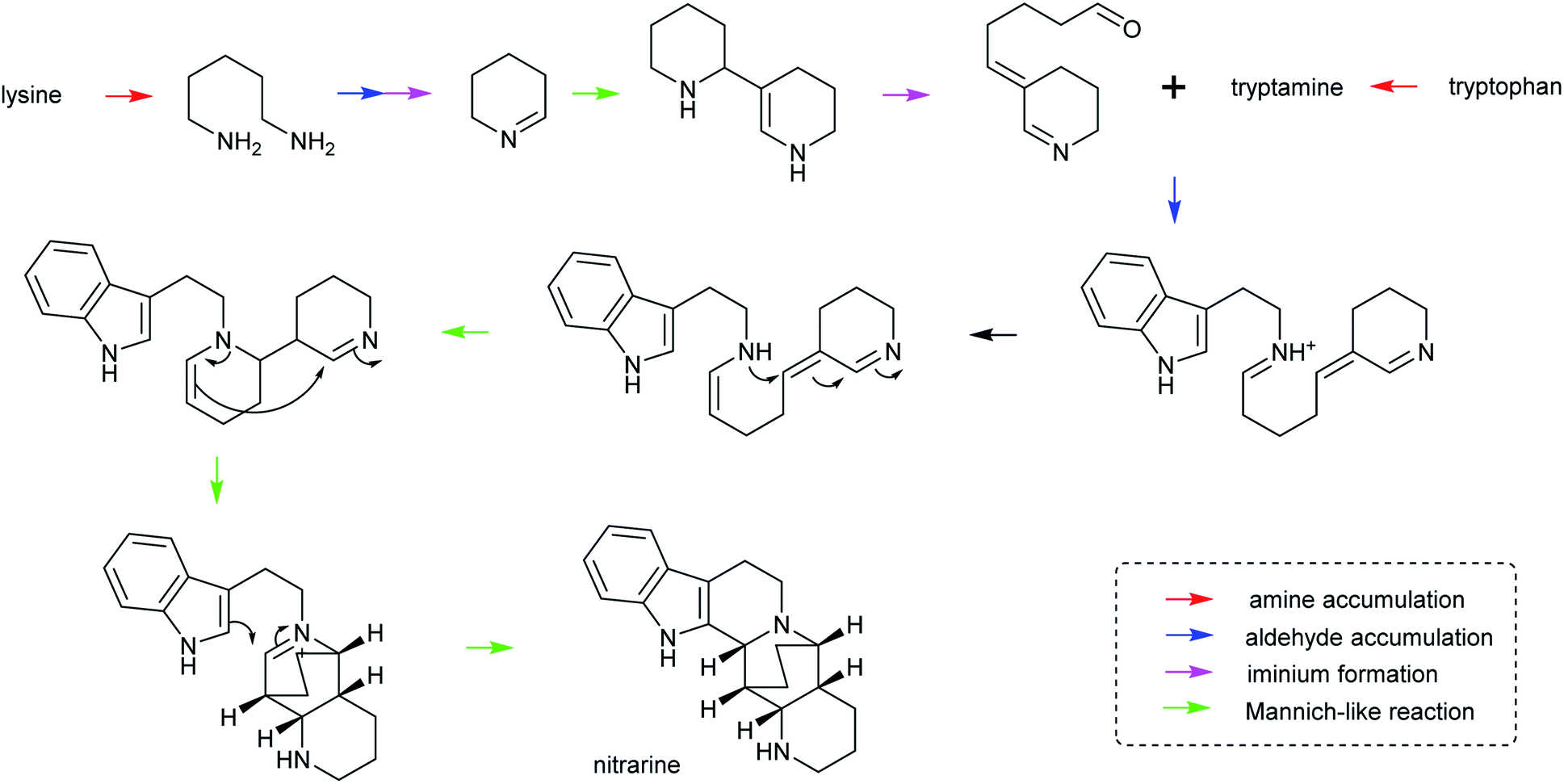 | ||
| Scheme 28 Hypothetical biosynthesis of nitrarine from Nitraria. This route highlights how the themes of amine/aldehyde accumulation, iminium formation and Mannich-like reactions are modular and lead to chemical complexity.164,217 | ||
8. Conflicts of interest
There are no conflicts to declare.9. Acknowledgements
The author acknowledges support from a UKRI Future Leaders Fellowship (MR/S01862X/1).10. References
- K. Fester, in Encyclopedia of Life Sciences (ELS), 2010 Search PubMed.
- B. Debnath, W. S. Singh, M. Das, S. Goswami, M. K. Singh, D. Maiti and K. Manna, Mater. Today Chem., 2018, 9, 56–72 CrossRef CAS.
- V. Amirkia and M. Heinrich, Phytochem. Lett., 2014, 10, 48–53 CrossRef.
- T. Dugé de Bernonville, N. Papon, M. Clastre, S. E. O'Connor and V. Courdavault, Trends Pharmacol. Sci., 2020, 41, 142–146 CrossRef.
- B. R. Lichman, G. T. Godden and C. R. Buell, Curr. Opin. Plant Biol., 2020, 55, 74–83 CrossRef CAS.
- T. Winzer, V. Gazda, Z. He, F. Kaminski, M. Kern, T. R. Larson, Y. Li, F. Meade, R. Teodor, F. E. Vaistij, C. Walker, T. A. Bowser and I. A. Graham, Science, 2012, 336, 1704–1708 CrossRef CAS.
- H. W. Nützmann, A. Huang and A. Osbourn, New Phytol., 2016, 211, 771–789 CrossRef.
- S. A. Kautsar, H. G. Suarez Duran, K. Blin, A. Osbourn and M. H. Medema, Nucleic Acids Res., 2017, 45, W55–W63 CrossRef CAS.
- H. Kries and S. E. O'Connor, Curr. Opin. Chem. Biol., 2016, 31, 22–30 CrossRef CAS.
- R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol., 2020, 55, 69–76 CrossRef CAS.
- S. Brown, M. Clastre, V. Courdavault and S. E. O'Connor, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3205–3210 CrossRef CAS.
- S. Galanie, K. Thodey, I. J. Trenchard, M. F. Interrante and C. D. Smolke, Science, 2015, 349, 1095–1100 CrossRef CAS.
- S. Li, Y. Li and C. D. Smolke, Nat. Chem., 2018, 10, 395–404 CrossRef CAS.
- K. M. Frick, L. G. Kamphuis, K. H. M. Siddique, K. B. Singh and R. C. Foley, Front. Plant Sci., 2017, 8, 87 Search PubMed.
- S. Funayama and G. A. Cordell, Alkaloids, Elsevier, 2015 Search PubMed.
- M. Wink, Alkaloids, 1993, 43, 1–118 CAS.
- M. F. Roberts, D. Strack and M. Wink, Biosynthesis of Alkaloids and Betalains, 2010, vol. 40 Search PubMed.
- G. Anarat-Cappillino and E. S. Sattely, Curr. Opin. Plant Biol., 2014, 19, 51–58 CrossRef CAS.
- B. Daniel, B. Konrad, M. Toplak, M. Lahham, J. Messenlehner, A. Winkler and P. Macheroux, Arch. Biochem. Biophys., 2017, 632, 88–103 CrossRef CAS.
- S. Larsson and N. Rønsted, Curr. Top. Med. Chem., 2014, 14, 274–289 CrossRef CAS.
- S. Bunsupa, M. Yamazaki and K. Saito, Mini-Rev. Med. Chem., 2017, 17, 1002–1012 CrossRef CAS.
- N. Kim, O. Estrada, B. Chavez, C. Stewart and J. C. D'Auria, Molecules, 2016, 21, 1–25 Search PubMed.
- A. Singh, I. M. Menéndez-Perdomo and P. J. Facchini, Phytochem. Rev., 2019, 240, 19–32 Search PubMed.
- J. M. Hagel and P. J. Facchini, Plant Cell Physiol., 2013, 54, 647–672 CrossRef CAS.
- Z. Jin and G. Yao, Nat. Prod. Rep., 2019, 36, 1462–1488 RSC.
- T. Hotchandani and I. Desgagné-Penix, Curr. Top. Med. Chem., 2017, 17, 418–427 CrossRef CAS.
- M. B. Kilgore and T. M. Kutchan, Phytochem. Rev., 2016, 15, 317–337 CrossRef CAS.
- S. Schramm, N. Köhler and W. Rozhon, Molecules, 2019, 24, 498 CrossRef.
- K. L. Kohnen-Johannsen and O. Kayser, Molecules, 2019, 24, 796 CrossRef.
- G. Polturak and A. Aharoni, Molecular Plant, 2018, 11, 7–22 CrossRef CAS.
- A. Timoneda, T. Feng, H. Sheehan, N. Walker-Hale, B. Pucker, S. Lopez-Nieves, R. Guo and S. Brockington, New Phytol., 2019, 224, 71–85 CrossRef.
- J. P. Michael, in The Alkaloids: Chemistry and Biology, Elsevier, 2016, vol. 75 Search PubMed.
- F. F. Zenkner, M. Margis-Pinheiro and A. Cagliari, Tobacco Science, 2019, 56, 1–9 CrossRef.
- V. De Luca, V. Salim, A. Thamm, S. A. Masada and F. Yu, Curr. Opin. Plant Biol., 2014, 19, 35–42 CrossRef CAS.
- D. Chen, Q. Shao, L. Yin, A. Younis and B. Zheng, Front. Plant Sci., 2019, 9, 1945 CrossRef.
- L. A. E. Erland, C. E. Turi and P. K. Saxena, in Serotonin: The Mediator that Spans Evolution, Elsevier Inc., 2018, pp. 23–46 Search PubMed.
- J. Graça, Front. Chem., 2015, 3, 62 Search PubMed.
- S. Ishiai, H. Kondo, T. Hattori, M. Mikami, Y. Aoki, S. Enoki and S. Suzuki, Physiol. Mol. Plant Pathol., 2016, 96, 94–100 CrossRef CAS.
- H. A. Maeda, Front. Plant Sci., 2019, 10, 881 CrossRef.
- G. D. Moghe and R. L. Last, Plant Physiol., 2015, 169, 1512–1523 CAS.
- K. Urano, T. Hobo and K. Shinozaki, FEBS Lett., 2005, 579, 1557–1564 CrossRef CAS.
- D. Ober and T. Hartmann, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14777–14782 CrossRef CAS.
- A. J. Michael, Biochem. J., 2016, 473, 2315–2329 CrossRef CAS.
- A. L. Jancewicz, N. M. Gibbs and P. H. Masson, Front. Plant Sci., 2016, 7, 870 Search PubMed.
- Y. S. Lee and Y. D. Cho, Biochem. J., 2001, 360, 657–665 CrossRef CAS.
- T. Zhao, C. Wang, F. Bai, S. Li, C. Yang, F. Zhang, G. Bai, M. Chen, X. Lan and Z. Liao, Front. Plant Sci., 2019, 10, 229 CrossRef.
- T. Docimo, M. Reichelt, B. Schneider, M. Kai, G. Kunert, J. Gershenzon and J. C. D'Auria, Plant Mol. Biol., 2012, 78, 599–615 CrossRef CAS.
- C. Hanfrey, S. Sommer, M. J. Mayer, D. Burtin and A. J. Michael, Plant J., 2001, 27, 551–560 CrossRef CAS.
- C. Illingworth, M. J. Mayer, K. Elliott, C. Hanfrey, N. J. Walton and A. J. Michael, FEBS Lett., 2003, 549, 26–30 CrossRef CAS.
- A. J. Michael, Biochem. J., 2017, 474, 2277–2299 CrossRef CAS.
- H. L. Dalton, C. K. Blomstedt, A. D. Neale, R. Gleadow, K. D. Deboer and J. D. Hamill, J. Exp. Bot., 2016, 67, 3367–3381 CrossRef CAS.
- T. Zhao, S. Li, J. Wang, Q. Zhou, C. Yang, F. Bai, X. Lan, M. Chen and Z. Liao, ACS Synth. Biol., 2020, 9, 437–448 CrossRef CAS.
- S. Xu, T. Brockmöller, A. Navarro-Quezada, H. Kuhl, K. Gase, Z. Ling, W. Zhou, C. Kreitzer, M. Stanke, H. Tang, E. Lyons, P. Pandey, S. P. Pandey, B. Timmermann, E. Gaquerel and I. T. Baldwin, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6133–6138 CrossRef CAS.
- S. Bunsupa, K. Katayama, E. Ikeura, A. Oikawa, K. Toyooka, K. Saito and M. Yamazakia, Plant Cell, 2012, 24, 1202–1216 CrossRef CAS.
- S. Bunsupa, K. Hanada, A. Maruyama, K. Aoyagi, K. Komatsu, H. Ueno, M. Yamashita, R. Sasaki, A. Oikawa, K. Saito and M. Yamazaki, Plant Physiol., 2016, 171, 2432–2444 CrossRef CAS.
- B. Xu, L. Lei, X. Zhu, Y. Zhou and Y. Xiao, Phytochemistry, 2017, 136, 23–30 CrossRef CAS.
- M. Yang, W. You, S. Wu, Z. Fan, B. Xu, M. Zhu, X. Li and Y. Xiao, BMC Genomics, 2017, 18, 245 CrossRef.
- A. Reimann, N. Nurhayati, A. Backenköhler and D. Ober, Plant Cell, 2004, 16, 2772–2784 CrossRef CAS.
- A. Imai, T. Matsuyama, Y. Hanzawa, T. Akiyama, M. Tamaoki, H. Saji, Y. Shirano, T. Kato, H. Hayashi, D. Shibata, S. Tabata, Y. Komeda and T. Takahashi, Plant Physiol., 2004, 135, 1565–1573 CrossRef CAS.
- D. Ober and T. Hartmann, Plant Mol. Biol., 2000, 44, 445–450 CrossRef CAS.
- E. Kaltenegger, E. Eich and D. Ober, Plant Cell, 2013, 25, 1213–1227 CrossRef CAS.
- T. Livshultz, E. Kaltenegger, S. C. K. Straub, K. Weitemier, E. Hirsch, K. Koval, L. Mema and A. Liston, New Phytol., 2018, 218, 762–773 CrossRef CAS.
- M. P. Torrens-Spence, T. Pluskal, F. S. Li, V. Carballo and J. K. Weng, Molecular Plant, 2018, 11, 205–217 CrossRef CAS.
- V. De Luca, C. Marineau and N. Brisson, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 2582–2586 CrossRef CAS.
- P. J. Facchini, K. L. Huber-Allanach and L. W. Tari, Phytochemistry, 2000, 54, 121–138 CrossRef CAS.
- M. P. Torrens-Spence, Y. Chiang, T. Smith, M. A. Vicent, Y. Wang and J.-K. Weng, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 10806–10817 CrossRef CAS.
- M. P. Torrens-Spence, M. Lazear, R. Von Guggenberg, H. Ding and J. Li, Phytochemistry, 2014, 106, 37–43 CrossRef CAS.
- R. Wang, X. Han, S. Xu, B. Xia, Y. Jiang, Y. Xue and R. Wang, PeerJ, 2019, 7, e6729 CrossRef.
- P. J. Facchini and V. De Luca, Plant Cell, 1995, 7, 1811–1821 CrossRef CAS.
- P. J. Facchini and V. De Luca, Phytochemistry, 1995, 38, 1119–1126 CrossRef CAS.
- H. Wang, J. Yu, Y. Satoh, Y. Nakagawa, R. Tanaka, K. Kato and M. Yao, Biochem. Biophys. Res. Commun., 2020, 523, 500–505 CrossRef CAS.
- T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Chem. Commun., 2011, 47, 3242–3244 RSC.
- T. Pesnot, M. C. Gershater, J. M. Ward and H. C. Hailes, Adv. Synth. Catal., 2012, 354, 2997–3008 CrossRef CAS.
- M. F. Roberts, D. Mccarthy, T. M. Kutchan and C. J. Cosciat, Arch. Biochem. Biophys., 1983, 222, 599–609 CrossRef CAS.
- T. M. Kutchan, M. Rush and C. J. Coscia, Plant Physiol., 1986, 81, 161–166 CrossRef CAS.
- J. M. Hagel, A. M. Weljie, H. J. Vogel and P. J. Facchini, Plant Physiol., 2008, 147, 1805–1821 CrossRef CAS.
- W. C. DeLoache, Z. N. Russ, L. Narcross, A. M. Gonzales, V. J. J. Martin and J. E. Dueber, Nat. Chem. Biol., 2015, 11, 465–471 CrossRef CAS.
- S. F. Brockington, Y. Yang, F. Gandia-Herrero, S. Covshoff, J. M. Hibberd, R. F. Sage, G. K. S. Wong, M. J. Moore and S. A. Smith, New Phytol., 2015, 207, 1170–1180 CrossRef CAS.
- G. Polturak, D. Breitel, N. Grossman, A. Sarrion-Perdigones, E. Weithorn, M. Pliner, D. Orzaez, A. Granell, I. Rogachev and A. Aharoni, New Phytol., 2016, 210, 269–283 CrossRef CAS.
- R. Sunnadeniya, A. Bean, M. Brown, N. Akhavan, G. Hatlestad, A. Gonzalez, V. V. Symonds and A. Lloyd, PLoS One, 2016, 11, e0149417 CrossRef.
- G. J. Hatlestad, R. M. Sunnadeniya, N. A. Akhavan, A. Gonzalez, I. L. Goldman, J. M. McGrath and A. M. Lloyd, Nat. Genet., 2012, 44, 816–820 CrossRef CAS.
- G. Polturak, U. Heinig, N. Grossman, M. Battat, D. Leshkowitz, S. Malitsky, I. Rogachev and A. Aharoni, Molecular Plant, 2018, 11, 189–204 CrossRef CAS.
- G. Polturak, N. Grossman, D. Vela-Corcia, Y. Dong, A. Nudel, M. Pliner, M. Levy, I. Rogachev and A. Aharoni, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9062–9067 CrossRef CAS.
- S. Lopez-Nieves, Y. Yang, A. Timoneda, M. Wang, T. Feng, S. A. Smith, S. F. Brockington and H. A. Maeda, New Phytol., 2018, 217, 896–908 CrossRef CAS.
- H. Maeda and N. Dudareva, Annu. Rev. Plant Biol., 2012, 63, 73–105 CrossRef CAS.
- S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532–547 RSC.
- Y. Yamazaki, H. Sudo, M. Yamazaki, N. Aimi and K. Saito, Plant Cell Physiol., 2003, 44, 395–403 CrossRef CAS.
- W. Liu, R. Chen, M. Chen, H. Zhang, M. Peng, C. Yang, X. Ming, X. Lan and Z. Liao, Planta, 2012, 236, 239–250 CrossRef CAS.
- M. Lopez-Meyer and C. L. Nessler, Plant J., 1997, 11, 1167–1175 CrossRef CAS.
- B. Rohde, J. Hans, S. Martens, A. Baumert, P. Hunziker and U. Matern, Plant J., 2008, 53, 541–553 CrossRef CAS.
- J. Li and R. L. Last, Plant Physiol., 1996, 110, 51–59 CrossRef CAS.
- J. Bohlmann, Y. De Luca, U. Eilert and W. Martin, Plant J., 1995, 7, 491–501 CrossRef CAS.
- J. Bohlmann, T. Lins, W. Martin and U. Eilert, Plant Physiol., 1996, 111, 507–514 CrossRef CAS.
- W. Wang, K. Paschalidis, J. C. Feng, J. Song and J. H. Liu, Front. Plant Sci., 2019, 10, 561 CrossRef.
- M. Naconsie, K. Kato, T. Shoji and T. Hashimoto, Plant Cell Physiol., 2014, 55, 436–444 CrossRef CAS.
- S. Biastoff, W. Brandt and B. Dräger, Phytochemistry, 2009, 70, 1708–1718 CrossRef CAS.
- A. Katoh, T. Shoji and T. Hashimoto, Plant Cell Physiol., 2007, 48, 550–554 CrossRef CAS.
- W. G. Heim, K. A. Sykes, S. B. Hildreth, J. Sun, R. H. Lu and J. G. Jelesko, Phytochemistry, 2007, 68, 454–463 CrossRef CAS.
- J. Jirschitzka, G. W. Schmidt, M. Reichelt, B. Schneider, J. Gershenzon and J. C. D'Auria, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10304–10309 CrossRef CAS.
- E. G. Minguet, F. Vera-Sirera, A. Marina, J. Carbonell and M. A. Blázquez, Mol. Biol. Evol., 2008, 25, 2119–2128 CrossRef CAS.
- A. Junker, J. Fischer, Y. Sichhart, W. Brandt and B. Dräger, Front. Plant Sci., 2013, 4, 260 Search PubMed.
- B. J. Shelp, G. G. Bozzo, C. P. Trobacher, A. Zarei, K. L. Deyman and C. J. Brikis, Plant Science, 2012, 193–194, 130–135 CrossRef CAS.
- A. Zarei, C. P. Trobacher and B. J. Shelp, FEBS Lett., 2015, 589, 2695–2700 CrossRef CAS.
- J. Sun, H. Morita, G. Chen, H. Noguchi and I. Abe, Bioorg. Med. Chem. Lett., 2012, 22, 5784–5790 CrossRef CAS.
- T. Yang, I. Nagy, D. Mancinotti, S. L. Otterbach, T. B. Andersen, M. S. Motawia, T. Asp and F. Geu-Flores, J. Exp. Bot., 2017, 68, 5527–5537 CrossRef CAS.
- C. Frölich, D. Ober and T. Hartmann, Phytochemistry, 2007, 68, 1026–1037 CrossRef.
- X. Liu, Y. Liu, P. Huang, Y. Ma, Z. Qing, Q. Tang, H. Cao, P. Cheng, Y. Zheng, Z. Yuan, Y. Zhou, J. Liu, Z. Tang, Y. Zhuo, Y. Zhang, L. Yu, J. Huang, P. Yang, Q. Peng, J. Zhang, W. Jiang, Z. Zhang, K. Lin, D. K. Ro, X. Chen, X. Xiong, Y. Shang, S. Huang and J. Zeng, Molecular Plant, 2017, 10, 975–989 CrossRef CAS.
- E.-J. Lee and P. J. Facchini, Plant Physiol., 2011, 157, 1067–1078 CrossRef CAS.
- Y. Kaminaga, J. Schnepp, G. Peel, C. M. Kish, G. Ben-Nissan, D. Weiss, I. Orlova, O. Lavie, D. Rhodes, K. Wood, D. M. Porterfield, A. J. L. Cooper, J. V. Schloss, E. Pichersky, A. Vainstein and N. Dudareva, J. Biol. Chem., 2006, 281, 23357–23366 CrossRef CAS.
- L. Christinet, F. X. Burdet, M. Zaiko, U. Hinz and J. P. Zrÿd, Plant Physiol., 2004, 134, 265–274 CrossRef CAS.
- H. H. Chung, K. E. Schwinn, H. M. Ngo, D. H. Lewis, B. Massey, K. E. Calcott, R. Crowhurst, D. C. Joyce, K. S. Gould, K. M. Davies and D. K. Harrison, Front. Plant Sci., 2015, 6, 499 Search PubMed.
- F. Terradad and H. Wyler, Helv. Chim. Acta, 1991, 74, 124–140 CrossRef.
- A. Bean, R. Sunnadeniya, N. Akhavan, A. Campbell, M. Brown and A. Lloyd, New Phytol., 2018, 219, 287–296 CrossRef CAS.
- H. Sheehan, T. Feng, N. Walker-Hale, S. Lopez-Nieves, B. Pucker, R. Guo, W. C. Yim, R. Badgami, A. Timoneda, L. Zhao, H. Tiley, D. Copetti, M. J. Sanderson, J. C. Cushman, M. J. Moore, S. A. Smith and S. F. Brockington, New Phytol., 2020, 227, 914–929 CrossRef CAS.
- L. Yang, J. Zhu, C. Sun, Z. Deng and X. Qu, Chem. Sci., 2020, 11, 364–371 RSC.
- N. J. Gallage, E. H. Hansen, R. Kannangara, C. E. Olsen, M. S. Motawia, K. Jørgensen, I. Holme, K. Hebelstrup, M. Grisoni and B. L. Møller, Nat. Commun., 2014, 5, 4037 CrossRef CAS.
- R. J. Parry, M. N. T. Chang, J. M. Schwab and B. M. Foxmanle, J. Am. Chem. Soc., 1980, 102, 1099–1111 CrossRef CAS.
- A. Nasreen, H. Gundlach and M. H. Zenk, Phytochemistry, 1997, 46, 107–115 CrossRef CAS.
- H. Abdelkafi and B. Nay, Nat. Prod. Rep., 2012, 29, 845–869 RSC.
- M. Ibdah, A. Berim, S. Martens, A. L. H. Valderrama, L. Palmieri, E. Lewinsohn and D. R. Gang, Phytochemistry, 2014, 107, 24–31 CrossRef CAS.
- H. Kasahara, Y. Jiao, D. L. Bedgar, S. J. Kim, A. M. Patten, Z. Q. Xia, L. B. Davin and N. G. Lewis, Phytochemistry, 2006, 67, 1765–1780 CrossRef CAS.
- K. Miettinen, L. Dong, N. Navrot, T. Schneider, V. Burlat, J. Pollier, L. Woittiez, S. van der Krol, R. Lugan, T. Ilc, R. Verpoorte, K.-M. Oksman-Caldentey, E. Martinoia, H. Bouwmeester, A. Goossens, J. Memelink and D. Werck-Reichhart, Nat. Commun., 2014, 5, 3606 CrossRef.
- J. Gershenzon and T. J. Mabry, Nord. J. Bot., 1983, 3, 5–34 CrossRef CAS.
- S. Dobler, G. Petschenka and H. Pankoke, Phytochemistry, 2011, 72, 1593–1604 CrossRef CAS.
- I. F. Makarevich, S. N. Kovalenko, T. D. Gusarova and Y. I. Gubin, Chem. Nat. Compd., 2009, 45, 40–44 CrossRef CAS.
- S. R. Jensen, in Ecological Chemistry and Biochemistry of Plant Terpenoids, Clarendon Press Oxford, 1991, pp. 133–158 Search PubMed.
- T. Nomura and T. M. Kutchan, J. Biol. Chem., 2010, 285, 7722–7738 CrossRef CAS.
- T. Nomura and T. M. Kutchan, Plant Signaling Behav., 2010, 5, 875–877 CrossRef CAS.
- W. De-Eknamkul, N. Suttipanta and T. M. Kutchan, Phytochemistry, 2000, 55, 177–181 CrossRef CAS.
- S. Irmler, G. Schröder, B. St-Pierre, N. P. Crouch, M. Hotze, J. Schmidt, D. Strack, U. Matern and J. Schröder, Plant J., 2000, 24, 797–804 CrossRef CAS.
- W. Prall, O. Hendy and L. E. Thornton, PLoS One, 2016, 11, e0163024 CrossRef.
- T. Dugé de Bernonville, E. Foureau, C. Parage, A. Lanoue, M. Clastre, M. A. Londono, A. Oudin, B. Houillé, N. Papon, S. Besseau, G. Glévarec, L. Atehortùa, N. Giglioli-Guivarc’h, B. St-Pierre, V. De Luca, S. E. O'Connor and V. Courdavault, BMC Genomics, 2015, 16, 619 CrossRef.
- R. Sadre, M. Magallanes-Lundback, S. Pradhan, V. Salim, A. Mesberg, A. D. Jones and D. Dellapenna, Plant Cell, 2016, 28, 1926–1944 CrossRef CAS.
- Y. Yang, W. Li, J. Pang, L. Jiang, X. Qu, X. Pu, G. Zhang and Y. Luo, ACS Chem. Biol., 2019, 14, 1091–1096 CrossRef CAS.
- G. A. Rather, A. Sharma, S. A. Pandith, V. Kaul, U. Nandi, P. Misra and S. K. Lattoo, Plant Mol. Biol., 2018, 96, 197–215 CrossRef CAS.
- G. A. Rather, A. Sharma, P. Misra, A. Kumar, V. Kaul and S. K. Lattoo, Protoplasma, 2020, 257, 391–405 CrossRef.
- D. J. Hupe, in New Comprehensive Biochemistry, Elsevier Inc, 1984, vol. 6, pp. 271–301 Search PubMed.
- S. Eldin and W. P. Jencks, J. Am. Chem. Soc., 1995, 117, 4851–4857 CrossRef CAS.
- S. Eldin, J. A. Digits, S. T. Huang and W. P. Jencks, J. Am. Chem. Soc., 1995, 117, 6631–6632 CrossRef CAS.
- P. N. Scheller, M. Lenz, S. C. Hammer, B. Hauer and B. M. Nestl, ChemCatChem, 2015, 7, 3239–3242 CrossRef CAS.
- J. H. Atherton, J. Blacker, M. R. Crampton and C. Grosjean, Org. Biomol. Chem., 2004, 2, 2567–2571 RSC.
- G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan and N. J. Turner, Nat. Chem., 2017, 9, 961–969 CrossRef CAS.
- A. Rouchaud and J. C. Braekman, Eur. J. Org. Chem., 2009, 5, 2666–2674 CrossRef.
- S. Takano, N. Ogawa and K. Ogasawara, Heterocycles, 1981, 16, 915–919 CrossRef CAS.
- L. Y. P. Luk, S. Bunn, D. K. Liscombe, P. J. Facchini and M. E. Tanner, Biochemistry, 2007, 46, 10153–10161 CrossRef CAS.
- J. J. Maresh, L. A. Giddings, A. Friedrich, E. A. Loris, S. Panjikar, B. L. Trout, J. Stöckigt, B. Peters and S. E. O'Connor, J. Am. Chem. Soc., 2008, 130, 710–723 CrossRef CAS.
- X. Sheng and F. Himo, J. Am. Chem. Soc., 2019, 141, 11230–11238 CrossRef CAS.
- B. R. Lichman, A. Sula, T. Pesnot, H. C. Hailes, J. M. Ward and N. H. Keep, Biochemistry, 2017, 56, 5274–5277 CrossRef CAS.
- E. Eger, A. Simon, M. Sharma, S. Yang, W. B. Breukelaar, G. Grogan, K. N. Houk and W. Kroutil, J. Am. Chem. Soc., 2020, 142, 792–800 CrossRef CAS.
- W. Schliemann, N. Kobayashi and D. Strack, Plant Physiol., 1999, 119, 1217–1232 CrossRef CAS.
- N. Chen, Z. H. Yu and X. G. Xiao, Front. Plant Sci., 2017, 8, 831 CrossRef.
- M. A. Bedewitz, A. D. Jones, J. C. D. Auria and C. S. Barry, Nat. Commun., 2018, 9, 5281 CrossRef CAS.
- J. P. Huang, C. Fang, X. Ma, L. Wang, J. Yang, J. Luo, Y. Yan, Y. Zhang and S. X. Huang, Nat. Commun., 2019, 10, 4036 CrossRef.
- Y. Ping, X. Li, W. You, G. Li, M. Yang, W. Wei, Z. Zhou and Y. Xiao, ACS Synth. Biol., 2019, 8, 1257–1262 CrossRef CAS.
- D. A. Restrepo, E. Saenz, O. A. Jara-Muñoz, I. F. Calixto-Botía, S. Rodríguez-Suárez, P. Zuleta, B. G. Chavez, J. A. Sanchez and J. C. D'Auria, Molecules, 2019, 24, 3788 CrossRef CAS.
- L. H. Yan, F. Dagorn, E. Gravel, B. Séon-Méniel and E. Poupon, Tetrahedron, 2012, 68, 6276–6283 CrossRef CAS.
- K. D. De Boer, J. C. Lye, C. D. Aitken, A. K.-K. Su and J. D. Hamill, Plant Mol. Biol., 2009, 69, 299–312 CrossRef CAS.
- M. Kajikawa, N. Hirai and T. Hashimoto, Plant Mol. Biol., 2009, 69, 287–298 CrossRef CAS.
- E. Leete and S. A. Slattery, J. Am. Chem. Soc., 1976, 98, 6326–6330 CrossRef CAS.
- M. Kajikawa, T. Shoji, A. Kato and T. Hashimoto, Plant Physiol., 2011, 155, 2010–2022 CrossRef CAS.
- J. Schachtsiek and F. Stehle, Plant Biotechnol. J., 2019, 17, 2228–2230 CrossRef.
- B. Cai, B. Siminszky, J. Chappell, R. E. Dewey and L. P. Bush, J. Biol. Chem., 2012, 287, 42804–42811 CrossRef CAS.
- S. Bunsupa, K. Komastsu, R. Nakabayashi, K. Saito and M. Yamazaki, Plant Biotechnol., 2014, 31, 511–518 CrossRef CAS.
- E. Poupon and E. Gravel, Chem.–Eur. J., 2015, 21, 10604–10615 CrossRef CAS.
- H. Sato, M. Uchiyama, K. Saito and M. Yamazaki, Metabolites, 2018, 8, 48 CrossRef.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 CrossRef.
- A. Calcaterra, L. Mangiardi, G. D. Monache, D. Quaglio, S. Balducci, S. Berardozzi, A. Iazzetti, R. Franzini, B. Botta and F. Ghirga, Molecules, 2018, 25, 414 CrossRef.
- K. Iwasa, M. Moriyasu, Y. Tachibana, H. S. Kim, Y. Wataya, W. Wiegrebe, K. F. Bastow, L. M. Cosentino, M. Kozuka and K. H. Lee, Bioorg. Med. Chem., 2001, 9, 2871–2884 CrossRef CAS.
- H. Minami, J.-S. Kim, N. Ikezawa, T. Takemura, T. Katayama, H. Kumagai and F. Sato, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 7393–7398 CrossRef CAS.
- N. Samanani, D. K. Liscombe and P. J. Facchini, Plant J., 2004, 40, 302–313 CrossRef CAS.
- D. K. Liscombe, B. P. Macleod, N. Loukanina, O. I. Nandi and P. J. Facchini, Phytochemistry, 2005, 66, 1374–1393 CrossRef CAS.
- E.-J. Lee and P. Facchini, Plant Cell, 2010, 22, 3489–3503 CrossRef CAS.
- L. Bourgeois, M. E. Pyne and V. J. J. Martin, ACS Synth. Biol., 2018, 7, 2675–2685 CrossRef CAS.
- H. Fernandes, K. Michalska, M. Sikorski and M. Jaskolski, FEBS J., 2013, 280, 1169–1199 CrossRef CAS.
- J. Li, E.-J. Lee, L. Chang and P. J. Facchini, Sci. Rep., 2016, 6, 39256 CrossRef CAS.
- L. Chang, Y. Yelpaala, J. Colbeck, H.-Y. Chen, P. J. Facchini, S. A. Shiigi, A. B. Ibáñez, J. E. Tucker, R. Estrada, G. Cottarel, J. M. Hagel, G. M. Vidanes, X. Chen and M. Enquist-Newman, Nat. Chem. Biol., 2018, 14, 738–743 CrossRef.
- M. Dastmalchi, X. Chen, J. M. Hagel, L. Chang, R. Chen, S. Ramasamy, S. Yeaman and P. J. Facchini, Nat. Chem. Biol., 2019, 15, 384–390 CrossRef CAS.
- K. Karppinen, E. Derzsó, L. Jaakola and A. Hohtola, Front. Plant Sci., 2016, 7, 526 Search PubMed.
- A. Casañal, U. Zander, C. Muñoz, F. Dupeux, I. Luque, M. A. Botella, W. Schwab, V. Valpuesta and J. A. Marquez, J. Biol. Chem., 2013, 288, 35322–35332 CrossRef.
- B. R. Lichman, G. T. Godden, J. P. Hamilton, L. Palmer, M. O. Kamileen, D. Zhao, B. Vaillancourt, J. C. Wood, M. Sun, T. J. Kinser, L. K. Henry, C. Rodriguez-Lopez, N. Dudareva, D. E. Soltis, P. S. Soltis, C. R. Buell and S. E. O'Connor, Sci. Adv., 2020, 6, eaba0721 CrossRef CAS.
- J. M. Hagel and P. J. Facchini, In Vitro Cell. Dev. Biol.: Plant, 2012, 48, 233–240 CrossRef CAS.
- N. H. Sherden, B. Lichman, L. Caputi, D. Zhao, M. O. Kamileen, C. R. Buell and S. E. O'Connor, Phytochemistry, 2018, 145, 48–56 CrossRef CAS.
- R. Roddan, G. Gygli, A. Sula, D. Méndez-Sánchez, J. Pleiss, J. M. Ward, N. H. Keep and H. C. Hailes, ACS Catal., 2019, 9, 9640–9649 CrossRef CAS.
- S. Vimolmangkang, X. Deng, A. Owiti, T. Meelaph, C. Ogutu and Y. Han, Sci. Rep., 2016, 6, 26323 CrossRef CAS.
- Y. Li, T. Winzer, Z. He and I. A. Graham, Plant Communications, 2020, 1, 100029 CrossRef.
- I. Menéndez-Perdomo and P. Facchini, Molecules, 2018, 23, 2899 CrossRef.
- W. De-Eknamkul, A. Ounaroon, T. Tanahashi, T. M. Kutchan and M. H. Zenk, Phytochemistry, 1997, 45, 477–484 CrossRef CAS.
- T. Nomura, A. L. Quesada and T. M. Kutchan, J. Biol. Chem., 2008, 283, 34650–34659 CrossRef CAS.
- T. M. Kutchan, FEBS Lett., 1989, 257, 127–130 CrossRef CAS.
- M. A. Hicks, A. E. Barber, L. A. Giddings, J. Caldwell, S. E. Connor and P. C. Babbitt, Proteins: Struct., Funct., Bioinf., 2011, 79, 3082–3098 CrossRef CAS.
- J. Stöckigt, L. Barleben, S. Panjikar and E. a Loris, Plant Physiol. Biochem., 2008, 46, 340–355 CrossRef.
- T. Zou, S. Li, M. Liu, T. Wang, Q. Xiao, D. Chen, Q. Li, Y. Liang, J. Zhu, Y. Liang, Q. Deng, S. Wang, A. Zheng, L. Wang and P. Li, Sci. Rep., 2017, 7, 6863 CrossRef.
- M. M. Sohani, P. M. Schenk, C. J. Schultz and O. Schmidt, Plant Biol., 2009, 11, 105–117 CrossRef CAS.
- L. H. Stevens, T. J. M. Blom and R. Verpoorte, Plant Cell Rep., 1993, 12, 573–576 CrossRef CAS.
- V. Courdavault, N. Papon, M. Clastre, N. Giglioli-Guivarc’h, B. St-Pierre and V. Burlat, Curr. Opin. Plant Biol., 2014, 19, 43–50 CrossRef CAS.
- A. Stavrinides, E. C. Tatsis, E. Foureau, L. Caputi, F. Kellner, V. Courdavault and S. E. O'Connor, Chem. Biol., 2015, 22, 336–341 CrossRef CAS.
- R. M. E. Payne, D. Xu, E. Foureau, M. I. S. Teto Carqueijeiro, A. Oudin, T. D. De Bernonville, V. Novak, M. Burow, C. E. Olsen, D. M. Jones, E. C. Tatsis, A. Pendle, B. A. Halkier, F. Geu-Flores, V. Courdavault, H. H. Nour-Eldin and S. E. O'Connor, Nat. Plants, 2017, 3, 16208 CrossRef.
- D. Zhao, J. P. Hamilton, G. M. Pham, E. Crisovan, K. Wiegert-Rininger, B. Vaillancourt, D. DellaPenna and C. Robin Buell, Gigascience, 2017, 6, 1–7 CrossRef.
- B. L. Manjunatha, H. R. Singh, G. Ravikanth, K. N. Nataraja, R. Shankar, S. Kumar and R. U. Shaanker, J. Biosci., 2016, 41, 119–131 CrossRef CAS.
- J. Berlin, C. Rugenhagen, N. Greidziak, I. N. Kuzovkina, L. Witte and V. Wray, Phytochemistry, 1993, 33, 593–597 CrossRef CAS.
- M. B. Kilgore, C. K. Holland, J. M. Jez and T. M. Kutchan, J. Biol. Chem., 2016, 291, 16740–16752 CrossRef CAS.
- A. Singh, M. A. Massicotte, A. Garand, L. Tousignant, V. Ouellette, G. Bérubé and I. Desgagné-Penix, BMC Plant Biol., 2018, 18, 338 CrossRef CAS.
- T. Hotchandani, J. De Villers and I. Desgagne-Penix, Genes, 2019, 10, 594 CrossRef CAS.
- A. Singh and I. Desgagné-Penix, Sci. Rep., 2017, 7, 17356 CrossRef.
- A. Reis, K. Magne, S. Massot, L. R. Tallini, M. Scopel, J. Bastida, P. Ratet and J. A. S. Zuanazzi, Sci. Rep., 2019, 9, 8471 CrossRef.
- T. Shoji, Front. Plant Sci., 2019, 10, 560 CrossRef.
- R. De Brito Francisco and E. Martinoia, Plant Cell Physiol., 2018, 59, 1326–1336 Search PubMed.
- N. Shitan, Biosci., Biotechnol., Biochem., 2016, 80, 1283–1293 CrossRef CAS.
- H. J. Byeon, K. H. Jung, G. S. Moon, S. K. Moon and H. Y. Lee, Sci. Rep., 2020, 10, 1057 CrossRef CAS.
- B. R. Lichman, S. E. O'Connor and H. Kries, Chem.–Eur. J., 2019, 25, 6864–6877 CrossRef CAS.
- L. B. Davin, H. Wang, A. L. Crowell, D. L. Bedgar, D. M. Martin, S. Sarkanen and N. G. Lewis, Science, 1997, 275, 362–366 CrossRef CAS.
- B. R. Lichman, M. O. Kamileen, G. R. Titchiner, G. Saalbach, C. E. M. Stevenson, D. M. Lawson and S. E. O. Connor, Nat. Chem. Biol., 2019, 15, 71–79 CrossRef CAS.
- L. Noda-garcia, W. Liebermeister and D. S. Tawfik, Annu. Rev. Biochem., 2018, 87, 187–216 CrossRef CAS.
- L. Caputi, J. Franke, S. C. Farrow, K. Chung, R. M. E. Payne, T. D. Nguyen, T. T. T. Dang, I. S. Teto Carqueijeiro, K. Koudounas, T. D. De Bernonville, B. Ameyaw, D. M. Jones, I. J. Curcino Vieira, V. Courdavault and S. E. O'Connor, Science, 2018, 360, 1235–1239 CrossRef CAS.
- P. Srinivasan and C. D. Smolke, Nat. Commun., 2019, 10, 3634 CrossRef.
- Mint Evolutionary Genomics Consortium, Molecular Plant, 2018, 11, 1084–1096 CrossRef.
- E. Gravel and E. Poupon, Nat. Prod. Rep., 2010, 27, 32–56 RSC.
| This journal is © The Royal Society of Chemistry 2021 |