
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCreating and screening natural product libraries
Brice A. P.
Wilson
 a,
Christopher C.
Thornburg
b,
Curtis J.
Henrich
c,
Tanja
Grkovic
b and
Barry R.
O'Keefe
*ad
a,
Christopher C.
Thornburg
b,
Curtis J.
Henrich
c,
Tanja
Grkovic
b and
Barry R.
O'Keefe
*ad
aMolecular Targets Program, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702-1201, USA. E-mail: okeefeba@mail.nih.gov
bNatural Products Support Group, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702-1201, USA
cBasic Science Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research, Frederick, Maryland 21702-1201, USA
dNatural Products Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Frederick, Maryland 21702-1201, USA
First published on 18th March 2020
Abstract
Covering: up to 2020
The National Cancer Institute of the United States (NCI) has initiated a Cancer Moonshot program entitled the NCI Program for Natural Product Discovery. As part of this effort, the NCI is producing a library of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 partially purified natural product fractions which are being plated into 384-well plates and provided to the research community free of charge. As the first 326000 of these fractions have now been made available, this review seeks to describe the general methods used to collect organisms, extract those organisms, and create a prefractionated library. Importantly, this review also details both cell-based and cell-free bioassay methods and the adaptations necessary to those methods to productively screen natural product libraries. Finally, this review briefly describes post-screen dereplication and compound purification and scale up procedures which can efficiently identify active compounds and produce sufficient quantities of natural products for further pre-clinical development.
000000 partially purified natural product fractions which are being plated into 384-well plates and provided to the research community free of charge. As the first 326000 of these fractions have now been made available, this review seeks to describe the general methods used to collect organisms, extract those organisms, and create a prefractionated library. Importantly, this review also details both cell-based and cell-free bioassay methods and the adaptations necessary to those methods to productively screen natural product libraries. Finally, this review briefly describes post-screen dereplication and compound purification and scale up procedures which can efficiently identify active compounds and produce sufficient quantities of natural products for further pre-clinical development.
 Brice A. P. Wilson | Brice Wilson is a Staff Scientist in the Molecular Targets Program (MTP) of the National Cancer Institute in Frederick, MD, USA. After receiving his Ph.D. in Pharmacology and Molecular Sciences from the Johns Hopkins University School of Medicine, he joined the MTP as a Post-Doctoral Fellow where he earned promotion to a Staff Scientist position. In the MTP, Brice develops high-throughput biochemical assays for novel cancer chemotherapeutic discovery from both natural product and synthetic libraries. He is also responsible for carrying out biochemical mechanism of action studies for lead molecules discovered through screening. |
 Christopher C. Thornburg | Christopher Thornburg obtained his Ph.D. in Pharmaceutical Sciences from Oregon State University, where he studied the isolation and structure elucidation of biologically active marine natural products under the guidance of Dr Kerry McPhail. He joined the Natural Products Support Group at the U.S. National Cancer Institute in Frederick, MD in 2013, where he is currently focused on the integration of natural products with high-throughput platforms and the discovery of natural product drug leads from microbial, marine and plant sources. |
 Curtis Henrich | Curtis Henrich received his Ph.D. in biochemistry from the University of Michigan. After postdoctoral research in biochemistry and cell biology and several years in the biotechnology industry, he moved to SAIC-Frederick (now Leidos Biomedical Research – research contractor for the National Cancer Institute (NCI) in Frederick, Maryland). He has worked for more than 15 years at NCI in development and application of assays for high-throughput screening (HTS), developing particular expertise in the design and adaptation of HTS assays for applications with natural products. |
 Tanja Grkovic | Tanja Grkovic obtained her PhD degree from the University of Auckland under the supervision of Professor Brent Copp. She then carried out postdoctoral research at the Molecular Targets Laboratory at the National Cancer Institute in Maryland, USA and the Eskitis Institute for Drug Discovery in Brisbane, Australia. She is currently a Senior Scientist in the Natural Products Support Group at the Frederick National Laboratory for Cancer Research where her research is focused on the generation of prefractionated natural product libraries as well as the isolation and structure elucidation of natural products sourced from marine, plant, and microbial biota. |
 Barry R. O'Keefe | Barry O'Keefe received a B.S in Botany from Michigan State University and a Ph.D. in Pharmacognosy from the University of Illinois at Chicago. Dr O'Keefe joined the National Cancer Institute's Laboratory of Drug Discovery Research and Development in 1994 to study novel proteins from natural products extracts. Dr O'Keefe currently leads the Protein Chemistry and Molecular Biology Section and is Acting Chief of the Molecular Targets Program at the Center for Cancer Research, NCI, NIH which specializes in the isolation and identification of novel bioactive proteins and the development of novel assay systems for the evaluation and screening of natural products against biochemical targets. Dr O'Keefe is also Chief of the Natural Products Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis at the NCI which is responsible for the collection, extraction, pre-fractionation and discovery of bioactive natural products. |
1 Introduction
The earliest forms of medicine utilized by Homo sapiens were natural products. Humans have continued to look to nature for more chemicals that can be made into drugs with continually improving technologies and methods. Due to these advances, natural products and their derivatives still make up a significant percentage of approved drugs worldwide.1 Despite this track record of success however, natural products make up only a small number of the samples utilized for high throughput screening as shown by the percentage of published manuscripts on the results of drug screens which include natural products, Fig. 1. If researchers are to include natural product samples in their screens, they must become knowledgeable in several areas of science. To aid in that effort, this review details many of the necessary components for a modern high throughput screening program utilizing natural products as sources of chemical diversity.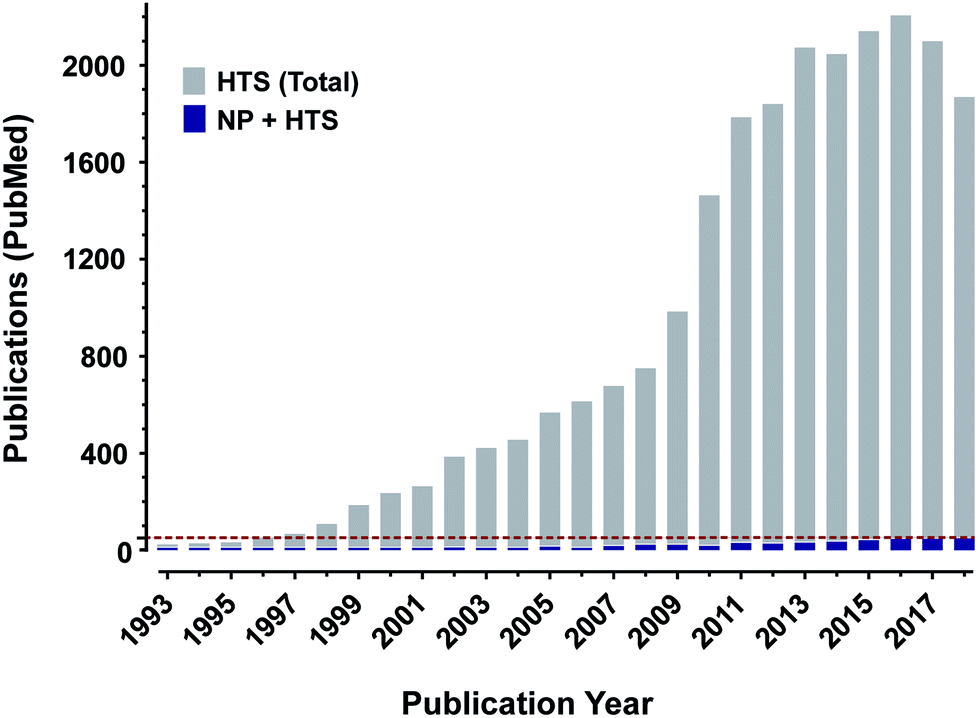 | ||
| Fig. 1 Natural products may be under-utilized in high throughput screening. A count of publications available on pubmed (https://www.ncbi.nlm.nih.gov/pubmed/) using either “natural products and high throughput screening” (green boxes) or “high throughput screening alone” (red boxes) reveals a profound disparity in publication counts. Green dashed line is at the 50 publication point. | ||
To ethically and effectively assess biodiversity for new drug development there are many necessary considerations. First and foremost is the observance of national and international regulations on access to and benefit sharing from natural product source organisms.2 Without the appropriate permissions to collect organisms and agreements for planned benefit sharing with host countries where collections are to take place (and, where applicable, with local indigenous populations), no researcher should engage in the collection of source organisms. Once these necessary agreements are in place, it will be important to properly annotate all collections, ideally including voucher specimens, so that the maximum scientific benefit can be achieved from the collections. Extraction and, if applicable, prefractionation procedures will also need to be tested and optimized for individual classes of source organism (i.e. plant, marine and microbial). The resultant library will then need to be tested against targets of interest in assays (both molecularly-targeted and phenotypic) that have been optimized to provide reliable results in the presence of natural product samples. Finally, compound isolation, identification/structure elucidation, and resupply will be necessary to be able to move individual bioactive compounds towards potential drug development.
The goal of this review is to highlight recent strategies used to efficiently create natural product-based libraries for drug discovery as well as both biochemical and cell-based screening strategies for these natural product samples. Finally, we conclude with an examination of technologies used “post-screening” to rapidly dereplicate identified activities and resupply isolated active compounds in quantities sufficient for the initial stages of development. This review is meant to lead the reader through some of the processes necessary to develop a modern natural product-based drug discovery program by summarizing the methods and strategies used to create and screen natural product libraries.
2 Creating natural product libraries
2.1 Collection and conservation of biological diversity
Natural product libraries generally comprise extracts of plants, marine invertebrates, and/or microorganisms, which may be diversified through collections made on both temporal and geographical scales, often in biologically diverse regions.3,4 Importantly, with collecting biota internationally, access to, and the use of biological resources should be on mutually agreed upon terms with each participating source country and follow the objectives outlined in the United Nations Convention on the Law of the Sea (UNCLOS) and the United Nations Convention on Biological Diversity (CBD), which advocates the conservation and sustainable use of biological diversity.2,5 Furthermore, a supplementary agreement to the CBD: the Nagoya Protocol on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising from their Utilization (ABS), provides a legal framework to share equitably the benefits arising from the use of genetic resources (i.e. plants, animals and microorganisms that are used for research and development).6 At present (2020), the CBD and Nagoya Protocol on ABS have been either ratified, accorded to, approved or accepted by 196 and 123 countries, respectively. Regardless of the acceptance of these protocols, programs involved in the biodiscovery process are still encouraged to adhere to the CBD principles. Notably, samples collected through the NCI Natural Products Collection program have been acquired through collection agreements based on the NCI Letter of Collection (LOC), which predates the CBD and stipulates equitable benefit sharing from commercial products derived from discoveries, irrespective of whether or not a formal agreement has been signed by each participating source country or their representatives.7For academic and industry researchers, newer regulations on accessing and developing international sources of biota, including the time required to obtain all the necessary permits, such as visas, collecting, shipping and export permits, may restrict broad access to collections from biodiversity-rich source countries. Alternatively, research on local biota is simpler and, considering the enormous biodiversity of prokaryotes (archaea and bacteria) and fungi (∼1012 species),8 as well as their continued impact on the development of antimicrobial and antitumor compounds,9,10 relatively small collections of soil or water could be potentially useful for drug discovery efforts. To this end, several academic groups have recently employed crowdsourcing as a mechanism to obtain soil samples from the personal property of citizen scientists who, in turn, agree to the release of all intellectual property (IP) rights generated from their respective sample with the understanding that their contribution may have a meaningful impact on the project or cause.11,12 Although citizen science programs are granted permission from property owners, permits for institutions to receive materials still need to be acquired by the recipient institution from all necessary local state and federal departments. In the United States, federal legislation and agencies such as the Centers for Disease Control and Prevention (CDC), US Department of Agriculture (USDA) and US Department of Transportation regulate the possession, use and transfer of substances having the potential to pose a threat to public health and safety and/or agricultural consequences. As a final note on generating source organism libraries, it is essential to collect voucher specimens, accurately tag (e.g. barcoded labels) and document each collection with the collecting institution, collector(s), taxonomy and taxonomist(s), location coordinates, date and time, and any relevant field notes. Ideally, these vouchers would be available to researchers to encourage efforts to keep the categorization and naming of samples current with changes in taxonomy. Collection of metadata such as this is central to the establishment of a database for sample tracking, possible recollection of sourced material, as well as the conservation and understanding of biological diversity.
2.2 Natural product libraries for high-throughput screening
Natural product samples have been used for decades in a variety of screening programs throughout both industry and academia. As assay systems became more advanced, more target-oriented and higher throughput, the utility of crude natural product extracts was diminished. This led to the increased use of partially-purified or “prefractionated” natural product libraries in screening programs. As detailed below, a variety of techniques and sampling algorithms have been reported for prefractionation. In general, these libraries have performed better in modern molecularly-targeted assay systems. It should be noted, however, that experimental methods that result in the reproducible production of well-defined, weighed samples with the total number of fractions optimized to provide the separation and concentration of active compounds, the sequestration of common nuisance compounds, and the restraint of downstream assay costs should be prioritized during method development.000 unique extracts derived from plant, marine and microbial organisms that have been collected from biodiverse regions throughout the world. Notably, during peak periods of production, starting from approximately 1 kg of organism, between 15000 and 20000 extracts were generated per year using high-throughput extraction processing methods described by McCloud.20 Alternatively, throughput can be significantly increased by decreasing the initial scale of collected material (e.g. <1 g) used to generate each extract. However, additional extractions may be required to provide enough material for screening in multiple campaigns and downstream processes such as the isolation, identification and verification of active compounds.
000 fractions (Table 1). Importantly, prefractionated natural product samples typically show improved screening performance (often observed as a higher confidence in observed hit rates), enhanced biological activity due to the concentration of active components present as only minor metabolites, sequestration of common nuisance compound classes, as well as streamlined downstream processes for dereplication and the isolation of bioactive components.21,27,29,40
| Company/institute | Sample type (number) | Number of screening samples | Ref. | ||
|---|---|---|---|---|---|
| Extracts (sample source #) | Fractions (extract source #) | Compounds (type) | |||
| a B = bacteria; F = fungi; MB = marine bacteria; MI = marine invertebrates; P = plant; NP = pure natural products; SS = semi-synthetic (NP-based); na = data not available. b Acquired from MerLion Pharmaceuticals (Singapore). c No cost for materials, recipients only cover shipping charges. d The current Natural Products Set IV was selected from the DTP Open Repository Collection of >4500 pure natural product compounds. e Former Merck and Schering-Plough Natural Product Libraries. f Accessed through InterLink Biotechnologies.222 | |||||
| Albany Molecular Research, Inc. (AMRI) | B/F and P (>190000) |
102000 (23375) |
209000 (12349) |
— | 205 |
| AnalytiCon Discovery | B/F (na); P (na); SS (>25000) |
— | — | >25000 (SS); >5000 (NP) |
206 |
| Bioinformatics Institute Singapore (BII)—A*STAR Natural Product Library | B/F (>120000); P (>37000) |
∼270000 (>157000) |
∼70000 (na) |
2600 (NP) | 207 |
| Developmental Therapeutics Program—The National Cancer Institute | MI (>20000); B/F (>25000); P (>80000) |
>230000 (>108000) |
326000 (46570) |
419 (NP set IV)d | 26 |
| Fondazione Ricerca per la Vita (FIIRV) | B/F (>15000) |
166000 (15000) |
— | — | 208 |
| Fundación MEDINA | B/F (190000) |
>130000 (na) |
— | — | 209 |
| Griffith Institute for Drug Discovery (GRIDD)—Nature Bank | MI and P (30000) |
10000 (10000) |
50000 (10000) |
210 | |
| InterBioScreen (IBS) | MI (na); B/F (na) and P (na) | — | — | >67000 (NP) |
211 |
| Magellan BioScience Group, Inc. | MB (10000); F (55000) |
>15000 (na) |
— | — | 212 |
| Mycosynthetix | F (>55000) |
55000 (na) |
— | — | 213 |
| Natural Products Discovery Institute (NPDI) | B/F (>30000); P (>20000) |
80000 (na) |
— | — | 214 |
| PharmaMar | MI (>118000); MB (>100000) |
100000 (na) |
— | — | 215 |
| PhytoPharmacon | P (4000) | 4000 (4000) | 25000 (4000) |
500 (NP) | 216 |
| RIKEN Natural Products Repository (NPDepo) | B/F (na) and P (na) | — | — | 8000 (NP) | 217 |
| The Institut de Chimie des Substances Naturelles (ICSN) | MI, B/F, and P (>7000) | 14000 (7000) |
— | — | 218 |
| The Natural Products Library Initiative at the Scripps Research Institute (Florida) | B (>5500) | 8500 (na) | 3400 (na) | 450 (NP) | 219 |
| The University of Mississippi—National Center for Natural Products Research | MI and F (>2000); P (>18000) |
>20000 (>20000) |
>43000 (>3400) |
∼700 (NP) | 220 |
| Unigen (PhytoLogix Library) | P (8000) | 9000 (na) | 200000 (na) |
— | 221 |
Similar to the development of large natural product extract libraries, techniques to generate subsequent fraction libraries should balance the retention of maximal chemical diversity with throughput and cost relative to the amount of extract used, number of fractions produced, solvent scheme, drying, weighing, long-term storage, and formatting for HTS (Fig. 2). The mass of extract required ultimately depends on the number of expected assays to support, the test concentration planned, and the number of fractions generated. Each fraction should ideally contain enough mass to support a larger number of HTS campaigns, as well as subsequent dereplication, compound isolation and structure elucidation efforts. In this regard, a smaller set of fractions (5 to 10) generated per extract requires less starting material and, as shown in Table 1, optimizes the coverage of chemical and biological space of the screening library. Automated weighing stations and liquid handling systems that can not only solubilize samples, but also integrate with SPE columns and generate assay plates can significantly increase sample throughput and reproducibility (Fig. 1).26 Finally, method validation and proof-of-principle studies should be performed and can include challenge sets containing known compounds,28 comparison of bioassay readouts in several assay systems, mass recovery and distribution from parallel or repeated processes, and analytical (LCMS or NMR) quality control measures.
 | ||
| Fig. 2 Overview of automated and high-throughput processes developed at the NCI National Program for Natural Products Discovery (NPNPD) to facilitate the production of a natural product-based screening library. (1) Since 1986, more than 80000 samples have been acquired through collection agreements based on the NCI letter of collection with each participating source country or their representatives, which stipulates equitable benefit sharing from commercial products derived from discoveries made through these collections. (2) Extracts in the NCI NPR are prepared in a high-throughput manner using both an aqueous and organic solvent extraction process, resulting in two sequential extracts per collected specimen/sample.27 At present, the US National Cancer Institute's (NCI) natural product repository contains over 230000 unique extracts derived from plant, marine and microbial organisms. (3) Extracts (n = 88) are prefractionated on a customized Positive Pressure Solid Phase Extraction workstation (PPSPE) with two robotic arms working in parallel to produce seven fractions per extract (3.5 h; n = 616 fractions). (4) Fractions are dried using high-capacity centrifugal evaporation systems (18 h; n = 2304), and the final mass of each fraction is determined on an automated weighing station (5). (6) An automated sample management system with the capacity to store 1.1 million 2D-barcoded tubes (10 ml) is integrated with robotic systems designed to generate 384-well microtiter plates for HTS and 96-well plates for secondary HPLC-based fractionation of active primary fractions. | ||
000 natural products reported, only a small portion are commercially available.46 Ultimately, the compilation of a complementary collection of diverse source organisms resulting in well-annotated natural product extracts and fractions, should yield more structurally-diverse pure compounds from HTS for further evaluation as potential drug leads.
3 Cell-based HTS for natural product discovery
The category of cell-based HTS includes a wide variety of targets and detection technologies which have been the subjects of a number of recent reviews,47–56 including those detailing use for natural product discovery.47,52,57 While there are many examples of non-mammalian cell-based assays that have been utilized for identification and characterization of anti-infective agents in natural product extracts, including anti-fungal,54,58 anti-parasitic,59–62 anti-bacterial63–65 as well as in model organism-based cellular assay platforms including yeast,66,67Xenopus oocytes,68 zebra fish69 and C. elegans,70 this section will focus on screens involving human cells. Mammalian and/or human cell-based natural products HTS has been employed in a wide range of disease areas and cellular phenomena. A few examples include immunomodulation,71,72 nuclear export,73 and metabolomics,74 and disease areas including diabetes75 and cystic fibrosis76 among many active target areas.77 Cell-based assays for natural product discovery have been particularly plentiful in cancer research.49,77 Due to the lengthy history of cell-based anticancer screening of natural product extracts at the National Cancer Institute, and the current focus of the Molecular Targets Program within the NCI's Center for Cancer Research, the examples in the following section will be drawn largely from cancer-focused cell-based HTS efforts used in the discovery of active natural products.3.1 Phenotypic screening in human cells
The success in any HTS campaign (natural products or otherwise) is dependent on the quality and relevance of the cells, assays, readouts, and screening libraries utilized. Target validation and the selection and substantiation of specific cellular models for HTS have been extensively reviewed elsewhere (e.g., ref. 48 and 50) and all of the cellular characteristics important to general HTS campaigns apply to natural product-focused HTS as well. Cell-based techniques are often depicted as phenotypic or molecularly targeted. The phenotypic approach allows for the identification of active compounds that effect cells by unidentified interactions, potentially leading to the discovery of novel mechanisms of action. This approach requires more extensive down-stream mechanistic studies than a targeted approach such as a specific reporter-based assay. Targeted assays have the advantage of focusing activities on a defined cellular target (generally a protein or pathway) which enables more selective screening and more rapid post-assay functional studies. Alternatively, a “hybrid” approach can be taken by measuring a phenotypic endpoint that is dependent on the expression or activity of a specific molecular target. All of these have distinct advantages and disadvantages and each assay type has been applied to screens of natural products libraries. Examples of these assay systems and aspects important to the development of robust assays suitable for natural product screening are discussed below.A second level of phenotypic screening has been called “mechanism-informed”48 phenotypic screening, the most common of which are reporter gene assays. In this case, cells are engineered to contain a construct that can be easily measured (usually a fluorescent protein or luciferase) as a readout of specific transcription factor activity. This approach does not target an individual macromolecule, but instead is focused on a specific signaling pathway (or pathways). As with broader cellular phenotypes, reporter gene assays can be affected by directly or indirectly modulating any of a number of potential molecular targets in the pathway and can sometimes identify novel mechanisms of pathway regulation. For example, a recent cell-based HTS of natural products utilized a reporter gene construct to identify inhibitors of the EWS-FLI1 transcription factor, a fusion protein that drives development of the rare Ewing's sarcoma (EWS) tumor. The assay utilized a construct containing the NROB1 promoter (a target of EWS-FLI1) driving luciferase allowing for measurement of transcriptional activity engendered by this fusion protein.92 In this screen, an active natural product extract (from the plant Phyllanthus engleri) yielded the compound englerin A which affected the binding of the EWS-FLI1 transcription factor to DNA via modulation of intracellular calcium and PKC activity.93 The discovery of an indirect modulation of this transcription factor as a novel mechanism of action highlights the diversity of possible outcomes from mechanistically informed phenotypic screening. Other reporter assays for modulation of gene expression by natural products have identified a significant number of natural compounds in eye cancers94 providing new insights into molecular targets, pathways, and mechanisms. Fluorescent reporter proteins can also be used to assess other cellular mechanisms. For example, screening of natural product extracts for inhibition of nuclear export via imaging of a fluorescent biosensor protein95 resulting in a natural product that covalently bound to a nuclear export protein.
In another example of a targeted phenotypic assay, the effect of test samples on the phenotype requires the expression or activity of a specific molecular target. For example, many renal carcinoma cells (RCCs) are resistant to the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), so a cell-based HTS assay was configured to identify natural products able to sensitize RCCs to TRAIL by assessing sample-induced cell death in the presence and absence of TRAIL.96 Treatment with active samples resulted in cell death only in the presence of TRAIL. An active extract (from the plant Physalis peruviana) was identified and yielded a series of withanolides which were able to sensitize cells to TRAIL-induced apoptosis by stimulating increased degradation of cFLIP, a regulator of TRAIL signaling.97 Mechanism-informed phenotypic screening thus led to novel activities and new insights into the cellular phenotypes and their regulation, as well as identification of potential new molecular targets.
Although molecularly-targeted HTS is sometimes thought of as comprising only biochemical assays, it is often possible to configure a targeted cell-based assay to address a specific molecular target in the cellular milieu. One such example is the discovery of natural products able to inhibit drug efflux, a significant contributor to drug resistance in a variety of cancer cells.98 In order to assess the ability of compounds to block drug efflux via the ABCG2 multidrug resistance protein, a cell line expressing only that transporter was selected and accumulation of a fluorescent ABCG2 substrate was monitored. The substrate only accumulated in the cells when the transporter was inhibited and was easily measured on a fluorescence plate reader. The selected substrate, pheophorbide a (PhA), was chosen based on its relative specificity for ABCG2 as well as its fluorescence properties. In particular, PhA has a large Stokes shift (excitation and emission at 395 and 670 nm respectively) thus minimizing the probability of interference by fluorescent compounds common in natural product extracts. Among the active natural product extracts was an organic extract of the sponge Botryllus tyreus which yielded a series of botryllamides, some with very specific activity at inhibiting only the ABCG2 transporter while others had broader activity profiles against efflux transporters (i.e. p-glycoprotein).99 One of the botryllamides has shown efficacy in an animal model and is now in pre-clinical development for enhancing drug uptake.
3.2 Assay optimization for screening natural products in cell-based screens
An important factor in the success of cell-based HTS is the quality and diversity of the libraries screened (see, e.g.ref. 100). Synthetic compound libraries have been developed based on a variety of criteria and are widely available. In recent years, increasing numbers of pure natural products, either synthetic or isolated, have become available providing unique and valuable resources for drug discovery. Purified known natural products can be obtained from a number of commercial, academic, and government entities (including for example the NCI Developmental Therapeutics Program). As such, pure natural products are included in many HTS screening libraries, although they tend to be under-represented.100 As pure compounds, they are handled in the same way and with the same general advantages and disadvantages as synthetic compounds with regard to cell-based HTS techniques.51,52,55,77,101 By contrast, the discovery of new bioactive natural products requires the use of extract libraries which offer unique challenges for cell-based screening. Natural product extracts are typically complex mixtures of known and unknown compounds in unknown concentrations. In addition, they tend to be rich in pan-assay interference compounds (“PAINS”102), including fluorescent molecules and fluorescence quenchers, colored compounds, redox-active compounds, aggregators, and surfactants (like saponins and fatty acids, etc.) which can affect both cell viability and assay readouts.47 Cell-based HTS readouts include flow cytometry, imaging, fluorescence (intensity, FRET, or TRF), luminescence, InCell westerns, and the use of colorimetric substrates all of which can be affected by natural product PAINS.Although it is sometimes the case that screens configured for use with pure compound libraries can be deployed without significant modification for screening natural product extract libraries; assays typically must be re-optimized, and sometimes completely re-developed, for compatibility with natural product samples. These modifications do not often find their way into publications describing the assays or the active molecules discovered from their activity in these modified assays. In general, the first step in transitioning from pure compounds to extracts is a re-assessment of assay acceptability criteria in a pilot study using representative natural product extract samples. In particular, optimal assay conditions may be altered by extracts as compared to pure compounds. Therefore, all assay variables must be re-assessed at the apparent optimal level and at both higher and lower levels in the presence of extracts as well as assessing any effects of extracts on the assay visualization endpoint. These variables include cell number, incubation time, order of addition, cell substrate, cell growth conditions, and general assay interference (e.g. loss of signal due to inhibition of detection reagents, or increased signal due to the presence of colored or fluorescent molecules, etc.). It is not uncommon for a plate washing step to be required to reduce quenching and/or increase signal in fluorescent assays due to quenchers or intrinsically fluorescent compounds found in many natural product extracts and sometimes exclusion of categories of particularly problematic extracts is necessary. Both of these approaches were used in modifying the ABCG2 inhibition assay for application to natural product extracts in order to reduce false positives (i.e., increases in cell-associated fluorescence due to extracts containing fluorescent compounds).98,99 Eliminating extract samples from screens is obviously not ideal since those extracts could well also contain ABCG2 inhibitors. Often there is no good option except to completely reconfigure the assay, in some cases including re-engineering cells or choosing different cells or assay readouts. Parallel and/or secondary assays can often be used to identify and eliminate false positives (in this case by assessing the inherent fluorescence of apparent hits). Interestingly, the prevalence of fluorescent molecules in natural product extracts has also provided an opportunity for development of new chemical probes with novel fluorophores.103
Another potential challenge posed in cell-based HTS is the presence of non-specific cytotoxic compounds. Although it is difficult to find solid corroboration, anecdotally there tends to be an expectation that cytotoxic extracts may be chemically and biologically more diverse than innocuous extracts. As an example of the extent of the problem as applied to cell-based HTS and some approaches to address it, an assay for substances able to specifically induce growth inhibition/cytotoxicity in mast cells expressing constitutively active mutant c-KIT receptor tyrosine kinase was used to assess crude natural product extracts.104 In preliminary assays with a selection of extracts representative of the total library, 22% of samples tested at a single test concentration reduced target cell survival. A hit rate this high can impede HTS implementation, particularly in moderately resourced research environments, and therefore further optimization was necessary. Two commonly used adaptations were made to allow this growth inhibition/cytotoxicity assay to be fruitful. First, a second cell line (same lineage, expressing wild type cKIT) was assessed in parallel to identify samples that differentially affected the two cell lines (i.e., cytotoxicity dependent on the mutant protein). The vast majority of the active extracts also affected wild type cell survival and were therefore deprioritized for further study. In cancer research, this approach, selection based on differential cytotoxicity, goes back to the origins of the NCI-60 cell assay80 and has been applied extensively ever since. The second adaptation was to perform the assay at multiple extract concentrations. Together with standard hit confirmation and secondary assays, the screen resulted in identification of ∼30 differentially active extracts (from >135000 screened) and subsequently led to the characterization of several interesting molecules.104
Cell toxicity and other non-specific effects can interfere with other cell-based assays as well. As noted, reporter gene assays for transcriptional activation of target expression programs are very commonly used in cell-based HTS. Although not always the case, reporter gene assays are often configured to find substances that inhibit specific gene expression. As a result, toxic compounds and non-specific inhibitors of transcription or translation could “look like” inhibitors by reducing the signal and result in false positives. In this case as well, parallel assays using control cells (e.g., expressing reporters under the control of constitutively active transcription factors) can help identify false positive test samples. For example, it became immediately apparent that constituents of natural product extracts would provide significant non-specific interference in an assay for inhibitors of HIF2α-induced gene expression.105 Identification of specifically active extracts required parallel or sequential analysis of expression by a constitutively active reporter as well as a growth inhibition/cytotoxicity assay, thus controlling for both toxicity and non-specific effects on transcription, translation, or assay readout (such as luciferase enzyme stabilization or luminescence interference). An assay that measures the increase in signal (like the ABCG2 example above) can circumvent some of the challenges related to natural product extract screening in cell-based systems. For example, a reporter assay was developed to measure stabilization of the tumor suppressor protein Pdcd4.106 A luciferase-Pdcd4 fusion protein was responsive to conditions that would induce Pdcd4 degradation so active test samples (stabilizers) would increase the luciferase signal under these conditions (TPA treatment in the assay). In this case, a toxic sample or one that inhibits the reporter (luciferase) would not be identified as a hit. However, the presence of cytotoxic extract components could easily mask possible active compounds by eliminating the signal entirely. Parallel or sequential assay of controls is, of course, important in HTS of pure compound libraries, but is even more significant when assessing natural product extracts.
Unfortunately, these approaches are insufficient for finding underlying biological activities that may be masked by cytotoxic and/or other generally interfering components in extracts. Re-optimization of an assay for application to extracts (e.g., cells less susceptible to cytotoxicity, re-cloned reporters, cell washing, detection reagents, etc.) can reduce interference by extracts,98,106 but the problem cannot always be eliminated in this way. Similarly, assaying at multiple sample concentrations can be useful for addressing this issue or for prioritization of hit extracts with unknown individual compound concentrations. However, crude natural product extracts themselves remain a challenge. As discussed in Section 2 of this review, partial purification, or prefractionation, of crude natural product extracts can be highly useful in removing or sequestering problematic compounds while providing increased test concentrations of potentially active compounds (ideally in different fractions) as well as providing less complex mixtures which can result in higher confirmation rates.107 The transition from crude to prefractionated extracts for cell-based HTS has been validated in HTS campaigns at the NCI.106,107Fig. 3 shows an example of the power of partial purification of natural product extracts to significantly improve identification of active extracts in a cell-based assay. A dual luciferase reporter HTS assay for modulators of NF1-mutant astrocytoma cells provided simultaneous measurement of cell proliferative activity and toxicity.29 Based on statistical analysis of results, a hit was defined as a sample that reduced proliferative activity to <20% of untreated controls with minimal toxic effect (cell health index > 50% of control). This assay was then applied to crude and prefractionated natural product extracts (5 fractions and crude tested for each extract – labeled A–E and crude for four examples in the figure). For active extracts, in the vast majority of cases (86%), the crude extract was inactive or toxic while one or more of the fractions showed activity and minimal toxicity (examples in panels A and D respectively). In only 3.5% of hits were crudes active but fractions inactive (panel C). <10% of the extracts showed activity in both the crude and one or more fraction (panel B). Thus for this assay, most of the hits would not have been identified in crude extracts and those that were active in the crude were also active in fractions. This clearly illustrates the power of partial purification to increase effective concentrations of active components and/or sequester toxic compounds.
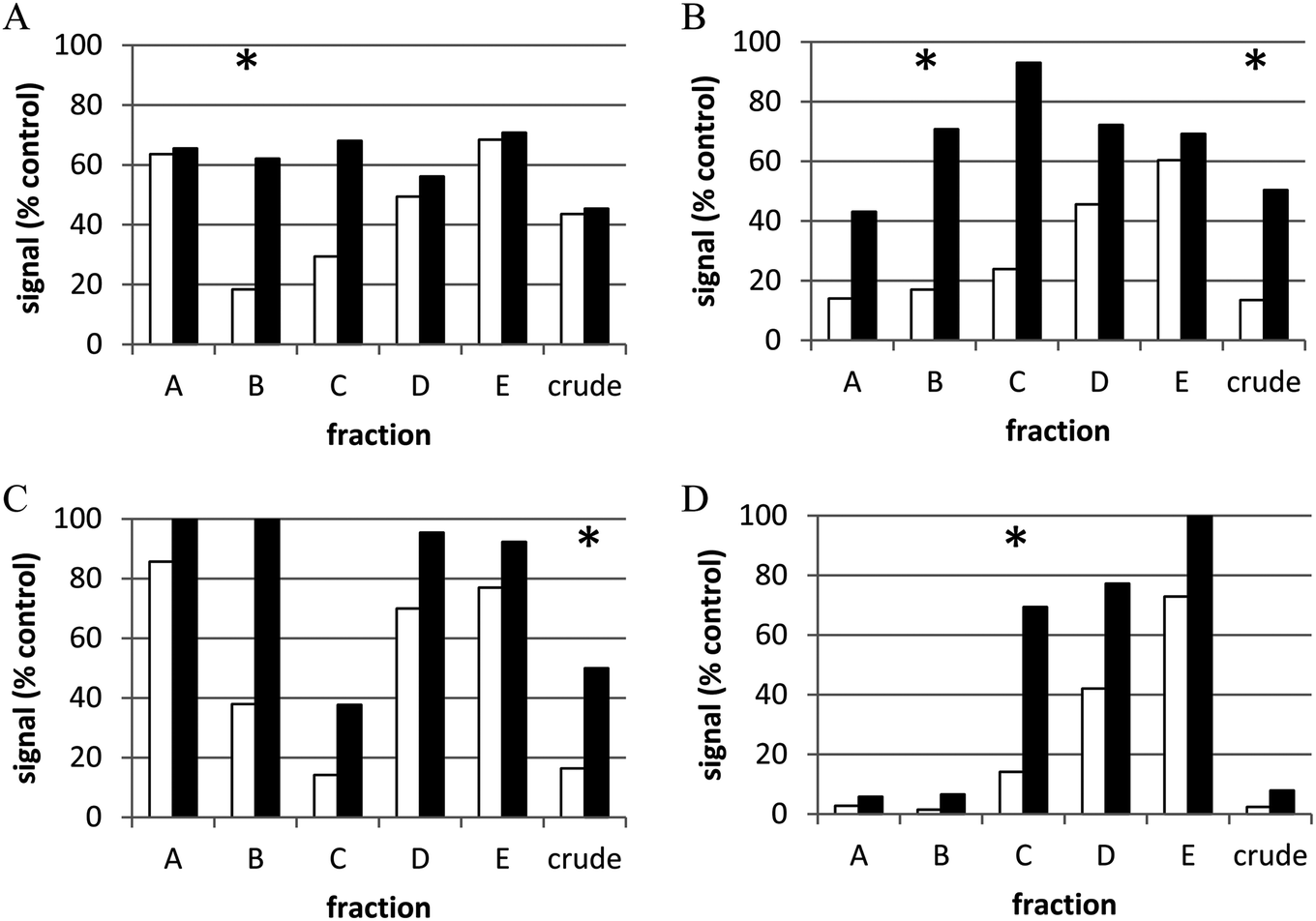 | ||
| Fig. 3 Representative examples of types of active extract samples. NF1-mutant astrocytoma cells expressing luciferase reporters for proliferation and cell viability were treated for 48 h with crude or partially purified (“prefractionated”) natural product extracts (10 μg ml−1). Open bars represent green signal (i.e., proliferative index), black bars represent red signal (i.e., cell health index). (A) Active fraction and inactive crude extract; (B) fraction and crude extract are active; (C) only crude extract is active; (D) toxic crude extract and active fraction *active sample (i.e., >80% reduction in cell proliferative index, <50% reduction in cell health index). Reprinted with permission from C. J. Henrich, L. K. Cartner, J. A. Wilson, R. W. Fuller, A. E. Rizzo, K. M. Reilly, J. B. Mcmahon and K. R. Gustafson, J. Nat. Prod., 2015, 78, 2776–2781. Copyright 2015, American Chemical Society. | ||
3.3 Active sample prioritization
Conversely, cell-based assays also complement biochemical HTS. For example, a cell-free protein–protein interaction assay was developed to screen for substances able to disrupt the binding interaction between HIF1α and the transcriptional co-activator P300.109 Among the major challenges of cell-free assays, particularly with regard to natural product extracts, is the inability to measure or predict either the ability of active compounds to access the intracellular environment or whether a given hit sample might be toxic to target cells. As a result, moderate throughput cell-based assays were included to confirm disruption of HIF1α/P300-driven transcription and to identify cytotoxic/growth inhibitory extracts.
The development of lead molecules from screening hits derived from natural products is aided by the analysis of structural analogs and establishment of SAR for active compounds often produced in the same source organism.99,103,109 Analog development for isolated natural products can also often be addressed by the development of synthetic methods.52,100,101 Rocaglamide, from Aglaia extracts, was found to be able to sensitize TRAIL-resistant cells to TRAIL-induced apoptosis. Advances in natural product synthetic chemistry approaches as well as purification of additional rocaglates from extracts allowed for development and analysis of rocaglamide and 55 analogs for establishment of SAR as TRAIL sensitizers and as protein synthesis inhibitors in renal carcinoma cells.110 Similarly, another group of TRAIL sensitizers, the withanolides, were initially isolated from active extracts97 and subsequently further development of much larger numbers of synthetic and semi-synthetic analogs.111 Identification of mithramycin from the EWS-FLI1 screen92 led to synthesis and evaluation of a large number of analogs, one of which is in pre-clinical development.112 Similarly, initial isolation of botryllamides from natural product extracts as inhibitors of ABCG2 (ref. 99) led to development of a synthetic method for generating increased quantities of analogs for SAR analysis and further pre-clinical development.113,114 Thus, while hit-to-lead progression is always a challenge, active molecules identified by cell-based HTS, advances in natural product chemistry and in synthetic methodology have made it much more feasible for natural products, a trend likely to continue.
3.4 Future considerations for the cell-based screening of natural products
Many of the natural products identified as modulators of the hallmarks of cancer were identified via cell-based assessment of extracts from small numbers of organisms; as were many of the molecular targets successfully exploited for cancer chemotherapy. A cursory look at the recent literature identifies dozens if not hundreds of phenotypic or cell-based assays applied to a only few extracts, often derived from traditional medicine(s).51 However, in order to access the broader chemical diversity in nature, it would be extremely valuable to adapt many of these assays to HTS and to apply them to larger libraries of natural product extracts. Within individual natural products discovery programs, screening of extracts against multiple targets and phenotypes can result in significantly increased understanding of extract characteristics. For example, identification of promiscuously active growth inhibitory and/or cytotoxic samples or modulators of gene expression across a variety of cells and assay platforms can allow for annotation of promiscuously active extracts and fractions. As large libraries of prefractionated extracts become available to a larger number of screening laboratories,26,108 over time it should become possible to annotate both extracts and fractions with reported activities in cell-based HTS, allowing for identification of problematic samples (i.e. extracts containing “PAINS”) and for data mining to increase the efficiency of isolation and structure elucidation efforts.A repeated criticism of cell-based assays in general is that they are often based on established cell lines in 2D culture which have far from in vivo characteristics after long term adaptation to cell culture.48,50,53,56 Many of the emerging technologies increasingly employed in cell-based screening are designed to make screening more physiologically relevant and include 3D spheroid culture, multi-cell models (e.g., tumor cells and tumor-associated fibroblasts in 2D or 3D culture as models of growth, migration, invasion), induced pluripotent stem cells, cancer stem cells, patient-derived cells (especially tumor cells), and “tissue on a chip” and “organ on a chip” technology to name a few.47–53,55,56 Unfortunately, there tends to be a trade-off between physiological relevance and throughput, so although some of these models are amenable to HTS, many are more suitable to secondary screening. These technologies can also be particularly problematic for application to crude natural product extracts due to some of the challenges discussed in this section. So, at this point, higher-order cell culture models do not appear to be widely used in natural products drug discovery. However, as illustrated above, prefractionation can substantially alleviate many of the problems due to toxicity, off-target effects on cell attachment, morphology, and migration, etc., and assay interference often realized when screening crude natural product extracts. Such efforts should make natural product extracts more amenable to cell-based screening in general, but particularly useful in assays utilizing novel approaches.
4 Natural product screening using cell-free assay technologies
In contrast to cell-based screening approaches, cell-free screening technologies enable the a priori restriction of potential molecular targets to a limited number of macromolecules (i.e. proteins, RNA or DNA) included in the assay. This allows for immediate orthogonal studies that can describe the kinetic, thermodynamic or structural basis for macromolecule–ligand interactions. Though these assays make for a more targeted approach to drug discovery, in the case of natural product samples, especially mixtures, they are prone to assay interference from many common “nuisance” compounds found in natural product extracts. The result is that natural product discovery in the context of a biochemical screen can be both a rewarding and a formidable endeavor. The former is evident in the preponderance of natural products that have gained approval from regulatory agencies across the globe, and the latter is acknowledged in the number of natural product scaffolds that have apparent non-specific activity across a wide array of biochemical assays.1,47,115 This duality highlights that although amenable to the same biochemical targets, assays, automation, and miniaturization as screens of synthetic libraries, biochemical screening campaigns of crude extracts for natural product discovery (NPD) are a distinct undertaking from pure compound screening campaigns.In contrast to a pure compound screen, in a NPD campaign substances screened range from a crude extract of a whole organism to an extract fraction separated by some chemical property (most commonly polarity).29 Given this complexity, single chemical agents arising from the primary NPD screen are most often isolated through an iterative process commonly referred to as assay guided fractionation (AGF). During AGF natural products chemists work toward isolating the single chemical entity responsible for assay activity by identifying the most potent fractions. AGF is oriented toward purity-based activity relationship (PAR) experiments whereby the observed potency of the tested substance increases as purity goes up. AGF creates a collaborative screening environment at the interface of chemistry and biology and highlights both a strength and weakness in NPD screens: the need for both highly trained biochemists for assay development and execution, as well as expert natural products chemists for compound isolation and structure elucidation. Despite the unique attributes of biochemical NPD screening campaigns, any assay established for a pure compound screen can generally be adapted for use in a NPD campaign. The balance of this section is oriented towards concepts, considerations and best practices for establishing or adapting biochemical screens for natural products discovery.
4.1 Target selection
The foundation of any biochemical screen is the target itself, and significant consideration about what positive and negative target modulation would look like in the context of the screening assay format should be taken prior to assay development.116 The particular validity of any one biochemical target to a given biological outcome (carcinogenesis, viability, senescence, etc.) is entirely dependent on the quality of the basic science research through which the target was identified, and is beyond the scope of this review. However, there is an increasing understanding that while a small molecule modulator may be found for any biological target, not all targets are equally accessible to small molecule binding.117,118 To this end, when considering a new target for biochemical assay development some consideration of “ligandability” (or druggability) is warranted. Ligandability is the concept that that there may be ways to assess whether a target will be easily accessible to common chemotypes and thus is likely to result in the productive discovery of a small molecule modulator.119,120 This concept has been recently described by the work of Edfeldt and colleagues at AstraZeneca who have retrospectively examined more than 30 biochemical high throughput screening campaigns and attempted to develop tools to better predict a priori which campaigns were likely to yield productive drug candidates.120,121 Originally this was done using NMR based fragment screening of small libraries (<2000 substances) of simple chemical scaffolds (<200 Da) and scoring which targets bound the most number of substances at a screening concentration of 1 mM.119,120 Under these constraints, targets were assigned low, medium and high ligandability based on the percent of compounds which bound the target. Retrospectively examining the AstraZeneca HTS outcomes, all targets assigned low ligandability failed to yield an actionable HTS lead while greater than 70% of targets identified with medium or high ligandability progressed from HTS into the AstraZeneca drug development pipeline. Subsequently, Edfelt and colleagues extended this observation to show a similar outcome when carrying out fragment based ligandability experiments using a thermal shift assay (discussed in detail below) rather than an NMR based approach.121 Adoption of this kind of biochemical target assessment in the field of natural products can be seen in the recent extension of ligandability methodology to “native mass spectroscopy” experiments from the research group of R. J. Quinn.122An additional body of literature assessing which classes of both biochemical targets and chemical scaffolds have been most and least successful for HTS development has also recently emerged.121,123–125 These “target-class” assessments highlight potential limitations of a target or scaffold; but may not predict the behavior of either a novel target or a well-annotated target in a novel assay system. The emergent nature of both ligandability and target-class assessments suggests that due diligence prior to undertaking assay development for a biochemical screen is critical.
4.2 Assay selection and development
Depending on the nature of the target, a decision must be made as to whether the screening assay will have an enzymatic (for enzyme targets) or a biophysical readout, useful for both enzymes and many other biomolecules. This choice is most often dictated by the resources and experience of the research group; however, the strengths, limitations, and a few of the options available for each assay type are discussed below.4.3 Enzymatic assays
As our understanding of molecular biology has advanced, enzymes and enzymatic assays have become a significant driver of drug discovery campaigns.41 Indeed, with advances in technology, the paradigm of drug discovery has evolved from one of “a drug for every disease” to a drug for every gene variant or mutation.126 Translational biotechnology has for the most part kept pace with advances in basic science through the rise of recombinant protein expression systems in a variety of different host organisms.127 This has greatly expanded the biological space of tractable enzymatic targets for assay development.Enzymatic assays are pervasive, productive, and proven to generate clinically useful drugs.41,128 However, clinical approval of natural products (or their derivatives) whose activity originated in a biochemical high throughput screen has recently lagged that of leads derived from screening synthetic libraries.129–131 One possible explanation for this decrease is an understanding that biochemical NPD screens must be deliberately designed to counter nonspecific interactions with substances from the source organism. These substances have come to be known informally in the literature as Pan Assay INterference compoundS (PAINS).47,102,115 False negatives rarely fall into the PAINS class, and arise primarily from the absence of a reagent from the assay (misdelivery of the substance via a clogged tip/air bubble for example), which is largely a matter of chance and is therefore difficult to account for in assay design and execution. False positives, compounds for which SAR cannot be developed, on the other hand generally operate through several well annotated mechanisms of action whose presence should be accounted for during assay design. Several of these mechanisms and recommendations to specifically account for them are described in detail below.
An alternative excipient to consider alone or in combination with glutathione is an assay-independent protein that can be added in vast excess to analytes while minimally affecting the robustness of the assay.135 The addition of albumin, casein, gelatin, or another protein at a saturating concentration without affecting enzymatic catalytic parameters can be beneficial in providing “biological decoys” for the interaction with either aggregators or electrophilic substances. The effect of optimizing screening buffers by the addition of small amounts of detergents and excipient proteins has been well documented for use in pure compound screens.139 Consistent with observations for pure compound screening, we have found that incorporating both excipient proteins and non-ionic detergents has dramatically reduced our primary screening hit rate while allowing for the discovery of legitimate enzymatic activity modulators.107
An alternative to prereading a library prior to assay execution is to specifically interrogate primary screening leads against the fluorophore/chromophore used in the assay of interest. If the library component diminishes the observed signal from the fluorophore/chromophore alone, it is very likely due to signal interference rather than modulation of actual assay activity.141 As shown in Fig. 4, we have recently used this method to ensure that a recently reported natural product protease inhibitor discovered in a fluorescence-based assay did not substantial interfere with the assay readout over the relevant IC50 range. Fig. 4 clearly shows that while the enzymatic activity is significantly reduced at the IC50 (32 μM), the fluorescence of the fluorophore itself (red line) at the assay concentration is unaffected by the presence of the inhibitor. In the context of biochemical NPD screens, it is worth giving thought to the absorbance profile of the chromophore/fluorophore of interest. There are very likely to be strongly absorptive substances in a natural products extract, indeed many fluorophores/chromophores were initially identified from natural products. However, there are now several recently developed red-shifted chromophore/fluorophores whose bathochromic shift is better suited to natural products discovery than many of those used historically.142
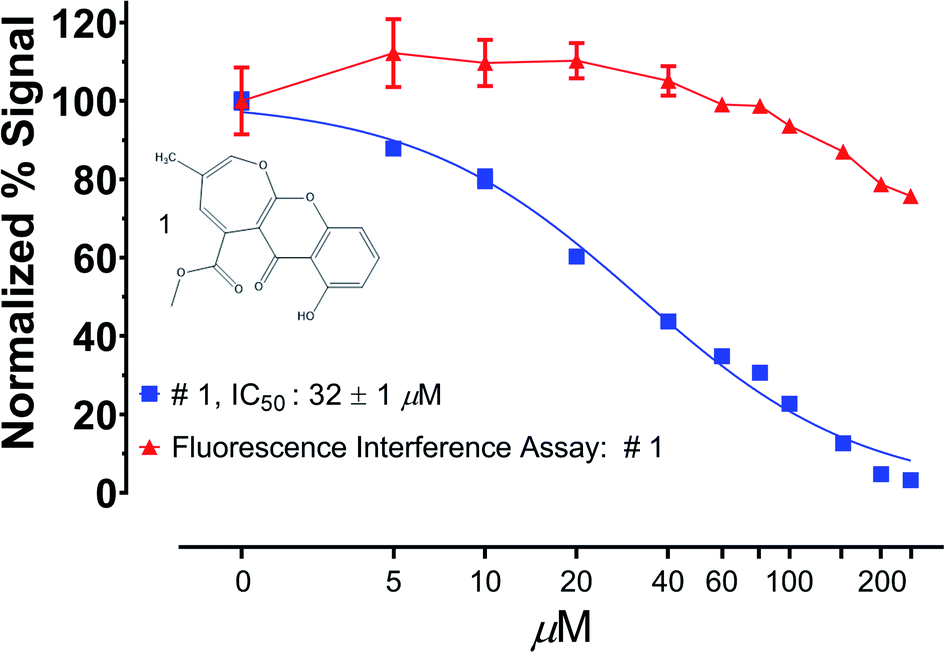 | ||
| Fig. 4 Fluorescence interference assay. Compound 1 was isolated from a fluorescence based protease inhibitor assay and was then rescreened in dose response against the fluorophore alone (7-aminomethylcoumarin) and found not to interfere with fluorescence detection throughout the IC50 range of compound 1. Reprinted with permission from: T. D. Tran, B. A. P. Wilson, C. J. Henrich, L. M. Staudt, L. R. H. Krumpe, E. A. Smith, J. King, K. L. Wendt, A. M. Stchigel, A. N. Miller, R. H. Cichewicz, B. R. O'keefe and K. R. Gustafson, J. Nat. Prod., 2019, 82, 154–162. Copyright 2019 American Chemical Society. | ||
A final option, particularly well-suited to AGF during natural products discovery, is to monitor the assay specific wavelengths during isolation and purification steps of the potential active component. Many modern HPLC instruments come equipped with a UV-vis diode array that makes monitoring multiple wavelengths during purification a tractable option for gaining insight in the spectroscopic profile of potential lead molecules.
4.4 Orthogonal screening
The use of a specific orthogonal assay, particularly a cell-based one in the context of a biochemical primary screen, can provide clarity about the viability of further development of a potential lead.142 For example, aggregators and thiol reactive compounds could be anticipated to be competed away by the presence of high concentrations of soluble protein found in many tissue culture media formulations; while the different assay readout would most likely identify any compounds whose initial activity was largely due to signal interference. More importantly, a well-designed cell-based assay should be able to discriminate between non-specific activities associated with PAINS compounds and specific activities necessary for progression through a drug development pipeline.An orthogonal target-specific secondary assay is essential for increasing confidence in a potential lead molecule. However there can be tremendous value in establishing secondary assays against unrelated enzyme classes.134 For many natural products discovery programs there may already be a database of annotated activities from prior screening campaigns that can act as an early indicator of potential target specificity. When this is not the case, a second unrelated assay to counter-screen leads identified in the primary assay can be helpful. It has been our experience that an excellent counter-screening assay is that of the β-lactamase AmpC.134,143 We have found that we can isolate large quantities of stable active enzyme from the spent Lysogeny Broth (LB) media used to grow ampicillin resistant bacteria for recombinant protein expression. Using spent media as an enzyme source and a straightforward chromatography purification protocol, we can isolate active enzyme which can then be assayed against a variety of commercially available chromogenic β-lactamase substrates.144 Beyond insight into target specificity, this assay has the added benefit of being able to provide information about potential aggregators. The AmpC β-lactamase can tolerate levels of non-ionic detergent that can readily disperse compound aggregates.134 Therefore, if a compound is active against AmpC only in the absence of detergent it is likely an aggregator and can be deprioritized for further development.
4.5 Mass-based screening approaches
A significant limitation for enzymatic screens is the detection reagents themselves. This can be a product-specific label (fluorophore/chromophore/radioisotope), primary antibody (for ELISA based detection), or a coupled enzymatic assay system. Emerging technologies have sought to address this by establishing label free methods of enzymatic turnover detection. Directly measuring the enzymatic conversion of substrate to product is the ideal assay readout.145 This would improve assays by reducing artifact generation due to indirect measurements, reducing the false positive rate due to non-enzymatic signal interference (quenching), and reducing costs of assay specific detection reagents. With the exception of isomerases, all other classes of enzymes catalyze reactions resulting in a change in mass in order to generate the product; therefore, a generalizable label-free enzymatic assay would involve monitoring mass with time, an observation ideally suited to mass spectrometry (MS). The potential impact of mass spectrometry applied to enzymatic high throughput screening has been realized in the field for years and yet progress towards its implementation has been slow. Inherent in the idea of high-throughput screening is that assays are optimized to read the most number of samples in the smallest amount of time. However, most mass spectrometers require a liquid chromatography (LC) step prior to sample ionization which can become an insurmountable time-sink during large screening campaigns. An initial MS solution to this problem acknowledged that in the context of high throughput screening the central role of chromatography was not analyte separation but rather buffer exchange to eliminate assay buffer components that are incompatible with electrospray ionization (ESI). Therefore, traditional gradient based LC programs were eliminated in favor of rapid solid phase extraction (SPE) buffer exchange programs. This lead Agilent to develop the RapidFire MS system, which when using an autosampler can reduce sample read times to ten seconds per sample from a previous average of one minute per sample.146,147 However, with many screening campaigns on the order of >500000 samples, the RapidFire MS is most useful for selective library screening, SAR development, and orthogonal assay confirmation applications.148 More recently, HTS by MS has been undertaken through the use of acoustic dispensing directly into the ionizer itself, further reducing the per sample screening time to 3 samples per second.149 However, it should be noted that in addition to a direct injector acoustic dispenser, care must be taken to both optimize the assay buffers to an ESI compatible system as well as to empirically determine the ionization efficiency of both the product and the reactant ions in order to appropriately account and correct for the effect of differential ionization on interpretations of enzymatic turnover.149–151 At present acoustic dispensing ESI-MS for HTS has not been commercialized but will likely see further progress for new label free HTS assay development.
An alternative to ESI-MS driven HTS is to switch the ionization format to matrix assisted laser desorption ionization mass spectroscopy (MALDI). Progress in automation has facilitated the use of MALDI for HTS in 384, 1536, and most recently 6144 formats.150–152 As with any assay, HTS by MALDI does require significant optimization to ensure that catalytically relevant turnover is being detected as well as a quantitative understanding of the degree of ionization of both the substrate and product, often accomplished through the inclusion, titration, and calibration of isotopically labeled substrates and products as standards (both during assay development and often as an internal control during the HTS screening campaign).150–152 Despite the need for optimization, the reduction in reagent costs make MALDI an attractive potential screening format.152
4.6 Non-enzymatic biochemical screens
In addition to enzymatic biochemical screens, new cell-free technologies have recently emerged that allow for the direct interrogation of the interaction between biomolecules and small molecules in the context of a high throughput NPD screen.153–155 For many disease-associated molecular targets that are not enzymes but do have structural features that can be probed for small molecule binding biophysical screening techniques are useful. Since these newly developed HTS technologies do not rely on biochemically catalyzed reactions but rather on a physical interaction between the small molecule and the target protein/nucleic acid, they are considered biophysical high throughput assays.A few technologies, like fluorescence polarization (FP), can be utilized for both enzymatic biochemical assays and interaction dependent biophysical assays.156 Based on the observation that fluorescence emission originating from a polarized light source is depolarized during the course of emission and that this depolarization is related to the geometric volume of the fluorescent species, FP can be used to measure alterations in this volume due to ligand binding or enzymatically catalyzed reactions which result in a mass change of the fluorescent species.156 FP calculations are accomplished by measuring fluorescent emission at two angles, one perpendicular and one parallel to the excitation plane. These two measurements are then used to calculate the difference in fluorescence intensity between the two angles. Measurements made in the presence of ligand (small molecule, biomolecule, enzymatic substrate, etc.) for the fluorescent species can be compared to a reference reading without the ligand to infer a change in the molecular volume of the target due to ligand binding. As with many fluorescent homogenous phase assays, this technique was rapidly adopted into MWP format, and has be employed for a wide range of drug discovery campaigns.157 Since the FP measurement itself can be affected by the chemical characteristics of the fluorophore, the nature and location of the fluorophore itself on the target molecule is crucial.156,157 For example, FP measurements are dependent on the excited state lifetime of the fluorophore (τ), and in general large target molecules will require fluorophores with large (τ); however, for most FP dependent screening campaigns it will be necessary to empirically determine which fluorophores (and linker lengths) are best suited to the assay. Interestingly because FP depends on a relational ratio between fluorescence at the same wavelength, it suffers less from fluorescence interference (since quenching affects both angles of measurement equally). However, fluorescence amplification due to the fluorescence of screening substances can make comparisons to controls difficult. Despite these limitations, well-developed FP assays can yield clinically relevant drug candidates.156,157
Another biophysical assay that has been adopted for HTS assays is the thermal shift assay (TSA).153,155 This assay measures the thermal stability of a target biomolecule (proteins initially but more recently nucleic acids) across a temperature gradient by monitoring the unfolding dependent binding of a fluorogenic dye.153,155 As the biomolecule slowly unfolds with increasing temperature, more of the dye is incorporated into the structure so that a characteristic melting curve is generated and a specific melting temperature is calculated (TM). Comparing the deviations of the TM in the presence of a test substance has been shown to indicate specific interactions (stabilizing or destabilizing) between the substance and the target. The availability of MWP compatible optical thermocyclers has facilitated the adoption of this biophysical screening technique. Although implementation of this screening modality is both convenient and relatively cheap, it does have a few potential limitations.153,158 As it depends on fluorescence detection, a TSA can suffer from fluorescent interference from test compounds. In addition, deflections of the TM are not necessarily indicative of binding affinities (greater changes in TM do not equate to higher or lower affinity). To accurately model the thermal stability of the target fluorescence data must be collected over the entire temperature range of the experiment which can generate large amounts of raw data requiring significant data processing capabilities. Finally, the suitability of the target to the TSA must first be assessed to ensure the measured control TM is in a range compatible with dye binding. Despite these variables, the TSA has seen increasing utility for diverse targets across a number of commercial and academic drug discovery centers.153,155,159
Among the earliest high throughput biophysical drug discovery assays was the scintillation proximity assay (SPA).142,154 An early iteration of this microbead based assay used target protein coated scintillant microbeads and radiolabeled ligands to detect substances that interfered with the target–ligand interaction, detectible as a decrease in bead (target) dependent scintillation. As the field moved away from radiolabeled ligands the SPA was replaced with Förster Resonance Energy Transfer (FRET) based bead dependent assays, such as the LANCE format, whereby a lanthanide containing microbead coated with one member of an interacting pair was incubated with a complimentary FRET acceptor labeled interactor and then probed with a chemical library to find substances that competed with this interaction. The AlphaScreen is a related modification on this format.160 One partner has a microbead whose excitation causes the emission of a singlet oxygen species whose excited state electron can be accepted by the acceptor bead ligated to the bound partner.160 Again, any reduction in energy transfer in the presence of screening substance could be attributable to disruption of the pairwise binding interaction. While the above bead-based assays were designed specifically to be compatible with MWP based formats, the Luminex bead-based system offers a flow cytometric multiplex bead array-based readout. A bead itself is fluorogenically encoded to indicate which target molecule it carries and the bead is then probed with a potential binding partner that is itself fluorescently labeled or can be secondarily labeled with a fluorescent antibody.161 The target beads, in solution, can then be siphoned into a fluorescence activated sorter and both target and binding partner fluorescence can be measured, indicating which target and which ligand are interacting. Perturbations to the interaction between the binding pair in presence of a substance may be indicative of the substance specifically binding to one member of the pair. An advantage of the Luminex bead based format is the ability to probe multiple targets in a single well (related kinases for example) due to the fluorescence encoding of the target bead.162 For each of these assays there have been modifications to broaden their applicability and ease of use but, as they all are fluorescent assays, the chance for false positive identification due to fluorescence interference is present. In the realm of natural products discovery this has proven to be true even in the context of the AlphaScreen, where the ability of natural products to scavenge singlet oxygen has been observed as a source of false positive discovery.115,142 A recent iteration on bead based biophysical assays is the use of quantum dots in place of microbeads, these dots can function in the same manner as microbeads, but are smaller, allowing for further assay miniaturization, and have been found to be more “tunable” to wavelengths that are red shifted and less likely to suffer from interference from natural products.163,164 Their small size necessitates the use of laser induced fluorescence (LIF) which, while an exciting technological innovation, has yet to reach peak commercialization.
In addition to the screening formats identified above, which have published papers indicating the ability to process >8000 samples per day, there are emerging technologies for establishing biophysical screens that may become accessible for HTS in the coming years. Among these techniques are biolayer interferometry (BLI) and microscale thermophoresis (MST). BLI is a technology for quantitating the interaction between a target and a ligand by measuring the degree of photometric interference as light transverses an optically clear probe that has a target of interest attached to the end.154,165,166 In a MWP format, the degree of interaction between the target coated probe and anything in the well (like a natural product) can be assessed by the effect on light reflected to the detector (relative to control wells). While there are currently both 96 and 384 well compatible systems, the visualization optics is limited to 8 and 16 wells, respectively; requiring column by column progression across a plate and increasing the read time per plate. At the moment BLI based screening is largely limited to selective library subsets, primary hit triage, and secondary screening.165,166
MST is an emerging technology that measures the transit of a fluorescent analyte across a temperature gradient.153,154,167 The interaction of a ligand with the analyte will slow the migration time across this gradient, generating a signal indicative of a binding event. Currently a 96 well MST instrument is commercially available which uses capillaries to sample all wells across the plate. A laser is used to generate a thermal gradient within an individual capillary and the migration of the analyte is measured. Unfortunately, measuring a well-controlled gradient requires approximately 20 seconds per sample, limiting its utility.167
An interesting hybrid assay format is micro/nanocapillary electrophoresis (CE), which continues to be an HTS format that is frequently employed for drug discovery.168 Improvements in the optics of both excitation and emission using LIF has allowed increasingly small reaction volumes to be deployed in the context of an HTS screening campaign. This has spurred the exploration of microfluidics, or even nanofluidics, in the context of high throughput drug discovery.169,170 To this end the use of both microfluidic capillary electrophoresis (CE) and nanofluidic droplet based assays have both been recently used as the assays of choice for novel drug screens.171 Currently, the single greatest impediment to implementation for these assays is the pervasive adoption of MWP formats for chemical library generation, storage, and utilization. Microfluidic based assays are dependent on the movement of an analyte through a sample window and were not initially developed with MWP formats in mind, which means that the assay itself often has to be reformatted from a MWP format prior to introduction into the detection system. This reformatting can both lower throughput and necessitate additional capital outlays which has been a hinderance to both commercialization and adoption in the drug discovery community.
4.7 Multiplexed biochemical assays
Reducing the cost of biochemical NPD screening is often the first priority in assay development. Advances in target expression systems and reagent delivery (robotic liquid handling and now acoustic ejection) have begun to bring these costs down.172 Acoustic ejection for reagent delivery is particularly useful in the context of NPD screens because nanoliter delivery volumes are more efficient when screening libraries of natural products for which resupply can be challenging.1,142A creative approach to reducing the cost of a single assay is to make the screening campaign more data rich through multiplexing. In the context of biochemical NPD screens, multiplexing can encompass several approaches during assay execution: (1) including two or more discrete biochemical targets in a single well; (2) using multiple distinct substrates so that MWP can be pooled and read simultaneously (reducing read time but maintaining the same number of screened wells); (3) pooling substance plates to reduce the total number of wells screened while maintaining the total number of substances screened. The ability to maximize a single extract/fraction for multiple assay endpoints is an ideal though infrequently utilized technique to assess both the potency and specificity of a given library component. An example of this approach was published in 2011 where the authors measured deubiquitinase, deSUMOylation, and ISG15 removal (another small ubiquitin like protein, deISGylase) simultaneously in the presence of three non-overlapping fluorescent activity probes to discover potent and specific small molecule modulators of these enzymes.173 While that publication utilized enzymes from similar enzymatic classes, proteases, this need not always be the case. It may be possible to establish multiplex assays for non-redundant collections of enzymes as long as the ultimate readout of each class gives a discrete indicator of enzymatic activity. One common approach to multiplexing is to assay multi-enzyme complexes (the entire E1/E2/E3 ubiquitin ligase cascade for example) in a manner that allows for interpretation of where in the given cascade there is a productive interaction between the library substance and the target cascade.174 This approach can have the added benefit of the ability to substitute related family members (different E3's ubiquitin ligases for example) in order to elucidate and quantify specificity among related targets. Additionally, multiplex immunoassays (several distinct antibodies specific for different targets spotted at the bottom of a MWP) are commercially available (Meso Scale Discovery for example) which allow for the interrogation for multiple assay products in a single well; however, these formats are limited by both the specificity of the antibodies used and often the bespoke manufacture by a single commercial vendor, limiting their accessibility.
An alternative multiplexing approach has been applied to decrease the assay read time for HTS-MS.175 In this case four distinct (by mass) substrates for a lysine demethylase were created and optimized. Each probe was used to screen one-fourth of the total library and then four discrete assay plates could be combined and screened simultaneously by RapidFire M.S. (Agilent Technologies). The extent of enzymatic inhibition for each substance could then be read by correlating it's well location to the probe type used for that plate.175 While this example does require more data processing, it is an automatable process and did increase overall throughput by 4-fold. A final iteration on multiplexing HTS is library pooling.176,177 In this strategy, the screening library itself is pooled and screened as normal and then any potential leads are then rescreened as individual components during secondary follow-up. This approach is most amenable to compound screening with assays that have very low hit rates and thus attempts to address the “dark-matter” problem (the majority of screened wells are inactive) encountered in large screening campaigns.178,179 Some implementations of this approach will pool samples more than once throughout the screening process in order to speed hit identification (a legitimate lead should reproduce regardless of its pooled constituents, and a false positive should not be reproducible upon rescreening) at a cost of expanding the total number of wells screened. While it is not clear that this multiplexing approach is well suited to natural products discovery, where library components are infrequently single compounds, one could envision pooling parts of libraries that are infrequently found to contain leads in order to reduce overall screening costs.
4.8 Recommendations for biochemical assay development for natural products discovery
Our experience with various biochemical NPD screening campaigns has compelled us to establish what we call PAINS Aware Assay Development (PAAD), summarized in Fig. 5. These are rudimentary suggestions for how one might design and develop a biochemical assay in anticipation of and incorporate countermeasures against the appearance of PAINS compounds during library screening, this section is intended to be a detailed description of the PAAD workflow depicted in Fig. 5. | ||
| Fig. 5 Pains aware assay development workflow. A schematic of a generalized assay development workflow in anticipation of and incorporating countermeasures against pan assay interference compounds (pains). | ||
All assay development of course begins with pre-development consideration of the assay target, both in terms of concepts introduced above (Section 4.1) as well as practical considerations like the observed signal-to-noise ratio prior to PAAD implementation. This can be important because many of the PAAD recommendations can reduce the overall signal, and so beginning with a usable signal-to-noise ratio (generally at least 5:1) is important. In the context of photometric assays, potentially absorptive natural products as screening components must be taken into consideration and we recommend using red-shifted assay readouts. All assay actives will eventually need to be validated in an orthogonal assay, preferably in a format distinct from the screening assay (cell based for biochemical primary screening assays and vice versa for cell-based screening assays); therefore, it is critical that this assay be fully considered (and ideally already validated) prior to beginning primary screening. Although it may not possible for many biochemical targets, having a positive control (even with low potency) to help delineate the likely dynamic range of an assay under screening conditions can be extremely beneficial at both validating the assay readout and orienting the data analysis to the detection of assay actives. The hit rate of a novel assay is unknowable a priori; however, some consideration of how either end of the hit rate spectrum will be handled during the course of screening is an important factor to consider. For example, if during screening it becomes apparent that fractions derived from certain source types tend to behave discretely from others, are there methods for rapidly assessing this behavior or triaging their follow-up? Is there a hit rate that is likely to overload the follow-up capacity of the orthogonal assays, and if so by what metrics will you prioritize some leads over others? The greater consideration given to screening outcomes before screening begins, the better executed the screening campaign is likely to be.
Once the assay has been shown to be functional, it can often be useful to screen a small but representative portion of the natural products library, if resources permit. This will provide insight into the upper end of the hit rate range, a baseline for the effect of further PAAD implementation, and a reference for how PAAD has impacted assay performance. Depending on the assay target, some consideration for the actual pH buffering components (phosphate vs. tris base for example) will be necessary, but that will largely be dictated by the idiosyncrasies of the target itself and is beyond the scope of this review. However, there are a number of additional buffer additives, described more fully above and summarized below, that we recommend for consideration. We recommend determining maximum assay tolerance (indicated by the effect on the signal) for non-ionic detergents and excipient proteins (“decoy” or molecular “sponge” proteins). Inclusion of these as buffer components can dramatically reduce false positive detection due to library component aggregation.180 While it is necessary for many biochemical assay targets to remain in a fully reduced state for physiologically relevant screening (thus necessitating the inclusion of reducing agents in the buffer), the reductive capacity of the reducing agents may also facilitate the non-specific attack of the target by nucleophilic library components.136 Therefore, it is worth empirically determining the performance of the assay with diminishing concentrations of reducing agents. Following assay optimization along these vectors, a rescreening of the initial pilot library will hopefully reveal increased assay robustness in the form of a reduced hit rate while maintaining an acceptable signal-to-noise ratio and z-factor.
At this point in the PAAD process, primary screening should be initiated. Following primary screening, we recommend first establishing reproducibility leads at the screening concentration at least in quadruplicate with a maximum potency drift of 10 to 15%. Following reproducibility confirmation, an assessment of dose response is warranted. We recommend examining all dose response curves for both unusual plateaus in potency and for steepness of the Hill slope either of which has been shown to represent a likely indicator of assay interference or non-specificity.181 Having now established that leads are both reproducible and have an acceptable dose response profile, we now recommend re-examining the potential for photometric assay interference in dose response. The execution of this counter-screen will vary based on the assay type, but within our research group it is typically carried out by incubating the potential lead agent with the signal generator itself (fluorophore/chromophore) at a concentration that generates a signal commensurate with the raw values of the assay itself, Fig. 4.141 Since the potential lead is not likely to be spectrophotometrically identical to the signal generator, it is important that the concentration of the generator be titrated to match the raw readout of the screening assay and not used at the same concentration as in the screening assay itself. For example, if an assay is run at probe concentration of 10 μM with a 10% turnover into the signal, then we would run the spectrophotometric interference assay at a concentration of 0.1 μM of the free signal generator. While there is no accepted standard for what level of photometric interference is allowable, we typically deprioritized leads if a photometric signal reduction of more than 10% at the estimated EC50 of the potential lead molecule is observed. Finally, should a potential screening lead continue to hold up throughout the battery of countermeasures described so far, we would finally recommend that the lead be screened against the target in dose response in the presence of a thiol scavenger like glutathione.182 This of course assumes that the assay itself is tolerant of the presence of millimolar quantities of glutathione (we frequently use 5 mM) during assay execution. Shifts in potency solely attributable to the presence of a thiol scavenger suggest that the behavior of the lead is dependent on the assay conditions themselves and not on a tractable interaction between the lead and the target. At this point in the lead assessment process we would consider remaining leads of interest to be validated as assay actives and would then assess their target specificity in our previously established orthogonal assay. The point of the PAAD process is to subject early screening leads to an exhaustive battery of potential counter-screens in order to eliminate non-specific or nuisance compounds as well as to generate as much data as possible to validate on-target potency. One beneficial outcome of this rigorous screening is that leads which at this point fail to remain active in the subsequent orthogonal assay (generally for reasons of cell permeability or other pharmacokinetic parameters) have been thoroughly vetted as biochemical bioprobes whose chemistry can be further optimized for bioavailability and whose target binding sites can likely be characterized through further biophysical means (crystallography, molecular docking, etc.). All of which is to say we have found that by employing PAAD during our screening process we have been able to prioritize our research efforts on lead discovery of molecules which are likely to be of use to both the clinical and the research communities.
4.9 Emerging trends in biochemical assays for natural products discovery
Very few of the numerous published biochemical screens include natural products discovery as part of the campaign.142 However, a review of the literature in the last 5 years indicates that NPD campaigns have been successfully executed using all of the biochemical and biophysical screening technologies identified above.183–190 Of the published NPD screens, biophysical screening technologies, like TSA and FP are more frequently employed than other technologies. This may be due to the fact that these formats generally require only a single input (the target, rather than substrates and cofactors) and are largely agnostic to the state of the target (active as well as inactive enzymes are suitable if they are appropriately folded). However, the observation that all screening technologies appear to be amenable to NPD screening suggests that the dearth of published NPD campaigns in the last five years is not due to a technological limitation, but rather a more fundamental structural deficit. One explanation is that the skillsets necessary to establish a relevant biochemical assay and to fully elucidate any natural products that emerge from that assay are too widely divergent for a single research group to effectively master both. This limitation most likely underlies the observation that the majority of NPD campaigns involve collaborations between independent biochemical and natural products discovery groups. This collaborative imperative in NPD campaigns suggests that for natural products to continue to be a rich source of both chemical and pharmacologic diversity, specific collaborative programs and initiatives should be encouraged and funded across institutional, governmental, and industrial research organizations.A second explanation for the underrepresentation of biochemical NPD campaigns in the literature is that the utilization of natural products is proportional to their distribution within the research community itself. The conscious uncoupling of natural products research from industrial pharmaceutical research and development has been comprehensively covered elsewhere; however, the number of academic centers with natural products discovery capabilities has remained relatively constant, see Fig. 6. While it is heartening to see that the total number of discrete natural product screening grants has not decreased significantly in the last 10 years, a period marked by global fiscal austerity, it is worth noting that though a significant percent of approved pharmaceuticals are derived from natural product scaffolds, the number of grants awarded for the NPD campaigns is consistently 5-fold lower than non-natural product discovery efforts, Fig. 6.1 Collaboration here too may be a way to increase the number of biochemical NPD screening campaigns as there is a recently announced initiative from the National Cancer Institute to widely distribute a large novel library of prefractionated natural product extracts to any organization for use in NPD programs.26 Optimistically this can serve as template for other large natural products discovery groups to more widely distribute their natural products collections in order to facilitate both comprehensive coverage of global organismal diversity as well as to inspire biochemical screening groups to implement novel and productive NPD campaigns.
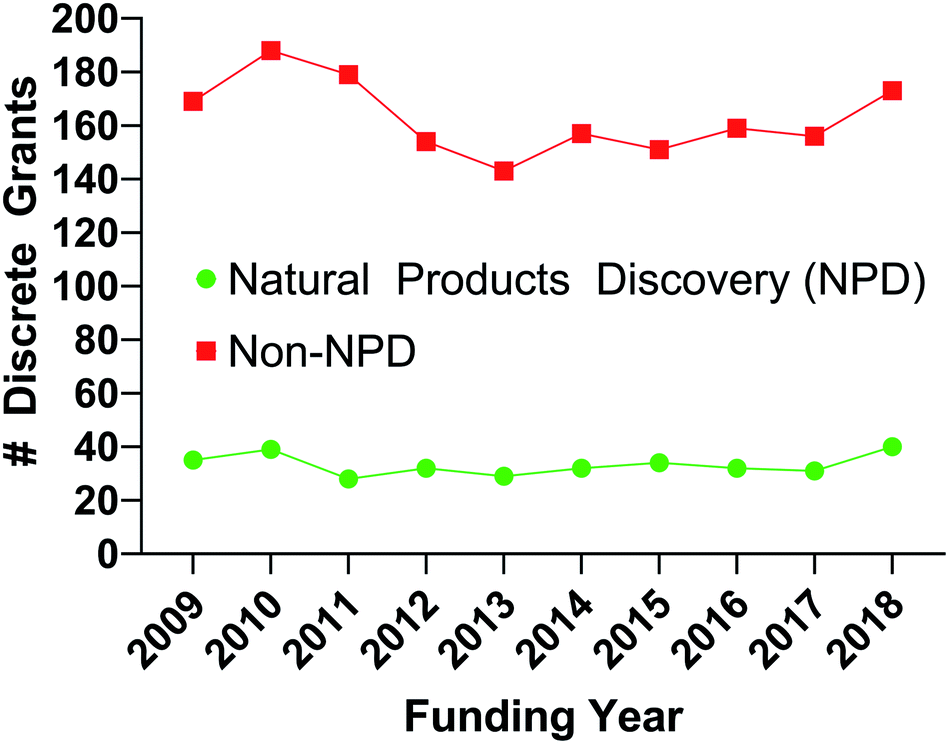 | ||
| Fig. 6 Funding for High Throughput Screening (HTS) for Natural Products Discovery (NPD) has remained stable but 5-fold lower than other HTS campaigns. Compilation of NIH reporter data (https://projectreporter.nih.gov/reporter.cfm) for specific search terms within awarded NIH grant abstracts (green: “high throughput” and “natural products”; red: “high throughput” and “drug discovery”) reveals dramatically fewer grants awarded specifically for NPD. | ||
5 Post-assay natural products discovery
Post screening, the task of isolating and identifying active compounds is guided by the fact that natural product extracts represent a mixture of secondary metabolites and can number up to hundreds of individual small molecules. Traditionally natural product-based AGF processes include several iterations of fractionation and secondary screening which significantly increases hit identification timelines. Moreover, knowledge of structure and thus novelty, drug- and lead-likeness comes at the very end, a disadvantage for NPD efforts compared to those with pure compound libraries. To overcome this isolation bottleneck and improve on the speed of hit identification, several approaches are now commonly employed by natural product chemists including prefractionation of crude extracts, small-scale dereplication for the identification of known compounds, and use of automated chromatography instrumentation in the isolation workflow.5.1 Benefits of prefractionation post-HTS
The benefits of prefractionation for screening efforts have been discussed previously in this review. Following HTS, the use of prefractionated libraries significantly reduces the number of compounds present in active samples and simplifies the isolation and structure elucidation efforts. For example, the NPNPD prefractionated library, comprised of seven fractions per crude extract, was shown to contain anywhere between 2 to 28 compounds per fraction when analyzed by LC-MS-ELSD detectors.26 This compared to estimates of 100+ natural products per crude extract results in a substantial reduction of follow-up isolation procedures. Part of the NPNPD methods development involved demonstrating that structurally diverse natural products, isolated from plant, marine invertebrate, and microbial sources can be isolated in a rapid two-step procedure. Fig. 7 shows four biologically active natural products that were isolated in only two chromatographic steps, which included initial SPE-based prefractionation and a single HPLC separation as the second step. The two-step procedure yielded sufficient material for complete structural confirmation (including 13C NMR spectral acquisition) as well as NCI-60 analysis. | ||
| Fig. 7 Structures and NCI-60 human tumor cell lines screen activity of natural products isolated in two steps from a prefractionated library. | ||
5.2 Dereplication
Dereplication is herein defined as the identification known natural products from extracts identified as active in a bioassay by means of spectral fingerprint data combined with library searching. Incorporating a dereplication step early-on in the isolation workflow decreases the chances of re-isolating known or nuisance compounds and improves efficiency post HTS. Dereplication usually requires an analytical step where extracts or fractions are subjected to LC separation which can be coupled to multiple detectors such as UV-vis, mass spectrometry, light scattering, as well as capillary-flow NMR. The analytical data collected can then be used for in-house library matching as well as comparison against databases of known natural products such as the Dictionary of Natural Products,191 AntiBase192 or MarinLit.193 Dereplication methods based on LC-MS provide sensitivity as well as limited structural information in the form of molecular weight and formula. Tandem MS/MS spectra add an additional fragmentation fingerprint which, when combined with multivariate statistical analyses such as molecular networking, can improve the dereplication of known compounds and identification of close structural analogues.194 Tandem MS-based analyses have found particular value with web-based platforms such as the Global Natural Products Social Molecular Networking (GNPS), a publicly-accessible database of MS/MS spectra that allows users to deposit and analyze data, as well as search existing data to annotate compounds and identify putative analogues.195 Complementary to MS-based methods is the NMR analysis as this analytical technique has the potential to detect all natural products with paramagnetic nuclei and therefore analyze a much larger range of structural classes, significantly increasing confidence in dereplication results. NMR spectral acquisition can either be coupled to LC workflow using a capillary flow NMR instrument or performed offline. On-flow LC-NMR analyses have found limited use in post-HTS efforts on natural products as they require deuterated solvents for chromatography, water suppression pulse sequences, and short experimental acquisition times; all of which have a detrimental effect on the sensitivity of the signal. An alternative is to use stop-flow LC-NMR where the pumps are paused to allow for a longer acquisition time including two dimensional NMR experiments.196 The most common practice however is to generate replicate LC samples and conduct the NMR analysis offline. Then structural information from the NMR experiment is incorporated into a database search,197 or used to generate a library of NMR fingerprints that can be analyzed to define structural uniqueness and novelty.198 One of the disadvantages of NMR compared to MS-based dereplication is a significant loss of sensitivity, however advances of micro-cryoprobe and capillary probe NMR technology as well as the development of 1.7 and 1.3 mm cryo-probes have now enabled not only nano-gram dereplication but also full structural elucidation of either small-scale isolations or low yielding natural products.199,200 Ultimately, post-HTS dereplication efforts have the potential to rapidly detect known compounds and identify crude extract or fraction hits with identical and similar chemotypes, which can aid in the identification of projects that might be of further interest for follow-up isolation work and secondary screening.5.3 Natural product isolation and resupply
The field of small-scale dereplication and structural elucidation of natural products is rapidly advancing to require smaller amounts of the initial extract and of the purified compound. This translates to less time pursuing known structures with well identified targets and mechanisms of action and more time in isolating new and novel biologically active natural products. Although these advances have significantly increased the efficiency of efforts to identify new compounds of interest and decreased natural product-based isolation timelines, they have not addressed the need to supply adequate material for follow-up confirmation and mechanistic studies. While sub-milligram quantities of a natural product may be adequate for structure elucidation and initial biological activity assessment; re-confirmation, target identification, mechanism of action studies, and animal testing require significantly more material.Post-HTS natural product isolation and compound resupply can therefore greatly benefit from standardized, scalable, and automated isolation procedures. Most HPLC column vendors currently offer column technologies in a range of column dimensions and particle sizes with the ability to scale up from UHPLC to preparative HPLC. Other traditionally preparative-scale techniques such as flash chromatography, supercritical fluid chromatography, and counter-current chromatography have become more accessible, the equipment automated and sold as benchtop instruments with a relatively small footprint. In addition to automating chromatography, use of large capacity liquid handlers and fraction collectors is showing potential to significantly speed up the natural product isolation bottleneck. Some of the largest prefractionated natural product libraries, such as Nature Bank (202983 fractions),201 Bioinformatics Institute Singapore prefractionated library (120000 fractions),202 and the NPNPD prefractionated library (326000 fractions as of January 2020)26 have been generated with automated, liquid handler-equipped LC- or SPE-based instrumentation.
Ultimately, for a natural product to progress to preclinical and clinical trials, multi-gram scale isolations are warranted. For such efforts, standard natural product laboratory equipment simply cannot do the task in a short timeframe where customized processes are required. To supply sufficient material for early preclinical development of bryostatin 1, a 13000 kg (wet weight) collection of the bryozoan Bugula neritina was collected off the coast of Southern California and a large-scale isolation under GMP (Good Manufacturing Practices) performed in NCI facilities yielded 18 g of pure bryostatin 1 in only seven isolation steps.203
Where possible, microbes represent a more sustainable and economical source of biologically active natural products. Modifications in growth media as well as manipulation of the biosynthetic gene clusters have the potential to enhance the yield as well as simplify the isolation procedures. Recently media optimization studies combined with biosynthetic engineering and gene deletion enhanced the production of a spliceostatin analogue thailanstatin A 40-fold compared to the wild-type producing organism, to a yield of 2.5 g L−1.204 In addition, the crude extract was shown to be 55% thailanstatin A which simplified purification to a single chromatographic step and provided adequate material for preclinical development.
The rapid development and automation of dereplication and isolation procedures following the identification of active natural product samples in high-throughput screens is facilitating an increased usage of natural product libraries in screening. As detailed above, the ability to subsequently acquire sufficient quantities of active compounds for pre-clinical studies is improving for amounts less than 1 g. Large-scale purification of active compounds from natural sources, such as that of bryostatin 1, is still a challenge which usually requires individual optimization and processes. The use of scalable technologies in both library creation and post-screen purification methods should bring benefits for later large-scale re-isolation efforts.
6 Conclusions
The field of pharmacognosy, or natural products chemistry, is dependent upon a series of allied efforts resulting in advances in library generation, cell-free and cell-based screening technologies, rapid dereplication methods and that ability to quickly isolate, identify and re-supply active compounds to researchers investigating the utility of compounds from nature. This review has detailed some of the recent advances in those technologies and shown how, taken together, they can facilitate an efficient process for the discovery of novel bioactive natural products. Though not detailed in this section, the underpinnings for all these processes is the rigorous annotation of organisms, their derived samples and the database, bioinformatics and quality control infrastructure to record and access taxonomic, geographic, genomic, biological and chemical details derived from those samples. Standards for this annotation have changed over the years. This has sometimes made “legacy” collections from the last century difficult to adapt to the modern realities of the equitable and reproducible creation and screening of natural product libraries. We hope that this review will encourage researchers to consider all of these aspects when creating the screening libraries of the future and that some of the technologies herein described will prove useful in their efforts.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E and by the National Cancer Institute's Cancer MoonshotSM. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and by the NCI Division of Cancer Treatment and Diagnosis Developmental Therapeutics Program. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.9 References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- Convention on Biological Diversity, Rio de Janeiro, 22nd May 1992, entered into force 29th Dec. 1993, https://www.cbd.int, accessed 2018 Search PubMed.
- E. Esquenazi, A. C. Jones, T. Byrum, P. C. Dorrestein and W. H. Gerwick, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5226–5231 CrossRef CAS PubMed.
- J. Koricheva and K. E. Barton, in The Ecology of Plant Secondary Metabolites: From Genes to Global Processes, ed. G. R. Iason, M. Dicke and S. E. Hartley, Cambridge University Press, Cambridge, 2012, pp. 34–55 Search PubMed.
- United Nations Convention on the Law of the Sea, Montego Bay, 10th Dec. 1982, entered into force 16th Nov. 1994, http://www.un.org/depts/los/index.htm, accessed 2018 Search PubMed.
- The Nagoya Protocol on Access to Genetic Resources and the Fair and Equitable Sharing of Benefits Arising from their Utilization to the Convention on Biological Diversity, 29th Oct. 2010, entered into force 12th Oct. 2014, https://www.cbd.int/abs/, accessed 2018 Search PubMed.
- E. C. Brown and D. J. Newman, J. Environ. Monit., 2006, 8, 800–805 RSC.
- C. Magnabosco, L. H. Lin, H. Dong, M. Bomberg, W. Ghiorse, H. Stan-Lotter, K. Pedersen, T. L. Kieft, E. van Heerden and T. C. Onstott, Nat. Geosci., 2018, 11, 707–717 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Front. Chem., 2015, 3, 34 Search PubMed.
- L. Du, A. J. Robles, J. B. King, D. R. Powell, A. N. Miller, S. L. Mooberry and R. H. Cichewicz, Angew. Chem., Int. Ed., 2014, 53, 804–809 CrossRef CAS PubMed.
- Z. Charlop-Powers, J. G. Owen, B. V. Reddy, M. A. Ternei, D. O. Guimaraes, U. A. de Frias, M. T. Pupo, P. Seepe, Z. Feng and S. F. Brady, eLife, 2015, 4, e05048 CrossRef PubMed.
- S. Fox, S. Farr-Jones, L. Sopchak, A. Boggs, H. W. Nicely, R. Khoury and M. Biros, J. Biomol. Screening, 2006, 11, 864–869 CrossRef PubMed.
- B. E. Richter, B. A. Jones, J. L. Ezzell, N. L. Porter, N. Avdalovic and C. Pohl, Anal. Chem., 1996, 68, 1033–1039 CrossRef CAS.
- T. A. Johnson, M. V. C. Morgan, N. A. Aratow, S. A. Estee, K. V. Sashidhara, S. T. Loveridge, N. L. Segraves and P. Crews, J. Nat. Prod., 2010, 73, 359–364 CrossRef CAS PubMed.
- F. Chemat, N. Rombaut, A.-G. Sicaire, A. Meullemiestre, A.-S. Fabiano-Tixier and M. Abert-Vian, Ultrason. Sonochem., 2017, 34, 540–560 CrossRef CAS PubMed.
- C.-H. Chan, R. Yusoff, G.-C. Ngoh and F. W.-L. Kung, J. Chromatogr. A, 2011, 1218, 6213–6225 CrossRef CAS PubMed.
- M. Herrero, A. d. P. Sánchez-Camargo, A. Cifuentes and E. Ibáñez, TrAC, Trends Anal. Chem., 2015, 71, 26–38 CrossRef CAS.
- W. P. Jones and A. D. Kinghorn, in Natural Products Isolation, ed. S. D. Sarker and L. Nahar, Humana Press, Totowa, NJ, 2012, pp. 341–366 Search PubMed.
- T. G. McCloud, Molecules, 2010, 15, 4526–4563 CrossRef CAS PubMed.
- M. S. Butler, F. Fontaine and M. A. Cooper, Planta Med., 2014, 80, 1161–1170 CAS.
- V. Seidel, in Natural Products Isolation, ed. S. D. Sarker and L. Nahar, Humana Press, Totowa, NJ, 2012, pp. 27–41 Search PubMed.
- C. J. Henrich and J. A. Beutler, Nat. Prod. Rep., 2013, 30, 1284–1298 RSC.
- J. H. Cardellina, M. H. G. Munro, R. W. Fuller, K. P. Manfredi, T. C. McKee, M. Tischler, H. R. Bokesch, K. R. Gustafson, J. A. Beutler and M. R. Boyd, J. Nat. Prod., 1993, 56, 1123–1129 CrossRef CAS PubMed.
- I. Schmid, I. Sattler, S. Grabley and R. Thiericke, J. Biomol. Screening, 1999, 4, 15–25 CrossRef CAS PubMed.
- C. C. Thornburg, J. R. Britt, J. R. Evans, R. K. Akee, J. A. Whitt, S. K. Trinh, M. J. Harris, J. R. Thompson, T. L. Ewing, S. M. Shipley, P. G. Grothaus, D. J. Newman, J. P. Schneider, T. Grkovic and B. R. O'Keefe, ACS Chem. Biol., 2018, 13, 2484–2497 CrossRef CAS PubMed.
- M. Månsson, R. K. Phipps, L. Gram, M. H. G. Munro, T. O. Larsen and K. F. Nielsen, J. Nat. Prod., 2010, 73, 1126–1132 CrossRef PubMed.
- D. Camp, R. A. Davis, M. Campitelli, J. Ebdon and R. J. Quinn, J. Nat. Prod., 2012, 75, 72–81 CrossRef CAS PubMed.
- C. J. Henrich, L. K. Cartner, J. A. Wilson, R. W. Fuller, A. E. Rizzo, K. M. Reilly, J. B. McMahon and K. R. Gustafson, J. Nat. Prod., 2015, 78, 2776–2781 CrossRef CAS PubMed.
- S. Wu, L. Yang, Y. Gao, X. Liu and F. Liu, J. Chromatogr. A, 2008, 1180, 99–107 CrossRef CAS PubMed.
- J. B. Friesen, J. B. McAlpine, S. N. Chen and G. F. Pauli, J. Nat. Prod., 2015, 78, 1765–1796 CrossRef CAS PubMed.
- Y. Tu, C. Jeffries, H. Ruan, C. Nelson, D. Smithson, A. A. Shelat, K. M. Brown, X.-C. Li, J. P. Hester, T. Smillie, I. A. Khan, L. Walker, K. Guy and B. Yan, J. Nat. Prod., 2010, 73, 751–754 CrossRef CAS PubMed.
- G. R. Eldridge, H. C. Vervoort, C. M. Lee, P. A. Cremin, C. T. Williams, S. M. Hart, M. G. Goering, M. O'Neil-Johnson and L. Zeng, Anal. Chem., 2002, 74, 3963–3971 CrossRef CAS PubMed.
- D. R. Appleton, A. D. Buss and M. S. Butler, CHIMIA International Journal for Chemistry, 2007, 61, 327–331 CrossRef CAS.
- T. S. Bugni, B. Richards, L. Bhoite, D. Cimbora, M. K. Harper and C. M. Ireland, J. Nat. Prod., 2008, 71, 1095–1098 CrossRef CAS PubMed.
- T. A. Johnson, J. Sohn, W. D. Inman, S. A. Estee, S. T. Loveridge, H. C. Vervoort, K. Tenney, J. Liu, K. K. Ang, J. Ratnam, W. M. Bray, N. C. Gassner, Y. Y. Shen, R. S. Lokey, J. H. McKerrow, K. Boundy-Mills, A. Nukanto, A. Kanti, H. Julistiono, L. B. Kardono, L. F. Bjeldanes and P. Crews, J. Nat. Prod., 2011, 74, 2545–2555 CrossRef CAS PubMed.
- M. M. Wagenaar, Molecules, 2008, 13, 1406–1426 CrossRef CAS PubMed.
- M. Herrero, J. A. Mendiola, A. Cifuentes and E. Ibáñez, J. Chromatogr. A, 2010, 1217, 2495–2511 CrossRef CAS PubMed.
- R. P. F. F. da Silva, T. A. P. Rocha-Santos and A. C. Duarte, TrAC, Trends Anal. Chem., 2016, 76, 40–51 CrossRef CAS.
- A. Cutignano, G. Nuzzo, A. Ianora, E. Luongo, G. Romano, C. Gallo, C. Sansone, S. Aprea, F. Mancini, U. D'Oro and A. Fontana, Mar. Drugs, 2015, 13, 5736–5749 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- W. R. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 CrossRef PubMed.
- D. E. Scott, A. G. Coyne, S. A. Hudson and C. Abell, Biochemistry, 2012, 51, 4990–5003 CrossRef CAS PubMed.
- J. Hert, J. J. Irwin, C. Laggner, M. J. Keiser and B. K. Shoichet, Nat. Chem. Biol., 2009, 5, 479–483 CrossRef CAS PubMed.
- Y. Chen, C. de Bruyn Kops and J. Kirchmair, J. Chem. Inf. Model., 2017, 57, 2099–2111 CrossRef CAS PubMed.
- J. Bisson, J. B. McAlpine, J. B. Friesen, S. N. Chen, J. Graham and G. F. Pauli, J. Med. Chem., 2016, 59, 1671–1690 CrossRef CAS PubMed.
- N. P. Coussens, J. C. Braisted, T. Peryea, G. S. Sittampalam, A. Simeonov and M. D. Hall, Pharmacol. Rev., 2017, 69, 479–496 CrossRef CAS PubMed.
- M. K. Ediriweera, K. H. Tennekoon and S. R. Samarakoon, J. Appl. Toxicol., 2019, 39, 38–71 CrossRef CAS PubMed.
- P. Horvath, N. Aulner, M. Bickle, A. M. Davies, E. D. Nery, D. Ebner, M. C. Montoya, P. Östling, V. Pietiäinen, L. S. Price, S. L. Shorte, G. Turcatti, C. von Schantz and N. O. Carragher, Nat. Rev. Drug Discovery, 2016, 15, 751–769 CrossRef CAS PubMed.
- M. Isgut, M. Rao, C. Yang, V. Subrahmanyam, P. C. G. Rida and R. Aneja, Med. Res. Rev., 2018, 38, 504–524 CrossRef PubMed.
- L. Laraia and H. Waldmann, Drug Discovery Today: Technol., 2017, 23, 75–82 CrossRef PubMed.
- J. G. Moffat, F. Vincent, J. A. Lee, J. Eder and M. Prunotto, Nat. Rev. Drug Discovery, 2017, 16, 531–543 CrossRef CAS PubMed.
- N. Robbins, M. Spitzer, W. Wang, N. Waglechner, D. J. Patel, J. S. O'Brien, L. Ejim, O. Ejim, M. Tyers and G. D. Wright, Cell Chem. Biol., 2016, 23, 1383–1394 CrossRef CAS PubMed.
- A. Roy, High-Throughput, 2018, 7, 4 CrossRef PubMed.
- M. J. Wildey, A. Haunso, M. Tudor, M. Webb and J. H. Connick, in Annual Reports in Medicinal Chemistry, ed. R. A. Goodnow, Academic Press, 2017, vol. 50, pp. 149–195 Search PubMed.
- B. Izar, J. Rotow, J. Gainor, J. Clark and B. Chabner, Pharmacol. Rev., 2013, 65, 1351–1395 CrossRef CAS PubMed.
- N. C. Parsley, C. L. Kirkpatrick, C. M. Crittenden, J. G. Rad, D. W. Hoskin, J. S. Brodbelt and L. M. Hicks, Phytochemistry, 2018, 152, 61–70 CrossRef CAS PubMed.
- F. Annang, G. Pérez-Moreno, R. García-Hernández, C. Cordon-Obras, J. Martín, J. R. Tormo, L. Rodríguez, N. de Pedro, V. Gómez-Pérez, M. Valente, F. Reyes, O. Genilloud, F. Vicente, S. Castanys, L. M. Ruiz-Pérez, M. Navarro, F. Gamarro and D. González-Pacanowska, J. Biomol. Screening, 2015, 20, 82–91 CrossRef CAS PubMed.
- J. J. Chen, C. A. Tsu, J. M. Gavin, M. A. Milhollen, F. J. Bruzzese, W. D. Mallender, M. D. Sintchak, N. J. Bump, X. Yang, J. Ma, H.-K. Loke, Q. Xu, P. Li, N. F. Bence, J. E. Brownell and L. R. Dick, J. Biol. Chem., 2011, 286, 40867–40877 CrossRef CAS PubMed.
- K. T. Lim, Z. Zahari, A. Amanah, Z. Zainuddin and M. I. Adenan, Exp. Parasitol., 2016, 162, 49–56 CrossRef CAS PubMed.
- G. Pérez-Moreno, J. Cantizani, P. Sánchez-Carrasco, L. M. Ruiz-Pérez, J. Martín, N. e. Aouad, I. Pérez-Victoria, J. R. Tormo, V. González-Menendez, I. González, N. d. Pedro, F. Reyes, O. Genilloud, F. Vicente and D. González-Pacanowska, PLoS One, 2016, 11, e0145812 CrossRef PubMed.
- R. Gupta, M. Netherton, T. F. Byrd and K. H. Rohde, Front. Microbiol., 2017, 8, 2204 CrossRef PubMed.
- A. Mishra, S. V. Dobritsa, M.-L. Crouch, J. Rabenstein, J. X. Y. Lee and S. Dhakshinamoorthy, J. Microbiol. Methods, 2015, 118, 173–175 CrossRef CAS PubMed.
- A. E. Sikora, R. Tehan and K. McPhail, in Vibrio Cholerae: Methods and Protocols, ed. A. E. Sikora, Springer New York, New York, NY, 2018, pp. 135–146 Search PubMed.
- R. A. Lewis, J. Li, N. E. E. Allenby, J. Errington, J. Hayles and P. Nurse, J. Cell Sci., 2017, 130, 3173–3185 CrossRef CAS PubMed.
- G. M. Walter, A. Raveh, S.-A. Mok, T. J. McQuade, C. J. Arevang, P. J. Schultz, M. C. Smith, S. Asare, P. G. Cruz, S. Wisen, T. Matainaho, D. H. Sherman and J. E. Gestwicki, Chem. Biol. Drug Des., 2014, 83, 440–449 CrossRef CAS PubMed.
- Q. Wang, T. Grkovic, J. Font, S. Bonham, R. H. Pouwer, C. G. Bailey, A. M. Moran, R. M. Ryan, J. E. J. Rasko, M. Jormakka, R. J. Quinn and J. Holst, ACS Chem. Biol., 2014, 9, 1369–1376 CrossRef CAS PubMed.
- D. Copmans, M. Rateb, J. N. Tabudravu, M. Pérez-Bonilla, N. Dirkx, R. Vallorani, C. Diaz, J. Pérez del Palacio, A. J. Smith, R. Ebel, F. Reyes, M. Jaspars and P. A. M. de Witte, ACS Chem. Neurosci., 2018, 9, 1652–1662 CrossRef CAS PubMed.
- C. Wang, H. Niederstrasser, P. M. Douglas, R. Lin, J. Jaramillo, Y. Li, N. W. Oswald, A. Zhou, E. A. McMillan, S. Mendiratta, Z. Wang, T. Zhao, Z. Lin, M. Luo, G. Huang, R. A. Brekken, B. A. Posner, J. B. MacMillan, J. Gao and M. A. White, Nat. Commun., 2017, 8, 2270 CrossRef PubMed.
- J. Pérez del Palacio, C. Díaz, M. de la Cruz, F. Annang, J. Martín, I. Pérez-Victoria, V. González-Menéndez, N. de Pedro, J. R. Tormo, F. Algieri, A. Rodriguez-Nogales, M. E. Rodríguez-Cabezas, F. Reyes, O. Genilloud, F. Vicente and J. Gálvez, J. Biomol. Screening, 2016, 21, 567–578 CrossRef PubMed.
- Z. Zhao, J. A. deMayo, A. M. West, M. J. Balunas and A. Zweifach, J. Biomol. Screening, 2016, 21, 556–566 CrossRef CAS PubMed.
- B. Cautain, N. de Pedro, F. Reyes and W. Link, in Membrane Trafficking, ed. B. L. Tang, Springer New York, New York, NY, 2nd edn, 2015, pp. 307–319 Search PubMed.
- K. L. Kurita, E. Glassey and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11999–12004 CrossRef CAS PubMed.
- S. B. Park, J. S. Park, W. H. Jung, A. Park, S. R. Jo, H. Y. Kim, S. D. Rhee, S. Y. Ryu, H. G. Jeong, S. Park, H. Lee and K. Y. Kim, Pharmacol. Res., 2015, 102, 245–253 CrossRef CAS PubMed.
- Y. Zhang, B. Yu, Y. Sui, X. Gao, H. Yang and T. Ma, PLoS One, 2014, 9, e94302 CrossRef PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- J. M. Cassady, W. M. Baird and C.-J. Chang, J. Nat. Prod., 1990, 53, 23–41 CrossRef CAS PubMed.
- P. Cozzi, N. Mongelli and A. Suarato, Curr. Med. Chem.: Anti-Cancer Agents, 2004, 4, 93–121 CrossRef CAS PubMed.
- R. H. Shoemaker, Nat. Rev. Cancer, 2006, 6, 813–823 CrossRef CAS PubMed.
- C. Kim and B. Kim, Nutrients, 2018, 10, 1021 CrossRef PubMed.
- Y. Suzuki-Karasaki, M. Suzuki-Karasaki, M. Uchida and T. Ochiai, Front. Oncol., 2014, 4, 128 Search PubMed.
- Y. Wang, J. Zhong, J. Bai, R. Tong, F. An, P. Jiao, L. He, D. Zeng, E. Long, J. Yan, J. Yu and L. Cai, Curr. Drug Metab., 2018, 19, 739–749 CrossRef CAS PubMed.
- S.-A. Park and Y.-J. Surh, Ann. N. Y. Acad. Sci., 2017, 1401, 65–74 CrossRef PubMed.
- M. S. Sarwar, H. J. Zhang and S. W. Tsang, Curr. Med. Chem., 2018, 25, 5057–5087 CrossRef CAS PubMed.
- X. Bai, L. Yao, X. Ma and X. Xu, Mini-Rev. Med. Chem., 2018, 18, 1151–1157 CrossRef CAS PubMed.
- J.-J. Qin, X. Li, C. Hunt, W. Wang, H. Wang and R. Zhang, Genes Dis., 2018, 5, 204–219 CrossRef CAS PubMed.
- M. Gaestel, A. Kotlyarov and M. Kracht, Nat. Rev. Drug Discovery, 2009, 8, 480–499 CrossRef CAS PubMed.
- A. R. Guerra, M. F. Duarte and I. F. Duarte, J. Agric. Food Chem., 2018, 66, 10663–10685 CrossRef CAS PubMed.
- S. Lascano, M. Lopez and P. B. Arimondo, Chem. Rec., 2018, 18, 1854–1876 CrossRef CAS PubMed.
- N. M. Peñaranda Fajardo, C. Meijer and F. A. E. Kruyt, Biochem. Pharmacol., 2016, 118, 1–8 CrossRef PubMed.
- P. J. Grohar, G. M. Woldemichael, L. B. Griffin, A. Mendoza, Q.-R. Chen, C. Yeung, D. G. Currier, S. Davis, C. Khanna, J. Khan, J. B. McMahon and L. J. Helman, JNCI, J. Natl. Cancer Inst., 2011, 103, 962–978 CrossRef CAS PubMed.
- V. Caropreso, E. Darvishi, T. J. Turbyville, R. Ratnayake, P. J. Grohar, J. B. McMahon and G. M. Woldemichael, J. Biol. Chem., 2016, 291, 10058–10066 CrossRef CAS PubMed.
- M. G. Zhang, J. Y. Lee, R. A. Gallo, W. Tao, D. Tse, R. Doddapaneni and D. Pelaez, Pharmacol. Res., 2018, 129, 365–374 CrossRef CAS PubMed.
- B. Cautain, N. de Pedro, V. Murillo Garzón, M. Muñoz de Escalona, V. González Menéndez, J. R. Tormo, J. Martin, N. El Aouad, F. Reyes, F. Asensio, O. Genilloud, F. Vicente and W. Link, J. Biomol. Screening, 2014, 19, 57–65 CrossRef CAS PubMed.
- N. L. Booth, T. J. Sayers, A. D. Brooks, C. L. Thomas, K. Jacobsen, E. I. Goncharova, J. B. McMahon and C. J. Henrich, Cancer Immunol. Immunother., 2009, 58, 1229–1244 CrossRef PubMed.
- C. J. Henrich, A. D. Brooks, K. L. Erickson, C. L. Thomas, H. R. Bokesch, P. Tewary, C. R. Thompson, R. J. Pompei, K. R. Gustafson, J. B. McMahon and T. J. Sayers, Cell Death Dis., 2015, 6, e1666 CrossRef CAS PubMed.
- C. J. Henrich, H. R. Bokesch, M. Dean, S. E. Bates, R. W. Robey, E. I. Goncharova, J. A. Wilson and J. B. McMahon, J. Biomol. Screening, 2006, 11, 176–183 CrossRef CAS PubMed.
- C. J. Henrich, R. W. Robey, K. Takada, H. R. Bokesch, S. E. Bates, S. Shukla, S. V. Ambudkar, J. B. McMahon and K. R. Gustafson, ACS Chem. Biol., 2009, 4, 637–647 CrossRef CAS PubMed.
- C. J. Gerry and S. L. Schreiber, Nat. Rev. Drug Discovery, 2018, 17, 333–352 CrossRef CAS PubMed.
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531–541 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- A. J. Singh, A. P. Gorka, H. R. Bokesch, A. Wamiru, B. R. O'Keefe, M. J. Schnermann and K. R. Gustafson, J. Nat. Prod., 2018, 81, 2750–2755 CrossRef CAS PubMed.
- C. J. Henrich, E. I. Goncharova, J. A. Wilson, R. S. Gardella, T. R. Johnson, J. B. McMahon, K. Takada, H. R. Bokesch and K. R. Gustafson, Chem. Biol. Drug Des., 2007, 69, 321–330 CrossRef CAS PubMed.
- G. M. Woldemichael, J. R. Vasselli, R. S. Gardella, T. C. McKee, W. M. Linehan and J. B. McMahon, J. Biomol. Screening, 2006, 11, 678–687 CrossRef CAS PubMed.
- K. M. Ruocco, E. I. Goncharova, M. R. Young, N. H. Colburn, J. B. McMahon and C. J. Henrich, J. Biomol. Screening, 2007, 12, 133–139 CrossRef CAS PubMed.
- A. Bermingham, E. Price, C. Marchand, A. Chergui, A. Naumova, E. L. Whitson, L. R. H. Krumpe, E. I. Goncharova, J. R. Evans, T. C. McKee, C. J. Henrich, Y. Pommier and B. R. O'Keefe, SLAS DISCOVERY: Advancing Life Sciences R&D, 2017, 22, 1093–1105 Search PubMed.
- A. L. Harvey, Drug Discovery Today, 2008, 13, 894–901 CrossRef CAS PubMed.
- A. K. L. Goey, C. H. Chau, T. M. Sissung, K. M. Cook, D. J. Venzon, A. Castro, T. R. Ransom, C. J. Henrich, T. C. McKee, J. B. McMahon, T. Grkovic, M. M. Cadelis, B. R. Copp, K. R. Gustafson and W. D. Figg, J. Nat. Prod., 2016, 79, 1267–1275 CrossRef CAS PubMed.
- A. D. Nalli, L. E. Brown, C. L. Thomas, T. J. Sayers, J. A. Porco and C. J. Henrich, Sci. Rep., 2018, 8, 17519 CrossRef PubMed.
- Y.-M. Xu, A. D. Brooks, E. M. K. Wijeratne, C. J. Henrich, P. Tewary, T. J. Sayers and A. A. L. Gunatilaka, J. Med. Chem., 2017, 60, 3039–3051 CrossRef CAS PubMed.
- C. L. Osgood, N. Maloney, C. G. Kidd, S. Kitchen-Goosen, L. Segars, M. Gebregiorgis, G. M. Woldemichael, M. He, S. Sankar, S. L. Lessnick, M. Kang, M. Smith, L. Turner, Z. B. Madaj, M. E. Winn, L. E. Nunez, J. Gonzalez-Sabin, L. J. Helman, F. Moris and P. J. Grohar, Clin. Cancer Res., 2016, 22, 4105–4118 CrossRef CAS PubMed.
- K. Takada, N. Imamura, K. R. Gustafson and C. J. Henrich, Bioorg. Med. Chem. Lett., 2010, 20, 1330–1333 CrossRef CAS PubMed.
- J. D. Strope, C. J. Peer, T. M. Sissung, O. M. Hall, P. A. Huang, E. M. Harris, K. R. Gustafson, C. J. Henrich, D. M. Sigano, G. T. Pauly, J. P. Schneider, S. E. Bates and W. D. Figg, Cancer Biol. Ther., 2020, 21, 223–230 CrossRef CAS PubMed.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS PubMed.
- M. G. Acker and D. S. Auld, Perspect. Sci., 2014, 1, 56–73 CrossRef.
- C. V. Dang, E. P. Reddy, K. M. Shokat and L. Soucek, Nat. Rev. Cancer, 2017, 17, 502–508 CrossRef CAS PubMed.
- L. N. Makley and J. E. Gestwicki, Chem. Biol. Drug Des., 2013, 81, 22–32 CrossRef CAS PubMed.
- P. J. Hajduk, J. R. Huth and S. W. Fesik, J. Med. Chem., 2005, 48, 2518–2525 CrossRef CAS PubMed.
- F. N. B. Edfeldt, R. H. A. Folmer and A. L. Breeze, Drug Discovery Today, 2011, 16, 284–287 CrossRef CAS PubMed.
- M. Chilton, B. Clennell, F. Edfeldt and S. Geschwindner, J. Med. Chem., 2017, 60, 4923–4931 CrossRef CAS PubMed.
- H. Vu, L. Pedro, T. Mak, B. McCormick, J. Rowley, M. Liu, A. Di Capua, B. Williams-Noonan, N. B. Pham, R. Pouwer, B. Nguyen, K. T. Andrews, T. Skinner-Adams, J. Kim, W. G. J. Hol, R. Hui, G. J. Crowther, W. C. Van Voorhis and R. J. Quinn, ACS Infect. Dis., 2018, 4, 431–444 CrossRef CAS PubMed.
- A. Bender, D. Bojanic, J. W. Davies, T. J. Crisman, D. Mikhailov, J. Scheiber, J. L. Jenkins, Z. Deng, W. A. G. Hill, M. Popov, E. Jacoby and M. Glick, Curr. Opin. Drug Discovery Dev., 2008, 11, 327–337 CAS.
- I. Coma, D. Bandyopadhyay, E. Diez, E. A. Ruiz, M. T. de los Frailes and G. Colmenarejo, J. Biomol. Screening, 2014, 19, 749–757 CrossRef PubMed.
- K. D. Barnash, L. I. James and S. V. Frye, Nat. Chem. Biol., 2017, 13, 1053–1056 CrossRef CAS PubMed.
- J. Tsai, J. T. Lee, W. Wang, J. Zhang, H. Cho, S. Mamo, R. Bremer, S. Gillette, J. Kong, N. K. Haass, K. Sproesser, L. Li, K. S. M. Smalley, D. Fong, Y.-L. Zhu, A. Marimuthu, H. Nguyen, B. Lam, J. Liu, I. Cheung, J. Rice, Y. Suzuki, C. Luu, C. Settachatgul, R. Shellooe, J. Cantwell, S.-H. Kim, J. Schlessinger, K. Y. J. Zhang, B. L. West, B. Powell, G. Habets, C. Zhang, P. N. Ibrahim, P. Hirth, D. R. Artis, M. Herlyn and G. Bollag, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3041–3046 CrossRef CAS PubMed.
- B. R. Glick and C. L. Patten, Molecular biotechnology: principles and applications of recombinant DNA, ASM Press, Washington, DC, 5th edn, 2017 Search PubMed.
- E. Perola, J. Med. Chem., 2010, 53, 2986–2997 CrossRef CAS PubMed.
- E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discovery Today, 2016, 21, 204–207 CrossRef CAS PubMed.
- M. A. Skinnider and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E6271–E6272 CrossRef CAS PubMed.
- C. R. Pye, M. J. Bertin, R. S. Lokey, W. H. Gerwick and R. G. Linington, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5601–5606 CrossRef CAS PubMed.
- B. Y. Feng, A. Simeonov, A. Jadhav, K. Babaoglu, J. Inglese, B. K. Shoichet and C. P. Austin, J. Med. Chem., 2007, 50, 2385–2390 CrossRef CAS PubMed.
- S. L. McGovern, E. Caselli, N. Grigorieff and B. K. Shoichet, J. Med. Chem., 2002, 45, 1712–1722 CrossRef CAS PubMed.
- B. Y. Feng and B. K. Shoichet, Nat. Protoc., 2006, 1, 550–553 CrossRef CAS PubMed.
- H. B. Brooks, S. Geeganage, S. D. Kahl, C. Montrose, S. Sittampalam, M. C. Smith and J. R. Weidner, in Assay Guidance Manual, ed. G. S. Sittampalam, N. P. Coussens, K. Brimacombe, A. Grossman, M. Arkin, D. Auld, C. Austin, J. Baell, B. Bejcek, J. M. M. Caaveiro, T. D. Y. Chung, J. L. Dahlin, V. Devanaryan, T. L. Foley, M. Glicksman, M. D. Hall, J. V. Haas, J. Inglese, P. W. Iversen, S. D. Kahl, S. C. Kales, M. Lal-Nag, Z. Li, J. McGee, O. McManus, T. Riss, O. J. Trask, J. R. Weidner, M. J. Wildey, M. Xia and X. Xu, Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda (MD), 2004 Search PubMed.
- J. L. Dahlin, J. W. M. Nissink, J. M. Strasser, S. Francis, L. Higgins, H. Zhou, Z. Zhang and M. A. Walters, J. Med. Chem., 2015, 58, 2091–2113 CrossRef CAS PubMed.
- M. Gersch, J. Kreuzer and S. A. Sieber, Nat. Prod. Rep., 2012, 29, 659–682 RSC.
- G. M. Rishton, Am. J. Cardiol., 2008, 101, S43–S49 CrossRef PubMed.
- D. S. Auld, J. Inglese and J. L. Dahlin, in Assay Guidance Manual, ed. G. S. Sittampalam, A. Grossman, K. Brimacombe, M. Arkin, D. Auld, C. P. Austin, J. Baell, B. Bejcek, J. M. M. Caaveiro, T. D. Y. Chung, N. P. Coussens, J. L. Dahlin, V. Devanaryan, T. L. Foley, M. Glicksman, M. D. Hall, J. V. Haas, S. R. J. Hoare, J. Inglese, P. W. Iversen, S. D. Kahl, S. C. Kales, S. Kirshner, M. Lal-Nag, Z. Li, J. McGee, O. McManus, T. Riss, P. Saradjian, O. J. Trask Jr, J. R. Weidner, M. J. Wildey, M. Xia and X. Xu, Bethesda, MD, 2004 Search PubMed.
- J. Baell and M. A. Walters, Nature, 2014, 513, 481–483 CrossRef CAS.
- T. D. Tran, B. A. P. Wilson, C. J. Henrich, L. M. Staudt, L. R. H. Krumpe, E. A. Smith, J. King, K. L. Wendt, A. M. Stchigel, A. N. Miller, R. H. Cichewicz, B. R. O'Keefe and K. R. Gustafson, J. Nat. Prod., 2019, 82, 154–162 CrossRef CAS PubMed.
- C. J. Henrich and J. A. Beutler, Nat. Prod. Rep., 2013, 30, 1284–1298 RSC.
- G. S. Weston, J. Blazquez, F. Baquero and B. K. Shoichet, J. Med. Chem., 1998, 41, 4577–4586 CrossRef CAS PubMed.
- G. A. Jacoby, Clin. Microbiol. Rev., 2009, 22, 161–182 CrossRef CAS PubMed.
- R. A. Copeland, Methods Biochem. Anal., 2005, 46, 1–265 Search PubMed.
- M. E. Swartz, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 1253–1263 CrossRef CAS.
- A. K. Quercia, W. A. Lamarr, J. Myung, C. C. Özbal, J. A. Landro and K. J. Lumb, J. Biomol. Screening, 2007, 12, 473–480 CrossRef CAS PubMed.
- M. Rohman and J. Wingfield, in High Throughput Screening: Methods and Protocols, ed. W. P. Janzen, Springer New York, New York, NY, 2016, pp. 47–63 Search PubMed.
- I. Sinclair, R. Stearns, S. Pringle, J. Wingfield, S. Datwani, E. Hall, L. Ghislain, L. Majlof and M. Bachman, J. Lab. Autom., 2016, 21, 19–26 CrossRef PubMed.
- M. Winter, R. Ries, C. Kleiner, D. Bischoff, A. H. Luippold, T. Bretschneider and F. H. Büttner, SLAS Technol., 2019, 24, 209–221 CrossRef PubMed.
- K. Beeman, J. Baumgärtner, M. Laubenheimer, K. Hergesell, M. Hoffmann, U. Pehl, F. Fischer and J.-C. Pieck, SLAS DISCOVERY: Advancing Life Sciences R&D, 2017, 22, 1203–1210 Search PubMed.
- C. Haslam, J. Hellicar, A. Dunn, A. Fuetterer, N. Hardy, P. Marshall, R. Paape, M. Pemberton, A. Resemannand and M. Leveridge, J. Biomol. Screening, 2016, 21, 176–186 CrossRef CAS PubMed.
- C. C. Genick and S. K. Wright, Expert Opin. Drug Discovery, 2017, 12, 897–907 CrossRef CAS PubMed.
- S. Choi and K.-Y. Choi, Expert Opin. Drug Discovery, 2017, 12, 293–303 CrossRef CAS PubMed.
- J.-P. Renaud, C.-w. Chung, U. H. Danielson, U. Egner, M. Hennig, R. E. Hubbard and H. Nar, Nat. Rev. Drug Discovery, 2016, 15, 679–698 CrossRef CAS PubMed.
- M. D. Hall, A. Yasgar, T. Peryea, J. C. Braisted, A. Jadhav, A. Simeonov and N. P. Coussens, Methods Appl. Fluoresc., 2016, 4, 022001 CrossRef PubMed.
- C. Vinegoni, P. F. Feruglio, I. Gryczynski, R. Mazitschek and R. Weissleder, Adv. Drug Delivery Rev., 2019, 151–152, 262–288 CrossRef CAS PubMed.
- D. E. Scott, C. Spry and C. Abell, in Fragment-based Drug Discovery Lessons and Outlook, John Wiley & Sons, Ltd, 2016, pp. 139–172 Search PubMed.
- A. Simeonov, Expert Opin. Drug Discovery, 2013, 8, 1071–1082 CrossRef CAS PubMed.
- R. M. Eglen, T. Reisine, P. Roby, N. Rouleau, C. Illy, R. Bossé and M. Bielefeld, Curr. Chem. Genomics, 2008, 1, 2–10 CrossRef CAS PubMed.
- B. S. Edwards and L. A. Sklar, J. Biomol. Screening, 2015, 20, 689–707 CrossRef CAS.
- Z. Surviladze, A. Waller, Y. Wu, E. Romero, B. S. Edwards, A. Wandinger-Ness and L. A. Sklar, J. Biomol. Screening, 2010, 15, 10–20 CrossRef CAS PubMed.
- J. Yao, P. Li, L. Li and M. Yang, Acta Biomater., 2018, 74, 36–55 CrossRef CAS PubMed.
- O. Kovtun, X. Arzeta-Ferrer and S. J. Rosenthal, Nanoscale, 2013, 5, 12072–12081 RSC.
- C. A. Wartchow, F. Podlaski, S. Li, K. Rowan, X. Zhang, D. Mark and K. S. Huang, J. Comput.-Aided Mol. Des., 2011, 25, 669–676 CrossRef CAS.
- Y. Yu, S. Mitchell, H. Lynaugh, M. Brown, R. P. Nobrega, X. Zhi, T. Sun, I. Caffry, Y. Cao, R. Yang, I. Burnina, Y. Xu and P. Estep, J. Biomol. Screening, 2016, 21, 88–95 CrossRef CAS PubMed.
- J. M. Rainard, G. C. Pandarakalam and S. P. McElroy, SLAS Discovery, 2018, 23, 225–241 CrossRef CAS PubMed.
- K. Zlotkowski, W. M. Hewitt, P. Yan, H. R. Bokesch, M. L. Peach, M. C. Nicklaus, B. R. O'Keefe, J. B. McMahon, K. R. Gustafson and J. S. Schneekloth Jr, Org. Lett., 2017, 19, 1726–1729 CrossRef CAS PubMed.
- M. Sesen, T. Alan and A. Neild, Lab Chip, 2017, 17, 2372–2394 RSC.
- A. Kulesa, J. Kehe, J. E. Hurtado, P. Tawde and P. C. Blainey, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6685–6690 CrossRef PubMed.
- C. M. Ouimet, C. I. D'amico and R. T. Kennedy, Expert Opin. Drug Discovery, 2017, 12, 213–224 CrossRef CAS PubMed.
- B. Hadimioglu, R. Stearns and R. Ellson, J. Lab. Autom., 2016, 21, 4–18 CrossRef.
- X. Tian, N. S. Isamiddinova, R. J. Peroutka, S. J. Goldenberg, M. R. Mattern, B. Nicholson and C. Leach, Assay Drug Dev. Technol., 2010, 9, 165–173 CrossRef.
- C. A. Sasiela, D. H. Stewart, J. Kitagaki, Y. J. Safiran, Y. Yang, A. M. Weissman, P. Oberoi, I. V. Davydov, E. Goncharova, J. A. Beutler, J. B. McMahon and B. R. O'Keefe, J. Biomol. Screening, 2008, 13, 229–237 CrossRef CAS.
- M. Leveridge, R. Buxton, A. Argyrou, P. Francis, B. Leavens, A. West, M. Rees, P. Hardwicke, A. Bridges, S. Ratcliffe and C.-w. Chung, J. Biomol. Screening, 2014, 19, 278–286 CrossRef PubMed.
- R. M. Kainkaryam and P. J. Woolf, Curr. Opin. Drug Discovery Dev., 2009, 12, 339–350 CAS.
- L. L. Elkin, D. G. Harden, S. Saldanha, H. Ferguson, D. L. Cheney, S. N. Pieniazek, D. P. Maloney, J. Zewinski, J. O'Connell and M. Banks, J. Biomol. Screening, 2015, 20, 577–587 CrossRef CAS PubMed.
- R. Macarron, Nat. Chem. Biol., 2015, 11, 904–905 CrossRef CAS PubMed.
- A. M. Wassermann, E. Lounkine, D. Hoepfner, G. Le Goff, F. J. King, C. Studer, J. M. Peltier, M. L. Grippo, V. Prindle, J. Tao, A. Schuffenhauer, I. M. Wallace, S. Chen, P. Krastel, A. Cobos-Correa, C. N. Parker, J. W. Davies and M. Glick, Nat. Chem. Biol., 2015, 11, 958–966 CrossRef CAS PubMed.
- N. P. Coussens, G. S. Sittampalam, R. Guha, K. Brimacombe, A. Grossman, T. D. Y. Chung, J. R. Weidner, T. Riss, O. J. Trask, D. Auld, J. L. Dahlin, V. Devanaryan, T. L. Foley, J. McGee, S. D. Kahl, S. C. Kales, M. Arkin, J. Baell, B. Bejcek, N. Gal-Edd, M. Glicksman, J. V. Haas, P. W. Iversen, M. Hoeppner, S. Lathrop, E. Sayers, H. Liu, B. Trawick, J. McVey, V. P. Lemmon, Z. Li, O. McManus, L. Minor, A. Napper, M. J. Wildey, R. Pacifici, W. W. Chin, M. Xia, X. Xu, M. Lal-Nag, M. D. Hall, S. Michael, J. Inglese, A. Simeonov and C. P. Austin, Clin. Transl. Sci., 2018, 11, 461–470 CrossRef PubMed.
- B. K. Shoichet, J. Med. Chem., 2006, 49, 7274–7277 CrossRef CAS PubMed.
- M. K. Matlock, T. B. Hughes, J. L. Dahlin and S. J. Swamidass, J. Chem. Inf. Model., 2018, 58, 1483–1500 CrossRef CAS PubMed.
- C. A. Hassig, F. Y. Zeng, P. Kung, M. Kiankarimi, S. Kim, P. W. Diaz, D. Zhai, K. Welsh, S. Morshedian, Y. Su, B. O'Keefe, D. J. Newman, Y. Rusman, H. Kaur, C. E. Salomon, S. G. Brown, B. Baire, A. R. Michel, T. R. Hoye, S. Francis, G. I. Georg, M. A. Walters, D. B. Divlianska, G. P. Roth, A. E. Wright and J. C. Reed, J. Biomol. Screening, 2014, 19, 1201–1211 CrossRef PubMed.
- N. B. Videira, F. A. H. Batista, A. Torres Cordeiro and A. C. M. Figueira, PPAR Res., 2018, 2018, 3681590 CrossRef PubMed.
- F. Olivon, L. F. Nothias, V. Dumontet, P. Retailleau, S. Berger, G. Ferry, W. Cohen, B. Pfeiffer, J. A. Boutin, E. Scalbert, F. Roussi and M. Litaudon, J. Nat. Prod., 2018, 81, 1610–1618 CrossRef CAS PubMed.
- F. Li, Y. Zhang, D. Qiu and J. Kang, J. Chromatogr. A, 2015, 1400, 117–123 CrossRef CAS PubMed.
- G. L. Daino, A. Frau, C. Sanna, D. Rigano, S. Distinto, V. Madau, F. Esposito, E. Fanunza, G. Bianco, O. Taglialatela-Scafati, L. Zinzula, E. Maccioni, A. Corona and E. Tramontano, Biochemistry, 2018, 57, 6367–6378 CrossRef CAS PubMed.
- A. Tripathi, M. M. Schofield, G. E. Chlipala, P. J. Schultz, I. Yim, S. A. Newmister, T. D. Nusca, J. B. Scaglione, P. C. Hanna, G. Tamayo-Castillo and D. H. Sherman, J. Am. Chem. Soc., 2014, 136, 1579–1586 CrossRef CAS PubMed.
- A. Ochoa, E. Álvarez-Bohórquez, E. Castillero and L. F. Olguin, Anal. Chem., 2017, 89, 4889–4896 CrossRef CAS PubMed.
- Y. Hu, M. Keniry, S. O. Palmer and J. M. Bullard, Antimicrob. Agents Chemother., 2016, 60, 4820–4829 CrossRef CAS PubMed.
- Dictionary of Natural Products, http://dnp.chemnetbase.com, accessed January 2020 Search PubMed.
- H. Laatsch, AntiBase 2012: The Natural Compound Identifiers, Wiley-VCH Search PubMed.
- MarinLit, http://pubs.rsc.org/marinlit/, accessed January 2020 Search PubMed.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crusemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. P. Boya, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. Van Leer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828–837 CrossRef CAS PubMed.
- R. Brkljaca and S. Urban, J. Nat. Prod., 2015, 78, 1486–1494 CrossRef CAS PubMed.
- G. Lang, N. A. Mayhudin, M. I. Mitova, L. Sun, S. van der Sar, J. W. Blunt, A. L. J. Cole, G. Ellis, H. Laatsch and M. H. G. Munro, J. Nat. Prod., 2008, 71, 1595–1599 CrossRef CAS PubMed.
- T. Grkovic, R. H. Pouwer, M.-L. Vial, L. Gambini, A. Noel, J. N. A. Hooper, S. A. Wood, G. D. Mellick and R. J. Quinn, Angew. Chem., Int. Ed., 2014, 53, 6070–6074 CrossRef CAS PubMed.
- J.-F. Hu, E. Garo, H.-D. Yoo, P. A. Cremin, L. Zeng, M. G. Goering, M. O'Neil-Johnson and G. R. Eldridge, Phytochem. Anal., 2005, 16, 127–133 CrossRef CAS PubMed.
- T. F. Molinski, Nat. Prod. Rep., 2010, 27, 321–329 RSC.
- D. Camp, M. Campitelli, A. R. Carroll, R. A. Davis and R. J. Quinn, Chem. Biodiversity, 2013, 10, 524–537 CrossRef CAS PubMed.
- D. R. Appleton, A. D. Buss and M. S. Butler, Chimia, 2007, 61, 327–331 CrossRef CAS.
- D. E. Schaufelberger, M. P. Koleck, J. A. Beutler, A. M. Vatakis, A. B. Alvarado, P. Andrews, L. V. Marzo, G. M. Muschik and J. Roach, et al. , J. Nat. Prod., 1991, 54, 1265–1270 CrossRef CAS PubMed.
- A. S. Eustaquio, L.-P. Chang, G. L. Steele, C. J. O'Donnell and F. E. Koehn, Metab. Eng., 2016, 33, 67–75 CrossRef CAS PubMed.
- Albany Molecular Research Inc: Natural Product Libraries, https://www.amriglobal.com, accessed 2018.
- AnalytiCon Discovery: Libraries and Collections, https://ac-discovery.com, accessed 2018.
- Bioinformatics Institute (BII): A*STAR Natural Product Library, http://www.bii.a-star.edu.sg, accessed 2018.
- Fondazione Instituto Insubrico Ricerca per la Vita (FIIRV): Scientific Asset, http://www.ricercaperlavita.it/en, accessed 2018.
- Fundación MEDINA: Natural Product Libraries, http://www.medinadiscovery.com, accessed 2018.
- Griffith Institute for Drug Discovery (GRIDD): Nature Bank, https://www.griffith.edu.au/institute-drug-discovery, accessed 2018.
- InterBioScreen: Natural Compound (NC) Collection, https://www.ibscreen.com, accessed 2018.
- Magellan BioScience Group Inc: Oceans of Possibilities, http://www.magellanbioscience.com, accessed 2018.
- Mycosynthetix Inc: The Healing Power of Nature, http://www.mycosynthetix.com, accessed 2018.
- Natural Products Discovery Institute (NPDI): A Division of The Baruch S. Blumberg Institute, http://www.npdi-us.org, accessed 2018.
- PharmaMar: Marine Compound Library. https://www.pharmamar.com, accessed 2018.
- PhytoPharmacon: Natural Product Library, http://www.phytopharmacon.com, accessed 2018.
- RIKEN: Natural Products Depository (NPDepo), http://www.riken.jp/dmp/english/index_en.html, accessed 2018.
- A. Marinetti, The Institut de Chimie des Substances Naturelles (ICSN): Past and Present, Eur. J. Org. Chem., 2018,(42), 5774–5776 CrossRef CAS.
- The Scripps Research Institute (TSRI): The Natural Products Library (NPL) at TSRI, https://www.scripps.edu, accessed 2018.
- The Univeristiy of Mississippi National Center for Natural Products Research, https://pharmacy.olemiss.edu/ncnpr/, accessed 2019.
- Unigen: PhytoLogix, https://unigen.net, accessed 2018.
- InterLink Biotechnologies: Natural Products, http://www.interlinkbiotech.com, accessed 2018.
| This journal is © The Royal Society of Chemistry 2020 |