Occurrence, synthesis and applications of natural and designed [3.3.3]propellanes
Alicia M.
Dilmaç
a,
Tim
Wezeman
a,
Robin M.
Bär
a and
Stefan
Bräse
*ab aInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu bInstitute of Toxicology and Genetics (ITG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
Received
10th October 2018
First published on 29th May 2019
Abstract
Covering: 1978 to 2019
The synthetically challenging [3.3.3]propellane core has caught a lot of attention over the last 50 years. This comprehensive review details all synthetic strategies reported in the period 1978–2019 to facilitate the synthesis of carbocyclic [3.3.3]propellanes. The described strategies span from acid-catalyzed rearrangements and photo-mediated cycloadditions of ketones, heteropropellanes and dispiroundecanes to thermal rearrangements of acetylenes and alkenes. Other approaches, such as radical reactions with halogenated alkenes, domino cyclizations, the smart use of epoxide-carbonyl rearrangements and intramolecular palladium-catalyzed ring contractions are discussed as well. A special section is dedicated to triptindanes, a subclass of [3.3.3]propellanes which are of interest to material sciences.
A. M. Dilmaç
Alicia M. Dilmaç graduated in chemistry at the Ecole Nationale Supérieure de Chimie de Clermont-Ferrand in France in 2008. The very same year, she obtained a Master's degree from the University of Salford with distinction. She did her PhD under the supervision of Prof. Marie-Christine Lallemand at the Pharmacognosy Laboratory of the Paris Descartes University in 2012. In two postdoctoral stays she worked with Prof. Stefan Bräse (KIT) and in Prof. Pannecoucke's group (supervised by Prof. Jaubault & Prof. Poisson) at the COBRA Laboratory in Rouen. Her main topics of interest are bioconjugation, propellanes and fluorine chemistry.
T. Wezeman
Tim Wezeman obtained his PhD from the Karlsruhe Institute of Technology in 2016 under guidance of Prof. Stefan Bräse, for his work on heterocyclic reaction methodologies. He then moved to Switzerland to join Prof. Kay Severin at the EPFL as a Marie Curie “EPFL Fellow” to study the chemical activation of nitrous oxide and explore the reactivity of alkynyl triazenes. In summer of 2019 Tim will join Prof. Luc Brunsveld at the Laboratory of Chemical Biology at the Eindhoven University of Technology as a Marie Curie Individual Fellow, working in the field of supramolecular chemical biology with a strong focus on cucurbiturils.
R. M. Bär
Robin M. Bär obtained his BSc in Chemical Biology at the Karlsruhe Institute of Technology (KIT) in 2014. After an internship in medicinal chemistry at Bayer Pharma AG he finished his MSc in Chemical Biology in 2016 at the KIT. Subsequently, he started as a PhD student in the group of Prof. S. Bräse working on propellanes and their application in the synthesis of bioactive compounds.
S. Bräse
Stefan Bräse studied in Göttingen, Bangor (UK), and Marseille and received his PhD in 1995 with Armin de Meijere in Göttingen. After postdoctoral research at Uppsala University (Jan E. Bäckvall) and The Scripps Research Institute (K. C. Nicolaou), he began his independent research career at the RWTH Aachen in 1997 (associated with Dieter Enders). In 2001, he finished his Habilitation and moved to the University of Bonn as Professor for organic chemistry. Since 2003, he has been Professor at the Karlsruhe Institute of Technology (KIT) and since 2012 also Director of the Institute of Toxicology and Genetics (ITG) at the KIT. His research interests include asymmetric processes, combinatorial methods of biologically active compounds and synthetic functional materials.
1 Introduction
Propellanes are polycyclic compounds in which three ring systems share a single bond (Fig. 1). As such, propellanes constitute an exciting class of tricyclo[m.n.p.0x,y]alkanes1 that covers a wide array of natural and synthetic products. Among them are [1.1.1]propellanes, which have a unique inverted tetrahedral geometry and therefore possess unconventional reactivity. These propellanes have raised a special interest in the scientific community, becoming the subject of numerous studies, publications and reviews.2–5 They hold promise to “escape the flatland” in a way that compounds with rigid three-dimensional saturated structures can lead to improved properties, e.g. solubility, compared to aryl systems.6–8 Although propellanes with larger ring sizes, such as the intriguing [3.3.3]propellanes do not share the same properties of their counterparts with smaller rings,9–11 they have equally fascinated many organic chemists by the originality of their structures and by the challenge that their syntheses represent.
Fig. 1 Examples for the propellane nomenclature, introduced by Ginsburg in 1966.1 The numbers in the brackets indicate the number of atoms in one ring additionally to the central bond (bold).
Indeed, since the discovery of the natural [3.3.3]propellane modhephene (1) in 1978, numerous synthetic strategies have been reported to access [3.3.3]propellanes. While most of these approaches aim at the formal or total synthesis of modhephene and its derivatives, several of the recently reported synthetic strategies give access to new synthetic structures, some of which have found interesting applications in the area of material sciences. In particular, triptindanes have recently proven to be of high interest in the construction of 2D materials and hold great promise for further applications in this field. Similarly, the recent isolation of a new taxane bearing a unique [3.3.3]propellane-type structure provides a new ambitious synthetic target. Despite this increasing amount of new [3.3.3]propellanes being reported and the promises that these structures hold in terms of future applications, no reviews have been dedicated to the detailed account of the synthetic strategies and applications of this family of compounds. Thus, our aim is to provide a comprehensive overview of the [3.3.3]propellanes known to date, of their occurrence in nature and of the various synthetic strategies reported until now to access such propellanic structures. In this regard, this review covers the synthetic efforts made to access carbocyclic [3.3.3]propellanes and derivatives thereof between 1978 and 2019, with a strong focus on reported formal and total syntheses of natural products. For better clarity, the synthetic strategies are sorted based on the type of the propellane forming step, i.e. the formation of the tricyclic core. Information on other classes of [m.n.p]propellanes can be found in a variety of other reviews.5,12,13
2 Isolation of naturally occurring [3.3.3]propellanes and biosynthetic aspects
2.1 Modhephene
The most studied natural product containing a [3.3.3]propellane core is the propellane triquinane modhephene (1) (Fig. 2). Zalkow et al.14 were the first to isolate this sesquiterpene in 1978 from Isocoma wrightii, Asteraceae (Rayless Goldenrod). Since then, it was found in different plants of the Asteraceae family, such as in the roots of Silphium perfoliatum (L.),15 in the rhizomes of Echinops giganteus var. lelyi (C. D. Adams)16 and in many species of the Berkheya genus (e.g. in Berkheya radula).17 The intriguing molecular structure of modhephene was elucidated by X-ray crystallography of its diol derivative 2 (Fig. 2). Thanks to the enantioselective synthesis of modhephene and optical rotation comparisons, the absolute configuration of (−)-modhephene as found in Isocoma wrightii was determined to be 1R,5S,8R.18–20 The same absolute configuration was confirmed later by Fitjer et al. for modhephene in Otanthus maritimus (L.) Hoffmanns. & Link, Asteraceae (cotton weed).21 They also showed that this triquinane was produced enantiopure by this plant.
Fig. 2 Modhephene (1) and the molecular structure of the diol derivative 2 as reported by Zalkow et al.14
The biosynthesis commonly admitted for modhephene (1) is depicted in Scheme 1.
Scheme 1 Proposed biosynthetic routes to modhephene (1) and derivatives in plants.
When isolating 1 for the first time, Zalkow et al. proposed a biosynthesis that would involve the main sesquiterpenoid found in Isocoma wrightii, caryophyllene (6), as a common precursor to all triquinanes isolated from this plant.14 Soon after, Bohlmann et al. proposed a mechanism based on the formation in few steps of the presilphiperfolan-8-yl cation (8) from 6.15 This key carbocation would then rearrange through 1,2- and 1,3 bond shifts to yield different sesquiterpenes, including 1. Noteworthy, this hypothesis was later supported by works of Fitjer et al., who showed the possibility to access different triquinanes, including 1, through the acid-catalyzed rearrangement via a common cationic precursor, similar to 10, and through successive 1,2-bond shifts.22 These works are part of the synthetic section of this review.
This hypothetic pathway has also been strengthened by works of Coates and coworkers, who achieved the acid-catalyzed rearrangement of alcohol 12 into angular triquinanes,23 but also observed the formation of traces of 1 in the solvolysis of mesylated silphin-1α and -1β-yl mesylates 13 and 14 (Scheme 1).24
Noteworthy, an equivalent pathway was proposed by Hanson and coworkers for the biosynthesis of dihydrobotrydial (15) and botrydial (16), which are found in the fungus Botrytis cinerea.25–27 In this case, the proposed route was also based on the cationic rearrangement of caryophillenyl cation 4. It was suggested that this cation would be formed from farnesyl pyrophosphate (FPP) by cyclization. This hypothesis was then corroborated with NMR studies of the metabolites produced by Botrytis cinerea that had been fed with labelled precursors.25–27
Among the many studies on the subject, one of them showed the implication of the gene BcBOT2 in the biosynthesis of botrydial.28 This gene encodes for a terpene synthase that was shown to catalyze the cyclization of FPP into 12. Thus, given the abovementioned results, one can fairly hypothetize that enzymes with similar function would be involved in the biosynthesis of 1 and its derivatives in other natural sources.
For full details about historical and recent works concerning the biosynthesis of 1 and other triquinanes, the reader is invited to consult the nice review from Stoltz and coworkers on the bio- and chemical synthesis of presilphiperfolanol natural products, as well as the references therein.29
2.2 Simple modhephene derivatives
Other natural products that have a [3.3.3]propellane core include modhephene derivatives (Fig. 3) that were isolated from different sources. The epoxide derivative 17 was isolated from the aerial parts of Grindelia paludosa (Asteraceae) and was found amongst a wide array of diterpenes.30 Only 1 mg of the modhephenepoxide was isolated from 900 g of aerial plant material. Its structure was confirmed by comparison of NMR and TLC data of a synthetic modhephenepoxide, prepared using mCPBA under basic conditions in dichloromethane.30 Another modhephene derivative 18 was isolated in 2003 from the essential oil extracted from fresh Psiadia anchusifolia (Asteraceae) leaves.31 The structure of this novel hydroxide derivative of modhephene was elucidated using modern NMR and X-ray diffraction techniques.31 13-Acetoxymodhephene (19) was isolated in 1979 from the roots of Liabum eggersii Hieron (Asteraceae),32 while rel-(1S,2S,5S,8S)-modhephan-3-one (20) was isolated from the essential oil of Pulicaria mauritanica Coss (Asteraceae) roots.33 The 14- and 15-acetoxy derivatives (not shown) were also isolated from the roots of Pluchea sericea (Asteraceae)34 for the former, and from the alpine plant Leontopodium alpinum (Asteraceae)35 for the latter. Noteworthy, all the natural sources of modhephene and derivatives thereof mentioned above are part of the Asteraceae family.
Fig. 3 Several isolated derivatives of modhephene.
2.3 Canataxapropellane and other taxane-derived propellanes
Although there is a limited number of [3.3.3]propellane natural products reported to date, efforts from Zamir and co-workers led to the isolation of canataxapropellane from the needles of the yew Taxus canadensis (Taxaceae) in 2003.36 Two similar [3.3.2]propellanes with resembling chemical structures were also isolated from the same species.37,38 The complex structure of the taxane-derived canataxapropellane was elucidated using high resolution mass spectrometry and 2D NMR studies. A plausible biosynthetic pathway was proposed based on taxinine A (21), which was previously isolated from the same species. The proposed biosyntheses of canataxapropellane (28) and of three other taxane-derived di-propellanes (29–31), isolated from the same natural source, heavily rely on a series of rearrangements, as depicted in Scheme 2.36,39
Scheme 2 Proposed biosynthetic routes to taxane-derived propellanes from taxinine A (21).36,39
2.4 Dichrocephones A and B
Dichrocephones A (32) and B (33) (Fig. 4) were found in 2013 in the plant Dichrocephala benthamii (Asteraceae).40,41 Noteworthy, both compounds showed cytotoxicity against three cancer cells lines: human cervical cancer cells (HeLa), human oral squamous carcinoma cells (KB) and non-small-cell lung carcinoma cells (A549). However, dichrocephone B drastically out competed dichrocephone A with IC50 values as low as 4.1, 3.3 and 4.5 μM, versus 29.5, 23.2 and 28.5 μM for dichrocephone A, and 5.9, 6.1 and 6.9 μM for 5-fluorouracil (positive control).40,41 The initially assigned structures of both dichrocephone A and B were proven wrong by the total synthesis approach of Christmann et al.42 With the correctly assigned structure it seems plausible that dichrocephones share a biosynthetic pathway with the modhephene family.
Fig. 4 Dichrocephones A and B, pulicaral and pulicaric acid.
2.5 Pulicaral
Pulicaral (34) (Fig. 4) was first isolated together with some of its derivatives, including the acid 35 (Fig. 4), from the aerial parts of Pulicaria paludosa (Asteraceae) commonly known as the Spanish false fleabane.43 Its structure was elucidated using conventional mass spectrometry and 2D NMR techniques. In total, seven derivatives were isolated, of which four are glycosides.43
3 Synthesis of [3.3.3]propellanes
The combination of the challenging propellane core with the stereochemical requirements has fascinated many chemists and driven several groups to undertake synthetic efforts towards these compounds. Below, the various synthetic strategies to modhephene and modhephene-type structures that have been reported to date are presented. In the first paragraph, the only semi-synthesis of modhephene (1) reported to date is presented. The different total syntheses are then presented separately. The different strategies have been sorted according to the chemistry used to build the tricyclic propellane core, and by research groups. Fig. 5 summarizes the different synthetic approaches and key precursors that are presented in the following chapters.
Fig. 5 Overview of the different strategies towards modhephene (1) and derivatives with key precursors.
3.1 Semi-synthesis
The works reported by Morales-Ríos et al. showed a way to obtain (−)-modhephene ((−)-1): the semi-synthesis of the target compound from the naturally occurring derivative 14-hydroxymodhephene (36). Their semi-synthesis starts with an extraction of 36 from Pluchea sericea roots. Next, this natural product was subjected to an oxidation reaction (Scheme 3). (−)-Modhephene ((−)-1) was then obtained through the sulfurization and reductive desulfurization of the resulting aldehyde.44
Scheme 3 Semi-synthesis of (−)-modhephene from 14-hydroxymodhephene (Pluchea sericea).44
3.2 Acid-catalyzed rearrangements
3.2.1 Synthesis by Smith and co-workers. One of the first total syntheses of (±)-modhephene ((±)-1) was reported by Smith et al. in the 1980s.45–47 Their approach took advantage of previous works from Cargill et al. on the acid-catalyzed rearrangement of strained β,γ-unsaturated ketones, known as the Cargill rearrangement (as shown in Scheme 4).48–51
Scheme 4 The Cargill rearrangement towards a [3.3.3]propellane core.48–51
Smith et al.'s synthesis of modhephene (Scheme 5) employed a Cargill rearrangement and built the [3.3.3]propellane core from a β,γ-unsaturated tricyclic ketone 39. The required ketone was prepared by a photo-activated [2 + 2]-cycloaddition of 1,2-dichloroethene to a bicyclic enone (Scheme 5). This first cycloaddition step was found to be crucial, as the placement of the ethane bridge anti to the methyl group would yield the required stereochemistry of C8. Although different syn/anti ratios were obtained depending on the starting enones (43a, 43b or 43c) used, this step always yielded a mixture of epimers. After the synthesis of the strained enones 45a–b from the corresponding dichlorinated [4.3.2]propellanones 44a–c, a Cargill rearrangement in presence of p-toluenesulfonic acid (p-TsOH) gave access to the [3.3.3]propellane 46. A few more steps finally afforded a mixture of (±)-modhephene ((±)-1) and its epimer (±)-49. Enones 45a and 45b could both be rearranged into an epimeric mixture of the same products 46a–b, however with different syn/anti ratios. Only the synthesis from 44b provided an epimeric ratio favoring the epimer anti of 45b that is needed to access (±)-modhephene (1).
Scheme 5 Synthesis of (±)-modhephene ((±)-1) and (±)-epimodhephene ((±)-49) by Smith (s/a = syn/anti ratio).45,46 *Based on recovered material.
When the synthesis was carried out on epimeric mixtures until the very end, modhephene and epimodhephene were obtained as a mixture and could not be separated. However, each could be obtained as a pure compound from their respective precursors 48a and 48b after separation of the latter using preparative vapor-phase chromatography. When using this additional purification step, (±)-modhephene ((±)-1) and (±)-epimodhephene ((±)-49) were obtained as pure compounds from 43a and 43b, with overall yields of 10% and 7%, respectively.45,46
3.2.2 Synthesis by Kakiuchi and co-workers. Kakiuchi and co-workers have reported in-depth studies on the acid-catalyzed rearrangement of [m.n.2]propellanones. Such a rearrangement can proceed via the 1,2-alkyl shift of either the central propellane bond, or of one of the cyclobutane external bonds, like in the Cargill rearrangement. Through these studies they have highlighted how the mechanism and outcome of the rearrangements are influenced by the ring size of the tricyclic substrates and by the reaction conditions (presence or absence of a nucleophile).52–54 For example, they showed that in non-nucleophilic conditions, 50 rearranges into [3.3.3]propellane 51via an acid-catalyzed 1,2-alkyl shift of the cyclobutane external bond.55 However, in presence of sulphuric acid or acetic acid, the same substrate rearranges primarily to the tricyclo[4.3.2.01,5]undecane-type products 52 or 53, as illustrated in Scheme 6.52,53
Scheme 6 Rearrangement of [4.3.2]propellanone 50 in non-nucleophilic and nucleophilic conditions.52,53
As part of these works, Kakiuchi et al. also described the synthesis of 57 from the reductive rearrangement of compound 54, of the endo-alcohol 55 or of the [3.3.3]propellane 56 (Scheme 7). In this case, 57 was obtained together with other tricyclo-undecanes type products 58–61, one of which is actually a rearrangement product of 57 itself, as well as other unidentified products.54 Additionally, the same [3.3.3]propellane 57 was also obtained for the acid-catalyzed rearrangement of 56 with H2SO4 in pentane under reflux (Scheme 7).54
Scheme 7 Access to propellane 57 from the rearrangement of 54, 55 and 56.54
Several years later, Kakiuchi et al. reported the syntheses of the α-methylene [3.3.3]propellanone derivatives 64–67, which were obtained from the functionalization of [3.3.3]propellanone (51) (Fig. 6).56
Fig. 6 α-Methylene [3.3.3]propellanones synthesized from 51.
The aim of their syntheses was to determine the impact of the polycyclic structure around the α-methylene cyclopentanone moiety on the antiproliferative activity of such compounds. Therefore, the bioactivity of these [3.3.3]propellanes were assessed and compared to the ones of various other polyquinanes (not shown).56 Interestingly, the α-methylene [3.3.3]propellanone 68 showed the best antiproliferative activity against P388, L1210, 3LL and LY cancer cell lines, with IC50 values of 0.11, 0.11, 0.06 and 0.45 μM, respectively.56
Given these promising results, Kakiuchi and co-workers reported a second study on the synthesis of additional [3.3.3]propellanone derivatives (Scheme 8).57 [3.3.3]Propellanone 42 was obtained from 70 using their acid-catalyzed rearrangement strategy. Further functionalization of 42 permitted the synthesis of 71 in six additional steps. Similarly, [3.3.3]propellanones 74 and 75 were synthetized from 73, which was itself obtained from 50 in three steps. Interestingly, the rearrangement of 72 into 73 did not take place with concentrated H2SO4 in benzene. However, a screening of various acidic conditions showed that this rearrangement proceeds with acceptable yield in the presence of tin(IV) chloride in nitromethane.57 Two more [3.3.3]propellanone derivatives, 76 and 77, were then obtained by deprotection of the hydroxyl group of 74 and 75, respectively.
Scheme 8 Synthesis of [3.3.3]propellanone derivatives through acid-catalyzed rearrangement of [4.3.2]propellanones by Kakiuchi and co-workers.57
To the best of our knowledge, the antiproliferative activities of the [3.3.3]propellanone derivatives 71, 74, 75, 76 and 77 have not been reported yet.
3.2.3 Mundy and co-workers. Mundy and co-workers58,59 reported another synthetic route to (±)-modhephene ((±)-1) based on the intermediate 47a. In their approach, propellane 47a was obtained through the acid-catalyzed rearrangement of the hetero[3.3.3]propellane 80 (Scheme 9), which was obtained by treatment of the diester 79 with methyllithium. It is worth noting that the characterization of the rearrangement product 47a was done by comparison to a sample received from Smith et al., from whom Mundy and co-workers also received the authentic NMR spectra.58,59 Yet, in 1990 Curran and Jasperse reported an inversion of spectral assignments in Smith's experimental section for 47a and 47b,60 which was also confirmed by Smith et al. themselves (see ref. 70 in ref. 60). Therefore, it is unknown whether Mundy and Wilkening reanalyzed the reference samples or compared their spectra to the ones that had been provided. In either case, and given this intricate situation, it remains unclear whether Mundy and Wilkening did obtain the described product.13
Scheme 9 Synthesis of (±)-modhephene ((+)-1) by Mundy and co-workers.58,59
An alternative route to modhephene was reported from intermediate 81.59 This compound also resulted from the treatment of 79 with methyllithium and was isolated after the acidic treatment of the aqueous phase of the extraction step. It was then used to complete the total synthesis of modhephene. Yet, the fact that the final product was only characterized by HRMS analysis, combined with the aforementioned uncertainties on the characterization of the synthetic intermediates, left the actual nature of the product uncertain.59
3.2.4 Fitjer and co-workers. As part of their works on the rearrangement of dispiroundecanes into triquinanes,61–63 Fitjer et al. also reported the synthesis of the natural product modhephene, starting from dispiroundecane 82 (Scheme 10),21,22,64 which is accessible in eight steps from isopropylidenecyclobutane22,64 and in nine steps from acetone.22 Fitjer et al. showed that when heated at 70 °C under acidic conditions, 82 undergoes a series of rearrangements that yield two kinetic products: (±)-modhephene ((±)-1) and compound 86.22,64 However, prolonged heating leads to further rearrangement of these two kinetic products into a range of thermodynamic products, two of which are triquinane 91 and isocomene 90.22,64 The rearrangement of 82 into (±)-modhephene ((±)-1) proceeds through an initial dehydration step and successive 1,2-shifts, as shown in Scheme 10.
Scheme 10 Rearrangement of (±)-dispiroundecane ((+)-82) into (±)-modhephene ((+)-1).21,22,64
Fitjer et al. used this discovery to report one of the first enantiospecific syntheses of (−)-modhephene and (+)-modhephene.21 As shown in Scheme 11, this approach was based on the treatment of dispiroundecane ketone 92 with (+)-(S)-N,S-dimethyl-S-phenylsulfoximine 93, which allowed the separation of 94a from 94b, as 94a precipitated and could easily be filtrated. Both sulfoximines were then heated at 80 °C to afford ketones (−)-92 and (+)-92, respectively. Then, the enantiospecific methylation of each isomer gave access to (+)-82 (Scheme 10) on the one hand, and to (−)-82 on the other. Finally, the kinetically controlled rearrangement of these two intermediates yielded optically pure (+)-(1S,5R,8S)-modhephene ((+)-1) and (−)-(1R,5S,8R)-modhephene ((−)-1) (95% ee).21
Scheme 11 Enantioselective synthesis of (+)-modhephene ((+)-1) and (−)-modhephene ((−)-1) through the rearrangement of a (±)-dispiroundecane precursor.21
3.3 Photochemical reactions
3.3.1 Mehta and co-workers, Singh and co-workers. Other routes towards the synthesis of (±)-modhephene employ photochemical pathways, such as a photo-mediated oxa di-π-methane rearrangement, as used by Mehta and Subrahmanyam in 1985.65–67 Metha and co-workers found that the strained tetracyclic compound 97 was accessible from an oxa di-π-methane rearrangement of the β,γ-unsaturated ketone 96 (Scheme 12).66–68
Scheme 12 Mehta and co-workers’ total synthesis of (±)-modhephene and (±)-epimodhephene.65–67 PCC = pyridium chlorochromate, LHMDS = lithium bis(trimethylsilyl)amide, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
After the core the structure 97 was formed, introduction of the gem-dimethyl group at position C4 and subsequent reductive C1–C9 bond cleavage gave access to the propellane 99, which presents the main [3.3.3]propellane core of (±)-modhephene.65–67 Intermediate 101b, which exhibits the desired stereochemistry at C8 with regard to the synthesis of (±)-modhephene ((±)-1), was then easily obtained after reduction of 100 and separation from its epimer 101avia column chromatography. Finally, a dehydration step from 101b gave access to (±)-modhephene ((±)-1), while dehydration of 101a led to (±)-epimodhephene ((±)-49).69,70
A similar procedure was used by Singh et al. in 2007 to access different [4.3.3] and [3.3.3]propellanes.71 Indeed, after synthesizing 102 and 105 from aromatic precursors (not shown), they used the same photochemical approach as Mehta and coworkers to access 103 from 102 and 106 from 105, in 51% and 30% yield respectively. The stereoselective synthesis of [3.3.3]propellanes 104 and 107 was then accomplished by treatment of these tetracyclic structures with tributyltin hydride and azobisisobutyronitrile (AIBN) (Scheme 13).71
Scheme 13 Syntheses of [3.3.3]propellanones 104 and 107 by Singh and co-workers, using a photochemical strategy.71 AIBN = azobisisobutyronitrile.
3.3.2 Indane-vinyl acetate photocycloaddition: Wender and Dreyer, Mehta and co-workers. As illustrated in Scheme 14, the short synthesis of (±)-modhephene ((±)-1) reported in 1982 by Wender and Dreyer was also based on a tetracyclic intermediate.72 Compound 110, which structurally resembles compound 97, was obtained from a single photo-initiated reaction between indane 108 and vinyl acetate 109. It was then converted into 111, which gave access to (±)-modhephene ((±)-1) in three straightforward steps. The methyl group at C8 in modhephene was introduced with the required configuration through a stereoselective 1,5-addition on 112.
Scheme 14 (±)-Modhephene ((±)-1) synthesis by Wender and Dreyer.72
The cyclopropane ring-opening reaction of tetracyclic structures such as 112 using lithium cuprates was also investigated by Cook and co-workers to access [3.3.3]propellanes (see section Anionic approaches).73,74
Interestingly, Mehta and co-workers also used the indane-vinyl acetate photocycloaddition strategy with an additional reduction step of the Δ6,7 isomer of 111 to form the [3.3.3]propellane 114. The latter was then used as a substrate in a novel cationic rearrangement toward the total synthesis of a biologically active quadrane named decarboxyquadrone (117) (Scheme 15).69,70
Scheme 15 Use of the indane-vinyl acetate photocycloaddition in the total synthesis of decarboxyquadrone.46,69,70
As part of a subsequent study on the effects of alkyl substituents in this cationic rearrangement of [3.3.3]propellanones, Mehta and Subrahmanyam also synthetized several methylated derivatives of 114 (not shown).70
3.4 Thermal rearrangements
3.4.1 Karpf and co-workers. Through the thermal rearrangement of alkynone 119 at 620 °C, Karpf and Dreiding75 synthesized the known modhephene intermediate 46a (Scheme 16) that is also encountered in the synthesis of Smith et al. (see section Acid-catalyzed rearrangements). This rearrangement was the key step of their ten-step synthesis of (±)-modhephene ((±)-1), which they reported independently to Smith and co-workers' works.13
Scheme 16 Synthesis of (±)-modhephene ((±)-1) by Karpf and Dreiding.75
3.4.2 Paquette and co-workers. Similarly, Paquette and Schostarez76,77 also based their total synthesis of (±)-modhephene ((±)-1) on the thermal rearrangement of an acetylenic intermediate 127 (Scheme 17), a procedure adapted from earlier works reported by Conia and co-workers78–81 (routes A and B in Scheme 17).78,79,81–83 The rearrangement used for the synthesis of (±)-modhephene ((±)-1) proceeded through an intramolecular hydrogen transfer from the enol tautomer of the cyclic ketone of 127 to the alkyne functional group (route C, Scheme 17).76,77,83 After the rearrangement of 127 into 128, a Wittig olefination step was carried out, followed by an epoxidation with mCPBA. Subsequent isomerization led to 129. This isomerization occurred in a highly regio- and stereoselective manner and ensured the correct stereochemistry of the methyl group at C8, as required for a facile synthesis of (±)-modhephene ((±)-1) in two more steps. (±)-Epimodhephene ((±)-49) was also synthesized in a similar and expeditive way, using an intramolecular ene cyclization of 130 (route D, Scheme 17).76,77 Several years later, Paquette and Usui published a chemically related route to [3.3.3]propellanes using a radical procedure (see section Radical cyclizations).84
Scheme 17 Conia et al.'s (A & B)78,79,81–83 and Paquette et al.'s (C & D)76,77,83 investigations into the synthesis of the [3.3.3]propellane core and subsequent modhephene and epimodhephene syntheses. mCPBA = meta-chloroperoxybenzoic acid.
Interestingly, intermediate 126 was also used by these research teams,83 as well as Simmons and co-workers,85 to access other ring-functionalized [3.3.3]propellanes, like the ones represented in Fig. 7. The intriguing 137 was obtained through the thermal or acid-catalyzed rearrangement of 133 or 134.83,86,87 The rearrangement of 132 was also attempted by Paquette and co-workers leading to a product with alleged structure 138.83
Fig. 7 Other functionalized [3.3.3]propellanes resulting from the functionalization of 126.
3.4.3 Oppolzer and co-workers. Shortly after Paquette and co-workers reported their synthesis of modhephene, Oppolzer and Marazza reported an additional stereoselective synthesis of (±)-modhephene ((±)-1), also based on the thermal cyclization of a bicyclic precursor 140 (Scheme 18).88 This strategy relied on the fact that the cis-positioning of the H-donor relative to the enophilic site would induce the correct positioning of the methyl group.88,89 The same strategy allowed Oppolzer and Bättig to report an alternative route to the same natural product the very same year, which was based on the ene cyclization of 143.89
Scheme 18 Syntheses of (±)-modhephene ((±)-1) via thermal rearrangements.88–90
3.4.4 Barczak and Jarvo. In 2011 Barczak and Jarvo reported the formal total synthesis of modhephene, by preparing intermediate 143 in a mere two steps from 3-methylbutanoyl chloride, using a Kondako–Darzens acylation reaction followed by a Nazarov cyclization, as depicted in Scheme 18.90
3.4.5 Mash and co-workers. A few years after the synthesis of modhephene by Paquette et al., Mash and Nelson adapted their own methodology for the diastereoselective cyclopropanation of bicyclic enones91–93 to the synthesis of enantiomerically enriched precursor 151 (Scheme 19). Then, following a synthetic strategy equivalent to the one of Paquette et al., Mash and co-workers described the first enantioselective synthesis of (−)-modhephene ((−)-1).18–20 In this case, this product was obtained as an inseparable 5:1 mixture with epimodhephene 49.
Scheme 19 Mash et al.'s enantioselective synthesis of (−)-modhephene.20 TMS = trimethylsilyl.
3.5 Radical cyclizations
Radical cyclizations constitute another popular approach to the construction of the [3.3.3]propellane cores, either via an intramolecular radical cyclization from a cyclopentane or diquinane core bearing appropriate side chains60,84,94–102 or by means of a domino transannular radical cyclization of a substituted cyclooctane derivative.103–105
3.5.1 Curran and co-workers. In 1990, Curran and Jasperse developed a synthesis of the natural product (±)-modhephene ((±)-1) based on a series of radical cyclizations.60 Their first synthesis relied on the cyclization of vinyl stannane 156 to 157 (Scheme 20). The alkylation of the latter in a few synthetic steps permitted a second radical cyclization and the formation of the propellane core from 46a. Based on this strategy, (±)-modhephene ((±)-1) was synthesized in eleven steps with more than 16% overall yield.60
Scheme 20 Modhephene synthesis using a series of radical cyclizations.60 LDA = lithium diisopropylamide, HMPA = hexamethylphosphoramide, AIBN = azobisisobutyronitrile, DPPE = 1,2-bis(diphenylphosphino)ethane.
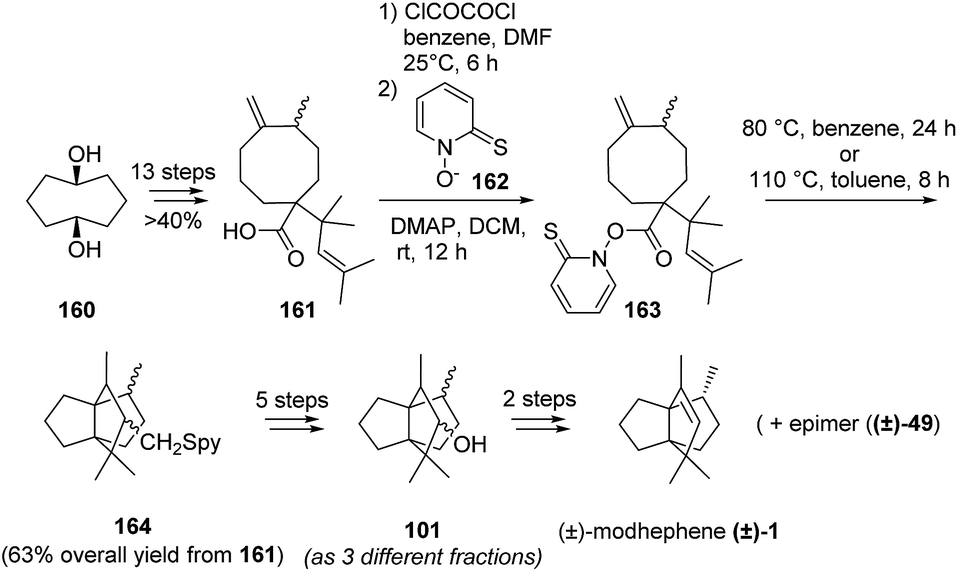
A few years later, Curran and Shen103 also reported the synthesis of (±)-modhephene ((±)-1) based on a domino transannular radical cyclization of an exo(methylene)cyclooctane. This synthetic route started with the synthesis of 161 (1:1 isomeric mixture), which involved an Ireland–Claisen rearrangement (Scheme 21). Then, decarboxylation of 161 using the Barton thiohydroxamate method106–114 allowed the radical formation that permitted the domino transannular radical cyclization to occur. However, since 101 was obtained as a mixture of diastereomers that were difficult to separate, the access to pure (±)-modhephene ((±)-1) was rendered possible only from one of the three fractions obtained after chromatographic separation of 101. Subsequent reaction with the other fractions led either to a mixture of the desired product together with its epimer ((±)-49) or to decomposition. Overall, although very attractive at first glance, this approach gave access to (±)-modhephene ((±)-1) and its epimer in 6% overall yield over 21 steps.103
Scheme 21 Synthesis of modhephene using a domino transannular radical cyclization of an exo(methylene)cyclooctane intermediate.103 DMAP = 4-dimethylaminopyridine.
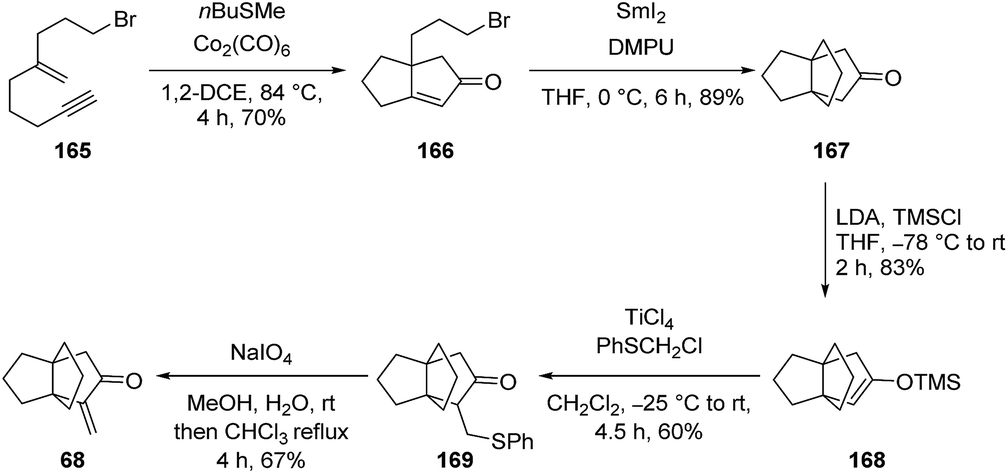
3.5.2 Synthesis of α-methylenene propellanone by Kerr and co-workers. A more recent application of some of Curran and Wolin's early works115 into radical cyclizations has been adopted by Kerr et al.102 to prepare the biologically active56,57 α-methylene propellanone 68 (Scheme 22). The original procedure by Curran and Wolin used 2.5 equivalents of samarium diiodide in DMPU and THF to form bicyclo[3.3.0]octan-3-ones from halogenated cyclopent-2-en-1-ones in a reductive manner.115 Kerr and co-workers were able to employ intramolecular Pauson–Khand reactions to synthesize the 5,5-fused bicycle 166 with ease and subsequently use the samarium diiodide-induced intramolecular conjugate addition procedure to form [3.3.3]propellanones 167 (see Scheme 22).102 Four subsequent steps led to the synthesis of the target [3.3.3]propellane, α-methylene propellanone 68 with an overall yield of 11% over twelve steps. This product was previously identified as a potent cytotoxic agent effective against a range of tumor cell lines (see section Acid-catalyzed rearrangements).56,57
Scheme 22 Synthesis of biologically active [3.3.3]propellane 68 using SmI2 by Kerr and co-workers.102 DMPU = 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone, LDA = lithium diisopropylamide, TMS = trimethylsilyl.
3.5.3 Uyehara and co-workers. Uyehara et al.'s approach to [3.3.3]propellanes 99a and 99b is based on their methodology to introduce substituents at both bridge heads of bicyclo[2.2.2]oct-5-en-2-ones and on a photo-mediated rearrangement of the substituted β,γ-unsaturated ketone into tricyclic structures like 172 (Scheme 23).94,95,116 In this way, all the methyl substituents required for the synthesis of (±)-modhephene ((±)-1) are installed prior to the construction of the propellane core. A straightforward radical cyclization of bromide 173 yielded a mixture of propellanes 99a and 99b, which were isolated by using HPLC.95,116
Scheme 23 Formal synthesis of (±)-modhephene ((±)-1) via strained tetracyclic intermediates.94,95 TBS = tert-butyldimethylsilyl, AIBN = azobisisobutyronitrile.
3.5.4 Towards capped propellanes: Paquette and Usui. Besides their work on thermal rearrangements, Paquette and Usui also investigated the synthesis of vinyl silane [3.3.3]propellane 178.84 They found that the thermal rearrangements they had previously relied on gave them direct access to the triene 177. Therefore, they developed a radical-based procedure to access the silane 178 (Scheme 24).84 The aim of these works was to propose a possible pathway toward capped [3.3.3]propellanes (179) (Scheme 24b). Yet, to the best of our knowledge, such [3.3.3]propellanes have not been reported to date.
Scheme 24 Synthesis of vinyl silane [3.3.3]propellanes using radical chemistry, towards capped propellanes.84 AIBN = azobisisobutyronitrile.
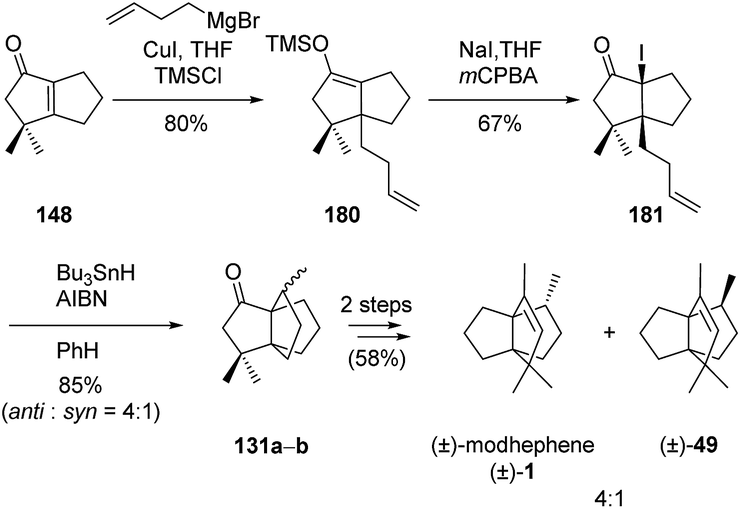
3.5.5 Sha and co-workers. Sha et al. based their synthesis of (±)-modhephene ((±)-1)96 on the stereoselective synthesis of α-iodoketone 181, which was submitted to an intramolecular radical cyclization to form intermediate 131a–b (Scheme 25). Few more synthetic steps yielded the desired product (±)-1 in mixture with its epimer (±)-49, in a 4:1 ratio.96
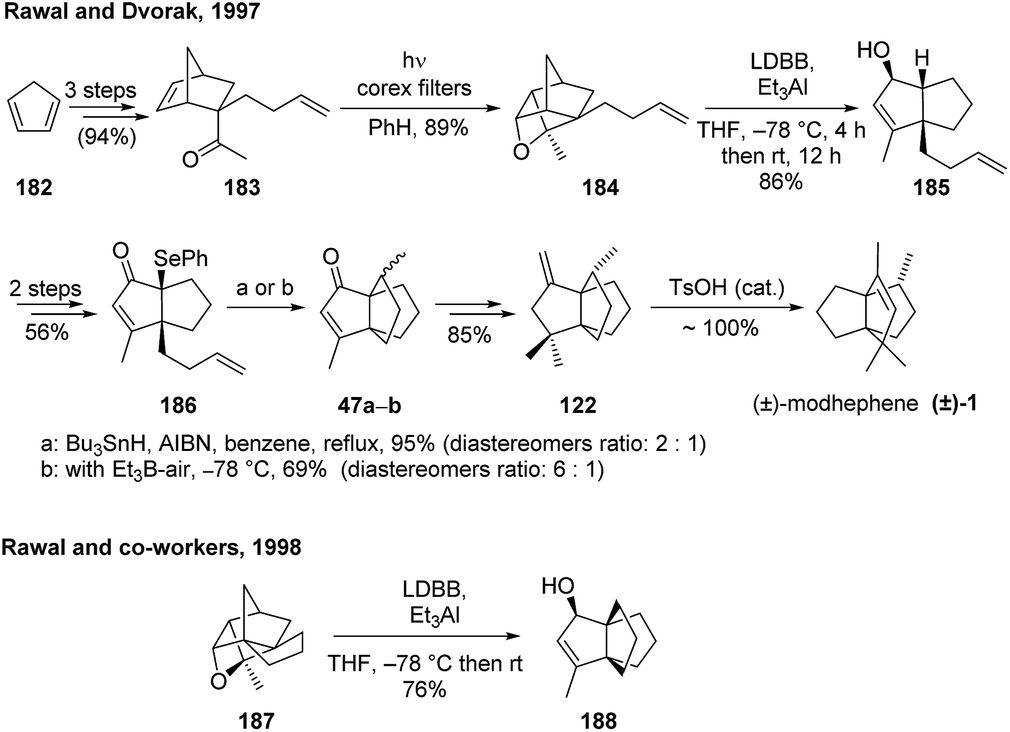
3.5.6 Rawal and co-workers. Rawal and Dvorak98 proposed another interesting stereoselective synthesis of (±)-modhephene ((±)-1), which first used a photocycloaddition-fragmentation sequence to construct the diquinane core 185 (Scheme 26). The third ring of the [3.3.3]propellane core was then built by another intramolecular radical cyclization step of a tethered side chain. Following this strategy, the team reported an 11-step synthesis of modhephene with 21% overall yield, a remarkable feat by itself.98 Notably, the major diastereomer with the desired stereochemistry at C8 was isolated by MPLC, just before isomerization of the propellane 122 to (±)-1. Interestingly, the scope of this strategy on the fragmentation of Paterno–Büchi reaction products, published a year later, showed how it was successfully used for the synthesis of a range of di-and triquinanes, including the [3.3.3]propellane 188 (Scheme 26).117 However, it must be noted that in this case, the propellane was built directly from the fragmentation of oxetane 187, without the need of an additional radical cyclization step.117
Scheme 26 Syntheses of (±)-modhephene and other [3.3.3]propellanes using a radical cyclization to form the [3.3.3]propellane core.98,117 AIBN = azobisisobutyronitrile, LDBB = lithium di-tert-butylbiphenylide.
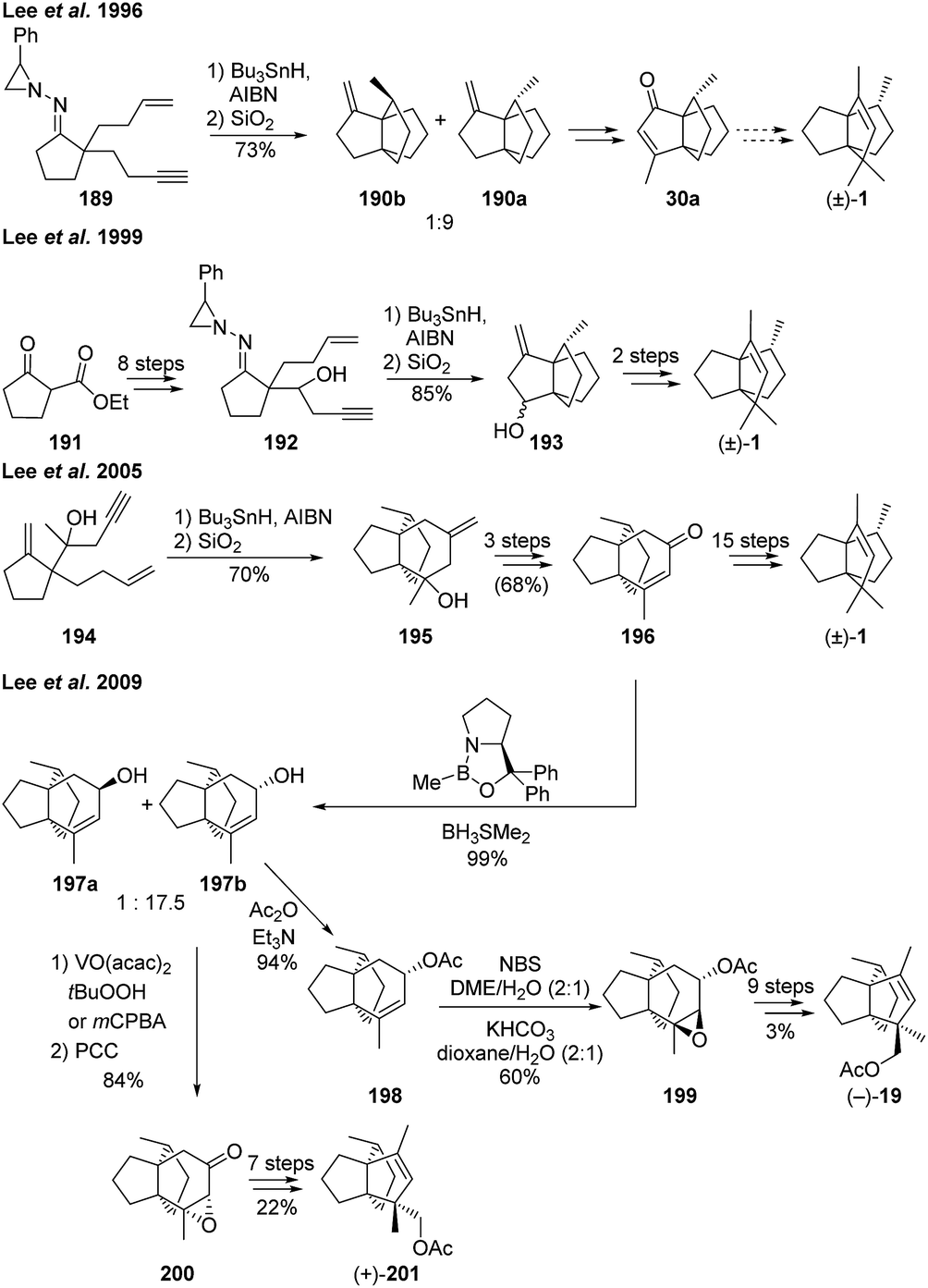
3.5.7 Lee and co-workers. Lee et al. managed to form two out of the three required rings for the [3.3.3]propellane core of modhephene in a single step, by using radical-mediated domino cyclizations.97,99 The required gem-diradical species were generated from a N-aziridinyl imine precursor, which also contained one vinyl radical precursor (189) (Scheme 27) or bromoalkene moiety (not shown) and one alkene functional group on tethered side-chains. Using this strategy, Lee et al. were able to prepare 190a,97 which, after an ozonolysis step, was easily converted to the known (±)-modhephene precursor [3.3.3]propellane 30a, according to the procedure from Oppolzer and Bättig.89 The stereochemistry that is desired at C8 of modhephene was obtained thanks to the stereoselectivity of the cyclization step, which depends on the substrate used. The use of an alkyne as the vinyl radical precursor led to a 9:1 mixture of the desired isomer (Scheme 27).
Scheme 27 Domino radical cyclization strategies to synthesize (±)-modhephene ((±)-1)97,99,100 and the enantioselective synthesis of (−)-13-acetoxymodhephene and (+)-14-acetoxymodhephene.101 AIBN = azobisisobutyronitrile, mCPBA = meta-chloroperoxybenzoic acid, PCC = pyridium chlorochromate, NBS = N-bromosuccinimide.
In 1999, Lee et al. reported the application of this strategy to the synthesis of intermediate 193, in order to achieve a new total synthesis of (±)-1.99
Several years later, the same team proposed a similar route to (±)-modhephene ((±)-1), based on the domino free radical cyclization of a dieneyne intermediate 194. This cyclization step resulted in the initial formation of the [4.3.3]propellane intermediate 195 (with 10:1 stereoselectivity towards the desired configuration), which could subsequently be modified into (±)-1via intermediate 196.100
More recently, this domino cyclization methodology was adapted to the enantioselective synthesis of (−)-13-acetoxy-modhephene ((−)-5) and (+)-14-acetoxymodhephene ((+)-201).101 An asymmetric borane reduction of ketone 196, derived from the domino cyclization, led to alcohols 197a and 197b with epimeric ratio of 1:17.5 and the desired [4.3.3] propellane 197b as major product. The respective stereochemistry at the carbon bearing the acetoxymethyl group in the final product were then obtained by choosing appropriate epoxidation conditions at this stage. The use of either a vanadium complex and a peroxide or mCPBA allowed for the synthesis of epoxide 200, which was a suitable intermediate to access (+)-201 over seven synthetic steps. Alternatively, treatment of the acetate-protected allyl alcohol 198 with NBS in water/DME, and then with KHCO3, gave access to the epoxide 199, with opposite stereochemistry. This epoxide was then used to synthesize (−)-19, over nine more steps.101
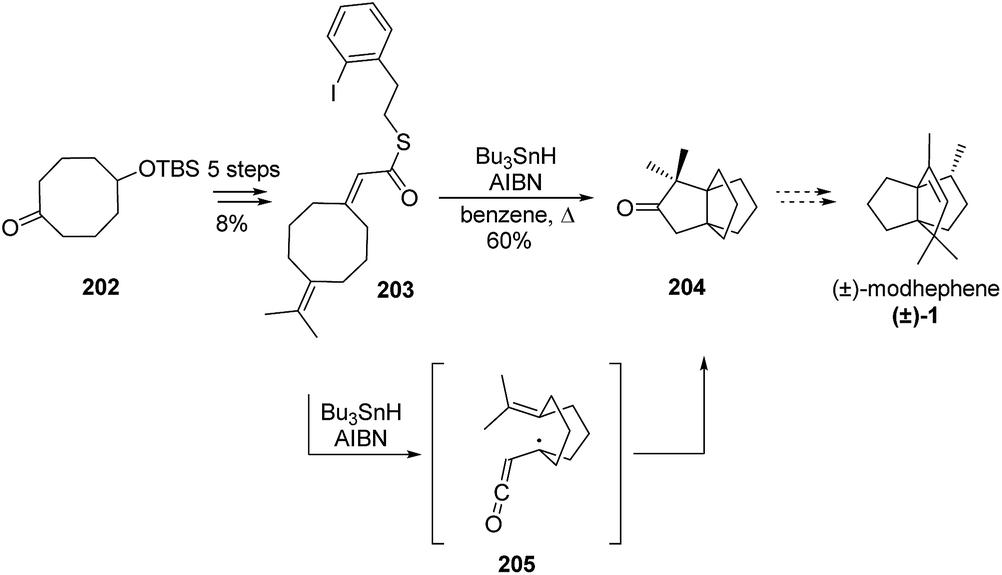
3.5.8 Pattenden and co-workers. Pattenden and co-workers also proposed a formal synthesis of (±)-modhephene ((±)-1) based on the domino transannular radical cyclization of a cyclooctenyl substituted α-ketenyl radical 205,104,105 itself generated by heating thioester 203 in presence of Bu3SnH and AIBN, as depicted in Scheme 28. This strategy allowed them to report a synthesis of [3.3.3]propellane 204, which is a precursor of (±)-modhephene ((±)-1).
Scheme 28 Formal synthesis of (±)-modhephene ((±)-1) using a domino transannular radical cyclization of an exo(methylene)cyclooctane intermediate.104,105 AIBN = azobisisobutyronitrile.
3.6 Anionic approaches
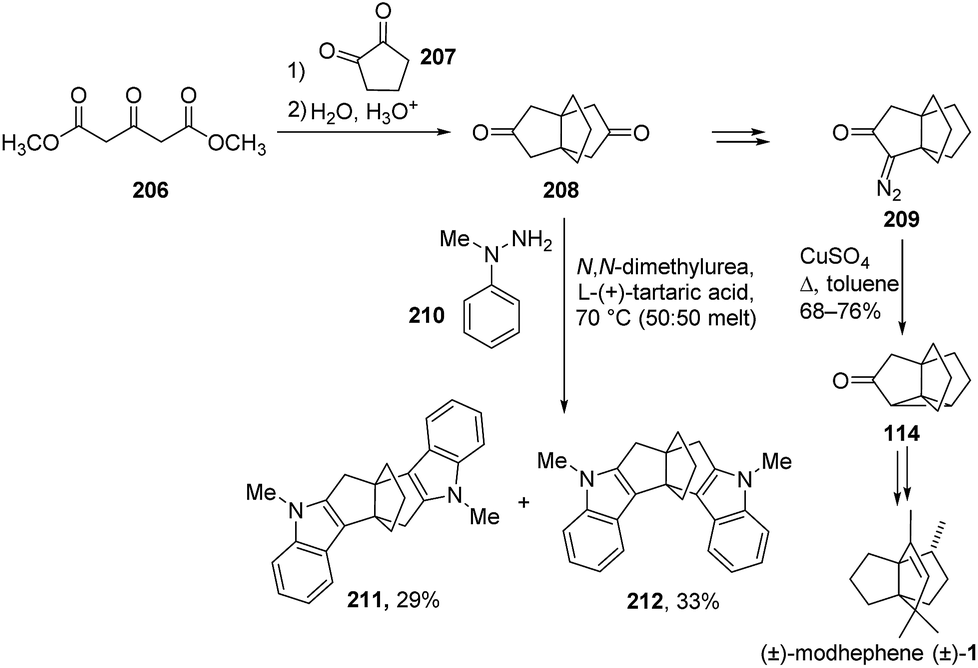
3.6.1 The Weiss reaction: Cook and co-workers. The strategy applied by Cook et al. involved a Weiss reaction118–121 between dimethyl-1,3-acetonedicarboxylate (206) and cyclopentane-1,2-dione (207), which yielded the dione 208 (Scheme 29).73 This dione was then converted into the diazo propellanone 209, which could then be used to synthesize the desired [3.3.3]propellane (±)-1, via the strained tetracyclic intermediate 114. This tetracyclic structure could only be opened with a copper reagent after the introduction of a second activating group (not shown).73 Indeed, as shown by the same research team one year later through a study on the opening of strained cyclopropanes, a two-fold activation is generally required to open this kind of tetracyclic structures that present no double bonds.74
Scheme 29 Synthesis of (±)-modhephene using a Weiss reaction and of a tetracyclic intermediate by Cook et al.,73 followed by a double Fischer indole cyclization to diindole propellanes.122
This strategy of carrying out a diazotation and subsequent annulation with copper sulfate to a tetracyclic intermediate was also used by Pattenden and de Boeck in one of their (±)-modhephene syntheses, as shown above (see section Radical cyclizations).104
In more recent years, a two-fold Fischer indole cyclization on diketone propellane 208 was reported by Kotha and Chinnam.122 They found that this diketone, which was easily available on a decagram scale, could undergo a double Fischer indole cyclization with 1-methyl-1-phenylhydrazine (210) in the presence of molten N,N′-dimethylurea and L-(+)-tartaric acid, leading to the formation of a mixture of the diindole propellanes 211 and 212 (Scheme 29).
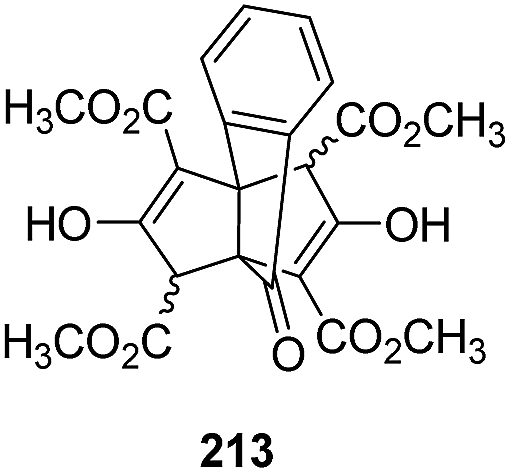
It is worth noting that the Weiss reaction strategy was also found to work reliably for a range of polycyclic compounds, including a range of [3.3.n]propellanes.123,124 For example, compound 213 (Fig. 8) could be synthesized from ninhydrin using this condensation reaction.124
Fig. 8 Product of the Weiss reaction between ninhydrin and dimethyl 1,3-acetonedicarboxylate.123
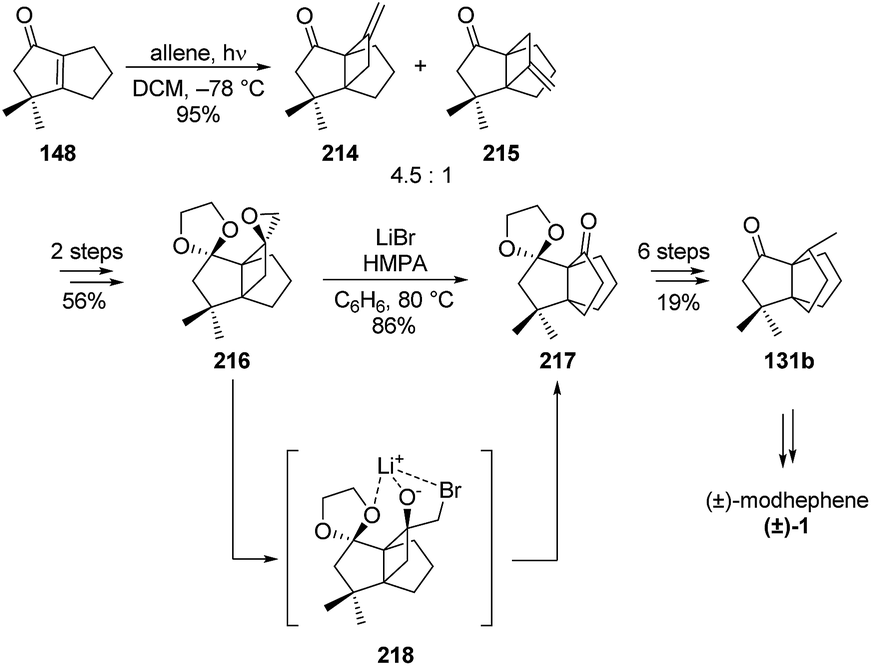
3.6.2 Synthesis by Tobe and co-workers. In 1984, Tobe et al. accessed (±)-modhephene ((±)-1) in a stereo-controlled manner by using a regioselective ring enlargement.53 Their formal synthesis of (±)-modhephene ((±)-1) using this strategy started with the head-to-head photo-mediated [2 + 2] cycloaddition of allene to the α,β-unsaturated ketone 148 (Scheme 30). After protection of its carbonyl functional group, the resulting bicyclic compound was then subjected to an epoxidation to yield three epoxy acetals, from which 216 was isolated in 61% yield (56% yield in two steps from the mixture of 214 and 215).53 The chelation-controlled epoxide-carbonyl rearrangement was then performed in the presence of lithium bromide and hexamethylphosphoramide (HMPA). At this critical step, the desired stereochemistry was asserted to result from the chelation of the lithium cation by the acetal oxygen atoms, as depicted in Scheme 30.53 This would also explain the unusual preferred migration of the less substituted carbon over the higher substituted one.53 Intermediate 217 gives an easy access to propellane 131b, a known precursor to (±)-modhephene ((±)-1) with the desired stereochemistry at C8.45,75,89
Scheme 30 Formal total synthesis of (±)-modhephene ((±)-1) by Tobe and co-workers.53 HMPA = hexamethylphosphoramide.
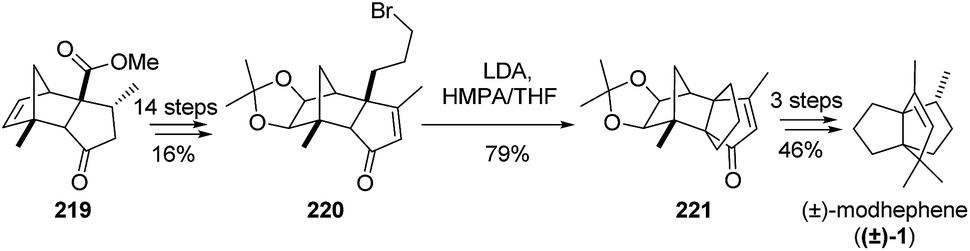
3.6.3 Synthesis by Suri. An additional synthesis of (±)-modhephene ((±)-1) involving a cyclization reaction for the construction of the [3.3.3]propellane core is the synthesis by Suri.125 Its key step is the cyclization of the bromide 220 to 221 by an intramolecular alkylation reaction (Scheme 31). Although this was an original approach, the main disadvantage of this synthetic path probably lies in its length of 18 steps it requires to access (±)-modhephene ((±)-1).
Scheme 31 Synthesis of modhephene ((±)-1) based on an intramolecular alkylation reaction.125 LDA = lithium diisopropylamide, HMPA = hexamethylphosphoramide.
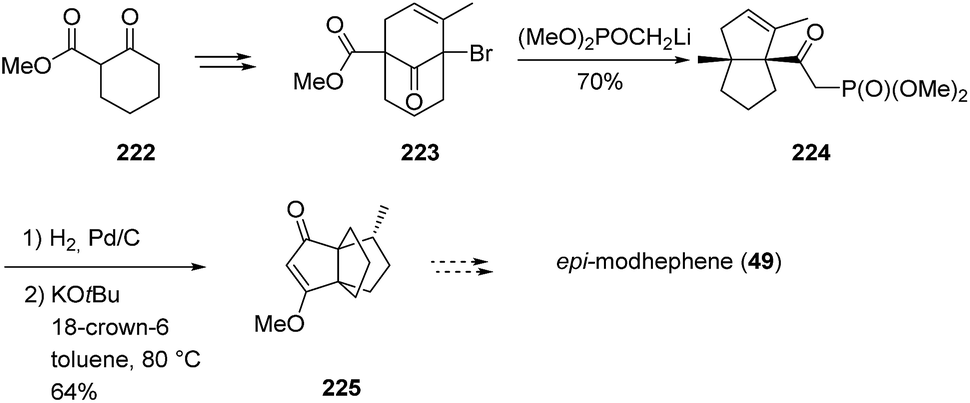
3.6.4 Synthesis by Kraus and Shi. Alternatively, addition-rearrangement reactions of bridgehead bromides have been proposed as synthetic routes toward [3.3.3]propellanes. And indeed, in 1990, Kraus and Shi126 showed that the addition of a nucleophile such as a dimethyl methyl phosphonate anion to the bromide 223 led to a ring contraction of the resulting alkoxide to form bicyclic structures such as 224. After a few modifications, these could be converted into the propellane 225, a precursor of (±)-epimodhephene ((±)-49).127 Product 223 was prepared by bromination of keto ester 222, followed by a Michael addition with methyl vinyl ketone. As depicted in Scheme 32, the third ring of the propellane core was then built in a hydrogenation step, followed by a base-mediated cyclization.126 A comparison of the analytical data showed that their product 225 was identical to an intermediate of the (±)-modhephene ((±)-1) synthesis of Mundy et al. Thus, given the aforementioned uncertainty about the stereochemistry of Mundy's products, the stereochemistry of 225 may also be questioned. Yet, Kraus and Shi showed that modifying their reduction conditions to an iridium-catalyzed directed hydrogenation of alkenes allowed them to access a product different from propellane 225.127 This compound was identified as its C8 epimer, which provided identical NMR spectra to Curran and Jasperse's intermediate.60 Thus, the team claimed that the stereochemical misassignment in the reported work by Mundy et al. would be the result of an inversion in Smith's NMR data and that their work allowed to resolve this uncertainty,127 although this assertion is still being debated.13
Scheme 32 Synthesis of epimodhephene (49) by Kraus and Shi.126
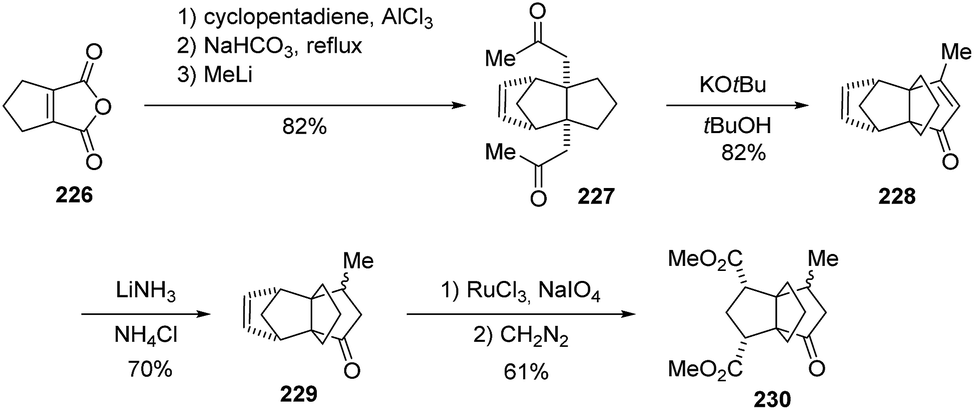
Efforts by Ghosh et al.128 to construct strained systems such as [3.3.3]propellanes revealed that easily accessible bicyclo[2.2.1]heptanes, such as the diketone 227, can serve as excellent precursors (Scheme 33). Diketone 227 was found to be very suitable to undergo potassium tert-butoxide-mediated ring closure to the highly functionalized [3.3.3]propellane 228. After the propellane formation and a reduction step, the bicyclo[2.2.1] bridge in 229 could be used conveniently to install two methyl ester groups in moderate yield.
Scheme 33 Synthesis of [3.3.3]propellanes from easily accessible bicyclo[2.2.1]heptanes.128
3.7 Metal-catalyzed reactions
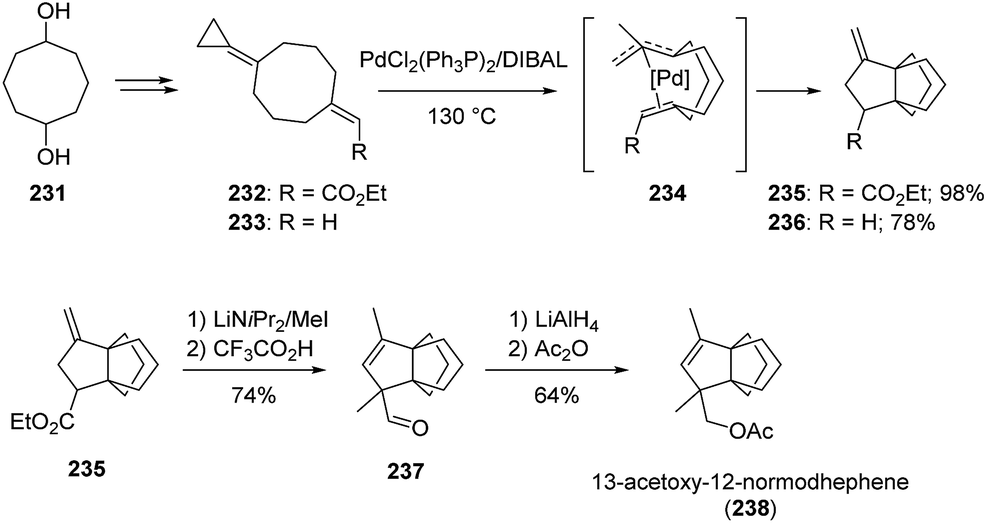
3.7.1 Transition metal-catalyzed intramolecular cyclizations: Yamago and Nakamura. Transition metal-catalyzed intramolecular cyclizations of the modified cyclooctanes 232–233, accessible in four steps from commercially available cyclooctane-1,5-diol (231), have been found to allow the facile construction of [3.3.3]propellane cores (Scheme 34). Both palladium and nickel complexes were found to be efficient catalysts, although isolated yields with the palladium-based catalyst were superior.129,130 Propellane 235 was subsequently transformed into 13-acetoxy-12-normodhephene 238 using a series of standard transformations.
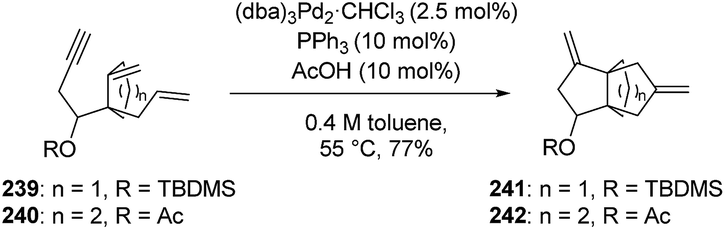
3.7.2 Palladium-catalyzed zipper reaction: Trost and Shi. While working on intramolecular polyolefinic cycloisomerization reactions, Trost and Shi developed a palladium-catalyzed zipper reaction.131,132 Although the primary use of this zipper reaction was to make polyspiro compounds, they found that the reaction was versatile enough to cyclize an array of different polyolefinic substrates, including 239 and 240, resulting in the corresponding [3.3.3] and [4.3.3]propellanes 241 and 242 (Scheme 35).
Scheme 35 Use of a Pd-catalyzed zipper reaction to form propellanes.131,132 dba = dibenzylideneacetone, TBDMS = tert-butyldimethylsilyl.
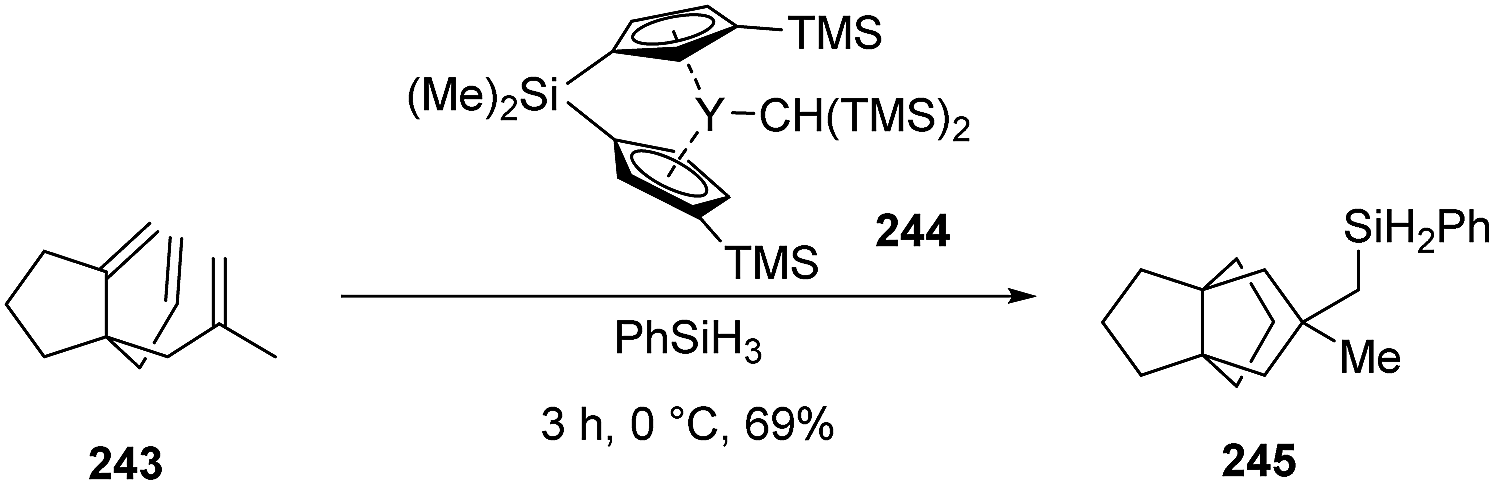
3.7.3 Organolanthanide-catalyzed cyclizations: Molander and co-workers. Alternatively, organolanthanide-catalyzed procedures have been reported to facilitate a cyclization/silylation reaction of hindered trienes, preparing several [4.3.3]propellanes with reasonable yields, as well as allowing for the synthesis of 245 in 69% yield (Scheme 36).133
Scheme 36 Use of organolanthanides to cyclize trienes.133 TMS = trimethylsilyl.
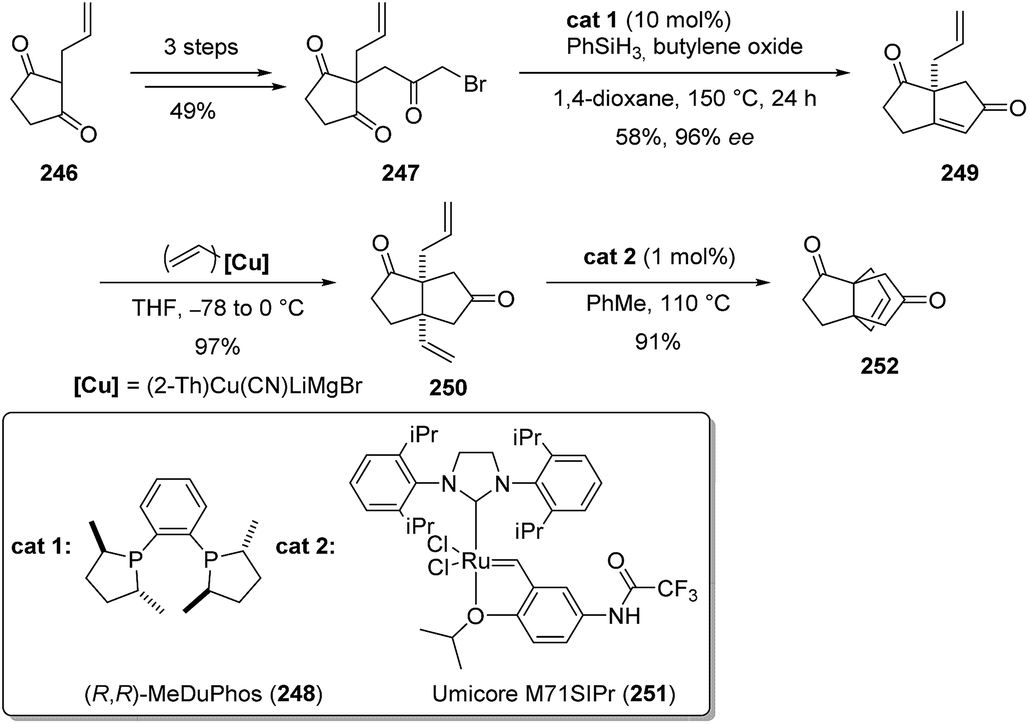
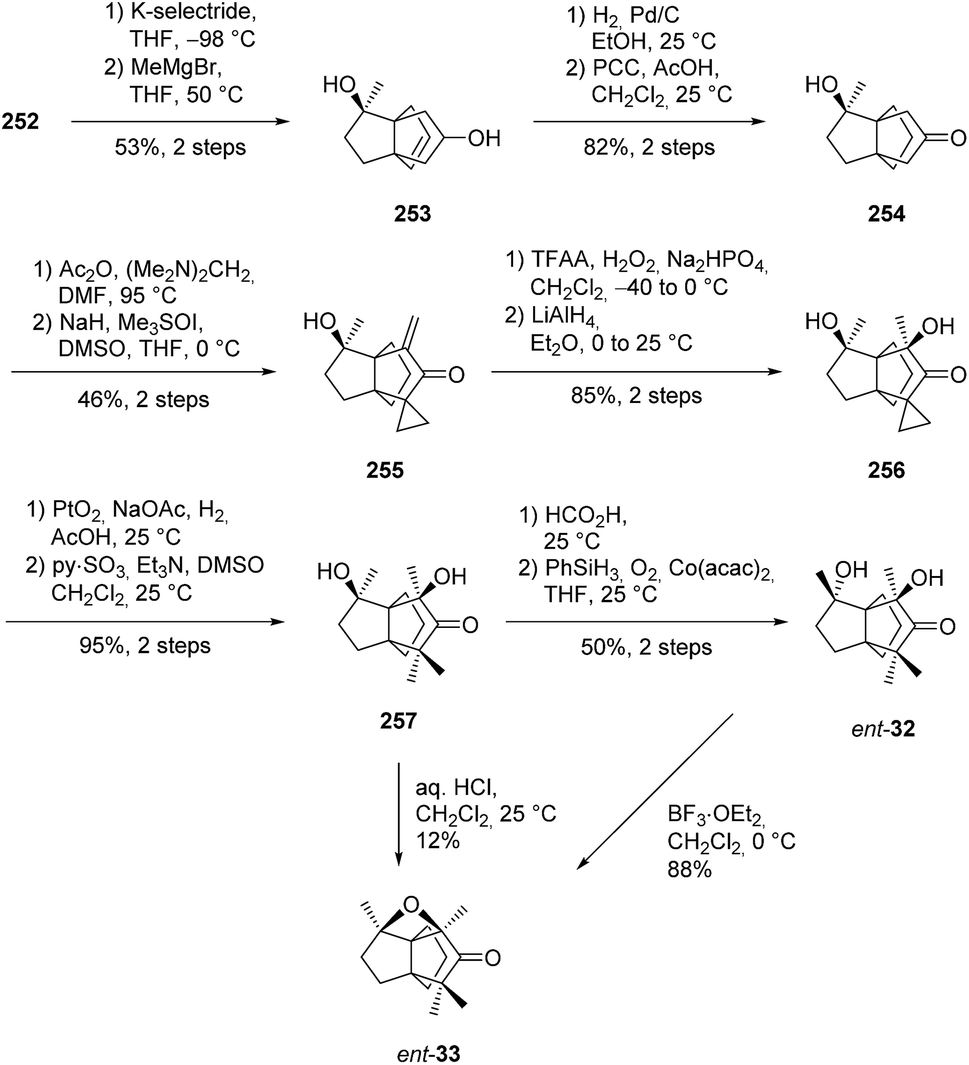
3.7.4 Metathesis: Christmann and co-workers. In a recent study, Christmann and co-workers reported a modular synthesis of functionalized propellanes, including the [3.3.3]propellanedione 252 (Scheme 37).134 In this strategy, prochiral triketones like 247 were catalytically desymmetrized to access bicylic structures such as 249. Then, diastereoselective addition of alkenyl-cuprates gave access to dienic structures such as 250, which were cyclized to propellanes using olefin metathesis. For this reaction, a commercially available ruthenium catalyst 251 was used (Scheme 37). The absolute configuration of 252 was determined by comparison of IR and VCD spectra with calculated spectra, after structure optimization.
Scheme 37 Synthesis of [3.3.3]propellane 252via metathesis of diene 250.134
The same group applied this strategy in a total synthesis approach of dichrocephone A and B (Scheme 38).42 However, during their study they found that the reported structure of dichrocephone A did not match the spectroscopic results of their compound 257. The C8-epimer ent-32 was found to be in accordance with the reported spectra, but not with the optical rotations of the isolated natural products. Consequently, the synthesized compounds ent-32 and ent-33 are the enantiomers of the natural products dichrocephone A and B, respectively.
Scheme 38 Synthesis of ent-32 and ent-33 from the propellane core 252.42
3.8 Synthesis and applications of triptindanes
Triptindanes are a special subclass of [3.3.3]propellanes, with often intriguing applications in the fields of gas storage, gas separation and charge transfer complexes. As their synthetic approaches differ greatly from the procedures described above, these syntheses have been grouped separately in the following section. We believe that this section is complementary to the previous chapters and necessary to gain a complete view on the topic of carbocyclic [3.3.3]propellanes. As such, we think that it could inspire researchers from the field of natural product synthesis, help them imagine novel approaches in this very field and envisage new applications for these fascinating molecules.
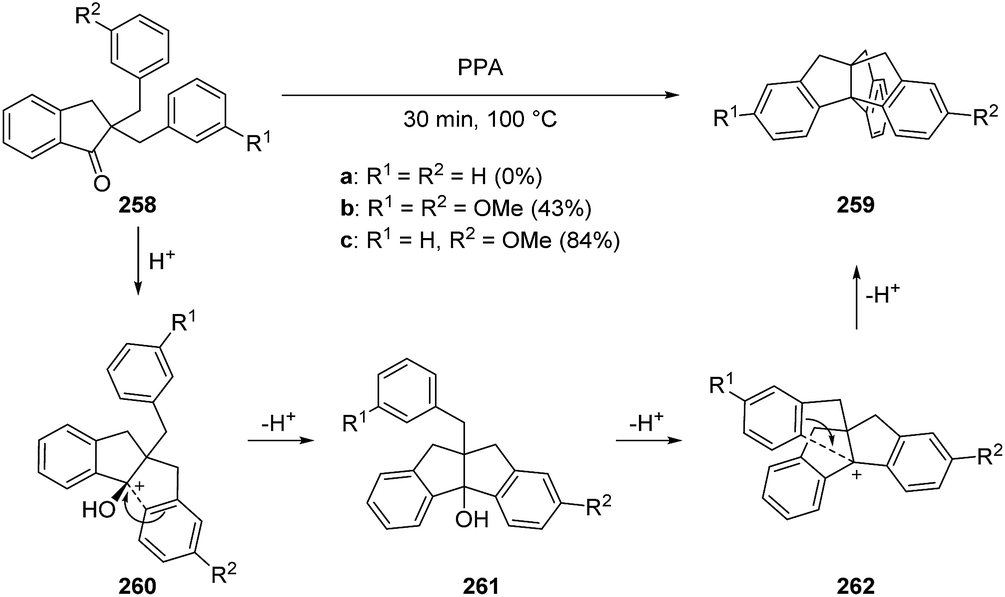
3.8.1 First synthesis of triptindane by Thompson. Triptindanes represent a class of synthetic tribenzo-[3.3.3]propellanes that have drawn interest for the last 50 years. The highly symmetrical structure and rigidity of this type of molecule motivated Thompson to synthesize the parent compound 259a in the 1960s. Furthermore, he investigated homoconjugative interactions between the isolated π-systems.135,136
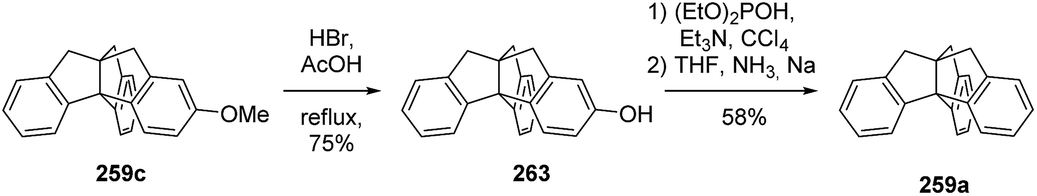
The first approach to the triptindane core included the acid-catalyzed double cyclization of the dibenzylindanone 258 (Scheme 39). However, the reaction proceeded only with activating groups like methoxy moieties present on the benzyl rings (258b, 258c).135,136 The insolubility of dibenzylindanone 258a in polyphosphoric acid (PPA) also hindered the reaction.135 The desired unsubstituted triptindane (259a) could finally be obtained by deoxygenation of 259c in 44% overall yield (Scheme 40).136
Scheme 39 First synthesis of the triptindane core by Thompson.135,136 PPA = polyphosphoric acid.
Scheme 40 Deoxygenation of methoxytriptindane 259c to the parent compound triptindane (259a).
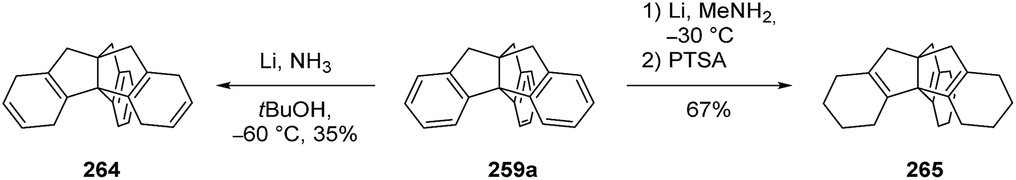
Kuck and Eckrich showed that triptindane (259a) could be converted to different derivatives by carefully choosing the reduction conditions (Scheme 41). While a Birch reduction led to the formation of the corresponding hexadiene 264, a Benkeser reduction was found to reduce triptindane even further to 265.137
Scheme 41 Reductions of triptindane (259a) under different conditions.137 PTSA = para-toluenesulfonic acid.
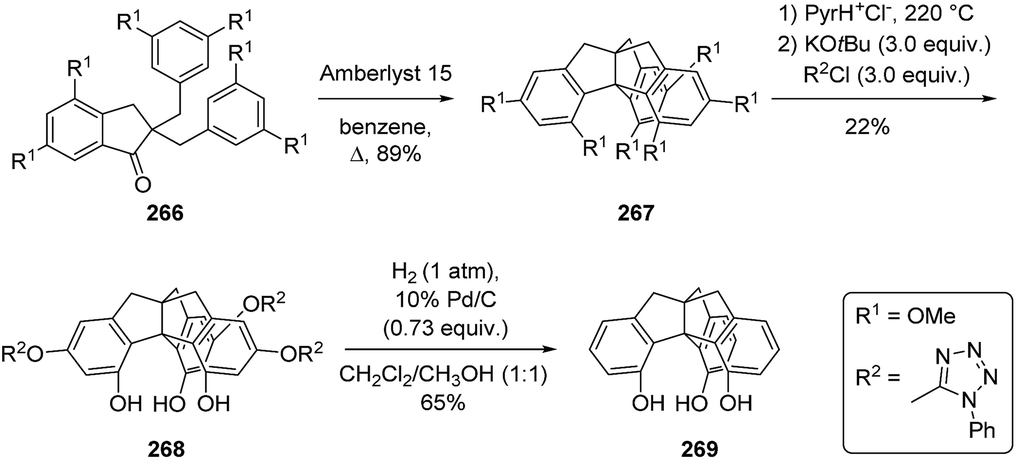
3.8.2 Derivatives of triptindane. An interesting feature of triptindanes is the molecular cavity in the structure. Kuck and co-workers synthesized the derivative 269 with hydroxyl groups exclusively in endo-position of the molecule (Scheme 42).138 The substitution of the catalyst PPA with the acidic resin Amberlyst 15 in non-polar solvents provided a facile approach to the methoxy-substituted triptindane 267. After cleavage of all ether groups, the exo-hydroxy groups could selectively be converted to tetrazolyl ether groups (268) by carefully controlling the stoichiometry of the reaction. Subsequent direct hydrogenolysis resulted in the isolation of 4,5,15-trihydroxytriptindane (269). The attempt to synthesize endo-trimethyltriptindane failed due to steric effects.139
Scheme 42 Synthesis of 4,5,15-trihydroxytriptindane (269) by Kuck et al.138
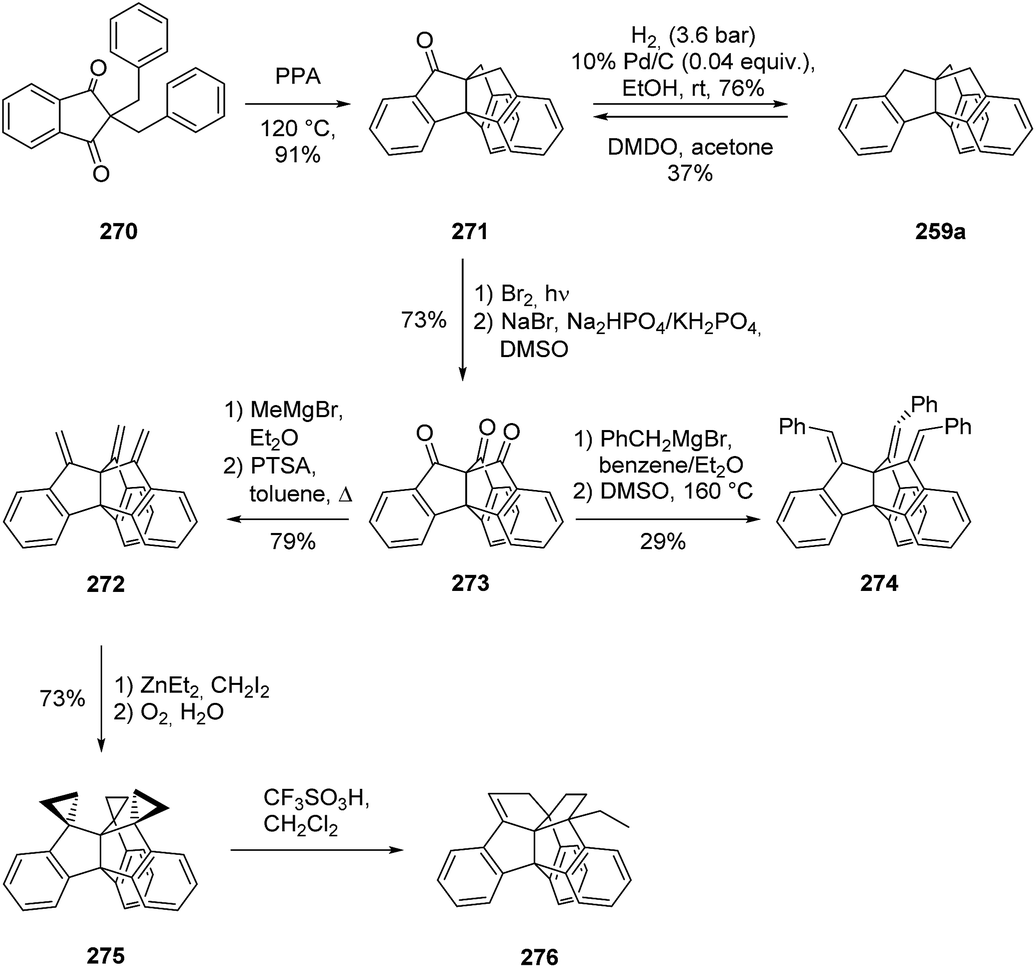
Besides strategies to modify the cavity of triptindane, the opposite side of the molecule was also addressed in synthetic approaches. The use of dibenzyl-1,3-indandione (270) as starting material provided not only a new route to triptindane, but also to the ketone derivative 272 as a useful building block (Scheme 43). This ketone was used to access several triptidane derivatives, including the trimethylenetriptindanes (272, 274), triptindane-9,10,11-trispirocyclopropane (275) and tribenzocentropentaquinene (276).140 An alternative route to the triptindane ketone (271) was found by oxidizing triptindane (259a) with dimethyldioxirane (DMDO).141
Scheme 43 New route to triptindane (259a) and to the ketone derivative 271 as a useful building block by Kuck and co-workers.140,141 DMDO = dimethyldioxirane, PPA = polyphosphoric acid, PTSA = para-toluenesulfonic acid.
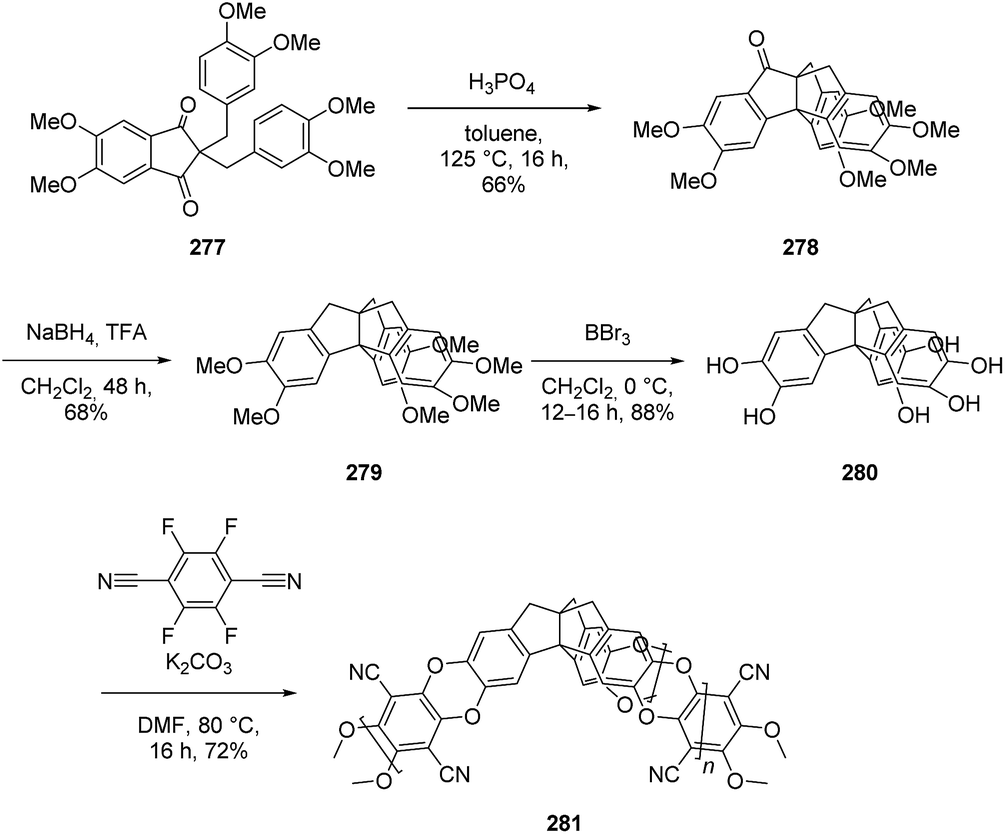
3.8.3 Microporous polymers based on triptindanes. The use of the 1,3-indanedione core and of methoxy groups on the benzene moieties made the compound 279 accessible under milder conditions. This compound was demethylated to the monomer 280, which was used for polymers of intrinsic microporosity (PIMs) (Scheme 44). These polymers showed large apparent Brunauer–Emmett–Teller (BET) surface areas and have potential for gas storage and gas separation.142
Scheme 44 Synthesis and polymerization of 280 to form microporous polymers.142 TFA = trifluoroacetic acid.
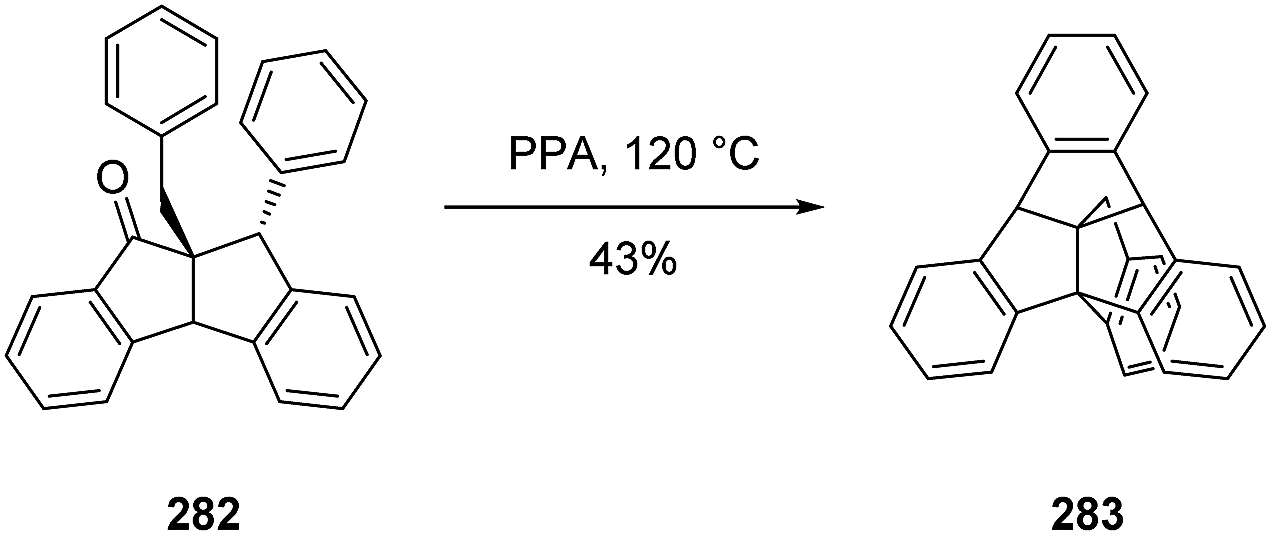
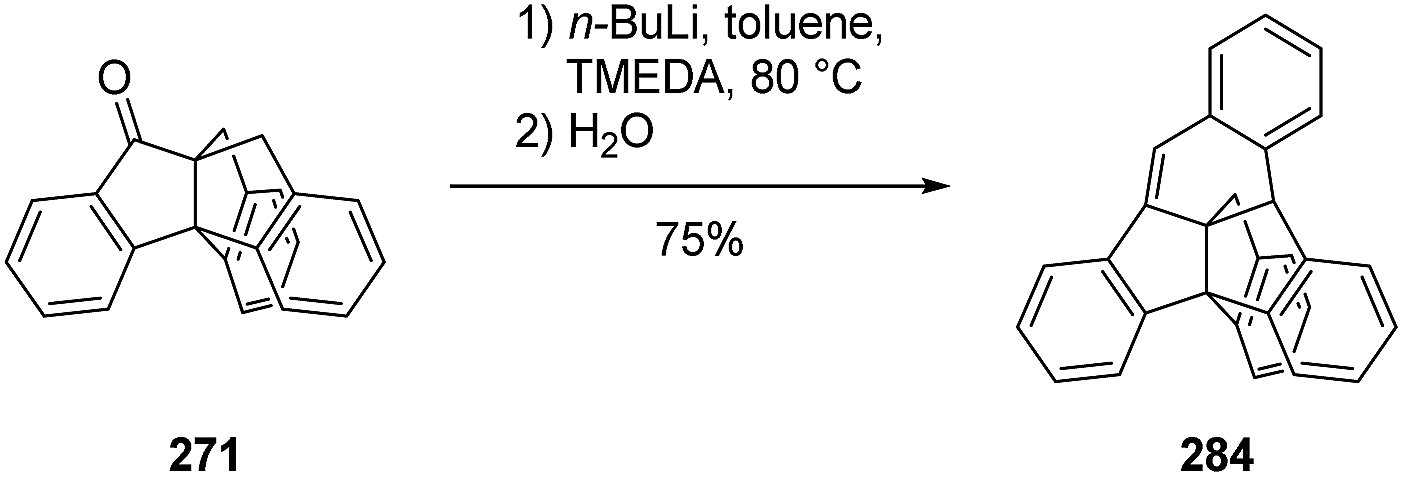
3.8.4 Bridged triptindanes synthesized by Kuck and co-workers. The bridged triptindane compound 283 was obtained from acid-catalyzed cyclization of phenylindanone 282 (Scheme 45).143 Alternatively, treatment of triptindane ketone 271 with n-BuLi and tetramethylethylenediamine (TMEDA) in toluene could be employed to construct a bridged triptindane 284 (Scheme 46).144
Scheme 45 The first bridged triptindane 283 prepared from the phenylindanone 282.143 PPA = polyphosphoric acid.
Scheme 46 Bridging of the keto triptindane 271.144 TMEDA = tetramethylethylenediamine.
3.8.5 Stability of triptindanes. Several studies investigating the stability and the chemical behavior of triptindanes have been performed. Jaworski et al.145 investigated the properties of radical anions and dianions of triptindane and its ketone derivatives. They found that the electronic communication between the carbonyl groups was two times smaller than in indanetrione. The groups of Ceccon and Kuck146 formed mono-, bis- and tris-Cr(CO)3 complexes of 259a and showed weak attractive electronic interactions bonds between the carbonyl oxygen and the polarized face of the benzene rings.146
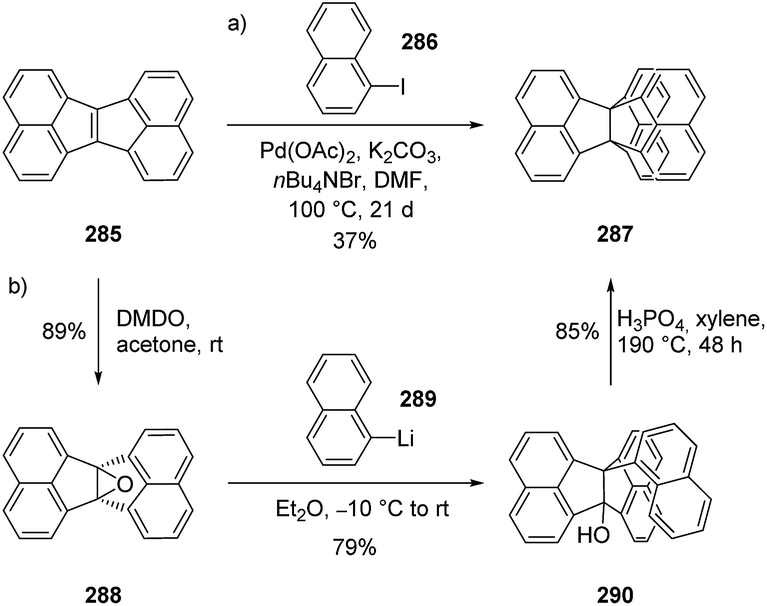
3.8.6 Trisnaphtho[3.3.3]propellanes (TNPs). The expansion of the triptindane family with bigger aryl groups started with the synthesis of trisnaphtho[3.3.3]propellane (TNP) (287).147 TNP was obtained in one step via a palladium-catalyzed reaction between hexacyclic alkene 285 and 1-iodonaphthalene (286) (Scheme 47a). The intermolecular Heck-type reaction at a tetrasubstituted double bond was observed for the first time in this study by Dyker et al.147 A few years later, the same group proposed a six-membered palladacycle as the key intermediate in this reaction.148,149 However, this reaction was not optimized, as it required prolonged reaction times of up to three weeks and a large excess (30 equiv.) of the iodonaphthalene. An improved synthesis relied on the epoxidation of 285 with dimethyldioxirane (DMDO), the addition of the organolithium analog 289 of iodonaphthalene (286) and the subsequent cyclization to TNP. Using this synthetic route, TNP was obtained with an overall yield of 59% (Scheme 47b).150
Scheme 47 Synthesis of trisnaphtho[3.3.3]propellane (TNP) (287) in (a) one step via a Pd-catalyzed reaction147 and (b) through epoxidation, addition of 1-naphthalenyllithium and cyclization.150 DMDO = dimethyldioxirane
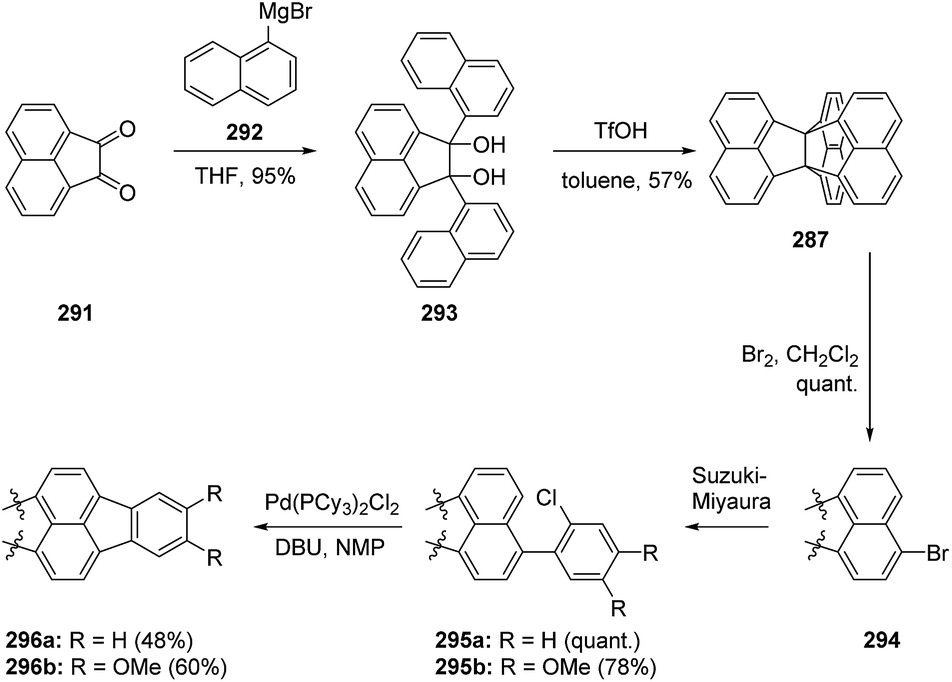
Given the symmetry and wide π-surface of TNP, Kubo et al. were interested in using this compound as a building block for 2D honeycomb lattices. Their synthesis was straightforward and involved Grignard chemistry and the acid-catalyzed cyclization of 293 (Scheme 48). Subsequent bromination, Suzuki–Miyaura coupling and direct arylation yielded the final functionalized TNPs, which, upon mixing with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4-TCNQ), were found to form charge transfer complexes, as well as the desired 2D honeycomb lattices.151
Scheme 48 Synthesis of trinaphtho[3.3.3]propellanes for the use in 2D honeycomb lattices.151 DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
3.8.7 Triphenanthro[3.3.3]propellanes. Phenanthrene derivatives of triptindane were synthesized by different approaches. Dibenzophenanthro[3.3.3]propellane (299) and benzodiphenanthro[3.3.3]propellane (301) could be obtained by photocyclodehydrogenation of the corresponding benzylidenetriptindanes (Scheme 49). Triphenanthro-[3.3.3]propellane (302) was only accessible in small yields by metal-catalyzed cyclodehydrogenation at high temperatures (Scheme 50).152
Scheme 49 Synthesis of mono- (299) and diphenanthrene (301) derivatives of triptidane by photocyclodehydrogenation.
Scheme 50 Metal-catalyzed cyclodehydrogenation of (Z,Z,Z)-274 to triphenanthro[3.3.3]propellane (302).
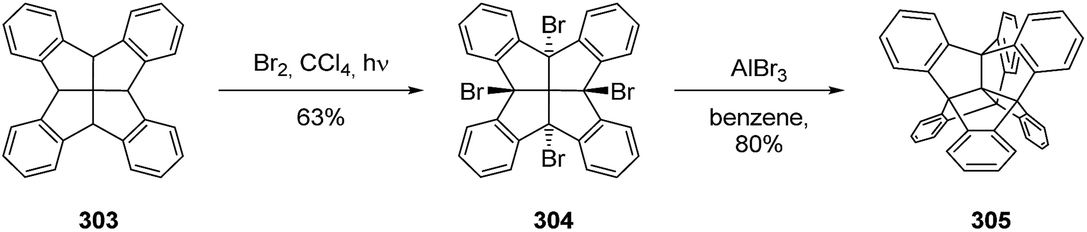
3.8.8 Centrohexaindanes. Centrohexaindanes represent a unique class of hydrocarbons with six fused indane units. They combine the [3.3.3]propellane core with fenestrane units. The first synthesis of this Td symmetrical compound (305) was reported 1988 by Kuck and Schuster (Scheme 51).153 Several improvements of the synthesis and alternative routes154–156 to 305 and to some methoxy-substituted157,158 and nitrated159 derivatives have been reported. The closely related centropentaindane was also synthesized by Kuck et al.160 However, as centrohexaindanes and centropentaindane constitute separate classes on their own, discussing all relevant details lies beyond the scope of this manuscript. Kuck published a summarizing article in 2006 about the structure and synthesis of centropolyindanes.161
Scheme 51 First synthesis of centrohexaindane (305) from fenestrindane (303).
4 Conclusions
For many years, the synthesis of carbocyclic [3.3.3]propellanes has posed a great challenge that has fascinated many organic chemists. Since the discovery of natural modhephene in 1978, many different strategies have been developed to access this intriguing compound and its derivatives, which span from the use of acid-catalysis, thermal rearrangements and photo-mediated reactions, to radical procedures, anionic rearrangements and transition metal-catalyzed reactions. Besides modhephene derivatives however, other new and original [3.3.3]propellanes have also been synthetized, some of which have even shown very interesting biological or physical properties and are of high potential for future applications in various fields. However, the amount of biological studies of these compounds is scarce and we encourage researchers to pursue such studies. Three-dimensional compounds like propellanes are gaining more and more interest in modern research, as they provide sought-after possibility to escape the flatland and to rigid linkers, not only in medicinal chemistry, but also in material sciences. In particular, tripindanes constitute a fascinating subclass of [3.3.3]propellanes with interesting electronic properties and hold great promises in the design of new materials. Considering the natural [3.3.3]propellanes that have been discovered over the last few years, the challenging synthesis and biological evaluation of these tricyclic structures will inspire many chemists in the future.
5 Conflicts of interest
There are no conflicts to declare.
6 Acknowledgements
We acknowledge continuous funding through the DFG (BR 1750) and the Helmholtz association. T. W. would like to acknowledge the Marie-Curie ITN ECHONET (grant No. 316379). R. M. B. acknowledges the SFB 1176 funded by the German Research Council (DFG) in the context of projects B3 and A4 for funding. We also acknowledge the contribution of Willi Sturz to the graphical abstract.
7 References
J. Altman, E. Babad, J. Itzchaki and D. Ginsburg, Tetrahedron, 1966, 22, 279–304 CrossRef.
K. B. Wiberg, Chem. Rev., 1989, 89, 975–983 CrossRefCAS.
K. B. Wiberg and S. T. Waddell, J. Am. Chem. Soc., 1990, 112, 2194–2216 CrossRefCAS.
M. D. Levin, P. Kaszynski and J. Michl, Chem. Rev., 2000, 100, 169–234 CrossRefCAS.
A. M. Dilmaç, E. Spuling, A. de Meijere and S. Bräse, Angew. Chem., Int. Ed., 2017, 56, 5684–5718 CrossRefPubMed.
F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRefCASPubMed.
G. M. Locke, S. S. R. Bernhard and M. O. Senge, Chem.–Eur. J., 2019, 25, 4590–4647 CrossRefCAS.
A. I. Ioffe, V. A. Svyatkin and O. M. Nefedov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1988, 37, 1827–1836 CrossRef.
T. E. Shubina, P. A. Gunchenko, T. S. Vigovskaya, P. R. Schreiner, A. G. Yurchenko and A. A. Fokin, Theor. Exp. Chem., 2003, 38, 229–236 CrossRef.
V. Polo, J. Andres and B. Silvi, J. Comput. Chem., 2007, 28, 857–864 CrossRefCAS.
G. Mehta and A. Srikrishna, Chem. Rev., 1997, 97, 671–720 CrossRefCAS.
A. J. Pihko and A. M. P. Koskinen, Tetrahedron, 2005, 61, 8769–8807 CrossRefCAS.
L. H. Zalkow, R. N. Harris III and D. Van Derveer, J. Chem. Soc., Chem. Commun., 1978, 420–421 RSC.
F. Bohlmann and J. Jakupovic, Phytochemistry, 1980, 19, 259–265 CrossRefCAS.
P. Weyerstahl, H. Marschall, I. Seelmann and J. Jakupovic, Eur. J. Org. Chem., 1998, 1205–1212 CrossRefCAS.
F. Bohlmann, N. Le Van, T. V. C. Pham, J. Jacupovic, A. Schuster, V. Zabel and W. H. Watson, Phytochemistry, 1979, 18, 1831–1834 CrossRefCAS.
E. A. Mash, S. K. Math and C. J. Flann, Tetrahedron Lett., 1988, 29, 2147–2150 CrossRefCAS.
E. A. Mash, S. K. Math and C. J. Flann, Tetrahedron Lett., 1989, 30, 2 CrossRef.
E. A. Mash, S. K. Math and C. J. Flann, Tetrahedron, 1989, 45, 4945–4950 CrossRefCAS.
L. Fitjer, H. M. Oltra and M. Noltemeyer, Angew. Chem., Int. Ed., 1991, 30, 1492–1494 CrossRef.
L. Fitjer, M. Majewski and H. Monzó-Oltra, Tetrahedron, 1995, 51, 8835–8852 CrossRefCAS.
R. M. Coates, Z. Ho, M. Klobus and S. R. Wilson, J. Am. Chem. Soc., 1996, 118, 9249–9254 CrossRefCAS.
R. M. Coates, J. Z. Ho, M. Klobus and L. Zhu, J. Org. Chem., 1998, 63, 9166–9176 CrossRefCAS.
A. P. W. Bradshaw, J. R. Hanson and R. Nyfeler, J. Chem. Soc., Perkin Trans. 1, 1981, 1469–1472 RSC.
A. P. W. Bradshaw, J. R. Hanson, R. Nyfeler and I. H. Sadler, J. Chem. Soc., Chem. Commun., 1981, 649–650 RSC.
A. P. W. Bradshaw, J. R. Hanson and I. H. Sadler, J. Chem. Soc., Chem. Commun., 1981, 1169–1170 RSC.
C. Pinedo, C.-M. Wang, J.-M. Pradier, B. Dalmais, M. Choquer, P. Le Pêcheur, G. Morgant, I. G. Collado, D. E. Cane and M. Viaud, ACS Chem. Biol., 2008, 3, 791–801 CrossRefCAS.
A. Y. Hong and B. M. Stoltz, Angew. Chem., Int. Ed., 2014, 53, 5248–5260 CrossRefCAS.
F. Bohlmann, M. Ahmed, N. Borthakur, M. Wallmeyer, J. Jakupovic, R. M. King and H. Robinson, Phytochemistry, 1982, 21, 167–172 CrossRefCAS.
A. Gauvin, J. Susperregui, P. Barth, R. Louis, G. Déléris and J. Smadja, Phytochemistry, 2004, 65, 897–901 CrossRefCAS.
F. Bohlmann, C. Zdero, R. Bohlmann, R. M. King and H. Robinson, Phytochemistry, 1980, 19, 579–582 CrossRefCAS.
T. Xu, M. Gherib, C. Bekhechi, F. Atik-Bekkara, H. Casabianca, F. Tomi, J. Casanova and A. Bighelli, Flavour Fragrance J., 2015, 30, 83–90 CrossRefCAS.
B. Reyes-Trejo and P. Joseph-Nathan, Phytochemistry, 1999, 51, 75–78 CrossRefCAS.
N. Comey, A. I. Grey, I. L. Hook, P. James and H. Sheridan, Phytochemistry, 1999, 50, 1057–1060 CrossRefCAS.
Q. W. Shi, F. Sauriol, O. Mamer and L. O. Zamir, Chem. Commun., 2003, 68–69 RSC.
C.-H. Huo, X.-H. Su, Y.-F. Wang, X.-P. Zhang, Q.-W. Shi and H. Kiyota, Tetrahedron Lett., 2007, 48, 2721–2724 CrossRefCAS.
M.-L. Zhang, M. Dong, C.-H. Huo, L.-G. Li, F. Sauriol, Q.-W. Shi, Y.-C. Gu, H. Makabe and H. Kiyota, Eur. J. Org. Chem., 2008, 5414–5417 CrossRefCAS.
Q. W. Shi, F. Sauriol, A. Lesimple and L. O. Zamir, Chem. Commun., 2004, 544–545 RSC.
X. Tian, L. Li, Y. Hu, H. Zhang, Y. Liu, H. Chen, G. Ding and Z. Zou, RSC Adv., 2013, 3, 7880–7883 RSC.
Z. Zhongmei, T. Xinhui, D. Gang, Z. Hongwu and H. Yanbao, Peop. Rep. China Pat., CN103664559A, 2014.
V. M. Schmiedel, Y. J. Hong, D. Lentz, D. J. Tantillo and M. Christmann, Angew. Chem., Int. Ed., 2018, 57, 2419–2422 CrossRefCASPubMed.
A. San Feliciano, M. Medarde, M. Gordaliza, E. Del Olmo and J. M. M. Del Corral, J. Nat. Prod., 1988, 51, 1153–1160 CrossRefCAS.
P. Joseph-Nathan, B. Reyes-Trejo and M. S. Morales-Rios, J. Org. Chem., 2006, 71, 4411–4417 CrossRefCAS.
A. B. Smith and P. J. Jerris, J. Am. Chem. Soc., 1981, 103, 194–195 CrossRef.
A. B. Smith and P. J. Jerris, J. Org. Chem., 1982, 47, 1845–1855 CrossRefCAS.
A. B. Smith, B. A. Wexler, C. Y. Tu and J. P. Konopelski, J. Am. Chem. Soc., 1985, 107, 1308–1320 CrossRefCAS.
R. L. Cargill and J. W. Crawford, Tetrahedron Lett., 1967, 8, 169–171 CrossRef.
R. L. Cargill and J. W. Crawford, J. Org. Chem., 1970, 35, 356–359 CrossRefCAS.
R. L. Cargill, D. M. Pond and S. O. LeGrand, J. Org. Chem., 1970, 35, 359–363 CrossRefCAS.
R. L. Cargill, T. E. Jackson, N. P. Peet and D. M. Pond, Acc. Chem. Res., 1974, 7, 106–113 CrossRefCAS.
K. Kakiuchi, Y. Hato, Y. Tobe and Y. Odaira, J. Chem. Soc., Chem. Commun., 1982, 6–7 RSC.
Y. Tobe, S. Yamashita, T. Yamashita, K. Kakiuchi and Y. Odaira, J. Chem. Soc., Chem. Commun., 1984, 1259–1260 RSC.
K. Kakiuchi, M. Ue, I. Wakaki, Y. Tobe, Y. Odaira, M. Yasuda and K. Shima, J. Org. Chem., 1986, 51, 281–287 CrossRefCAS.
K. Kakiuchi, K. Itoga, T. Tsugaru, Y. Hato, Y. Tobe and Y. Odaira, J. Org. Chem., 1984, 49, 659–665 CrossRefCAS.
K. Kakiuchi, M. Ue, M. Takeda, T. Tadaki, Y. Kato, T. Nagashima, Y. Tobe, H. Koike, N. Ida and Y. Odaira, Chem. Pharm. Bull., 1987, 35, 617–631 CrossRefCASPubMed.
K. Kakiuchi, M. Ue, T. Nagashima, M. Kinugawa, K. Kobiro, Y. Tobe and Y. Odaira, Chem. Pharm. Bull., 1988, 36, 542–550 CrossRefCAS.
D. Wilkening and B. P. Mundy, Tetrahedron Lett., 1984, 25, 4619–4622 CrossRefCAS.
B. P. Mundy, D. Wilkening and K. B. Lipkowitz, J. Org. Chem., 1985, 50, 5727–5731 CrossRefCAS.
C. P. Jasperse and D. P. Curran, J. Am. Chem. Soc., 1990, 112, 5601–5609 CrossRefCAS.
L. Fitjer, A. Kanschik and M. Majewski, Tetrahedron Lett., 1985, 26, 5277–5280 CrossRefCAS.
L. Fitjer, M. Majewski, A. Kanschik, E. Egert and G. M. Sheldrick, Tetrahedron Lett., 1986, 27, 3603–3606 CrossRefCAS.
L. Fitjer, B. Rissom, A. Kanschik and E. Egert, Tetrahedron, 1994, 50, 10879–10892 CrossRefCAS.
L. Fitjer, A. Kanschik and M. Majewski, Tetrahedron Lett., 1988, 29, 5525–5528 CrossRefCAS.
G. Mehta and D. Subrahmanyam, J. Chem. Soc., Chem. Commun., 1985, 768–769 RSC.
G. Mehta and D. Subrahmanyam, J. Chem. Soc., Perkin Trans. 1, 1991, 395–401 RSC.
G. Mehta and D. Subrahmanyam, J. Chem. Soc., Perkin Trans. 1, 1991, 2289–2290 CAS.
G. Mehta, D. Subrahmanyam, G. S. R. S. Rao and K. Pramod, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1985, 24, 797–798 Search PubMed.
G. Mehta, K. Pramod and D. Subrahmanyam, J. Chem. Soc., Chem. Commun., 1986, 247–248 RSC.
G. Mehta and D. Subrahmanyam, Tetrahedron Lett., 1989, 30, 2709–2712 CrossRefCAS.
V. Singh, P. K. Sahu, R. B. Singh and S. M. Mobin, J. Org. Chem., 2007, 72, 10155–10165 CrossRefCAS.
P. A. Wender and G. B. Dreyer, J. Am. Chem. Soc., 1982, 104, 5805–5807 CrossRefCAS.
J. Wrobel, K. Takahashi, V. Honkan, G. Lannoye, J. M. Cook and S. H. Bertz, J. Org. Chem., 1983, 48, 139–141 CrossRefCAS.
S. H. Bertz, G. Dabbagh, J. M. Cook and V. Honkan, J. Org. Chem., 1984, 49, 1739–1743 CrossRefCAS.
M. Karpf and A. S. Dreiding, Tetrahedron Lett., 1980, 21, 4569–4570 CrossRefCAS.
H. Schostarez and L. A. Paquette, Tetrahedron, 1981, 37, 4431–4435 CrossRefCAS.
H. Schostarez and L. A. Paquette, J. Am. Chem. Soc., 1981, 103, 722–724 CrossRefCAS.
F. Leyendecker, J. Drouin and J. M. Conia, Tetrahedron Lett., 1974, 15, 2931–2934 CrossRef.
J. Drouin, F. Leyendecker and J. M. Conia, Tetrahedron Lett., 1975, 16, 4053–4056 CrossRef.
T. Prange, J. Drouin, F. Leyendecker and J. M. Conia, J. Chem. Soc., Chem. Commun., 1977, 430–431 RSC.
J. Drouin and M. A. Boaventura, Tetrahedron Lett., 1987, 28, 3923–3926 CrossRefCAS.
L. A. Paquette and M. Vazeux, Tetrahedron Lett., 1981, 22, 291–294 CrossRefCAS.
L. A. Paquette, R. V. Williams, M. Vazeux and A. R. Browne, J. Org. Chem., 1984, 49, 2194–2197 CrossRefCAS.
L. A. Paquette and S. Usui, Tetrahedron Lett., 1999, 40, 3499–3502 CrossRefCAS.
J. E. Maggio, H. E. Simmons, III and J. K. Kouba, J. Am. Chem. Soc., 1981, 103, 1579–1581 CrossRefCAS.
H. E. Simmons, III and J. E. Maggio, Tetrahedron Lett., 1981, 22, 287–290 CrossRef.
S. A. Benner, J. E. Maggio and H. E. Simmons, III, J. Am. Chem. Soc., 1981, 103, 1581–1582 CrossRefCAS.
W. Oppolzer and F. Marazza, Helv. Chim. Acta, 1981, 64, 1575–1578 CrossRefCAS.
W. Oppolzer and K. Bättig, Helv. Chim. Acta, 1981, 64, 2489–2491 CrossRefCAS.
N. T. Barczak and E. R. Jarvo, Chem.–Eur. J., 2011, 17, 12912–12916 CrossRefCAS.
E. A. Mash and K. A. Nelson, J. Am. Chem. Soc., 1985, 107, 8256–8258 CrossRefCAS.
E. A. Mash and K. A. Nelson, Tetrahedron Lett., 1986, 27, 1441–1444 CrossRefCAS.
E. A. Mash and K. A. Nelson, Tetrahedron, 1987, 43, 679–692 CrossRefCAS.
T. Uyehara, K. Osanai, M. Sugimoto, I. Suzuki and Y. Yamamoto, J. Am. Chem. Soc., 1989, 111, 7264–7265 CrossRefCAS.
T. Uyehara, T. Murayama, K. Sakai, K. Onda, M. Ueno and T. Sato, Bull. Chem. Soc. Jpn., 1998, 71, 231–242 CrossRefCAS.
C. K. Sha, S. J. Tsong and D. C. Wang, Tetrahedron Lett., 1990, 31, 3745–3748 CrossRefCAS.
H.-Y. Lee, D.-I. Kim and S. Kim, Chem. Commun., 1996, 1539–1540 RSC.
C. A. Dvorak and V. H. Rawal, Chem. Commun., 1997, 2381–2382 RSC.
H.-Y. Lee, D. Kim, II and S. Kim, Bull. Korean Chem. Soc., 1999, 20, 269–270 CAS.
H.-Y. Lee, D. K. Moon and J. S. Bahn, Tetrahedron Lett., 2005, 46, 1455–1458 CrossRefCAS.
H.-Y. Lee, R. N. Murugan and D. K. Moon, Eur. J. Org. Chem., 2009, 5028–5037 CrossRefCAS.
W. J. Kerr, A. J. Morrison and L. C. Paterson, Tetrahedron, 2015, 71, 5356–5361 CrossRefCAS.
D. P. Curran and W. Shen, Tetrahedron, 1993, 49, 755–770 CrossRefCAS.
B. De Boeck and G. Pattenden, Tetrahedron Lett., 1998, 39, 6975–6978 CrossRefCAS.
B. De Boeck, N. M. Harrington-Frost and G. Pattenden, Org. Biomol. Chem., 2005, 3, 340–347 RSC.
D. H. R. Barton, D. Crich and G. Kretzschmar, Tetrahedron Lett., 1984, 25, 1055–1058 CrossRefCAS.
D. H. R. Barton, D. Crich and W. B. Motherwell, J. Chem. Soc., Chem. Commun., 1983, 939–941 RSC.
D. H. R. Barton, D. Crich and W. B. Motherwell, Tetrahedron Lett., 1983, 24, 4979–4982 CrossRefCAS.
D. H. R. Barton and G. Kretzschmar, Tetrahedron Lett., 1983, 24, 5889–5892 CrossRefCAS.
D. H. R. Barton, D. Crich and W. B. Motherwell, Tetrahedron, 1985, 41, 3901–3924 CrossRefCAS.
D. H. R. Barton, D. Crich and G. Kretzschmar, J. Chem. Soc., Perkin Trans. 1, 1986, 39–53 RSC.
D. H. R. Barton, D. Bridon, I. Fernandaz-Picot and S. Z. Zard, Tetrahedron, 1987, 43, 2733–2740 CrossRefCAS.
D. Crich and L. Quintero, Chem. Rev., 1989, 89, 1413–1432 CrossRefCAS.
D. H. R. Barton, Tetrahedron, 1992, 48, 2529–2544 CrossRefCAS.
D. P. Curran and R. L. Wolin, Synlett, 1991, 317–318 CrossRefCAS.
T. Uyehara, T. Murayama, K. Sakai, M. Ueno and T. Sato, Tetrahedron Lett., 1996, 37, 7295–7298 CrossRefCAS.
C. A. Dvorak, C. Dufour, S. Iwasa and V. H. Rawal, J. Org. Chem., 1998, 63, 5302–5303 CrossRefCAS.
U. Weiss and J. M. Edwards, Tetrahedron Lett., 1968, 9, 4885–4887 CrossRef.
S. Yang-Lan, M. Mueller-Johnson, J. Oehldrich, D. Wichman, J. M. Cook and U. Weiss, J. Org. Chem., 1976, 41, 4053–4058 CrossRefCAS.
R. W. Weber and J. M. Cook, Can. J. Chem., 1978, 56, 189–192 CrossRefCAS.
R. Mitschka, J. Oehldrich, K. Takahashi, J. M. Cook, U. Weiss and J. V. Silverton, Tetrahedron, 1981, 37, 4521–4542 CrossRefCAS.
S. Kotha and A. K. Chinnam, Synthesis, 2014, 46, 301–306 CrossRef.
S. H. Bertz, J. M. Cook, A. Gawish and U. Weiss, Org. Synth., 1986, 64, 27–38 CrossRefCAS.
H. Quast, H. Roeschert, E. M. Peters, K. Peters and H. G. Von Schnering, Chem. Ber., 1989, 122, 523–531 CrossRefCAS.
S. C. Suri, Tetrahedron Lett., 1993, 34, 8321–8324 CrossRefCAS.
G. A. Kraus and J. Shi, J. Org. Chem., 1990, 55, 5423–5424 CrossRefCAS.
G. A. Kraus and J. Shi, J. Org. Chem., 1991, 56, 4147–4151 CrossRefCAS.
S. Ghosh, S. S. Roy and A. Bhattacharya, Synth. Commun., 1989, 19, 3191–3197 CrossRefCAS.
S. Yamago and E. Nakamura, J. Chem. Soc., Chem. Commun., 1988, 1112–1113 RSC.
S. Yamago and E. Nakamura, Tetrahedron, 1989, 45, 3081–3088 CrossRefCAS.
B. M. Trost and Y. Shi, J. Am. Chem. Soc., 1991, 113, 701–703 CrossRefCAS.
B. M. Trost and Y. Shi, J. Am. Chem. Soc., 1993, 115, 9421–9438 CrossRefCAS.
G. A. Molander, E. D. Dowdy and H. Schumann, J. Org. Chem., 1998, 63, 3386–3396 CrossRefCAS.
L. M. Schneider, V. M. Schmiedel, T. Pecchioli, D. Lentz, C. Merten and M. Christmann, Org. Lett., 2017, 19, 2310–2313 CrossRefCAS.
H. W. Thompson, Tetrahedron Lett., 1966, 7, 6489–6494 CrossRef.
H. W. Thompson, J. Org. Chem., 1968, 33, 621–625 CrossRefCAS.
R. Eckrich and D. Kuck, Synlett, 1993, 1993, 344–347 CrossRef.
D. Kuck, B. Paisdor and H. F. Gruetzmacher, Chem. Ber., 1987, 120, 589–595 CrossRefCAS.
B. Paisdor, H.-F. Grützmacher and D. Kuck, Chem. Ber., 1988, 121, 1307–1313 CrossRefCAS.
B. Paisdor and D. Kuck, J. Org. Chem., 1991, 56, 4753–4759 CrossRefCAS.
D. Kuck, A. Schuster, C. Fusco, M. Fiorentino and R. Curci, J. Am. Chem. Soc., 1994, 116, 2375–2381 CrossRefCAS.
J. Vile, M. Carta, C. G. Bezzu, B. M. Kariuki and N. B. McKeown, Polymer, 2014, 55, 326–329 CrossRefCAS.
D. Kuck and M. Seifert, Chem. Ber., 1992, 125, 1461–1469 CrossRefCAS.
T. Hackfort, B. Neumann, H.-G. Stammler and D. Kuck, Eur. J. Org. Chem., 1999, 2879–2884 CrossRefCAS.
J. S. Jaworski, A. Kosson, S. Filipek, M. Kolinski and D. Kuck, J. Phys. Chem. C, 2009, 113, 7436–7442 CrossRefCAS.
A. Ceccon, A. Gambaro, F. Manoli, A. Venzo, P. Ganis, G. Valle and D. Kuck, Chem. Ber., 1993, 126, 2053–2060 CrossRefCAS.
G. Dyker, J. Körning, P. G. Jones and P. Bubenitschek, Angew. Chem., Int. Ed., 1993, 32, 1733–1735 CrossRef.
G. Dyker, J. Körning, F. Nerenz, P. Siemsen, S. Sostmann, A. Wiegand, P. G. Jones and P. Bubenitschek, Pure Appl. Chem., 1996, 68, 323–326 CAS.
G. Dyker, J. Körning, P. Bubenitschek and P. G. Jones, Liebigs Ann./Recl., 1997, 203–209 CrossRefCAS.
G. Dyker, T. Kerl, J. Körning, P. Bubenitschek and P. G. Jones, Tetrahedron, 2000, 56, 8665–8668 CrossRefCAS.
T. Kubo, S. Miyazaki, T. Kodama, M. Aoba, Y. Hirao and H. Kurata, Chem. Commun., 2015, 51, 3801–3803 RSC.
T. Hackfort and D. Kuck, Eur. J. Org. Chem., 1999, 2867–2878 CrossRefCAS.
D. Kuck and A. Schuster, Angew. Chem., Int. Ed., 1988, 27, 1192–1194 CrossRef.
D. Kuck, A. Schuster, B. Paisdor and D. Gestmann, J. Chem. Soc., Perkin Trans. 1, 1995, 721–732 RSC.
D. Kuck, B. Paisdor and D. Gestmann, Angew. Chem., Int. Ed., 1994, 33, 1251–1253 CrossRef.
D. Kuck, R. A. Krause, D. Gestmann, F. Posteher and A. Schuster, Tetrahedron, 1998, 54, 5247–5258 CrossRefCAS.
J. Tellenbroeker, D. Barth, B. Neumann, H.-G. Stammler and D. Kuck, Org. Biomol. Chem., 2005, 3, 570–571 RSC.
M. Harig and D. Kuck, Eur. J. Org. Chem., 2006, 1647–1655 CrossRefCAS.
D. Kuck, J. Linke, L. C. Teichmann, D. Barth, J. Tellenbroker, D. Gestmann, B. Neumann, H.-G. Stammler and H. Bogge, Phys. Chem. Chem. Phys., 2016, 18, 11722–11737 RSC.
D. Kuck, A. Schuster, D. Gestmann, F. Posteher and H. Pritzkow, Chem.–Eur. J., 1996, 2, 58–67 CrossRefCAS.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Robin M.
Bär
a,
Robin M.
Bär




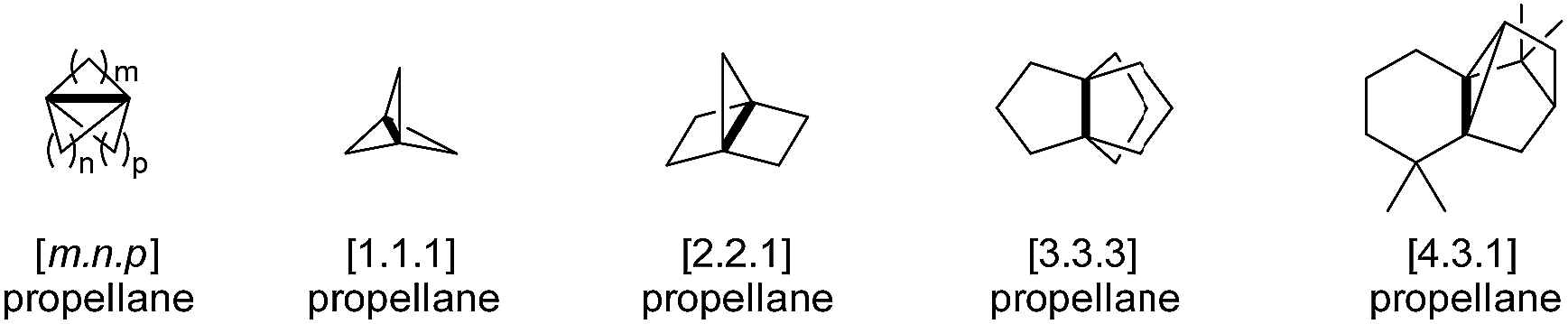
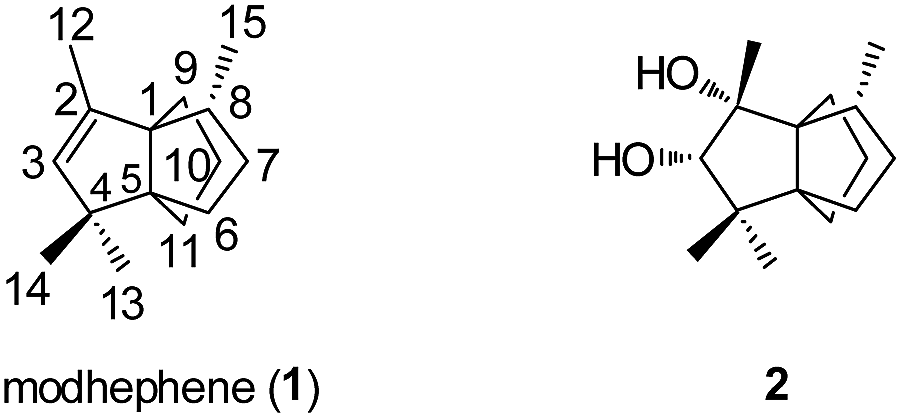

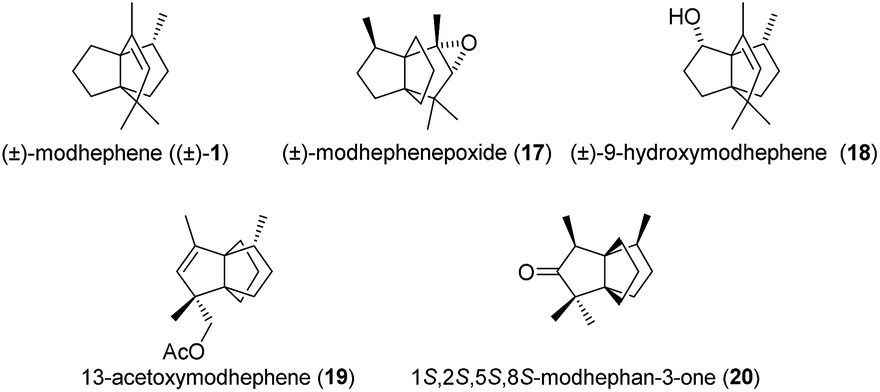
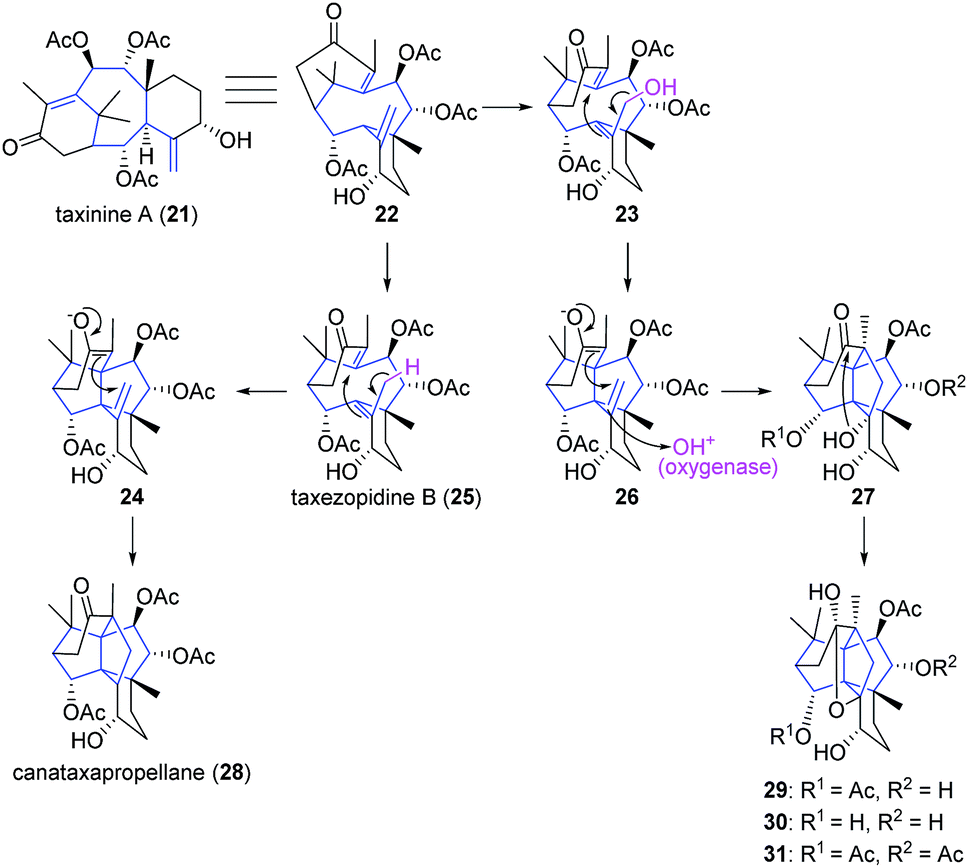
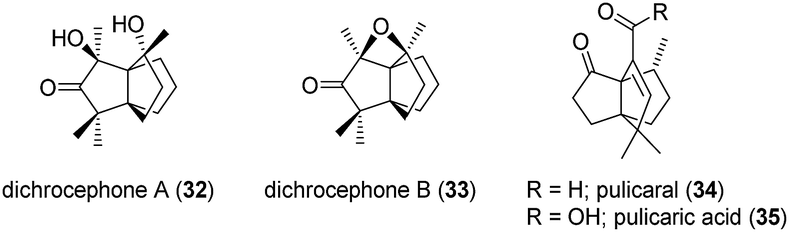
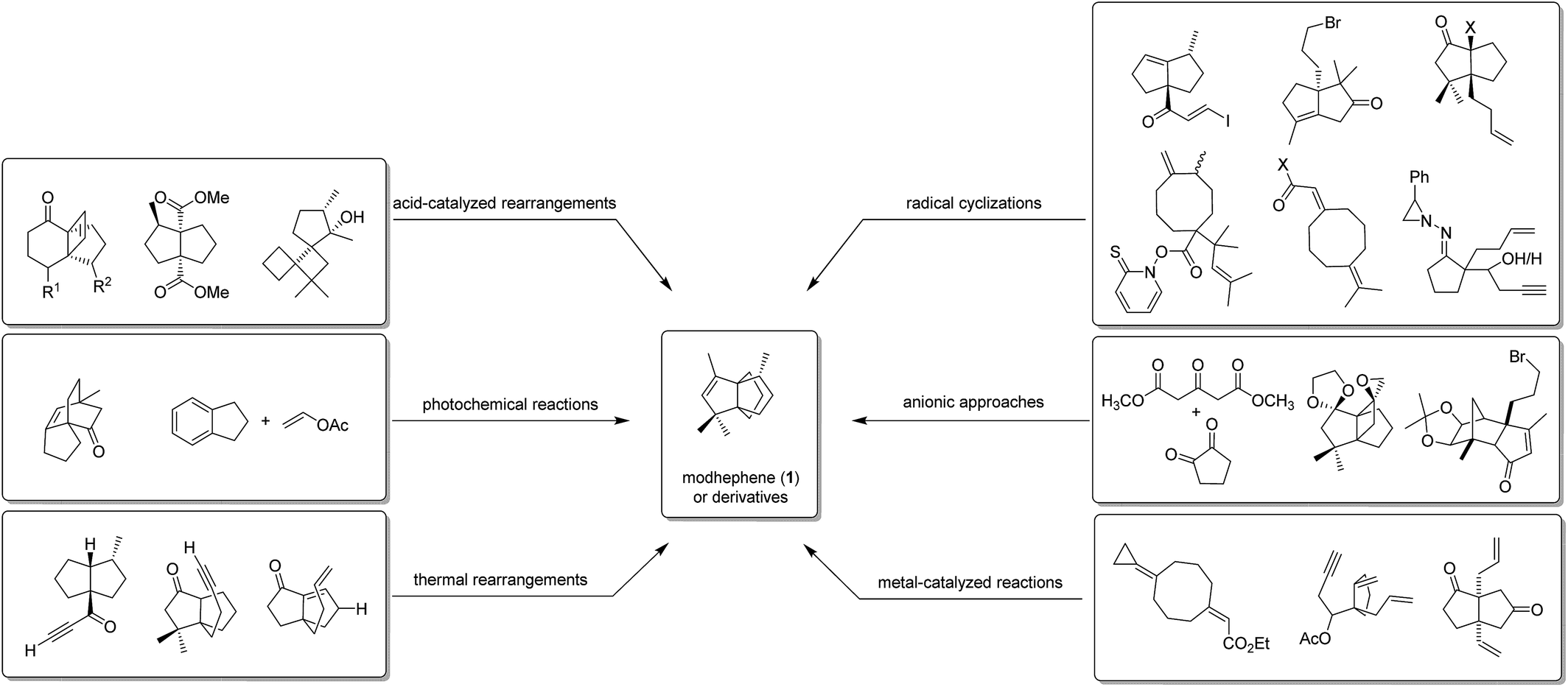
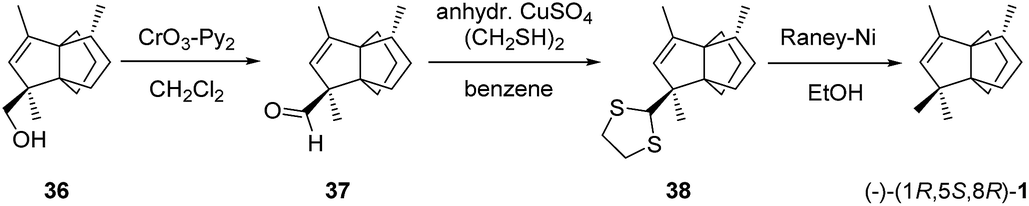
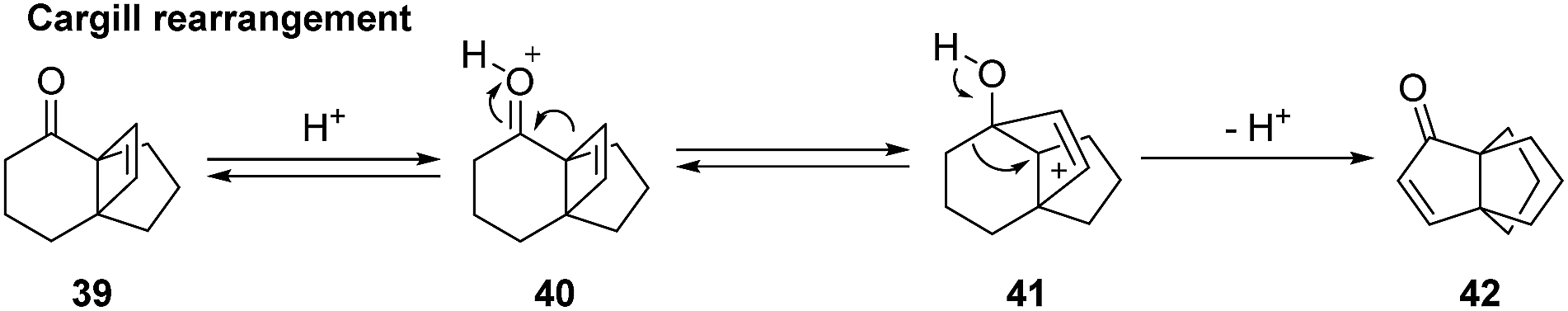
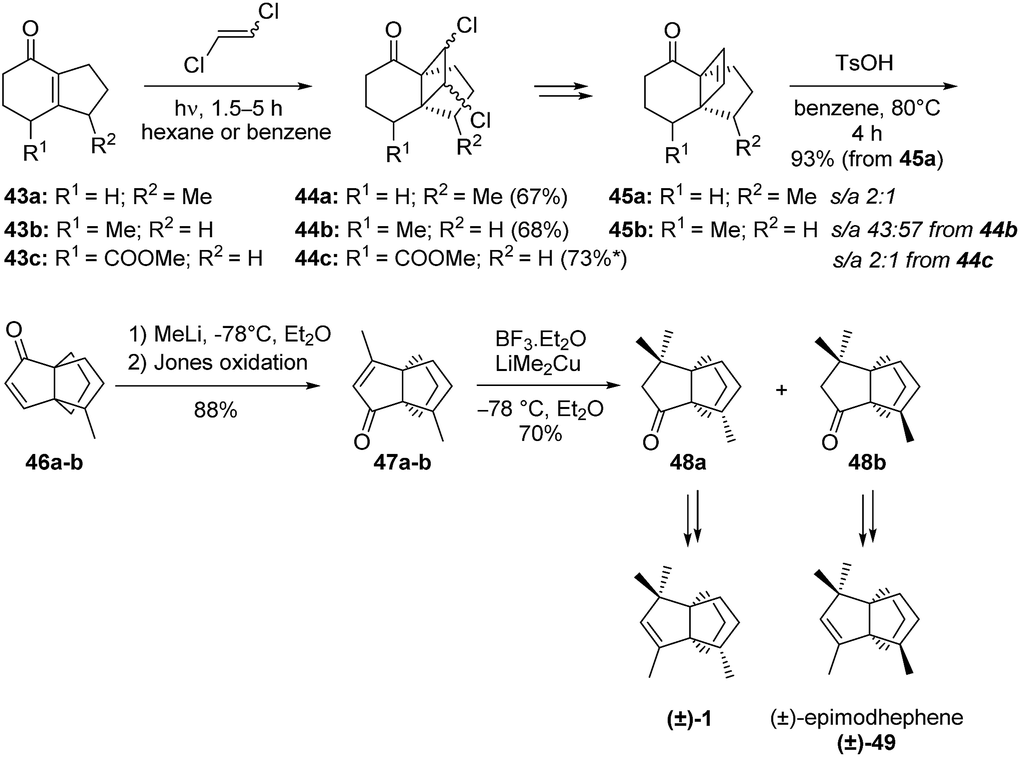
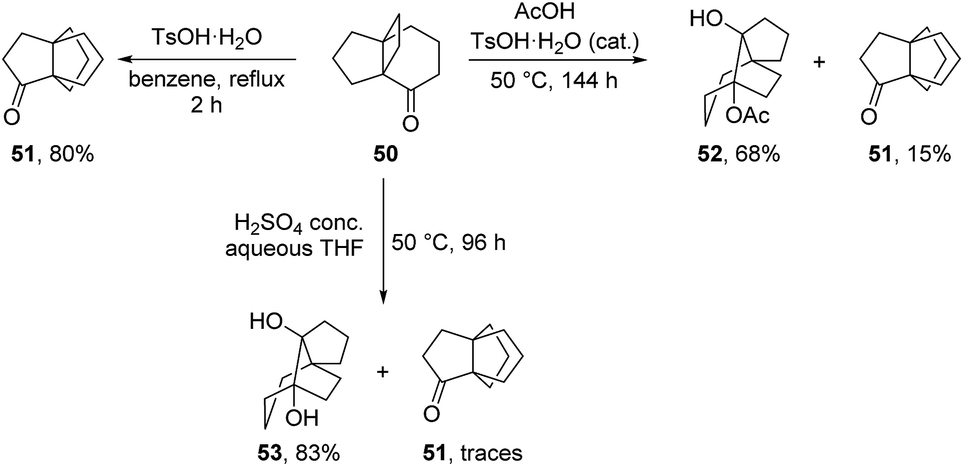
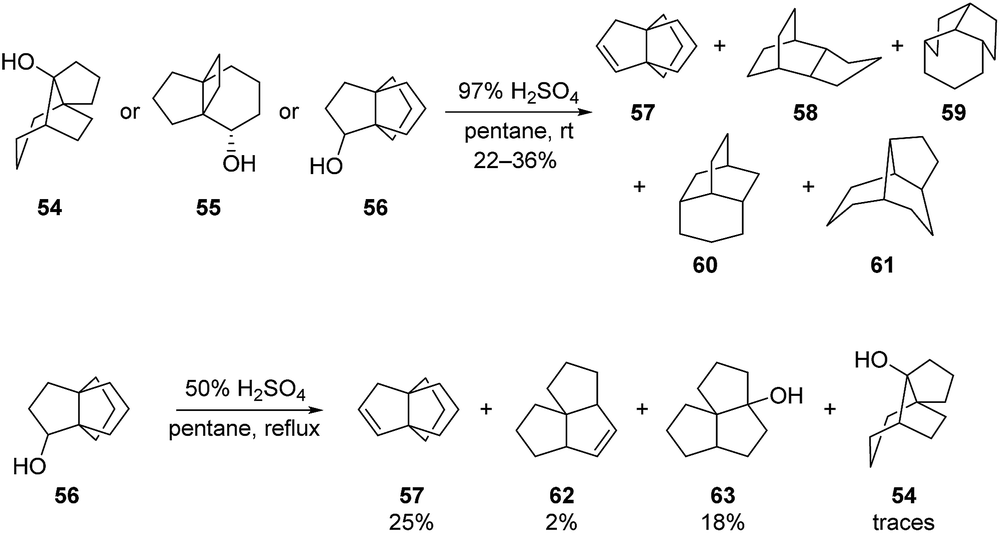
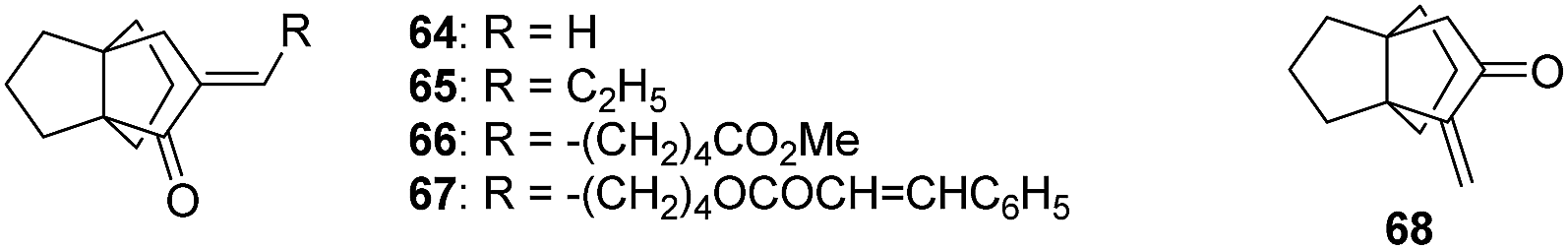
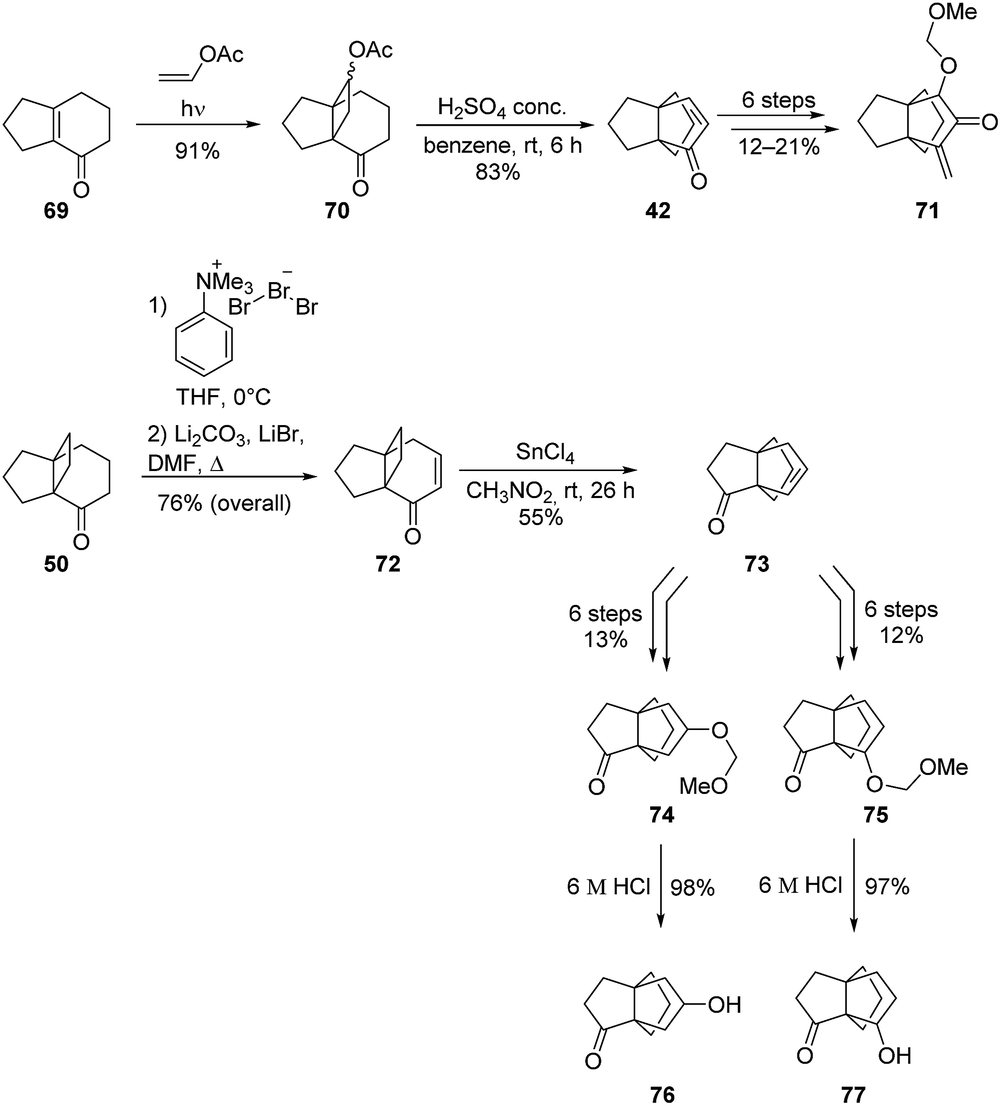
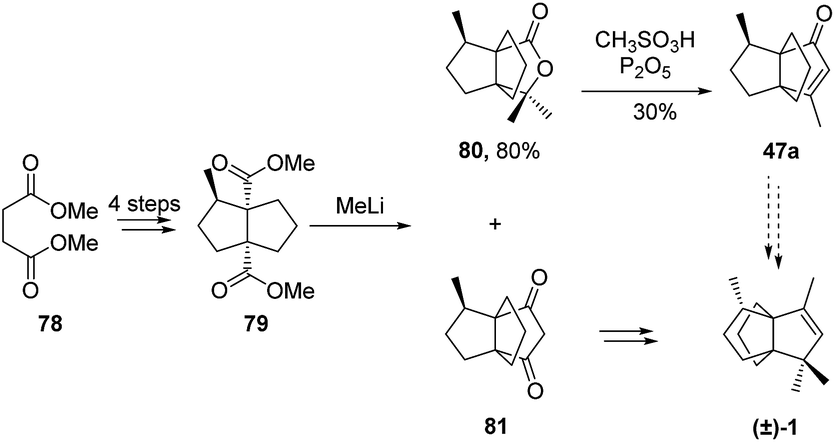
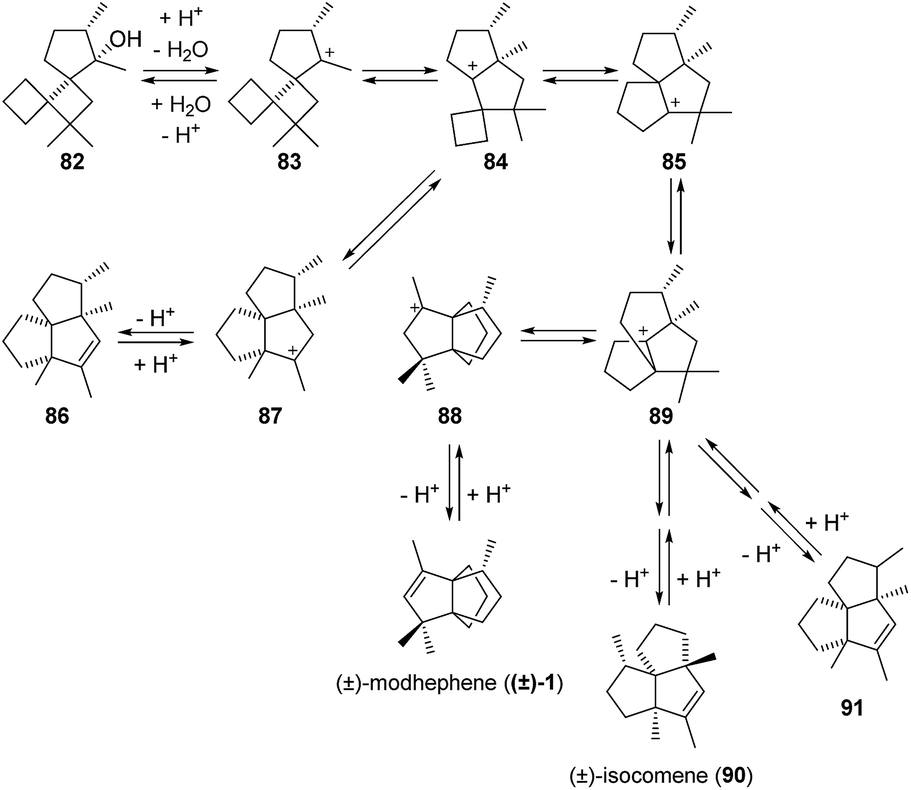

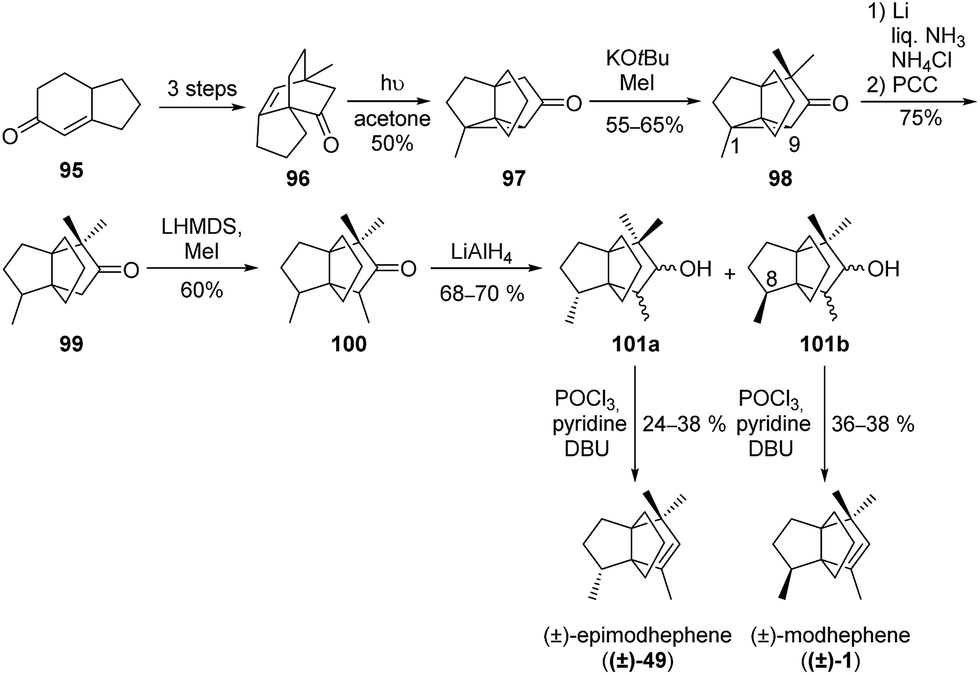
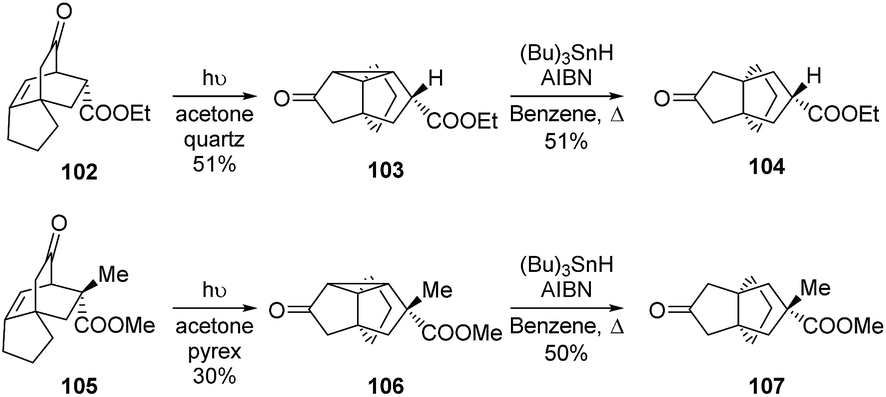
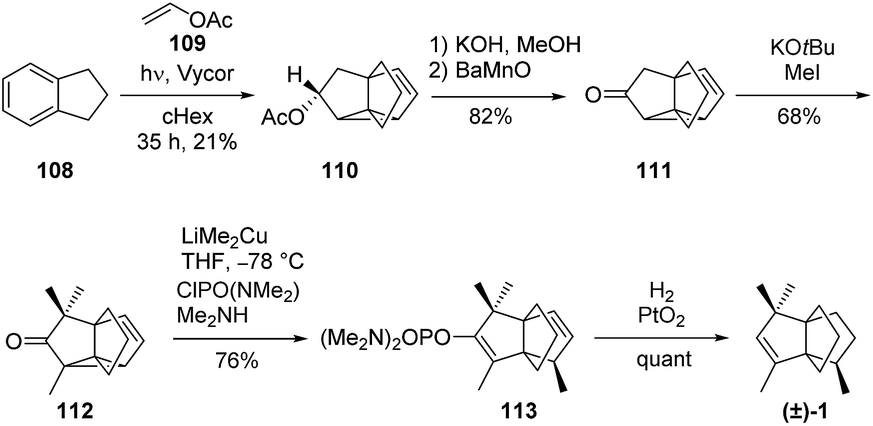
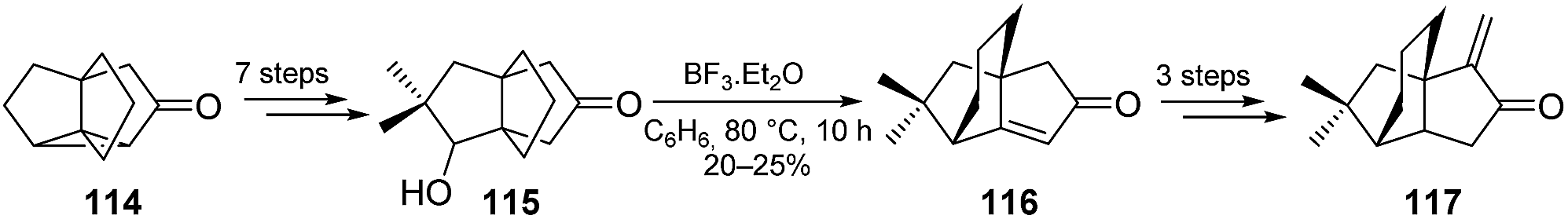
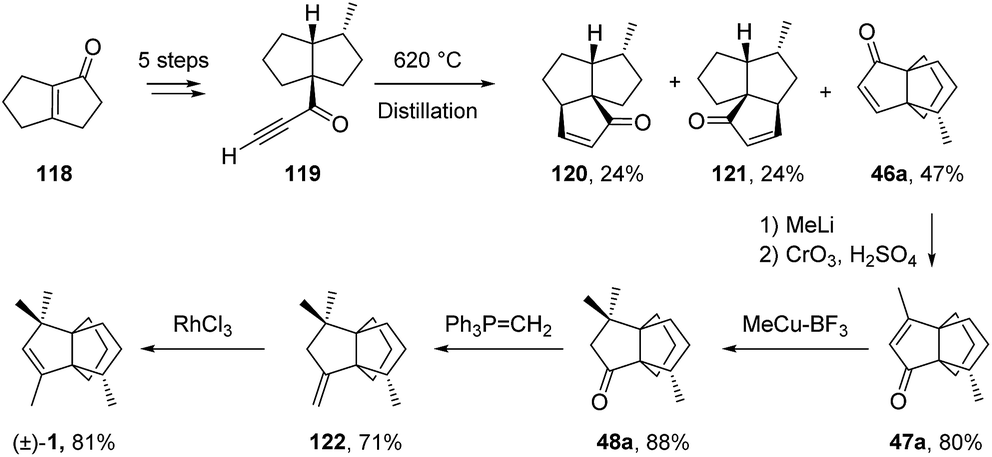
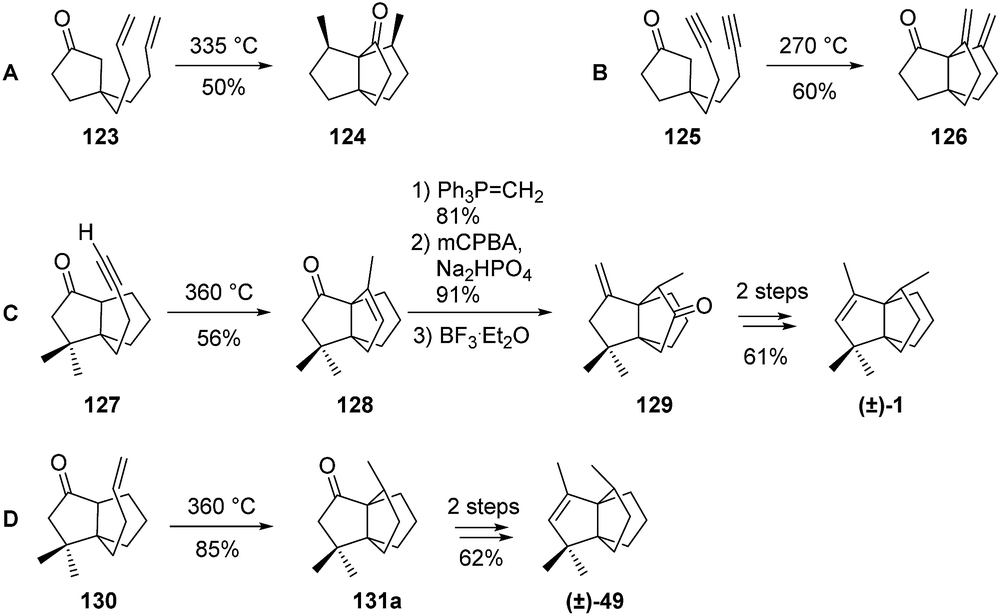
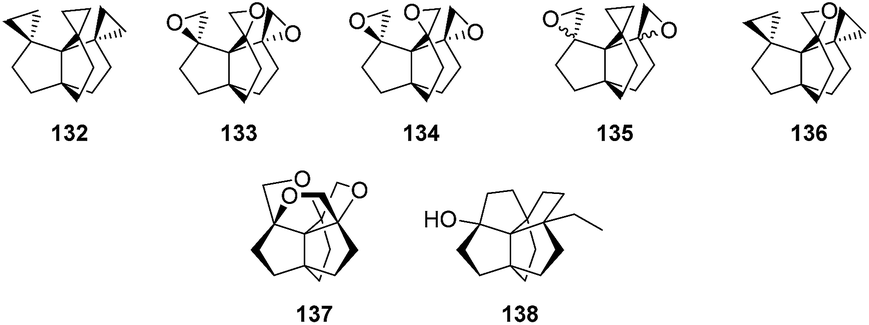

![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: