
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Making natural products from renewable feedstocks: back to the roots?
Jonas
Kühlborn†
,
Jonathan
Gro߆
and
Till
Opatz
 *
*
Institute of Organic Chemistry, Johannes Gutenberg University, Duesbergweg 10–14, 55128 Mainz, Germany. E-mail: opatz@uni-mainz.de
First published on 18th October 2019
Abstract
Covering: up to mid-2019
This review highlights the utilization of biomass-derived building blocks in the total synthesis of natural products. An overview over several renewable feedstock classes, namely wood/lignin, cellulose, chitin and chitosan, fats and oils, as well as terpenes, is given, covering the time span from the initial beginning of natural product synthesis until today. The focus is put on the origin of the employed carbon atoms and on the nature of the complex structures that were assembled therefrom. The emerging trend of turning away from petrochemically derived starting materials back to bio-based resources, just as seen in the early days of total synthesis, shall be demonstrated.
1. Introduction
1.1. Total synthesis – a driving force for new developments
The synthesis of organic compounds of natural origin from simple starting materials – the so-called total synthesis – has attracted and fascinated chemists ever since the successful synthesis of “organic” urea from inorganic silver cyanate and ammonium chloride by Wöhler in 1828.1 It should not be forgotten that the earlier isolation of pure morphine from opium by Sertürner in 1806![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 and his preparation of morphine salts in 18173 were further defining moments in organic chemistry. Total synthesis has since emerged to be one of the most challenging and prestigious disciplines among the chemical sciences, as underlined by the Nobel prizes awarded to R. B. Woodward and E. J. Corey.
2 and his preparation of morphine salts in 18173 were further defining moments in organic chemistry. Total synthesis has since emerged to be one of the most challenging and prestigious disciplines among the chemical sciences, as underlined by the Nobel prizes awarded to R. B. Woodward and E. J. Corey.
The fascination with this field arises from the diverse opportunities available in the application of natural products, combined with the possibility to provide these natural products independent of their natural source and to modify their structures at will.
Natural products have always served as an inspiration for the development of new pharmaceuticals, pesticides and herbicides or dyes – and total synthesis is often the only way to provide access to such products in sufficient quantities for extensive investigation, let alone commercial endeavours.4–6 Therefore, total synthesis, natural product isolation and structure elucidation have close ties. The search for new compounds with attractive biological activities or chemical structures has fueled the development of new analytical methods and synthetic chemists were never shy to tackle any attractive target molecule virtually regardless of its size. In contrast, the more challenging the target molecules were, the more effort was put into the development of new methods, technologies and theoretical concepts to make their preparation possible.7,8 This has made total synthesis an ideal proving ground for the utility of new synthetic developments. Furthermore, the ab initio synthesis serves to verify or correct a proposed structure and the structures of numerous compounds had to be revised when total synthesis proved the originally proposed structure to be incorrect, often in terms of relative or absolute stereochemistry.9–20
Working on total synthesis projects provides excellent training for synthetic chemists and is generally appreciated by future employers in the chemical and pharmaceutical sector because of the profound experience gained.21 The extensive and diverse challenges of organic synthesis provide a “feeling” for the reactivity of chemical compounds and profound knowledge on how to extract the literature to solve the numerous problems encountered on the way to the target. Although the number of total synthesis publications dwelling on the detours and problems encountered which contribute to the unmatched challenge of this discipline seems to be, unfortunately, declining, the successful pursuit of a total synthesis project is frequently associated with high skills in problem-solving and high levels of tolerance for frustration by the practitioner.22
1.2. Requirements of eco-friendly synthesis
The increasing awareness of sustainability in our modern society has also led to reverberations in chemical research and industry which culminated in the Rio Declaration on Environment and Development from The United Nations Conference on Environment and Development in 1992.23 Based on this declaration, Anastas and Warner have developed their twelve principles of green chemistry in 1998,24 just as Anastas and Zimmermann phrased the related twelve principles of green engineering five years later.25 Both catalogues are meant to be guidelines for the development of more eco-friendly syntheses, methodologies, technologies as well as processes and these are applicable to numerous facets of chemical research and production. However, the considerable potential of biocatalysis for “green” organic synthesis is not discussed therein. The present review will mainly focus on the use of renewable resources for the production of chemical building blocks and their value for organic synthesis. The reader shall also be referred to the rich body of existing literature providing more detailed information on the essence of green chemistry.26–52Furthermore, during the course of this paradigm shift in the world of chemistry, a number of terms and metrics to describe the extent of sustainability and “greenness” of certain reaction or process have been introduced.53 The first metric that has been introduced is the Atom Economy concept of Trost, soon followed by the Environmental (E) factor of Sheldon. The former is defined as the ratio of the molecular mass of the desired product and the sum total of the molecular masses of all substances produced according to the stoichiometric equation.54,55 The latter, introduced in 1992, is the mass ratio of total waste and product and therefore indicates the efficiency and the environmental impact of a given process.56–58 These two and further metrics have been widely applied and discussed in detail elsewhere.59–61 Researchers of GlaxoSmithKline have recently developed the carbon efficiency, which is the percentage of carbon atoms remaining in the product relative to all carbon atoms present in the entirety of reactants.62,63 Since organic synthesis is about constructing carbon skeletons, the origin and fate of carbon atoms significantly contributes to the sustainability of a certain organic product. Thus, the renewability of (starting) materials is a major aspect of green chemistry.
With few exceptions, chemical raw materials are currently produced from the fossil resources such as natural gas, coal and petroleum. Depletion of underground deposits, growing ecological risks associated with production as well as the carbon imbalance in the ecosphere resulting from the usage of these feedstocks makes them less favorable. Furthermore, the chemical diversity initially available from these sources is small and this has led to sometimes lengthy production processes but also to a rather limited primary product portfolio comprising alkanes, alkenes, alkynes and arenes. Any further functionalization such as the introduction of heteroatoms like oxygen, nitrogen, sulfur, phosphorus or halogens to generate advanced building blocks requires additional synthetic steps, often in the form of multistep cascades, which lead to additional waste production and energy consumption. In spite of these shortcomings, there was little incentive so far to change this traditional and irreversible chemical carbon flow.
In contrast, biomass-derived renewable starting materials often carry an appreciable degree of functionalization (heteroatoms, stereocenters) and can therefore represent useful advanced building blocks for synthesis. Because of their biogenetic relation or at least their structural proximity to nature-derived or -inspired target molecules, the step count required to convert them into suitable synthetic building blocks can be shorter than that available in the usage of their petrochemical counterparts. In addition to the renewability aspect and the option to close the carbon cycle, this is another advantage of bio-based starting materials. Here, we will mainly focus on the origin of the starting materials required to construct the carbon skeleton of natural products in order to illustrate their utility in total synthesis and organic synthesis of complex molecules in general.
Along with the utilization of biomass for fuel and energy production, the search for new sources of chemical feedstocks is a growing field in current chemical research. Numerous recent publications deal with the production of low molecular weight compounds from biomass and the valorization of these renewable feedstocks for synthetic purposes, although commodities such as adipic acid, acrylic acid or butanol currently still dominate the field.64–67
1.3. Atom origin: historical developments
The concept of producing starting materials from natural resources is by no means new. In fact, before petroleum oil and coal deposits were discovered and exploited, the only available sources for pure organic compounds such as camphor, ethanol, methanol, hexadecanol, acetic acid, benzoic acid or benzaldehyde were microorganisms, plants and animals. Therefore, the young field of organic synthesis was initially restricted to these natural feedstocks and chemists have sought out to expand this portfolio. Nevertheless, early-day chemists managed to synthesize natural products – some remarkable examples like von Baeyer's alizarin and quinalizarin68 as well as indigo synthesis,69–71 Ladenburg's coniine synthesis72,73 or the synthesis of tyrian purple by Sachs and Sichel74–76 are depicted in Schemes 1–4.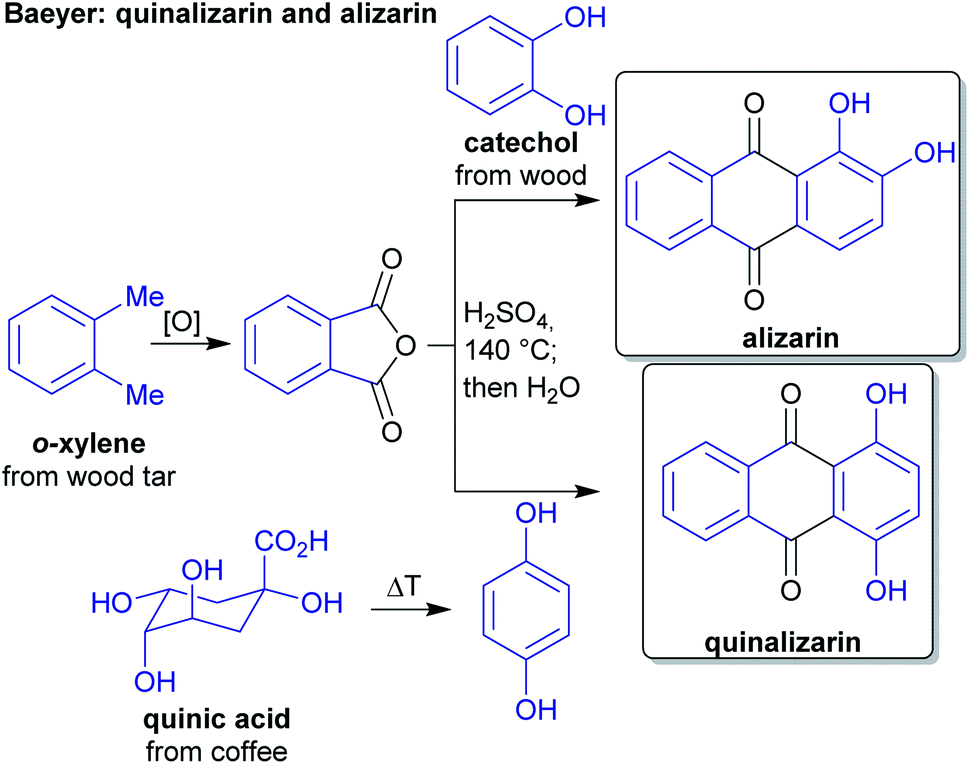 | ||
| Scheme 1 Baeyer's alizarin and quinalizarin synthesis from phthalic anhydride and catechol/hydroquinone. | ||
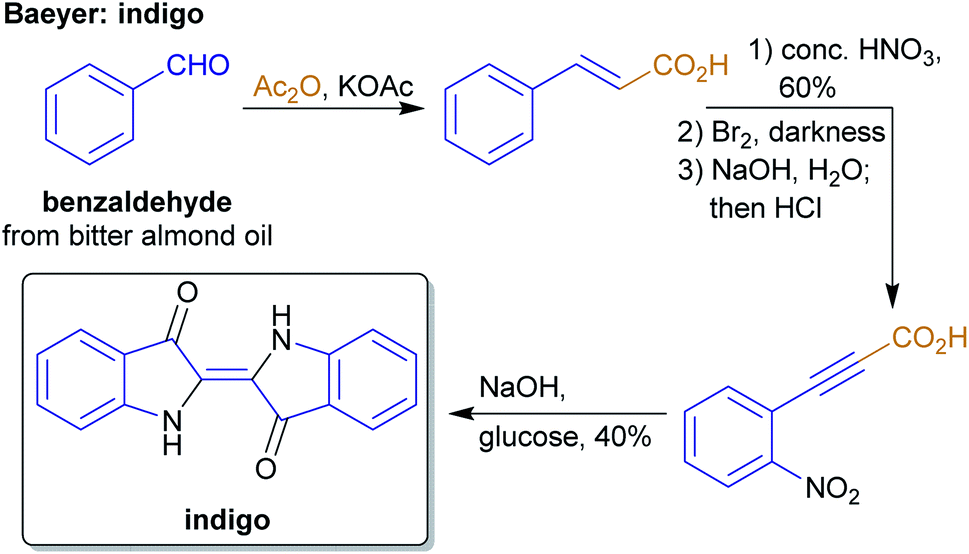 | ||
| Scheme 2 Baeyer's indigo synthesis from cinnamic acid. | ||
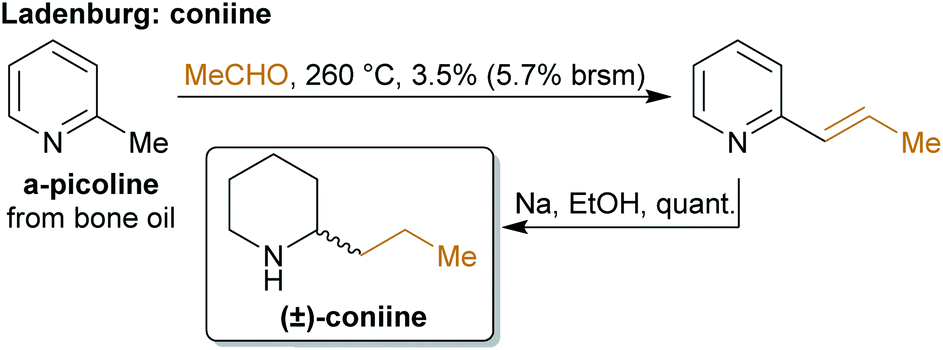 | ||
| Scheme 3 Ladenburg's coniine synthesis from α-picoline. | ||
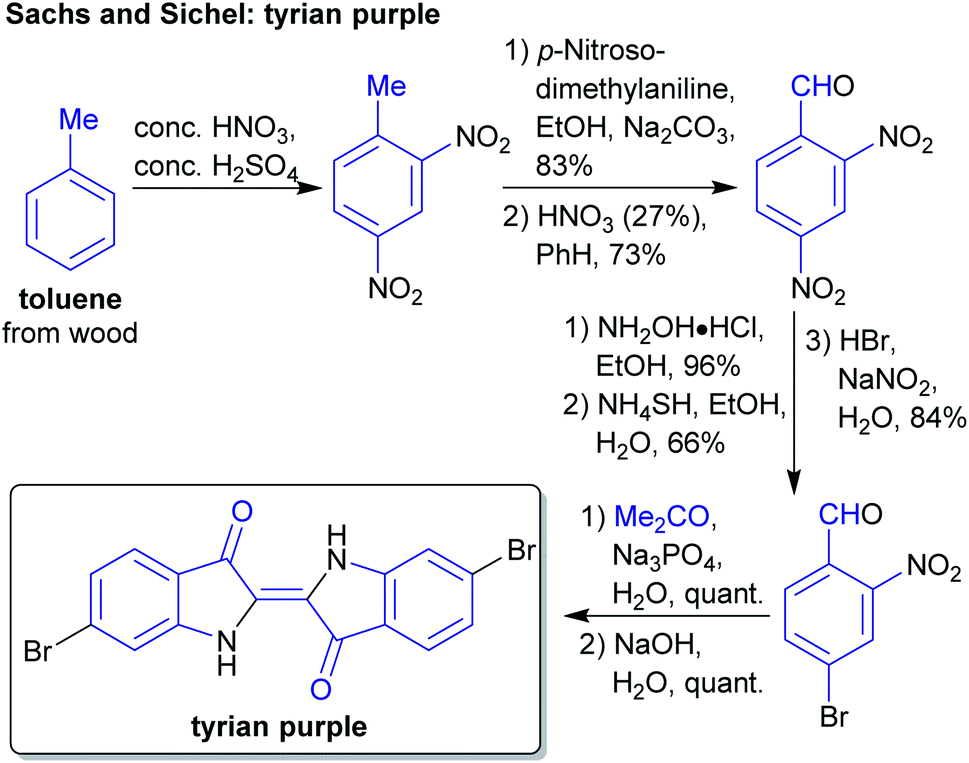 | ||
| Scheme 4 Sachs' and Sichel's synthesis of tyrian purple from xylochemical toluene. | ||
During this period, industrial organic chemistry was intensely focused on the production of dyes and pigments and the demand was growing for synthetic approaches instead of reliance on natural sources of these products. Therefore, productive sources of raw materials were needed and the industrial production of petroleum oil and coal provided the required carbon sources. These petrochemicals were available in quantities sufficient to satisfy the needs of the expanding chemical industry. Synthetic routes and products were planned according to the starting materials available from those sources. A prime example is the manufacturing of indigo which was at first obtained from plant sources at an annual rate of 19000 t in 1897 which dropped to 1000 t in 1914 as synthetic methods based on petrochemicals became available.77 Around the same time, the industrial production of synthetic pharmaceuticals gained importance, e.g. the industrial scale production of acetylsalicylic acid78 and of salvarsan,79 which were both already produced from petrochemicals. The chemical industry developed to an important basis for economic growth in Western countries.
Despite the shift towards petrochemicals as new starting materials, biomass-derived feedstocks never were completely eliminated. The chiral pool, which is the collection of chiral terpenes, amino acids and carbohydrates and other chiral compounds available from nature,80 was for many decades the only source of enantiopure catalysts and building blocks.81 The chiral pool was crucial for the synthesis of many natural products and other chiral nonracemic organic compounds.82–87
The anthropogenic rise in atmospheric CO2 levels casts shadows upon the continued extensive use of petrochemical resources.88 With the largest fraction of fossil fuels being used for heating and/or cooling and transportation, the opinion that the use of fossil resources for chemistry will always be possible in future economic settings is still widespread. This is at least in part based on the assumption that mankind will acknowledge the overriding importance of synthetic chemistry and that this area will remain unaffected by any future changes in our carbon economy, hence making any adaption of feedstocks towards a higher sustainability obsolete. On the other hand, it is entirely possible that this belief will suffer the same fate as the presumption that chlorofluorocarbons or tetrachloromethane will always be available at a reasonable price for laboratory-scale applications or that the number of chemicals available from commercial suppliers cannot ever decrease. It thus makes sense that researchers are now reviving the use of natural feedstocks in organic synthesis which had moved in the background more than a century ago. Although outstanding achievements have already been made in the valorization of biomass as a chemical feedstock, the full substitution of petrochemicals is still a distant dream.
2. Natural product syntheses using renewable feedstocks
The main intention of the present review is the appreciation of syntheses that were performed based on renewable carbon sources and that follow some of the concepts of sustainable chemistry. The potential of those approaches is highlighted to provide inspiration to fellow researchers. In contrast to the synthesis of polymers89–91 or other functional materials,92–96 the concept of total synthesis using renewable carbon sources is not widespread. Total synthesis is already operating under very stringent conditions and the way of accomplishing the ultimate goal is often subordinated, with the application of any suitable method and effort being justified, at least in an academic setting. To compare several functioning approaches to the same target molecule, aspects like step count and overall yield are commonly applied, yet softer or less well-defined aspects such as “elegance” also play a role.The following syntheses demonstrate that the challenge of avoiding fossil resources can be tackled at the same time. The key criterion for the inclusion of total syntheses in this review is the origin of carbon atoms and the renewability of the starting materials used. Other aspects of sustainable chemistry such as the use of benign solvents, of catalytic methods instead of stoichiometric transformations, the avoidance of protecting groups and critical reagents or the step efficiency will be addressed and discussed where relevant. Furthermore, we primarily selected examples where starting materials were used in a so-called “class-transcendent” fashion (i.e. the product belongs to a different class of compounds than the starting material) to demonstrate the full potential of the starting materials and the capability of combining different sources. We pay tribute to the fact that a synthesis of a saccharide from a terpene is more challenging and remarkable than a synthesis from another saccharide. In fact, (oligo)peptide, (oligo)saccharide and (oligo)nucleotide natural products will not be covered, as the transcendence criterion is not fulfilled in these cases and the natural origin of the starting materials is rather obvious. For a better visualization, we use a color coding for the carbon atoms of each moiety derived from the respective biomass resources presented herein.
Wood- (or lignin-) derived carbons are colored in blue, (hemi)cellulose-as well as carbohydrate-based moieties are shown in green, compounds obtainable from fats and oils are colored in orange, purple was chosen for chitin- and chitosan-derived groups, and terpene and terpenoids are shown in red.
Heteroatoms are also colored in this code if they are introduced from the respective bio-based material. Otherwise, inserted heteroatoms will be black. These five classes of renewable starting materials were selected as they represent the most intensely investigated and used sources for starting materials to date. Starting materials of natural origin not covered by the above list specified in this review (e.g. amino acids) are also colored black. Petrochemistry-derived carbon atoms that remain in the final product will not occur in this review, but for the sake of simplicity, carbons of this kind introduced transiently will also be depicted in black.
2.1. Wood/lignin
A major component of lignocellulose is lignin, the largest source of aromatics on earth, as wood-derived biomass consists of up to 35% of lignin.97 Lignin is an amorphous cross-linked biopolymer that, in combination with cellulose and hemicelluloses, confers structural stability to plants.98 The complexity of its structure and its chemical stability make this biopolymer difficult to break down into useful building blocks.99,100 Nevertheless, the benefits of its use would be its carbon-neutrality and the lack of competition with food production (not considering the competition for potentially arable land).101 Therefore, it is a promising alternative to petroleum resources.102,103 Lignin can be derived from wood pulp and is a waste product of paper production. It is biosynthetically derived from three phenylpropanoid monolignol monomers, differing only in their oxygenation pattern (Scheme 5).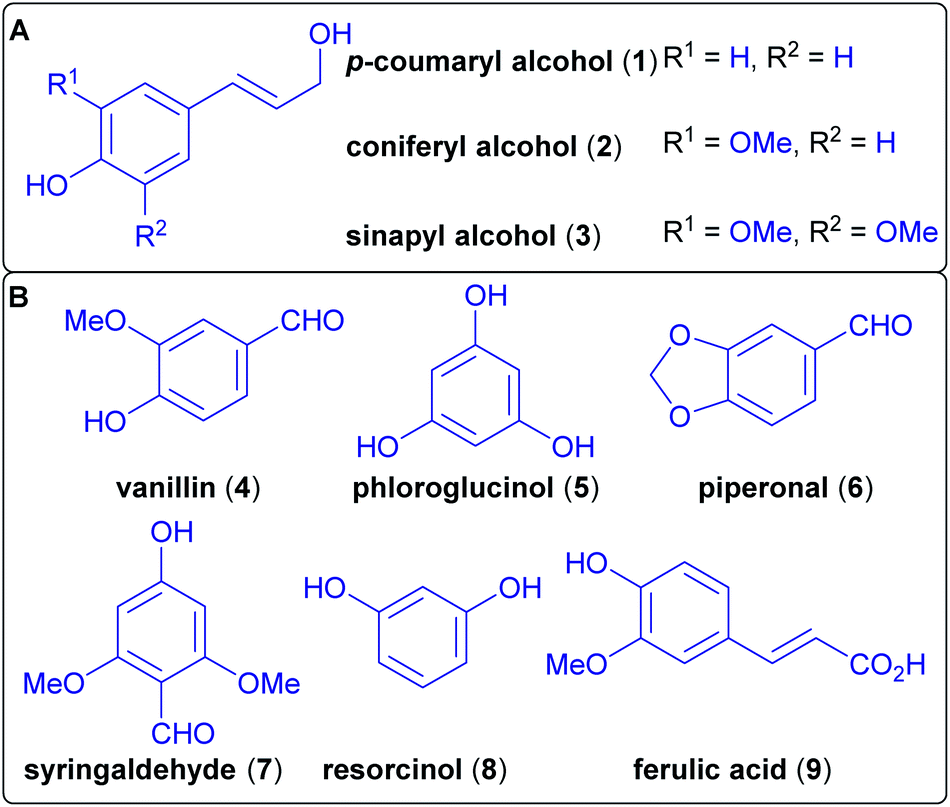 | ||
| Scheme 5 (A) Biosynthetic monomers of lignin. (B) Several wood-derived compounds used in natural product synthesis. | ||
More advanced strategies for lignin valorization are the focus of ongoing research and promise to convert wood-derived industrial waste or residues from agriculture into carbon-neutral, renewable building blocks. However, numerous issues such as lignin repolymerisation, low overall efficiency, structural variability and problems with product separation and purification need to be addressed.100 The reader may be referred to further references on utilizing wood or lignin as the source of platform chemicals and their acquisition.99,100,108,111–113 Arduengo and Opatz have coined the term “xylochemistry” for the synthesis of organic compound exclusively from wood-derived building blocks (vide infra).110,114–116
2.1.2.1. (±)-Usnic acid. Barton et al. published a two-step synthesis of (±)-usnic acid ((±)-12) by oxidative coupling of acetylated phloroglucinol 10 using potassium ferricyanide and subsequent acid-catalyzed dehydration (Scheme 6). Acetophenone 10 can be synthesized in two steps from natural phloroglucinol (5)117 through Vilsmeier–Haack reaction followed by aldehyde reduction and subsequent acetylation with acetic anhydride.118,119
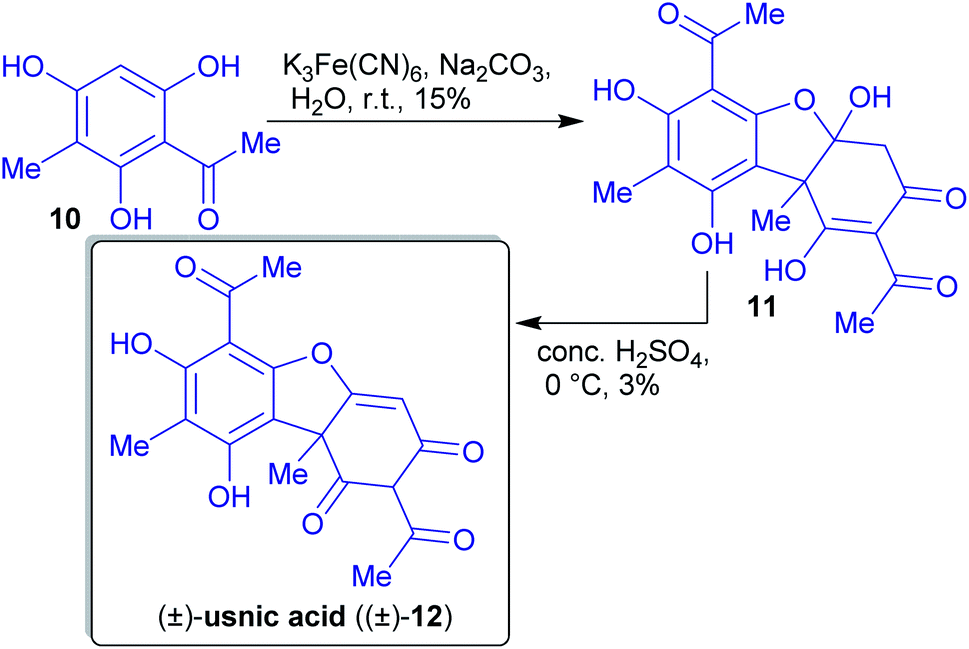 | ||
| Scheme 6 Synthesis of (±)-usnic acid ((±)-12) via oxidative coupling of acetophenone moieties 10. | ||
An advantage of this synthesis is the use of the environmentally benign solvents. Ferricyanide is a green oxidant and no carbon atom is “lost” during the synthesis (high atom economy). The reported yields are unfortunately low.
2.1.2.2. (+)-Garcibracteatone. George et al. reported the first enantioselective synthesis of the non-natural (+)-garcibracteatone ((+)-20) in 2014, which also allowed the determination of the absolute configuration of the naturally occuring enantiomer (Scheme 7).120,121 Benzoylphloroglucinol (17) can be derived from natural phloroglucinol (5) and benzoic acid via Friedel–Crafts acylation.117,122–125 Phloroglucinol derivative 17 was first reacted with prenyl bromide (14), accessible from the natural product prenol, and subsequent C-alkylation with iodide (−)-16 affording the dearomatized phloroglucinol 19.126–128 Oxidative radical cyclization completed the synthesis of (+)-garcibracteatone ((+)-20).
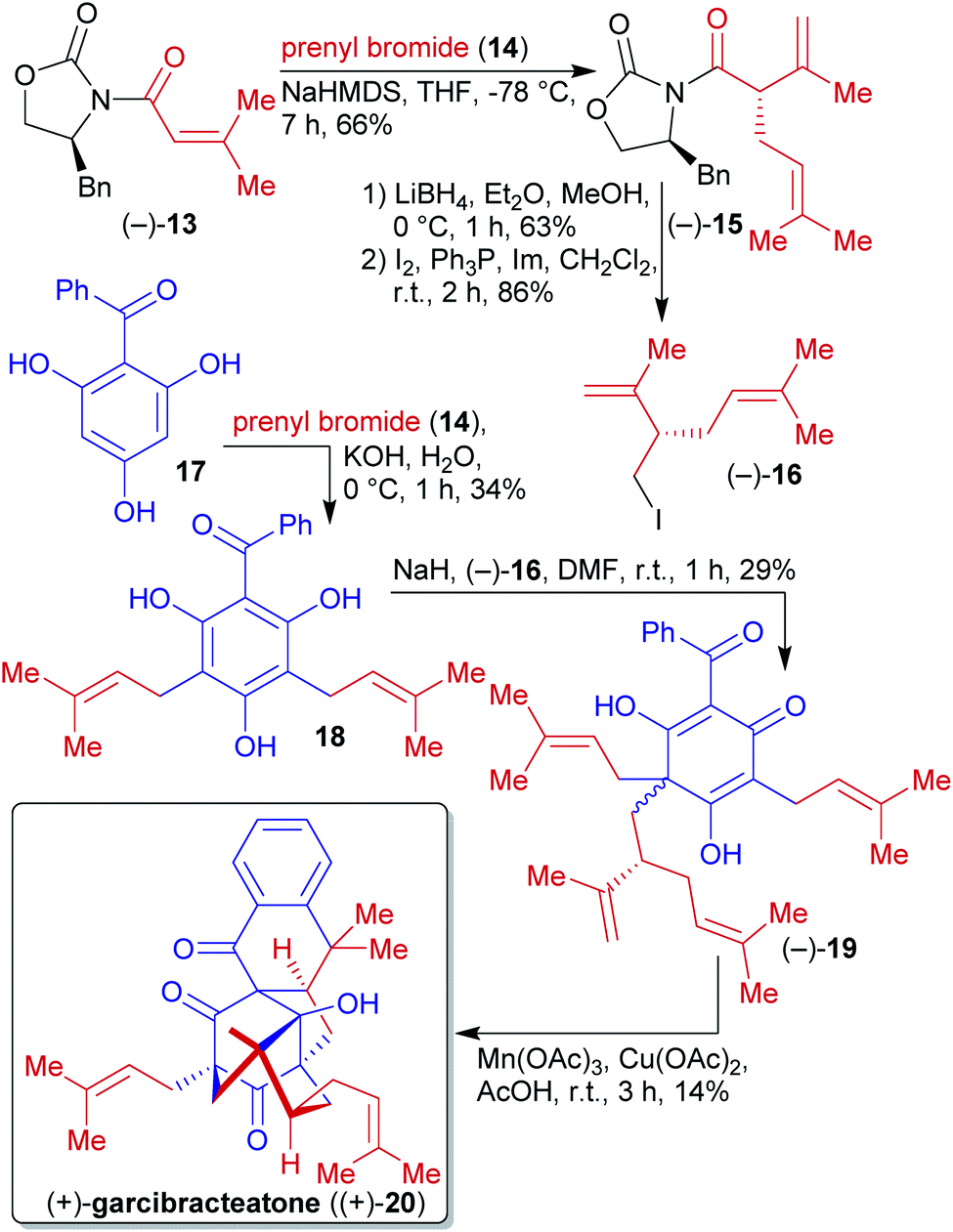 | ||
| Scheme 7 Enantioselective synthesis of (+)-garcibracteatone ((+)-20) mimicking biosynthesis. | ||
N-Acyloxazolidinone 13 could be prepared from biomass-derived 3-methylcrotonic acid129 and the phenylalanine-derived Evans auxiliary.130 Subsequent α-alkylation with prenyl bromide (14) and reduction followed by iodination delivered the enantioenriched fragment (−)-16.
The avoidance of protecting groups and the construction of a complex molecular scaffold from simple starting materials in a short route are the highlights of this sequence.
2.1.2.3. Lupinalbin H. The flavonoid lupinalbin H (28) was first isolated by Tahara et al. from the methanolic extract of the roots of yellow lupin (Lupinus luteus cv Topaz).131 Its first synthesis in 2011 by van Heerden et al. used a Suzuki–Miyaura reaction, followed by an oxidative cyclohydrogenation and a final 6π-electrocyclization (Scheme 8).132
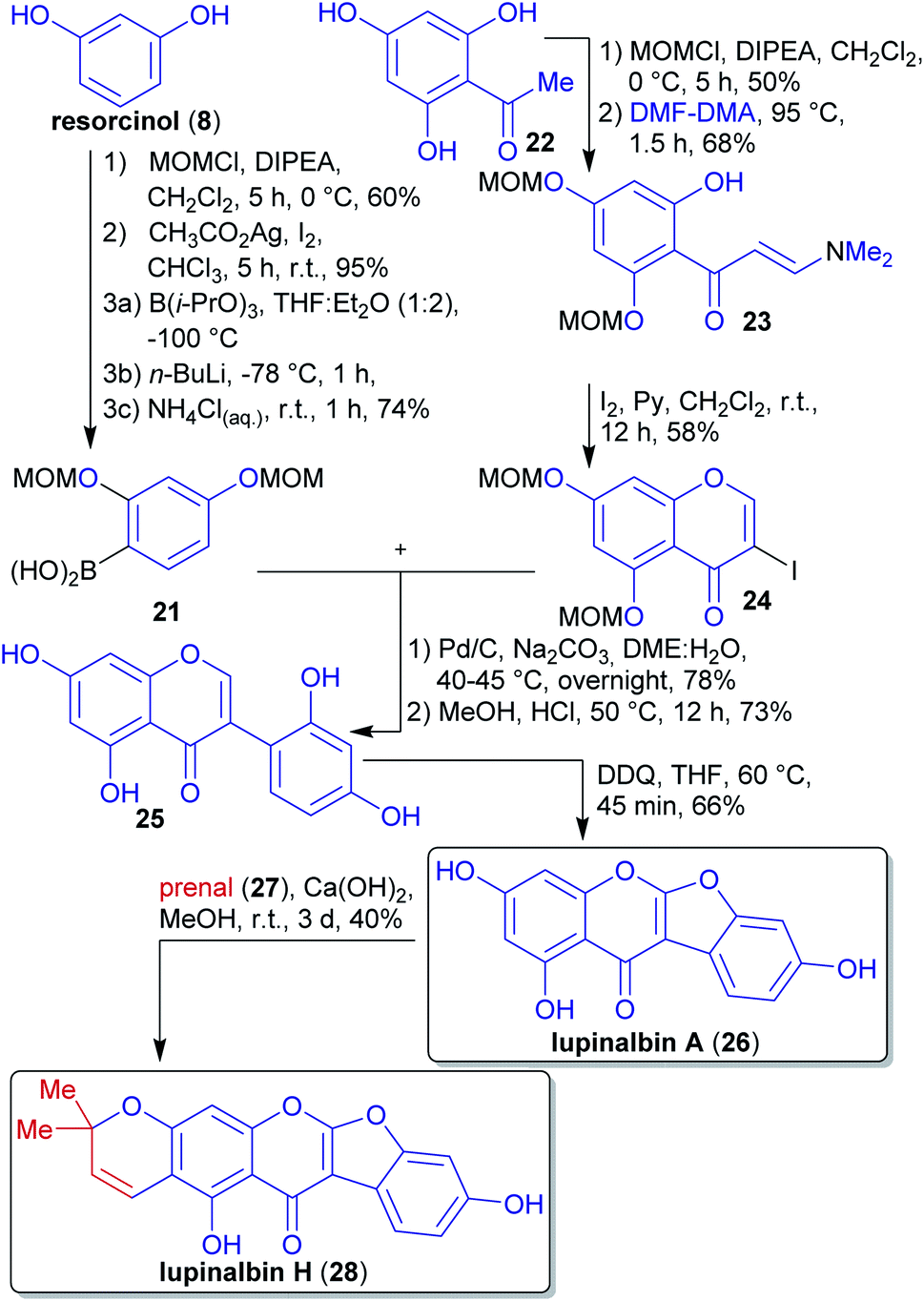 | ||
| Scheme 8 Synthesis of lupinalbin H (28) from fragments 21 and 24via Suzuki–Miyaura coupling. | ||
Boronic acid 21 was prepared from resorcinol (8), which can be isolated from various plant species.133–135 The hydroxy groups were protected and the arene regioselectively iodinated at C-5 to select the site of borylation.
The synthesis of fragment 24 was reported in 2010 by the same group starting from trihydroxyacetophenone 22.136 The isolation of the latter from plants has been reported.137,138 Reaction with DMF-dimethyl acetal, theoretically accessible from the wood-derived renewables DMF139 and (MeO)2SO2 (prepared by reaction of MeOH with SO3),140 and subsequent iodination led to fragment 24.141
By coupling of both precursors in the presence of a palladium catalyst, isoflavone 25 was obtained, and after deprotection, oxidation furnished lupinalbin A (26). This naturally-occurring phytoestrogen was condensed with prenal (27) (accesible via catalytic aerobic oxidation of prenol)128,142 to complete the synthesis of 28.143 The authors were able to formally derive all carbon- and heteroatoms from renewable sources. The choice of solvents and reagents (e.g. DDQ) was however traditional and less compatible with the principles of green chemistry.
2.1.2.4. (±)-Tylophorine. Opatz et al. reported a short synthesis of the phenanthroindolizine alkaloid (±)-tylophorine ((±)-36)144 with a Stevens rearrangement as the key step and devoid of any protecting group manipulations (Scheme 9).145 Three of five overall steps can be performed in a one-pot procedure and no chromatographic purification was required, which is in accordance with “green” principles of pollution prevention.24
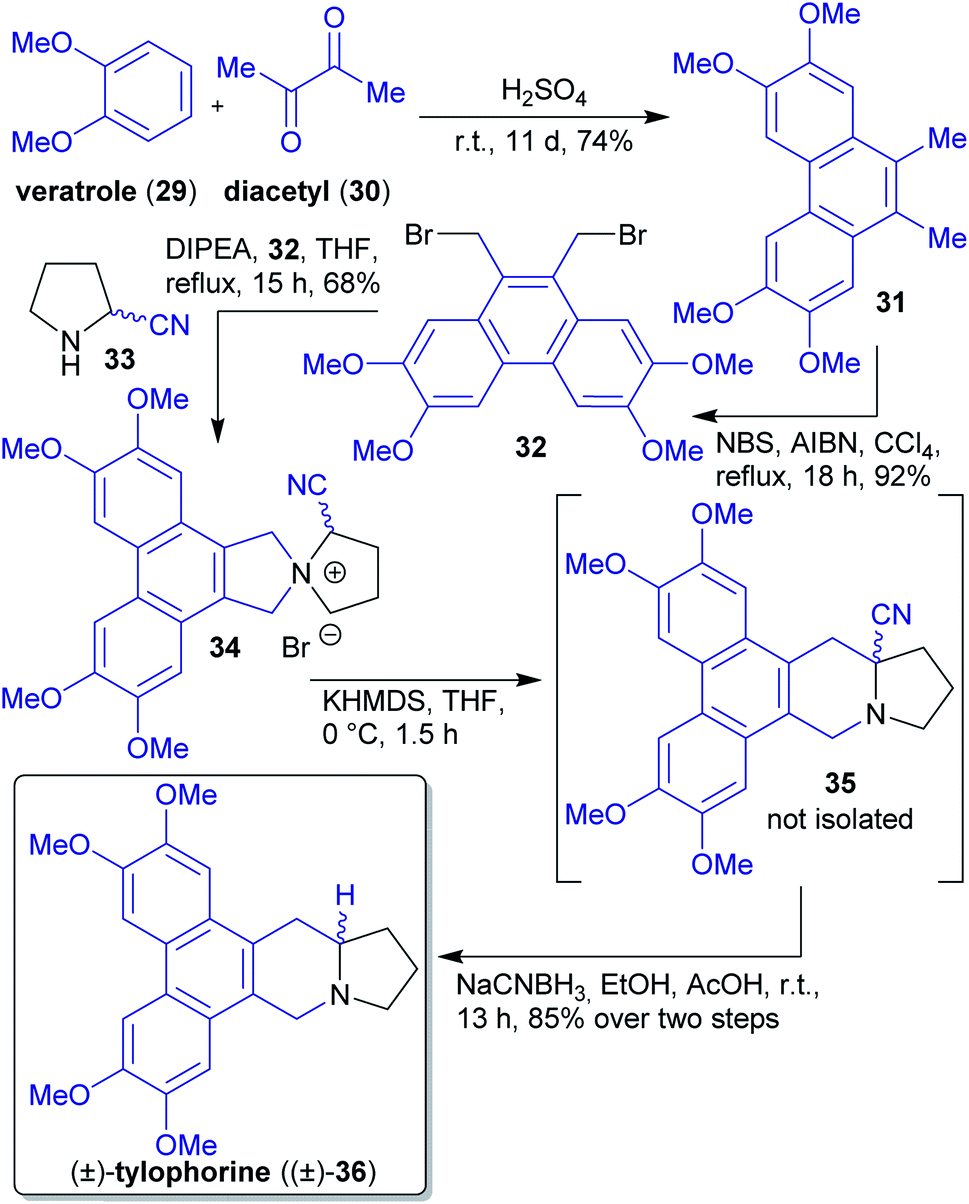 | ||
| Scheme 9 Synthesis of (±)-tylophorine ((±)-36) following xylochemical principles. | ||
Starting materials veratrole (29) and diacetyl (30) can be obtained from biomass146,147 and were subjected to an acid catalyzed reaction furnishing phenanthrene derivative 31, which was brominated under free radical conditions. Reaction with α-amino nitrile 33 afforded spirocyclic compound 34, which underwent a Stevens rearrangement to furnish natural (±)-tylophorine ((±)-36) after reduction with NaCNBH3.145
The α-amino nitrile 33 was synthesized from pyrrolidine and sodium cyanide, the former can be derived from proline e.g. via a Pd-catalyzed decarboxylation reaction.145,148
Drawbacks of the route are the use of toxic solvents and reagents, which are to be avoided according to “green” principles. It should be noted that natural tylophorine is almost racemic.149
2.1.2.5. (±)-Gracilamine. The natural product (±)-gracilamine ((±)-45) was isolated in 2005
150 and its first synthesis was reported in 2012 by Ma et al.151
The key step in the formation of spirocyclic intermediates 38a and 38b was an intramolecular oxidative phenol coupling (Scheme 10).152 The starting materials for this reaction, piperonal (6) and tyramine (37), can be obtained from renewable sources.153–155 Reduction of spirocyclic compound 38a and 38b with LiAlH4 followed by protection with TBDPSCl, ring opening using TrocCl gave benzyl alcohols 39a and 39b after treating with AgNO3 in the presence of H2O.
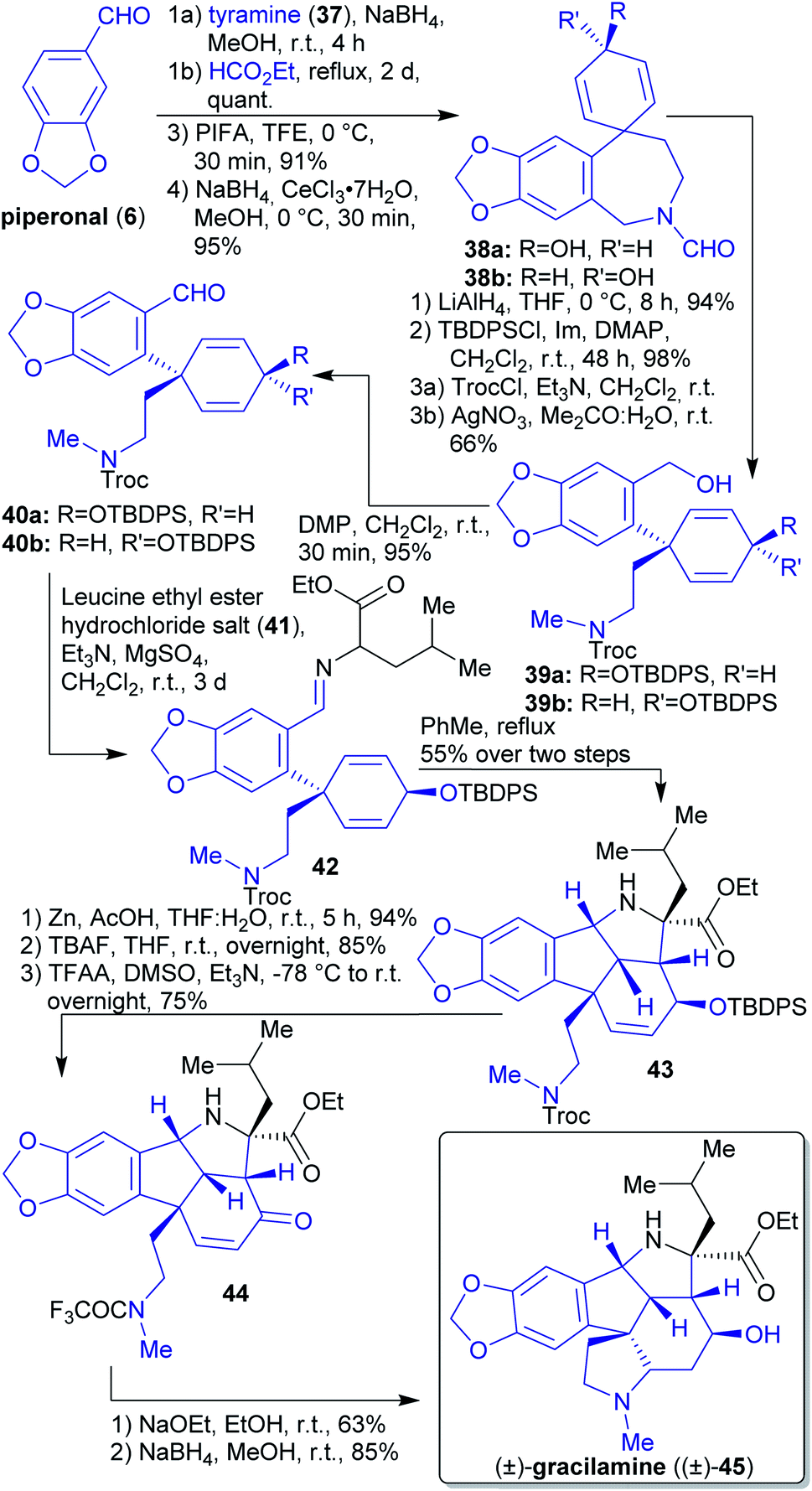 | ||
| Scheme 10 Synthesis of (±)-gracilamine ((±)-45) from piperonal (6). | ||
Alcohols 39a and 39b were oxidized to aldehydes 40a and 40b. Through the condensation reaction of aldehyde 40a and leucine ethyl ester (41), imine 42 was formed, which reacted in a [3 + 2]-cycloaddition with the corresponding azomethine ylide under formation of the natural product scaffold 43 containing all of the carbon atoms required. The final product 45 was obtained by deprotection, cyclization and reduction using NaBH4.151
In this synthesis, all carbon- and hetero atoms can be obtained from renewable sources.
2.1.2.6. Cochinchinenone. The first total synthesis of the natural product cochinchinenone (48)156 in only five steps and 58% overall yield was reported from Carreño et al.157 The synthesis commenced with the Mukaiyama aldol condensation of syringaldehyde (7) with PMB-protected 4-hydroxy-acetophenone (46), both derivable from lignin (Scheme 11).158,159 To obtain chalcone 48 with a p-quinol moiety in ring A, the corresponding ketone 47 was dearomatized oxidatively. To this end, the choice of the right protective group was crucial since derivatives such as OMOM, OTBDMS, OTBDPS, OTHP, and OBn led to negative results during the synthesis. Furthermore, the dearomatization did not proceed with a free OH-group because of a competing Baeyer–Villiger reaction taking place instead.157 Only a single protective group is required and there is no loss of carbon atoms throughout the synthesis.
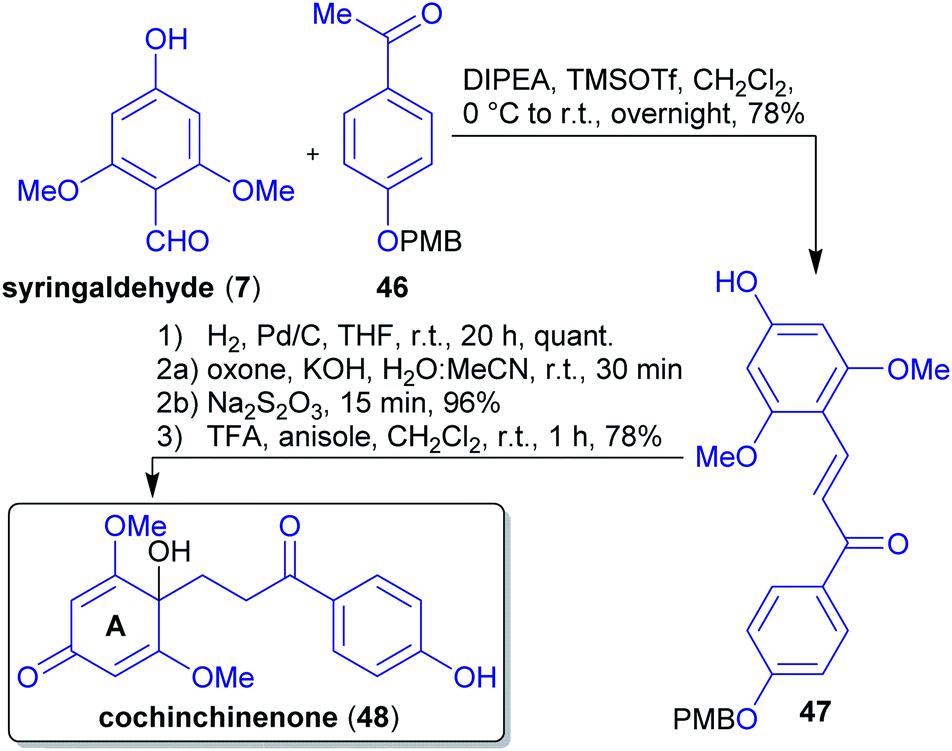 | ||
| Scheme 11 Concise and first synthesis of cochinchinenone (48) based on lignin derived starting materials. | ||
2.1.2.7. Taiwaniaquinones and taiwaniaquinols. The total synthesis of the racemic taiwaniaquinoids was reported by Li et al. in 2013.160
The synthesis commenced with the preparation of common intermediate 54 with a an trans A/B ring junction on a gram scale (Scheme 12). 1,2,4-Trimethoxybenzene is available from renewable resources and can be transformed into benzaldehyde 49 in several steps.161,162 This was then subjected to a Wittig olefination, followed by a Pd-catalyzed Suzuki–Miyaura coupling with iododiene 51 to afford diene 52. Through a Bi(OTf)3-catalyzed cationic cyclization and a Wolff-type ring contraction as the key steps, intermediate 54 was obtained. From there on, the natural products taiwaniaquinones A (57) and F (58), and taiwaniaquinols B (59) and D (60) were prepared in 2–3 steps in racemic form.160
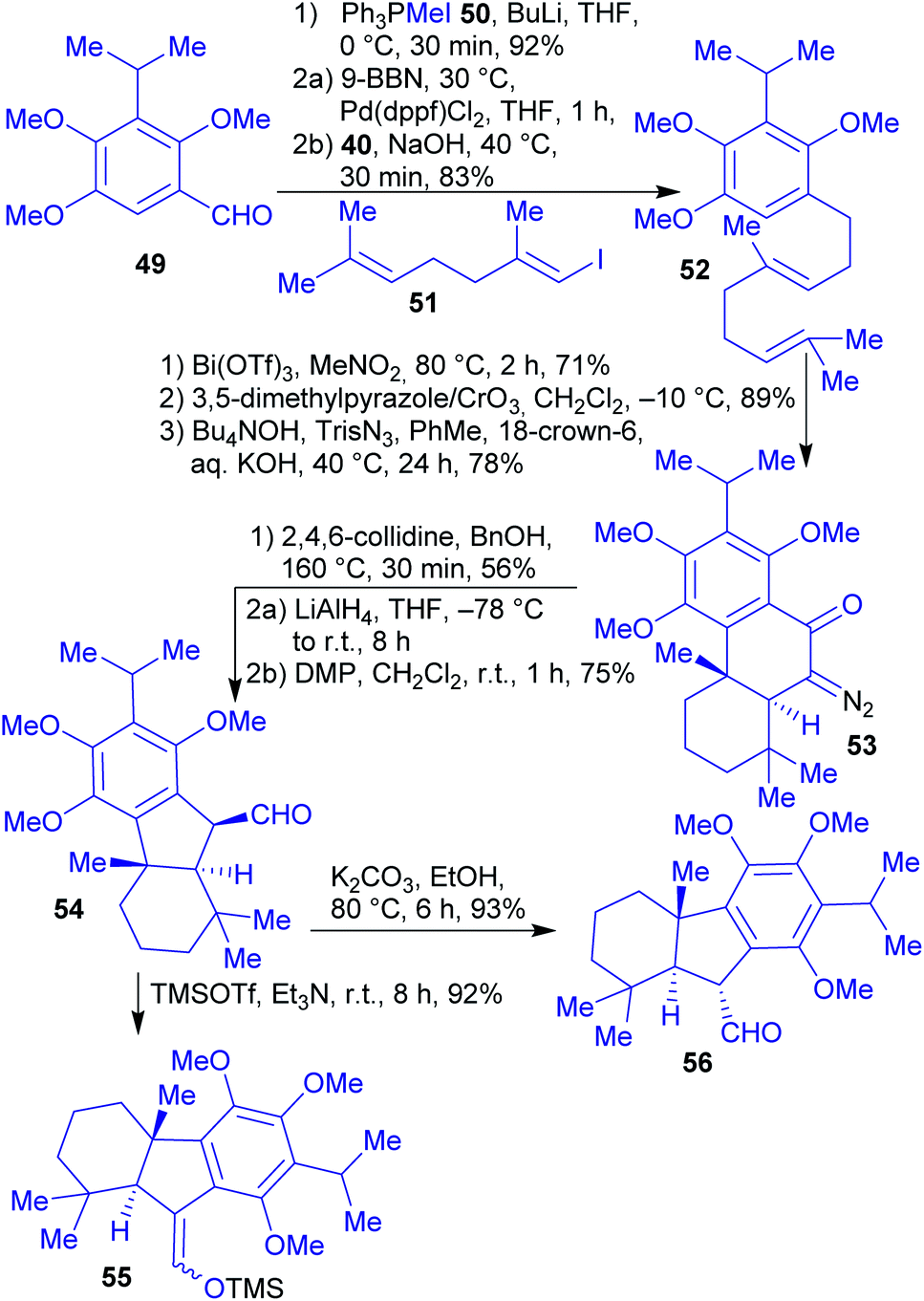 | ||
| Scheme 12 Synthesis of intermediate 54 and subsequent intermediates for the synthesis of taiwaniaquinones and taiwaniaquinols, accessible via a Wolff-type ring contraction. | ||
Quinones A 57 and F 58 were accessed by epimerization of aldehyde 54 followed by oxidation (taiwaniaquinone F (58)) or by oxidation and O-demethylation (taiwaniaquinone A (57), Scheme 13).
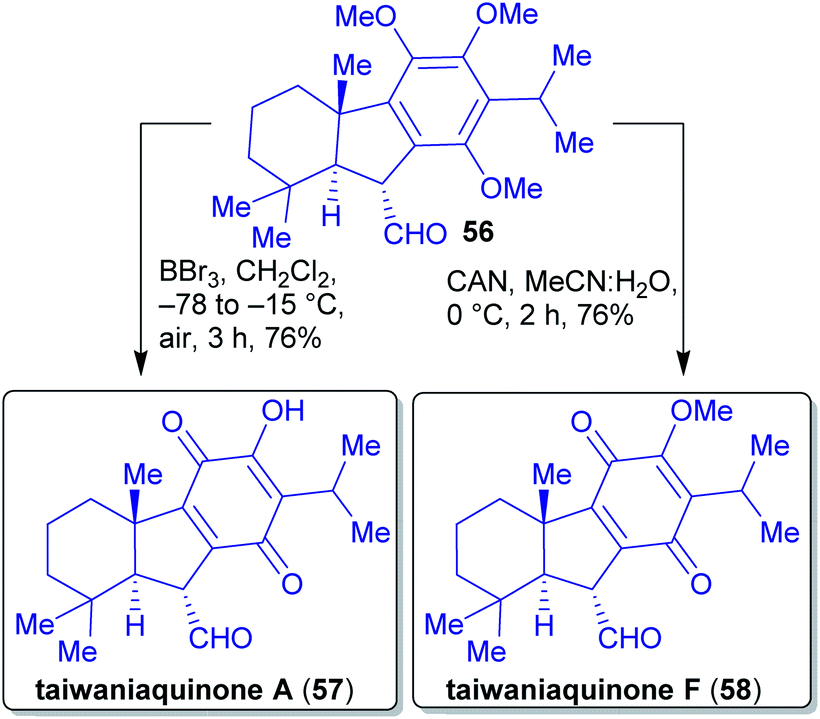 | ||
| Scheme 13 Closing synthesis of taiwaniaquinone A (57) and F (58). | ||
Quinols B 59 and D 60 were obtained from the silyl enol ether derived from intermediate 54. Via a sequence of Saegusa-Ito oxidation, demethylation and oxidation, quinol D 60 was furnished. Subjecting the silyl enol ether of 54 to dihydroxylation conditions, a demethylation and an oxidation reaction led to quinol B 59 (Scheme 14).
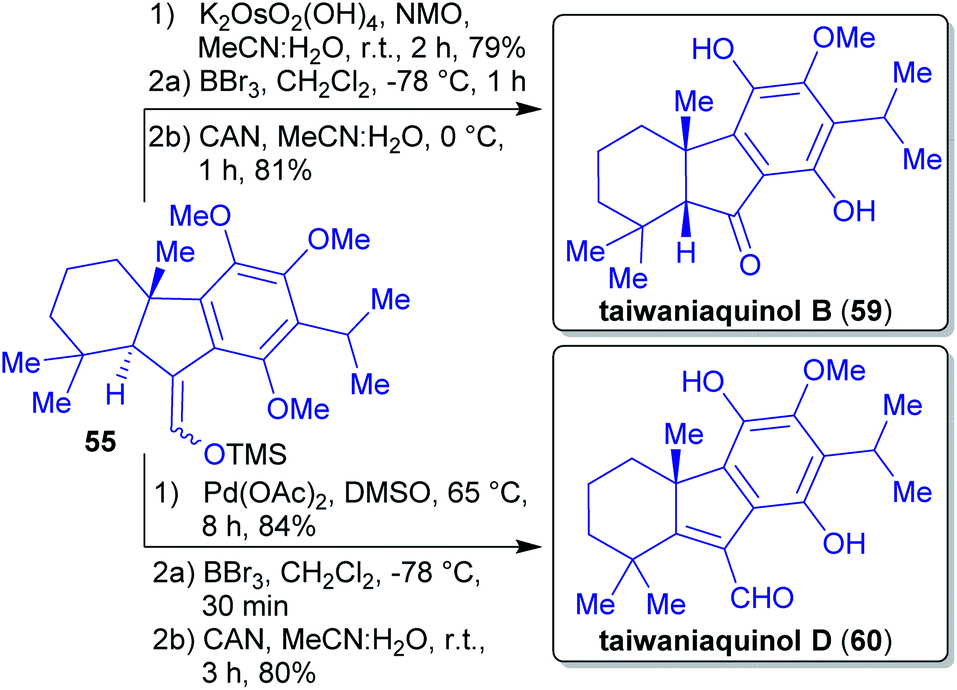 | ||
| Scheme 14 Closing synthesis of taiwaniaquinol B (59) and D (60). | ||
All four natural products synthesized occur in the same plant species.163
Methylenetriphenylphosphorane (50) was prepared from triphenylphosphine and iodomethane,164 available from methanol (wood spirit) and HI.165
Iododiene 51 can be prepared in three steps from 6-methylhept-5-en-2-one, which can be isolated from several plant species.166,167
2.1.2.8. (−)-Surinamensinol B. The first enantioselective synthesis of (−)-surinamensinol B ((−)-70) was achieved by Sudalai et al. in 2015.184
Enantiopure fragment 67 could be prepared in several steps from 61 (Scheme 15).173 Diol 63 was obtained by Wittig olefination, dihydroxylation, reduction and subsequent tosylation afforded compound 64. After epoxidation and O-protection, compound (±)-65 was subjected to a hydrolytic kinetic resolution using a cobalt catalyst to obtain enantiopure syn-epoxide 66 in 96% ee. A regioselective reductive ring-opening led to fragment 67.
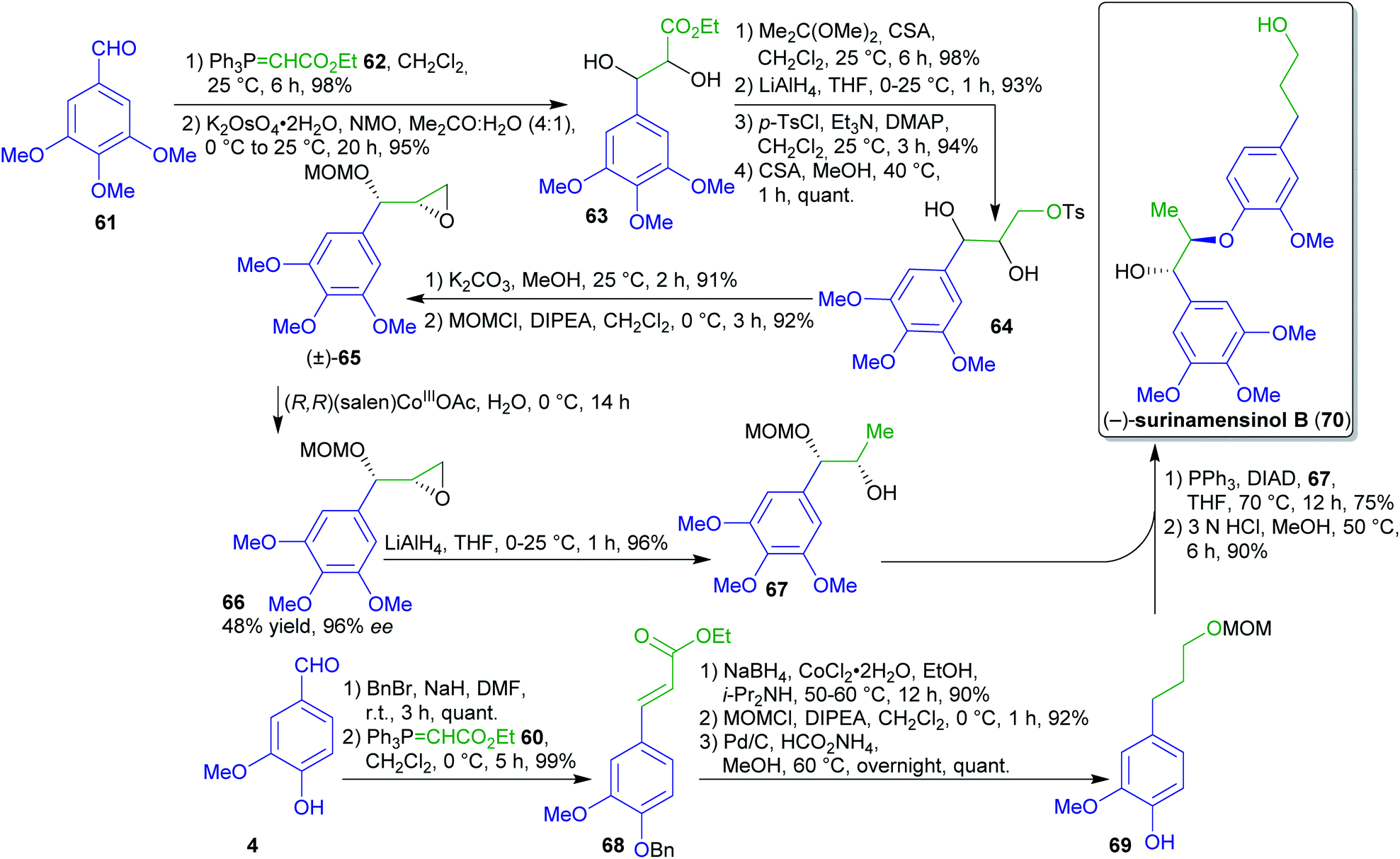 | ||
| Scheme 15 First enantioselective synthesis of surinamensinol B (70). CSA = camphor-10-sulfonic acid. | ||
The synthesis of the natural product surinamensinol B (70)185 commenced with O-benzylation of vanillin (4) followed by Wittig olefination and reduction to arylpropanol 69. The latter was coupled with fragment 67 and the natural product 70 was obtained after acid-catalyzed deprotection.184
The Wittig phosphonium ylide 62 employed twice in this sequence can be prepared from PPh3 and ethyl bromoacetate accessible from acetic acid or malonic acid via bromination186 and subsequent esterification.187 While the synthesis of PPh3 from biomass should be feasible but likely is lengthier than the classical petrochemical approach, the recycling from the oxide through the dichloride is well-known.188
While “green” solvents are employed in several places, the extensive use of protecting groups and the hydrolytic kinetic resolution step with a maximum yield of 50% are less favorable.
2.1.2.9. (+)-Monocerin. A concise asymmetric total synthesis of (+)-monocerin ((+)-76) was achieved by the group of She et al. via a Lewis acid-mediated stereoselective cyclization reaction (Scheme 16).168 The synthesis commenced with the conversion of 3,4,5-trimethoxybenzaldehyde (61) into an allylic alcohol. Enantioselective epoxidation followed by O-protection afforded compound 72.169 This was reacted with lithiated dithiane 73, followed by reduction and deprotection to furnish triol 74.
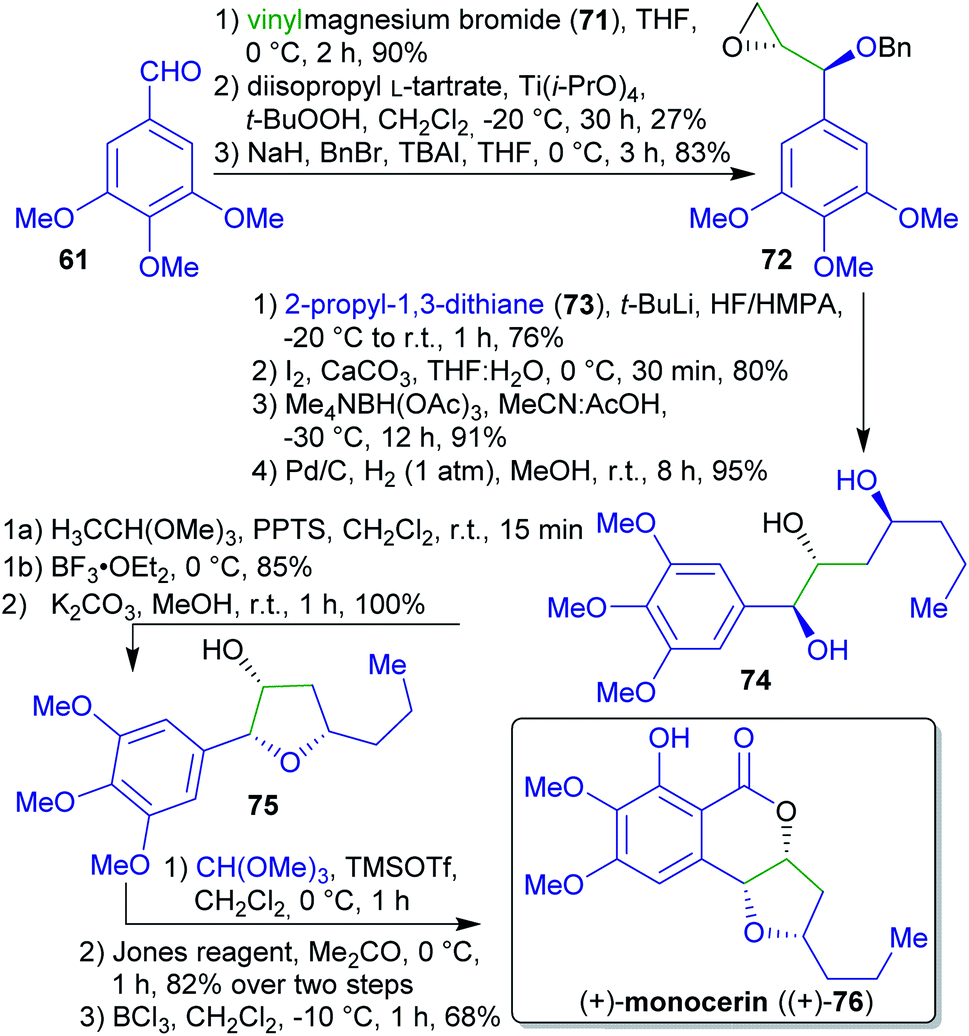 | ||
| Scheme 16 Synthesis of (+)-monocerin ((+)-76) from the renewable starting material trimethoxybenzaldehyde 61. | ||
2-Propyl-1,3-dithiane (73) is accessible via a condensation between potentially renewable butyraldehyde (1-butanol is a common fermentation product),170 and 1,3-propanedithiol.171 The synthesis was concluded by a Lewis acid-mediated cyclization reaction, followed by an oxa-Pictet–Spengler reaction, Jones oxidation and chelate-controlled regioselective O-demethylation affording the natural product (+)-monocerin ((+)-76).172
3,4,5-Trimethoxybenzaldehyde (61) is accesible from a renewable source.173 Vinylmagnesium bromide (71) can be synthesized in two steps from acrylic acid,174–176 derivable from lactic acid.177–181 Reaction of sodium methanolate with CHCl3 in the presence of DMF leads to trimethyl orthoformate.182 The required chloroform can e.g. be prepared by chlorination of biogenic methane or via reaction of methanol with FeCl3 but its general avoidance would be desirable.183 Considering “green” chemistry, particularly the use of toxic reagents (Jones reagent, HF/HMPA, BCl3) appears problematic.
2.1.2.10. (+)-Oxycodone. In 2019, Hudlicky et al. reported a synthesis of (+)-oxycodone ((+)-87), the non-natural enantiomer of this opioid.189 Its natural antipode190 is widely applied in pain management.191 The synthesis commenced with the microbial dihydroxylation of phenethyl acetate (81) (available through fermentation of corn, barley and sweet molasses),192 selective hydrogenation of the less hindered C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-double bond and a Mitsunobu reaction with iodophenol 80 (Scheme 17). The latter compound can be derived from natural isovanillin (77) via iodination,193 Wittig olefination with (methoxymethyl)triphenylphosphonium chloride (79) and reaction with methanolic HCl.194 Wittig salt 79 is theoretically accessible from the reaction of formaldehyde with MeOH and HCl, affording chloromethyl methyl ether,195 and subsequent reaction with PPh3.196 Intramolecular Heck reaction of ether 83 followed by dihydroxylation furnished diol 84. Through mesylation and DBU-catalyzed elimination, a ketone was obtained. Subsequent pinacol-type coupling with deprotected aldehyde and protection yielded carbonate 85. Methanolysis of the acetate followed by Mitsunobu coupling, carbonate hydrolysis and two-step dehydration led to tosylamide 86. The synthesis was completed via Parker radical amination and oxidation of deprotected alcohol yielding (+)-oxycodone ((+)-87).189 Tosylmethylamine can be prepared from methanol and tosylamide197 (product of toluene-derived TsCl with sodium cyanate or ammonia).198,199
C-double bond and a Mitsunobu reaction with iodophenol 80 (Scheme 17). The latter compound can be derived from natural isovanillin (77) via iodination,193 Wittig olefination with (methoxymethyl)triphenylphosphonium chloride (79) and reaction with methanolic HCl.194 Wittig salt 79 is theoretically accessible from the reaction of formaldehyde with MeOH and HCl, affording chloromethyl methyl ether,195 and subsequent reaction with PPh3.196 Intramolecular Heck reaction of ether 83 followed by dihydroxylation furnished diol 84. Through mesylation and DBU-catalyzed elimination, a ketone was obtained. Subsequent pinacol-type coupling with deprotected aldehyde and protection yielded carbonate 85. Methanolysis of the acetate followed by Mitsunobu coupling, carbonate hydrolysis and two-step dehydration led to tosylamide 86. The synthesis was completed via Parker radical amination and oxidation of deprotected alcohol yielding (+)-oxycodone ((+)-87).189 Tosylmethylamine can be prepared from methanol and tosylamide197 (product of toluene-derived TsCl with sodium cyanate or ammonia).198,199
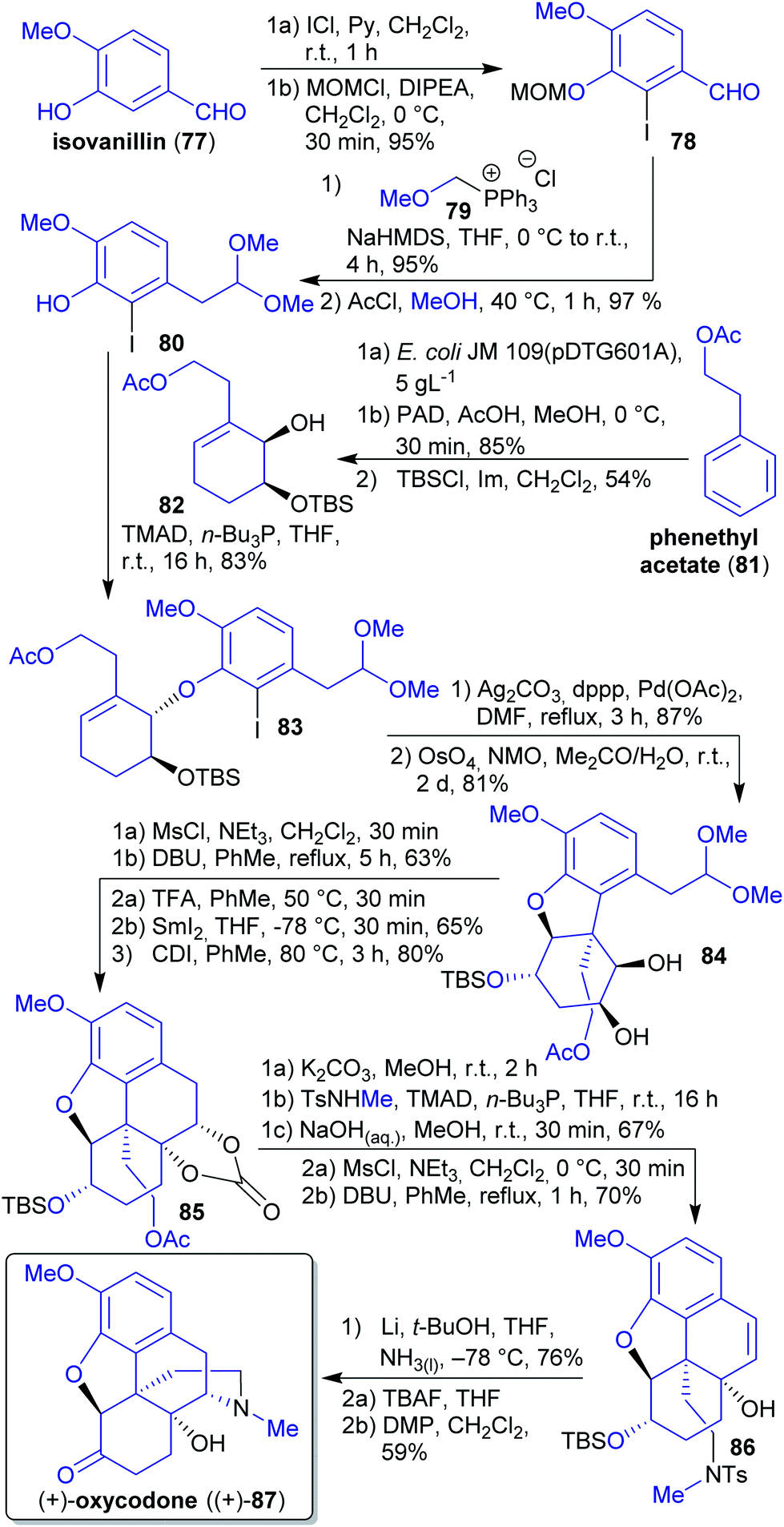 | ||
| Scheme 17 Synthesis of (+)-oxycodone ((+)-87) from phenethyl acetate (81) and isovanillin (77). PAD = potassium azodicarboxylate. | ||
The authors could have derived all carbon atoms in the product from renewable resources. The use of a highly stereoselective microbial dihydroxylation and of several “green” solvents (MeOH, Me2CO, H2O) are an advantage, yet toxic reagents and the use of non-green protecting groups had to be included.
2.1.2.11. Ilicifoline B. Opatz et al. reported the first total synthesis of the dimeric protoberberine-type alkaloid ilicifoline B (95) in 2015.114
The synthesis of 95200 commenced with methylation and hydrogenation of the wood-derivable natural product ferulic acid (9),201 followed by a Bischler–Napieralski cyclization and addition of in situ generated HCN202 to furnish α-amino nitrile 91 (Scheme 18). In a cascade reaction with dibromide 92, berberine alkaloid pseudopalmatine (93) is formed, subsequent oxidation and dimerization furnished alkaloid ilicifoline B (95).114 Dibromide 92 is accessible from veratrole (29), a pyrolysis product of wood.203 Formaldehyde and dimethyl sulfate can be obtained from methanol.140,204
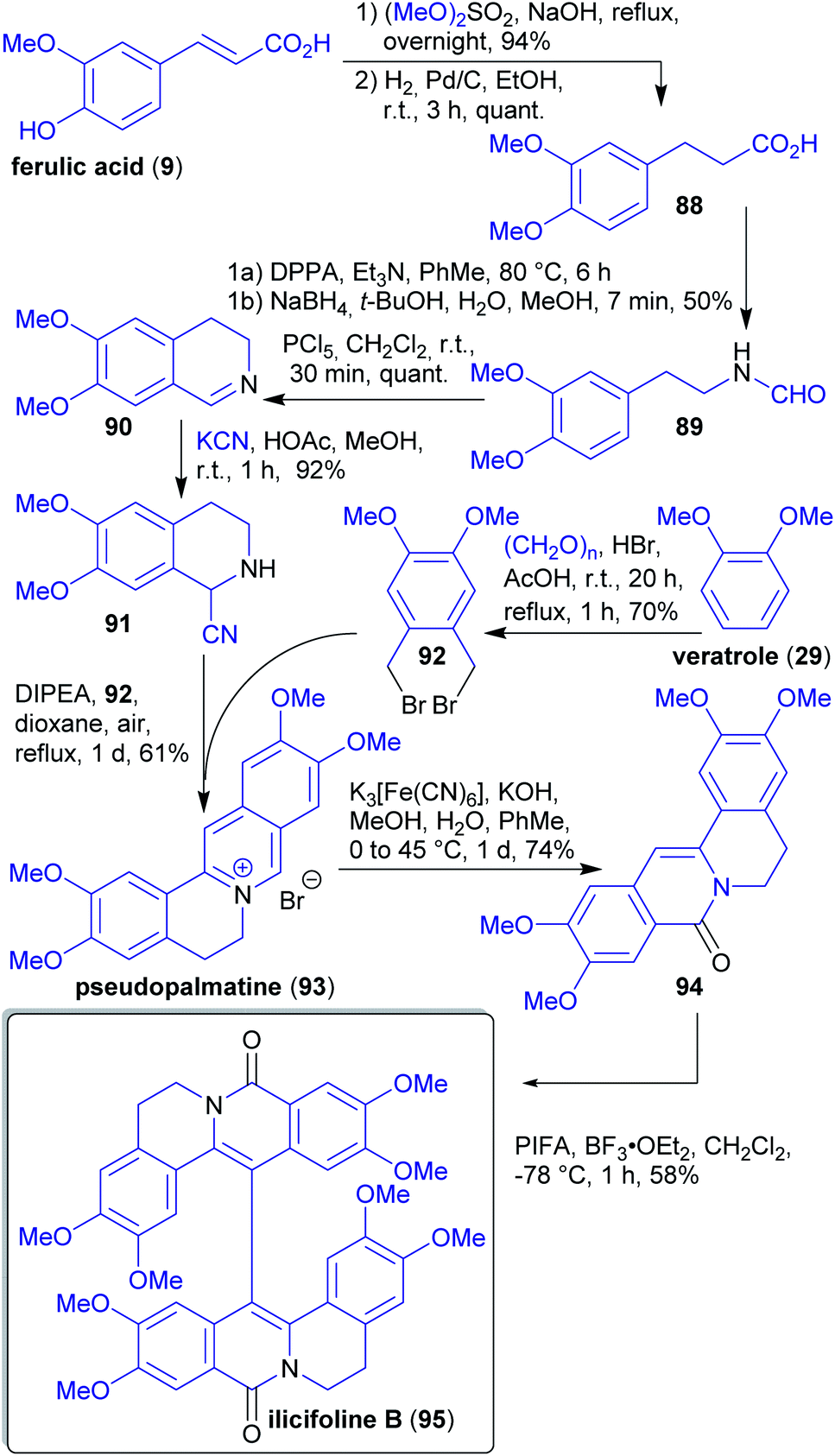 | ||
| Scheme 18 Synthesis of ilicifoline B (95) from wood derived starting materials 9 and 29. | ||
The authors used entirely wood-derivable building blocks, so-called xylochemicals, instead of conventional petrochemicals for the construction of the natural product carbon scaffold. In the light of “green” chemistry, the use of a non-toxic cyanide source and solvents like toluene, MeOH, H2O or EtOH is positive, yet undesired solvents like dioxane, CH2Cl2 should generally be avoided.
2.1.2.12. (−)-Viridin and (−)-viridiol. In 2017, Guerrero et al. reported the first enantioselective synthesis of the natural products (−)-viridin ((−)-111) and (−)-viridiol ((−)-112).205–207
The authors pursued the convergent approach of coupling two achiral fragments and employing an enantioselective intramolecular Heck reaction to set the absolute stereochemical configuration of an all-carbon quaternary stereocenter in the synthesis of 111 and 112.205–207 Indanone 99 was synthesized starting from plant-derivable 2,6-dihydroxybenzoic acid (96)208 by protection of all hydroxy groups,209,210 Heck alkenylation and subsequent hydrogenation furnishing dihydrocinnamic acid 98 (Scheme 19). This intermediate is converted into indanone 99 in three steps.
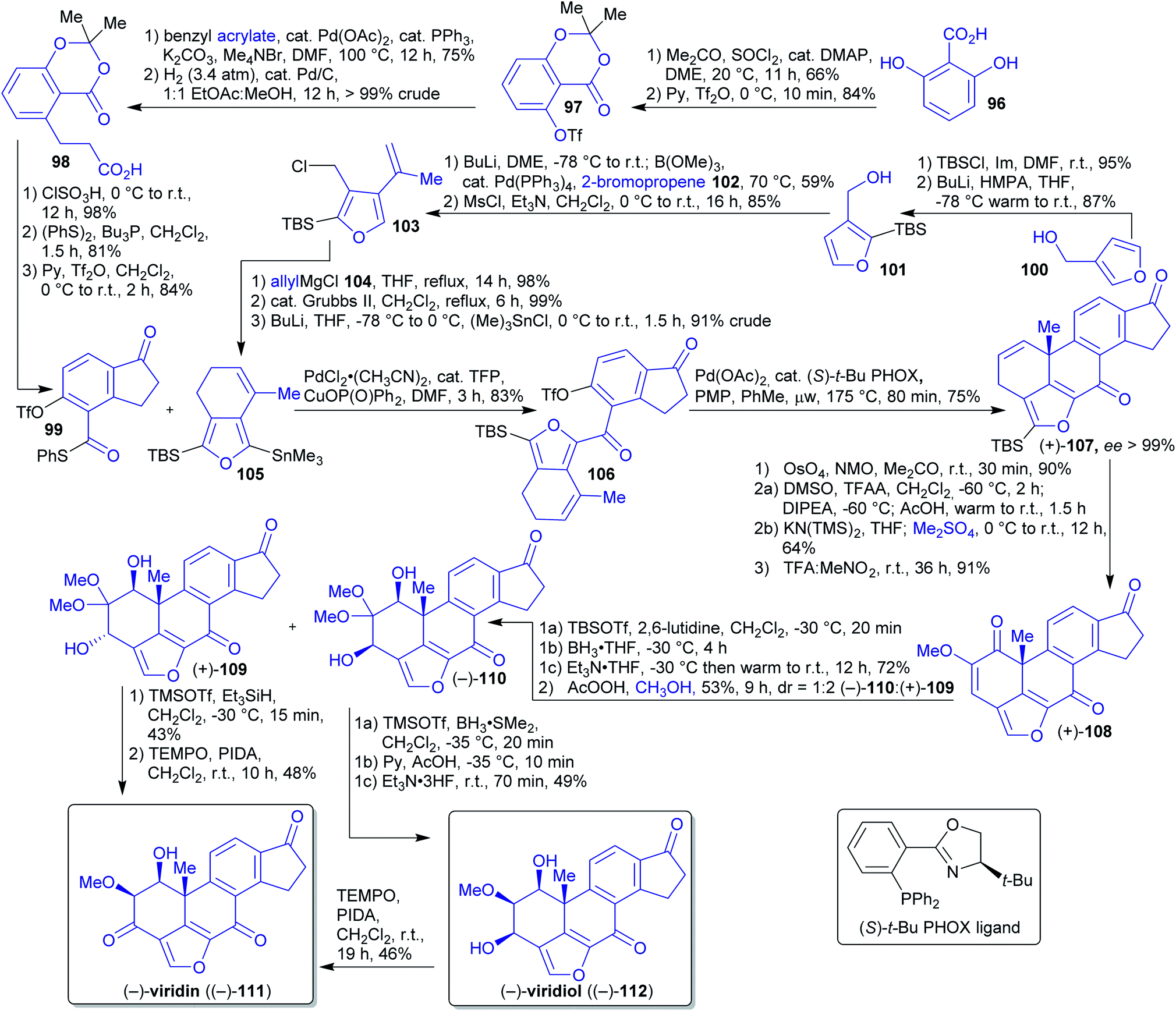 | ||
| Scheme 19 First enantioselective synthesis of (−)-viridin ((−)-111) and (−)-viridiol ((−)-112). | ||
The starting material 3-hydroxymethylfuran (100) for the synthesis of the second fragment can be obtained by reduction of naturally occurring 3-furoic acid.211,212 Subsequent silylation, reaction with 2-bromopropene (102) and chlorination led to compound 103. 2-Bromopropene (87) is accessible from wood-derived acetone213 in two steps via reaction with hydrazine and subsequent bromination.214,215 Compound 103 was reacted with allylmagnesium chloride 104, followed by ring closing metathesis and stannylation reaction affording second fragment 105. Allylmagnesium chloride (104) can be obtained from allylic alcohol (available from biomass)216,217via chlorination and reaction with magnesium.218,219
Enantiopure compound (+)-107 already bears the complete carbon skeleton of viridin (111) and was obtained from fragments 99 and 105via Liebeskind coupling and Heck cyclization. Upjohn dihydroxylation, double Swern oxidation and O-methylation yielded methoxy enone 108. Site- and diastereoselective reduction followed by treatment with AcOOH and MeOH delivered a mixture of hydroxy ketal diastereomers (+)-109 and (−)-110.
Reduction of ketal (+)-109 with Et3SiH and TEMPO-catalyzed oxidation led to viridin (−)-111. Alternatively, ketal (−)-110 was reduced with dimethyl borane to afford natural product viridiol (−)-112 and via same oxidative conditions as before viridin (−)-111.
Unfortunately, numerous highly toxic or otherwise problematic reagents had to be used (e.g. HMPA, OsO4, TFA, TFAA).
2.1.2.13. (±)-Latifine and (±)-cherylline. The hydrobenzoin substrates 124a and 124b were synthesized via a Wittig epoxidation/ring-opening one-pot protocol from substituted benzaldehydes accessible from biomass.109,201,220,221 The authors prepared 2,2-diarylacetaldehyde 125a and 125bvia a pinacol rearrangement using an eco-friendly catalyst and microwave irradiation under solvent-free conditions (Scheme 20). This key step was followed by one-pot reductive amination, Pictet–Spengler and hydrogenation reactions and only one chromatographic purification to afford the desired isoquinoline alkaloids 129 and 130.220 (±)-Cherylline (129) and (±)-latifine (130) are both secondary metabolites of Crinum latifolium L (Amaryllidaceae).222
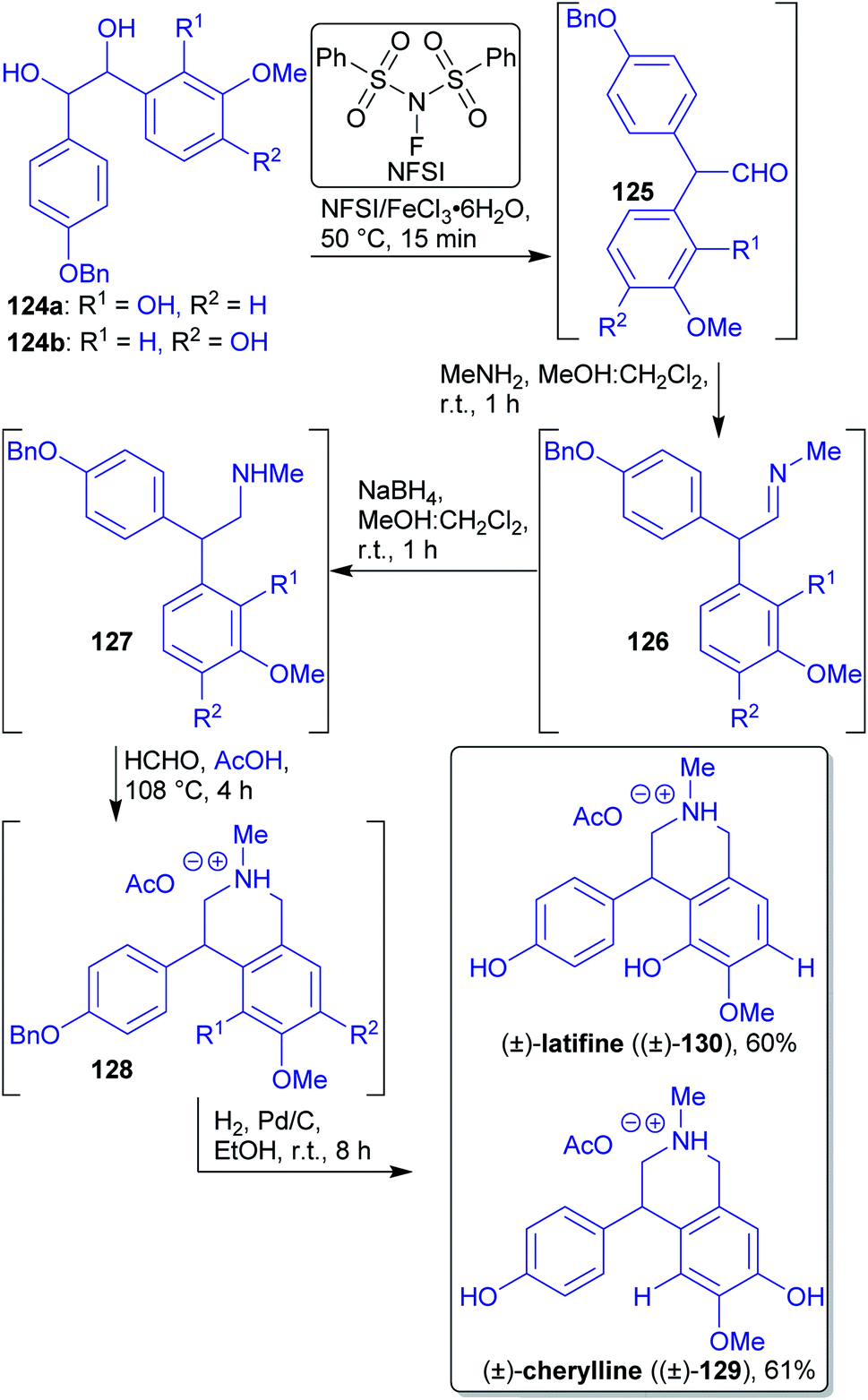 | ||
| Scheme 20 One-pot synthesis of (±)-latifine ((±)-130) and (±)-cherylline ((±)-129) utilizing a catalyzed, solvent free pinacol rearrangement. | ||
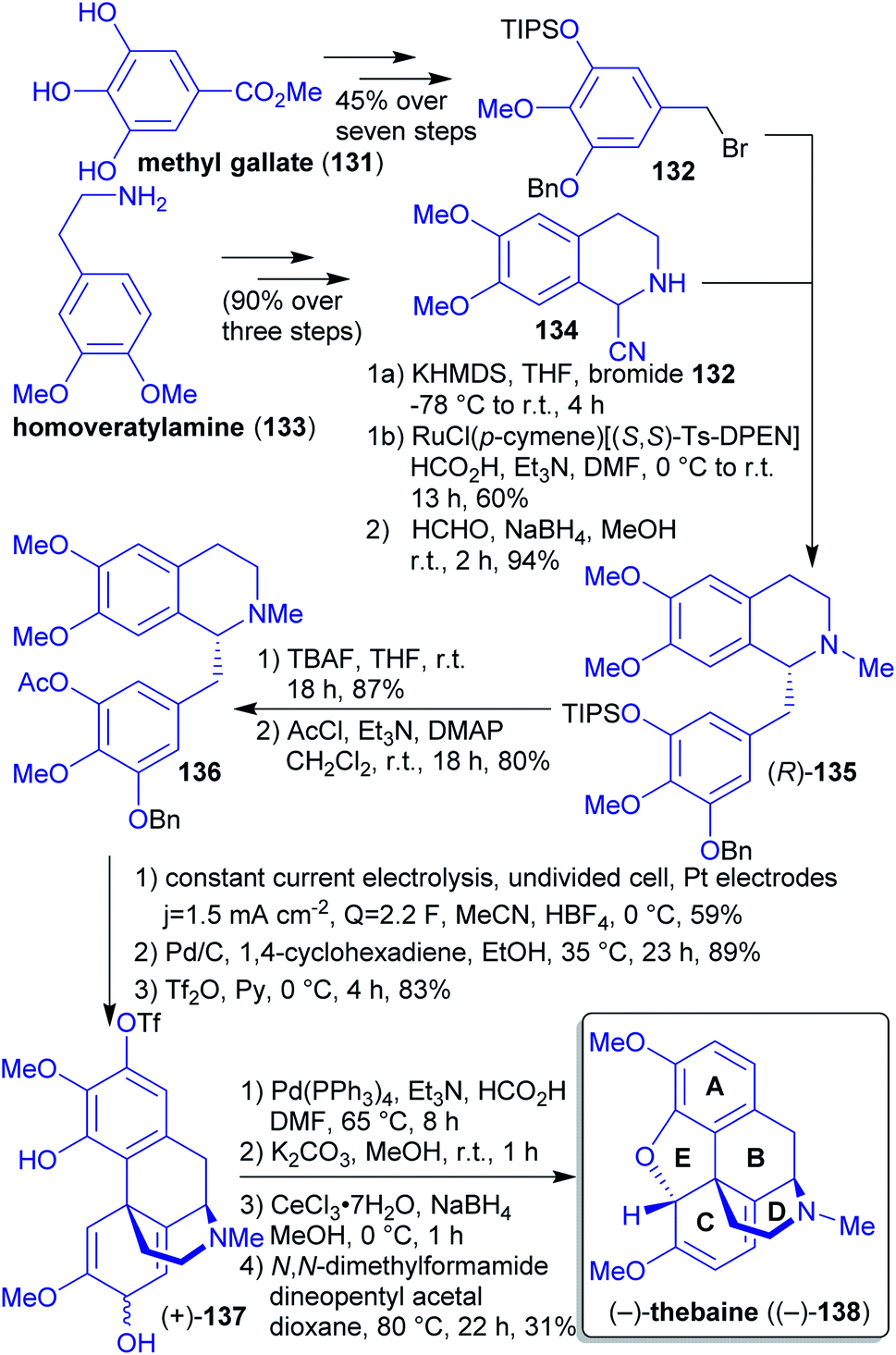 | ||
| Scheme 21 Synthesis of (−)-thebaine ((−)-138) from renewable starting materials. | ||
The authors provided an efficient and solvent-economical route in which half of the reactions operate at ambient temperature so that no additional heating is required. In general, one-pot approach are also favorable in the light of “green” chemistry.
2.1.2.14. (−)-Thebaine. So far, all synthetic approaches towards (−)-thebaine ((−)-138) using stoichiometric oxidants to mimic the biosynthetic oxidative phenol coupling delivered only low yields. The group of Opatz et al. reported the first electrochemical access to natural (−)-thebaine ((−)-138) via regio- and diastereoselective anodic coupling (Scheme 22).223,224
 | ||
| Scheme 22 Valorization of cellulose for selected platform chemicals. | ||
Homoveratrylamine (133) and methyl gallate (131) are both accessible from biomass225,226 and were reacted in several steps to furnish compounds 134 and 132. Via a deprotonation/alkylation/reduction sequence, tetrahydroisoquinoline 135 was formed. The anodic coupling was performed after a less favorable but inevitable switch of protecting groups, yielding intermediate 137. Subsequent deacetylation, Luche reduction and closure of the E-ring through conjugate nucleophilic substitution afforded (−)-thebaine ((−)-138).
Based on this procedure, the authors were also able to synthesize the natural opioid (−)-oxycodone ((−)-87), wheras the synthesis of its optical antipode was achieved by Hudlicky et al. along a different synthetic route (vide supra).189,227
Under “green” aspects, switching of protecting groups is not ideal. Furthermore, several of undesired solvents (DMF, THF, CH2Cl2) as well as toxic or hazardous reagents (1,4-cyclohexadiene, DMAP, HCO2H, Et3N) had to be used.
2.2. Cellulose
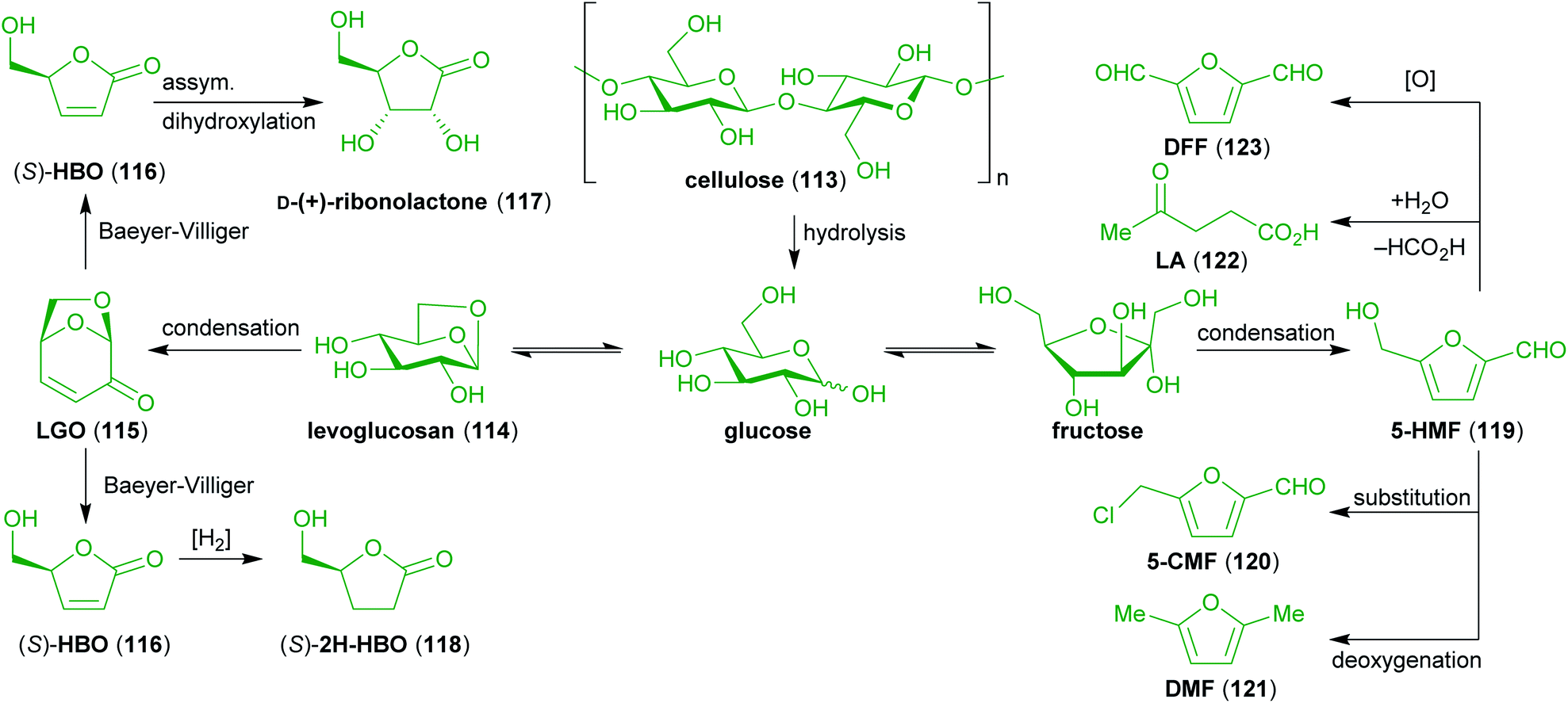
Cellulose (113) and hemicelluloses account for up to 80% of the dry biomass of plants in which they form the cell walls. While the chemical structures of hemicelluloses are very heterogeneous, cellulose consists exclusively of β(1 → 4) linked poly-D-glucose, making it a non-edible carbohydrate source for most animals including humans. It is a very abundant and promising sustainable feedstock for chemical raw materials.One of the most important small molecules obtainable from cellulose (as well as of hexoses in general) in high yields is 5-hydroxymethylfurfural (5-HMF, 119)234–236 and related compounds that can either be obtained from 5-HMF (119) or directly from cellulose. Those are 5-(chloromethyl)furfural (5-CMF, 120),237,238 2,5-diformylfuran (DFF, 123),239,240 2,5-dimethylfuran (DMF, 121, not to be confused with dimethylformamide commonly abbreviated in the same way)241,242 and levulinic acid (LA, 122).243–246 Another interesting raw material is levoglucosenone (LGO, 115), which bears several useful functional groups in the form of an enone and an acetal moiety in its chiral bicyclic skeleton.247–250 It is derived from levoglucosan (114), a product of the pyrolysis of cellulose.251–253 Furthermore, it can be transformed to other useful compounds like (S)-γ-hydroxymethyl-α,β-butenolide (HBO, 116) via Baeyer–Villiger oxidation,254,255 the respective saturated derivative (2H-HBO, 118),255 and D-(+)-ribono-1,4-lactone (117).256
Although glucose is an important compound obtainable from cellulose that has elaborately been used for the natural product synthesis, those syntheses are only cursory covered in this review due to the wealth of literature already available.257–262
2.2.2.1. (−)-Hongconin. The cardioprotective agent hongconin ((−)-145)263–265 was synthesized from (−)-levoglucosenone ((−)-115) via Hauser–Kraus-annulation with cyanophthalide 141 by Swenton et al. (Scheme 23).266 The latter can be synthesized from the xylochemical 3-methoxybenzoic acid (139).267 Acetal reduction, Appel reaction, radical reduction and methylation furnished naphthopyran 144. Deprotonation and treatment with iodomethane in the presence of DMPU (avoiding the more common but highly toxic and carcinogenic HMPA) gave a 4
:1-mixture in favor of the desired trans-product which was converted to the respective quinone with AgO and subsequently treated with sodium dithionite to obtain (−)-hongconin ((−)-145).
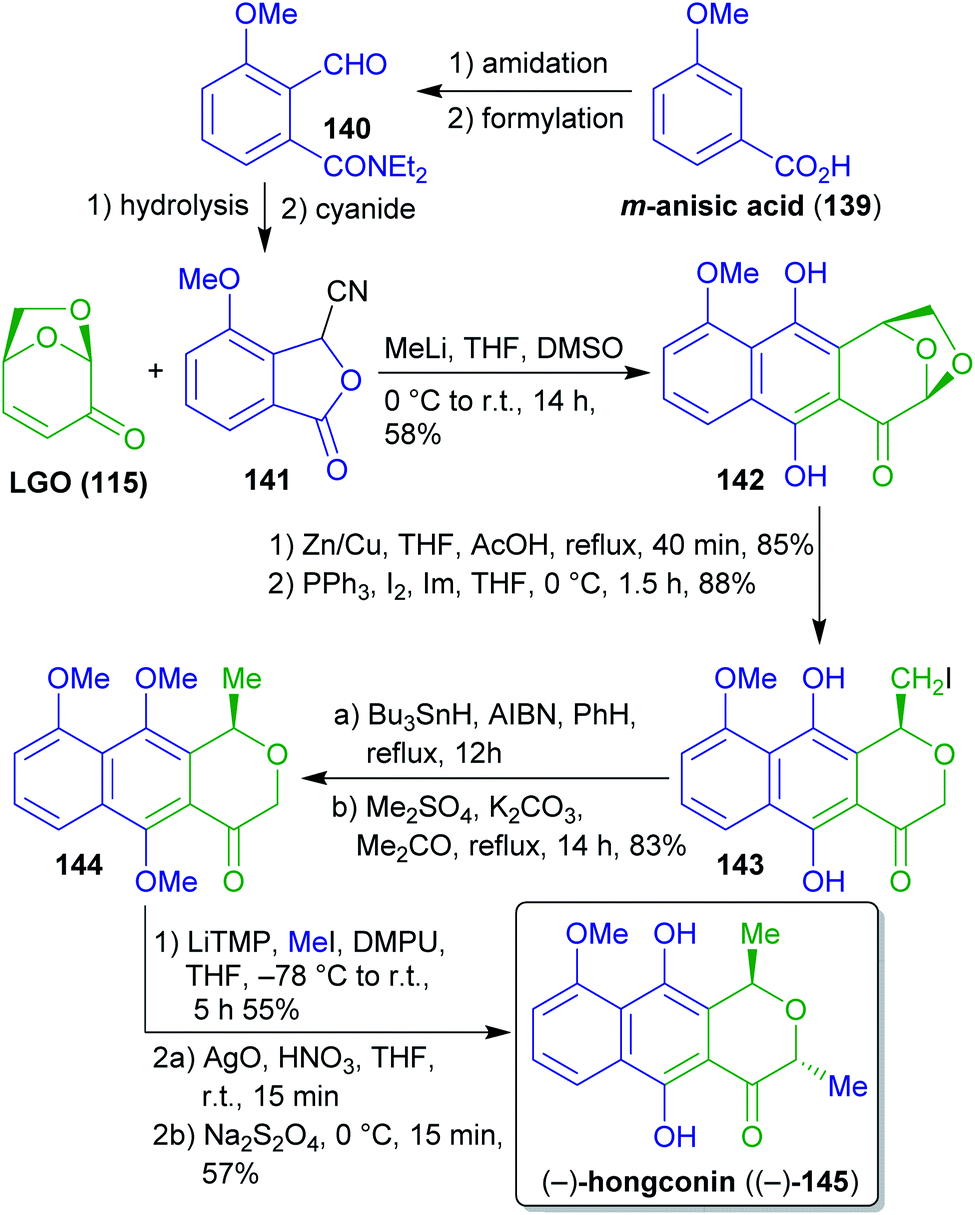 | ||
| Scheme 23 Synthesis of (−)-hongconin ((−)-145) from levoglucosenone (139). | ||
This very straightforward approach provides access to enantiopure (−)-hongconin (145) in a short sequence utilizing cellulose-derived 115 as a chiral synthon and wood-derived 139 as an aromatic building block. Although the procedures used are based on less green methods, only one protecting group transformation had to be performed.
2.2.2.2. (+)-Dairy lactone. A concise synthesis of the flavoring compound dairy lactone (149), named after its natural occurrence in cow milk as well as its dairy-like odor and flavor,268,269 was presented in 2016 using the levoglucosenone-derived γ-lactone 2H-HBO (118).270 To create an electrophilic species from alcohol 118, it was converted to epoxide 146via the respective tosylate and alkaline transesterification (Scheme 24). Reaction with lithiated 1-heptyne (147) and acidic transesterification furnished lactone 148, which was hydrogenated to (+)-149 using a Lindlar catalyst. Alkyne 147 can be produced from any ω-6 fatty acid derivative (e.g. linoleic acid) by cross metathesis with ethylene,271 bromination and dehydrobromination.272
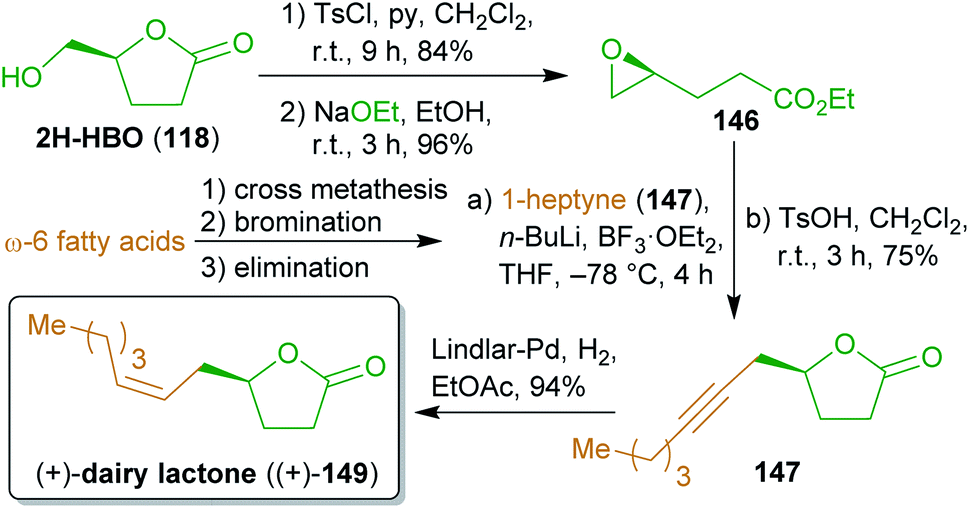 | ||
| Scheme 24 Synthesis of (+)-dairy lactone ((+)-149) from 2H-HBO (118). | ||
2.2.2.3. (+)-Herbarumins. Another levoglucosenone-derived starting material, δ-ribonolactone (117), was used as its acetonide protected derivative 150 by Fürstner et al.273 for the synthesis of phytotoxic herbarumins I and II (158 and 159, Scheme 25).274 Similar to the synthesis of 149, the lactone was converted to an epoxide through ring transformation, which was then transformed into lactol 152 by nucleophilic attack of ethylmagnesium bromide and reduction. The hemiacetal was subjected to a Steglich esterification with hexenoic acids 155a–b to furnish 156a–b. 5-Hexenoic acid (155a) as well as the 2-methoxymethyl derivative 155b, which was synthesized from 155a using Evans aldol methodology, are available from eicos-5-enoic acid (153) via cross metathesis with ethylene.275 Esters 156a–b were subjected to olefin metathesis using catalyst 157 to ensure E-selectivity. Final deprotection furnished enantiopure (+)-158 and (+)-159.
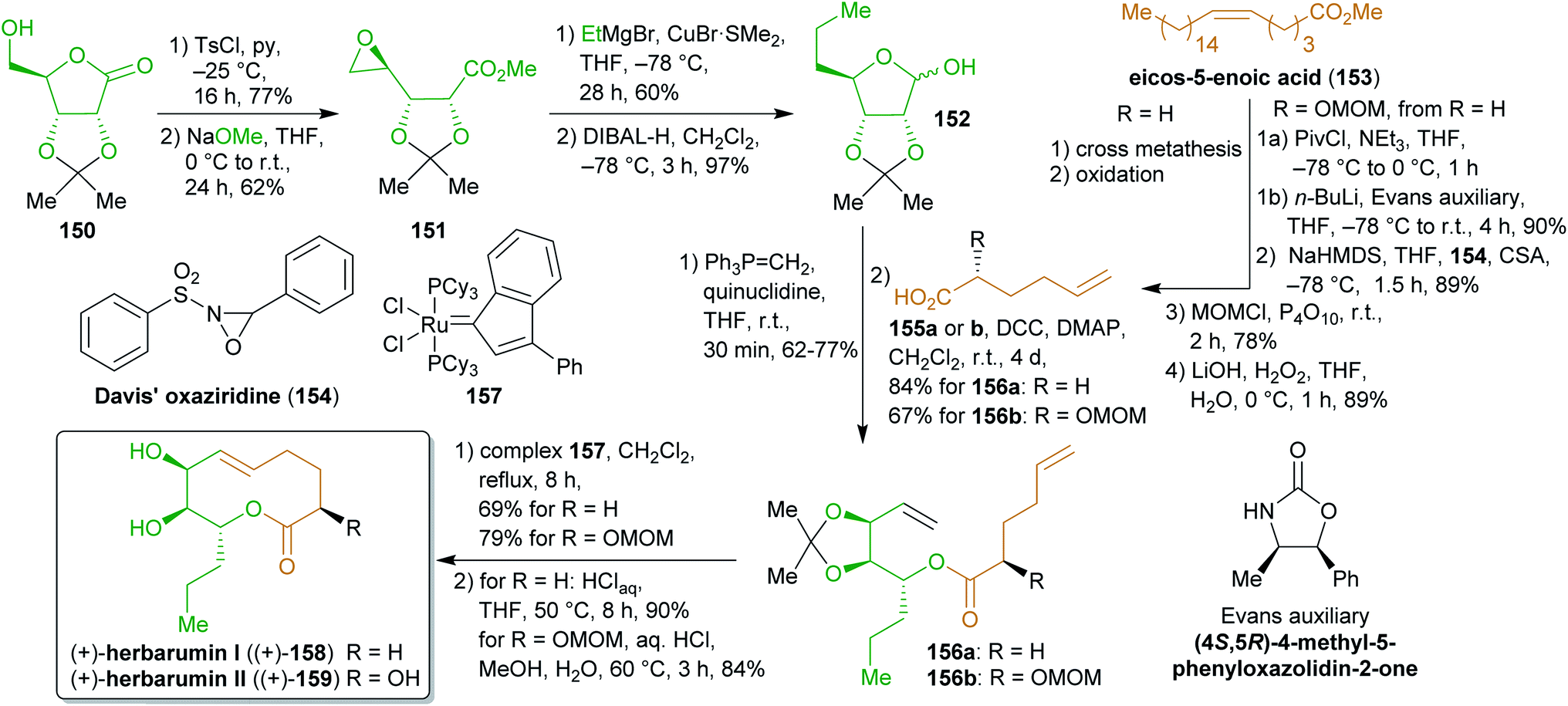 | ||
| Scheme 25 Synthesis of herbarumins from ribonolactone (117) and eicos-5-enoic acid (153). | ||
Although the solvents used during these syntheses are ecologically less favorable, the number of protecting group manipulations is minimal and makes this route a straightforward approach to two natural products from bio-based starting materials.
2.2.2.4. (−)-Jiadifenolide. Jiadifenolide (168), a neurotrophic sesquiterpenoid,284,285 was synthesized by Theodorakis et al.286 from cyclopentadienone (161) which is available from methyl levulinate (160) in a single step (Scheme 26).287 Allylation with allyl acetate and Michael addition to methyl vinyl ketone (MVK) furnished intermediate 162,288,289 which was converted to enantioenriched diketone 163via organocatalysis with D-prolinamide in high enantiopurity. Allyl acetate as an ester of allyl alcohol is available from glycerol217 while MVK is produced from acetone (available from wood by pyrolysis)290,291 and formaldehyde (available from methanol) via aldol condensation or Mannich reaction.292,293 Regio- and stereoselective reduction, silyl protection, carboxylation with methyl magnesium carbonate (MMC) and trapping with Meerwein's salt as well as methylation via the TMS-enolate gave intermediate 164 as a single isomer. MMC is made from magnesium methanolate and carbon dioxide and can therefore be considered to be renewable.294 Global reduction, TBS protection of the primary alcohol and oxidation of the secondary alcohol restored the carbonyl group, which was converted to the respective vinyl triflate to perform carbomethoxylation. After desilylation, spontaneous lactonization occurred to form 165, which was oxidized to the respective epoxide. Oxidative cleavage of the terminal olefin and oxidation of the resulting aldehyde triggered “6-exo-tet” epoxide opening to form the desired lactone 166 after TBS-deprotection. For this sequence, a direct RuIII-based oxidation of the terminal alkene to the carboxylic acid was investigated but was unsuccessful and led to decomposition. Therefore, the more circumstantial two-step sequence had to be used. Directed epoxidation and direct treatment of the acidic solution with DMP led to the α,β-unsaturated ketone, acid-catalyzed epoxide opening and transesterification to the thermodynamically favored 5-membered lactone. After hydrogenation of the double bond and TES protection, intermediate 167 was obtained. Next, the remaining carbonyl group should be transformed into a methyl group. This seemingly simple transformation proved challenging and the conventional methylenation approaches (Wittig reaction, Ti- and Zn-based) failed. Therefore, the vinyl triflate was prepared with Comins' reagent and Pd0-catalyzed cross coupling with AlMe3 (accessible from Al and iodomethane)295 gave the desired product. A final three-step one-pot sequence of hydrogenation, hydroxylation via the enolate and oxaziridine 154 as well as Jones oxidation gave (−)-jiadifenolide ((−)-168) in 1.5% overall yield over 25 steps, representing the first total synthesis of this natural product.
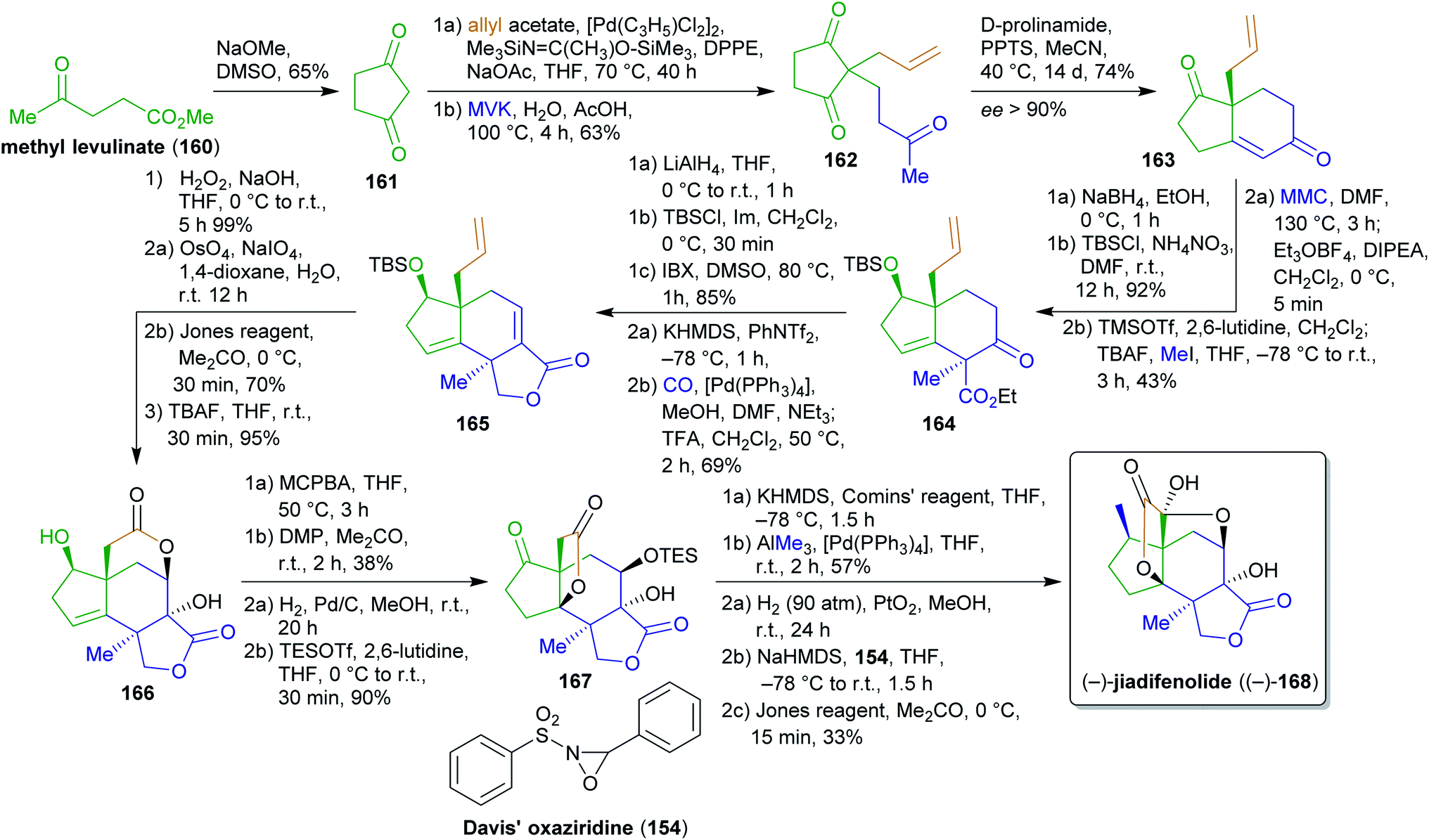 | ||
| Scheme 26 Synthesis of (−)-jiadifenolide ((−)-168) by Theodorakis et al. | ||
The foregoing synthesis is an excellent example of constructing a fairly complex and stereochemically demanding natural product from simple and bio-derivable building blocks. Inevitable detours had to be taken, which unfortunately decreased the eco-friendliness of the approach, yet the entire carbon backbone was constructed from renewable starting materials.
2.2.2.5. (+)-Chloriolide. In 2014, Schobert et al. synthesized the fungal 12-membered macrolide chloriolide (177)276 from levoglucosenone (115) and (+)-lactic acid via a Wittig-type macrocyclization (Scheme 27).277 Starting from 115, ketone reduction, acetal hydrolysis and acetonide protection led to alcohol 169, which was converted to the C1-homologated aldehyde 170via transformation into a leaving group, nucleophilic substitution with cyanide and reduction. Sodium cyanide is currently produced by the Andrussow process from methane and ammonia and subsequent reaction with lye.278,279 Considering that synthetic natural gas and methane derived from agricultural waste and manure are widely established concepts,280 even methane-derived NaCN could be produced on the basis of renewables. Another route to biomass-based HCN has been mentioned earlier.281 The cumulated ylide 175 which is prepared from the respective alkoxycarbonyl-methylenephosphorane and therefore from an α-haloacetic acid derivative282,283 was used to synthesize ylide ester 176 after TBS deprotection. Acetonide cleavage and Wittig-cyclization gave enantiopure (+)-177. The presented work makes use of simple chiral bio-based starting materials to build a fairly complex natural product.
 | ||
| Scheme 27 Synthesis of (+)-chloriolide ((+)-177) from levoglucosenone (115). | ||
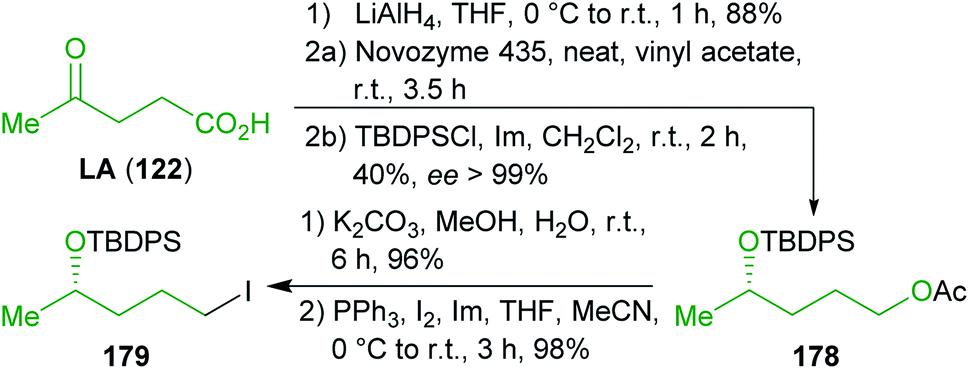 | ||
| Scheme 28 Synthesis of levulinic acid (122) derived intermediate 179 for the synthesis of aspergillides A (187) and B (186). | ||
2.2.2.6. (−)-Aspergillides. For a formal synthesis of the cytotoxic aspergillides A and B (186 and 187)296–298 Loh and Koh selected 5-HMF (119) and levulinic acid (122) as biomass-derived starting materials (Scheme 29).299 The tetrahydropyran moiety was synthesized from 119 which was benzyl protected and subjected to an aldol reaction. Oxidation of the racemic product and asymmetric transfer hydrogenation furnished β-hydroxyester 180. Although the non-stereoselective aldol reaction/oxidation/asymmetric reduction sequence does not look very eco-friendly at first glance, it turned out to be the only suitable way to access enantiopure 180. Asymmetric Mukaiyama aldol reactions as well as dynamic enzymatic kinetic resolutions of racemic 180 were attempted but neither of them provided the desired outcome in terms of yield and enantiomeric excess.
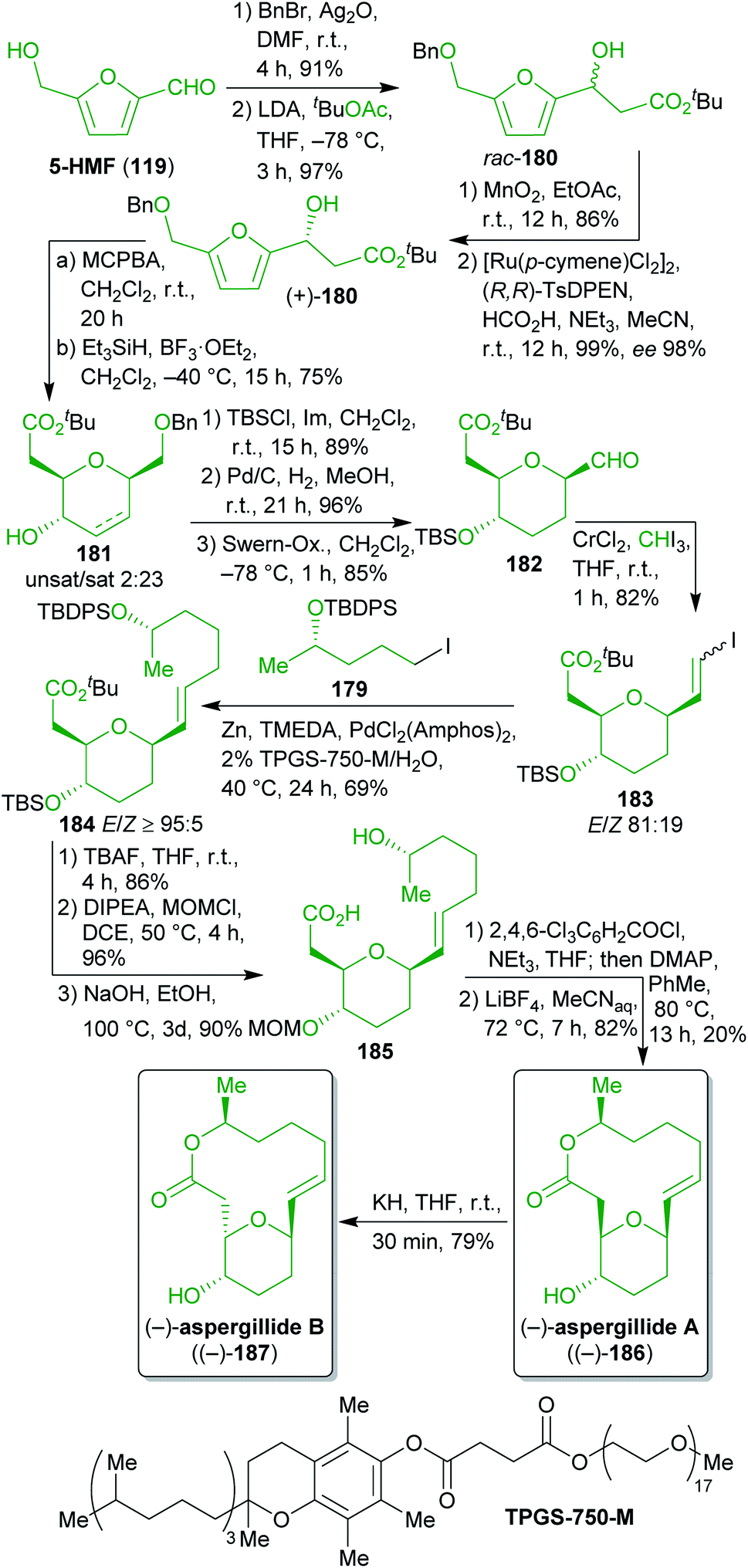 | ||
| Scheme 29 Synthesis of aspergillides A ((−)-187) and B ((−)-186) from 5-HMF (119) and LA (122). | ||
Achmatowicz rearrangement and reductive deoxygenation gave a mixture of dihydro- and tetrahydropyran 181. After O-protection, hydrogenation removed the double bond and the benzyl group. The resulting primary alcohol was subjected to Swern oxidation and the following Takai iodoolefination (iodoform is e.g. available from the reaction of ethanol with I2 in alkaline medium)300 furnished the first 5-HMF-derived building block 183.
On the other hand, levulinic acid (122) was reduced to the respective racemic diol, which was subjected to dynamic enzymatic kinetic resolution and the desired enantioenriched alcohol was TBDPS protected to obtain 184. Ester hydrolysis and iodination furnished the second building block 179 in excellent yield and ee (Scheme 28). Both building blocks were coupled by means of a Neigishi reaction with high selectivity in favor of the desired E-isomer. An eco-friendly micelle-based variant using the commercially available amphiphile TPGS-750 M301 in water and in situ-generation of the organozinc reagent was employed.302–304 After cleavage of the silyl protecting groups, a MOM group was installed and the ester was saponified to yield seco-acid 185 which was already used by Fuwa et al. as an intermediate and was converted to (−)-186 by Yamaguchi esterification.305 Epimerization with potassium hydride gave (−)-187.306
Throughout this remarkable sequence, a number of steps were intentionally conducted in accordance with the principles of “green” chemistry. As already pointed out, the Neigishi coupling of 183 and 179 is an example of “greener” chemistry and the dynamic enzymatic kinetic resolution also ensures a high overall yield.
2.2.2.7. (−)-Bissetone and (−)-palythazine. For the first total syntheses of the marine natural products bissetone (190)307 and palythazine (193)308 Lichtenthaler and coworkers309–311 used glucose as a biomass-derivable starting material in a class-transcendent fashion (Scheme 30). Both sequences proceed via the dihydropyranone 189, which can be prepared from D-glucose on a molar scale in six steps via the hydroxyglucal ester 188.312–314 Addition of the lithium enolate of (potentially xylochemical) acetone and subsequent protecting group cleavage furnished enantiopure (−)-bissetone ((−)-190). The nucleophilic attack is favored from the axial side and therefore leads to the product with the desired relative configuration.
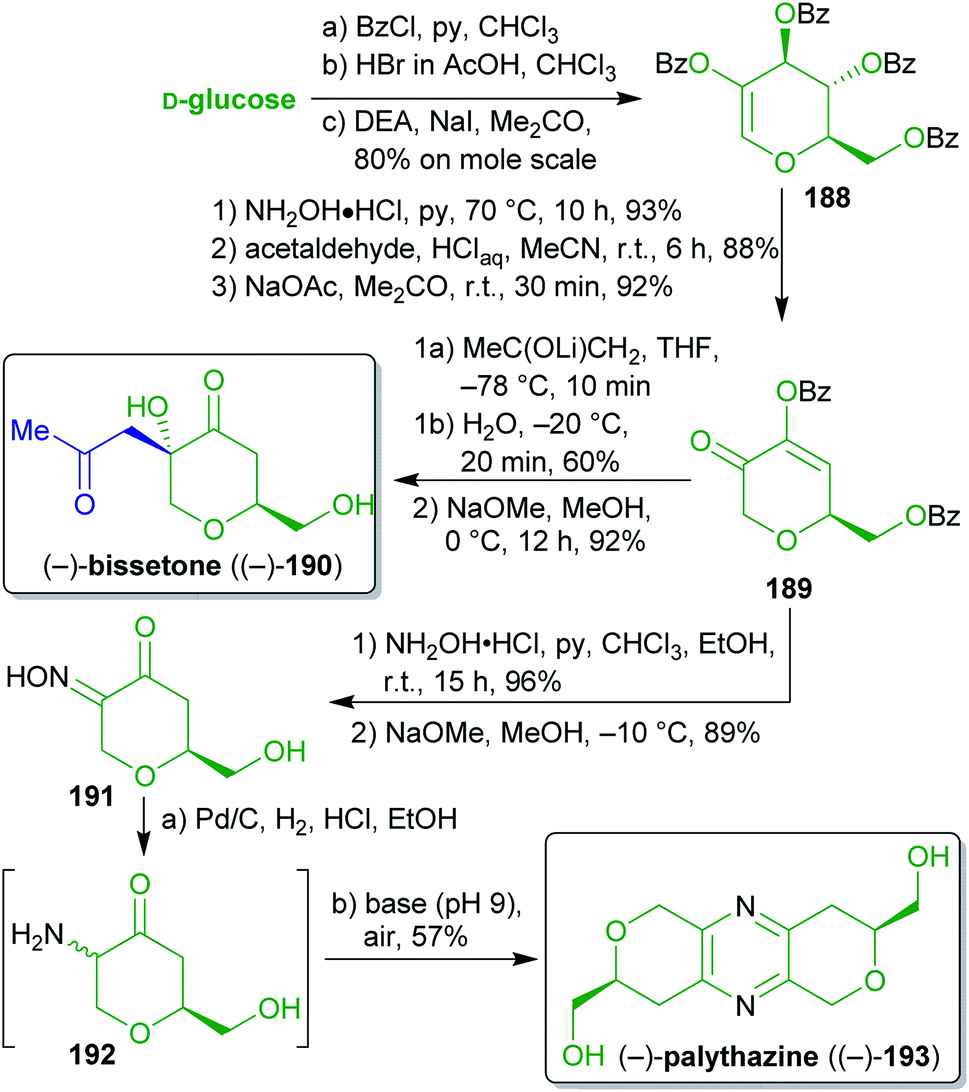 | ||
| Scheme 30 First total syntheses of (−)-bissetone ((−)-190) and (−)-palythazine ((−)-193) from glucose. | ||
For the synthesis of (−)-193, the carbonyl group of 189 was converted into the oxime 191 which spontaneously underwent oxidative dimerization into enantiopure (−)-palythazine ((−)-193) upon exposure to air after global deprotection and oxime reduction. The absolute configuration of (−)-palythazine ((−)-193) was proven through this stereospecific synthesis. These concise and straightforward syntheses make excellent use of biomass-based D-glucose as an enantiopure starting material (ex-chiral pool strategy) to prepare non-carbohydrate natural products. Acetone290,291 was used as a potentially renewable C3-synthon. Additionally, in contrast to classic carbohydrate chemistry, only a single protecting group transformation was performed throughout the entire sequence.
2.2.2.8. (+)-Castanospermine. Mootoo and coworkers315,316 utilized the hydroxyl substitution pattern of D-glucose for the synthesis of polyhydroxyindolizidine alkaloid (+)-castanospermine ((+)-198).317 The synthesis of this plant-derived natural product commenced with the allylation of aldehyde 194 which is readily available from glucose (Scheme 31).318–320 The desired threo-epimer was the major product (9
:1 selectivity) and could be separated by chromatography. After benzylation, treatment with iodonium dicollidine perchlorate (IDCP), reductive elimination and Swern-oxidation furnished ketone 196. Ozonolysis and acetal hydrolysis gave key tricarbonyl intermediate 197a in the lactol form 197b, which was subjected to triple reductive amination. During this reaction only 5% of the undesired C8a-epimer was formed. Global deprotection furnished enantiopure (+)-castanospermine ((+)-198) with a high yield of 22% over nine steps starting from 194.
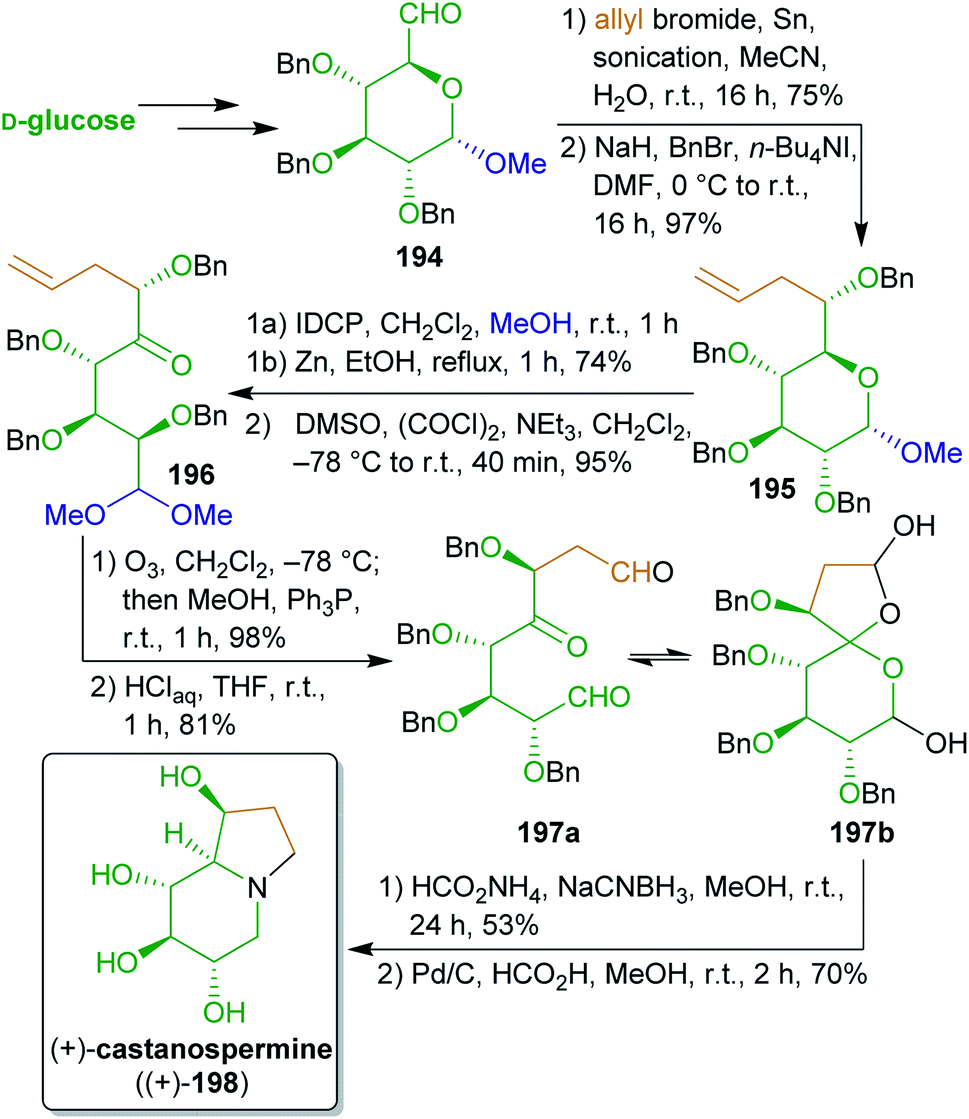 | ||
| Scheme 31 Synthesis of polyhydroxindolizidine alkaloid castanospermine (198). | ||
This remarkable work makes formidable use of the innate stereoinformation of bio-based glucose to synthesize this stereochemically demanding natural product.
2.3. Chitin and chitosan
Chitin 199 and chitosan 200 are the chemical constituents of the exoskeleton of crustaceans and insects as well as of cell walls of molluscan organs and fungi. This makes it the second most abundant biopolymer (after cellulose)321 and the most abundant nitrogen containing biopolymer on earth.322 Similar to the structure of cellulose, chitin is a linear polysaccharide, but unlike cellulose it is composed of β(1 → 4)-linked 2-acetamido-2-deoxy-D-glucopyranose (N-acetylglucosamine, GlcNHAc 201) monomers (Scheme 32). As depicted in Scheme 32, the C-2-substituent of the sugar-derived monomer is either an acetamido- or a free amino group. The term chitin refers to the material with >50% of acetylated amino groups while the material with a lower degree of N-acetylation is called chitosan.323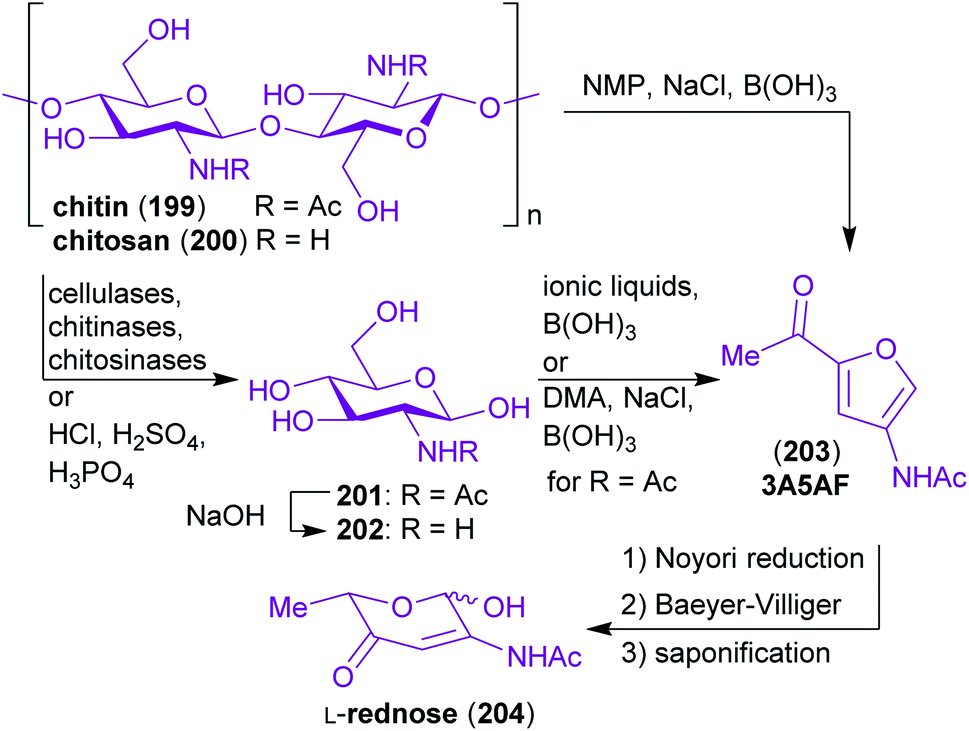 | ||
| Scheme 32 The chemical structure of chitin/chitosan and examples of starting materials derived thereof. | ||
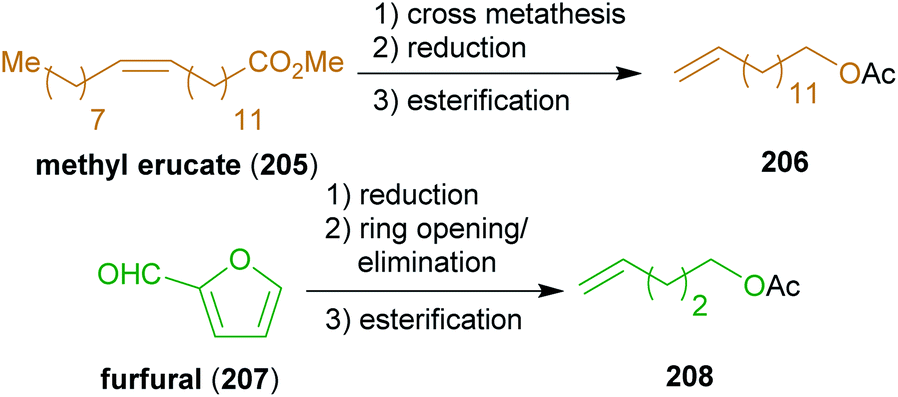 | ||
| Scheme 33 Schematic synthesis of terminal olefins 206 and 208 for the synthesis of rhizochanilin C (217). | ||
The natural material in the form of shrimp, crab or lobster shells has to be digested by grinding, deproteinization, demineralization, discoloration and drying prior to depolymerization.333 The prepared chitin and chitosan can be cleaved directly into the corresponding amino sugars N-Ac-glucosamine (GlcNHAc, 201) or 2-amino-2-deoxy-glucose (GlcNH2, 202) either enzymatic or with inorganic acids (Scheme 32).324,334–337 GlcNHAc can be deacetylated to yield GlcNH2 or be converted to two highly promising N-containing starting materials that are challenging to make from other resources.
The furan derivative 3-acetamido-5-acetylfuran (3A5AF, 203) can easily be prepared from 201 in a single step338–340 or directly from 199.338,341 3A5AF (203) can in turn be converted to L-rednose (204), which contains a β-aminoenone moiety rarely seen in carbohydrates, in a three step sequence.342
2.3.2.1. (−)-Rhizochalinin C. Molinski's and Ko's synthesis of rhizochalinin C (217)343,344 is a good example for the potential of the combinatorial use of biomass derived starting materials.345 Their approach to construct this L-threo, α,ω-bifunctionalized sphingoid base combines the use of D-glucosamine from chitin/chitosan as an element of the chiral pool for extracting the stereocenters and fats/oils as well as hemicellulose derived starting materials to build the largest part of the carbon backbone. The key achievement of this work is the conversion of 202 into the useful synthons for L-threo sphingoid bases 209 and 210 by a sequence starting with a Barbier reaction with allyl bromide, followed by N-protection, glycol cleavage, reduction and diol protection (Scheme 34). The allyl bromide used for the Barbier reaction is derivable from glycerol which is produced on a megaton scale by hydrolysis of fats and oils346 through allyl alcohol as an intermediate.217,347
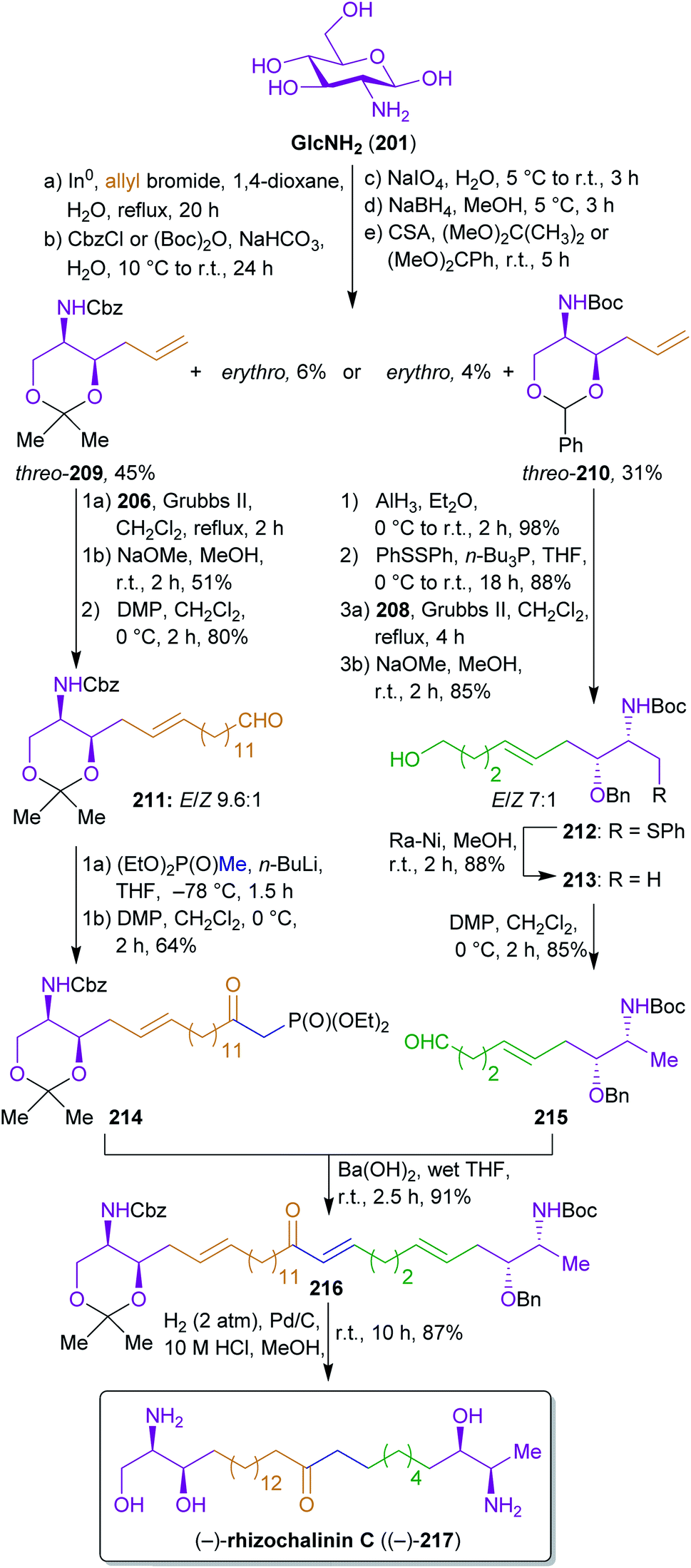 | ||
| Scheme 34 Synthesis of sphingoid base synthons and (−)-rhizochanilin C ((−)-217) from 202. | ||
The respective threo-diastereomers were subjected to olefin metathesis and further functionalized to furnish the respective Western 214 and Eastern halves 215, which were coupled by a Horner reaction. Prior to the olefin metathesis, a switch of the protecting groups was conducted to prepare the required deoxygenation.
The reactant for cross-metathesis with threo-209, tetradec-13-enyl acetate (206), can be easily obtained by cross metathesis of methyl erucate (205) with ethylene,348,349 followed by reduction and esterification (Scheme 33). Likewise, the olefin 208 could be prepared from hemicellulose via furfural 207 which is hydrogenated to tetrahydrofurfuryl alcohol.350 A ring opening/elimination sequence gave pent-4-ene-1-ol351 which was esterified to 208. The synthesis of the phosphonate 214 is conducted with diethyl methylphosphonate which in turn can be synthesized from methanol and triethyl phosphite.352 Hydrogenation under strongly acidic conditions ensured global deprotection and saturation of the Horner product 216 in a single step to furnish stereopure (−)-rhizochanilin C ((−)-217). This synthesis makes excellent use of a variety of sustainable building blocks to construct a rather demanding natural product.
Unfortunately, frequently used methods such as glycol cleavage, cross metathesis or Horner reactions are not very atom economic.
2.3.2.2. (−)-Pochonicine. The total synthesis of fungal polyhydroxylated pyrrolizidine (−)-pochonicine hydrochloride ((−)-226·HCl)353 from GlcNHAc (201) by Takahashi et al.354 is one of the numerous examples where the originally proposed absolute configuration of the natural product had to be revised based on synthetic work.
Starting from monothiolacetal 218, which was prepared from 201,355 the configuration of the hydroxyl group at C-3 and the N-protecting group were switched (Scheme 35). Reductive acetal opening and nucleophilic attack of the nitrogen at C-5 led to pyrrolidine 221. After debenzylation and oxidation, a nucleophilic allylation was performed through attack of a Grignard reagent. Both diastereomers (threo- and erythro-222) were formed in this step. The synthesis of (−)-pochonicine ((−)-226) was conducted with the erythro-isomer but the threo-isomer was also advanced to obtain the respective C-1 and C-3 epimers of 226. The structure shown in Scheme 35 corresponds to the originally proposed configuration353 but comparison of the analytical data proved to be inconsistent with that of the natural product. Dihydroxylation of the terminal olefin again led to a mixture of diastereomers 224, whereas the 7-(S)-epimer led to (−)-226 and the 7-(R)-epimer to the respective C-3-epimer (not shown). After conversion to the hydrochloride, the observed optical rotation of the synthetic product proved to be opposite to the one of the isolated compound.
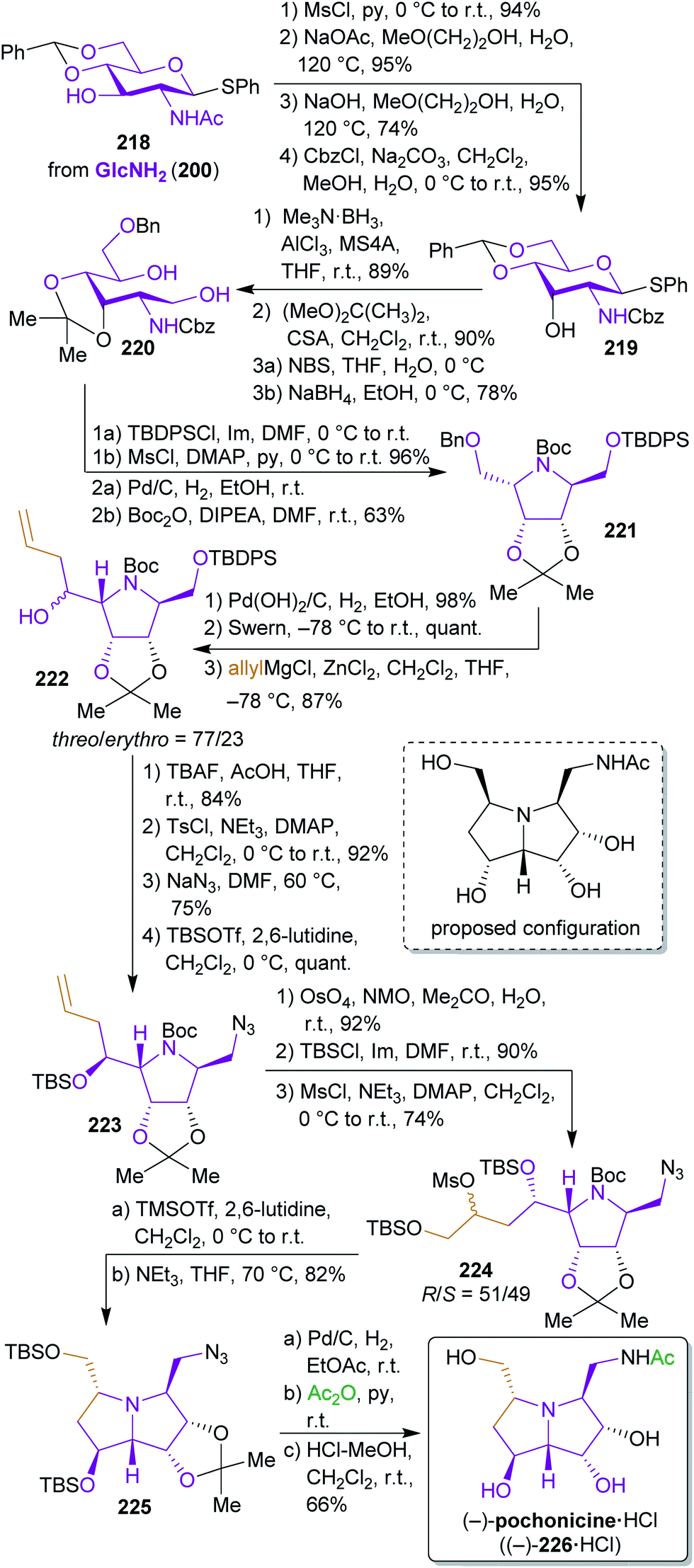 | ||
| Scheme 35 Synthesis of (−)-pochonicine hydrochloride ((−)-226·HCl) from GlcNHAc. | ||
In this noteworthy work, four different diastereomers were synthesized from two common precursors to clarify the absolute configuration of this stereochemically demanding natural product. Unfortunately, the use of protecting group chemistry was inevitable due to the high degree of functionalization of the intermediates and final product.
2.3.2.3. (−)-Allosamizoline. As mentioned earlier, a major advantage of bio-based starting materials is the often high degree of functionalization, in particular with respect to stereogenic centers. Therefore, GlcNHAc and GlcNH2 are ideal templates for the synthesis of stereochemically demanding natural products. Several groups have utilized GlcNH2 (201) in this respect for the synthesis of (−)-allosamizoline ((−)-231),356–361 a bacterial pseudo-aminosugar with chitinase inhibitory activity.362–365 For this potentially biomimetic approach,366 two main routes emerged for the crucial intramolecular ring closure to form the cyclopentane ring (Scheme 36). While Simpkins and Whittle used a radical ring closure of thiocarbamate 228 and an oxime ether 229,358,359 the Tatsuta and Kitahara groups opted for an oxidative cycloaddition to generate isoxazoline intermediate 235.356,360
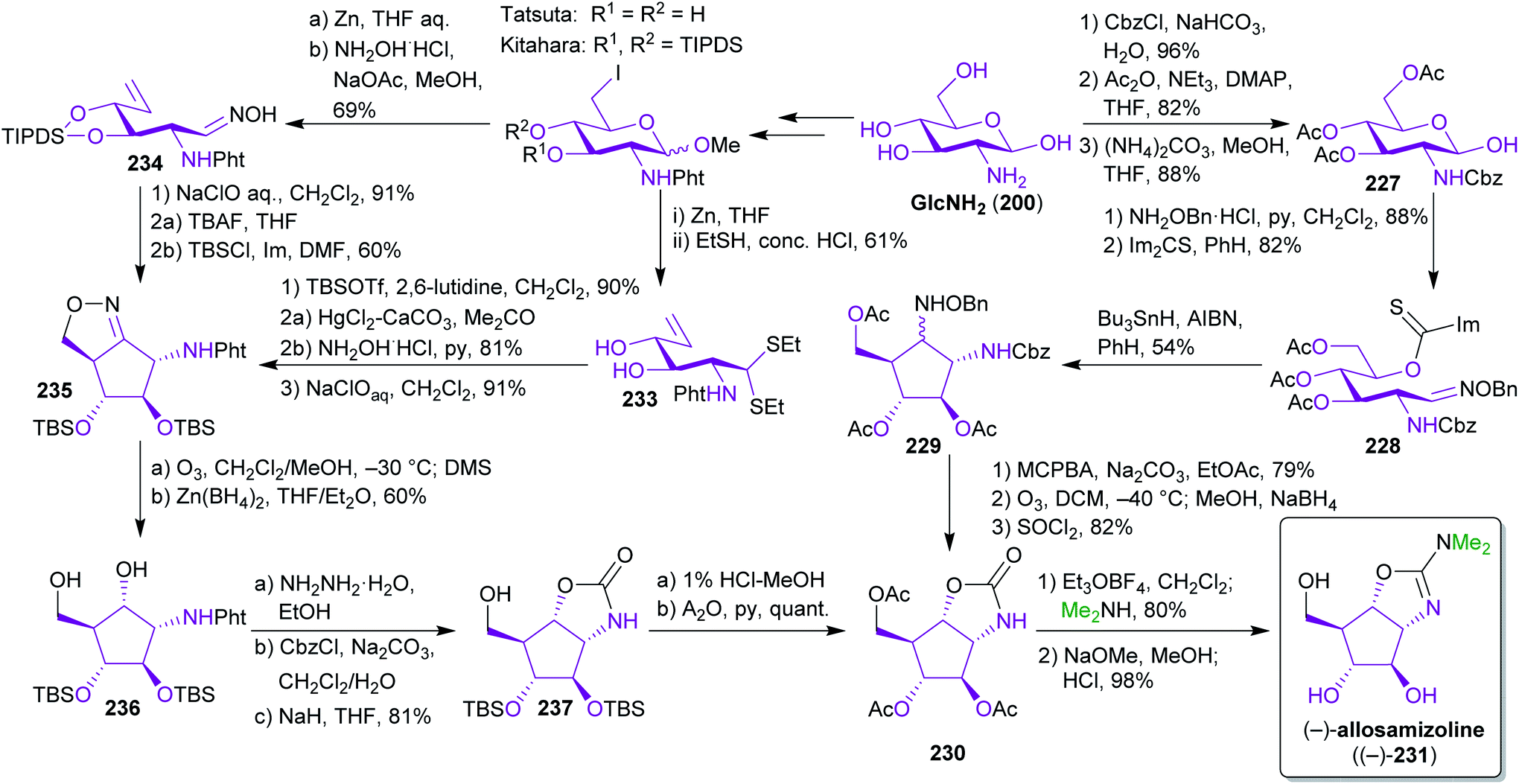 | ||
| Scheme 36 Synthesis of (−)-allosamizoline ((−)-231) from GlcNH2. | ||
All three ways converge into the same carbamate intermediate 230 and they rely on the same carbon sources to construct 231. This work highlights how simple biomass-based building blocks, namely GlcNH2 and dimethylamine (available from methanol and ammonia)367 can be transformed into valuable natural products without the use of fossil carbon sources.
2.3.2.4. Proximicin A. The nitrogen-containing furan derivative 3A5AF (203) was utilized by Sperry and coworkers for a proof-of-concept synthesis of proximicin A (241),368 a potential chemotherapeutic compound.369,370 It is the first natural product synthesis that uses this unique bio-based starting material, which appears to be perfectly suited for the synthesis of 241. Proximicin A was produced in a seven-step sequence from chitin via the key intermediates 238 and 239 (Scheme 37) simultaneously obeying the fundamentals of green chemistry (Scheme 38). In this remarkable work, only “green” solvents ([BMim]Cl – 1-butyl-3-methylimidazolium chloride, MeOH, H2O, DMC) and procedures were used while it still compared well with former “non-green” syntheses of 241 in terms of step count and yields.369,370 The non-toxic dimethyl carbonate (DMC), which can be made directly from methanol and carbon monoxide in the presence of CuCl and oxygen,371–373 was used for the introduction of the carbamate moiety374 instead of the conventional methyl chloroformate. Furthermore, the amide coupling of the intermediates 238 and 239 was conducted with the uronium-based coupling reagent COMU375((1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate) instead of the conventional, potentially explosive 1-hydroxybenzotriazole derivatives (e.g. TBTU, HATU, HBTU).
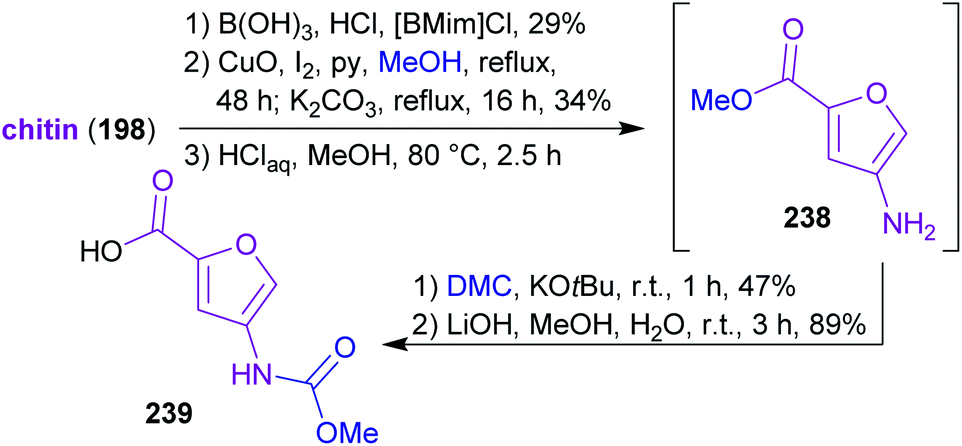 | ||
| Scheme 37 Synthesis of key intermediates 238 and 239 for proximicin A (241) from chitin via 3A5AF (203). | ||
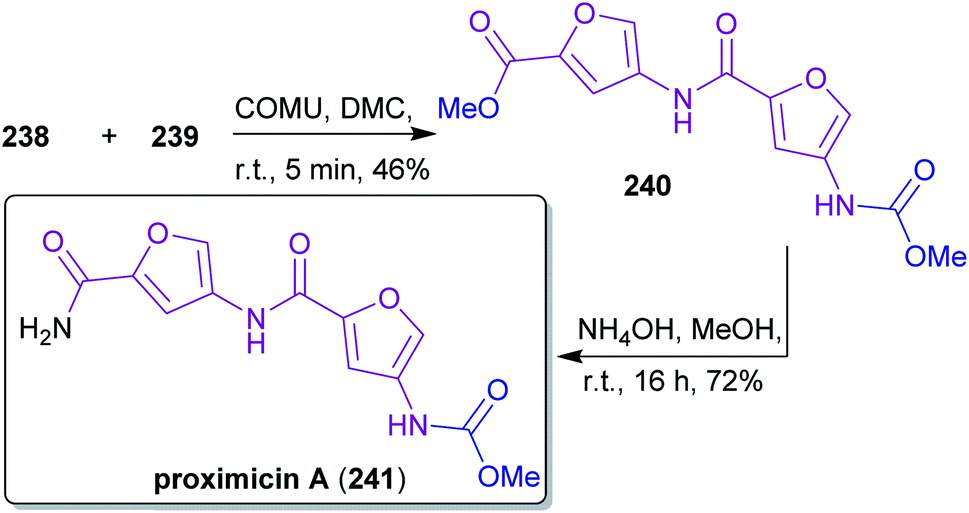 | ||
| Scheme 38 Closing amide coupling and amide synthesis for proximicin A (241). | ||
2.4. Fats and oils
Fats and oils, either of plant or animal origin, occur in the form of tri-, di- and monoglycerides with varying compositions of fatty acids depending on their origin. The annual production reaches almost 200 Mt and has been increasing over the last decades.376,377 Although this figure indicates a very large industrial product class, one has to keep in mind that the largest fraction of this amount is used for food and feed. Furthermore, with the advent of biodiesel, the ratio of utilization of fats and oils for food/feed versus industry was slowly shifting from 86:14 to 80:20 over the past few decades.378 If this ethical dilemma could be solved, fats and oils would represent a structurally ideal renewable feedstock not only for the production of chemical raw materials and synthetic chemistry but also for fuel and polymer production.
After saponification (the production of soap in this process is the origin of this word), the free fatty acids can be processed in various ways leading to different raw materials depending on the chain length, the degree of unsaturation and the position of the double bonds of the fatty acid. Glycerol is produced on a large scale through saponification of fats and oils346 and e.g. can serve for the production of glyceraldehyde,386,387 allylic alcohol,217 acrolein,388 and acrylic acid179 as well as of polymers of the latter. The main methods to valorize free fatty acids for chemical production are esterification, reduction, allylic oxidation, addition, dihydroxylation, epoxidation, cycloaddition, metathesis and ozonolysis. The latter two methods permit the simple variation of the length of the carbon chain. For example, erucic acid (docos-13-enoic acid), which is very abundant in rapeseed and mustard oil,389 can be converted to brassylic acid (tridecanedioic acid) derivatives by ozonolysis390 or to 13-tetradecenoic acid derivatives by olefin metathesis (see the synthesis of rhizochalinin, Scheme 34).348,349 Likewise, 9-decenoic acid is available from the abundant oleic acid391,392 whereas meadowfoam seed oil, rich in eicos-5-enoic acid (153),393,394 can be converted to 1-pentadecanal (276) by ozonolysis.395 In natural product synthesis, starting materials derived from fats and oils usually serve for the introduction of alkyl chains and generally have to be combined with other building blocks to introduce heteroatoms and stereoinformation.
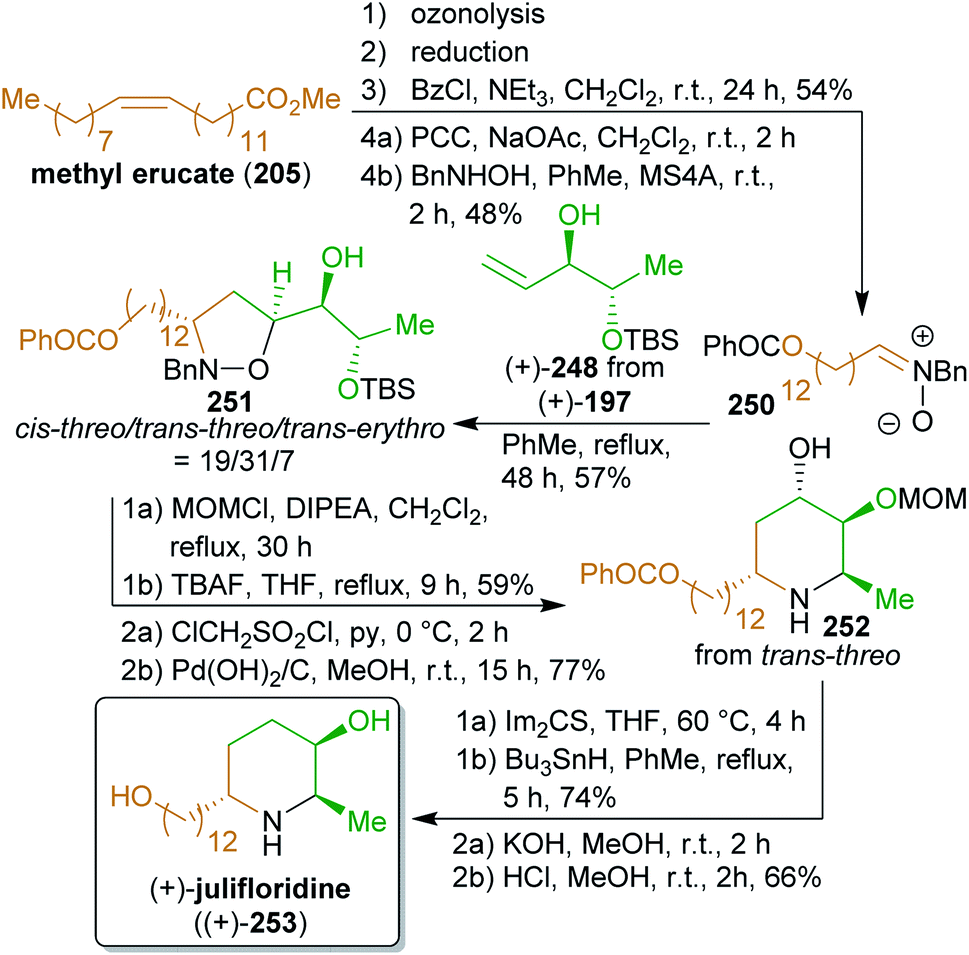
2.4.2.1. (+)-Azimic acid and (+)-julifloridine. Following this approach, the Naito group has accomplished the total synthesis of two 3-piperidinol alkaloids, (+)-azimic acid ((+)-249) and (+)-julifloridine ((+)-253),396,397 using methyl lactate (243) as the chiral template and eicos-5-enoic acid (153) as well as erucic acid derived building blocks (Schemes 39 and 40). Both enantiomers of lactic acid are readily available by fermentation of almost any type of carbohydrate biomass,398 whereas vinyl bromide is accessible from ethanol via ethylene (the so-called bio-ethylene) and 1,1- or 1,2-dibromoethane.
 | ||
| Scheme 39 Synthesis of (+)-azimic acid ((+)-249). | ||
 | ||
| Scheme 40 Synthesis of (+)-julifloridine ((+)-253). | ||
Methyl esters (+)-321 and (−)-321 were converted to the respective enantiopure olefins ((+)-245 and (−)-245)399 which served as the dipolarophiles in a 1,3-dipolar cycloaddition with the nitrones 242 and 250. Both are accessible from fatty acid starting materials. The terminal olefin obtained from cross-metathesis of methyl ester of 153 with ethylene349 can be converted to 242 by hydroformylation400,401 and reaction with N-benzylhydroxylamine. Ozonolysis of methyl erucate (205) and reduction of the product gives tridecan-1,13-diol, which was converted to nitrone 250. The cycloadditions produced three different diastereomers, of which the cis-threo- and the trans-erythro-form could be converted to (+)-249.
Because of the different configuration of julifloridine (253), only the trans-threo diastereomer (shown in Scheme 40) was converted to (+)-253. The foregoing work accomplished a very step-efficient synthesis of two stereochemically challenging natural products from simple building blocks.
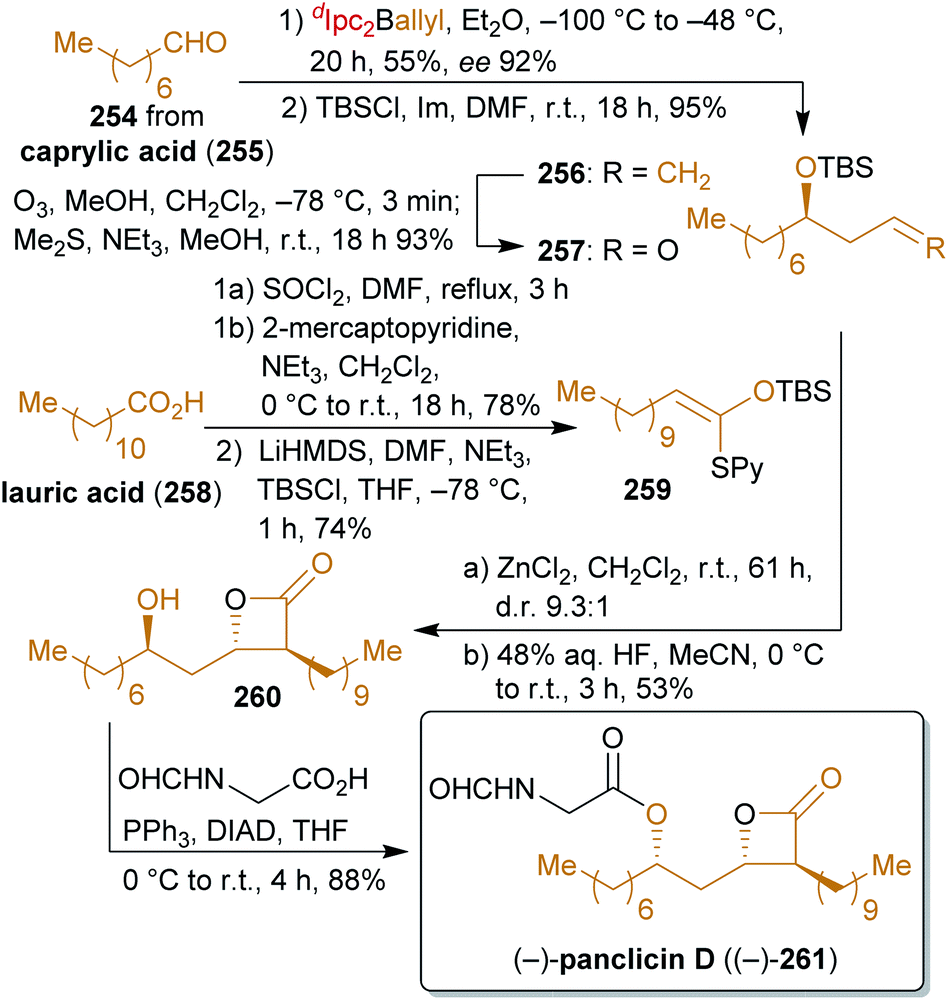
2.4.2.2. (−)-Panclicin D. Romo's concise synthesis402,403 of the β-lactone panclicin D (261) with pancreatic lipase inhibiting activity404,405 exclusively uses fat- and oil-derived starting materials in the form of lauric acid (258) and caprylic acid (255) as the main carbon sources as well as allyl diisopinocampheylborane as a terpene/glycerol-derivable chiral allylating agent (Scheme 41). While lauric acid is very abundant in laurel oil (hence the name), in coconut milk and oil as well as in palm kernel oil, caprylic acid can also be found in coconut oil, albeit in much lower concentration.406,407 Caprylic acid can be produced by de novo synthesis from bio-engineered yeast or E. coli strains408–410 or by anaerobic microbial fermentation from ethanol and acetate via chain elongation.411,412
 | ||
| Scheme 41 Synthesis of (−)-panclicin D ((−)-261) by Romo et al. | ||
Lauric acid is converted to the ketene thioacetal 259 which is subjected to a tandem Mukaiyama aldol-lactonization with aldehyde 257 to form the β-lactone ring. For the synthesis of 257, methyl caprylate is converted to octanal (254)413 followed by enantioselective addition of an allyl group, protection and ozonolysis. After desilylation, N-formyl glycine was attached by a Mitsunobu reaction to furnish (−)-panclicin D ((−)-261). The use of little functionalized starting materials for the formation of this interesting natural product makes this synthesis remarkable.
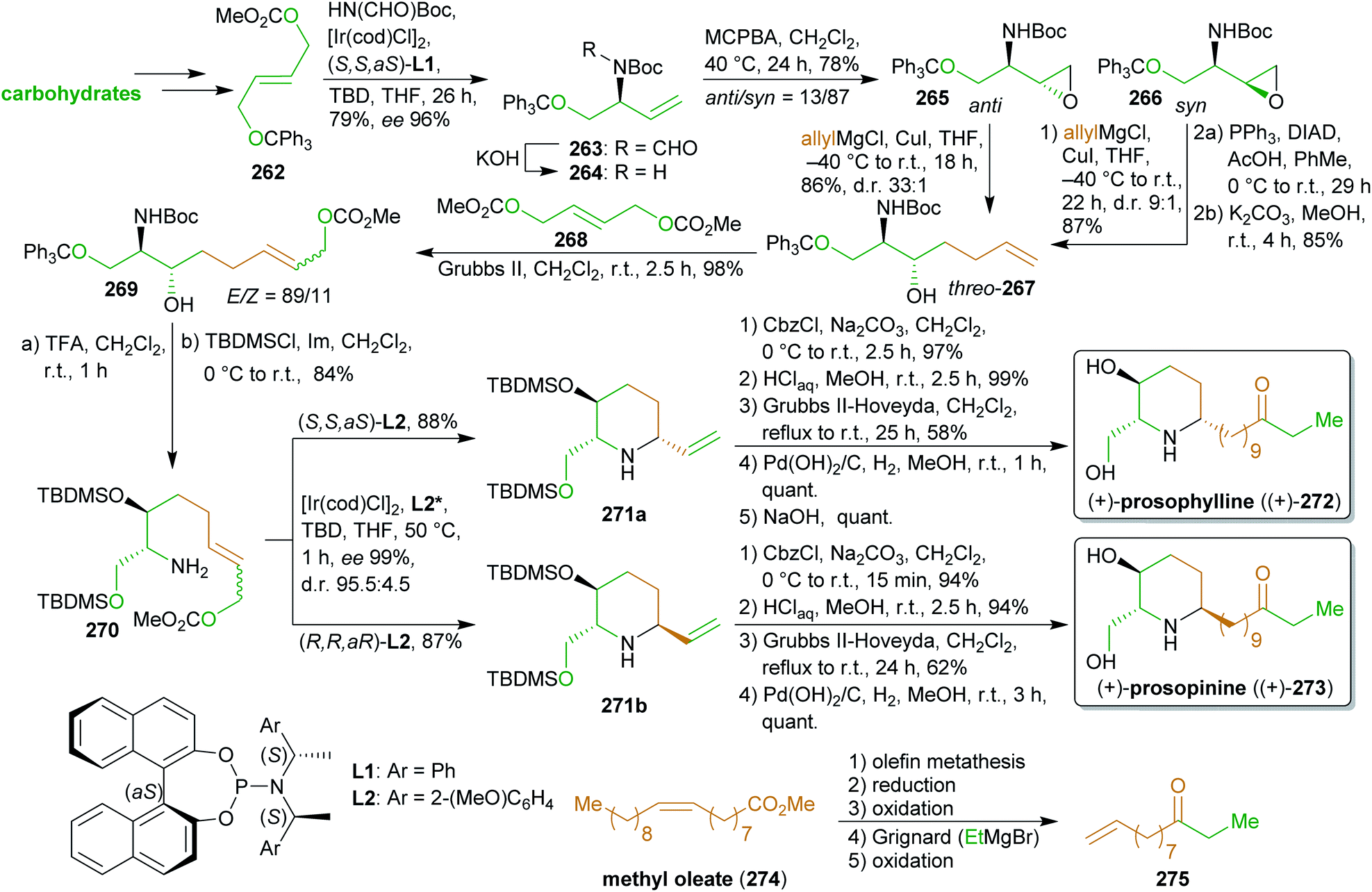
2.4.2.3. (+)-Prosophylline and (+)-prospinine. The antibiotic piperidine alkaloids (+)-prosophylline ((+)-272)414 and (+)-prosopinine ((+)-273)415,416 were synthesized by Helmchen et al. via an iridium-catalyzed allylic substitution (Scheme 42).417,418 This key reaction was used twice during the synthesis to introduce and modify the required stereochemistry. The first application of this methodology was the conversion of the trityl-protected carbonate 262 available from dimethyl fumarate419 to allylic amine 264. Dimethyl fumarate can be obtained from malic acid or furfural (207),420–422 both available from cellulose. After epoxidation, Grignard reaction and cross metathesis with a biscarbonate likewise accessible from dimethyl fumarate, the nitrogen was deprotected to enable the ring closure, while the free hydroxy group was protected. Both diastereomers from the epoxidation reaction were convergently converted to the same threo-amino aclohol 267 with the desired configuration. Again, allylic substitution with both enantiomers of the chiral ligand (L2) led to the respective piperidine diastereomers 271a, b with excellent ee and high dr which is the result of extensive optimization studies. N-protection, olefin metathesis with dodec-1-en-10-one (275), hydrogenation and Cbz-removal led to the desired natural products 272 and 273. Ketone 275
423 is produced from the cross metathesis product of methyl oleate (274) with ethylene.275
 | ||
| Scheme 42 Synthesis of (+)-prosophylline ((+)-272) and (+)-prospinine ((+)-273). TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene. | ||
This synthesis is characterized by the use of powerful methodology for the catalytic introduction of chiral information in combination with the utilization of various sustainable carbon sources to produce two alkaloids through a common precursor.
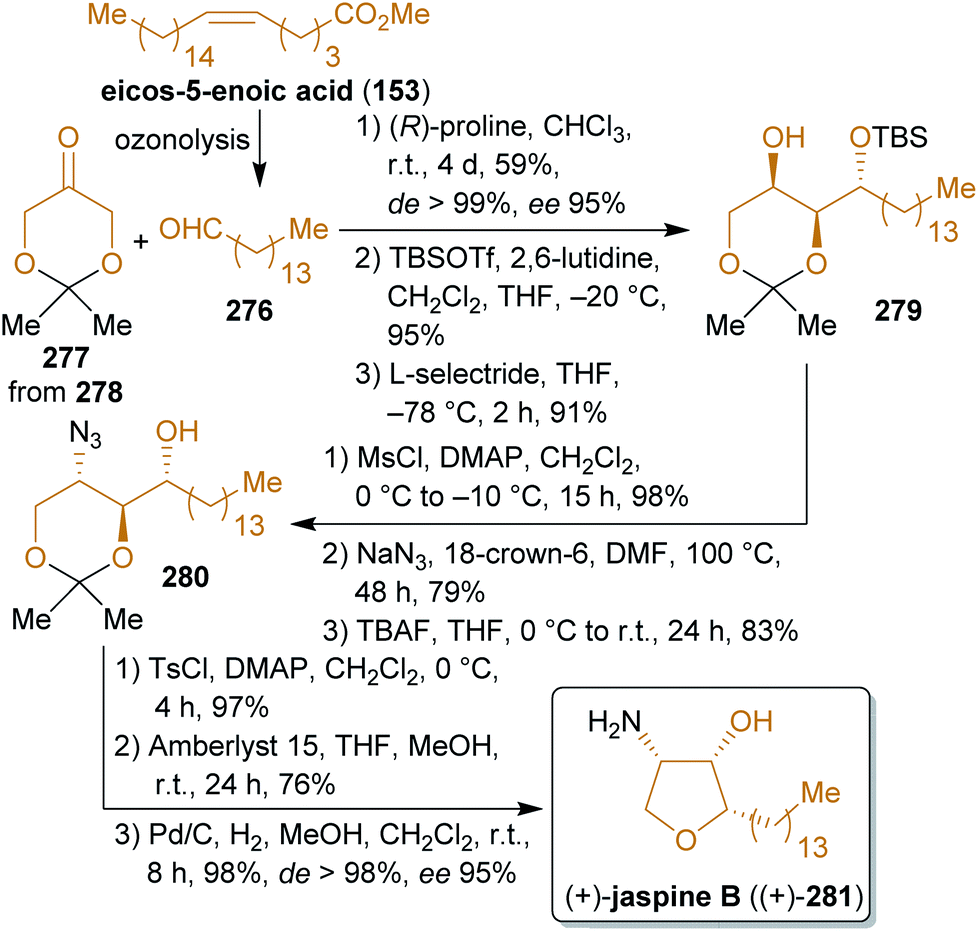
2.4.2.4. (+)-Jaspine B. The cytotoxic marine natural product jaspine B (281) received great attention since its isolation and elucidation in 2002
424,425 and numerous synthetic approaches have been published in the past decade426–436 due to its remarkably low IC50-values against a variety of tumor cell lines.424 Among these syntheses, the concise procedure of Enders and coworkers stands out, which starts from protected dihydroxyacetone 277 and 1-pentanal (276). Dihydroxyacetone (278) is produced on a large scale from glycerol by fermentation.437 The acetonide 277 is subjected to an organocatalyzed aldol reaction with 276 (Scheme 43). After stereoselective reduction of the keto group, the generated hydroxy group was converted to an azide moiety under inversion of the configuration. The same sequence was used for the ring closure to furnish the tetrahydrofuran ring with the required configuration. After hydrogenation of the azide group, (+)-jaspine B ((+)-281) was obtained in high stereoselectivity.
 | ||
| Scheme 43 (+)-Jaspine B ((+)-281) synthesis. | ||
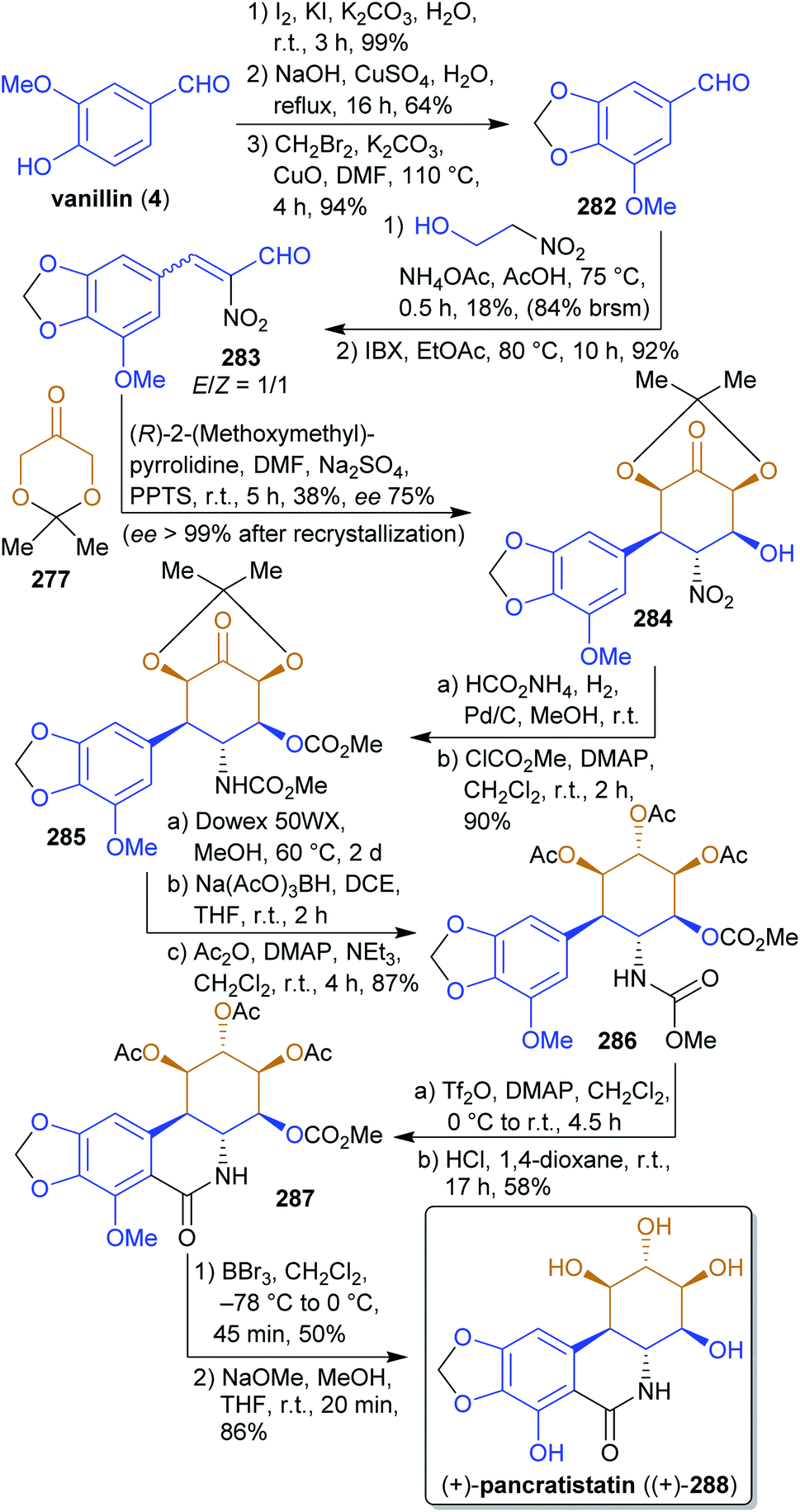
2.4.2.5. (+)-Pancratistatin. The antiproliferative isocarbostyril alkaloid pancratistatin (288)438,439 was synthesized by Alonso et al.440 from 277 and the xylochemical vanillin (4). The correct stereochemistry is established by the organocatalyzed reaction of 277 with nitroenal 283
441 during which five stereocenters are formed (Scheme 44). Nitro group reduction, carbamate formation and reduction of the keto group gave the precursor 286 for the Bischler–Napieralski reaction, after which global deprotection led to (+)-pancratistatin ((+)-288). 2-Nitroethanol can be considered a xylochemical since it is produced from nitromethane (itself available in a single operation from methanol)442 and formaldehyde.443 The presented approach is very straightforward and uses renewable inexpensive starting materials as well as simple synthetic methods.
 | ||
| Scheme 44 Total synthesis of (+)-pancratitstatin ((+)-288) by Alonso et al. | ||
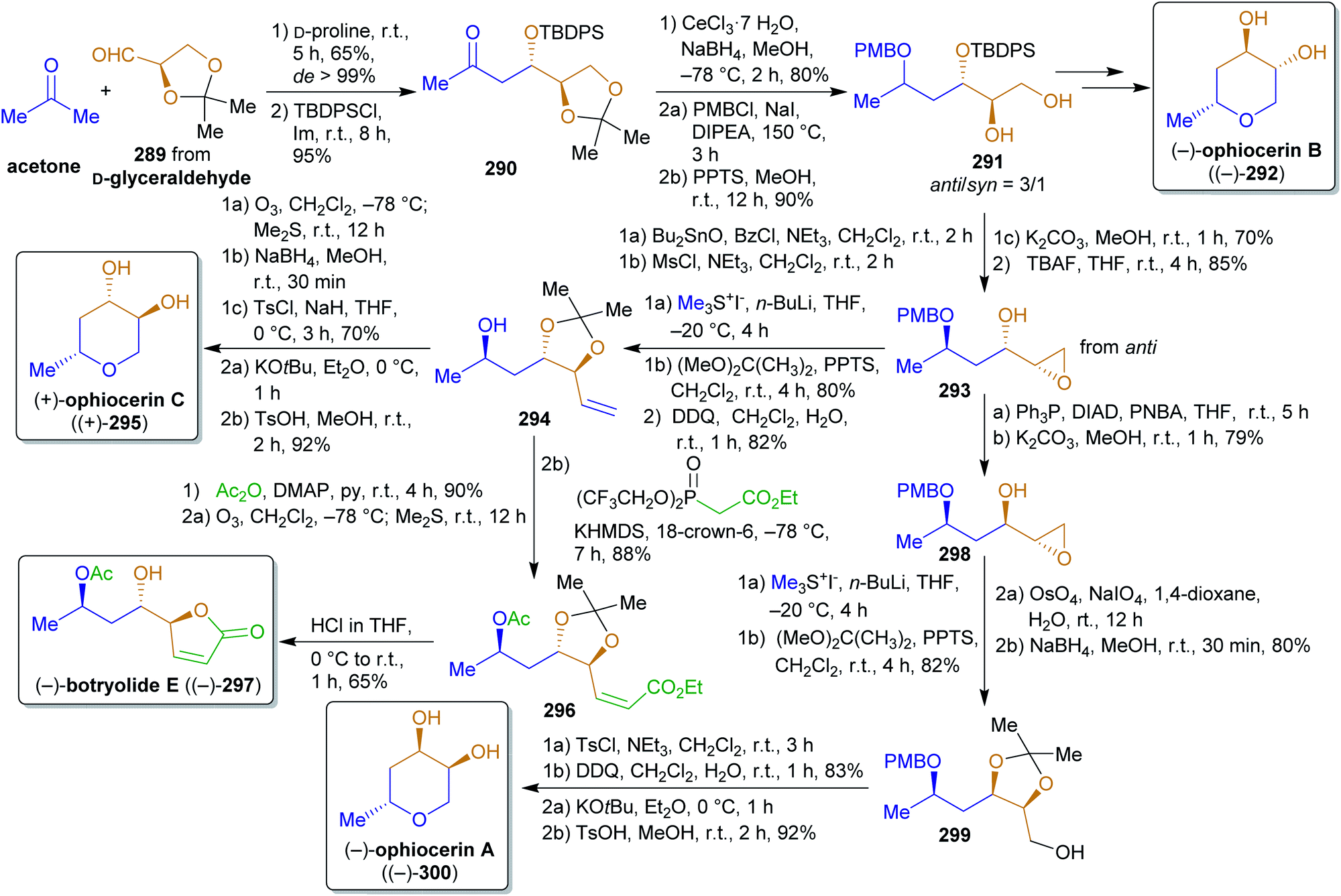
2.4.2.6. Ophiocerins and (−)-botryolide E. The following synthesis of the ophiocerins A–C (300, 292 and 295) and botryolide E (297)444 is an excellent example of the combination of a biomass-derived chiral precursor (isopropylideneglyceraldehyde (289)), other renewables-derived simple building blocks and organocatalysis for the formation of a variety of natural products (Scheme 45). Like dihydroxyacetone, glyceraldehyde can be generated from glycerol, e.g. by aerial oxidation over platinum catalysts,386 or directly by fermentation of carbohydrate biomass.445 Proline-catalyzed aldol reaction of acetonide-protected aldehyde 289 and acetone led to the respective β-hydroxy ketone 290 in excellent de which was protected with a TBDPS group. Before the cumene process was introduced, acetone was produced by Weizmann's acetone–butanol–ethanol (ABE) fermentation of carbohydrates through Clostridium acetobutylicum.446 Another source of acetone is the pyrolysis and dry distillation of wood and therefore it is considered a xylochemical.290,291 Reduction, PMB-protection and acetonide cleavage led to the pivotal intermediate 291 and all four natural products that were mentioned earlier were synthesized from this anti-diastereomer. For the synthesis of ophiocerins A and C (300 and 295), the secondary hydroxy group was converted into a leaving group and the syn-epoxy alcohol 293 was formed by nucleophilic substitution. For 300, the inversion of alcohol, epoxide opening, acetonide protection, Lemiuex–Johnson oxidation and reduction were performed. Trimethylsulfonium iodide was used for the olefin synthesis from epoxide 293 (available from dimethylsulfide and iodomethane,447 both methanol-derived). After PMB cleavage, the primary alcohol was converted into a leaving group and the tetrahydropyran ring was closed by nucleophilic substitution. Final acetonide cleavage furnished (−)-ophiocerin A ((−)-300). Ophiocerin C (295) was synthesized by essentially the same sequence except that no inversion was conducted and that PMB deprotection was performed prior to olefin cleavage by ozonolysis to obtain a suitable intermediate for the synthesis of botryolide E (297). Likewise, ophiocerin B (292) was prepared via the respective anti-epoxy alcohol (sequence not shown).
 | ||
| Scheme 45 Synthesis of ophiocerins A–C (300, 292 and 295) and botryolide E (297). | ||
Starting from the olefin 294, botryolide E was synthesized by ester synthesis, ozonolysis and the Still–Gennari modification of the Horner reaction to obtain E-olefin 296. The required phosphonate can be produced from an α-haloacetic acid derivative and is therefore accessible by fermentation of carbohydrates through acetic acid.347,448,449 Acid catalyzed acetonide cleavage and lactone formation produced (−)-297.
The use of simple bio-based starting materials and pivotal intermediates for the stereochemically flexible synthesis of natural products are notable key features of this work.
2.5. Terpenes
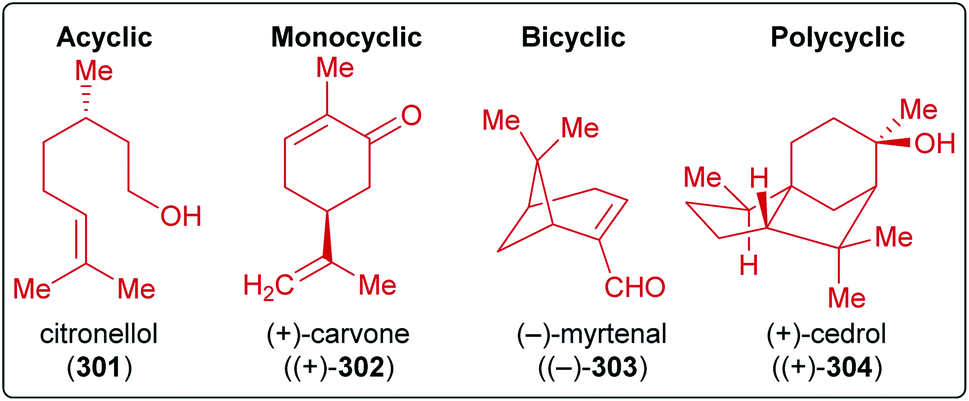
Terpenes represent abundant and renewable, inexpensive and versatile chiral starting materials and were employed in natural product synthesis ever since.80,450 Furthermore, terpenes are one of the largest and most diverse classes of plant produced organic compounds and do not directly compete with food production.451–454C-double bonds and having a limited degree of oxygenation. They can be divided into subgroups named after their carbon count, since isoprene units containing five C-atoms are the biosynthetic precursors of all terpenes:452 the monoterpenes (C10), the sesquiterpenes (C15), the diterpenes (C20), etc. They also differ in the arrangement of the isoprenoid units (acylic, mono- or polycyclic) and in their oxidation state (Scheme 46). Despite the enormous advances in asymmetric synthesis in the 20th century, terpenes are still widely used as chiral starting materials.458 Furthermore, they can be useful as potential fuels,459 agents for the chemical communication of plants, flavor enhancement and pesticides,460,461 or as a source of chirality in catalysts.81
 | ||
| Scheme 46 Different classes of terpenes. | ||
The major sources of monoterpenes are turpentine oil (a waste product of paper pulp industry, contains mainly α- and β-pinene),462 and citrus oil (contains mainly (+)-limonene),463 a co-product of citrus juice production.452 Terpenes can also be transformed and functionalized by biotechnological methods or biotransformations.452,464–466 For further information about sources of terpenes, their use in total synthesis and utilization as chiral building blocks, the reader may be referred to the literature.80,450,452
2.5.2.1. Thymol. Phillips et al. published a five-step synthesis of natural thymol (311) in 1920.467 The synthesis commenced with nitration of natural p-cymene (305)468 followed by Béchamp reduction. Sulfonation gave two isomeric products 309 and 308. Subsequent diazotation/reduction led to sulfonic acid 310. Fusion with NaOH finally produced thymol (311)469 (Scheme 47).467
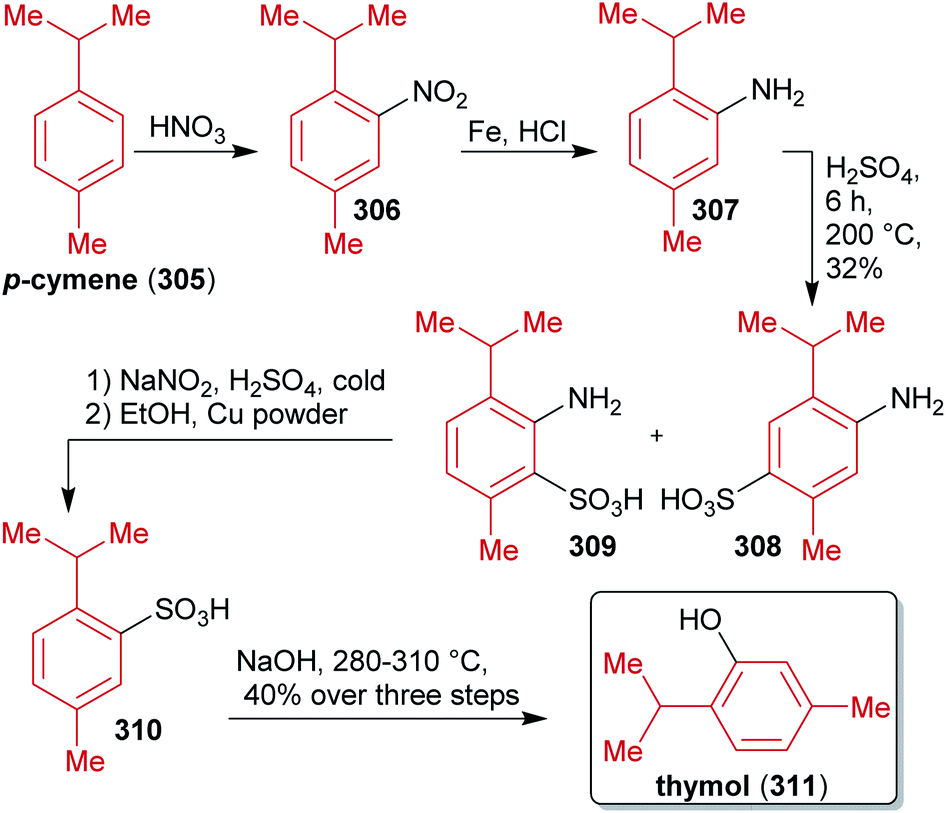 | ||
| Scheme 47 Synthesis of thymol (311) from p-cymene (305). | ||
2.5.2.2. (+)-Grandisol. In 1992, Mori et al. reported a synthesis of the natural product (+)-grandisol ((+)-315) in eighteen steps and ≥98% ee (analyzed by GC).470
The synthesis commenced with epoxidation of (−)-carvone ((−)-302), accessible from biomass,471,472 which is ring-opened oxidatively in a four-step sequence to yield acetal ester 312. The latter was reduced, tosylated, and subjected to a nucleophilic substitution by iodide furnishing iodoester 313. Subsequent cyclization, methylation and iodolactonization led to 314 (Scheme 48). The enantioselective synthesis was concluded by multiple reduction reactions, followed by tosylation, cyanation and further reductions affording the natural product (+)-grandisol ((+)-315).470,473
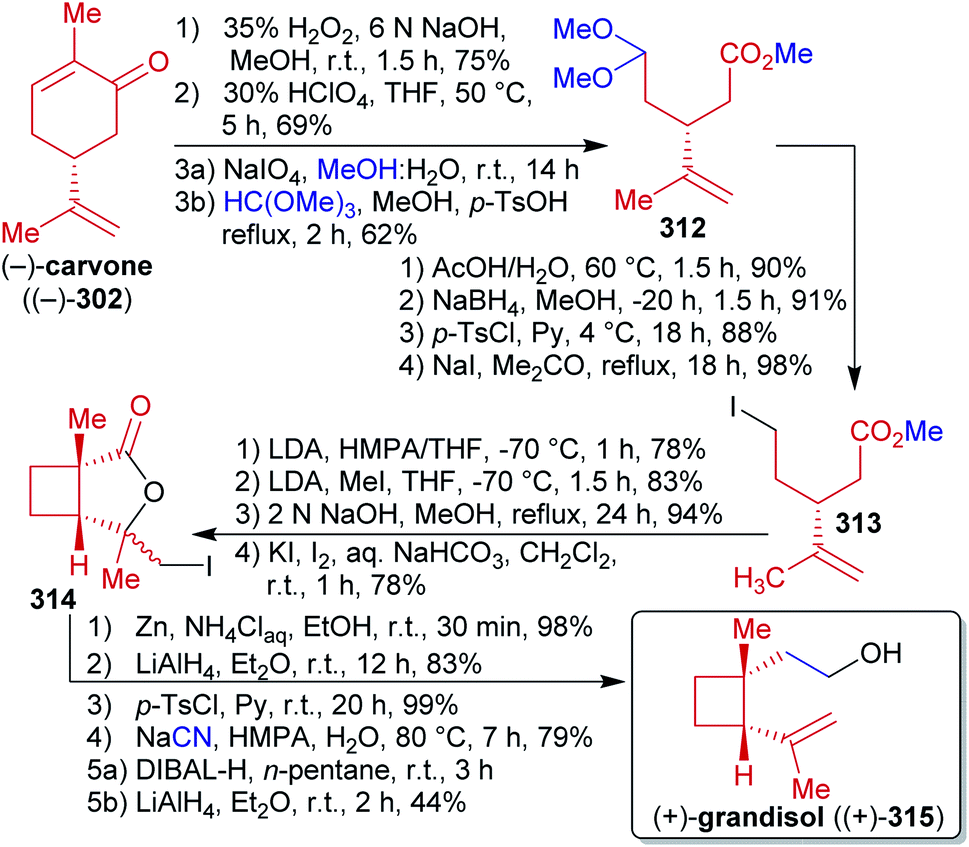 | ||
| Scheme 48 Synthesis of (+)-grandisol ((+)-315) from (−)-carvone ((−)-302). | ||
Sodium cyanide is currently produced by the Andrussow process from methane and ammonia278,279 and a subsequent reaction with lye producing only water as the co-product.474 Considering that synthetic natural gas is a widely established concept,280 NaCN is a potentially bio-based carbon source.281 Trimethyl orthoformate is accessible by the reaction of sodium methanolate with chloroform, another methane- or methanol-derived chemical (vide supra).182
The synthesis is not perfectly carbon-atom efficient as atoms are lost in the oxidation process of starting material 302.
A replacement for NaCN with a non-toxic source for cyanide-anions would be favorable regarding “green” aspects.281,475
2.5.2.3. (−)-Majucin and (−)-jiadifenoxolane A. Maimone et al. reported the first synthetic route to complex majucin-type natural products (−)-majucin ((−)-320) and (−)-jiadifenoxolane A ((−)-321) starting from the abundant feedstock (+)-cedrol ((+)-304).284,476–479 The synthesis commenced with a Suárez oxidation, followed by a hydroboration/double oxidation sequence and a NaBH4 reduction yielding alcohol 316. The C-4 methine position was oxidized under Suárez conditions to afford ether 317. Oxidation with in situ-generated RuO4, Riley oxidation and treatment with L-selectride led to the tetracyclic enol lactone 318. The latter was transformed via DMDO-oxidation to an α-hydroxyketone, bond reorganization by heating in trifluorotoluene, selective reduction of the α-ketol group using Me4NBH(OAc)3 and treatment with acid to furnish the δ-lactone 319. (−)-Majucin ((−)-320) was obtained via an enolate oxidation using Vedejs' MoOPH reagent, subsequent epimerization via Ru-catalyzed transfer hydrogenation and final dihydroxylation. Majucin (320) was converted into (−)-jiadifenoxolane A ((−)-321) via an intramolecular etherification promoted by regioselective mesylation and nucleophilic displacement (Scheme 49).
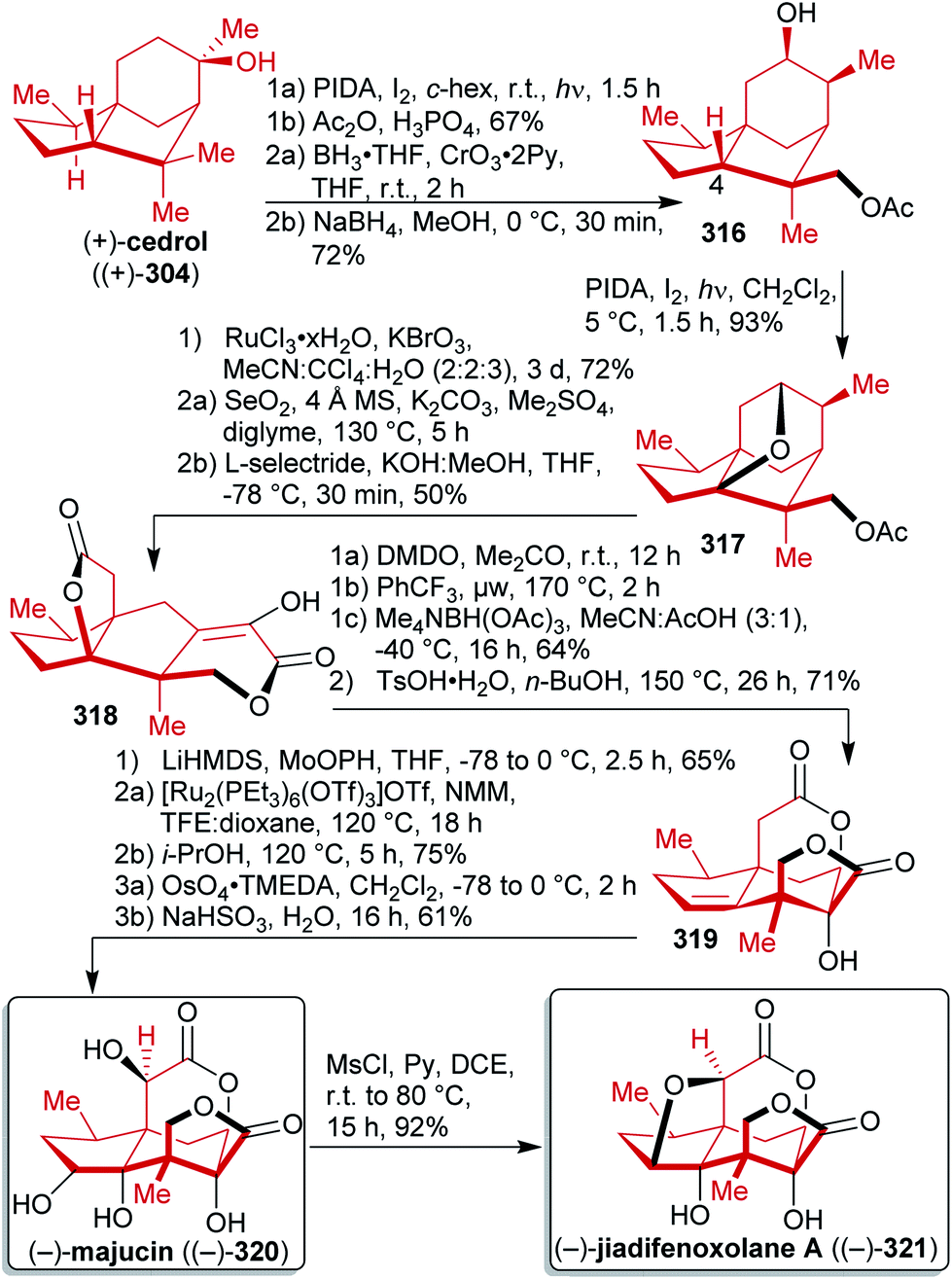 | ||
| Scheme 49 Synthesis of (−)-majucin ((−)-320) and (−)-jiadifenoxolane A ((−)-321) from (+)-cedrol ((+)-304). | ||
Overall, 13 oxidations were employed while three reduction steps were necessary to achieve the correct oxidation state and for stereochemical adjustments. Favorable aspects in the light of “green” chemistry are the use of a photocatalyzed reaction, and of green solvents (acetone, H2O, MeOH), yet undesired solvents like CCl4 or PhCF3 also occur.
2.5.2.4. (+)-Mikanokryptin. Maimone et al. reported the first gram-scale total synthesis of a guaianolide natural product, (+)-mikanokryptin ((+)-326). The guaianolides are a major subgroup of sesquiterpene lactones and were investigated from both medicinal and synthetic perspectives.480
The synthesis commenced with a one-pot chlorination and Luche reduction of the renewable (+)-carvone ((+)-302).481,482 This was followed by O-silylation and a regioselective ozonolysis of the trisubstituted CC-double bond. Reductive quenching led to an intramolecular aldol condensation affording enal 322. The first of two allylation reactions afforded compound 324 and utilized allylic bromide 323, which can be prepared from wood-based renewable resources methanol and glyoxal furnishing dimethoxyacetaldehyde.483,484 The latter can be reacted with methyl acrylate485 and subsequently brominated to obtain desired building block 323.480 The dehydration of lactic acid as a cheap and bio-based starting material for acrylic acid is of growing interest and could become a future source for this commodity chemical.177–181 With the second (intramolecular) allylation reaction, the seven-memberd ring was formed, completing the full guaianolide skeleton 325. Via a reduction using Adams catalyst, subsequent desilylation and allylic oxidation using MnO2, the natural product (+)-mikanokryptin ((+)-326) was obtained (Scheme 50).486
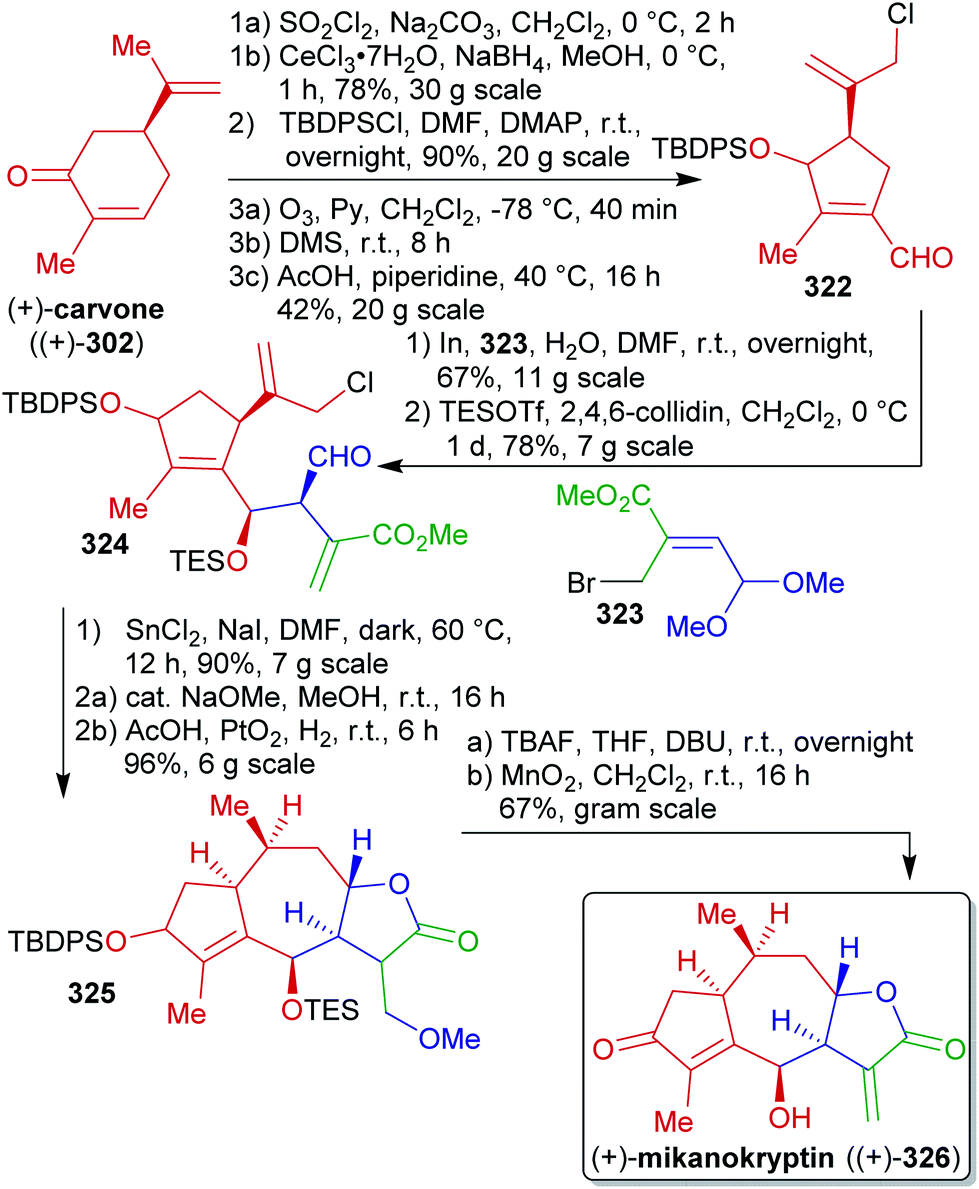 | ||
| Scheme 50 Synthesis of (+)-mikanokryptin ((+)-326) using terpene- and wood-derived building blocks. | ||
2.5.2.5. (+)-Pseudoanisatin. In 2016, Maimone et al. reported the conversion of sesquiterpene (+)-cedrol ((+)-304), an inexpensive terpene feedstock obtained from biomass,487 into the Illicium-sesquiterpene (+)-pseudoanisatin ((+)-333).488,489
The synthesis started with the remote oxidation of one of the geminal methyl groups of cedrol (304), methylation and elimination of the formed tetrahydrofuran ring using Meerwein's salt and proton sponge to furnish methoxycedrene 327 (Scheme 51). The CC-double bond was oxidatively cleaved, lactonization and subsequent lactone hydrolysis, followed by an α-ketol rearrangement then led to compound 329. This crucial intermediate already bears the correct stereochemistry for the final product. Silylation and C–H-oxidation using the terpene-derived homochiral iron catalyst 330 afforded lactone 331.
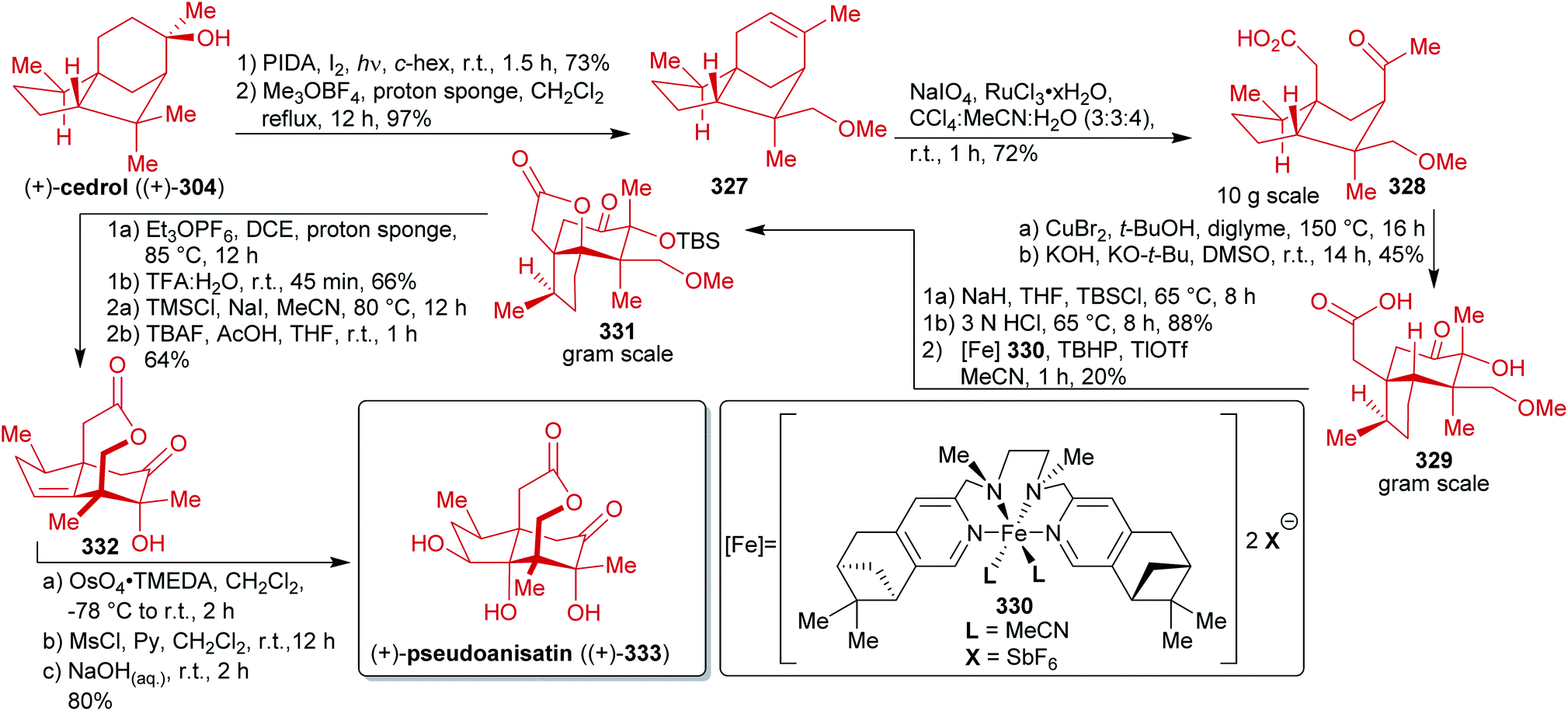 | ||
| Scheme 51 Use of chiral starting material (+)-cedrol ((+)-304) for the first synthesis of (+)-pseudoanisatin ((+)-333). | ||
Subsequently, 331 was O-ethylated and H2O eliminated. Through action of in situ-formed TMSI, the methyl ether was dealkylated and subsequent treatment with TBAF produced the ε-lactone characteristic of pseudoanisatin 333. Dihydroxylation and secondary alcohol inversion completed the enantioselective synthesis of natural (+)-pseudoanisatin ((+)-333).
2.5.2.6. (+)-Cardamom peroxide. Starting from natural (−)-myrtenal ((−)-303),490 the antimalarial terpene (+)-cardamom peroxide ((+)-335)491 was obtained in a four-step synthesis by Maimone et al. in 2014 (Scheme 52), proving the absolute configuration of the latter natural product.492
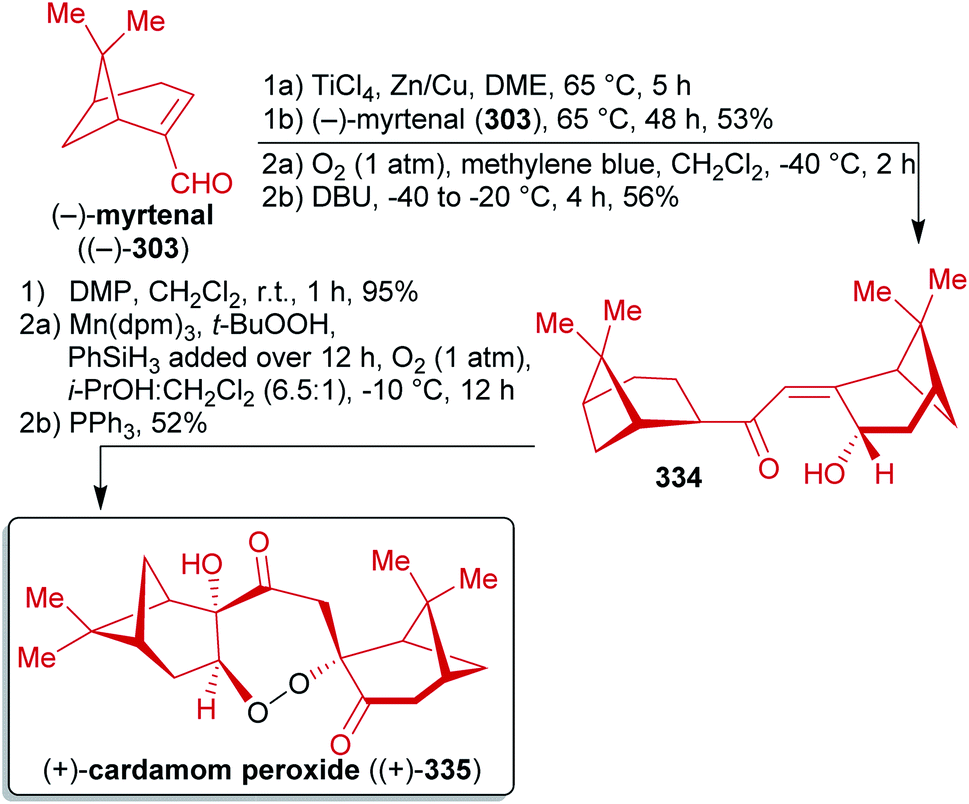 | ||
| Scheme 52 Synthesis of (+)-cardamom peroxide ((+)-335) from (−)-myrtenal ((−)-303). | ||
The synthesis started with a McMurry coupling of (−)-myrtenal ((−)-303), affording the dimeric C20 carbon skeleton of the final product, followed by a [4 + 2]-cycloaddition with singlet oxygen furnishing enone 334. Subsequent oxidation with DMP and manganese-catalyzed tandem hydroperoxidation completed the synthesis of (+)-cardamom peroxide ((+)-335). Highlights of the synthesis are the high atom-efficiency, the use of molecular oxygen and the application of a photocatalytic reaction.
2.5.2.7. (+)-Paeonisuffrone. In 2008, Bermejo et al. used (+)-carvone ((+)-302) for the synthesis of the non-natural enantiomer (+)-paeonisuffrone ((+)-339).493,494 Natural (+)-carvone ((+)-302) was converted into (+)-10-hydroxycarvone ((+)-336) following a known procedure (Scheme 53).495Via epoxidation and protection of the hydroxy function, the subsequent Ti(III)-promoted reaction was enabled. A stereoselective radical cyclization, initiated by reductive epoxide opening, produced triol pivalate 337. The synthesis was completed via protection of the diol moiety, an allylic oxidation using CrO3 and an oxa-Michael addition to the resulting enone to furnish compound 338. A Pd/C promoted reductive deprotection ultimately afforded (+)-paeonisuffrone ((+)-339). The first synthesis of the naturally occurring enantiomer, (−)-paeonisuffrone, was published by Hatakeyama et al. in 1995.494,496
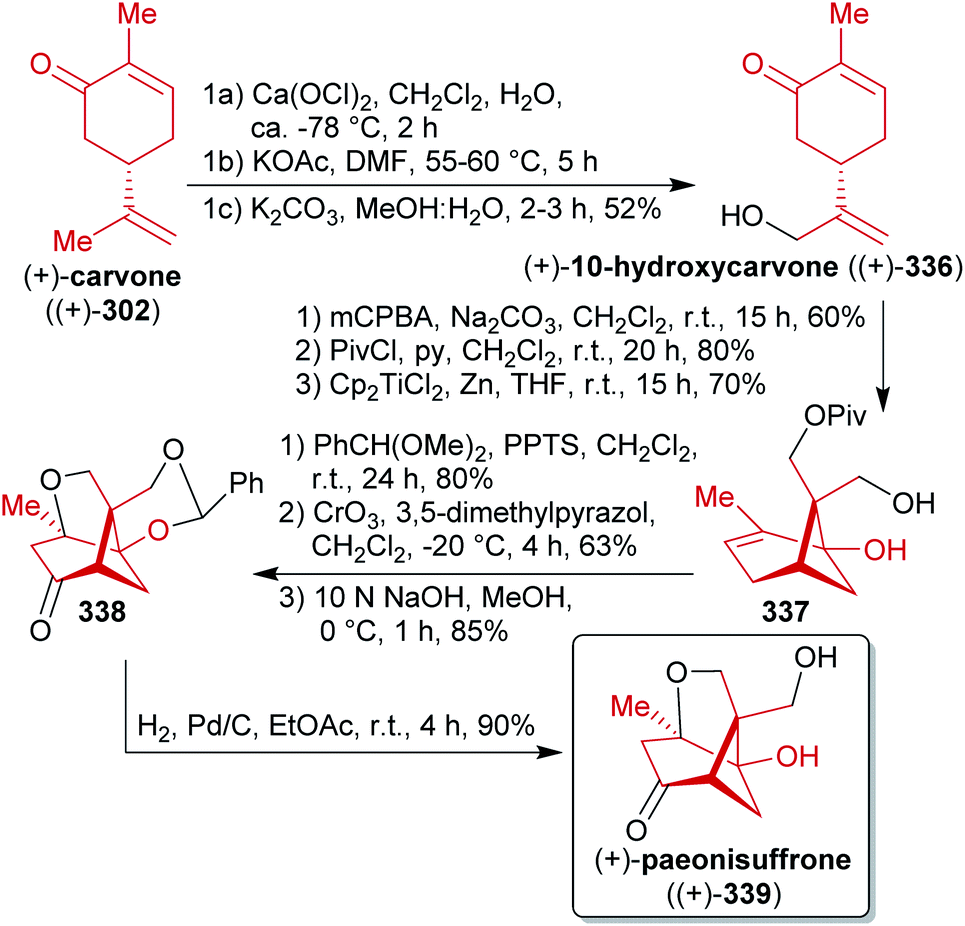 | ||
| Scheme 53 Synthesis of (+)-paeonisuffrone ((+)-339) employing a Ti(III)-promoted epoxide opening and Ti(III)-catalyzed reductive C–C formation as key step. | ||
2.5.2.8. (+)-Yingzhaosu A. The synthesis of the natural product (+)-yingzhaosu A ((+)-345), including the first evaluation of its antimalarial and cytotoxic activities, was reported by Bachi et al. in 2005.497 The β-sulfenyl endoperoxide 341 was prepared via a four component sequential free radical reaction of (−)-limonene ((−)-340) with thiophenol and oxygen, in which five bonds were formed. This was followed by in situ-reduction of the formed hydroperoxy group (Scheme 54).498 The starting material (−)-limonene ((−)-340) is accessible from biomass.499,500 Subsequently, compound 341 was dehydrated, the thioether functionality was selectively oxidized and subjected to a Pummerer rearrangement followed by hydrolysis of the formed hemithioacetal ester, to afford bicyclic aldehyde 342 after hydrogenation of the C
C-double bond.
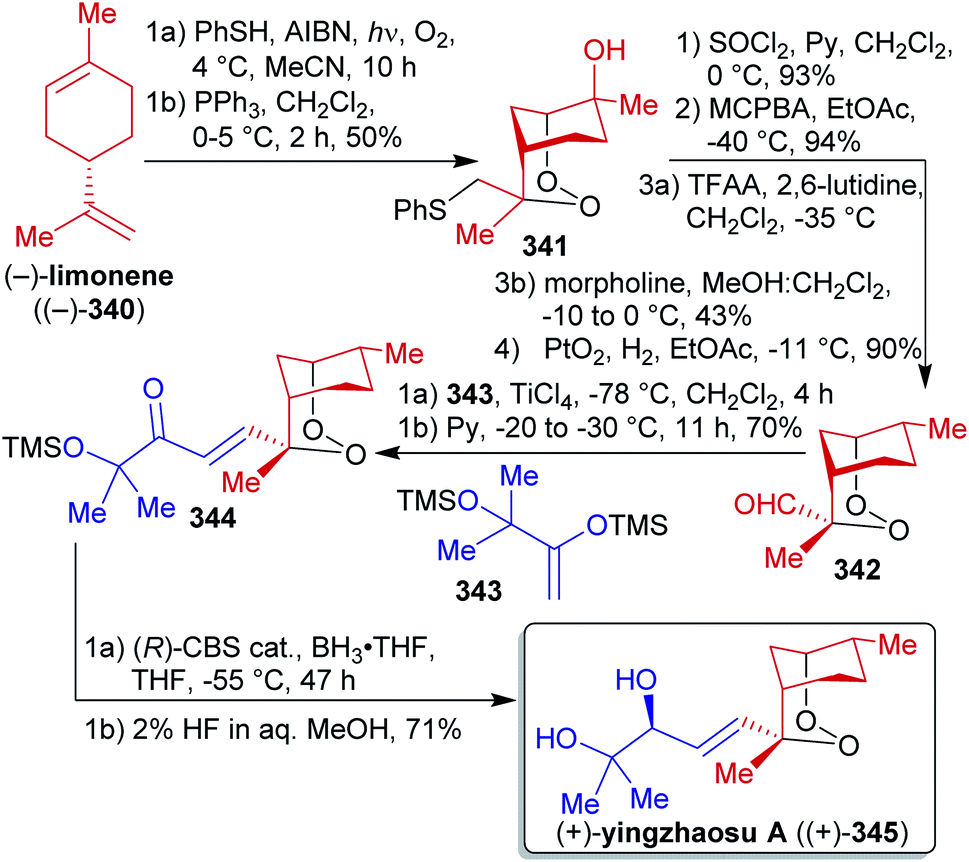 | ||
| Scheme 54 Synthesis of natural product and antimalarial agent (+)-yingzhaosu A ((+)-345). | ||
The second major synthon 343 can be synthesized in three steps from acetone and acetylene (accessible via electric arc pyrolysis of methane or from wood-derived charcoal through calcium carbide).501 First, a reaction to 2-methylbut-3-yn-2-ol is conducted,502 followed by oxidation503 and completed by enolization and protection of the afforded hydroxy groups.504
Both synthons were linked through a Mukaiyama aldol reaction followed by in situ base-induced dehydration affording the α,β-unsaturated ketone 344. The synthesis was completed by selective reduction of ketone 344 and acidic workup furnishing the natural product (+)-yingzhaosu A ((+)-345).505,506 The obtained product was then subjected to in vitro and in vivo tests.497
2.5.2.9. (+)-Omphadiol. The natural product (+)-omphadiol ((+)-351) was first isolated from the basidiomycete Omphalotus illudens507 and the first total synthesis was reported by the group of Romo in 2011 (Scheme 55).508 The key intermediate in the synthesis of (+)-omphadiol ((+)-351) is bicyclic β-lactone 347, which was prepared from (−)-carvone ((−)-302).452,471,472 The synthesis commenced with catalyzed hydration of the enone moiety, followed by an oxidative C
C-cleavage. An aldol lactonization afforded bicyclic β-lactone 347, which was then reduced to the corresponding diol, subjected to a one-pot tosylation/bromination sequence and a subsequent acylation reaction to furnish ester 348. Treatment with base and iodomethane formed two C–C-bonds in a single operation, affording δ-lactone 349. With in situ-formed allyllithium, a conversion into a ring-opened β,γ-unsaturated ketone was performed and an olefin isomerization/RCM sequence led to cycloheptenone 350. After reduction and cyclopropanation, the natural product (+)-omphadiol ((+)-351) was obtained. Diiodomethane is available from iodoform,509 the latter being a product of the well-known reaction from ethanol with I2 in alkaline medium.300 The synthesis proceeds in a highly efficient manner, using one-pot, sequential and tandem processes, and avoids the use of protecting groups.
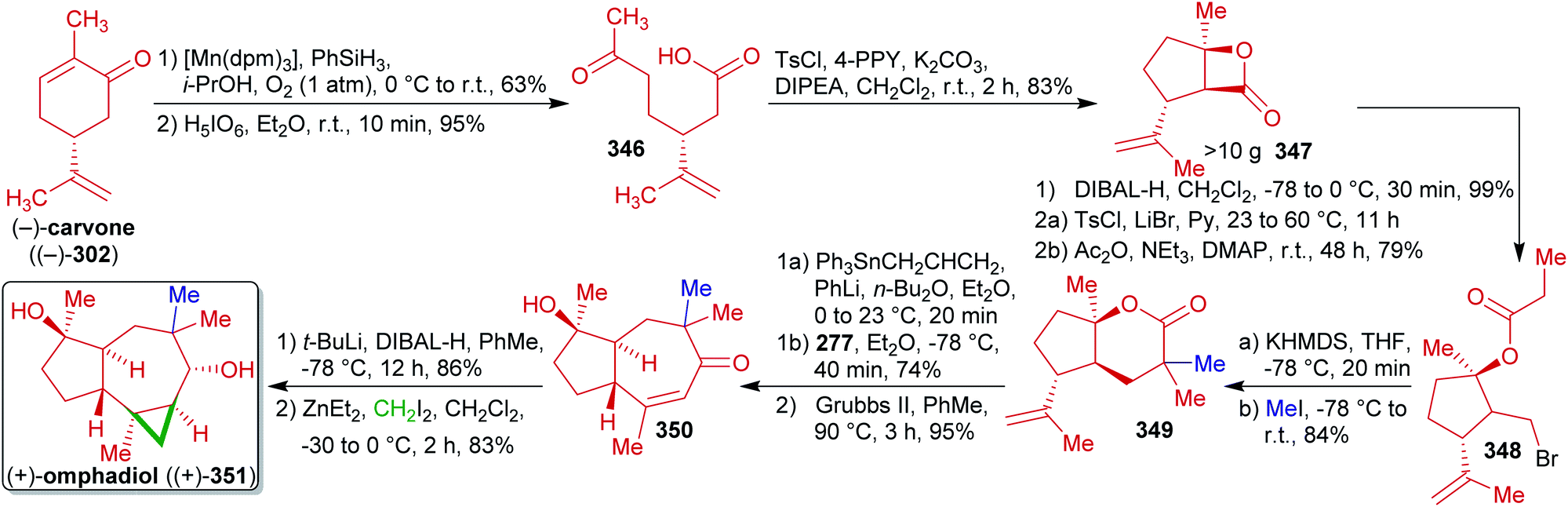 | ||
| Scheme 55 Synthesis of (+)-omphadiol ((+)-351) from (−)-carvone ((−)-302). 4-PPY = 4-(1-pyrrolidinyl)pyridine. | ||
2.5.2.10. (−)-Bolivianine and (+)-onoseriolide. In 2013, Liu et al. reported the first total synthesis of (−)-bolivianine ((−)-359) in 14 steps, including the synthesis of (+)-onoseriolide ((+)-357).510 The synthesis commenced with a Michael addition of vinyl reagent 244 to (+)-verbenone ((+)-352) (accessible via bioconversion of (+)-α-pinene),511 ring-opening of the cyclobutane moiety and formation of 1,3-dioxolane 353 under acidic conditions (Scheme 56). Through Riley oxidation and intermediate preparation of a tosylhydrazone, a diazoalkane was produced, which was subsequently subjected to a metal-catalyzed carbene insertion reaction affording cyclopropane 354. Acid-catalyzed deprotection enabled the reaction with functionalized pyruvate 355 to afford furan 356. Building block 355 could be prepared in two steps from natural methyl glycerate by silylation and oxidation.510,512 DIBAL-H reduction of furan 356 and modification of the furan ring furnished the natural product (+)-onoseriolide ((+)-357).513–515 By oxidizing the furan moiety, the formed electron-withdrawing group activates the dienophile by decreasing its LUMO energy. This enables a one-pot Diels–Alder/intramolecular hetero-Diels–Alder reaction cascade with natural β-(E)-ocimene (358)516,517 generating three rings, four C–C-bonds, and five stereogenic centers and ultimately furnishing the natural product (−)-bolivianine ((−)-359).510,518 Vinyl bromide (244) can be prepared from acrylic acid (available from biomass e.g. through lactic acid)174,177–181via ultrasonically assisted Vilsmeier–Haack reaction.175 The use of Diels–Alder reactions in the synthesis of natural products is quite attractive against the backdrop of “green” chemistry because of their flawless atom economy. The choice of solvents in the synthesis is exemplary in most cases (acetone, H2O, EtOAc).
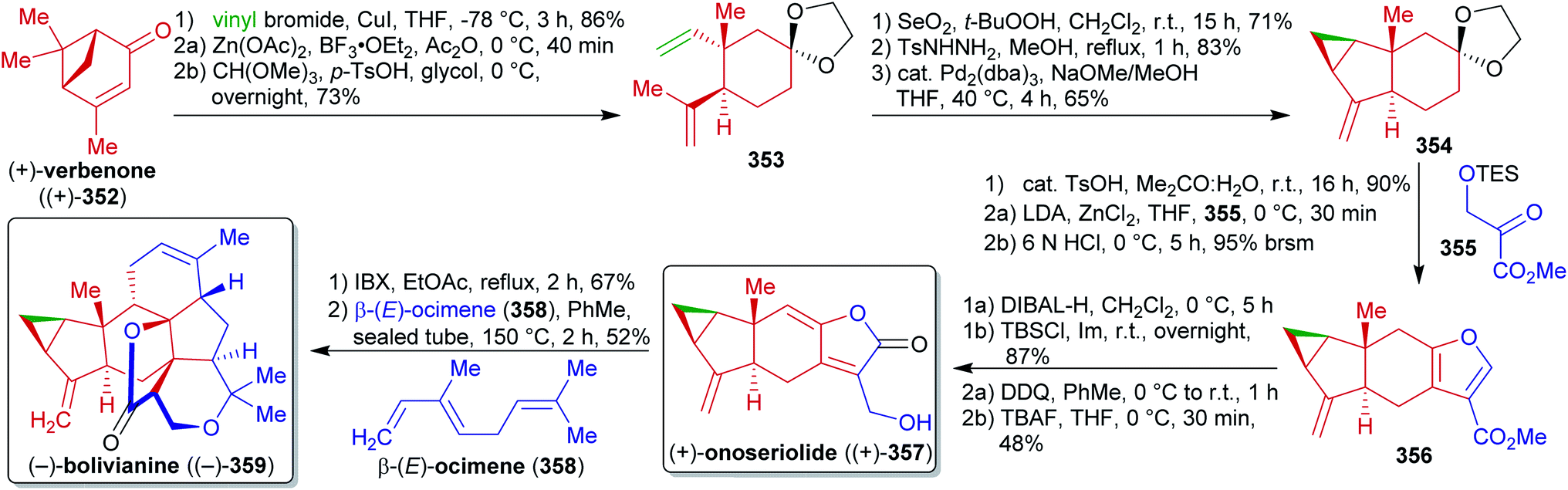 | ||
| Scheme 56 Synthesis of (−)-bolivianine ((−)-359) from (+)-verbenone ((+)-352). | ||
2.5.2.11. (+)-Welwitindolinone A, (−)-fischerindole I and G. In 2007, Baran et al. were able to synthesize several natural products in a protecting group-free synthesis starting from (−)-carvone oxide ((−)-360) (Scheme 57). Intermediate 363 was synthesized on a gram scale by vinylation and chlorination of (−)-carvone oxide ((−)-360) affording chloroketone 361,519 followed by coupling with indole (362) and an acid catalyzed Friedel–Crafts cyclization. Reduction of the ketone, mesylation and nucleophilic substitution with azide, followed by reduction afforded amine 364. The latter was formylated and dehydrated to furnish the natural product (−)-fischerindole G ((−)-365).519,520
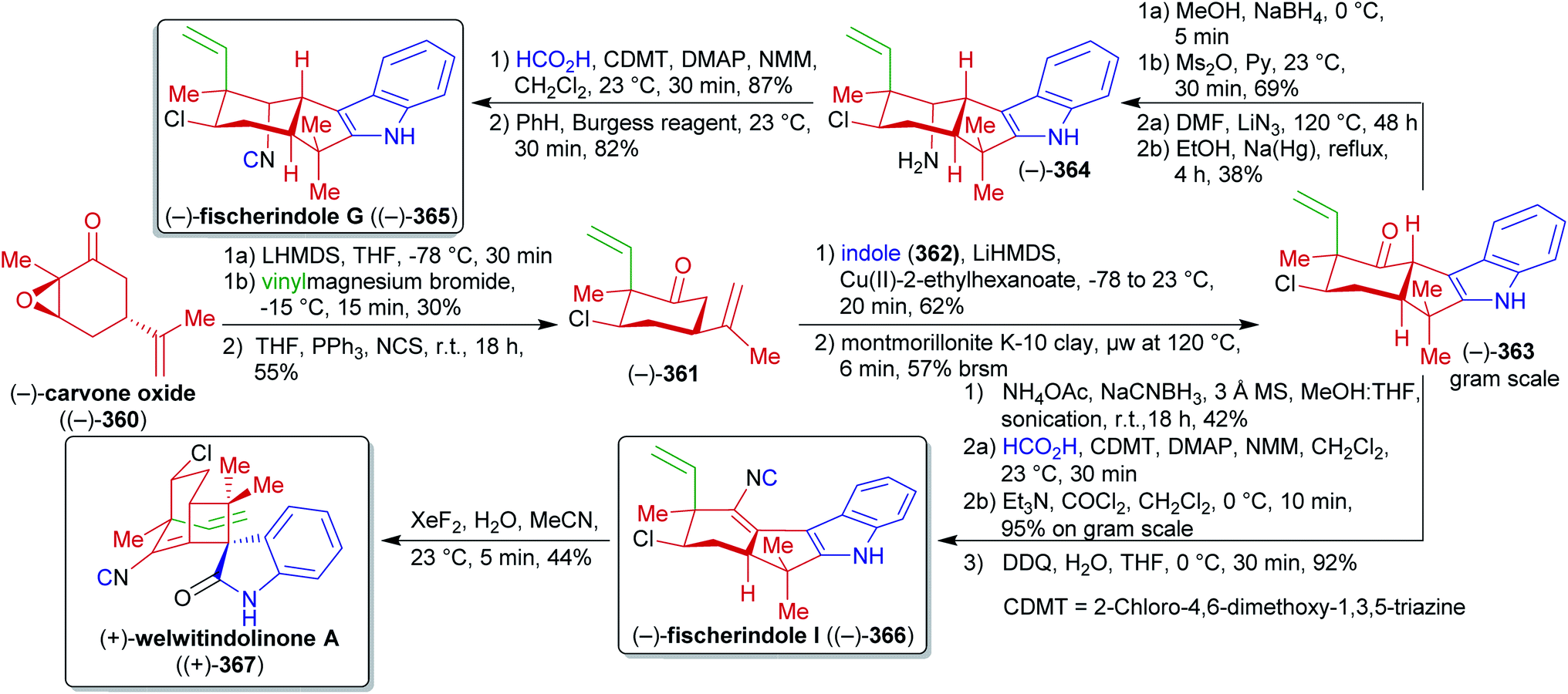 | ||
| Scheme 57 Synthesis of (−)-fischerindole G ((−)-365) and I ((−)-366) and (+)-welwitindolinone A ((+)-367). CDMT = 2-chloro-4,6-dimethoxy-1,3,5-triazine. | ||
Along the other path, a reductive amination of intermediate 363, followed by formylation, immediate dehydration with phosgene and an oxidation with DDQ in the presence of water led to (−)-fischerindole I ((−)-366). In a cascade reaction, the natural product 366 was converted to the spirocyclic, natural product (+)-welwitindolinone A ((+)-367).520 The reaction proceeded through electrophilic fluorination of the indole nucleus with XeF2 and trapping with H2O. Subsequently, fluoride was eliminated and a [1,5]-sigmatropic rearrangement took place.521
Vinylmagnesium bromide (71) can be synthesized from biomass-derived building blocks (vide supra). Indole (362) can e.g. be prepared from tryptophan or from indigo.522 (−)-Carvone oxide ((−)-360) can be prepared from (−)-carvone ((−)-302).523
The oxidation states of intermediates gradually escalated over the course of the synthesis with the sole exception of a stereoselective reductive amination.
2.5.2.12. (+)-Cubitene. Lindel et al. reported an enantioselective total synthesis of the diterpene (+)-cubitene ((+)-373).524 (+)-Carvone ((+)-302) was reacted first with aldehyde 368 and subsequently O-phosphorylated to furnish allyl phosphate 369 (Scheme 58).525 By treating the latter with SmI2, an intramolecular coupling reaction afforded an [8.2.2]-bicyclic compound, which was converted into an acyloin and subsequently reduced to diol 370. The latter was cleaved with H5IO6, followed by an oxidation under Pinnick conditions, affording macrocycle 371. Subsequently, 371 was subjected to a Wittig reaction, followed by deprotection and an oxidation of the hydroxyl group under Parikh–Doering conditions promoting a decarboxylation to ultimately form ketone 372. A three-step sequence, comprising a reduction of the keto group, silylation of the resulting hydroxy group and an allylic deoxygenation using Li/EtNH2, afforded the natural product (+)-cubitene ((+)-373).524,526
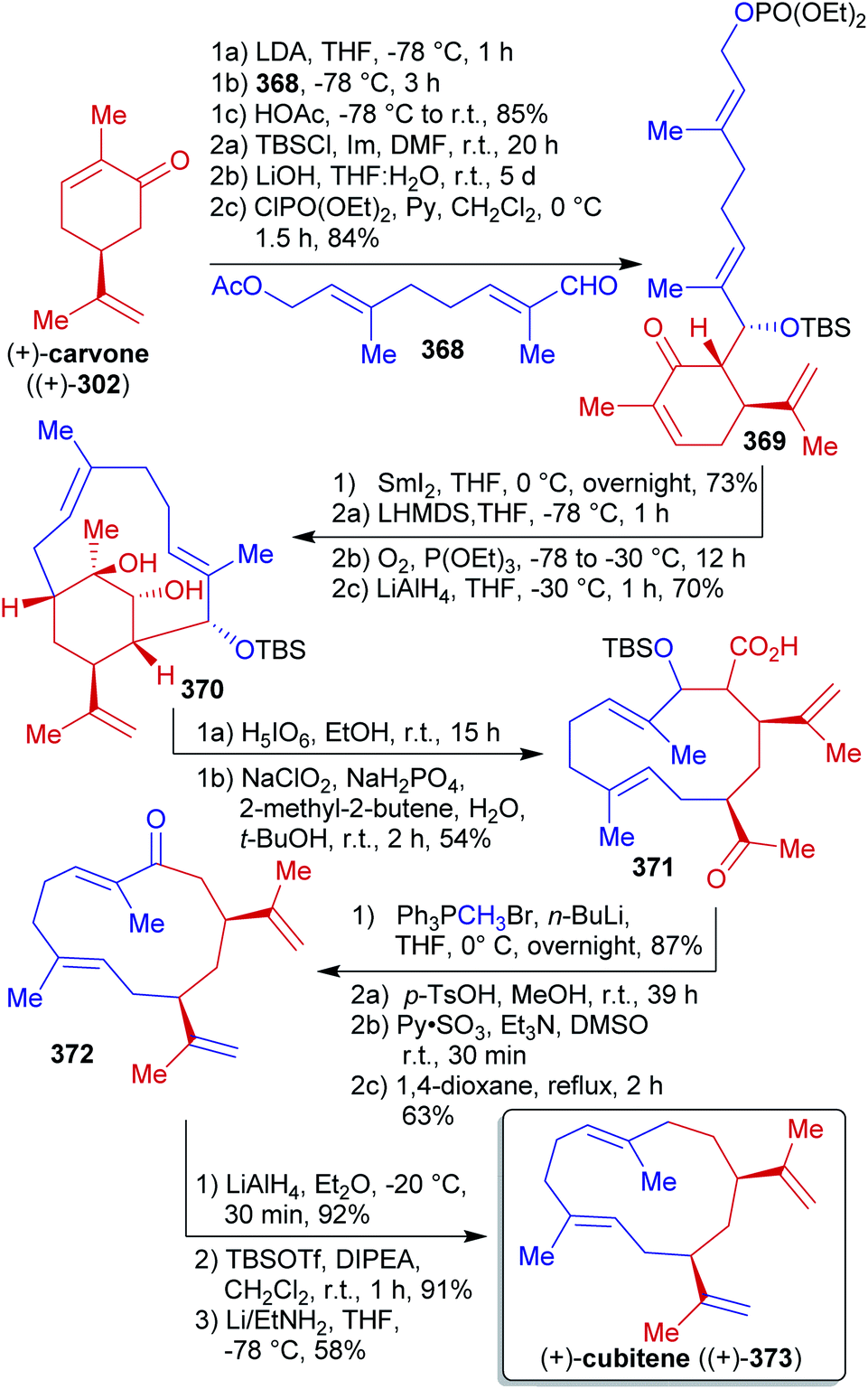 | ||
| Scheme 58 Synthesis of (+)-cubitene ((+)-373) from (+)-carvone ((+)-302) and geraniol derived aldehyde (368). | ||
Aldehyde 368 is available from natural geraniol via an allylic oxidation.479,527,528
2.6. Miscellaneous
Since natural products can be very complex molecules and the total synthesis often is already challenging, the construction of the entire carbon framework from bio-based starting materials may constitute a considerable challenge. In addition, it is not always possible to adhere to green synthetic methods and guidelines throughout the entire sequence so that conventional methodology has to be employed where the available eco-friendly alternatives failed. An efficient step in the right direction is the synthesis of fragments and to build at least as much as possible from biomass derived starting materials. | ||
| Scheme 59 Tetracycline natural products containing an L-rednose (L-204) moiety. | ||
 | ||
| Scheme 60 Anthracycline natural products containing an L-rednose (L-204) moiety. | ||
 | ||
| Scheme 61 First asymmetric synthesis of (+)-cis-nemorensic acid ((+)-386) based on cellulose derived 121. | ||
The eco-friendliness of this route is quite favorable regarding the carbon efficiency. Furthermore, almost every carbon atom introduced into the molecule during the synthesis is retained (except for the trifluoroethyl and methyl groups), further increasing the carbon efficiency. Further circumstantial protecting group operations are completely avoided.
542 which could be converted into the antibiotic glucolipsins (394a–b, Scheme 62)543 and cycloviracins (not shown).544–547 Although a number of total and formal syntheses which also use bio-based starting materials in the form of e.g. glucose547 have been published,548–553 we chose this work to showcase the value of levoglucosan as a less common starting material.
 | ||
| Scheme 62 Synthesis of macrocyclic building block 393 from levoglucosan (114) for formal synthesis of glucolipsins 394a, b. DMC = 2-chloro-1,3-dimethylimidazolinium chloride. | ||
After benzyl protection, 114 was transformed into the monocyclic trichloracetimidate 389, which was reacted with the dimethyl D-malate-derived building block 390 in a Schmidt glycosylation. Both ester groups were saponified and subsequent ring forming esterification furnished 393.
This approach makes exemplary use of enantiopure bio-based starting materials to furnish a pivotal building block for the syntheses of fairly complex natural products, but is hampered by extensive protecting group usage well known from carbohydrate chemistry.554
 | ||
| Scheme 63 CNSL constituents: anacardic acid (395), cardanol (396) and cardol (397). | ||
Magalhães and dos Santos570 managed to utilize CNSL-derived cardol (397) for the synthesis of antileukemic lasiodiplodin (402).571,572 Acetyl protected cardol was subjected to ozonolysis and following reductive treatment to yield a truncated alcohol (Scheme 64).
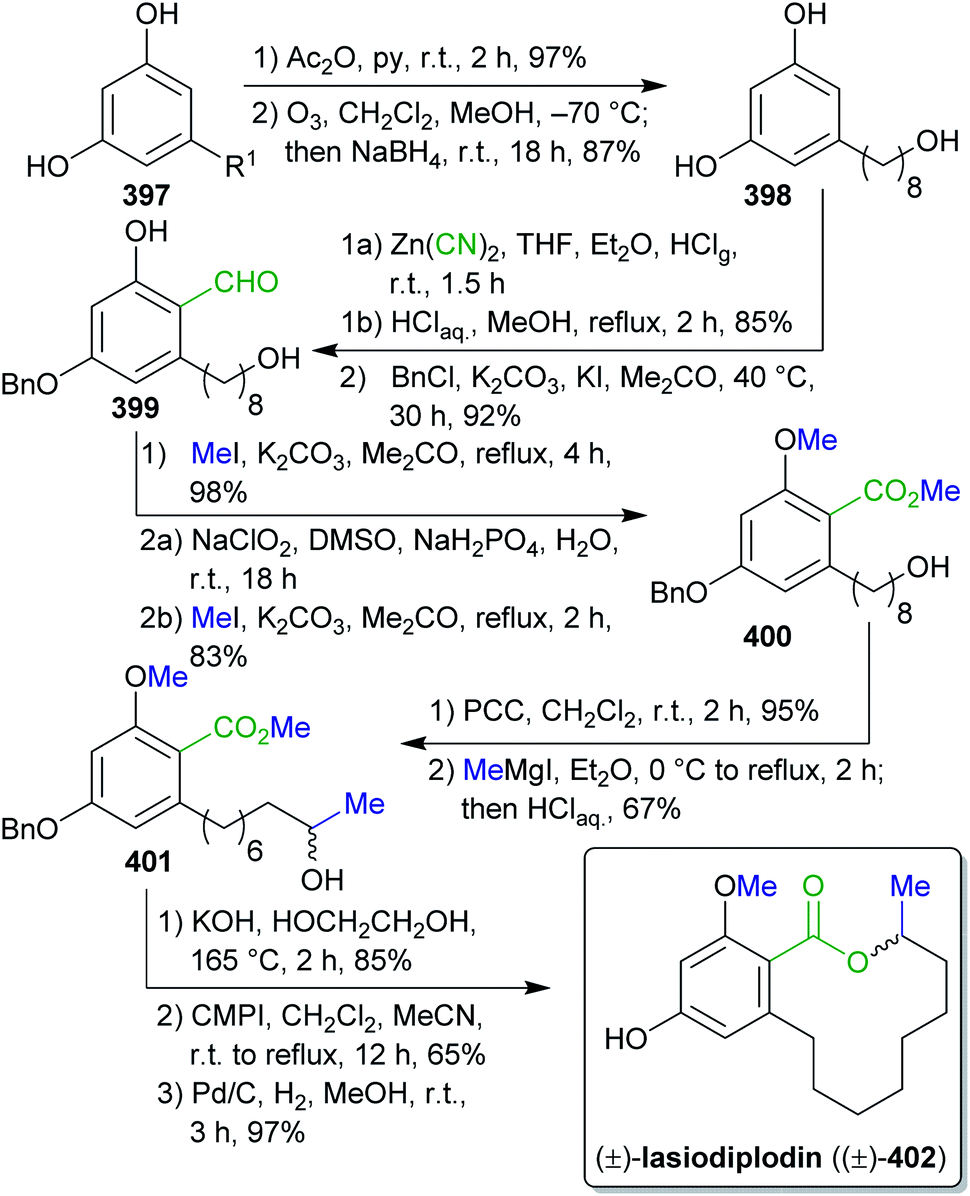 | ||
| Scheme 64 Lasiodiplodin (402) synthesis from cardol (397). | ||
After saponification of the acetates, formylation was performed by means of a Gattermann reaction and the p-hydroxy group was benzylated to furnish aldehyde 399. The second aromatic hydroxy group was converted to the methyl ether and aldehyde oxidation was achieved under Pinnick conditions. The respective carboxylic acid was O-alkylated with iodomethane and the remaining primary hydroxy group was oxidized to an aldehyde, which was subjected to a Grignard reaction with methylmagnesium iodide to yield secondary alcohol 401. After saponification and macrolactone formation through the action of 2-chloro-1-methylpyridinium iodide (CMPI), the benzyl group was cleaved to furnish racemic lasiodiplodin.
Although this very concise approach makes formidable use of this particular starting material, it also demonstrates an issue when used for total synthesis: The C15 moiety often has to be truncated so that a non-negligible part of the molecule (in this case about one third) is not retained in the desired final product. Therefore, the carbon and atom efficiency is decreased – however, the C7-fragment split off may e.g. find application in the synthesis of the fragrance jasmin aldehyde.573
3. Future challenges and outlook
The use of fossil carbon sources as the basis of the vast majority of current activities in synthetic organic chemistry is only rarely questioned by practitioners and scholars. Nevertheless, the current discussion on the necessity of CO2 reduction and the size of the doubtlessly finite underground deposits of fossil carbon brings up interesting challenges for current and future chemists. Atom awareness among the chemical community, i.e. the knowledge of the origin of the matter that we deal with in our work, certainly leaves some room for improvement. As outlined in the Introduction, chemists were never shy to accept new challenges, e.g. to utilize the seemingly unattractive, black and smelly coal tar to produce beautiful dyestuffs or life-saving drugs. The combination of starting materials from renewable feedstocks with eco-friendly solvents, reagents, and methodologies as well as a reduced energy consumption can ultimately lead to processes with minimal impact on our ecosystem. Therefore, existing methods, synthetic routes and processes need to be revisited constantly. A big part of whether or how quickly those concepts will be applied on a large scale is dependent on the economic competitiveness.Furthermore, the conception of bio-based starting materials has also reached other organic chemical fields like pharmaceutical and agrochemistry. The synthesis of the anti-ulcer drug ranitidine574 and the insecticide prothrin from CMF (120)575 as well as the synthesis of norfenefrine and fenoprofen from cashew nut shell liquid-derived cardanol576 are pioneering examples of this development.
Another attractive trend in organic chemistry in terms of sustainability is the conception of protecting group free sequences577–580 as already demonstrated for the synthesis of fischerindole G (365) and I (366) as well as of welwitindolinone A (367) (Scheme 57).521,577–580 This could particularly influence natural product synthesis, a field in which the extensive use of protecting group transformations is commonplace. Atom- and step-efficiency of synthetic routes could be significantly increased if alternative methods for achieving selectivity can be employed instead.
4. Conflicts of interest
There are no conflicts of interest to declare.5. Acknowledgements
We thank the members of the STANCE consortium for helpful discussions.6. Notes and references
- F. Wöhler, Ann. Phys., 1828, 87, 253–256 CrossRef.
- F. W. Serturner, Trommsdorffs J. Pharm., 1806, 14, 47–98 Search PubMed.
- F. W. Serturner, Ann. Phys., 1817, 25, 56–89 CrossRef.
- K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6–14 RSC.
- D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022–1037 CrossRef CAS PubMed.
- W. R. Roush, J. Am. Chem. Soc., 2008, 130, 6654–6656 CrossRef CAS.
- C. A. Kuttruff, M. D. Eastgate and P. S. Baran, Nat. Prod. Rep., 2014, 31, 419–432 RSC.
- E. M. Carreira, Isr. J. Chem., 2018, 58, 114–121 CrossRef CAS.
- As a counterevidence to this statement, we recommend the literature on putaminoxin, which has been synthesized several times with the assumed absolute configuration until, more than 20 years after its isolation, Pietruszka and coworkers noticed for the first time that the configuration of one of its two stereocenters had been missassigned: C. Bisterfeld, C. Holec, D. Böse, P. Marx and J. Pietruszka, J. Nat. Prod., 2017, 80, 1563–1574 CrossRef CAS.
- The reader may be referred to the story of patchouli alcohol, for which a wrong structure had been proposed and the natural product was accidentally formed in an unforeseen rearrangement of this (wrong) structure. Dunitz and coworkers only later discovered that the synthesis was not the proof of a correct structural assignment: G. Büchi, R. E. Erickson and N. Wakabayashi, J. Am. Chem. Soc., 1961, 83, 927–938 CrossRef.
- G. Büchi and W. D. MacLeod, J. Am. Chem. Soc., 1962, 84, 3205–3206 CrossRef.
- D. Dickmann, M. Diekmann, C. Holec and J. Pietruszka, Tetrahedron, 2019, 75, 689–696 CrossRef CAS.
- M. Dobler, J. D. Dunitz, B. Gubler, H. P. Weber, G. Büchi and J. Padilla, Proc. Chem. Soc., 1963, 357–392 Search PubMed.
- A. Evidente, R. Lanzetta, R. Capasso, A. Andolfi, A. Bottalico, M. Vurro and M. C. Zonno, Phytochemistry, 1995, 40, 1637–1641 CrossRef CAS.
- J. Fuchser and A. Zeeck, Liebigs Ann./Recl., 1997, 1997, 87–95 CrossRef.
- K. Götz, J. C. Liermann, E. Thines, H. Anke and T. Opatz, Org. Biomol. Chem., 2010, 8, 2123–2130 RSC.
- J. B. McAlpine, S.-N. Chen, A. Kutateladze, J. B. MacMillan, G. Appendino, A. Barison, M. A. Beniddir, M. W. Biavatti, S. Bluml, A. Boufridi, M. S. Butler, R. J. Capon, Y. H. Choi, D. Coppage, P. Crews, M. T. Crimmins, M. Csete, P. Dewapriya, J. M. Egan, M. J. Garson, G. Genta-Jouve, W. H. Gerwick, H. Gross, M. K. Harper, P. Hermanto, J. M. Hook, L. Hunter, D. Jeannerat, N.-Y. Ji, T. A. Johnson, D. G. I. Kingston, H. Koshino, H.-W. Lee, G. Lewin, J. Li, R. G. Linington, M. Liu, K. L. McPhail, T. F. Molinski, B. S. Moore, J.-W. Nam, R. P. Neupane, M. Niemitz, J.-M. Nuzillard, N. H. Oberlies, F. M. M. Ocampos, G. Pan, R. J. Quinn, D. S. Reddy, J.-H. Renault, J. Rivera-Chávez, W. Robien, C. M. Saunders, T. J. Schmidt, C. Seger, B. Shen, C. Steinbeck, H. Stuppner, S. Sturm, O. Taglialatela-Scafati, D. J. Tantillo, R. Verpoorte, B.-G. Wang, C. M. Williams, P. G. Williams, J. Wist, J.-M. Yue, C. Zhang, Z. Xu, C. Simmler, D. C. Lankin, J. Bisson and G. F. Pauli, Nat. Prod. Rep., 2019, 36, 35–107 RSC.
- K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 CrossRef CAS.
- G. Sabitha, K. Yadagiri, R. Swapna and J. S. Yadav, Tetrahedron Lett., 2009, 50, 5417–5419 CrossRef CAS.
- H.-D. Yoo, S.-J. Nam, Y.-W. Chin and M.-S. Kim, Arch. Pharmacal Res., 2016, 39, 143–153 CrossRef CAS PubMed.
- K. C. Nicolaou, Isr. J. Chem., 2018, 58, 104–113 CrossRef CAS.
- M. A. Sierra and M. C. de la Torre, Angew. Chem., Int. Ed., 2000, 39, 1538–1559 CrossRef CAS.
- M. Eissen, J. O. Metzger, E. Schmidt and U. Schneidewind, Angew. Chem., Int. Ed., 2002, 41, 414–436 CrossRef CAS.
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- P. T. Anastas and J. B. Zimmerman, Environ. Sci. Technol., 2003, 37, 94A–101A CrossRef.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- P. T. Anastas, Crit. Rev. Anal. Chem., 1999, 29, 167–175 CrossRef CAS.
- P. T. Anastas, M. M. Kirchhoff and T. C. Williamson, Appl. Catal., A, 2001, 221, 3–13 CrossRef CAS.
- J. H. Clark, R. Luque and A. S. Matharu, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 183–207 CrossRef CAS.
- A. B. Eldin, O. A. Ismaiel, W. E. Hassan and A. A. Shalaby, J. Anal. Chem., 2016, 71, 861–871 CrossRef CAS.
- J. H. Clark, Green Chem., 1999, 1, 1–8 RSC.
- S. C. Hammer, A. M. Knight and F. H. Arnold, Curr. Opin. Green Sustainable Chem., 2017, 7, 23–30 CrossRef.
- J. G. Ibanez, M. Hernandez-Esparza, C. Doria-Serrano, A. Fregoso-Infante and M. M. Singh, Environmental Chemistry: Fundamentals, Springer, New York, NY, 2007 Search PubMed.
- M. Koel, Green Chem., 2016, 18, 923–931 RSC.
- J. Li, J. Albrecht, A. Borovika and M. D. Eastgate, ACS Sustainable Chem. Eng., 2018, 6, 1121–1132 CrossRef CAS.
- J. A. Linthorst, Found. Chem., 2010, 12, 55–68 CrossRef CAS.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- P. A. Ramachandran, D. Shonnard, R. Hesketh, D. Fichana, C. Stewart Slater, A. Lindner, N. Nguyen and R. Engler, in Handbook of Industrial Chemistry and Biotechnology, ed. J. A. Kent, T. V. Bommaraju and S. D. Barnicki, Springer International Publishing, Cham, 2017, pp. 1921–1994 Search PubMed.
- N. Ran, L. Zhao, Z. Chen and J. Tao, Green Chem., 2008, 10, 361–372 RSC.
- P. A. Schulte, L. T. McKernan, D. S. Heidel, A. H. Okun, G. S. Dotson, T. J. Lentz, C. L. Geraci, P. E. Heckel and C. M. Branche, Environ. Health, 2013, 12, 31 CrossRef.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- R. A. Sheldon, in Green Biocatalysis, ed. R. N. Patel, Wiley-VCH, Delft, Johannesburg, Weinheim, 2016 Search PubMed.
- R. A. Sheldon, Curr. Opin. Green Sustainable Chem., 2019, 18, 13–19 CrossRef.
- R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Delft, Weinheim, 2007 Search PubMed.
- R. A. Sheldon and J. M. Woodley, Chem. Rev., 2018, 118, 801–838 CrossRef CAS.
- J. Sherman, B. Chin, D. T. H. Paul, R. Garcia-Valls and T. A. Hatton, Environ. Health Perspect., 1998, 106, 253–271 CAS.
- H. F. Sneddon, Green Chem., 2016, 18, 5082–5085 RSC.
- S. Stolte, S. Steudte, A. Igartua and P. Stepnowski, Curr. Org. Chem., 2011, 15, 1946–1973 CrossRef CAS.
- J. Tao and R. J. Kazlauskas, Biocatalysis for Green Chemistry and Chemical Process Development, Wiley-VCH, Weinheim, 2011 Search PubMed.
- N. P. Tarasova, F. I. Ingel' and A. S. Makarova, Russ. J. Phys. Chem. B, 2015, 9, 406–411 CrossRef CAS.
- A. Tiwari, A. M. Ramirez, R. Jain and A. Saxena, Key Eng. Mater., 2012, 517, 755–762 CAS.
- V. G. Zuin, M. L. Segatto and L. Z. Ramin, in Encyclopedia of Sustainability Science and Technology, ed. R. A. Meyers, Springer New York, New York, NY, 2018, pp. 1–24 Search PubMed.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471–1478 CrossRef CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- R. A. Sheldon, Chem. Ind., 1992, 903–906 CAS.
- R. A. Sheldon, Industrial Environmental Chemistry, ed. D. T. Sawyer and A. E. Martell, Plenum, New York, 1992 Search PubMed.
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC.
- J. Andraos, Org. Process Res. Dev., 2005, 9, 149–163 CrossRef CAS.
- P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452–1461 RSC.
- T. Hudlicky, D. A. Frey, L. Koroniak, C. D. Claeboe and L. E. Brammer Jr, Green Chem., 1999, 1, 57–59 RSC.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cunningham, Green Chem., 2001, 3, 1–6 RSC.
- N. Brun, P. Hesemann and D. Esposito, Chem. Sci., 2017, 8, 4724–4738 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- A. Nzihou, Waste Biomass Valorization, 2010, 1, 3–7 CrossRef.
- A. Baeyer and H. Caro, Ber. Dtsch. Chem. Ges., 1874, 7, 968–976 CrossRef.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1880, 13, 2254–2263 CrossRef.
- F. Beilstein and A. Kuhlberg, Justus Liebigs Ann. Chem., 1872, 163, 121–143 CrossRef.
- W. H. Perkin, J. Chem. Soc., 1877, 31, 388–427 RSC.
- A. Ladenburg, Ber. Dtsch. Chem. Ges., 1886, 19, 2578–2583 CrossRef.
- O. Lange, Ber. Dtsch. Chem. Ges., 1885, 18, 3436–3441 CrossRef.
- F. Beilstein and A. Kuhlberg, Justus Liebigs Ann. Chem., 1870, 155, 1–29 CrossRef.
- F. Sachs and R. Kempf, Ber. Dtsch. Chem. Ges., 1902, 35, 1224–1240 CrossRef CAS.
- F. Sachs and E. Sichel, Ber. Dtsch. Chem. Ges., 1904, 37, 1861–1874 CrossRef CAS.
- E. Steingruber, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2004 Search PubMed.
- F. Hoffmann, US Pat., US644077 (A), 1900.
- N. C. Lloyd, H. W. Morgan, B. K. Nicholson and R. S. Ronimus, Angew. Chem., Int. Ed., 2005, 44, 941–944 CrossRef CAS.
- Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753–11795 CrossRef CAS.
- H. U. Blaser, Chem. Rev., 1992, 92, 935–952 CrossRef CAS.
- D. Ager, Handbook of chiral chemicals, CRC Press and Taylor and Francis Group, Boca Raton, 2005 Search PubMed.
- J. M. Brown and S. G. Davies, Nature, 1989, 342, 631–636 CrossRef CAS.
- S. Hanessian, Total synthesis of natural products: The Chiron Approach, Pergamon Press, Elmsford, 1983 Search PubMed.
- T.-L. Ho, Enantioselective synthesis: natural products from chiral terpenes, Wiley, New York, 1992 Search PubMed.
- H. Kunz and K. Rück, Angew. Chem., Int. Ed. Engl., 1993, 32, 336–358 CrossRef.
- J. Mulzer, K.-D. Graske and B. Kirste, Liebigs Ann. Chem., 1988, 1988, 891–897 CrossRef.
- IPCC 2014, in Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. Core Writing Team, R. K. Pachauri and L. Meyer, IPCC, Geneva, Switzerland, 2014 Search PubMed.
- A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS.
- M. J. L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS.
- E. F. Gomez and A. J. Steckl, ACS Photonics, 2015, 2, 439–445 CrossRef CAS.
- F. Liebner and T. Rosenau, Functional Materials from Renewable Sources, American Chemical Society, Vienna, 2012 Search PubMed.
- K. Y. Sandhya, A. Saritha and K. Joseph, in Liquid Crystalline Polymers: Volume 1–Structure and Chemistry, ed. V. K. Thakur and M. R. Kessler, Springer International Publishing, Cham, 2016, pp. 273–306 Search PubMed.
- N. Thejo Kalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2015, 44, 319–347 CrossRef.
- M. Yusuf, Handbook of Renewable Materials for Coloration and Finishing, Wiley-VCH, Weinheim, 2018 Search PubMed.
- C. A. S. Hill, Wood Modification: Chemical, Thermal and Other Processes, Wiley, 2007 Search PubMed.
- M. Carrier, A. Loppinet-Serani, D. Denux, J.-M. Lasnier, F. Ham-Pichavant, F. Cansell and C. Aymonier, Biomass Bioenergy, 2011, 35, 298–307 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- C. Xu, R. A. D. Arancon, J. Labidi and R. Luque, Chem. Soc. Rev., 2014, 43, 7485–7500 RSC.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Science, 2012, 337, 695 CrossRef CAS.
- L. A. Lucia, BioResources, 2008, 3, 981–982 Search PubMed.
- J. J. Bozell, Science, 2010, 329, 522 CrossRef CAS.
- X. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS.
- K. Triantafyllidis, A. Lappas and M. Stöcker, The Role of Catalysis for the Sustainable Production of Bio-fuels and Bio-chemicals, Elsevier-Science, Newnes, 2013 Search PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- E. Feghali, G. Carrot, P. Thuéry, C. Genre and T. Cantat, Energy Environ. Sci., 2015, 8, 2734–2743 RSC.
- A. Demirbas, Energy Sources, Part A, 2009, 32, 1–9 CrossRef.
- F. Stecker, I. M. Malkowsky, A. Fischer, S. R. Waldvogel and C. Regenbrecht, US Pat. application, US20130040031A1, 2014.
- J. Kühlborn, A.-K. Danner, H. Frey, R. Iyer, A. J. Arduengo and T. Opatz, Green Chem., 2017, 19, 3780–3786 RSC.
- T.-Q. Yuan, F. Xu and R.-C. Sun, J. Chem. Technol. Biotechnol., 2013, 88, 346–352 CrossRef CAS.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS.
- K. McCormick and N. Kautto, Sustainability, 2013, 5, 2589–2608 CrossRef.
- D. Stubba, G. Lahm, M. Geffe, J. W. Runyon, A. J. Arduengo III and T. Opatz, Angew. Chem., Int. Ed., 2015, 54, 14187–14189 CrossRef CAS.
- J. Kühlborn, M. Konhäuser, J. Groß, P. R. Wich and T. Opatz, ACS Sustainable Chem. Eng., 2019, 7, 4414–4419 CrossRef.
- T. Opatz, J. Kühlborn, C. B. de Koning, K. J. Ngwira and Q. A. Mgani, South African provisional Pat., ZA2019/01514, 2019.
- C. T. Hammond and P. G. Mahlberg, Phytochemistry, 1994, 37, 755–756 CrossRef CAS.
- D. Barton, A. Deflorin and O. Edwards, J. Chem. Soc. Resumed, 1956, 530–534 RSC.
- K. Nakagawa-Goto and K.-H. Lee, Tetrahedron Lett., 2006, 47, 8263–8266 CrossRef CAS.
- O. Thoison, D. D. Cuong, A. Gramain, A. Chiaroni, N. V. Hung and T. Sévenet, Tetrahedron, 2005, 61, 8529–8535 CrossRef CAS.
- H. P. Pepper, S. J. Tulip, Y. Nakano and J. H. George, J. Org. Chem., 2014, 79, 2564–2573 CrossRef CAS PubMed.
- F. A. Ramos, Y. Takaishi, M. Shirotori, Y. Kawaguchi, K. Tsuchiya, H. Shibata, T. Higuti, T. Tadokoro and M. Takeuchi, J. Agric. Food Chem., 2006, 54, 3551–3557 CrossRef CAS PubMed.
- X. Wu, Y. Liu, W. Sheng, J. Sun and G. Qin, Planta Med., 1997, 63, 55–57 CrossRef CAS PubMed.
- A. J. Núñez Sellés, H. T. Vélez Castro, J. Agüero-Agüero, J. González-González, F. Naddeo, F. De Simone and L. Rastrelli, J. Agric. Food Chem., 2002, 50, 762–766 CrossRef.
- J. H. Boyce and J. A. Porco Jr, Angew. Chem., Int. Ed., 2014, 53, 7832–7837 CrossRef CAS.
- R. D. Hartley and C. H. Fawcett, Phytochemistry, 1968, 7, 1395–1400 CrossRef CAS.
- S. Y. Chow, H. J. Williams, Q. Huang, S. Nanda and A. I. Scott, J. Org. Chem., 2005, 70, 9997–10003 CrossRef CAS.
- K. Anjou and E. von SYDOW, Acta Chem. Scand., 1969, 23, 109–114 CrossRef CAS.
- Y.-C. Chien, C.-H. Lin, M. Y. Chiang, H.-S. Chang, C.-H. Liao, I.-S. Chen, C.-F. Peng and I.-L. Tsai, Phytochemistry, 2012, 80, 50–57 CrossRef CAS PubMed.
- D. A. Evans, T. C. Britton, R. L. Dorow and J. F. Dellaria, Tetrahedron, 1988, 44, 5525–5540 CrossRef CAS.
- S. Tahara, Y. Katagiri, J. L. Ingham and J. Mizutani, Phytochemistry, 1994, 36, 1261–1271 CrossRef CAS.
- M. A. Selepe, S. E. Drewes and F. R. van Heerden, Tetrahedron, 2011, 67, 8654–8658 CrossRef CAS.
- P. Reveglia, S. Savocchia, R. Billones-Baaijens, A. Cimmino and A. Evidente, J. Agric. Food Chem., 2018, 66, 1760–1764 CrossRef CAS.
- P. H. Dang, H. X. Nguyen, N. T. Nguyen, H. N. T. Le and M. T. T. Nguyen, Phytother. Res., 2014, 28, 1632–1636 CrossRef CAS PubMed.
- M. Royer, G. Herbette, V. Eparvier, J. Beauchêne, B. Thibaut and D. Stien, Phytochemistry, 2010, 71, 1708–1713 CrossRef CAS.
- M. A. Selepe, S. E. Drewes and F. R. van Heerden, J. Nat. Prod., 2010, 73, 1680–1685 CrossRef CAS.
- G. R. Nagarajan and V. S. Parmar, Phytochemistry, 1977, 16, 614–615 CrossRef CAS.
- E. Chosson, A. Chaboud, A. J. Chulia and J. Raynaud, Phytochemistry, 1998, 47, 87–88 CrossRef CAS.
- T. Milkova, G. Talev, R. Christov, S. Dimitrova-Konaklieva and S. Popov, Phytochemistry, 1997, 45, 93–95 CrossRef CAS.
- J. Guyot and L. J. Simon, C. R. Hebd. Seances Acad. Sci., 1919, 169, 795–797 CAS.
- D. Mesnard and L. Miginiac, J. Organomet. Chem., 1989, 373, 1–10 CrossRef CAS.
- L. Tonucci, M. Nicastro, N. d'Alessandro, M. Bressan, P. D'Ambrosio and A. Morvillo, Green Chem., 2009, 11, 816–820 RSC.
- S. B. Ateba, D. Njamen, S. Medjakovic, M. Zehl, H. Kaehlig, A. Jungbauer and L. Krenn, BMC Complementary Altern. Med., 2014, 14, 294 CrossRef.
- Y.-Z. Lee, C.-W. Huang, C.-W. Yang, H.-Y. Hsu, I.-J. Kang, Y.-S. Chao, I.-S. Chen, H.-Y. Chang and S.-J. Lee, Planta Med., 2011, 77, 1932–1938 CrossRef CAS.
- G. Lahm, A. Stoye and T. Opatz, J. Org. Chem., 2012, 77, 6620–6623 CrossRef CAS.
- J. Hussain, H. Khan, L. Ali, A. Latif Khan, N. Ur Rehman, S. Jahangir and A. Al-Harrasi, Helv. Chim. Acta, 2015, 98, 719–723 CrossRef CAS.
- B. Zierer, P. Schieberle and M. Granvogl, J. Agric. Food Chem., 2016, 64, 9515–9522 CrossRef CAS.
- J. Verduyckt, M. Van Hoof, F. De Schouwer, M. Wolberg, M. Kurttepeli, P. Eloy, E. M. Gaigneaux, S. Bals, C. E. A. Kirschhock and D. E. De Vos, ACS Catal., 2016, 6, 7303–7310 CrossRef CAS.
- A. Stoye, T. E. Peez and T. Opatz, J. Nat. Prod., 2013, 76, 275–278 CrossRef CAS PubMed.
- N. Ünver and G. İ. Kaya, Turk. J. Chem., 2005, 29, 547–553 Search PubMed.
- S. Tian, W. Zi and D. Ma, Angew. Chem., Int. Ed., 2012, 51, 10141–10144 CrossRef CAS.
- S. Kodama, H. Takita, T. Kajimoto, K. Nishide and M. Node, Tetrahedron, 2004, 60, 4901–4907 CrossRef CAS.
- S. Ghosal, Y. Kumar and S. Singh, Phytochemistry, 1984, 23, 1167–1171 CrossRef CAS.
- T. A. Wheaton and I. Stewart, Phytochemistry, 1969, 8, 85–92 CrossRef CAS.
- H. Matsuda, K. Ninomiya, T. Morikawa, D. Yasuda, I. Yamaguchi and M. Yoshikawa, Bioorg. Med. Chem., 2009, 17, 7313–7323 CrossRef CAS.
- Y. Zhu, P. Zhang, H. Yu, J. Li, M.-W. Wang and W. Zhao, J. Nat. Prod., 2007, 70, 1570–1577 CrossRef CAS.
- S. Barradas, G. Hernández-Torres, A. Urbano and M. C. Carreño, Org. Lett., 2012, 14, 5952–5955 CrossRef CAS.
- T. Mohn, I. Plitzko and M. Hamburger, Phytochemistry, 2009, 70, 924–934 CrossRef CAS.
- S. Agostini, J.-M. Desjobert and G. Pergent, Phytochemistry, 1998, 48, 611–617 CrossRef CAS.
- J. Deng, R. Li, Y. Luo, J. Li, S. Zhou, Y. Li, J. Hu and A. Li, Org. Lett., 2013, 15, 2022–2025 CrossRef CAS.
- J.-X. Zhu, J.-J. Qin, F. Zhang, R.-J. Chang, J. Ren, X.-R. Cheng, Q. Zeng, H.-Z. Jin and W.-D. Zhang, Chem. Nat. Compd., 2013, 49, 383–387 CrossRef CAS.
- A. Padwa, M. J. Chughtai, J. Boonsombat and P. Rashatasakhon, Tetrahedron, 2008, 64, 4758–4767 CrossRef CAS.
- C.-I. Chang, J.-Y. Chang, C.-C. Kuo, W.-Y. Pan and Y.-H. Kuo, Planta Med., 2005, 71, 72–76 CrossRef CAS.
- M. R. Crimmin and A. J. P. White, Chem. Commun., 2012, 48, 1745–1747 RSC.
- S. M. Klein, C. Zhang and Y. L. Jiang, Tetrahedron Lett., 2008, 49, 2638–2641 CrossRef CAS.
- L. R. Salgueiro, R. Vila, F. Tomi, X. Tomas, S. Cañigueral, J. Casanova, A. P. da Cunha and T. Adzet, Phytochemistry, 1997, 45, 1177–1183 CrossRef CAS.
- G. A. Burdock, Fenaroli's handbook of flavor ingredients, CRC press and Taylor and Francis Group, Boca Raton, 2016 Search PubMed.
- B. Fang, X. Xie, H. Li, P. Jing, J. Gu and X. She, Tetrahedron Lett., 2013, 54, 6349–6351 CrossRef CAS.
- A. Briot, C. Baehr, R. Brouillard, A. Wagner and C. Mioskowski, J. Org. Chem., 2004, 69, 1374–1377 CrossRef CAS.
- J. Ejlertsson, M. Alnervik, S. Jonsson and B. H. Svensson, Environ. Sci. Technol., 1997, 31, 2761–2764 CrossRef CAS.
- J. R. Vale, T. Rimpiläinen, E. Sievänen, K. Rissanen, C. A. M. Afonso and N. R. Candeias, J. Org. Chem., 2018, 83, 1948–1958 CrossRef CAS PubMed.
- K. Tianpanich, S. Prachya, S. Wiyakrutta, C. Mahidol, S. Ruchirawat and P. Kittakoop, J. Nat. Prod., 2011, 74, 79–81 CrossRef CAS PubMed.
- R. C. C. Martins, J. H. G. Lago, S. Albuquerque and M. J. Kato, Phytochemistry, 2003, 64, 667–670 CrossRef CAS.
- A. Benavides, C. Bassarello, P. Montoro, W. Vilegas, S. Piacente and C. Pizza, Phytochemistry, 2007, 68, 1277–1284 CrossRef CAS PubMed.
- M. S. Kumar, K. C. Rajanna, P. Venkanna, M. Venkateswarlu and V. Sudhakar Chary, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 642–646 CrossRef CAS.
- T. Holm, J. Chem. Soc., Perkin Trans. 2, 1981, 464–467 RSC.
- Z. Guo, D. S. Theng, K. Y. Tang, L. Zhang, L. Huang, A. Borgna and C. Wang, Phys. Chem. Chem. Phys., 2016, 18, 23746–23754 RSC.
- C. T. Lira and P. J. McCrackin, Ind. Eng. Chem. Res., 1993, 32, 2608–2613 CrossRef CAS.
- X. Xu, J. Lin and P. Cen, Chin. J. Chem. Eng., 2006, 14, 419–427 CrossRef CAS.
- J. Zhang, J. Lin and P. Cen, Can. J. Chem. Eng., 2008, 86, 1047–1053 CrossRef CAS.
- X. Zhang, L. Lin, T. Zhang, H. Liu and X. Zhang, Chem. Eng. J., 2016, 284, 934–941 CrossRef CAS.
- P. L. Anelli, M. Brocchetta, D. Copez, D. Palano, M. Visigalli and P. Paoli, Tetrahedron, 1997, 53, 15827–15832 CrossRef CAS.
- E. Lloyd, C. B. Brown, D. G. R. Bonnell and W. J. Jones, J. Chem. Soc. Resumed, 1928, 658–666 RSC.
- K. G. Lalwani and A. Sudalai, Eur. J. Org. Chem., 2015, 2015, 7344–7351 CrossRef CAS.
- K. H. Kim, E. Moon, H. K. Kim, J. Y. Oh, S. Y. Kim, S. U. Choi and K. R. Lee, Bioorg. Med. Chem. Lett., 2012, 22, 6155–6159 CrossRef CAS.
- L. H. Zhang, J. Duan, Y. Xu and W. R. Dolbier, Tetrahedron Lett., 1998, 39, 9621–9622 CrossRef CAS.
- A. H. G. Siebum, W. S. Woo and J. Lugtenburg, Eur. J. Org. Chem., 2003, 2003, 4664–4678 CrossRef.
- W. Reif and H. Grassner, Chem. Ing. Tech., 1973, 45, 646–652 CrossRef CAS.
- M. A. A. Endoma-Arias, M. Makarova, H. E. Dela Paz and T. Hudlicky, Synthesis, 2019, 51, 225–232 CrossRef CAS.
- A. Jakubska, D. Przado, M. Steininger, J. Aniol-Kwiatkowska and M. Kadej, Appl. Ecol. Environ. Res., 2005, 3, 29–38 CrossRef.
- H.-C. Lin, Z. Wang, C. Boyd, L. Simoni-Wastila and A. Buu, Addict. Behav., 2018, 76, 348–354 CrossRef PubMed.
- Y. Hirose, M. Ogawa and Y. Kusuda, Agric. Biol. Chem., 1962, 26, 526–531 CrossRef CAS.
- K. M. Markovich, V. Tantishaiyakul, A. Hamada, D. D. Miller, K. J. Romstedt, G. Shams, Y. Shin, P. F. Fraundorfer, K. Doyle and D. R. Feller, J. Med. Chem., 1992, 35, 466–479 CrossRef CAS.
- H. Koizumi, S. Yokoshima and T. Fukuyama, Chem.–Asian J., 2010, 5, 2192–2198 CrossRef CAS PubMed.
- M. P. Tsyurupa, Z. K. Blinnikova, M. M. Il’in, V. A. Davankov, O. O. Parenago, O. I. Pokrovskii and O. I. Usovich, Russ. J. Phys. Chem. A, 2015, 89, 2064–2071 CrossRef CAS.
- P. Xing, Z.-g. Huang, Y. Jin and B. Jiang, Synthesis, 2013, 45, 596–600 CrossRef CAS.
- F. Li, J. Xie, H. Shan, C. Sun and L. Chen, RSC Adv., 2012, 2, 8645–8652 RSC.
- D. Azarifar and F. Soleimanei, RSC Adv., 2014, 4, 12119–12126 RSC.
- E. V. Papp and J. Pogany, Angew. Chem., 1941, 54, 55 CrossRef.
- H. Itokawa, O. Shirota, H. Ikuta, H. Morita, K. Takeya and Y. Iitaka, Phytochemistry, 1991, 30, 3713–3716 CrossRef CAS.
- W. Wang, J. Guo, J. Zhang, J. Peng, T. Liu and Z. Xin, Food Chem., 2015, 171, 40–49 CrossRef CAS PubMed.
- A. M. Nauth, N. Otto and T. Opatz, Adv. Synth. Catal., 2015, 357, 3424–3428 CrossRef CAS.
- M. a. D. Guillén and M. a. J. Manzanos, Food Chem., 2002, 79, 283–292 CrossRef.
- T. R. Nunn, J. B. Howard, J. P. Longwell and W. A. Peters, Ind. Eng. Chem. Process Des. Dev., 1985, 24, 844–852 CrossRef CAS.
- M. Del Bel, A. R. Abela, J. D. Ng and C. A. Guerrero, J. Am. Chem. Soc., 2017, 139, 6819–6822 CrossRef CAS PubMed.
- J. R. Hanson, M. A. O'Leary, H. J. Wadsworth and L. Y. Boon, Phytochemistry, 1988, 27, 387–389 CrossRef CAS.
- P. W. Brian, P. J. Curtis, H. G. Hemming and J. C. McGowan, Ann. Appl. Biol., 1946, 33, 190–200 CrossRef CAS PubMed.
- J. M. Barbosa-Filho, M. Yoshida, O. R. Gottlieb, R. de C. S. B. C. Barbosa, A. M. Giesbrecht, M. Claudia and M. Young, Phytochemistry, 1987, 26, 2615–2617 CrossRef CAS.
- A. Hadfield, H. Schweitzer, M. P. Trova and K. Green, Synth. Commun., 1994, 24, 1025–1028 CrossRef CAS.
- A. Fürstner and I. Konetzki, Tetrahedron Lett., 1998, 39, 5721–5724 CrossRef.
- H.-P. Xiong, J.-L. Mi, J.-M. Le, Z.-J. Wu and W.-S. Chen, Chem. Nat. Compd., 2017, 53, 791–793 CrossRef CAS.
- B. A. Czeskis, P. Baeckström, A. M. Moiseenkov and T. Norin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 131–134 CrossRef.
- M. Miyazawa and H. Kameoka, Agric. Biol. Chem., 1988, 52, 1053–1055 CAS.
- B. Giese and U. Erfort, Angew. Chem., Int. Ed., 1982, 21, 130–131 CrossRef.
- Y. Leng, J. Liu and X. Feng, China Pat., CN105503923 (A), 2016.
- M. Grodzki and G. Kraemer, Ber. Dtsch. Chem. Ges., 1874, 7, 1492–1497 CrossRef.
- O. Kamm and C. Marvel, Org. Synth., 1921, 1, 15–17 CrossRef.
- J. Houben, J. Boedler and W. Fischer, Ber. Dtsch. Chem. Ges., 1936, 69, 1766–1788 CrossRef.
- M. V. Kazantseva, B. V. Timokhin, A. V. Rokhin, D. G. Blazhev, A. I. Golubin and Y. V. Rybakova, Russ. J. Gen. Chem., 2001, 71, 1233–1235 CrossRef CAS.
- H. Shi, C. Du, X. Zhang, F. Xie, X. Wang, S. Cui, X. Peng, M. Cheng, B. Lin and Y. Liu, J. Org. Chem., 2018, 83, 1312–1319 CrossRef CAS PubMed.
- M.-J. Cheng, I.-L. Tsai and I.-S. Chen, J. Chin. Chem. Soc., 2001, 48, 235–239 CrossRef CAS.
- S. Kobayashi, T. Tokumoto and Z. Taira, J. Chem. Soc., Chem. Commun., 1984, 1043–1044 RSC.
- H. Meshulam and D. Lavie, Phytochemistry, 1980, 19, 2633–2635 CrossRef CAS.
- A. Lipp, D. Ferenc, C. Gütz, M. Geffe, N. Vierengel, D. Schollmeyer, H. J. Schäfer, S. R. Waldvogel and T. Opatz, Angew. Chem., Int. Ed., 2018, 57, 11055–11059 CrossRef CAS.
- S. Ghosal and R. S. Srivastava, Phytochemistry, 1973, 12, 193–197 CrossRef CAS.
- B. Pfundstein, S. K. El Desouky, W. E. Hull, R. Haubner, G. Erben and R. W. Owen, Phytochemistry, 2010, 71, 1132–1148 CrossRef CAS.
- A. Lipp, M. Selt, D. Ferenc, D. Schollmeyer, S. R. Waldvogel and T. Opatz, Org. Lett., 2019, 21, 1828–1831 CrossRef CAS.
- L. Hu, Z. Qiuyun, R. Anders and Y. Song, Curr. Nanosci., 2015, 11, 1–14 CrossRef.
- W. Den, V. K. Sharma, M. Lee, G. Nadadur and R. S. Varma, Front. Chem., 2018, 6, 141 CrossRef.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- Z. Xue, Q. Liu, J. Wang and T. Mu, Green Chem., 2018, 20, 4391–4408 RSC.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2019, 7, 1826–1840 CrossRef CAS.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- Z. Xue, M.-G. Ma, Z. Li and T. Mu, RSC Adv., 2016, 6, 98874–98892 RSC.
- P. K. Rout, A. D. Nannaware, O. Prakash, A. Kalra and R. Rajasekharan, Chem. Eng. Sci., 2016, 142, 318–346 CrossRef CAS.
- F. Menegazzo, E. Ghedini and M. Signoretto, Molecules, 2018, 23, 2201 CrossRef PubMed.
- H. H. Szmant and D. D. Chundury, J. Chem. Technol. Biotechnol., 1981, 31, 205–212 CrossRef CAS.
- M. Mascal, ACS Sustainable Chem. Eng., 2019, 7, 5588–5601 CrossRef CAS.
- G. A. Halliday, R. J. Young and V. V. Grushin, Org. Lett., 2003, 5, 2003–2005 CrossRef CAS PubMed.
- Q. Girka, B. Estrine, N. Hoffmann, J. Le Bras, S. Marinković and J. Muzart, React. Chem. Eng., 2016, 1, 176–182 RSC.
- Y. Qian, L. Zhu, Y. Wang and X. Lu, Renewable Sustainable Energy Rev., 2015, 41, 633–646 CrossRef CAS.
- M. Braun and M. Antonietti, Green Chem., 2017, 19, 3813–3819 RSC.
- X. Zheng, X. Gu, Y. Ren, Z. Zhi and X. Lu, Biofuels, Bioprod. Biorefin., 2016, 10, 917–931 CrossRef CAS.
- S. Kang, J. Fu and G. Zhang, Renewable Sustainable Energy Rev., 2018, 94, 340–362 CrossRef CAS.
- A. M. R. Galletti, C. Antonetti, V. De Luise, D. Licursi and N. Nassi, BioResources, 2012, 7, 1824–1835 Search PubMed.
- C. Antonetti, D. Licursi, S. Fulignati, G. Valentini and A. M. Raspolli Galletti, Catalysts, 2016, 6, 196 CrossRef.
- M. S. Miftakhov, F. A. Valeev and I. N. Gaisina, Russ. Chem. Rev., 1994, 63, 869–882 CrossRef.
- J. He, M. Liu, K. Huang, T. W. Walker, C. T. Maravelias, J. A. Dumesic and G. W. Huber, Green Chem., 2017, 19, 3642–3653 RSC.
- M. B. Comba, Y.-h. Tsai, A. M. Sarotti, M. I. Mangione, A. G. Suárez and R. A. Spanevello, Eur. J. Org. Chem., 2018, 2018, 590–604 CrossRef CAS.
- A. V. Bridgwater, D. Meier and D. Radlein, Org. Geochem., 1999, 30, 1479–1493 CrossRef CAS.
- S. Maduskar, V. Maliekkal, M. Neurock and P. J. Dauenhauer, ACS Sustainable Chem. Eng., 2018, 6, 7017–7025 CrossRef CAS.
- M. S. Mettler, A. D. Paulsen, D. G. Vlachos and P. J. Dauenhauer, Energy Environ. Sci., 2012, 5, 7864–7868 RSC.
- X. Zhang, W. Yang and C. Dong, J. Anal. Appl. Pyrolysis, 2013, 104, 19–27 CrossRef CAS.
- B. T. Sharipov, A. N. Davidova, A. S. Ryabova, N. F. Galimzyanova and F. A. Valeev, Chem. Heterocycl. Compd., 2019, 55, 31–37 CrossRef CAS.
- A. L. Flourat, A. A. M. Peru, A. R. S. Teixeira, F. Brunissen and F. Allais, Green Chem., 2015, 17, 404–412 RSC.
- M. Moreaux, G. Bonneau, A. Peru, F. Brunissen, M. Janvier, A. Haudrechy and F. Allais, Eur. J. Org. Chem., 2019, 2019, 1600–1604 CrossRef CAS.
- Z. J. Witczak and K. Tatsuta, in ACS Symp. Ser., American Chemical Society, 2002, vol. 841, p. 228 Search PubMed.
- A. Tauss, T. M. Wrodnigg and A. E. Stütz, Recent Res. Dev. Org. Chem., 1999, 3, 319–342 CAS.
- K. Tatsuta and S. Hosokawa, Sci. Technol. Adv. Mater., 2006, 7, 397–410 CrossRef CAS.
- K. C. Nicolaou and H. J. Mitchell, Angew. Chem., Int. Ed., 2001, 40, 1576–1624 CrossRef CAS PubMed.
- T. Hudlicky, Chem. Rev., 1996, 96, 3–30 CrossRef CAS PubMed.
- N. Chida and T. Sato, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, pp. 207–239 Search PubMed.
- P. P. Deshpande, K. N. Price and D. C. Baker, Bioorg. Med. Chem. Lett., 1995, 5, 1059–1060 CrossRef CAS.
- C. Zhengxiong, H. Huizhu, W. Chengrui, L. Yuhui, D. Jianmi, U. Sankawa, H. Noguchi and Y. Iitaka, Heterocycles, 1984, 22, 691–694 CrossRef.
- C. Zhengxiong, H. Huizhu, W. Chengrui, L. I. Yuhui, D. Jianmi, U. Sankawa, H. Noguchi and Y. Iitaka, Chem. Pharm. Bull., 1986, 34, 2743–2746 CrossRef.
- J. S. Swenton, J. N. Freskos, P. Dalidowicz and M. L. Kerns, J. Org. Chem., 1996, 61, 459–464 CrossRef CAS PubMed.
- M. Dischmann, T. Frassetto, M. A. Breuning and U. Koert, Chem.–Eur. J., 2014, 20, 11300–11302 CrossRef CAS PubMed.
- European Food Safety Authority, EFSA J., 2005, 3, 246 CrossRef.
- A. Latrasse, E. Guichard, C. Piffaut, N. Fournier and L. Dufosse, Chirality, 1993, 5, 379–384 CrossRef CAS PubMed.
- A. A. M. Peru, A. L. Flourat, C. Gunawan, W. Raverty, M. Jevric, B. W. Greatrex and F. Allais, Molecules, 2016, 21, 988 CrossRef PubMed.
- M. Wang, M. Chen, Y. Fang and T. Tan, Biotechnol. Biofuels, 2018, 11, 30 CrossRef PubMed.
- G. B. Bachman and A. J. Hill, J. Am. Chem. Soc., 1934, 56, 2730–2732 CrossRef CAS.
- A. Fürstner, K. Radkowski, C. Wirtz, R. Goddard, C. W. Lehmann and R. Mynott, J. Am. Chem. Soc., 2002, 124, 7061–7069 CrossRef PubMed.
- J. Fausto Rivero-Cruz, G. García-Aguirre, C. M. Cerda-García-Rojas and R. Mata, Tetrahedron, 2000, 56, 5337–5344 CrossRef CAS.
- A. Nickel and R. L. Pederson, in Olefin Metathesis, ed. K. Grela, Wiley-VCH, Weinheim, 2014 Search PubMed.
- P. Jiao, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2006, 69, 636–639 CrossRef CAS PubMed.
- M. Ostermeier and R. Schobert, J. Org. Chem., 2014, 79, 4038–4042 CrossRef CAS PubMed.
- L. Andrussow, Ber. Dtsch. Chem. Ges., 1927, 60, 2005–2018 CrossRef.
- L. Andrussow, Angew. Chem., 1935, 48, 593–595 CrossRef CAS.
- A. Tilche and M. Galatola, Water Sci. Technol., 2008, 57, 1683–1692 CrossRef CAS PubMed.
- A. M. Nauth and T. Opatz, Org. Biomol. Chem., 2019, 17, 11–23 RSC.
- H. J. Bestmann and D. Sandmeier, Angew. Chem., Int. Ed. Engl., 1975, 14, 634 CrossRef.
- R. Schobert, R. K. Boeckman Jr and J. E. Pero, Org. Synth., 2005, 82, 140–143 CrossRef CAS.
- M. Kubo, C. Okada, J.-M. Huang, K. Harada, H. Hioki and Y. Fukuyama, Org. Lett., 2009, 11, 5190–5193 CrossRef CAS PubMed.
- L. Trzoss, J. Xu, M. H. Lacoske, W. C. Mobley and E. A. Theodorakis, Chem.–Eur. J., 2013, 19, 6398–6408 CrossRef CAS PubMed.
- J. Xu, L. Trzoss, W. K. Chang and E. A. Theodorakis, Angew. Chem., Int. Ed., 2011, 50, 3672–3676 CrossRef CAS PubMed.
- K. Shimizu and F. Matsushita, US Pat., US7897802B2, 2011.
- E. Lacoste, E. Vaique, M. Berlande, I. Pianet, J.-M. Vincent and Y. Landais, Eur. J. Org. Chem., 2007, 2007, 167–177 CrossRef.
- L. Trzoss, J. Xu, M. H. Lacoske, W. C. Mobley and E. A. Theodorakis, Org. Lett., 2011, 13, 4554–4557 CrossRef CAS PubMed.
- A. W. Schorger, Ind. Eng. Chem., 1925, 17, 944 CrossRef CAS.
- Pyrénéenne de Carburants et Solvants, France Pat., FR993861A, 1944.
- H. Siegel and M. Eggersdorfer, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed.
- A. L. Wilds, R. M. Nowak and K. E. McCaleb, Org. Synth., 1957, 37, 18 CrossRef CAS.
- S. H. Park and A. K. Bose, Bull. Chem. Soc. Jpn., 2001, 74, 1917–1925 CrossRef CAS.
- K. S. Pitzer and H. S. Gutowsky, J. Am. Chem. Soc., 1946, 68, 2204–2209 CrossRef CAS.
- S. M. Hande and J. i. Uenishi, Tetrahedron Lett., 2009, 50, 189–192 CrossRef CAS.
- K. Kito, R. Ookura, S. Yoshida, M. Namikoshi, T. Ooi and T. Kusumi, Org. Lett., 2008, 10, 225–228 CrossRef CAS PubMed.
- O. Ryuhei, K. Keijiro, S. Yota, K. Takenori and O. Takashi, Chem. Lett., 2009, 38, 384 CrossRef.
- P. F. Koh and T. P. Loh, Green Chem., 2015, 17, 3746–3750 RSC.
- A. Bouchardat, Ann. Pharm., 1837, 22, 225–236 CrossRef.
- B. H. Lipshutz, S. Ghorai, A. R. Abela, R. Moser, T. Nishikata, C. Duplais, A. Krasovskiy, R. D. Gaston and R. C. Gadwood, J. Org. Chem., 2011, 76, 4379–4391 CrossRef CAS PubMed.
- P. Klumphu and B. H. Lipshutz, J. Org. Chem., 2014, 79, 888–900 CrossRef CAS PubMed.
- A. Krasovskiy, C. Duplais and B. H. Lipshutz, Org. Lett., 2010, 12, 4742–4744 CrossRef CAS PubMed.
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS PubMed.
- H. Fuwa, H. Yamaguchi and M. Sasaki, Org. Lett., 2010, 12, 1848–1851 CrossRef CAS PubMed.
- M. Kanematsu, M. Yoshida and K. Shishido, Angew. Chem., Int. Ed., 2011, 50, 2618–2620 CrossRef CAS PubMed.
- J. H. Cardellina II, R. L. Hendrickson, K. P. Manfredi, S. A. Strobel and J. Clardy, Tetrahedron Lett., 1987, 28, 727–730 CrossRef.
- U. Daisuke, T. Yoshiaki, W. Ichiro and H. Yoshimasa, Chem. Lett., 1979, 8, 1481–1482 CrossRef.
- M. Brehm, W. G. Dauben, P. Köhler and F. W. Lichtenthaler, Angew. Chem., 1987, 1318–1319 CrossRef CAS.
- M. Brehm, V. H. Göckel, P. Jarglis and F. W. Lichtenthaler, Tetrahedron: Asymmetry, 2008, 19, 358–373 CrossRef CAS.
- P. Jarglis and F. W. Lichtenthaler, Angew. Chem., 1982, 94, 140–141 CrossRef CAS.
- R. J. Ferrier and G. H. Sankey, J. Chem. Soc. C, 1966, 2339–2345 RSC.
- H. Fletcher, Methods Carbohydr. Chem., 1963, 2, 226–228 Search PubMed.
- K. Maurer and R. Böhme, Ber. Dtsch. Chem. Ges. A/B, 1936, 1399–1410 CrossRef CAS.
- H. Zhao, S. Hans, X. Cheng and D. R. Mootoo, J. Org. Chem., 2001, 66, 1761–1767 CrossRef CAS PubMed.
- H. Zhao and D. R. Mootoo, J. Org. Chem., 1996, 61, 6762–6763 CrossRef CAS PubMed.
- L. D. Hohenschutz, E. A. Bell, P. J. Jewess, D. P. Leworthy, R. J. Pryce, E. Arnold and J. Clardy, Phytochemistry, 1981, 20, 811–814 CrossRef CAS.
- S. Czernecki, S. Horns and J.-M. Valery, J. Org. Chem., 1995, 60, 650–655 CrossRef CAS.
- H. Hashimoto, K. Asano, F. Fujii and J. Yoshimura, Carbohydr. Res., 1982, 104, 87–104 CrossRef CAS.
- A. Lipták, I. Jodál and P. Nánási, Carbohydr. Res., 1975, 44, 1–11 CrossRef.
- N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed.
- M. J. Hülsey, Green Energy & Environment, 2018, 3, 318–327 Search PubMed.
- X. Chen, Y. Gao, L. Wang, H. Chen and N. Yan, ChemPlusChem, 2015, 80, 1565–1572 CrossRef CAS.
- X. Chen, H. Yang and N. Yan, Chem.–Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- F. M. Kerton, Y. Liu, K. W. Omari and K. Hawboldt, Green Chem., 2013, 15, 860–871 RSC.
- R. A. Muzzarelli, C. Jeuniaux and G. W. Gooday, Chitin in nature and technology, Springer, Boston, 1986 Search PubMed.
- S. Salmon and S. M. Hudson, J. Macromol. Sci., Chem., 1997, 37, 199–276 Search PubMed.
- K. Y. Zhu, H. Merzendorfer, W. Zhang, J. Zhang and S. Muthukrishnan, Annu. Rev. Entomol., 2016, 61, 177–196 CrossRef CAS PubMed.
- M. N. V. R. Kumar, R. A. A. Muzzarelli, C. Muzzarelli, H. Sashiwa and A. J. Domb, Chem. Rev., 2004, 104, 6017–6084 CrossRef PubMed.
- K. Müller, C. Zollfrank and M. Schmid, Macromol. Mater. Eng., 2019, 1800760 CrossRef.
- Q. Yan, E. Hong and S. S. Fong, Appl. Microbiol. Biotechnol., 2017, 101, 7567–7578 CrossRef CAS PubMed.
- I. Younes and M. Rinaudo, Mar. Drugs, 2015, 13, 1133 CrossRef CAS PubMed.
- R. N. Tharanathan and F. S. Kittur, Crit. Rev. Food Sci. Nutr., 2003, 43, 61–87 CrossRef CAS PubMed.
- S. Hitoshi, F. Shizu, Y. Naoko, K. Norioki, N. Atsuyoshi, M. Einosuke and A. Sei-ichi, Chem. Lett., 2001, 30, 308–309 CrossRef.
- J. H. Yoon, Enzyme Microb. Technol., 2005, 37, 663–668 CrossRef CAS.
- W. Xia, P. Liu and J. Liu, Bioresour. Technol., 2008, 99, 6751–6762 CrossRef CAS PubMed.
- G. Sibi, K. Dhananjaya, K. Ravikumar, H. Mallesha, R. Venkatesha, D. Trivedi, K. Bhusal and K. Gowda, Am.-Eurasian J. Sci. Res., 2013, 8, 63–67 CAS.
- X. Chen, Y. Liu, F. M. Kerton and N. Yan, RSC Adv., 2015, 5, 20073–20080 RSC.
- M. W. Drover, K. W. Omari, J. N. Murphy and F. M. Kerton, RSC Adv., 2012, 2, 4642–4644 RSC.
- K. W. Omari, L. Dodot and F. M. Kerton, ChemSusChem, 2012, 5, 1767–1772 CrossRef CAS PubMed.
- X. Chen, S. L. Chew, F. M. Kerton and N. Yan, Green Chem., 2014, 16, 2204–2212 RSC.
- T. T. Pham, G. Gözaydın, T. Söhnel, N. Yan and J. Sperry, Eur. J. Org. Chem., 2019, 2019, 1355–1360 CrossRef CAS.
- T. N. Makarieva, V. A. Denisenko, V. A. Stonik, Y. M. Milgrom and Y. V. Rashkes, Tetrahedron Lett., 1989, 30, 6581–6584 CrossRef CAS.
- T. N. Makarieva, P. S. Dmitrenok, A. M. Zakharenko, V. A. Denisenko, A. G. Guzii, R. Li, C. K. Skepper, T. F. Molinski and V. A. Stonik, J. Nat. Prod., 2007, 70, 1991–1998 CrossRef CAS PubMed.
- J. Ko and T. F. Molinski, J. Org. Chem., 2013, 78, 498–505 CrossRef CAS PubMed.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- D. Yoffe, R. Frim, S. D. Ukeles, M. J. Dagani, H. J. Barda, T. J. Benya and D. C. Sanders, in Ullmann's Encyclopedia of Industrial Chemistry, 2013 Search PubMed.
- S. Warwel, P. Bavaj, M. R. g. Klaas and B. Wolff, in Perspektiven nachwachsender Rohstoffe in der Chemie, ed. H. Eierdanz, Wiley-VCH, Weinheim, 1996, pp. 119–135 Search PubMed.
- S. Warwel, F. Brüse, C. Demes, M. Kunz and M. R. g. Klaas, Chemosphere, 2001, 43, 39–48 CrossRef CAS PubMed.
- Y. Nakagawa and K. Tomishige, Catal. Today, 2012, 195, 136–143 CrossRef CAS.
- H. P. Thomas and C. L. Wilson, J. Am. Chem. Soc., 1951, 73, 4803–4805 CrossRef CAS.
- J. Cason and W. N. Baxter, J. Org. Chem., 1958, 23, 1302–1305 CrossRef CAS.
- H. Usuki, M. Toyo-oka, H. Kanzaki, T. Okuda and T. Nitoda, Bioorg. Med. Chem., 2009, 17, 7248–7253 CrossRef CAS PubMed.
- Y. Kitamura, H. Koshino, T. Nakamura, A. Tsuchida, T. Nitoda, H. Kanzaki, K. Matsuoka and S. Takahashi, Tetrahedron Lett., 2013, 54, 1456–1459 CrossRef CAS.
- L. Cai, W. Guan, M. Kitaoka, J. Shen, C. Xia, W. Chen and P. G. Wang, Chem. Commun., 2009, 2944–2946 RSC.
- M. Nakata, S. Akazawa, S. Kitamura and K. Tatsuta, Tetrahedron Lett., 1991, 32, 5363–5366 CrossRef CAS.
- S. Takahashi, H. Terayama and H. Kuzuhara, Tetrahedron Lett., 1991, 32, 5123–5126 CrossRef CAS.
- N. S. Simpkins, S. Stokes and A. J. Whittle, Tetrahedron Lett., 1992, 33, 793–796 CrossRef CAS.
- N. S. Simpkins, S. Stokes and A. J. Whittle, J. Chem. Soc., Perkin Trans. 1, 1992, 2471–2477 RSC.
- T. Kitahara, N. Suzuki, K. Koseki and K. Mori, Biosci., Biotechnol., Biochem., 1993, 57, 1906–1909 CrossRef CAS.
- T. J. Donohoe and C. P. Rosa, Org. Lett., 2007, 9, 5509–5511 CrossRef CAS PubMed.
- S. Sakuda, A. Isogai, S. Matsumoto, A. Suzuki and K. Koseki, Tetrahedron Lett., 1986, 27, 2475–2478 CrossRef CAS.
- S. Sakuda, A. Isogai, S. Matsumoto and A. Suzuki, J. Antibiot., 1987, 40, 296–300 CrossRef CAS PubMed.
- P. J. Somers, R. C. Yao, L. E. Doolin, M. J. Mcgowan, D. S. Fukuda and J. S. Mynderse, J. Antibiot., 1987, 40, 1751–1756 CrossRef CAS PubMed.
- Y. Nishimoto, S. Sakuda, S. Takayama and Y. Yamada, J. Antibiot., 1991, 44, 716–722 CrossRef CAS PubMed.
- S. Sakuda and A. Isogai, Agric. Biol. Chem., 1988, 52, 1615 CAS.
- I. Mochida, A. Yasutake, H. Fujitsu and K. Takeshita, J. Catal., 1983, 82, 313–321 CrossRef CAS.
- A. D. Sadiq, X. Chen, N. Yan and J. Sperry, ChemSusChem, 2018, 11, 532–535 CrossRef CAS PubMed.
- F. Brucoli, A. Natoli, P. Marimuthu, M. T. Borrello, P. Stapleton, S. Gibbons and A. Schätzlein, Bioorg. Med. Chem., 2012, 20, 2019–2024 CrossRef CAS PubMed.
- F. E. Wolter, K. Schneider, B. P. Davies, E. R. Socher, G. Nicholson, O. Seitz and R. D. Süssmuth, Org. Lett., 2009, 11, 2804–2807 CrossRef CAS PubMed.
- L. Cassar, La Chimica & L'Industria, 1989, 18–22 Search PubMed.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2–29 CrossRef CAS.
- P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219–2224 CrossRef CAS PubMed.
- A. El-Faham, R. S. Funosas, R. Prohens and F. Albericio, Chem.–Eur. J., 2009, 15, 9404–9416 CrossRef CAS PubMed.
- H. Baumann, M. Bühler, H. Fochem, F. Hirsinger, H. Zoebelein and J. Falbe, Angew. Chem., Int. Ed., 1988, 27, 41–62 CrossRef.
- U. Biermann, U. Bornscheuer, M. A. R. Meier, J. O. Metzger and H. J. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 3854–3871 CrossRef CAS PubMed.
- F. D. Gunstone, Lipid Technol., 2008, 20, 48 CrossRef.
- A. S. Berenblyum, V. Y. Danyushevsky, P. S. Kuznetsov, E. A. Katsman and R. S. Shamsiev, Pet. Chem., 2016, 56, 663–671 CrossRef CAS.
- H. Brännström, H. Kumar and R. Alén, BioEnergy Res., 2018, 11, 592–613 CrossRef.
- B. Zhang, J. Wu, C. Yang, Q. Qiu, Q. Yan, R. Li, B. Wang, J. Wu and Y. Ding, BioEnergy Res., 2018, 11, 689–702 CrossRef CAS.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- U. Biermann, W. Friedt, S. Lang, W. Lühs, G. Machmüller, J. O. Metzger, M. Rüsch gen. Klaas, H. J. Schäfer and M. P. Schneider, Angew. Chem., Int. Ed., 2000, 39, 2206–2224 CrossRef CAS PubMed.
- J. O. Metzger, Eur. J. Lipid Sci. Technol., 2009, 111, 865–876 CrossRef CAS.
- J. O. Metzger and M. A. R. Meier, Eur. J. Lipid Sci. Technol., 2008, 110, 787 CrossRef CAS.
- S. Chornaja, E. Sproge, K. Dubencovs, L. Kulikova, V. Serga, A. Cvetkovs and V. Kampars, Key Eng. Mater., 2014, 604, 138–141 Search PubMed.
- A. El Roz, P. Fongarland, F. Dumeignil and M. Capron, Front. Chem., 2019, 7, 1–9 CrossRef PubMed.
- A. Martin, U. Armbruster and H. Atia, Eur. J. Lipid Sci. Technol., 2012, 114, 10–23 CrossRef CAS.
- M. R. Sahasrabudhe, J. Am. Oil Chem. Soc., 1977, 54, 323–324 CrossRef CAS.
- H. Neischlag, I. Wolff, T. Manley and R. Holland, Ind. Eng. Chem. Prod. Res. Dev., 1967, 6, 120–123 CrossRef.
- D. A. Knauft and K. M. Moore, J. Hered., 1989, 80, 252–253 Search PubMed.
- J. E. Villarreal-Lozoya, L. Lombardini and L. Cisneros-Zevallos, Food Chem., 2007, 102, 1241–1249 CrossRef CAS.
- S.-P. Chang and J. A. Rothfus, J. Am. Oil Chem. Soc., 1977, 54, 549–552 CrossRef CAS.
- C. R. Smith, M. O. Bagby, T. K. Miwa, R. L. Lohmar and I. A. Wolff, J. Org. Chem., 1960, 25, 1770–1774 CrossRef CAS.
- T. Takagi, M. Kaneniwa, Y. Itabashi and R. G. Ackman, Lipids, 1986, 21, 558–565 CrossRef CAS.
- U. Ahmad Viqar, A. Basha and W. Haque, Z. Naturforsch., B: Chem. Sci., 1978, 33, 347–348 Search PubMed.
- T. M. Smalberger, G. J. H. Rall, H. L. de Waal and R. R. Arndt, Tetrahedron, 1968, 24, 6417–6421 CrossRef CAS PubMed.
- A. N. Collins, G. Sheldrake and J. Crosby, Chirality in Industry: The Commercial Manufacture and Applications of Optically Active Compounds, Wiley-VCH, Heidelberg, 1995 Search PubMed.
- M. J. Ford, J. G. Knight, S. V. Ley and S. Vile, Synlett, 1990, 1990, 331–332 CrossRef.
- P. Dydio, R. J. Detz and J. N. H. Reek, J. Am. Chem. Soc., 2013, 135, 10817–10828 CrossRef CAS PubMed.
- P. Dydio, W. I. Dzik, M. Lutz, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2011, 50, 396–400 CrossRef CAS PubMed.
- H. W. Yang and D. Romo, J. Org. Chem., 1997, 62, 4–5 CrossRef CAS PubMed.
- H. W. Yang, C. Zhao and D. Romo, Tetrahedron, 1997, 53, 16471–16488 CrossRef CAS.
- M. Mutoh, N. Nakada, S. Matsukuma, S. Ohshima, K. Yoshinri, J. Watanabe and M. Arisawa, J. Antibiot., 1994, 47, 1369–1375 CrossRef CAS PubMed.
- K. Yoshinari, M. Aoki, T. Ohtsuka, N. Nakayama, Y. Itezono, M. Mutoh, J. Watanabe and K. Yokose, J. Antibiot., 1994, 47, 1376–1384 CrossRef CAS PubMed.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinberner and A. Westfechtel, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- J. L. Beare-Rogers, A. Dieffenbacher and J. V. Holm, Pure Appl. Chem., 2001, 73, 685–744 CAS.
- J. Gajewski, R. Pavlovic, M. Fischer, E. Boles and M. Grininger, Nat. Commun., 2017, 8, 14650–14657 CrossRef PubMed.
- Z. Tan, J. M. Yoon, A. Chowdhury, K. Burdick, L. R. Jarboe, C. D. Maranas and J. V. Shanks, Biotechnol. Biofuels, 2018, 11, 87 CrossRef PubMed.
- J. Wu, X. Zhang, X. Xia and M. Dong, Metab. Eng., 2017, 41, 115–124 CrossRef CAS PubMed.
- L. A. Kucek, C. M. Spirito and L. T. Angenent, Energy Environ. Sci., 2016, 9, 3482–3494 RSC.
- L. A. Kucek, J. Xu, M. Nguyen and L. T. Angenent, Front. Microbiol., 2016, 7, 1892 Search PubMed.
- D. Webb and T. F. Jamison, Org. Lett., 2012, 14, 568–571 CrossRef CAS PubMed.
- Q. Khuonghuu, R. Goutarel, X. Monseur and G. Ratle, Bull. Soc. Chim. Belg., 1972, 81, 443–458 CrossRef.
- Q. Khuong-Huu, G. Ratle, X. Monseur and R. Goutarel, Bull. Soc. Chim. Belg., 1972, 81, 425–441 CrossRef.
- G. Ratle, X. Monseur, B. Das, J. Yassi, Q. Khuong-Huu and R. Goutarel, Bull. Soc. Chim. Fr., 1966, 9, 2945–2947 CAS.
- C. Gnamm, K. Brödner, C. M. Krauter and G. Helmchen, Chem.–Eur. J., 2009, 15, 10514–10532 CrossRef CAS PubMed.
- C. Gnamm, G. Franck, N. Miller, T. Stork, K. Brödner and G. Helmchen, Synthesis, 2008, 2008, 3331–3350 CrossRef.
- C. Nguyen, G. F. Ruda, A. Schipani, G. Kasinathan, I. Leal, A. Musso-Buendia, M. Kaiser, R. Brun, L. M. Ruiz-Pérez, B.-L. Sahlberg, N. G. Johansson, D. González-Pacanowska and I. H. Gilbert, J. Med. Chem., 2006, 49, 4183–4195 CrossRef CAS PubMed.
- N. A. Milas, Org. Synth., 1943, 2, 302 Search PubMed.
- S. Takkellapati, T. Li and M. A. Gonzalez, Clean Technol. Environ., 2018, 20, 1615–1630 CrossRef CAS PubMed.
- T. P. West, Fermentation, 2017, 3, 14 CrossRef.
- X. Qin, T. Tzvetkov, X. Liu, D.-C. Lee, L. Yu and D. C. Jacobs, J. Am. Chem. Soc., 2004, 126, 13232–13233 CrossRef CAS PubMed.
- I. Kuroda, M. Musman, I. I. Ohtani, T. Ichiba, J. Tanaka, D. G. Gravalos and T. Higa, J. Nat. Prod., 2002, 65, 1505–1506 CrossRef CAS PubMed.
- V. Ledroit, C. Debitus, C. Lavaud and G. Massiot, Tetrahedron Lett., 2003, 44, 225–228 CrossRef CAS.
- P. Bhaket, K. Morris, C. S. Stauffer and A. Datta, Org. Lett., 2005, 7, 875–876 CrossRef CAS PubMed.
- Y. Du, J. Liu and R. J. Linhardt, J. Org. Chem., 2006, 71, 1251–1253 CrossRef CAS PubMed.
- Y. Génisson, L. Lamandé, Y. Salma, N. Andrieu-Abadie, C. André and M. Baltas, Tetrahedron: Asymmetry, 2007, 18, 857–864 CrossRef.
- J. Liu, Y. Du, X. Dong, S. Meng, J. Xiao and L. Cheng, Carbohydr. Res., 2006, 341, 2653–2657 CrossRef CAS PubMed.
- M. Passiniemi and A. M. P. Koskinen, Tetrahedron Lett., 2008, 49, 980–983 CrossRef CAS.
- K. R. Prasad and A. Chandrakumar, J. Org. Chem., 2007, 72, 6312–6315 CrossRef CAS PubMed.
- C. V. Ramana, A. G. Giri, S. B. Suryawanshi and R. G. Gonnade, Tetrahedron Lett., 2007, 48, 265–268 CrossRef CAS.
- L. V. R. Reddy, P. V. Reddy and A. K. Shaw, Tetrahedron: Asymmetry, 2007, 18, 542–546 CrossRef CAS.
- C. Ribes, E. Falomir, M. Carda and J. A. Marco, Tetrahedron, 2006, 62, 5421–5425 CrossRef CAS.
- N. Sudhakar, A. R. Kumar, A. Prabhakar, B. Jagadeesh and B. V. Rao, Tetrahedron Lett., 2005, 46, 325–327 CrossRef CAS.
- T. Yakura, S. Sato and Y. Yoshimoto, Chem. Pharm. Bull., 2007, 55, 1284–1286 CrossRef CAS PubMed.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- A. Evidente and A. Kornienko, Phytochem. Rev., 2009, 8, 449–459 CrossRef CAS.
- L. Ingrassia, F. Lefranc, V. Mathieu, F. Darro and R. Kiss, Transl. Oncol., 2008, 1, 1–13 CrossRef PubMed.
- F. Cagide-Fagín, O. Nieto-García, H. Lago-Santomé and R. Alonso, J. Org. Chem., 2012, 77, 11377–11382 CrossRef PubMed.
- P. Martínez-Bescos, F. Cagide-Fagín, L. F. Roa, J. C. Ortiz-Lara, K. Kierus, L. Ozores-Viturro, M. Fernández-González and R. Alonso, J. Org. Chem., 2008, 73, 3745–3753 CrossRef PubMed.
- J. Liu, Liquid Explosives, Springer, Berlin, Heidelberg, 2015 Search PubMed.
- D. Roberge, C. Noti, E. Irle, M. Eyholzer, B. Rittiner, G. Penn, G. Sedelmeier and B. Schenkel, J. Flow Chem., 2013, 4, 26–34 CrossRef.
- K. Show and P. Kumar, Eur. J. Org. Chem., 2016, 2016, 4696–4710 CrossRef CAS.
- H. L. Ohrem and F. Westmeier, United States Pat., US5770411A, 1998.
- C. Weizmann, Great Britain Pat., GB191504845 (A), 1919.
- F. Mussgnug, Naturwissenschaften, 1941, 29, 256 CrossRef CAS.
- D. Hellwinkel and T. Kosack, Liebigs Ann. Chem., 1985, 1985, 226–238 CrossRef.
- W. C. Still and C. Gennari, Tetrahedron Lett., 1983, 24, 4405–4408 CrossRef CAS.
- M. R. Thomsett, T. E. Storr, O. R. Monaghan, R. A. Stockman and S. M. Howdle, Green Mater., 2016, 4, 115–134 CrossRef.
- M. N. Belgacem and A. Gandini, Monomers, polymers and composites from renewable resources, Elsevier-Science, Aveiro, 2011 Search PubMed.
- W. Schwab, C. Fuchs and F. C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3–8 CrossRef CAS.
- T. J. Maimone and P. S. Baran, Nat. Chem. Biol., 2007, 3, 396 CrossRef CAS PubMed.
- S. Zwenger and C. Basu, Biotechnol. Mol. Biol. Rev., 2008, 3, 1 Search PubMed.
- S.-M. Paek, M. Jeong, J. Jo, M. Y. Heo, T. Y. Han and H. Yun, Molecules, 2016, 21, 951 CrossRef PubMed.
- M. Nakata, in Glycoscience: Chemistry and Chemical Biology, ed. B. O. Fraser-Reid, K. Tatsuta and J. Thiem, Springer Berlin Heidelberg, Berlin, Heidelberg, 2008, pp. 957–994 Search PubMed.
- T. Money and M. K. C. Wong, in Stud. Nat. Prod. Chem., ed. R. Atta ur, Elsevier, 1995, vol. 16, pp. 123–288 Search PubMed.
- W. A. Nugent, T. V. RajanBabu and M. J. Burk, Science, 1993, 259, 479 CrossRef CAS PubMed.
- M. A. Rude and A. Schirmer, Curr. Opin. Microbiol., 2009, 12, 274–281 CrossRef CAS PubMed.
- E. Breitmaier, Terpenes: flavors, fragrances, pharmaca, pheromones, Wiley-VCH, Weinheim, 2006 Search PubMed.
- V. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman and J. D. Keasling, Nat. Biotechnol., 2003, 21, 796 CrossRef CAS PubMed.
- K. A. D. Swift, Top. Catal., 2004, 27, 143–155 CrossRef CAS.
- F. M. Kerton and R. Marriott, Alternative solvents for green chemistry, Royal Society of chemistry, St. John's, 2013 Search PubMed.
- J. Schrader, in Flavours and Fragrances: Chemistry, Bioprocessing and Sustainability, ed. R. G. Berger, Springer, Berlin, Heidelberg, 2007, pp. 507–574 Search PubMed.
- J. Schrader and R. G. Berger, in Biotechnology, ed. H. J. Rehm, G. Reeed, A. Pühler and P. Stadler, John Wiley & Sons, Ltd, Weinheim, 2001, pp. 373–422 Search PubMed.
- D.-K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS PubMed.
- M. Phillips and H. D. Gibbs, J. Int. Eng. Chem., 1920, 12, 733–734 CAS.
- M. S. Ali, M. Saleem and V. U. Ahmad, Z. Naturforsch., B: Chem. Sci.f, 1999, 54, 807–810 CAS.
- M. Kuhnt, A. Pröbstle, H. Rimpler, R. Bauer and M. Heinrich, Planta Med., 1995, 61, 227–232 CrossRef CAS PubMed.
- K. Mori and K. Fukamatsu, Liebigs Ann. Chem., 1992, 1992, 489–493 CrossRef.
- M. Ping Zhao, X. Chao Liu, D. Lai, L. Zhou and Z. Long Liu, Helv. Chim. Acta, 2016, 99, 90–94 CrossRef.
- A. El Mebtoul, M. Rouani, M. Chammache, H. Bouidida and A. Ilidrissi, Helv. Chim. Acta, 2011, 94, 433–437 CrossRef CAS.
- N. Hoffmann and H.-D. Scharf, Liebigs Ann. Chem., 1991, 1991, 1273–1277 CrossRef.
- A. Rubo, R. Kellens, J. Reddy, N. Steier and W. Hasenpusch, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- C. Grundke and T. Opatz, Green Chem., 2019, 21, 2362–2366 RSC.
- C.-S. Yang, I. Kouno, N. Kawano and S. Sato, Tetrahedron Lett., 1988, 29, 1165–1168 CrossRef CAS.
- M. L. Condakes, K. Hung, S. J. Harwood and T. J. Maimone, J. Am. Chem. Soc., 2017, 139, 17783–17786 CrossRef CAS PubMed.
- S.-S. Cheng, M.-J. Chung, C.-Y. Lin, Y.-N. Wang and S.-T. Chang, J. Agric. Food Chem., 2011, 60, 124–128 CrossRef PubMed.
- K. Bauer, D. Garbe and H. Surburg, Common Fragrance and Flavor Materials, Wiley-VCH, Weinheim, New York, 1997 Search PubMed.
- X. Hu, S. Xu and T. J. Maimone, Angew. Chem., Int. Ed., 2017, 56, 1624–1628 CrossRef CAS PubMed.
- Y.-r. Naves, Helv. Chim. Acta, 1949, 32, 967–968 CrossRef CAS PubMed.
- C. C. C. R. de Carvalho and M. M. R. da Fonseca, Food Chem., 2006, 95, 413–422 CrossRef CAS.
- Y. Shimizu, S. Matsuto, Y. Mizunuma and I. Okada, Agric. Biol. Chem., 1970, 34, 437–441 CAS.
- F. H. Sangsari, F. Chastrette and M. Chastrette, Synth. Commun., 1988, 18, 1343–1348 CrossRef CAS.
- J. Švenda and A. G. Myers, Org. Lett., 2009, 11, 2437–2440 CrossRef PubMed.
- W. Herz, A. Srińivasan and P. S. Kalyanaraman, Phytochemistry, 1975, 14, 233–237 CrossRef CAS.
- H. O. Yang, D.-Y. Suh and B. H. Han, Planta Med., 1995, 61, 37–40 CrossRef CAS PubMed.
- H. Jianmei and Y. Chunshu, Phytochemistry, 1996, 42, 1375–1376 CrossRef CAS.
- K. Hung, M. L. Condakes, T. Morikawa and T. J. Maimone, J. Am. Chem. Soc., 2016, 138, 16616–16619 CrossRef CAS PubMed.
- F. M. Couchman, A. R. Pinder and N. H. Bromham, Tetrahedron, 1964, 20, 2037–2045 CrossRef CAS.
- S. Kamchonwongpaisan, C. Nilanonta, B. Tarnchompoo, C. Thebtaranonth, Y. Thebtaranonth, Y. Yuthavong, P. Kongsaeree and J. Clardy, Tetrahedron Lett., 1995, 36, 1821–1824 CrossRef CAS.
- X. Hu and T. J. Maimone, J. Am. Chem. Soc., 2014, 136, 5287–5290 CrossRef CAS PubMed.
- M. Martín-Rodríguez, R. Galán-Fernández, A. Marcos-Escribano and F. A. Bermejo, J. Org. Chem., 2009, 74, 1798–1801 CrossRef PubMed.
- M. Yoshikawa, E. Hadrada, A. Kawaguchi, J. Yamahara, N. Murakami and I. Kitagawa, Chem. Pharm. Bull., 1993, 41, 630–632 CrossRef CAS.
- K. Weinges and G. Schwarz, Liebigs Ann. Chem., 1993, 1993, 811–814 CrossRef.
- S. Hatakeyama, M. Kawamura, Y. Mukugi and H. Irie, Tetrahedron Lett., 1995, 36, 267–268 CrossRef CAS.
- A. M. Szpilman, E. E. Korshin, H. Rozenberg and M. D. Bachi, J. Org. Chem., 2005, 70, 3618–3632 CrossRef CAS PubMed.
- E. E. Korshin, R. Hoos, A. M. Szpilman, L. Konstantinovski, G. H. Posner and M. D. Bachi, Tetrahedron, 2002, 58, 2449–2469 CrossRef CAS.
- M. Persson, K. Sjödin, A.-K. Borg-Karlson, T. Norin and I. Ekberg, Phytochemistry, 1996, 42, 1289–1297 CrossRef CAS.
- C. Suire, Y. Asakawa, M. Toyota and T. Takemoto, Phytochemistry, 1982, 21, 349–352 CrossRef CAS.
- K. Sennewald, E. Schallus and F. Pohl, Chem. Ing. Tech., 1963, 35, 1–6 CrossRef CAS.
- P. V. S. N. Vani, A. S. Chida, R. Srinivasan, M. Chandrasekharam and A. K. Singh, Synth. Commun., 2001, 31, 219–224 CrossRef CAS.
- Q.-W. Song, Q.-N. Zhao, J.-Y. Li, K. Zhang and P. Liu, Synthesis, 2019, 51, 739–746 CrossRef CAS.
- D. Limat and M. Schlosser, Tetrahedron, 1995, 51, 5799–5806 CrossRef CAS.
- X.-T. Liang, D.-Q. Yu, W.-L. Wu and H.-C. Deng, Acta Chim. Sin., 1979, 37, 215–230 CAS.
- F.-M. Xi, Y.-B. Liu, J. Qu, Y. Li, Z.-H. Tang, L. Li, Y.-H. Li, X.-G. Chen, S.-G. Ma and S.-S. Yu, Tetrahedron, 2017, 73, 571–582 CrossRef CAS.
- T. C. McMorris, R. Lira, P. K. Gantzel, M. J. Kelner and R. Dawe, J. Nat. Prod., 2000, 63, 1557–1559 CrossRef CAS PubMed.
- G. Liu and D. Romo, Angew. Chem., 2011, 123, 7679–7682 CrossRef.
- A. W. v. Hofman, Justus Liebigs Ann. Chem., 1860, 115, 267–269 CrossRef.
- C. Yuan, B. Du, L. Yang and B. Liu, J. Am. Chem. Soc., 2013, 135, 9291–9294 CrossRef CAS PubMed.
- R. P. Limberger, A. M. Aleixo, A. G. Fett-Neto and A. T. Henriques, Electron. J. Biotechnol., 2007, 10, 500–507 Search PubMed.
- H.-H. Ko, W.-L. Chang and T.-M. Lu, J. Nat. Prod., 2008, 71, 1930–1933 CrossRef CAS PubMed.
- L. Acebey, V. Jullian, D. Sereno, S. Chevalley, Y. Estevez, C. Moulis, S. Beck, A. Valentin, A. Gimenez and M. Sauvain, Planta Med., 2010, 76, 365–368 CrossRef CAS PubMed.
- F. Bohlmann, C. Zdero, R. M. King and H. Robinson, Phytochemistry, 1980, 19, 689–691 CrossRef CAS.
- F. Bohlmann, H. Suding, J. Cuatrecasas, R. M. King and H. Robinson, Phytochemistry, 1980, 19, 267–271 CrossRef CAS.
- N. J. Sadgrove, M. Gonçalves-Martins and G. L. Jones, Phytochemistry, 2014, 104, 60–71 CrossRef CAS PubMed.
- G. M. Petrović, J. G. Stamenković, I. R. Kostevski, G. S. Stojanović, V. D. Mitić and B. K. Zlatković, Chem. Biodiversity, 2017, 14, e1600367 CrossRef PubMed.
- L. Acebey, M. Sauvain, S. Beck, C. Moulis, A. Gimenez and V. Jullian, Org. Lett., 2007, 9, 4693–4696 CrossRef CAS PubMed.
- P. S. Baran and J. M. Richter, J. Am. Chem. Soc., 2005, 127, 15394–15396 CrossRef CAS PubMed.
- K. Stratmann, R. E. Moore, R. Bonjouklian, J. B. Deeter, G. M. Patterson, S. Shaffer, C. D. Smith and T. A. Smitka, J. Am. Chem. Soc., 1994, 116, 9935–9942 CrossRef CAS.
- P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS PubMed.
- A. Baeyer, Justus Liebigs Ann. Chem., 1866, 140, 295–296 CrossRef.
- H. Weinstabl, T. Gaich and J. Mulzer, Org. Lett., 2012, 14, 2834–2837 CrossRef CAS PubMed.
- K. Simon, J. Wefer, E. Schöttner and T. Lindel, Angew. Chem., Int. Ed., 2012, 51, 10889–10892 CrossRef CAS PubMed.
- E. Schöttner, M. Wiechoczek, P. G. Jones and T. Lindel, Org. Lett., 2010, 12, 784–787 CrossRef PubMed.
- G. D. Prestwich, D. F. Wiemer, J. Meinwald and J. Clardy, J. Am. Chem. Soc., 1978, 100, 2560–2561 CrossRef CAS.
- M. A. Umbreit and K. B. Sharpless, J. Am. Chem. Soc., 1977, 99, 5526–5528 CrossRef CAS.
- T. Takeo, Phytochemistry, 1981, 20, 2145–2147 CrossRef CAS.
- T. W. Doyle, D. E. Nettleton, R. E. Grulich, D. M. Balitz, D. L. Johnson and A. L. Vulcano, J. Am. Chem. Soc., 1979, 101, 7041–7049 CrossRef CAS.
- D. E. Nettleton Jr, D. M. Balitz, T. W. Doyle, W. T. Bradner, D. L. Johnson, F. A. O'Herron, R. H. Schreiber, A. B. Coon, J. E. Moseley and R. W. Myllymaki, J. Nat. Prod., 1980, 43, 242–258 CrossRef CAS PubMed.
- H. S. Kim, Y. H. Kim, O. J. Yoo and J. J. Lee, Biosci., Biotechnol., Biochem., 1996, 60, 906–908 CrossRef CAS PubMed.
- H.-S. Kim, Y.-S. Hong, Y.-H. Kim, O.-J. Yoo and J.-J. Lee, J. Antibiot., 1996, 49, 355–360 CrossRef CAS PubMed.
- S. M. Salem, S. Weidenbach and J. Rohr, ACS Chem. Biol., 2017, 12, 2529–2534 CrossRef CAS PubMed.
- K. A. Shaaban, T. A. Ahmed, M. Leggas and J. Rohr, J. Nat. Prod., 2012, 75, 1383–1392 CrossRef CAS PubMed.
- A. Klasek, P. Sedmera, A. Boeva and F. Šantavý, Collect. Czech. Chem. Commun., 1973, 38, 2504–2512 CrossRef CAS.
- A. Klásek, P. Sedmera, J. Vokoun, A. Boeva, S. Dvoráčková and F. Šantavý, Collect. Czech. Chem. Commun., 1980, 45, 548–558 CrossRef.
- N. T. Nghia, P. Sedmera, A. Klasek, A. Boeva, L. Drjanovska, L. Dolejš and F. Šantavý, Collect. Czech. Chem. Commun., 1976, 41, 2952–2963 CrossRef.
- A.-L. Pérez-Castorena, A. Arciniegas, A. Castro, J. L. Villaseñor, R. A. Toscano and A. Romo de Vivar, J. Nat. Prod., 1997, 60, 1322–1325 CrossRef.
- D. Robins, Nat. Prod. Rep., 1995, 12, 413–418 RSC.
- A. Romo de Vivar, A.-L. Pérez, A. Arciniegas, P. Vidales, R. Gaviño and J. L. Villaseñor, Tetrahedron, 1995, 51, 12521–12528 CrossRef CAS.
- J. Y. Sim, G.-S. Hwang, K. H. Kim, E. M. Ko and D. H. Ryu, Chem. Commun., 2007, 5064–5065 RSC.
- V. Bailliez, R. M. de Figueiredo, A. Olesker and J. Cleophax, Tetrahedron Lett., 2003, 44, 9151–9153 CrossRef CAS.
- J. Qian-Cutrone, T. Ueki, S. Huang, K. A. Mookhtiar, R. Ezekiel, S. S. Kalinowski, K. S. Brown, J. Golik, S. Lowe and D. M. Pirnik, J. Antibiot., 1999, 52, 245–255 CrossRef CAS PubMed.
- K. Tomita, Y. Hoshino and T. Miyaki, Int. J. Syst. Evol. Microbiol., 1993, 43, 297–301 CAS.
- M. Tsunakawa, N. Komiyama, O. Tenmyo, K. Tomita, K. Kawano, C. Kotake, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 1467–1471 CrossRef CAS PubMed.
- M. Tsunakawa, C. Kotake, T. Yamasaki, T. Moriyama, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 1472–1480 CrossRef CAS PubMed.
- S. Velarde, J. Urbina and M. R. Peña, J. Org. Chem., 1996, 61, 9541–9545 CrossRef CAS.
- A. Fürstner, Eur. J. Org. Chem., 2004, 2004, 943–958 CrossRef.
- A. Fürstner, M. Albert, J. Mlynarski and M. Matheu, J. Am. Chem. Soc., 2002, 124, 1168–1169 CrossRef PubMed.
- A. Fürstner, M. Albert, J. Mlynarski, M. Matheu and E. DeClercq, J. Am. Chem. Soc., 2003, 125, 13132–13142 CrossRef PubMed.
- A. Fürstner, J. Mlynarski and M. Albert, J. Am. Chem. Soc., 2002, 124, 10274–10275 CrossRef PubMed.
- A. Fürstner, J. Ruiz-Caro, H. Prinz and H. Waldmann, J. Org. Chem., 2004, 69, 459–467 CrossRef PubMed.
- D. V. Jarikote and P. V. Murphy, Eur. J. Org. Chem., 2010, 2010, 4959–4970 CrossRef.
- V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517 CrossRef CAS PubMed.
- R. Paramashivappa, P. P. Kumar, P. J. Vithayathil and A. S. Rao, J. Agric. Food Chem., 2001, 49, 2548–2551 CrossRef CAS PubMed.
- J. Mgaya, G. B. Shombe, S. C. Masikane, S. Mlowe, E. B. Mubofu and N. Revaprasadu, Green Chem., 2019, 21, 1186–1201 RSC.
- E. B. Mubofu, Pure Appl. Chem., 2016, 88, 17–27 CAS.
- C. N. Subbarao, K. Krishna Prasad and V. Prasad, The Pharma Research Journal, 2011, 6, 21–41 Search PubMed.
- E. A. Taiwo, in Advances in Petrochemicals, IntechOpen, Ile-Ife, 2015 Search PubMed.
- S. Kumar, P. Dinesha and M. A. Rosen, Energy Fuels, 2018, 32, 7237–7244 CrossRef CAS.
- M. C. Lubi and E. T. Thachil, Des. Monomers Polym., 2000, 3, 123–153 CrossRef CAS.
- A. S. Trita, L. C. Over, J. Pollini, S. Baader, S. Riegsinger, M. A. R. Meier and L. J. Gooßen, Green Chem., 2017, 19, 3051–3060 RSC.
- C. Voirin, S. Caillol, N. V. Sadavarte, B. V. Tawade, B. Boutevin and P. P. Wadgaonkar, Polym. Chem., 2014, 5, 3142–3162 RSC.
- L. Kisula, S. J. M. Mdachi, C. B. d. Koning and Q. A. Mgani, Tanz. J. Sci., 2015, 41, 27–37 Search PubMed.
- J. E. Mgaya, E. B. Mubofu, Q. A. Mgani, D. B. Cordes, A. M. Slawin and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2015, 117, 190–199 CrossRef CAS.
- J. A. Mmongoyo, Q. A. Mgani, S. J. M. Mdachi, P. J. Pogorzelec and D. J. Cole-Hamilton, Eur. J. Lipid Sci. Technol., 2012, 114, 1183–1192 CrossRef CAS.
- V. Bragoni, R. K. Rit, R. Kirchmann, A. S. Trita and L. J. Gooßen, Green Chem., 2018, 20, 3210–3213 RSC.
- P. Peungjitton, P. Sangvanich, S. Pornpakakul, A. Petsom and S. Roengsumran, J. Surfactants Deterg., 2009, 12, 85–89 CrossRef CAS.
- N. D. Ghatge and S. P. Vernekar, Die Angewandte Makromolekulare Chemie, 1971, 20, 165–174 CrossRef CAS.
- M. L. d. Santos and G. C. d. Magalhães, J. Braz. Chem. Soc., 1999, 10, 13–20 CrossRef.
- D. C. Aldridge, S. Galt, D. Giles and W. B. Turner, J. Chem. Soc. C, 1971, 1623–1627 RSC.
- K.-H. Leet, N. Hayashi, M. Okano, I. H. Hall, R.-Y. Wu and A. T. McPhailti, Phytochemistry, 1982, 21, 1119–1121 CrossRef.
- M. Pérez-Sánchez and P. D. de María, Catal. Sci. Technol., 2013, 3, 2732–2736 RSC.
- M. Mascal and S. Dutta, Green Chem., 2011, 13, 3101–3102 RSC.
- F. Chang, S. Dutta, J. J. Becnel, A. S. Estep and M. Mascal, J. Agric. Food Chem., 2014, 62, 476–480 CrossRef CAS PubMed.
- Y. Shi, P. C. J. Kamer and D. J. Cole-Hamilton, Green Chem., 2019, 21, 1043–1053 RSC.
- R. A. Fernandes, Protecting-Group-Free Organic Synthesis: Improving Economy and Efficiency, Wiley-VCH, Mumbai, Weinheim, 2018 Search PubMed.
- R. W. Hoffmann, Synthesis, 2006, 2006, 3531–3541 CrossRef.
- E. Roulland, Angew. Chem., Int. Ed., 2011, 50, 1226–1227 CrossRef CAS PubMed.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS PubMed.
Footnote |
| † Jonas Kühlborn and Jonathan Groß contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |