
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural products as modulators of eukaryotic protein secretion
Hendrik
Luesch
 *ab and
Ville O.
Paavilainen
*c
*ab and
Ville O.
Paavilainen
*c
aDepartment of Medicinal Chemistry, Center for Natural Products, Drug Discovery and Development (CNPD3), University of Florida, P.O. Box 100485, Gainesville, Florida 32610, USA. E-mail: luesch@cop.ufl.edu; Tel: +1-352-273-7738
bLee Kong Chian School of Medicine, Nanyang Technological University, 59 Nanyang Drive, 636921, Singapore
cInstitute of Biotechnology, University of Helsinki, Viikinkaari 1, Biocenter 3, Helsinki, 00014, Finland. E-mail: ville.paavilainen@helsinki.fi; Tel: +358-50-448-4600
First published on 18th February 2020
Abstract
Covering: up to the end of 2019
Diverse natural product small molecules have allowed critical insights into processes that govern eukaryotic cells' ability to secrete cytosolically synthesized secretory proteins into their surroundings or to insert newly synthesized integral membrane proteins into the lipid bilayer of the endoplasmic reticulum. In addition, many components of the endoplasmic reticulum, required for protein homeostasis or other processes such as lipid metabolism or maintenance of calcium homeostasis, are being investigated for their potential in modulating human disease conditions such as cancer, neurodegenerative conditions and diabetes. In this review, we cover recent findings up to the end of 2019 on natural products that influence protein secretion or impact ER protein homeostasis, and serve as powerful chemical tools to understand protein flux through the mammalian secretory pathway and as leads for the discovery of new therapeutics.
 Hendrik Luesch | Hendrik Luesch received his Diplom in Chemistry at the University of Siegen (Germany) in 1997. He studied marine natural products chemistry at the University of Hawaii at Manoa and obtained his PhD with Professor Richard E. Moore in 2002. He undertook three years of postdoctoral studies as an Irving S. Sigal Fellow at The Scripps Research Institute with Professor Peter G. Schultz in functional genomics and chemical biology. Since 2005 he has been faculty at the University of Florida (UF) and is currently Professor and Chair of the Department of Medicinal Chemistry. He holds the endowed Debbie and Sylvia DeSantis Chair in Natural Products Drug Discovery and Development and leads a multidisciplinary marine natural products program that integrates isolation, synthesis, pharmacology, mechanism of action and early development studies. He is UF's Director of the Center for Natural Products, Drug Discovery and Development (CNPD3). Since 2018, he is Visiting Professor at the Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore. |
 Ville O. Paavilainen | Ville Paavilainen is a Tenure Track Group Leader at the Institute of Biotechnology (HiLIFE) at the University of Helsinki. He received his PhD degree at the University of Helsinki where he studied regulation of the cellular actin cytoskeleton using biochemistry and structural biology. He then moved to University of California -San Francisco to work with Professor Jack Taunton as a Postdoctoral Fellow to develop covalent-reversible cysteine targeting kinase inhibitors and to study integral membrane protein membrane insertion using a substrate-selective inhibitor of ER protein import. His independent research group studies proteostasis modulating small molecules and specialized functions of organelle targeting polypeptides. He lives in Helsinki and is passionate about rock climbing, music and hanging out with his family. |
1. Introduction
Natural products (NPs) are a rich source of diverse bioactive molecules for discovery of therapeutic drugs.1–3 In addition, NPs serve as valuable probes to study the inner workings of the cell and have revealed a wealth of information about dynamic cellular processes such as mechanisms of protein synthesis, secretion, and regulated turnover.4 However, to gain relevant insights into biological systems and disease mechanisms, high quality probe molecules are required and many small molecules that are currently used do not fulfill the required criteria.5 Current technological development, including CRISPR-based chemogenomic screening in human cells, improved methods for identifying resistance-conferring point mutations in NP target genes, proteome-wide small molecule engagement approaches and new synthetic routes to affinity-based probe molecules such as photoaffinity probes have permitted new advances in identifying cellular targets of many natural product small molecules and to examine the degree to which the observed phenotypic changes can be attributed to on-target modulation.6–9 In this review, we will cover the discovery and recent literature of NPs that influence protein secretion by direct targeting of key factors influencing protein secretion or important homeostatic processes of the secretory pathway (Fig. 1), including different forms of ER stress, maintenance of calcium homeostasis and ER associated protein degradation.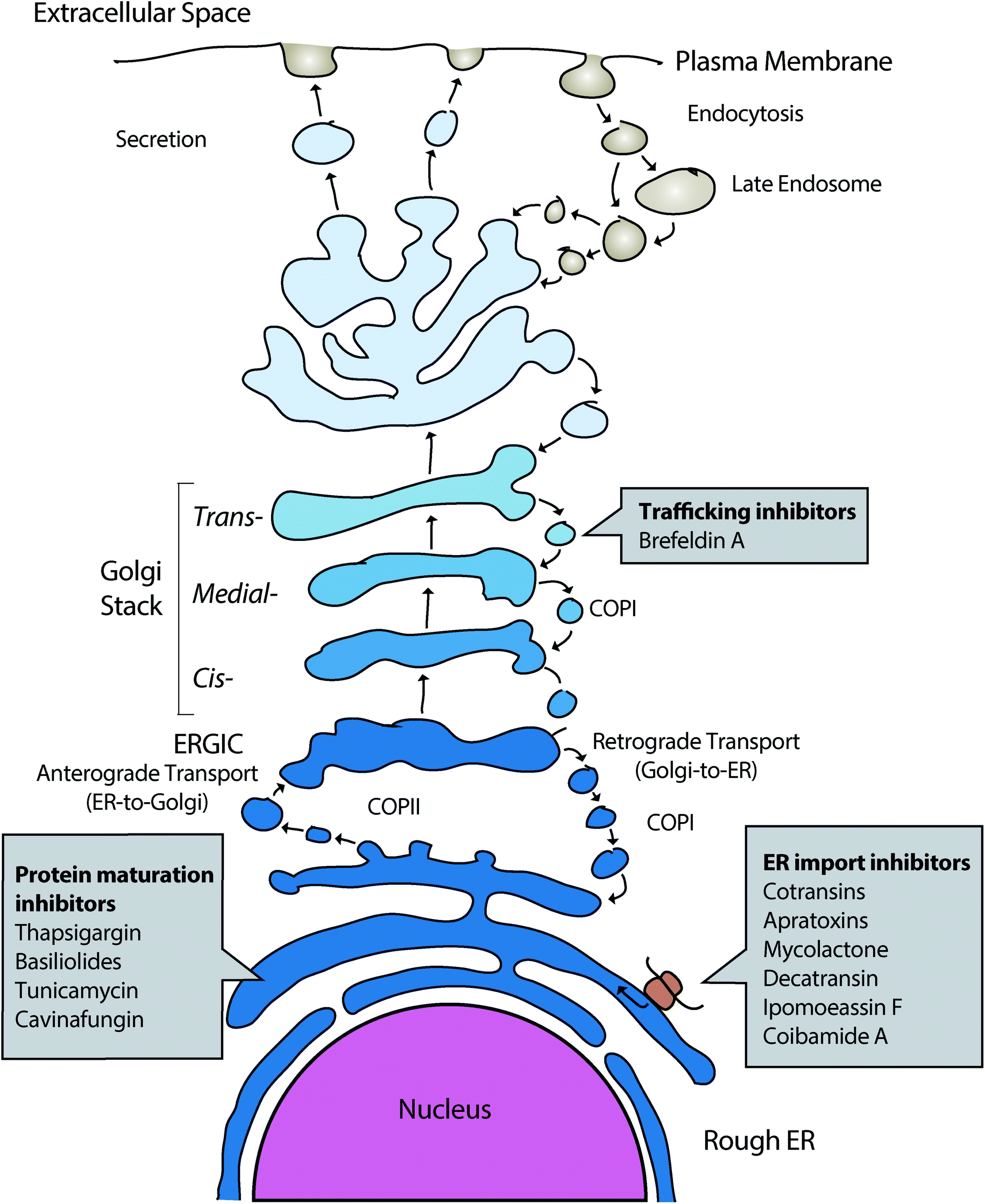 | ||
| Fig. 1 Outline of the mammalian protein secretory pathway and points of activity of different proteostasis modulating NPs. | ||
2. Inhibitors of ER protein insertion
Protein insertion into the lumen of the ER or insertion into the ER membrane for integral membrane proteins is the first step after protein translation on the protein secretion pathway. Recent work has now revealed a dizzying array of structurally diverse, potent natural products that act by preventing protein ER translocation (reviewed in ref. 10). These compounds are produced by a host of microorganisms ranging from endosymbiotic fungi to human pathogenic bacteria, marine cyanobacteria and even medicinal plants. Unexpectedly, it now appears that nearly all of these NPs act by directly targeting the central Sec61 protein translocation channel that is responsible for ER translocation of newly synthesized secretory and membrane proteins (Fig. 2). Here, we will review the discovery and current progress on understanding the mechanism of this diverse group of Sec61 modulating compounds.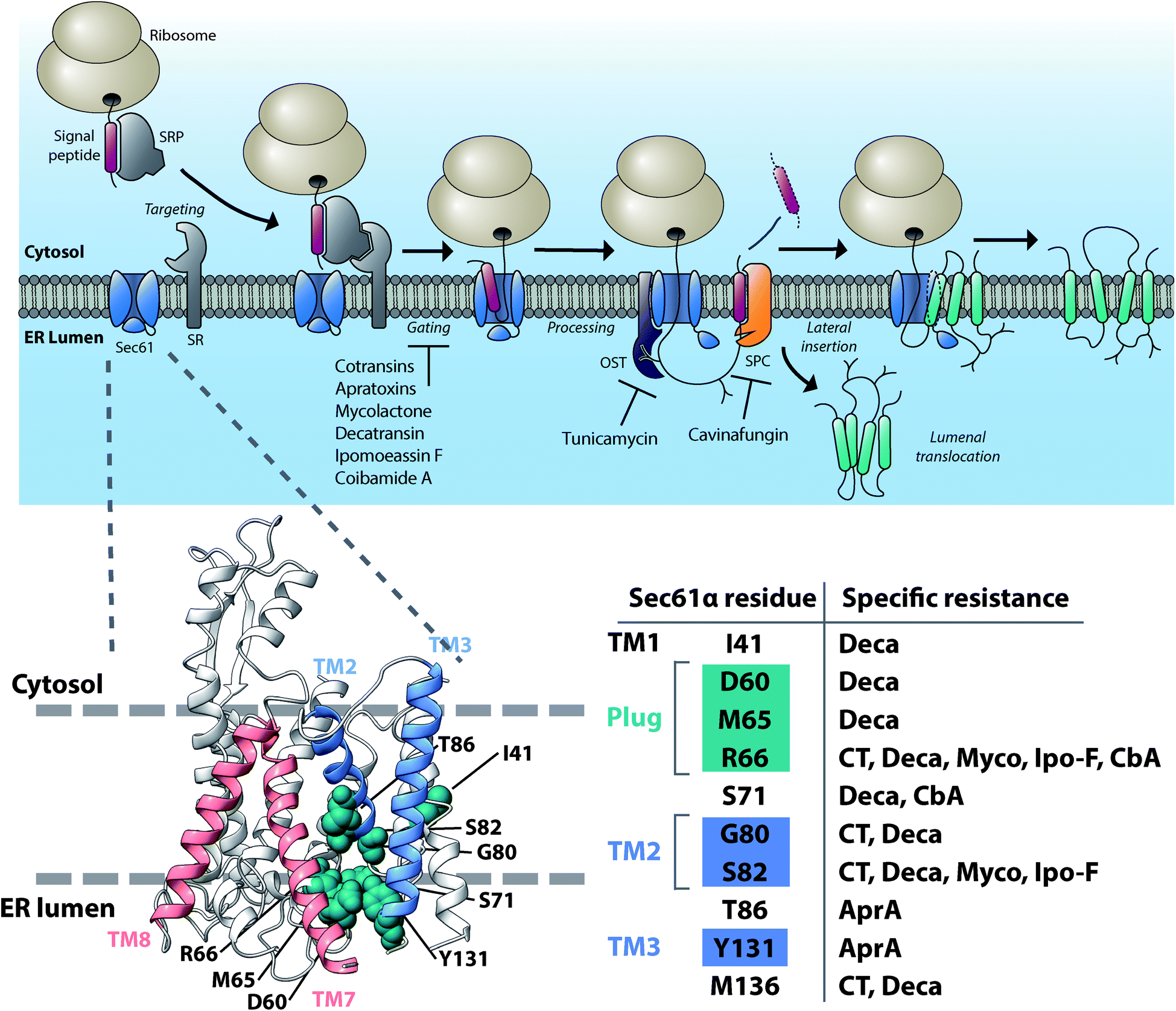 | ||
| Fig. 2 Mechanism of inhibition of protein ER import and maturation, and location of common resistance mutations within the structure of the Sec61 protein translocon. The pathway of cotranslational mammalian protein import into the ER (top). Inhibitors of Sec61 protein translocon prevent channel gating and insertion of secreted proteins into the ER lumen or diffusion of integral membrane proteins into the ER lipid bilayer. Tunicamycin is an inhibitor of cotranslational protein N-glycosylation, whereas cavinafungin prevents ER targeting signal peptide cleavage. In the structural model of mammalian Sec61 (bottom), the lateral gate helices of Sec61 are indicated in blue (TM2 and TM3) and red (TM7 and TM8) and the resistance mutations identified to confer strong resistance to most structurally diverse Sec61 inhibitors are shown in green and outline the putative NP-binding site on the lumenal end of the Sec61 lateral gate. SRP, signal recognition particle. OST, oligosaccharyl transferase complex. SPC, signal peptidase complex. CT, cotransin; Deca, decatransin; Myco, mycolactone; Ipo-F, ipomoeassin F; AprA, apratoxin A. | ||
2.1 Cotransins
A fungal natural product HUN-7293 (Fig. 3) was discovered as a potent suppressor of expression of the human vascular endothelial cell adhesion molecule I (VCAM-1)11 and a total synthesis of HUN-7293 was established enabling detailed SAR studies.12–14 Later, two independent studies revealed that HUN-7293 (later named cotransin) inhibits cell surface receptor expression by preventing ER translocation or membrane insertion of newly synthesized secreted or membrane proteins.15,16 Intriguingly, cotransin only inhibits biogenesis of a subset of the thousands of Sec61 substrate proteins in a manner dependent on the N-terminal ER targeting signal peptide or transmembrane segment.15,16 Photocrosslinking experiments with a photoactivatable analog of cotransin demonstrated that cotransin inhibits ER translocation by directly binding to the central pore-forming Sec61α subunit of the Sec61 translocon.17 Later, an unbiased screen to identify specific resistance-conferring point mutations, suggested that cotransin allosterically blocks Sec61 gating facilitated by nascent secretory polypeptides.18 Intriguingly, synthesis and testing of new cotransin analogs indicated that changes in the cotransin structure can alter the range of Sec61 substrate proteins it inhibits.19 A consensus sequence for cotransin-sensitive signal peptides or N-terminal transmembrane segments has not been identified, although specific point mutations that do not interfere with protein ER targeting or insertion, yet specifically modulate cotransin sensitivity of specific membrane proteins such as TNFα and AQP2, have been described.18,20 A quantitative proteomics study in human hepatocellular liver carcinoma cells using saturating (30 μM) concentration of cotransin, suggested that signal peptide-containing secreted proteins would be generally more sensitive to cotransin modulation than integral membrane proteins.20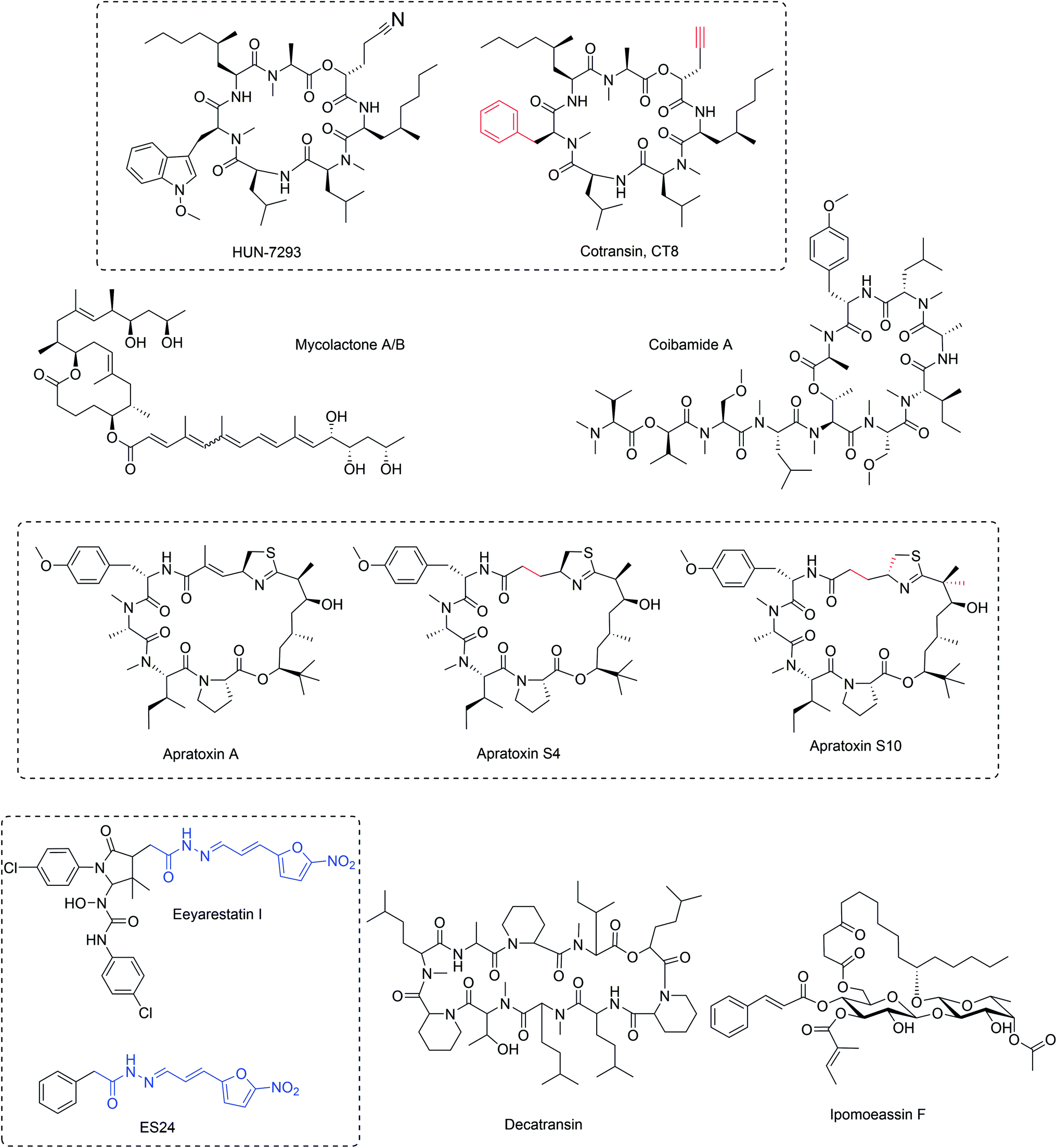 | ||
| Fig. 3 Structures of inhibitors of ER protein insertion. Their structural diversity translates into distinct interactions with Sec61 and differential pharmacological profiles. Red color indicates changes in synthetic compounds compared with the parent natural product. Blue indicates key fragment critical for compound activity. | ||
Another study identified CT8 (Fig. 3), a highly substrate-selective cotransin as a potent inhibitor of expression of the cancer-associated cell surface pseudokinase HER3, which was found to contain a highly cotransin-sensitive signal peptide.21 The reduced HER3 expression upon treatment of BT474 breast cancer cells with cotransin results from increased proteasomal turnover induced by the cytosolic displacement of the HER3 membrane protein. It is interesting that HER3 among the four signal peptide-containing HER family members is uniquely sensitive to CT8, and this opens up new therapeutic strategies to control HER activity in breast cancer. The authors further showed that CT8 efficiently enhances the efficiency of existing HER2 therapies in BT474 breast cancer cells.21 Further, many studies have identified importance of the role of the ER secretory pathway for replication of different flaviviruses and in one of these studies Sec61 was identified as a host factor required for growth and infectivity of several viruses including influenza, HIV and dengue.22 Treatment of cells with noncytotoxic concentrations of CT8 potently inhibited three different strains of HIV. The ability of CT8 to prevent ER trafficking of the HIV gp120 protein and therefore biogenesis and replication of the HI virus, suggests the possibility of targeting ER insertion with substrate-selective inhibitors as a viable antiviral strategy.22
2.2 Apratoxins
The natural secondary metabolite apratoxin A (Fig. 3) was originally discovered from a marine cyanobacterium Lyngbya majuscula (later identified/reclassified as Moorea bouillonii)23 and observed to potently inhibit cancer cell proliferation by inducing cell cycle arrest and apopotosis.24 Further, it was shown that treatment of cells with apratoxin A caused downregulation of expression of many cell surface receptors, particularly receptor tyrosine kinases and components of the endoplasmic reticulum.25 Further, it was demonstrated that apratoxin A inhibited protein translocation to the ER in biochemical experiments, suggesting that the direct cellular target may be an integral component of the ER machinery.25 Other cyanobacterial apratoxin family members showed similar anticancer activity, presumably by the same mechanism.26More recently, photocrosslinking competition, mutagenesis27 and co-immunoprecipitation experiments28 have directly identified Sec61 as the direct cellular binding target of apratoxin A. Biochemically, apratoxin A prevents protein ER translocation at an earlier stage than cotransin and appears to impact ER import of Sec61 substrate proteins in a substrate-nonselective fashion27 with the exception of so-called N-tail translocating integral membrane proteins that are initially inserted by the action of the EMC complex instead of Sec61.29 Intriguingly, the mutagenesis and competitive binding experiments indicate that similarly to cotransin, also apratoxin A binds Sec61α near the lumenal plug domain, suggesting that binding of different ligands to an at least partially overlapping binding site can result in inhibition of ER protein translocation with either a substrate-selective or a substrate-nonselective mechanism.
Several natural apratoxins and synthetic analogs have been synthesized, which provided clear structure–activity relationship information in cells and enabled the modulation of anticancer activity in vitro and in vivo through alteration of the ability to inhibit biogenesis of cell surface proteins and secreted factors,30–34 highlighting the potential for improving the currently narrow therapeutic window of apratoxins tested in in vivo cancer models.28,31,32 Specifically, removal of the Michael acceptor led to improved activity as apratoxin S4 (Fig. 3) demonstrated exquisite activity in a HCT116 colon cancer xenograft model.30 The next-generation apratoxin S10 (Fig. 3), possessing an inverted configuration of the thiazoline ring to improve potency and a gem-dimethyl group to prevent dehydration-induced deactivation of the compound, inhibited pancreatic cancer in an orthotopic PDX mouse model.31 Apratoxin S10 was able to modulate many but not all growth factors and cytokines in primary pancreatic cancer cells and tumor-associated stromal cells comprising the tumor microenvironment, suggesting the potential of apratoxin S10 to exert activity through dual inhibition of cancer growth and associated growth factor signaling but also through inhibition of growth-stimulating factors by the tumor microenvironment that are contributing to resistance. Furthermore, since VEGF and other proangiogenic factors appeared to be particularly sensitive to inhibition by apratoxins S4 and S10 in a cellular context and in vivo, these agents also have potent antiangiogenic activity,30,34 which is not only relevant for the inhibition of vascularized tumors but also for treating retinal angiogenic disorders. Consequently, apratoxin S4 was recently shown to inhibit retinal vascular cell activation by suppressing several angiogenic pathways and attenuated pathological ocular neovascularization in several animal models of ocular angiogenesis.35
2.3 Mycolactone
Mycolactone (Fig. 3) is produced by Mycobacterium ulcerans, the causative bacteria of a necrotising skin disease Buruli ulcers.36 For a comprehensive review on mycolactone mechanisms, see ref. 37. This bacteria harbors a megaplasmid that contains the polyketide synthetase genes required for mycolactone biosynthesis.38 Mycolactone is a macrolide that exerts an immunosuppressive effect at the site of bacterial infection, but also systemically prevents blood lymphocyte homing to draining lymph nodes.39 Because of the central role of mycolactone in mediating bacterial immune-evasion, its mechanism of action has attracted much interest.In immune cells, mycolactone potently inhibits cellular capacity to produce cytokines, chemokines and homing receptors, particularly inhibiting systemic production of IFN-γ.40 In monocytes and macrophages, mycolactone inhibited activation-induced production of cytokines and chemokines.40,41 Cellular and biochemical assays indicated that mycolactone inhibits a specific stage of ER insertion and differentially inhibits cotranslational and posttranslational protein translocation of secretory proteins.40,42
Unbiased resistance mutation mapping and competitive photocrosslinking experiments with photo-cotransin indicated that mycolactone targets Sec61α near its lumenal lateral gate, similarly as apratoxin and cotransin.43 Proteomic studies in mycolactone-treated CD4+ T lymphocytes, dendritic cells and dorsal root ganglion neurons support the notion that mycolactone broadly affects a broad range of Sec61 substrates including secreted proteins and diverse integral membrane proteins excluding tail anchored proteins and type III membrane proteins that are initially inserted through activity of the EMC complex.44 Further animal work demonstrated that Sec61 is the host receptor mediating mycolactone's diverse immunomodulatory effects,43 although interactions with other factors may also contribute to mycolactone's effects on the actin cytoskeleton45 and its analgesic properties.46 Displacement of secretory proteins into the cytosol by mycolactone triggers a cytosolic integrated stress response independently of the unfolded protein response in the ER.47,48 Because of its immunosuppressive properties, analogs of mycolactone are being pursued as potential anti-inflammatory agents.49
2.4 Eeyarestatin I
Eeyarestatin I (ESI, Fig. 3) was originally identified as a stabilizer of the known ERAD substrate MHC class I heavy chain.50 In this study, ESI was shown to stabilize two known ERAD substrates without general effects for proteasomal degradation of ubiquitinated substrates. Later treatment of cells with micromolar concentrations of ESI was shown to result in inhibition of ER insertion of a number of cotranslationally inserted substrate proteins, which was also recapitulated in biochemical ER insertion assays, however 250 μM ESI was required to detect effects in these in vitro experiments.51 A recent study demonstrated that ESI induces Ca2+ leakage from the lumen of the ER, which is apparently caused by ESI binding and inhibition of Sec61,52 suggesting that ESI binding to Sec61 might stabilize the channel in a partially open ion-conductive configuration and that Ca2+ leakage may be an important contributor to the cytotoxic effects of ESI. However, it should be noted that the direct binding partner of ESI has not been demonstrated by for example direct crosslinking or affinity chromatography approaches and therefore the exact mechanism of ER import inhibition by ESI remains incompletely understood. Furthermore, it has been suggested that ESI has dual targets (Sec61 and p97) and modulate ER insertion (via Sec61) and the ERAD pathway (via p97, see below).53 It appears that a truncated version, ES24 (Fig. 3) has Sec61 selectivity (over p97),52 although it remains currently unclear to what extent the observed phenotypic effects of ESI can be attributed to specific inhibition of Sec61.2.5 Decatransin
Decatransin (Fig. 3) is a novel decadepsipeptide isolated from the fungus Chaetosphaeria tulasneorum and found to inhibit growth of mammalian cells with nanomolar potency and also the yeast S. cerevisiae with an IC50 of approximately 2 μM.54 Chemogenetic screens in yeast suggested that decatransin would act by targeting the Sec61 complex in a similar manner as cotransin. Further, a genome-wide mutagenesis screen was carried out in yeast cells and identified several heterozygous point mutations in the SEC61 gene, encoding the Sec61α subunit of the Sec61 translocon. Even further, a similar mutagenesis screen in human cells identified yet more decatransin resistance mutations, establishing the Sec61 translocon as the direct decatransin target in fungal and mammalian cells with high certainty. It was shown that decatransin also inhibits co- and posttranslational ER translocation in both yeast and mammalian cells. This activity was shown to be independent of the sequence of the ER targeting signal peptide, which suggests that unlike cotransin, decatransin would act as a substrate-nonselective inhibitor of Sec61.54 Based on the ample mutagenesis data, it appears that binding of decatransin to Sec61 is similar, but distinct compared to cotransin.2.6 Ipomoeassin
Ipomoeassin F (Fig. 3) is one of the recent NPs described to specifically interfere with ER protein import. Ipomoeassins A–E are related amphiphilic glycolipids produced by the morning glory family plant Ipomoea squamosa, which were identified to inhibit proliferation of A2780 human ovarian cancer cells with a range of potencies.55 Later, a new member of the ipomoeassin family, ipomoeassin F was discovered56 and shown to be the most potent cytotoxin of the family with single-digit nanomolar inhibition against many cancer cell lines.57 Ipomoeassins contain a unique glycoconjugate structure, which prompted many groups to pursue its total synthesis.57,58 Having a facile synthesis method available allowed design of several ipomoeassin F probe molecules and attempts to directly identify its cellular target. Despite containing a potent Michael acceptor electrophile, it appears that ipomoeassin F does not bind its target covalently.59 However, immobilizing ipomoeassin F allowed attempts to identify its interaction partner by affinity chromatography, which revealed an approximately 40 kDa protein species which was identified as Sec61α.59 This finding was supported by competitive photo-cotransin binding experiments and biochemical experiments demonstrating that also ipomoeassin F inhibited protein ER import apparently in a substrate-nonselective manner. In cells, ipomoeassin F inhibits the production of secreted and glycosylated proteins without affecting production of cytosolic proteins. Heterozygous expression of earlier characterized Sec61α point mutations conferring resistance to cotransins, apratoxin and mycolactone18,27,43 revealed broad resistance to ipomoeassin F cytotoxicity demonstrating the connection between ER import inhibition and cytotoxic effect for ipomoeassin F. The observed difference in patterns of resistance mutations against ipomoeassin F and other Sec61 inhibitors suggests it binds a similar site on Sec61α, but in a distinct manner.2.7 Coibamide A
The latest NP to join the assorted collection of potent and specific inhibitors of Sec61 is coibamide A. Coibamide A is an N-methyl-stabilized lariat depsipeptide that was originally isolated from a Caldora species marine cyanobacteria from Panama.60 Coibamide A was observed to inhibit cell cycle progression of human glioblastoma cell lines and decrease their migratory and invasive capacity. Further, in xenograft models of glioblastoma, coibamide A was shown to inhibit tumor growth, although within a narrow therapeutic window.61 In addition, coibamide A induces mTOR-independent macroautophagy in mammalian cells.61,62 Development of several total synthesis methods have recently resulted in refinement of the absolute configuration of the natural product63–65 and allowed pursuing its cellular target.Coibamide A potently inhibits biogenesis of the integral membrane protein vascular endothelial growth factor receptor 2 (VEGFR-2) and its secreted ligand VEGF-A.61 Very recent work explored coibamide A structure–activity relationships to develop a potent coibamide A photoaffinity probe.66 Photocrosslinking experiments with this probe in isolated ER microsomes demonstrated specific crosslinking to a 40 kDa ER protein, which can be competed by known Sec61 ligands apratoxin A and mycolactone. Pulse-chase metabolic labeling experiments demonstrate that coibamide A potently inhibits cellular production of secreted and glycosylated proteins without impacting production of cytosolic proteins, consistent with substrate-nonselective inhibition of Sec61-mediated ER protein import. This notion was further supported by ER import inhibition of multiple classes of Sec61 substrate proteins in biochemical experiments using purified ER microsomes and in vitro translation.66
However, despite having apparently identical substrate-nonselective mechanisms for inhibiting Sec61 ER import, the three structurally diverse Sec61 inhibitors coibamide A, apratoxin A and mycolactone exhibit differential resistance to many Sec61 resistance mutations. Finally, a comparative analysis of the cytotoxic effects of these three Sec61 inhibitors towards cancer cell lines in the NCI-60 cancer cell line panel revealed a surprising difference in the cell types that were potently sensitive to the different Sec61 inhibitors.66 This is highly surprising considering that all of the compounds target Sec61 at the same site with a seemingly identical biochemical mechanism, and further suggests that modifications of Sec61 inhibitors may allow a surprising degree of cell type selectivity against different histological cancer cell types. In support of this notion, modifications to the natural structures of apratoxin A and coibamide A have already yielded new inhibitors with reduced general cytotoxic effects while retaining their original anti-tumor efficacy in tumor xenograft models.30,32,67
3. Inhibitors of secretory protein modification and maturation
3.1 Tunicamycin
The tunicamycin antibiotics (Fig. 4) were identified from Streptomyces lysosuperficus and exhibit potent antibacterial activity against various Gram-positive bacteria.68 Tunicamycins are nucleoside antibiotics that contain a uracil base attached to a C11 tunicamine sugar, glycosylated at C11 by a GlcNAc sugar and N-acylated at C10 by a C12-C15 fatty acid. Tunicamycins exhibit broad antimicrobial activity against a range of Gram-positive bacterium particularly Bacillus genus members (MIC 0.1–20 μg mL−1), but also exhibit significant cytotoxicity towards mammalian cells as a result of their ability to inhibit biogenesis of N-linked glycoproteins.69,70 Interest in tunicamycins later led to the discovery of their biosynthetic gene cluster.71 In bacteria, the direct target of tunicamycin, MraY, is a translocase, which catalyzes the first step during the biosynthesis cycle of bacterial peptidoglycan. Tunicamycin reversibly inhibits MraY with a Ki of 0.6 μM.72 Structure of tunicamycin bound to E. coli MrayY73 and its eukaryotic target enzyme, GlcNAc-1-phosphate transferase74,75 required for N-linked glycoprotein biosynthesis were reported. These structures revealed a clear difference in tunicamycin interactions between MraY and GPT and this allowed rapid structure-guided design of a modified tunicamyin analog with high selectivity for MraY over GPT (IC50 640 nM versus 15 μM, respectively),74 suggesting the possibility to develop tunicamycin as a selective antimicrobial agent.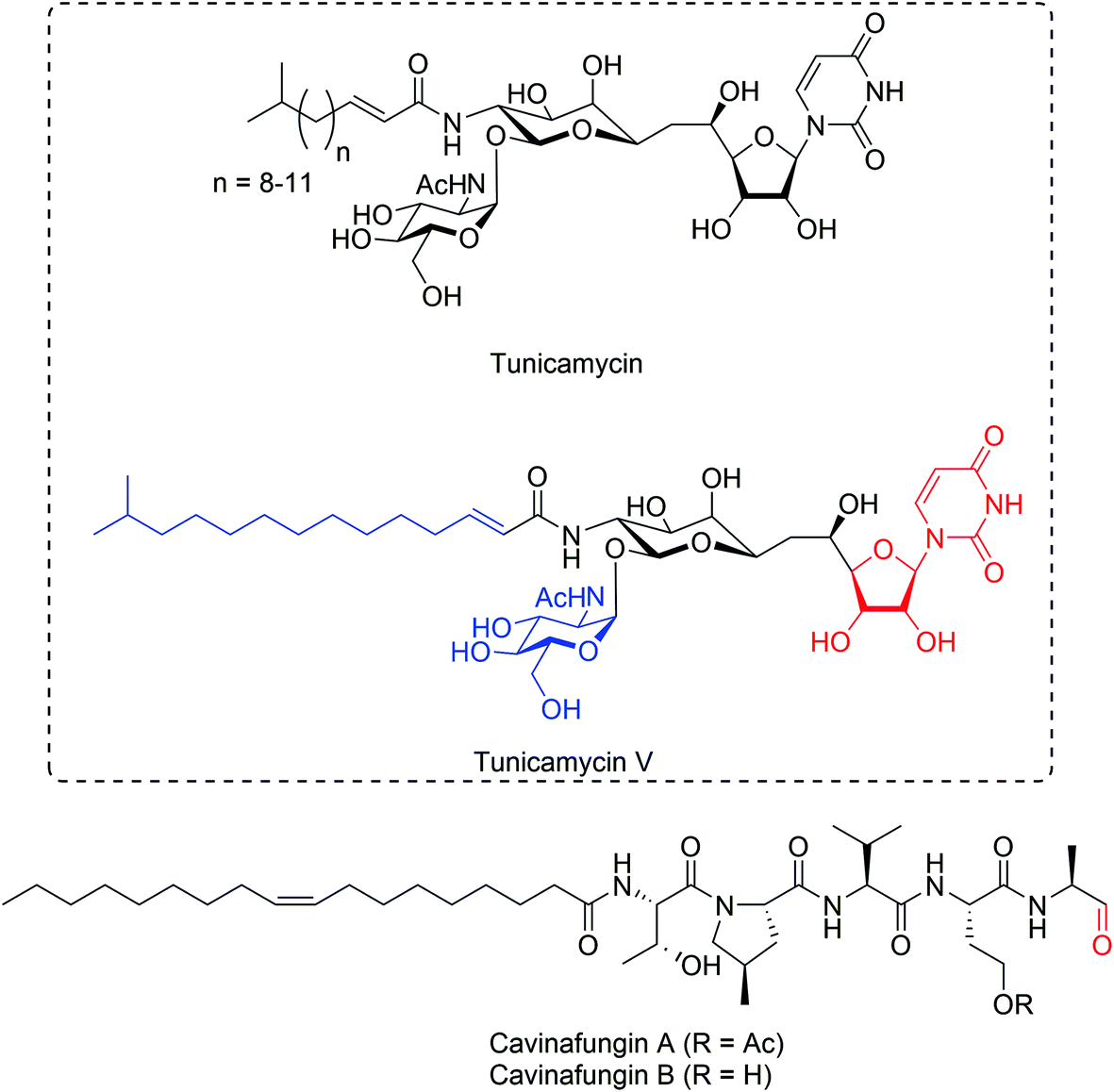 | ||
| Fig. 4 Inhibitors of secretory protein modification and maturation. Structures of N-glycosylation inhibitors, tunicamycins, and signal peptide processing inhibitors, cavinafungins. Red color indicates key moieties for activity. Structural features in blue are contributory to activity. | ||
Very recently, the total synthesis of tunicamycin V (Fig. 4) has been described,76 which led to synthesis of a series of tunicamycin analogs and generation of a structure–activity dataset for tunicamycin's antibacterial activity.77 Inhibition of N-glycosylation by tunicamycin treatment has been shown to overcome chemoresistance in a multidrug-resistant gastric cancer cell line.78 Because of its ability to robustly induce ER stress resulting from rapid misfolding of newly synthesized glycoproteins within the ER lumen, tunicamycin has become a powerful probe for studying ER protein misfolding and ER stress pathways.79
3.2 Cavinafungin
Cavinafungins (Fig. 4) were initially extracted from cultures of fungus Colispora cavincola isolated from plant matter from Argentina and demonstrated to exhibit antifungal activity against many fungal species.80 Determination of the absolute configuration demonstrated that cavinafungins are linear lipopeptides that contain a terminal aldehyde residue80 and exhibit broad antifungal activity against Candida species (MIC 0.4–4 μg mL−1) and a reduced potency against the filamentous fungi Aspergillus fumigatus (MIC 8 μg mL−1). Cavinafungins A and B are nearly identical lipopentapeptides, which only differ in acetylation of a serine sidechain (Fig. 4). A one-pot solid-phase total synthesis of cavinafungin B has recently been reported.81Cavinafungin A was discovered to inhibit replication of dengue and Zika viruses with a potency ranging from 4 to 400 nM IC50, and strongly depending on presence of the terminal aldehyde, suggesting a covalent mechanism of inhibition.82 A genome-wide siRNA screen in mammalian cells suggested the ER signal peptidase complex as a possible target of cavinafungin A and this was further supported by chemogenetic screens carried out in yeast cells. A mutagenesis approach focused on ER signal peptidase subunit genes revealed a range of mutations conferring cavinafungin A resistance exclusively in the catalytic signal peptidase subunit SEC11, whereas mutations in the other three subunits were not observed, suggesting that cavinafungin A directly targets the SEC11 subunit in the signal peptide binding cleft, possibly directly competing with substrate binding.82 Cavinafungin A prevents dengue virus replication at considerably lower (IC50 1–5 nM) concentrations compared to what is required to induce host cell cytotoxicity (CC50 800–18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 nM).82 Further, because most flaviviruses appear to depend on the ER signal peptidase activity for their replication, cavinafungins could be attractive lead compounds for development of broadly acting therapeutics against flavivirus infections.
000 nM).82 Further, because most flaviviruses appear to depend on the ER signal peptidase activity for their replication, cavinafungins could be attractive lead compounds for development of broadly acting therapeutics against flavivirus infections.
4. Inhibitors of protein trafficking
4.1 Brefeldins
Brefeldin A (BFA, Fig. 5) is a fungal natural product lactone that was originally isolated in the 1950s from Penicillium decumbens.83 More recently, several analogues have been identified from fungi of the genus Penicillium derived from soil and marine sediments, including the marine Penicillium spp. PSU-F44 (ref. 84) and DT-F29,85 and terrestrial Penicillium janthinellum.86 The recent synthesis and discovery of new brefeldin A analogs has been reviewed in detail.87 Numerous analogues were synthesized to improve drug-like properties and various prodrug strategies were considered as well (Fig. 5).87 | ||
| Fig. 5 Structures of brefeldin A, and natural and synthetic prodrug analogues that retained potent activity. Sites of modification of the core skeleton to develop prodrugs are numbered and modifications indicated in blue. | ||
BFA is a classical tool compound used in cell biology studies as a secretion inhibitor because of its action on the Golgi apparatus and endosomes.88,89 The elucidation of its mechanism of action led to the concept of interfacial inhibition as a way to stabilize protein–protein interactions (PPI) instead of inhibiting PPI90 (Fig. 6). BFA displays specific effects on membrane trafficking by affecting small G-proteins of the ADR-ribosylation factor (ARF) family, which regulate cellular traffic by GDP/GTP cycling and consequent capacity to interact with GTP-bound effectors. ARF proteins are N-myristoylated to promote membrane association when GTP bound.89 The activation of ARF is facilitated by Arf-guanine nucleotide exchange factors (ArfGEFs) which are characterized by a conserved catalytic domain consisting of ∼200 amino acids, termed Sec7 domain, involved in Arf binding.91 BFA is an uncompetitive inhibitor of ARF at the Golgi, blocking the ArfGEF mediated Arf-GDP/GTP guanine nucleotide exchange reaction.92
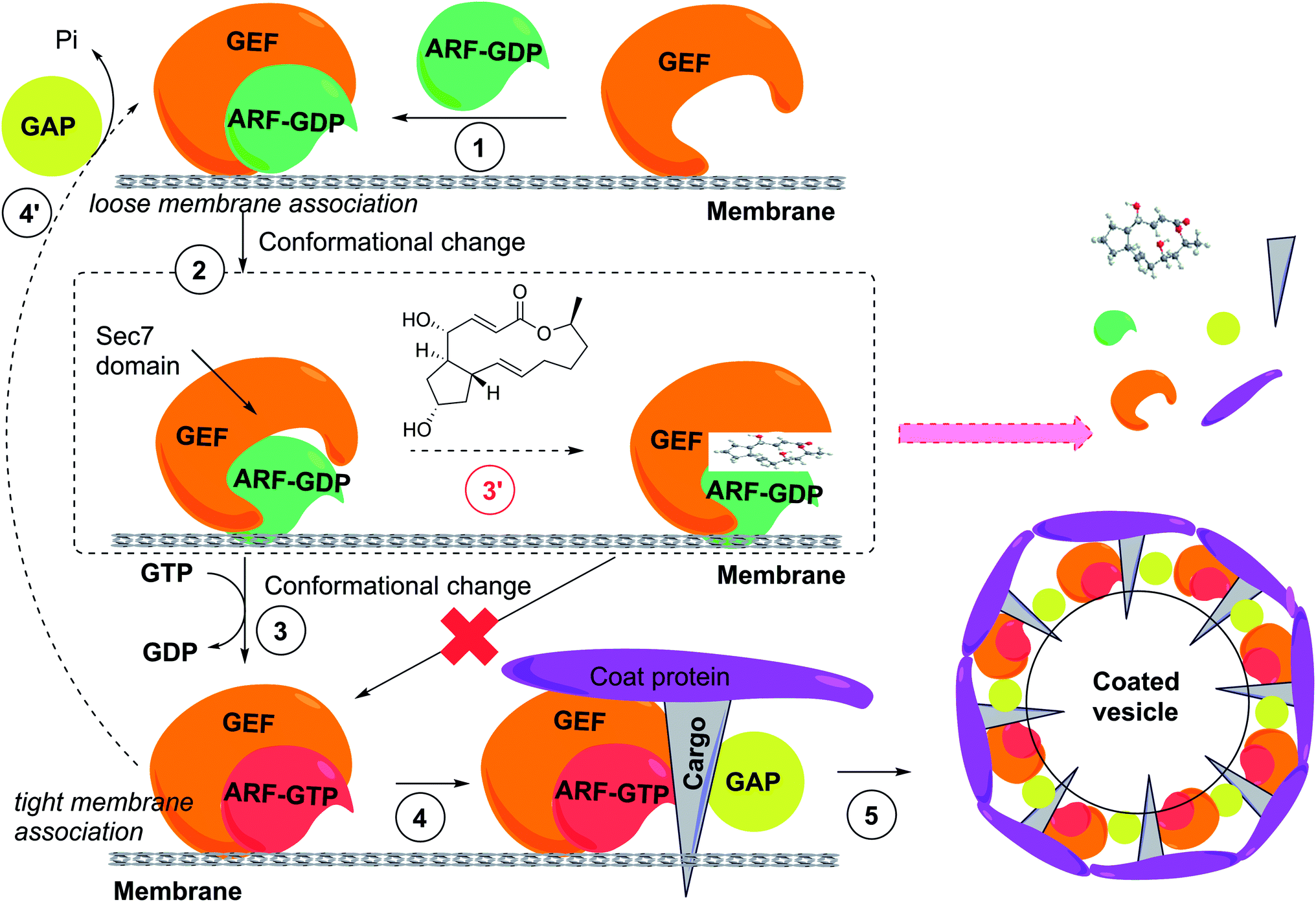 | ||
| Fig. 6 ARF GTPase activation by GDP/GTP exchange factors to regulate vesicle formation and unusual uncompetitive mechanism of action of BFA, which led to the concept of interfacial inhibition, where protein complexes are stabilized near interacting interfaces. (1) GDP-bound ARF associates with GEF. (2) Conformational change of ARF is required to stimulate GDP dissociation. (3) GEF catalyzes the activation of ARF through GDP/GTP exchange and the complex now tightly associates with the donor membrane. (4) ARF-GTP recruits effectors, including coat components (coat proteins, cargo) from the cytosol, and also GTPase activating protein (GAP), which usually catalyzes the inactivation of ARF-GTP, closing the GDP-GTP cycle (4′). (5) GAP appears to be inactive when bound to the coat-containing complex, presumably driving coat polymerization, budding and vesicle formation. GAP becomes active in this conformational complex state, leading to GTP hydrolysis coat and GAP release and the uncoated vesicle is able to fuse with an acceptor membrane (not shown). (3′) In the presence of BFA the low-affinity complex is trapped through binding of BFA at the interface of ARF-GDP and the Sec7 domain of GEF. Binding of BFA to the Arf-GDP-Sec7 domain complex blocks GDP/GTP exchange on ARFs and consequently vesicle coat recruitment and vesicle formation. The ultimate result is the release of coat components into the cytosol and prevention of membrane trafficking. | ||
BFA does not bind to GEF alone but to the Arf-GDP-Sec7 domain complex (Fig. 6), sequestering GEFs in an abortive conformation and acting as a “dominant negative”.93 In a remarkable mechanism, BFA inserts into the hydrophobic pocket transiently formed in the Arf-GDP-Sec7 complex, preventing conformational changes required for nucleotide exchange and requisite catalytic contact. The crystal structure has been solved,94,95 providing the basis for the discovery of novel GEF inhibitors and possibly modulate their specificity. Because BFA contacts both ARF-GDP and ArfGEF in this complex, BFA has specificity for a subset of both ArfGEFs and ARF GTPases.96 ArfGEF family members, 15 of which are encoded in the human genome, can be classified based on their BFA sensitivity.
GTP bound Arf recruits coat proteins including the COPI complex to induce budding of coated carrier vesicles (Fig. 6). ArfGEFs confer spatial and temporal specificity to vesicular transport.90 Phenotypically, BFA induces the release of vesicle coat proteins, including COPI and clathrin coat subunits, into the cytosol. BFA affects integrity of subcellular compartments including Golgi apparatus and endosomes, inhibiting the secretion and re-distributing Golgi membrane proteins to the endoplasmic reticulum (ER). Of note, several BFA effects are species specific, which requires caution when interpreting results in certain systems.88,89
4.2 Discovery of GEF and other secretory pathway inhibitors inspired by brefeldin A
BFA provide the inspiration for structure-based screening, which identified LM11 (Fig. 7) that targets BFA-insensitive GEFs, including ARNO (CYTH2).97 Both BFA and LM11 affect activation of class I ARFs (ARF1-3) and class II ARFs (ARF4 and 5) but not ARF6. Phenotypic screens of endocytic and secretory transports yielded Secin H3 (Fig. 7), which also bind to Sec7 of ArfGEFs. Secin H3 inhibits cytohesins, which leads to insulin resistance.98 Golgicide A (Fig. 7) was discovered to selectively inhibit GBF1, which is the ArfGEF that activates ARF1 and recruits COPI to the cis-Golgi membranes. The mechanism of action by BFA might be generally exploited to discover more selective drugs, and not only at the level of ARF and GEFs. Similarly, inhibitors of other GTPases besides ARF have been identified, including inhibitors of RhoGEFs activation. Ras GTPases might also be targeted in a similar fashion.96 | ||
| Fig. 7 BFA-inspired secretory pathway inhibitors via structure-based or phenotypic screening. | ||
In other phenotypic screens, Exo1 and Exo2 (Fig. 7) were identified using an image-based screens.99,100 Dynasore (Fig. 7), a cell permeable inhibitor of dynamin as probe for dynamin-dependent clathrin coat formation, was discovered to block synaptic vesicle endocytosis.101,102 Secramine (Fig. 7) was discovered as a potential new tool to dissect Cdc42 function from that of other Rho GTPases.103
5. Natural products that indirectly influence secretory capacity of the ER
5.1 SERCA inhibitors: thapsigargin and basiliolides
Thapsigargin (Fig. 8) is a structurally complex sesquiterpene lactone that was originally isolated in 1978 from roots and fruits of the plant Thapsia garganica and observed to potently liberate histamine from mast cells of the skin.104 In early 1990's several studies demonstrated the ability of thapsigargin to induce apoptosis in several cancer cells.105–108 However, later studies demonstrated that thapsigargin is similarly cytotoxic against malignant and normal cells, limiting its therapeutic potential. Thapsigargin induces cytotoxicity by elevating cytoplasmic Ca2+ levels at subnanomolar concentrations by inhibiting the sarco-endoplasmic reticulum Ca2+-ATPases (SERCAs).109,110 The X-ray crystal structure of SERCA bound to thapsigargin revealed the thapsigargin structural conformation and detailed interactions with SERCA.111 In the crystal structure, a large rearrangement of the SERCA channel was observed, providing a structural explanation for how thapsigargin binding induces a passive leakage of ER lumenal Ca2+ into the cytosol. Despite many challenges, several synthetic routes for synthesis of thapsigargin and analogues have been developed by different laboratories (reviewed in ref. 112).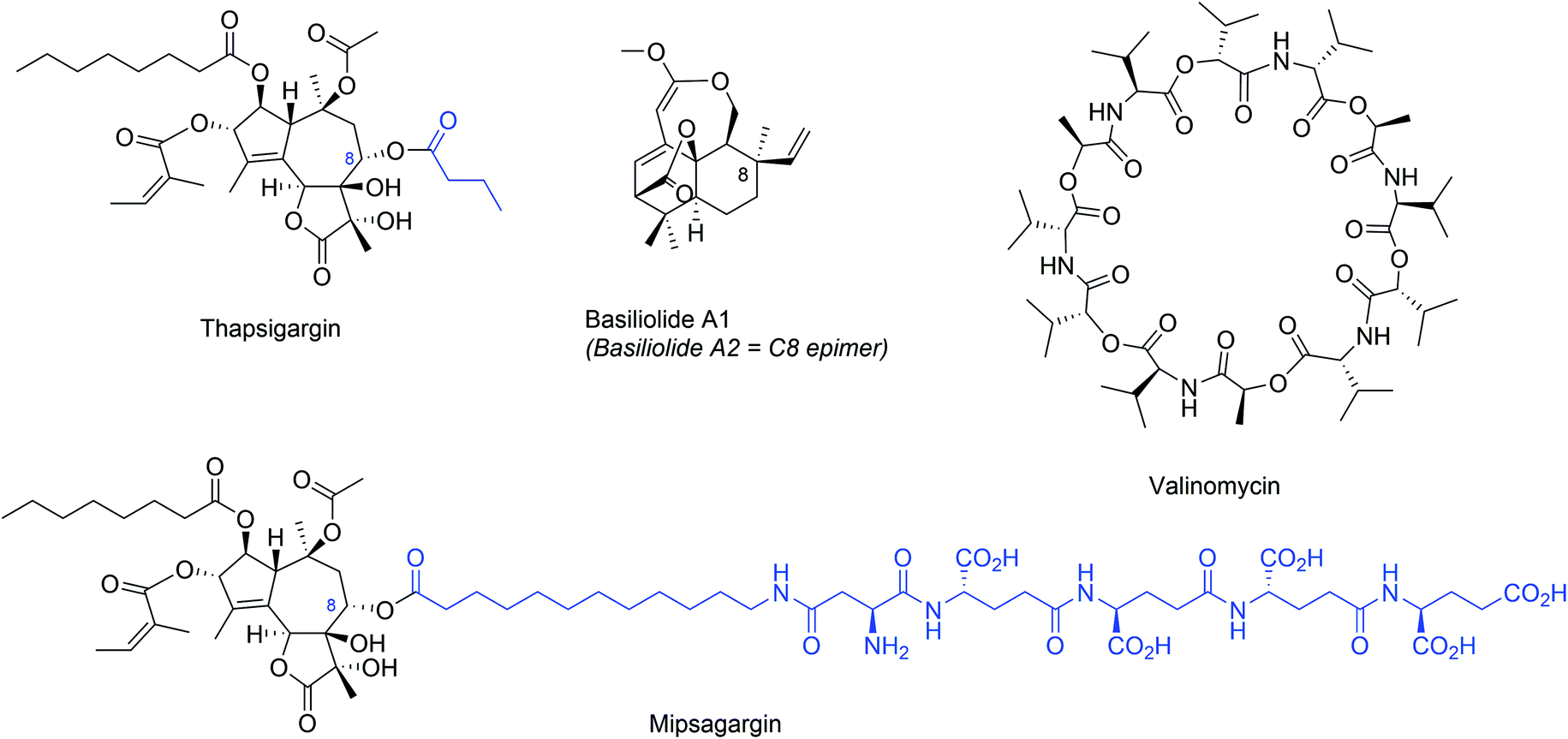 | ||
| Fig. 8 Structures of SERCA inhibitors (thapsigargin and its prodrug mipsagargin, and basiliolide A1) and the K+ ionophore valinomycin as indirect modulators of secretory capacity though cation import regulation. Structural differences between thapsigargin and mipsagargin are indicated in blue. | ||
The generally cytotoxic nature of thapsigargin has prevented its development as an anticancer agent, but recently its use as a cancer-targeting prodrug has been explored. In this strategy, a polypeptide was attached to the thapsigargin C8 position (Fig. 8), which targets the fusion compound to metastatic deposits of androgen independent prostate cancer.113 This fusion peptide is designed to be cleaved by a prostate tissue-specific protease (PSA), which is only produced in high levels in the extracellular space of metastatic prostate cancer cells. After extracellular cleavage, the released hydrophobic thapsigargin can enter the cancer cells and promote Ca2+-mediated cell death. Importantly, the prodrug mipsagargin (Fig. 8), is inactive against SERCA, which helps to minimize off-target cellular cytotoxicity. Mipsagargin is currently investigated in late stage clinical trials against prostate cancer, progressive glioblastoma and hepatocellular carcinoma.114
Later, basiliolides, a class of sesquiterpenes also isolated from T. garganica, were identified as structurally distinct SERCA inhibitors in sea urchin eggs115 and subsequently in mammalian cells.116 Despite triggering ER Ca2+ release, basiliolide A1 (Fig. 8) does not trigger apoptosis similarly as thapsigargin, suggesting it inhibits the SERCA pump with a distinct mechanism.116 A synthetic strategy for basiliolides has been reported.117
5.2 Potassium ionophore: valinomycin
Valinomycin (Fig. 8) is a cyclic dodecadepsipeptide isolated from several Streptomyces species.118,119 Valinomycin acts as a highly K+-specific ionophore120 making biological membranes selectively permeable for K+.121 Valinomycin was discovered to prevent upregulation of BIP expression as a response to ER stress.122 The valinomycin biosynthetic gene cluster has been identified.123,124 Interestingly, valinomycin was identified in a phenotypic screen for substrate-selective small molecule inhibitors of prion protein.125 In this study, valinomycin surprisingly influenced biogenesis of hamster prion protein without effects on biogenesis of human prion protein and this difference was attributed to differences in the sequence of the prion protein ER targeting signal peptide.125 However, how perturbation of cellular K+ homeostasis leads to signal peptide-dependent inhibition of prion protein ER insertion remains unknown.5.3 UPR modulators
At least some of the Sec61 inhibitors discussed above in Sections 2.1–2.7, which do not enter the ER, were shown to downregulate the UPR by depleting some of the key proteins (BIP/GRP78 and others). In turn, activation of the UPR upon accumulation of unfolded/misfolded proteins in the ER causes ER stress and also affects protein secretion, along with other processes, depending on the specific branch of UPR being modulated. In mammalian cells, there are three UPR signaling branches: (1) IRE1α-XBP1, (2) PERK-ATF4-CHOP, and (3) ATF6 cassette.126 GRP78 regulates all three branches (Fig. 9).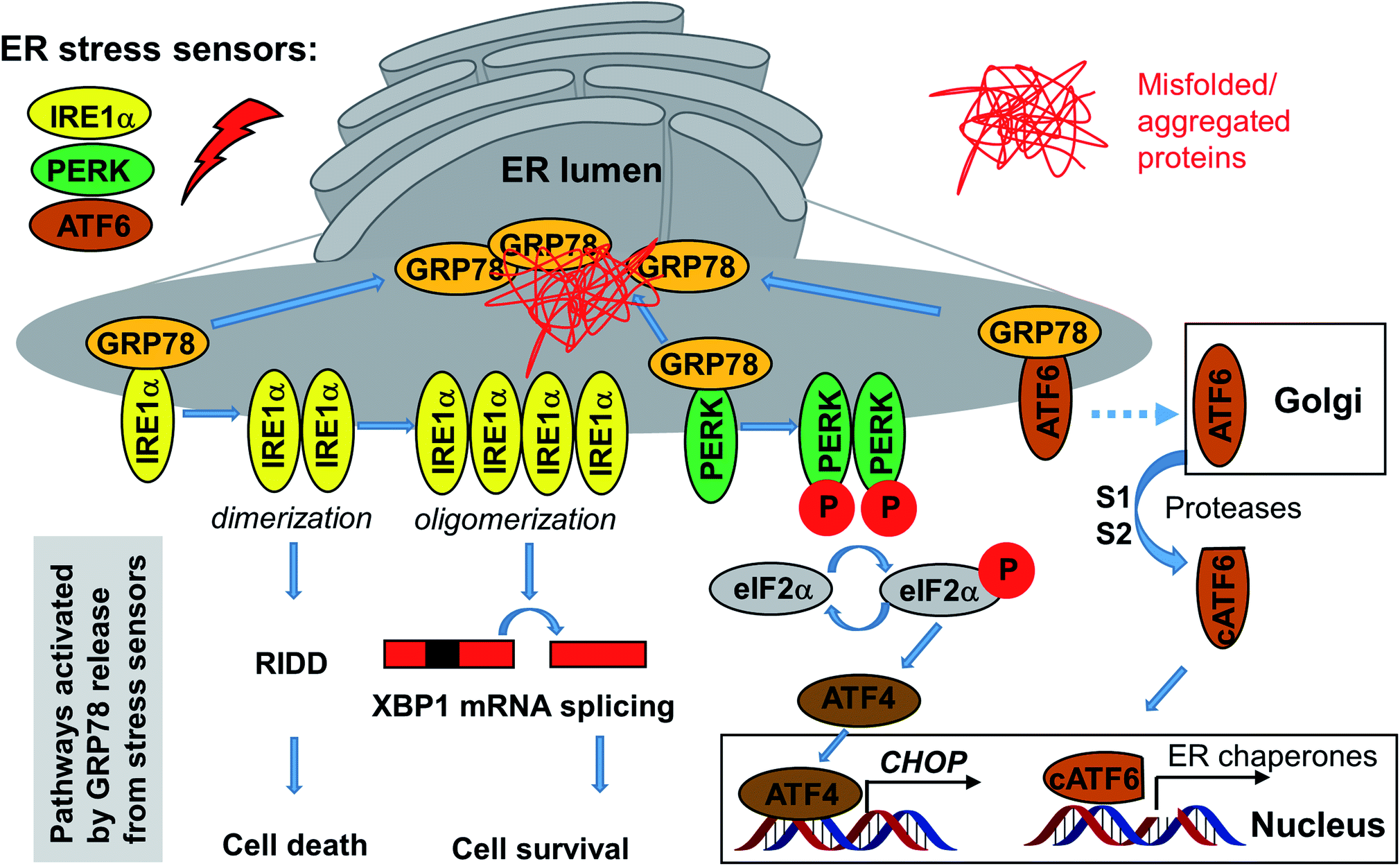 | ||
| Fig. 9 The branches of the ER stress pathways. Under unstressed conditions, GRP78 associates with the three membrane-bound ER stress sensors but upon activation by unfolded proteins GRP78 dissociates from them, which triggers parallel pathways that can be pharmacologically modulated, especially the IRE1α and PERK pathways (see text). | ||
There is a delicate balance whether UPR induction causes apoptosis or cellular protection, depending on pathway/branch and target but also extent/duration of activation etc., and modulators in both directions might be of therapeutic relevance.126
Using an XBP1-luciferase reporter screen in HeLa cells, trierixin and quinotrierixin (Fig. 10)128,129 were discovered from Streptomyces spp. as inhibitors of XBP1 activation by thapsigargin- and tunicamycin-induced ER stress.128,129 This activity was further validated by monitoring endogenous XBP1 splicing using RT-qPCR at nanomolar concentrations (IC50 30 ng mL−1). However, these and related compounds of the triene-ansamycin family of natural products possess pleiotropic biological activities, limiting their use as potential probes. Nevertheless, this activity might contribute to the antitumor properties and partially explain the inhibition of EGFR signaling.
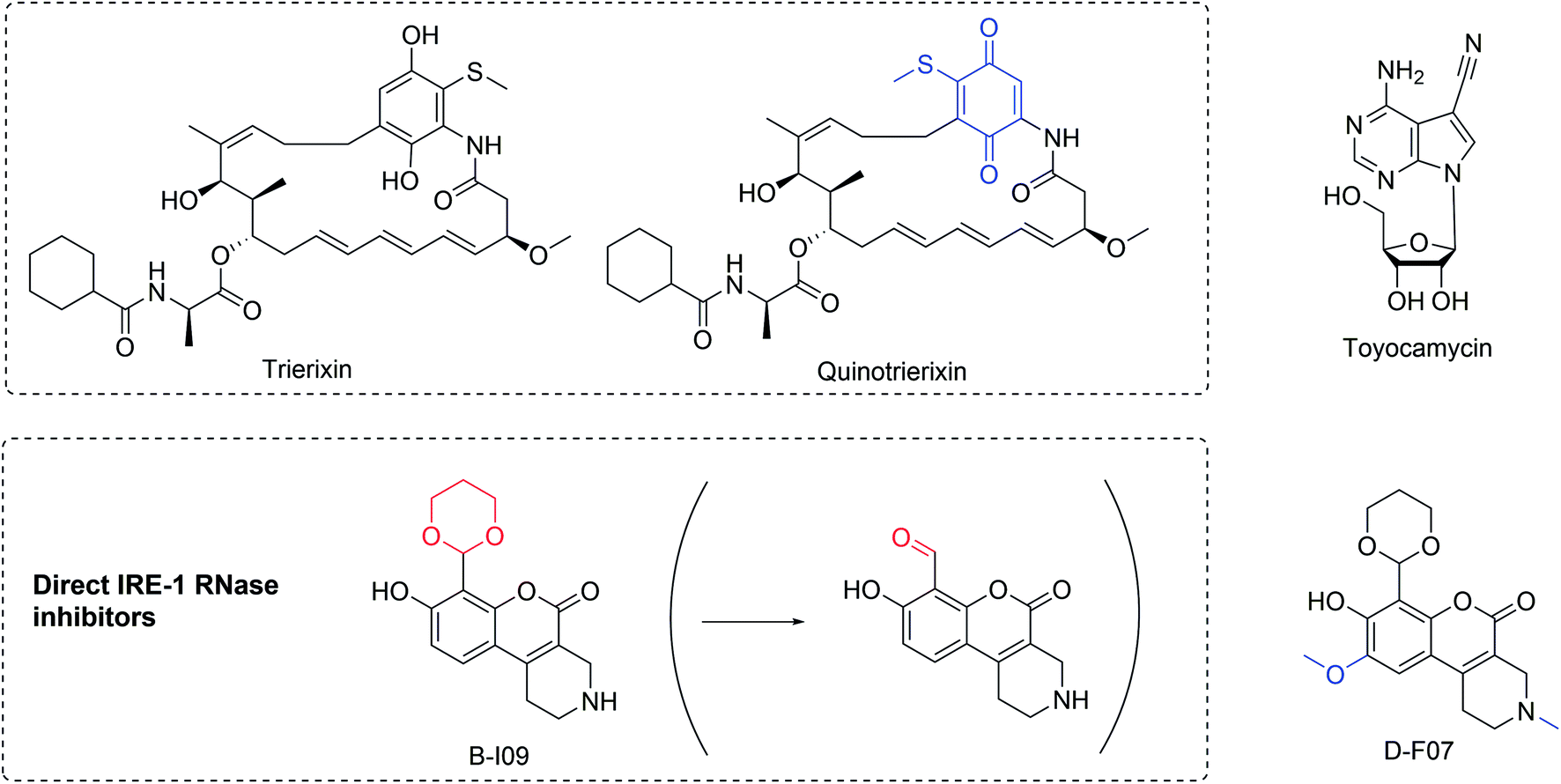 | ||
| Fig. 10 Inhibitors of XBP1 mRNA splicing. Structural changes relative to respective parent compounds are indicated in blue and the masked and reactive group for prodrug B-I09 in red. | ||
Toyocamycin (Fig. 10) is produced by an actinomycete strain and identified as an inhibitor of ER-stress induced XBP1 activation.127 This natural product selectively affects the IRE1α-XBP1 branch without impinging on the activation of PERK and ATF6. It also does not affect IRE1α phosphorylation, acting more downstream at the level of XBP1.127 While toyocamycin is also an RNA synthesis inhibitor, the XBP1 splicing inhibition occurs at ∼150 fold lower concentrations (IC50 12 μM vs. 0.08 μM in HeLa cells). SAR studies indicated the importance of the adenine moiety for the splicing inhibition. MM cells with higher levels of spliced XBP1 were particularly susceptible to toyocamycin.127
Recently, a potent covalent natural product-like IRE1α RNase inhibitor was developed based on a tricyclic chromenone scaffold, targeting the RNase domain of IRE1. This compound (B-I09) contains a masked aldehyde functionality acting as a prodrug to target lysine 907 in the RNase domain of IRE1α through specific Schiff base formation.130 The efficacy of B-I09 in B-cell cancer provided evidence that the IRE-1/XBP1 pathway reduces leukemic cell survival. In addition to multiple myeloma, these inhibitors show potential to treat mature B cell leukemia and lymphoma. Its application can further be extended to other c-Myc driven cancers.131 A fluorescent version of the inhibitor (D-F07) was recently reported that enables precise temporal control for the prodrug activation.132
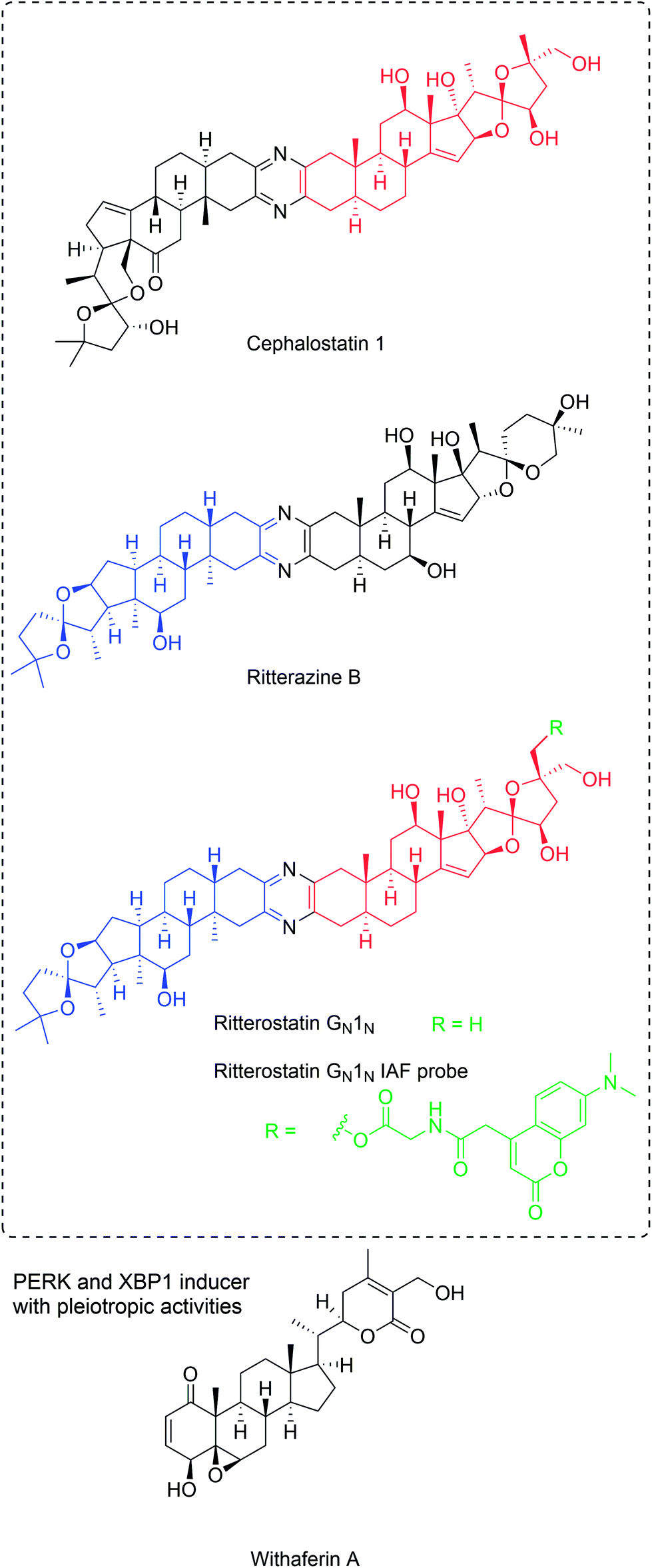 | ||
| Fig. 11 PERK pathway activators with differential selectivity. Cephalostatin-ritterazine hybrid was shown to have selectivity for the PERK branch, while withaferin A is non-selective. For ritterostatin GN1N, red and blue colors indicate cephalostatin 1 and ritterazine B fragments, respectively. Probe position and label are indicated in green. | ||
While these compounds have been found to target oxysterol-binding protein (OSBP) and related proteins,137 there is no obvious correlation to ER stress induction and apoptosis.136 Using immunoaffinity chromatography with ritterostatin GN1N (ritterazine-cephalostatin hybrid, Fig. 11), the ER-resident chaperone GRP78 (BIP) was identified as the dominant specific intracellular target of the compound, along with other HSP70 heat shock proteins to minor degrees.138 ITC with recombinant protein and hybrid GN1N (KD 190 nM) or cephalostatin (KD 679 nM) validated the direct interactions between GRP78 and this compound class. The binding site was identified to be the C-terminal domain by testing both domains of GRP78 individually. The N-terminal ATPase site was not targeted, consistent with the lack of inhibition of the ATPase activity. However, KD values for other HSP70 proteins are similar and GRP78 selectivity confirmed to be a result of the predominant localization in the ER.138 Obviously the relevant pharmacological effects are the result of a combination of affinity and subcellular localization.
GRP78 binding has been linked to UPR induction and the most affected branch was found to be the PERK arm since EIF2α phosphorylation was increased at lower concentration compared with downstream targets of the IRE1α arm (see above), including XBP1 and ATF4 (Fig. 9).138 GRP78 as a marker of the UPR was also strongly increased in HCT116 cells. While not confirmed yet, it is plausible that the induction of the UPR via the PERK arm is responsible for the antitumor and apoptotic activity of this compound class,138 although a net effect from multiple target interactions might also be likely.
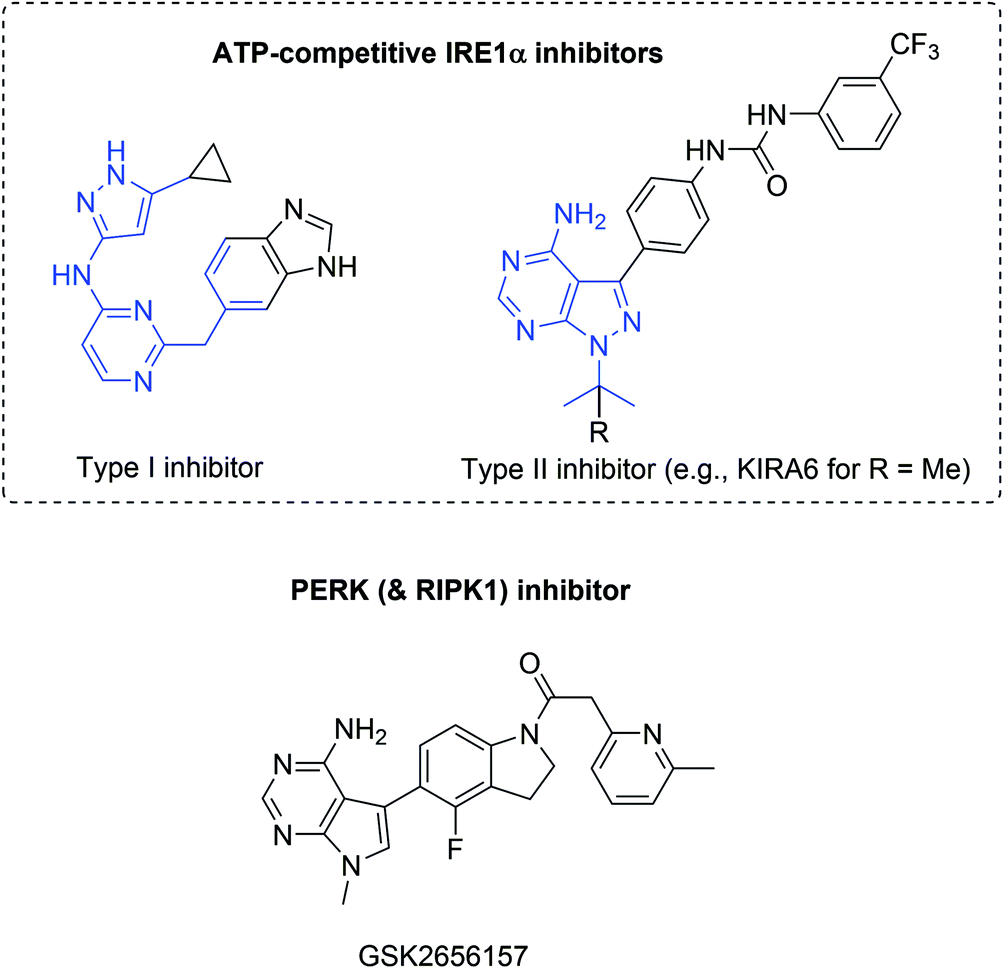 | ||
| Fig. 12 Representative ATP-competitive IRE1α and an advanced PERK inhibitor but with relevant RIPK1-inhibitory activity. Adenine-pocket interacting units (mimics) are indicated in blue. | ||
The PERK branch can be selectively inhibited by GSK2656157 and related compounds.142 These compounds prevent ER-stress induced autophosphorylation of PERK and consequently inhibit EIF2α substrate phosphorylation in vitro.143 However, in vivo data did not provide evidence of PERK inhibition; specificity is an issue with the current lead compound and RIPK1 inhibition is probably more relevant.144
In addition to natural products and natural product-like compounds, there are now numerous synthetic pharmacological agents that target the various branches of the UPR as positive and negative regulators, and their efficacy is being assessed in various disease states where unfolded protein accumulation plays a critical role.145
5.4 ERAD
ER-associated degradation (ERAD) alleviates ER stress and maintains ER homeostasis. It is a complementary mechanism to autophagy which can also clear protein aggregates generated by improper protein folding in the ER. Proteins commonly undergo N-glycosylation upon ER entry and this multi-step process requires the action by several enzymes. The resulting N-glycan structures promote glycoprotein folding, while mannose-trimmed structures are directed towards ERAD.126 In mammalian cells, ER degradation-enhancing α-mannosidase-like proteins (EDEMs), ER Mannosidase I (ER ManI) and Osteosarcoma amplified-9 (OS-9) ensure the routing of misfolded proteins, while p97 ATPase is required for functional ERAD.146The proteasome in the cytosol takes part in ERAD at a later stage by ubiquitin-dependent degradation. Several proteasome inhibitors are FDA approved, including carfilzomib based on epoxomicin produced by actinomycetes.147 In this brief section, however, we focus on the upstream ER events.
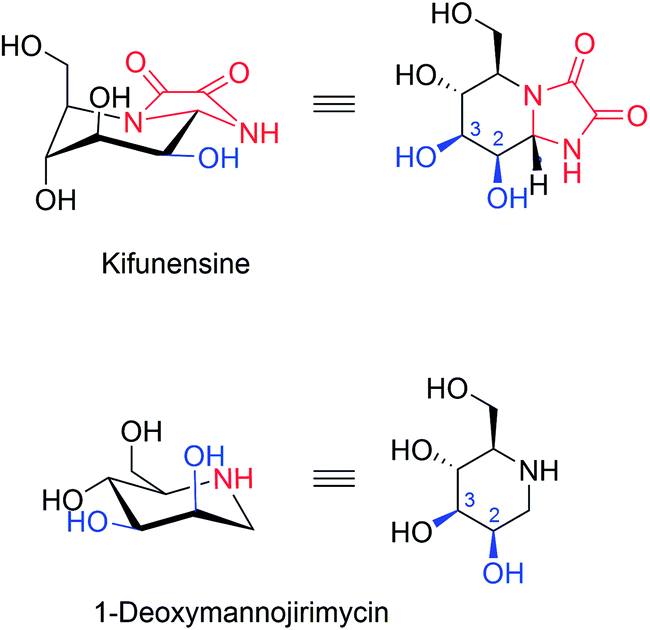 | ||
| Fig. 13 Structures of GH47 family α-mannosidase inhibitors as ERAD inhibitors. Kifunensine is 100-fold more potent than 1-deoxymannojirimycin. Structural differences are indicated in red. Hydroxy groups that coordinate the essential Ca2+ in the active site are indicated in blue. | ||
6. Therapeutic potential and opportunities for protein secretion modulation
The secretory pathway has emerged as an attractive yet complex target for therapeutic intervention, due to the multitude of secretory molecules and their disease involvement.145,154–156 Since protein secretion is an essential pathway for mammalian cell physiology, selective modulation of pathway components is required for ultimate translational success and for development of selective pharmacological tools to further understand the biology which is by no means fully illuminated yet. As we have described, in many cases natural products identified through phenotypic screening campaigns have provided the motivation and starting points for chemical biology approaches to identify and characterize the direct biological targets or target complexes. As one example, inhibitors of protein maturation and trafficking have historically been identified and have served as important chemical tools to provide insight into the pathways they inhibit, once their mechanisms had been successfully established. BFA is a prime example for the fact that studying the mechanism of action of natural products can yield unique mechanisms of action. Subsequently these natural products provide the inspiration for other screening and synthetic campaigns. Until recently the therapeutic development of secretion pathway modulators has not been perceived feasible, which has prevented their rigorous pursuit in part due to on-target or pleiotropic off-target toxic effects. However, in recent years increased efforts have been directed to more seriously interrogate the therapeutic potential of protein secretion modulators through analogue synthesis and prodrug strategies, while developing an increased understanding of the underlying biology. The availability of structural information at the protein level and refinement of screening technologies have enabled increasing compound specificity and/or identification of new chemical scaffolds. There is renewed interest in targeting components of the UPR network since we now are able to selectively target branches of the UPR that ultimately affect the pharmacological and toxicological profile of compounds, with promising XBP1 mRNA splicing inhibitors such as B-I09 as candidates for further development against multiple myeloma. The greatest level of activity in recent years has been arguably at the stage of ER insertion, with the availability of the first ER insertion inhibitors targeting Sec61, many of which appear to have differential pharmacological profiles due to binding to different sites and potentially conformations of Sec61 in a cell type-dependent manner. While cancer has been the predominant disease indication for all subsets of secretory pathway inhibitors, particularly the selective modulation of Sec61 gating opens up opportunities for application to other diseases where secretory substrates play a role, including ocular angiogenic diseases, as has been already demonstrated for specific synthetic apratoxins, whereas cotransins showed initial promise as antivirals.We predict that secretory pathway modulating compounds hold great therapeutic potential and that natural products will continue to deliver structurally diverse starting points to directly or indirectly modulate secretion. Natural products possess unique chemical and consequently biological properties as a result of evolutionary refinement, including unusual mechanisms of action and ability to stabilize or inhibit protein–protein interactions. The examples showcased in this review alone highlight the structural diversity of natural products targeting the secretory pathway at different stages, but even seemingly structurally distinct compounds were shown to interact with the same target in a different manner, such as Sec61. The continuous search for natural products and their rigorous pharmacological characterization will undoubtedly allow us to extend the druggable genome and validate previously unknown or undruggable targets or target complexes.
7. Conflicts of interest
H. L. is co-founder of Oceanyx Pharmaceuticals, Inc., which has licensed patents and patent applications related to compounds discussed in this article (apratoxins).8. Acknowledgements
This work was supported by a National Institutes of Health grant R01CA172310 (HL), National Institute of General Medical Sciences grant R01GM132649 (VOP), Academy of Finland grants 289737, 314672 and 330255 (VOP) and a grant from the Sigrid Juselius Foundation (VOP). We thank Dr Dale Tranter for helpful discussions and comments on the manuscript and the figures.9. References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- E. K. Davison and M. A. Brimble, Curr. Opin. Chem. Biol., 2019, 52, 1–8 CrossRef CAS PubMed.
- X. Liang, D. Luo and H. Luesch, Pharmacol. Res., 2019, 147, 104373 CrossRef PubMed.
- E. E. Carlson, ACS Chem. Biol., 2010, 5, 639–653 CrossRef CAS PubMed.
- C. H. Arrowsmith, J. E. Audia, C. Austin, J. Baell, J. Bennett, J. Blagg, C. Bountra, P. E. Brennan, P. J. Brown, M. E. Bunnage, C. Buser-Doepner, R. M. Campbell, A. J. Carter, P. Cohen, R. A. Copeland, B. Cravatt, J. L. Dahlin, D. Dhanak, A. M. Edwards, M. Frederiksen, S. V. Frye, N. Gray, C. E. Grimshaw, D. Hepworth, T. Howe, K. V. M. Huber, J. Jin, S. Knapp, J. D. Kotz, R. G. Kruger, D. Lowe, M. M. Mader, B. Marsden, A. Mueller-Fahrnow, S. Müller, R. C. O'Hagan, J. P. Overington, D. R. Owen, S. H. Rosenberg, B. Roth, R. Ross, M. Schapira, S. L. Schreiber, B. Shoichet, M. Sundström, G. Superti-Furga, J. Taunton, L. Toledo-Sherman, C. Walpole, M. A. Walters, T. M. Willson, P. Workman, R. N. Young and W. J. Zuercher, Nat. Chem. Biol., 2015, 11, 536–541 CrossRef CAS PubMed.
- M. Jost and J. S. Weissman, ACS Chem. Biol., 2018, 13, 366–375 CrossRef CAS PubMed.
- M. Schürmann, P. Janning, S. Ziegler and H. Waldmann, Cell Chem. Biol., 2016, 23, 435–441 CrossRef PubMed.
- H. Park, J. Ha and S. B. Park, Curr. Opin. Chem. Biol., 2019, 50, 66–72 CrossRef CAS PubMed.
- T. M. Kapoor and R. M. Miller, Trends Pharmacol. Sci., 2017, 38, 1100–1109 CrossRef CAS PubMed.
- V. Van Puyenbroeck and K. Vermeire, Cell. Mol. Life Sci., 2018, 75, 1541–1558 CrossRef CAS PubMed.
- C. A. Foster, M. Dreyfuss, B. Mandak, J. G. Meingassner, H. U. Naegeli, A. Nussbaumer, L. Oberer, G. Scheel and E. M. Swoboda, J. Dermatol., 1994, 21, 847–854 CrossRef CAS PubMed.
- Y. Chen, M. Bilban, C. A. Foster and D. L. Boger, J. Am. Chem. Soc., 2002, 124, 5431–5440 CrossRef CAS PubMed.
- E. P. Schreiner, M. Kern, A. Steck and C. A. Foster, Bioorg. Med. Chem. Lett., 2004, 14, 5003–5006 CrossRef CAS PubMed.
- I. Coin, M. Beerbaum, P. Schmieder, M. Bienert and M. Beyermann, Org. Lett., 2008, 10, 3857–3860 CrossRef CAS PubMed.
- J. L. Garrison, E. J. Kunkel, R. S. Hegde and J. Taunton, Nature, 2005, 436, 285–289 CrossRef CAS PubMed.
- J. Besemer, H. Harant, S. Wang, B. Oberhauser, K. Marquardt, C. A. Foster, E. P. Schreiner, J. E. de Vries, C. Dascher-Nadel and I. J. D. Lindley, Nature, 2005, 436, 290–293 CrossRef CAS PubMed.
- A. L. MacKinnon, J. L. Garrison, R. S. Hegde and J. Taunton, J. Am. Chem. Soc., 2007, 129, 14560–14561 CrossRef CAS PubMed.
- A. L. Mackinnon, V. O. Paavilainen, A. Sharma, R. S. Hegde and J. Taunton, Elife, 2014, 3, e01483 CrossRef PubMed.
- S. V. Maifeld, A. L. MacKinnon, J. L. Garrison, A. Sharma, E. J. Kunkel, R. S. Hegde and J. Taunton, Chem. Biol., 2011, 18, 1082–1088 CrossRef CAS PubMed.
- W. Klein, C. Westendorf, A. Schmidt, M. Conill-Cortés, C. Rutz, M. Blohs, M. Beyermann, J. Protze, G. Krause, E. Krause and R. Schülein, PLoS One, 2015, 10, e0120886 CrossRef PubMed.
- A. Ruiz-Saenz, M. Sandhu, Y. Carrasco, R. L. Maglathlin, J. Taunton and M. M. Moasser, Oncogene, 2015, 34, 5288–5294 CrossRef CAS PubMed.
- N. S. Heaton, N. Moshkina, R. Fenouil, T. J. Gardner, S. Aguirre, P. S. Shah, N. Zhao, L. Manganaro, J. F. Hultquist, J. Noel, D. Sachs, J. Hamilton, P. E. Leon, A. Chawdury, S. Tripathi, C. Melegari, L. Campisi, R. Hai, G. Metreveli, A. V. Gamarnik, A. García-Sastre, B. Greenbaum, V. Simon, A. Fernandez-Sesma, N. J. Krogan, L. C. F. Mulder, H. van Bakel, D. Tortorella, J. Taunton, P. Palese and I. Marazzi, Immunity, 2016, 44, 46–58 CrossRef CAS PubMed.
- H. Luesch, W. Y. Yoshida, R. E. Moore, V. J. Paul and T. H. Corbett, J. Am. Chem. Soc., 2001, 123, 5418–5423 CrossRef CAS PubMed.
- H. Luesch, S. K. Chanda, R. M. Raya, P. D. DeJesus, A. P. Orth, J. R. Walker, J. C. Izpisúa Belmonte and P. G. Schultz, Nat. Chem. Biol., 2006, 2, 158–167 CrossRef CAS PubMed.
- Y. Liu, B. K. Law and H. Luesch, Mol. Pharmacol., 2009, 76, 91–104 CrossRef CAS PubMed.
- H. Luesch, W. Y. Yoshida, R. E. Moore and V. J. Paul, Bioorg. Med. Chem., 2002, 10, 1973–1978 CrossRef CAS PubMed.
- A. O. Paatero, J. Kellosalo, B. M. Dunyak, J. Almaliti, J. E. Gestwicki, W. H. Gerwick, J. Taunton and V. O. Paavilainen, Cell Chem. Biol., 2016, 23, 561–566 CrossRef CAS PubMed.
- K.-C. Huang, Z. Chen, Y. Jiang, S. Akare, D. Kolber-Simonds, K. Condon, S. Agoulnik, K. Tendyke, Y. Shen, K.-M. Wu, S. Mathieu, H.-W. Choi, X. Zhu, H. Shimizu, Y. Kotake, W. H. Gerwick, T. Uenaka, M. Woodall-Jappe and K. Nomoto, Mol. Cancer Ther., 2016, 15, 1208–1216 CrossRef CAS PubMed.
- P. J. Chitwood, S. Juszkiewicz, A. Guna, S. Shao and R. S. Hegde, Cell, 2018, 175, 1507–1519.e16 CrossRef CAS PubMed.
- Q.-Y. Chen, Y. Liu and H. Luesch, ACS Med. Chem. Lett., 2011, 2, 861–865 CrossRef CAS PubMed.
- W. Cai, R. Ratnayake, M. H. Gerber, Q.-Y. Chen, Y. Yu, H. Derendorf, J. G. Trevino and H. Luesch, Invest. New Drugs, 2019, 37, 364–374 CrossRef CAS PubMed.
- Q.-Y. Chen, Y. Liu, W. Cai and H. Luesch, J. Med. Chem., 2014, 57, 3011–3029 CrossRef CAS PubMed.
- P. Wu, W. Cai, Q.-Y. Chen, S. Xu, R. Yin, Y. Li, W. Zhang and H. Luesch, Org. Lett., 2016, 18, 5400–5403 CrossRef CAS PubMed.
- W. Cai, Q.-Y. Chen, L. H. Dang and H. Luesch, ACS Med. Chem. Lett., 2017, 8, 1007–1012 CrossRef CAS PubMed.
- B. Qiu, A. Tan, A. B. Veluchamy, Y. Li, H. Murray, W. Cheng, C. Liu, J. M. Busoy, Q.-Y. Chen, S. Sistla, W. Hunziker, C. M. G. Cheung, T. Y. Wong, W. Hong, H. Luesch and X. Wang, Invest. Ophthalmol. Visual Sci., 2019, 60, 3254–3263 CrossRef PubMed.
- C. Demangel, T. P. Stinear and S. T. Cole, Nat. Rev. Microbiol., 2009, 7, 50–60 CrossRef CAS PubMed.
- C. Demangel and S. High, Biol. Cell., 2018, 110, 237–248 CrossRef CAS PubMed.
- T. P. Stinear, A. Mve-Obiang, P. L. C. Small, W. Frigui, M. J. Pryor, R. Brosch, G. A. Jenkin, P. D. R. Johnson, J. K. Davies, R. E. Lee, S. Adusumilli, T. Garnier, S. F. Haydock, P. F. Leadlay and S. T. Cole, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1345–1349 CrossRef CAS PubMed.
- L. Guenin-Macé, F. Carrette, F. Asperti-Boursin, A. Le Bon, L. Caleechurn, V. Di Bartolo, A. Fontanet, G. Bismuth and C. Demangel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12833–12838 CrossRef PubMed.
- B. S. Hall, K. Hill, M. McKenna, J. Ogbechi, S. High, A. E. Willis and R. E. Simmonds, PLoS Pathog., 2014, 10, e1004061 CrossRef PubMed.
- R. E. Simmonds, F. V. Lali, T. Smallie, P. L. C. Small and B. M. Foxwell, J. Immunol., 2009, 182, 2194–2202 CrossRef CAS PubMed.
- M. McKenna, R. E. Simmonds and S. High, J. Cell Sci., 2016, 129, 1404–1415 CrossRef CAS PubMed.
- L. Baron, A. O. Paatero, J.-D. Morel, F. Impens, L. Guenin-Macé, S. Saint-Auret, N. Blanchard, R. Dillmann, F. Niang, S. Pellegrini, J. Taunton, V. O. Paavilainen and C. Demangel, J. Exp. Med., 2016, 213, 2885–2896 CrossRef CAS PubMed.
- A. Guna, N. Volkmar, J. C. Christianson and R. S. Hegde, Science, 2018, 359, 470–473 CrossRef CAS PubMed.
- L. Guenin-Macé, R. Veyron-Churlet, M.-I. Thoulouze, G. Romet-Lemonne, H. Hong, P. F. Leadlay, A. Danckaert, M.-T. Ruf, S. Mostowy, C. Zurzolo, P. Bousso, F. Chrétien, M.-F. Carlier and C. Demangel, J. Clin. Invest., 2013, 123, 1501–1512 CrossRef PubMed.
- E. Marion, O.-R. Song, T. Christophe, J. Babonneau, D. Fenistein, J. Eyer, F. Letournel, D. Henrion, N. Clere, V. Paille, N. C. Guérineau, J.-P. Saint André, P. Gersbach, K.-H. Altmann, T. P. Stinear, Y. Comoglio, G. Sandoz, L. Preisser, Y. Delneste, E. Yeramian, L. Marsollier and P. Brodin, Cell, 2014, 157, 1565–1576 CrossRef CAS PubMed.
- J.-D. Morel, A. O. Paatero, J. Wei, J. W. Yewdell, L. Guenin-Macé, D. Van Haver, F. Impens, N. Pietrosemoli, V. O. Paavilainen and C. Demangel, Mol. Cell. Proteomics, 2018, 17, 1750–1765 CrossRef CAS PubMed.
- J. Ogbechi, B. S. Hall, T. Sbarrato, J. Taunton, A. E. Willis, R. C. Wek and R. E. Simmonds, Cell Death Dis., 2018, 9(3), 397 CrossRef PubMed.
- L. Guenin-Macé, L. Baron, A.-C. Chany, C. Tresse, S. Saint-Auret, F. Jönsson, F. Le Chevalier, P. Bruhns, G. Bismuth, S. Hidalgo-Lucas, J.-F. Bisson, N. Blanchard and C. Demangel, Sci. Transl. Med., 2015, 7, 289ra85 CrossRef PubMed.
- E. Fiebiger, C. Hirsch, J. M. Vyas, E. Gordon, H. L. Ploegh and D. Tortorella, Mol. Biol. Cell, 2004, 15, 1635–1646 CrossRef CAS PubMed.
- B. C. S. Cross, C. McKibbin, A. C. Callan, P. Roboti, M. Piacenti, C. Rabu, C. M. Wilson, R. Whitehead, S. L. Flitsch, M. R. Pool, S. High and E. Swanton, J. Cell Sci., 2009, 122, 4393–4400 CrossRef CAS PubMed.
- I. Gamayun, S. O'Keefe, T. Pick, M.-C. Klein, D. Nguyen, C. McKibbin, M. Piacenti, H. M. Williams, S. L. Flitsch, R. C. Whitehead, E. Swanton, V. Helms, S. High, R. Zimmermann and A. Cavalié, Cell Chem. Biol., 2019, 26, 571–583.e6 CrossRef CAS PubMed.
- Q. Wang, B. A. Shinkre, J.-G. Lee, M. A. Weniger, Y. Liu, W. Chen, A. Wiestner, W. C. Trenkle and Y. Ye, PLoS One, 2010, 5, e15479 CrossRef PubMed.
- T. Junne, J. Wong, C. Studer, T. Aust, B. W. Bauer, M. Beibel, B. Bhullar, R. Bruccoleri, J. Eichenberger, D. Estoppey, N. Hartmann, B. Knapp, P. Krastel, N. Melin, E. J. Oakeley, L. Oberer, R. Riedl, G. Roma, S. Schuierer, F. Petersen, J. A. Tallarico, T. A. Rapoport, M. Spiess and D. Hoepfner, J. Cell Sci., 2015, 128, 1217–1229 CrossRef CAS PubMed.
- S. Cao, R. C. Guza, J. H. Wisse, J. S. Miller, R. Evans and D. G. I. Kingston, J. Nat. Prod., 2005, 68, 487–492 CrossRef CAS PubMed.
- S. Cao, A. Norris, J. H. Wisse, J. S. Miller, R. Evans and D. G. I. Kingston, Nat. Prod. Res., 2007, 21, 872–876 CrossRef CAS PubMed.
- T. Nagano, J. Pospíšil and G. Chollet, Chem.–Eur. J., 2009, 15, 9697–9706 CrossRef CAS PubMed.
- A. Fürstner and T. Nagano, J. Am. Chem. Soc., 2007, 129, 1906–1907 CrossRef PubMed.
- G. Zong, Z. Hu, S. O'Keefe, D. Tranter, M. J. Iannotti, L. Baron, B. Hall, K. Corfield, A. O. Paatero, M. J. Henderson, P. Roboti, J. Zhou, X. Sun, M. Govindarajan, J. M. Rohde, N. Blanchard, R. Simmonds, J. Inglese, Y. Du, C. Demangel, S. High, V. O. Paavilainen and W. Q. Shi, J. Am. Chem. Soc., 2019, 141, 8450–8461 CrossRef CAS PubMed.
- R. A. Medina, D. E. Goeger, P. Hills, S. L. Mooberry, N. Huang, L. I. Romero, E. Ortega-Barría, W. H. Gerwick and K. L. McPhail, J. Am. Chem. Soc., 2008, 130, 6324–6325 CrossRef CAS PubMed.
- J. D. Serrill, X. Wan, A. M. Hau, H. S. Jang, D. J. Coleman, A. K. Indra, A. W. G. Alani, K. L. McPhail and J. E. Ishmael, Invest. New Drugs, 2016, 34, 24–40 CrossRef CAS PubMed.
- X. Wan, J. D. Serrill, I. R. Humphreys, M. Tan, K. L. McPhail, I. G. Ganley and J. E. Ishmael, Mar. Drugs, 2018, 16(3), 77 CrossRef PubMed.
- G. Yao, Z. Pan, C. Wu, W. Wang, L. Fang and W. Su, J. Am. Chem. Soc., 2015, 137, 13488–13491 CrossRef CAS PubMed.
- R. Nabika, T. L. Suyama, A. M. Hau, R. Misu, H. Ohno, J. E. Ishmael, K. L. McPhail, S. Oishi and N. Fujii, Bioorg. Med. Chem. Lett., 2015, 25, 302–306 CrossRef CAS PubMed.
- G. A. Sable, J. Park, H. Kim, S.-J. Lim, S. Jang and D. Lim, Eur. J. Org. Chem., 2015, 2015, 7043–7052 CrossRef CAS.
- D. Tranter, A. O. Paatero, S. Kawaguchi, S. Kazemi, J. D. Serrill, J. Kellosalo, W. K. Vogel, U. Richter, C. C. Thornburg, S. Oishi, K. L. Mcphail, J. E. Ishmael and V. O. Paavilainen, ChemRxiv, 2019, DOI:10.26434/chemrxiv.10092182.
- G. Yao, W. Wang, L. Ao, Z. Cheng, C. Wu, Z. Pan, K. Liu, H. Li, W. Su and L. Fang, J. Med. Chem., 2018, 61, 8908–8916 CrossRef CAS PubMed.
- A. Takatsuki, K. Arima and G. Tamura, J. Antibiot., 1971, 24, 215–223 CrossRef CAS.
- A. Heifetz, R. W. Keenan and A. D. Elbein, Biochemistry, 1979, 18, 2186–2192 CrossRef CAS PubMed.
- K. Olden, R. M. Pratt and K. M. Yamada, Cell, 1978, 13, 461–473 CrossRef CAS PubMed.
- J. M. Pemberton, K. M. Vincent and R. J. Penfold, Curr. Microbiol., 1991, 22, 355–358 CrossRef CAS.
- P. E. Brandish, K. I. Kimura, M. Inukai, R. Southgate, J. T. Lonsdale and T. D. Bugg, Antimicrob. Agents Chemother., 1996, 40, 1640–1644 CrossRef CAS PubMed.
- J. K. Hakulinen, J. Hering, G. Brändén, H. Chen, A. Snijder, M. Ek and P. Johansson, Nat. Chem. Biol., 2017, 13, 265 CrossRef CAS PubMed.
- J. Yoo, E. H. Mashalidis, A. C. Y. Kuk, K. Yamamoto, B. Kaeser, S. Ichikawa and S.-Y. Lee, Nat. Struct. Mol. Biol., 2018, 25, 217–224 CrossRef CAS PubMed.
- Y. Y. Dong, H. Wang, A. C. W. Pike, S. A. Cochrane, S. Hamedzadeh, F. J. Wyszyński, S. R. Bushell, S. F. Royer, D. A. Widdick, A. Sajid, H. I. Boshoff, Y. Park, R. Lucas, W.-M. Liu, S. S. Lee, T. Machida, L. Minall, S. Mehmood, K. Belaya, W.-W. Liu, A. Chu, L. Shrestha, S. M. M. Mukhopadhyay, C. Strain-Damerell, R. Chalk, N. A. Burgess-Brown, M. J. Bibb, C. E. Barry III, C. V. Robinson, D. Beeson, B. G. Davis and E. P. Carpenter, Cell, 2018, 175, 1045–1058.e16 CrossRef CAS PubMed.
- K. Yamamoto, F. Yakushiji, T. Matsumaru and S. Ichikawa, Org. Lett., 2018, 20, 256–259 CrossRef CAS PubMed.
- K. Yamamoto, A. Katsuyama and S. Ichikawa, Bioorg. Med. Chem., 2019, 27, 1714–1719 CrossRef CAS PubMed.
- J. Wu, S. Chen, H. Liu, Z. Zhang, Z. Ni, J. Chen, Z. Yang, Y. Nie and D. Fan, J. Exp. Clin. Cancer Res., 2018, 37, 272 CrossRef CAS PubMed.
- A. Abdullahi, M. Stanojcic, A. Parousis, D. Patsouris and M. G. Jeschke, Shock, 2017, 47, 506–513 CrossRef CAS PubMed.
- F. J. Ortiz-Lopez, M. C. Monteiro, V. Gonzalez-Menendez, J. R. Tormo, O. Genilloud, G. F. Bills, F. Vicente, C. Zhang, T. Roemer and S. B. Singh, et al. , J. Nat. Prod., 2015, 78, 468–475 CrossRef CAS PubMed.
- C. R. Zwick and H. Renata, Tetrahedron, 2018, 74, 6469–6473 CrossRef CAS.
- D. Estoppey, C. M. Lee, M. Janoschke, B. H. Lee, K. F. Wan, H. Dong, P. Mathys, I. Filipuzzi, T. Schuhmann, R. Riedl, T. Aust, O. Galuba, G. McAllister, C. Russ, M. Spiess, T. Bouwmeester, G. M. C. Bonamy and D. Hoepfner, Cell Rep., 2017, 19, 451–460 CrossRef CAS PubMed.
- V. L. Singleton, N. Bohonos and A. J. Ullstrup, Nature, 1958, 181, 1072–1073 CrossRef CAS PubMed.
- K. Trisuwan, V. Rukachaisirikul, Y. Sukpondma, S. Phongpaichit, S. Preedanon and J. Sakayaroj, Chem. Pharm. Bull., 2009, 57, 1100–1102 CrossRef CAS PubMed.
- Z.-F. Hu, L.-L. Qin, W.-J. Ding, Y. Liu and Z.-J. Ma, Nat. Prod. Res., 2016, 30, 2311–2315 CrossRef CAS PubMed.
- F. Zeng, C. Chen, A. A. Al Chnani, Q. Zhou, Q. Tong, W. Wang, Y. Zang, J. Gong, Z. Wu, J. Liu, J. Wang, H. Zhu and Y. Zhang, Bioorg. Chem., 2019, 86, 176–182 CrossRef CAS PubMed.
- S.-M. Paek, Mar. Drugs, 2018, 16(4), 133 CrossRef PubMed.
- H. R. Pelham, Cell, 1991, 67, 449–451 CrossRef CAS PubMed.
- N. Anders and G. Jürgens, Cell. Mol. Life Sci., 2008, 65, 3433–3445 CrossRef CAS PubMed.
- M. Zeghouf, B. Guibert, J.-C. Zeeh and J. Cherfils, Biochem. Soc. Trans., 2005, 33, 1265–1268 CrossRef CAS PubMed.
- H.-W. Shin and K. Nakayama, J. Biochem., 2004, 136, 761–767 CrossRef CAS PubMed.
- J. E. Casanova, Traffic, 2007, 8, 1476–1485 CrossRef CAS PubMed.
- P. Chardin and F. McCormick, Cell, 1999, 97, 153–155 CrossRef CAS PubMed.
- L. Renault, B. Guibert and J. Cherfils, Nature, 2003, 426, 525–530 CrossRef CAS PubMed.
- E. Mossessova, R. A. Corpina and J. Goldberg, Mol. Cell, 2003, 12, 1403–1411 CrossRef CAS PubMed.
- D. Vigil, J. Cherfils, K. L. Rossman and C. J. Der, Nat. Rev. Cancer, 2010, 10, 842–857 CrossRef CAS PubMed.
- J. Viaud, M. Zeghouf, H. Barelli, J.-C. Zeeh, A. Padilla, B. Guibert, P. Chardin, C. A. Royer, J. Cherfils and A. Chavanieu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10370–10375 CrossRef CAS PubMed.
- M. Hafner, A. Schmitz, I. Grüne, S. G. Srivatsan, B. Paul, W. Kolanus, T. Quast, E. Kremmer, I. Bauer and M. Famulok, Nature, 2006, 444, 941–944 CrossRef CAS PubMed.
- Y. Feng, S. Yu, T. K. R. Lasell, A. P. Jadhav, E. Macia, P. Chardin, P. Melancon, M. Roth, T. Mitchison and T. Kirchhausen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6469–6474 CrossRef CAS PubMed.
- Y. Feng, A. P. Jadhav, C. Rodighiero, Y. Fujinaga, T. Kirchhausen and W. I. Lencer, EMBO Rep., 2004, 5, 596–601 CrossRef CAS PubMed.
- E. Macia, M. Ehrlich, R. Massol, E. Boucrot, C. Brunner and T. Kirchhausen, Dev. Cell, 2006, 10, 839–850 CrossRef CAS PubMed.
- A. J. Newton, T. Kirchhausen and V. N. Murthy, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17955–17960 CrossRef CAS PubMed.
- H. E. Pelish, J. R. Peterson, S. B. Salvarezza, E. Rodriguez-Boulan, J.-L. Chen, M. Stamnes, E. Macia, Y. Feng, M. D. Shair and T. Kirchhausen, Nat. Chem. Biol., 2006, 2, 39–46 CrossRef CAS PubMed.
- U. Rasmussen, S. Brøogger Christensen and F. Sandberg, Acta Pharm. Suec., 1978, 15, 133–140 CAS.
- Y. Furuya, P. Lundmo, A. D. Short, D. L. Gill and J. T. Isaacs, Cancer Res., 1994, 54, 6167–6175 CAS.
- Y. Furuya and J. T. Isaacs, Prostate, 1994, 25, 301–309 CrossRef CAS PubMed.
- M. Lam, G. Dubyak and C. W. Distelhorst, Mol. Endocrinol., 1993, 7, 686–693 CAS.
- Y. Kaneko and A. Tsukamoto, Cancer Lett., 1994, 79, 147–155 CrossRef CAS PubMed.
- O. Thastrup, P. J. Cullen, B. K. Drøbak, M. R. Hanley and A. P. Dawson, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2466–2470 CrossRef CAS PubMed.
- Y. Sagara and G. Inesi, J. Biol. Chem., 1991, 266, 13503–13506 CAS.
- C. Toyoshima and H. Nomura, Nature, 2002, 418, 605–611 CrossRef CAS PubMed.
- S. Dey and S. O. Bajaj, Synth. Commun., 2018, 48, 1–13 CrossRef CAS.
- S. R. Denmeade, C. M. Jakobsen, S. Janssen, S. R. Khan, E. S. Garrett, H. Lilja, S. B. Christensen and J. T. Isaacs, J. Natl. Cancer Inst., 2003, 95, 990–1000 CrossRef CAS PubMed.
- D. Mahalingam, J. Peguero, P. Cen, S. P. Arora, J. Sarantopoulos, J. Rowe, V. Allgood, B. Tubb and L. Campos, Cancers, 2019, 11(6), 833 CrossRef PubMed.
- G. Appendino, S. Prosperini, C. Valdivia, M. Ballero, G. Colombano, R. A. Billington, A. A. Genazzani and O. Sterner, J. Nat. Prod., 2005, 68, 1213–1217 CrossRef CAS PubMed.
- C. Navarrete, R. Sancho, F. J. Caballero, F. Pollastro, B. L. Fiebich, O. Sterner, G. Appendino and E. Muñoz, J. Pharmacol. Exp. Ther., 2006, 319, 422–430 CrossRef CAS PubMed.
- W. Ma, X. Liang, W. Ye, Y. Wang, L. Min, S. He and C.-S. Lee, Eur. J. Org. Chem., 2018, 2018, 196–208 CrossRef CAS.
- Y. A. Ovchinnikov, Eur. J. Biochem., 1979, 94, 321–336 CrossRef CAS PubMed.
- B. C. Pressman, Annu. Rev. Biochem., 1976, 45, 501–530 CrossRef CAS PubMed.
- S. K. Berezin, J. Membr. Biol., 2015, 248, 713–726 CrossRef CAS PubMed.
- L. Rose and A. T. A. Jenkins, Bioelectrochemistry, 2007, 70, 387–393 CrossRef CAS PubMed.
- I.-J. Ryoo, H.-R. Park, S.-J. Choo, J.-H. Hwang, Y.-M. Park, K.-H. Bae, K. Shin-Ya and I.-D. Yoo, Biol. Pharm. Bull., 2006, 29, 817–820 CrossRef CAS PubMed.
- Y.-Q. Cheng, Chembiochem, 2006, 7, 471–477 CrossRef CAS PubMed.
- N. A. Magarvey, M. Ehling-Schulz and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 10698–10699 CrossRef CAS PubMed.
- J. Kim, I. Choi, J.-Y. Park and S.-W. Kang, Exp. Cell Res., 2013, 319, 2049–2057 CrossRef CAS PubMed.
- M. Wang, M. E. Law, R. K. Castellano and B. K. Law, Crit. Rev. Oncol. Hematol., 2018, 127, 66–79 CrossRef PubMed.
- M. Ri, E. Tashiro, D. Oikawa, S. Shinjo, M. Tokuda, Y. Yokouchi, T. Narita, A. Masaki, A. Ito, J. Ding, S. Kusumoto, T. Ishida, H. Komatsu, Y. Shiotsu, R. Ueda, T. Iwawaki, M. Imoto and S. Iida, Blood Cancer J., 2012, 2, e79 CrossRef CAS PubMed.
- E. Tashiro, N. Hironiwa, M. Kitagawa, Y. Futamura, S.-I. Suzuki, M. Nishio and M. Imoto, J. Antibiot., 2007, 60, 547–553 CrossRef CAS PubMed.
- T. Kawamura, E. Tashiro, K. Shindo and M. Imoto, J. Antibiot., 2008, 61, 312–317 CrossRef CAS PubMed.
- C.-H. A. Tang, S. Ranatunga, C. L. Kriss, C. L. Cubitt, J. Tao, J. A. Pinilla-Ibarz, J. R. Del Valle and C.-C. A. Hu, J. Clin. Invest., 2014, 124, 2585–2598 CrossRef CAS PubMed.
- H. Xie, C.-H. A. Tang, J. H. Song, A. Mancuso, J. R. Del Valle, J. Cao, Y. Xiang, C. V. Dang, R. Lan, D. J. Sanchez, B. Keith, C.-C. A. Hu and M. C. Simon, J. Clin. Invest., 2018, 128, 1300–1316 CrossRef PubMed.
- A. Shao, C. W. Kang, C.-H. A. Tang, C. F. Cain, Q. Xu, C. M. Phoumyvong, J. R. Del Valle and C.-C. A. Hu, J. Med. Chem., 2019, 62, 5404–5413 CrossRef CAS PubMed.
- M. Menna, Curr. Top. Med. Chem., 2014, 14, 207–223 CrossRef CAS PubMed.
- M. A. Iglesias-Arteaga and J. W. Morzycki, Alkaloids Chem. Biol., 2013, vol. 72, pp. 153–279 Search PubMed.
- B. R. Moser, J. Nat. Prod., 2008, 71, 487–491 CrossRef CAS PubMed.
- N. López-Antón, A. Rudy, N. Barth, M. L. Schmitz, G. R. Pettit, K. Schulze-Osthoff, V. M. Dirsch and A. M. Vollmar, J. Biol. Chem., 2006, 281, 33078–33086 CrossRef PubMed.
- A. W. G. Burgett, T. B. Poulsen, K. Wangkanont, D. R. Anderson, C. Kikuchi, K. Shimada, S. Okubo, K. C. Fortner, Y. Mimaki, M. Kuroda, J. P. Murphy, D. J. Schwalb, E. C. Petrella, I. Cornella-Taracido, M. Schirle, J. A. Tallarico and M. D. Shair, Nat. Chem. Biol., 2011, 7, 639–647 CrossRef CAS PubMed.
- A. J. Ambrose, E. A. Santos, P. C. Jimenez, D. D. Rocha, D. V. Wilke, P. Beuzer, J. Axelrod, A. Kumar Kanduluru, P. L. Fuchs, H. Cang, L. V. Costa-Lotufo, E. Chapman and J. J. La Clair, Chembiochem, 2017, 18, 506–510 CrossRef CAS PubMed.
- M. J. Choi, E. J. Park, K. J. Min, J.-W. Park and T. K. Kwon, Toxicol. In Vitro, 2011, 25, 692–698 CrossRef CAS PubMed.
- K. Ghosh, S. De, S. Mukherjee, S. Das, A. N. Ghosh and S. B. Sengupta, Toxicol. In Vitro, 2017, 44, 330–338 CrossRef CAS PubMed.
- L. Wang, B. G. K. Perera, S. B. Hari, B. Bhhatarai, B. J. Backes, M. A. Seeliger, S. C. Schürer, S. A. Oakes, F. R. Papa and D. J. Maly, Nat. Chem. Biol., 2012, 8, 982–989 CrossRef CAS PubMed.
- J. M. Axten, S. P. Romeril, A. Shu, J. Ralph, J. R. Medina, Y. Feng, W. H. H. Li, S. W. Grant, D. A. Heerding, E. Minthorn, T. Mencken, N. Gaul, A. Goetz, T. Stanley, A. M. Hassell, R. T. Gampe, C. Atkins and R. Kumar, ACS Med. Chem. Lett., 2013, 4, 964–968 CrossRef CAS PubMed.
- C. Atkins, Q. Liu, E. Minthorn, S.-Y. Zhang, D. J. Figueroa, K. Moss, T. B. Stanley, B. Sanders, A. Goetz, N. Gaul, A. E. Choudhry, H. Alsaid, B. M. Jucker, J. M. Axten and R. Kumar, Cancer Res., 2013, 73, 1993–2002 CrossRef CAS PubMed.
- D. Rojas-Rivera, T. Delvaeye, R. Roelandt, W. Nerinckx, K. Augustyns, P. Vandenabeele and M. J. M. Bertrand, Cell Death Differ., 2017, 24, 1100–1110 CrossRef CAS PubMed.
- C. Hetz, J. M. Axten and J. B. Patterson, Nat. Chem. Biol., 2019, 15, 764–775 CrossRef CAS PubMed.
- J. van den Boom and H. Meyer, Mol. Cell, 2018, 69, 182–194 CrossRef CAS PubMed.
- K. B. Kim and C. M. Crews, Nat. Prod. Rep., 2013, 30, 600–604 RSC.
- A. D. Elbein, J. E. Tropea, M. Mitchell and G. P. Kaushal, J. Biol. Chem., 1990, 265, 15599–15605 CAS.
- F. Vallee, K. Karaveg, A. Herscovics, K. W. Moremen and P. L. Howell, J. Biol. Chem., 2000, 275, 41287–41298 CrossRef CAS PubMed.
- A. Males, L. Raich, S. J. Williams, C. Rovira and G. J. Davies, Chembiochem, 2017, 18, 1496–1501 CrossRef CAS PubMed.
- Q. Wang, L. Li and Y. Ye, J. Biol. Chem., 2008, 283, 7445–7454 CrossRef CAS PubMed.
- Q. Wang, H. Mora-Jensen, M. A. Weniger, P. Perez-Galan, C. Wolford, T. Hai, D. Ron, W. Chen, W. Trenkle, A. Wiestner and Y. Ye, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2200–2205 CrossRef CAS PubMed.
- M.-O. Aletrari, C. McKibbin, H. Williams, V. Pawar, P. Pietroni, J. M. Lord, S. L. Flitsch, R. Whitehead, E. Swanton, S. High and R. A. Spooner, PLoS One, 2011, 6, e22713 CrossRef CAS PubMed.
- M. Wang and R. J. Kaufman, Nature, 2016, 529, 326–335 CrossRef CAS PubMed.
- N. Dejeans, S. Manié, C. Hetz, F. Bard, T. Hupp, P. Agostinis, A. Samali and E. Chevet, Trends Mol. Med., 2014, 20, 242–250 CrossRef CAS PubMed.
- S. C. Bell, M. A. Mall, H. Gutierrez, M. Macek, S. Madge, J. C. Davies, P.-R. Burgel, E. Tullis, C. Castaños, C. Castellani, C. A. Byrnes, F. Cathcart, S. H. Chotirmall, R. Cosgriff, I. Eichler, I. Fajac, C. H. Goss, P. Drevinek, P. M. Farrell, A. M. Gravelle, T. Havermans, N. Mayer-Hamblett, N. Kashirskaya, E. Kerem, J. L. Mathew, E. F. McKone, L. Naehrlich, S. Z. Nasr, G. R. Oates, C. O'Neill, U. Pypops, K. S. Raraigh, S. M. Rowe, K. W. Southern, S. Sivam, A. L. Stephenson, M. Zampoli and F. Ratjen, Lancet Respir. Med., 2020, 8(1), 65–124 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |