
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe first-principles study of the adsorption of NH3, NO, and NO2 gas molecules on InSe-like phosphorus carbide†
Andrey A.
Kistanov

Nano and Molecular Systems Research Unit, University of Oulu, 90014 Oulu, Finland. E-mail: andrey.kistanov@oulu.fi
First published on 12th May 2020
Abstract
The adsorption of environmental gas molecules, i.e., NH3, NO, and NO2 on the γ-PC surface has been studied using first-principles calculations. The lowest-energy configurations of these molecules on the γ-PC surface were found and the adsorption energies were calculated. The NH3, NO, and NO2 molecules were found to be physisorbed on the γ-PC surface. The analysis of the charge transfer between the molecules and the surface predicted NH3 and NO as donors to γ-PC, while NO2 acted as an acceptor to γ-PC. Remarkable changes in the band structure of γ-PC were found upon the adsorption of NO2 on its surface. In addition, significant modulations in the work function of γ-PC were observed after the adsorption of NH3 and NO.
Introduction
Ultrathin two-dimensional (2D) materials possess a high surface area-to-volume ratio and weak screening of electrons, which make them vulnerable to the influence of the ambient physical and chemical adsorbates.1,2 On the other hand, the advanced chemical activity of 2D materials offers the possibility to modify their electronic and chemical properties by the effect of ambient adsorbates.3 The high sensitivity of the 2D materials to environmental molecules and gases also makes them promising for gas adsorption and detection.4 For instance, first-principles calculations have predicted a strong binding of the NO and NO2 gas molecules to MoS2.5 The MoS2 monolayer produced by annealing sputtered Mo films in S vapor has shown excellent selectivity to NH3.6 Many investigations indicate potential for InSe in fabricating gas sensors.7,8 Particularly, the strong ability of InSe to interact with external NH3, NO2, and NO molecules has been found.9 It should be noted that the InSe surface degrades as a result of defect-photo-promoted oxidations, which may reduce its applications.10The discovery of new 2D materials, such as phosphorene, has shown that these materials may provide much stronger adsorption ability to ambient molecules and gases compared to MoS2 or InSe.9,11–13 The promoted activity of phosphorene has been reported for the NO, NO2, NH3, and CO molecules.14,15 In addition, the gas sensing ability of phosphorene has been improved by the doping of its surface by NH3 and NO2 gas molecules.16 However, such high chemical activity of phosphorene leads to its fast degradation in ambient conditions.17 In addition, the formation of defects on the phosphorene surface may lead to the degradation of its structure.18 Nevertheless, the adsorption of different adatoms on phosphorene including s and p valence metals, 3d transition metals, noble metals, H and O does not necessarily disturb its structural integrity.19,20
In the search of 2D materials that are highly sensitive to gases while remaining structurally stable in ambient conditions, considerable research has focused on the study of 2D hybrid materials.21 Phosphorus carbide (PC), a composite of graphene and phosphorene, is one of these materials.22,23 The latest studies show that PC has an anisotropic structure, wide band gap, and high charge carrier mobility.24 Furthermore, PC-based field-effect transistors have been fabricated recently via a novel carbon doping technique.25 More importantly, PC combines the high chemical activity existing in phosphorene and the structural stability inherent to graphene, which are the most important features of all gas sensors.24,26,27 Previous studies on the sensing mechanism of 2D layered materials, including monolayer MoS2,28–30 monolayer SnS2,31 and monolayer C3N,32 indicate that charge transfer plays an important role for the sensing of gas molecules.
Very recently, stable γ-PC with an InSe-like structure has been predicted theoretically.33 This PC allotrope possesses the promoted adsorption and low diffusion barrier of Li, which render its application in rechargeable nanodevices. However, to date, the possible effects of ambient adsorbates on the structure and electronic properties of γ-PC as well as the suitability of γ-PC for gas detection have not been investigated.
In this work, first-principles calculations have been performed for the adsorption of NH3, NO, and NO2 molecules on γ-PC. Their exact configurations on the surface and their preferential binding sites were determined by calculating their adsorption energy. In order to determine their effect on the electronic properties of γ-PC, the charge transfer and band structure analyses of molecule-adsorbed γ-PC were conducted.
Results and discussion
The top and side views of the unit cell of the optimized γ-InSe-like phosphorus carbide (γ-PC) are shown in Fig. 1a. The calculated lattice constants of γ-PC were a = 3.09 Å and b = 5.35 Å, and the lengths of the C–C and C–P bonds were 1.56 and 1.97 Å, respectively, which were comparable with those reported in the theoretical study of γ-PC.33 The band structure of γ-PC obtained by the GGA PBE (black) and HSE06 (red) methods is presented in Fig. 1b. Importantly, both methods predicted γ-PC to be an indirect band gap semiconductor. The GGA PBE approach only slightly underestimated the band gap value (1.51 eV) compared to that (2.58 eV) calculated using the HSE method, which was also consistent with previously reported results.33 Therefore, the less expensive computational method was used in this work. | ||
| Fig. 1 (a) The top and side views of the unit cell and (b) GGA PBE (the black line) and HSE (the red line) band structures of γ-PC. | ||
To determine the lowest-energy adsorption position of the NH3, NO, and NO2 molecules on γ-PC, several possible configurations of the molecules in all possible adsorption sites and aligned parallel, perpendicular or tilted to the surface plane of γ-PC were investigated. The calculated adsorption energy (Ea) values of NH3, NO, and NO2 on γ-PC, the charge transfer (Δq) between the adsorbed molecules and the γ-PC surface, the shortest distance (d) from the adsorbed molecules to the γ-PC surface, and the work function of the molecule-adsorbed γ-PC for the most stable adsorption configurations are given in Table 1. Several other examined configurations of the considered molecules on γ-PC with comparably lower Ea are shown in Fig. S1–S3 (see ESI†) and the Ea, d, and Δq values for these structures are presented in Table S1 (see ESI†).
| Molecule | E a, eV MoS2 | E a, eV InSe | E a, eV γ-PC | Role | Δq, e | d, Å | WF, eV |
|---|---|---|---|---|---|---|---|
| NH3 | −0.49 | −0.20 | −0.17 | Donor | −0.028 | 2.90 | 5.46 |
| NO | −0.54 | −0.13 | −0.20 | Donor | −0.003 | 3.01 | 4.71 |
| NO2 | −0.94 | −0.24 | −0.55 | Acceptor | 0.091 | 2.88 | 5.61 |
| No molecule | — | — | — | — | — | — | 5.61 |
NH3 adsorption
The lowest-energy configuration of an NH3 molecule on the γ-PC surface is shown in Fig. 2a. The molecule was located above the centre of the hollow hexagon with the three H atoms pointing away from the surface. After adsorption, the shortest distance (d) between the N atom of the molecule and the surface was 2.90 Å, and the H–N bonds elongated to 1.023 Å compared to those of a free NH3 gas molecule (1.01 Å). This elongation of bond lengths was mostly attributed to the transfer of electron density from the old bonds to the newly formed bonds between NH3 and γ-PC.35 The adsorption energy (Ea) of the NH3 molecule on the γ-PC surface was −0.17 eV, which suggested that the physisorption behaviour of NH3 on γ-PC was similar to that of NH3 on InSe (Ea = −0.20 eV);9 however, on MoS2, the NH3 molecule possessed about 2 times lower Ea (−0.49 eV).34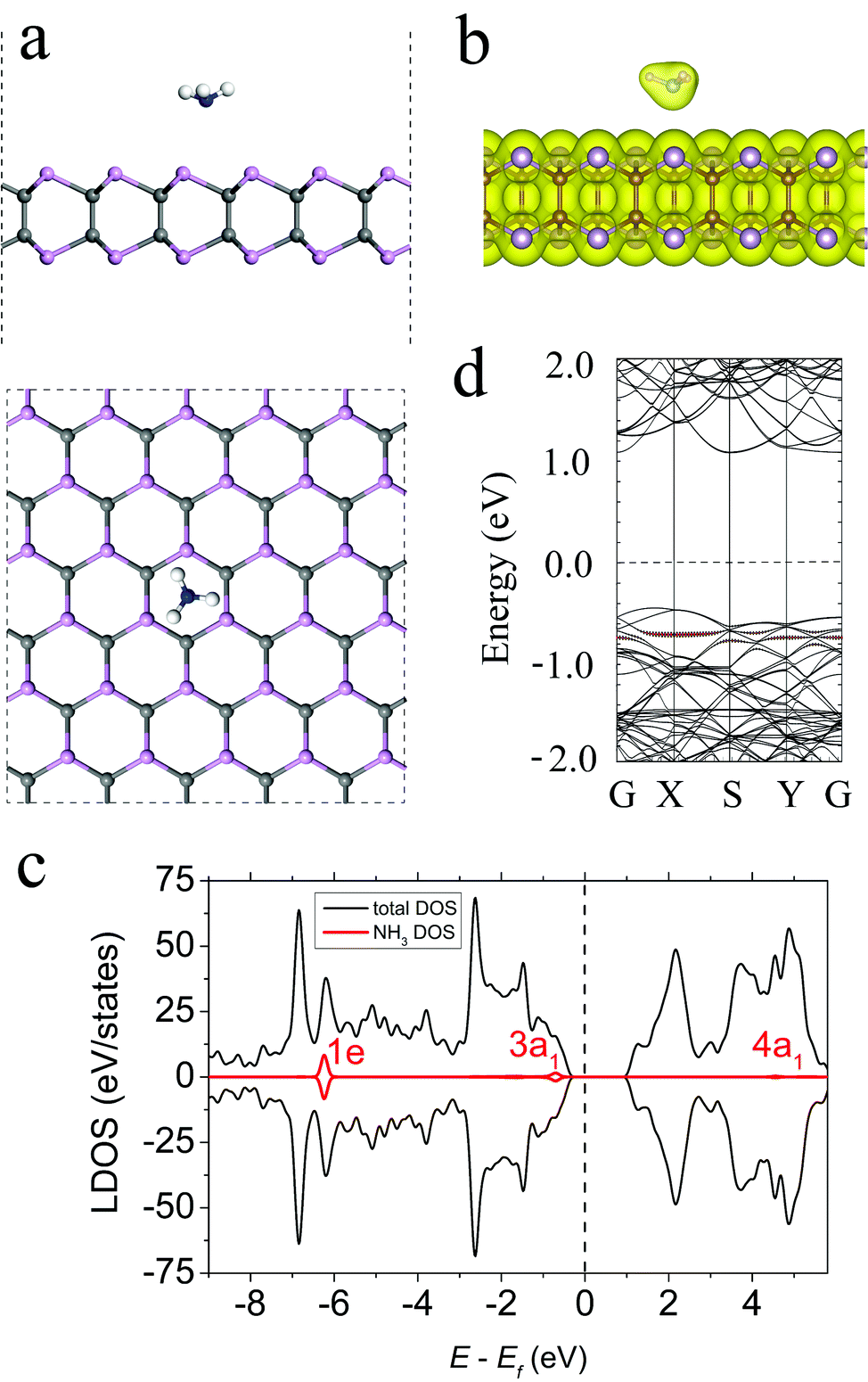 | ||
| Fig. 2 (a) The side and top views, (b) total electron density plot, (c) LDOS, and (d) band structure of NH3-adsorbed γ-PC. The black dashed line shows the Fermi level. The isosurface value is set to 0.05 e Å−3. The bands and DOS coloured in red correspond to the NH3 molecule. | ||
The Bader charge transfer analysis indicated that NH3 acted as an electron donor to γ-PC and the amount of charge transferred from the molecule to the surface was 0.028 e; however, on InSe9 and MoS2,34,36 the NH3 molecule played the role of an electron acceptor. Additional differential charge density (DCD) analysis (Fig. S4, ESI†) indicated a slight depletion of the electrons in the NH3 molecule and the accumulation of electrons at the P atoms near the surface. This is mainly due to the ionization of the lone pair of electrons on the N atom in the 3a1 state. The total electron density plot (Fig. 2b) indicates the zero-electron density in the interface region between the NH3 molecule and the γ-PC surface, which suggests non-covalent bonding between the molecule and the surface. In other considered configurations (Fig. S1, ESI†) also, NH3 is a weak donor to γ-PC (Table S1, ESI†).
The results for the total and local density of states (LDOS) of the NH3 molecule adsorbed on γ-PC are presented in Fig. 2c. The contribution of the molecule to the total DOS of γ-PC was far from the Fermi level, which confirmed the physisorption of NH3 on γ-PC. The non-bonding 3a1 state broadened and appeared slightly below the valence band maximum (VBM); this is also seen in the band structure in Fig. 2d and reflects the charge transfer. In addition, the adsorption of the NH3 molecule decreased the work function of γ-PC from 5.61 to 5.46 eV.
NO adsorption
For NO adsorption on γ-PC, the most energetically favourable structure is shown in Fig. 3a, where the N–O bond of the molecule is tilted to the surface and is located at d = 3.01 Å above the centre of the hexagon. The N–O bond of the molecule elongated compared to that of the free NO gas molecule from 1.010 to 1.167 Å after adsorption. This elongation of the bond is mostly attributed to the transfer of electron density from the old bond to the newly formed bond between NO and γ-PC.35 The Ea value is −0.20 eV for NO adsorbed on γ-PC, indicating a non-covalent interaction between the molecule and the surface, which is also confirmed by the zero-electron density in the interface region between the NO molecule and the γ-PC surface (Fig. 3b). It should be noted that the Ea of NO on γ-PC was lower than that of NO on InSe9 but was higher than that of NO on MoS2.34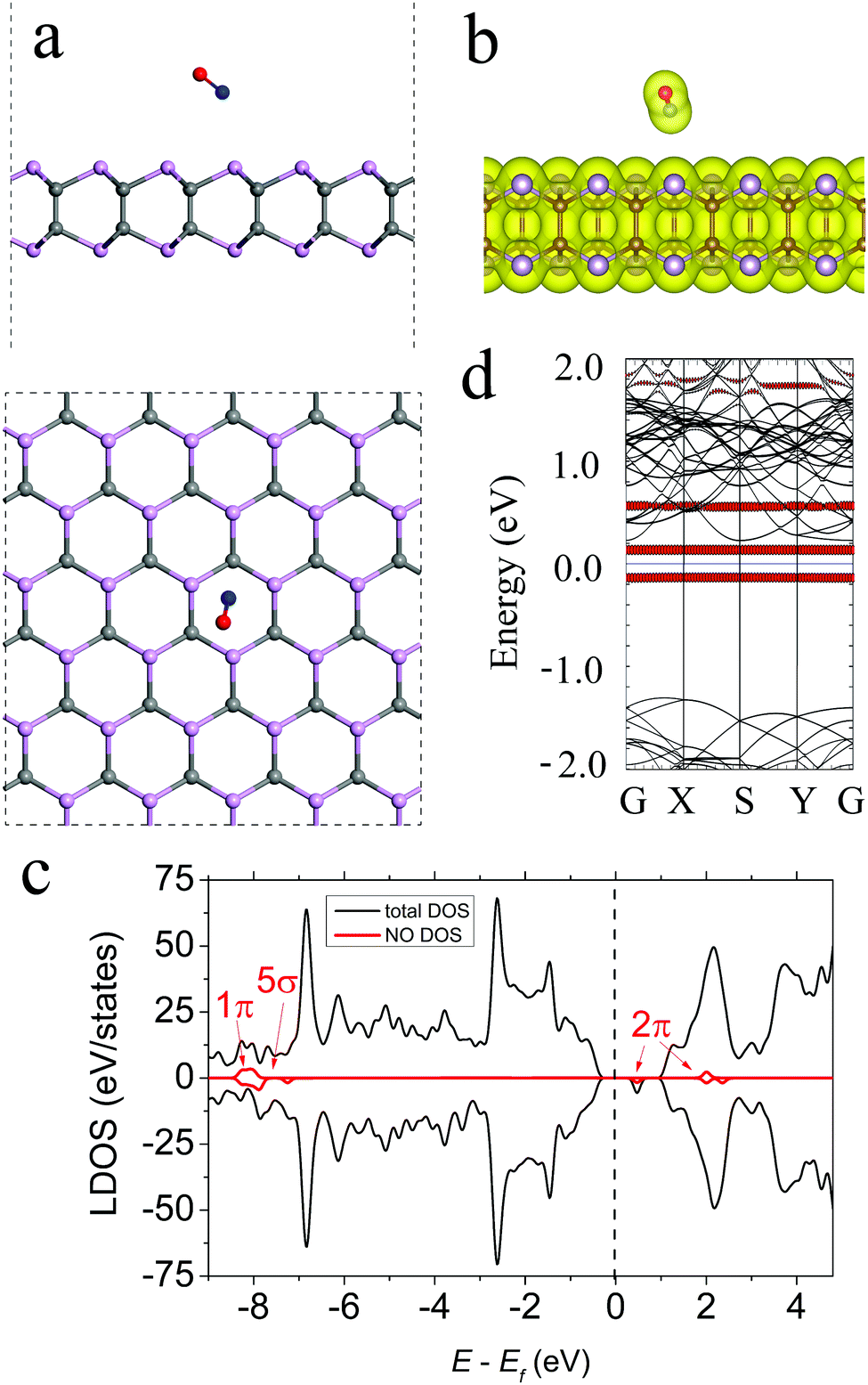 | ||
| Fig. 3 (a) The side and top views, (b) total electron density plot, (c) LDOS, and (d) band structure of NO-adsorbed γ-PC. The black dashed line shows the Fermi level. The isosurface value is set to 0.05 e Å−3. The bands and DOS coloured in red correspond to the NO molecule. | ||
The Bader analysis predicted NO to be a weak electron donor to γ-PC with the amount of charge transferred from the molecule to the surface being 0.003 e. Interestingly, on both InSe and MoS2, the NO molecule is an electron acceptor.9,34 To understand this specific sensitivity of γ-PC to NO the electron localization function of the NO-adsorbed γ-PC was plotted (Fig. S5a, ESI†). It was found that there was strong charge redistribution within the molecule, while a very little amount of charge was transferred to the nearest P atom at the surface. It was also found that the acceptor/donor role of the NO molecule to γ-PC mostly depended on its position on the surface. More details are presented in ESI.†
Fig. 3c presents the LDOS of the NO molecule adsorbed on the γ-PC surface. It can be seen that the half-filled 2π frontier state is doubly degenerated and the 2π down state is located between the Fermi level and the conduction band minimum (CBM); this is also depicted in the band structure plot in Fig. 3d. The presence of unoccupied states within the band gap of NO-adsorbed γ-PC but a weak charge flow between the molecule and the surface may explain the significant decrease in the work function of γ-PC from 5.61 to 4.71 eV upon NO adsorption.
NO2 adsorption
The most energetically preferable configuration of the NO2 molecule on the γ-PC surface is shown in Fig. 4a. The molecule was located above the C–P bond at d = 2.88 Å with the N–O bonds pointing towards the surface. These N–O bonds slightly elongated to 1.217 Å compared to those of the free NO2 gas molecule (1.20 Å). This elongation of bonds is mostly attributed to the transfer of electron density from the old bonds to the newly formed bonds between NO2 and γ-PC.35 The Ea value was −0.55 eV for NO2 on γ-PC; it was ∼2.3 times lower than that of NO2 on InSe (Ea = −0.24 eV)9 but was ∼1.7 times higher than that of NO2 on MoS2 (Ea = −0.94 eV).34 Despite the low Ea value of the NO2 molecule on the γ-PC surface, the zero-electron density in their interface region (Fig. 4b) designates the non-covalent nature of the interaction.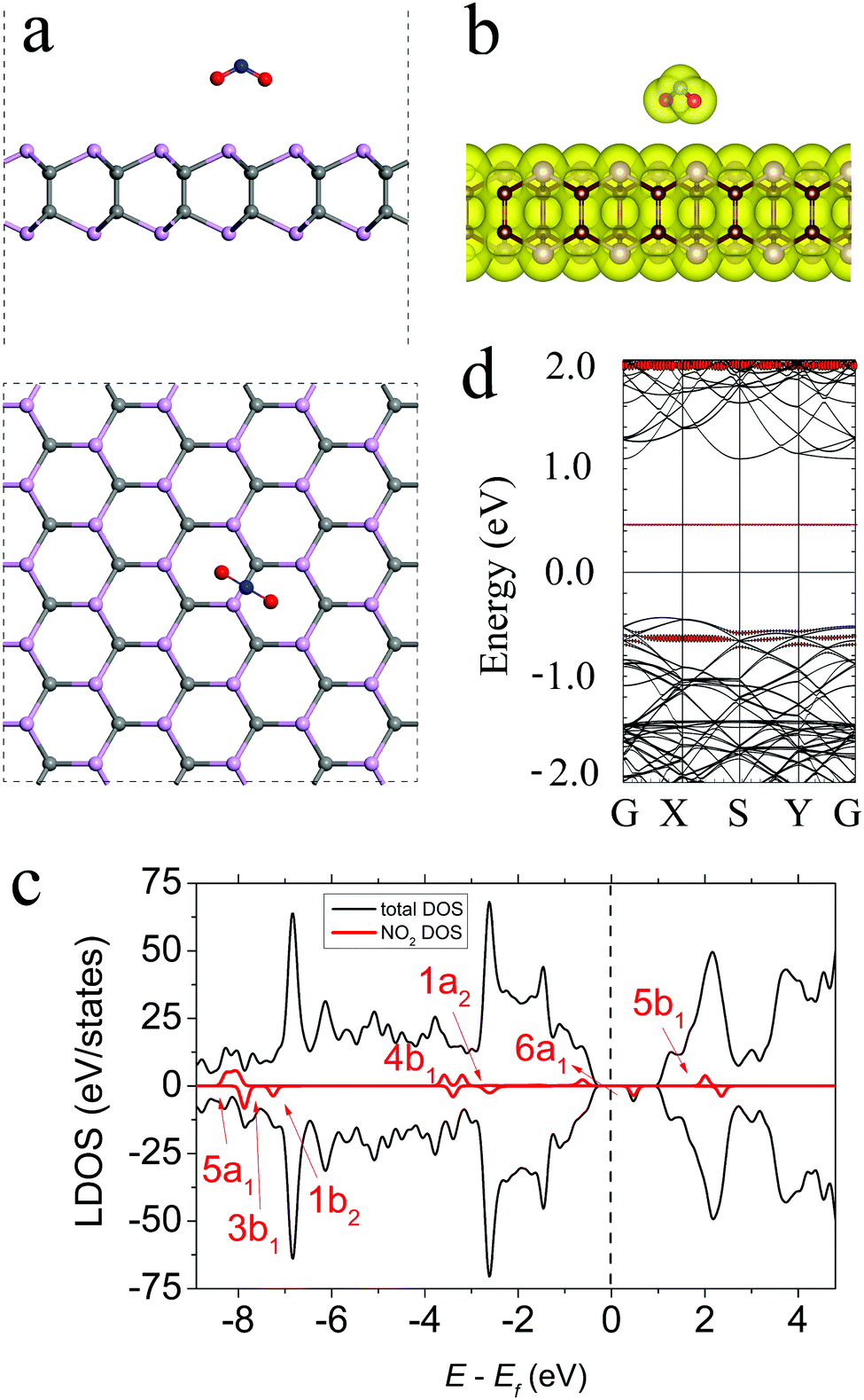 | ||
| Fig. 4 (a) The side and top views, (b) total electron density plot, (c) LDOS, and (d) band structure of NO2-adsorbed γ-PC. The black dashed line shows the Fermi level. The isosurface value is set to 0.05 e Å−3. The bands and DOS coloured in red correspond to the NO2 molecule. | ||
The electron localization function analysis (Fig. S5b, ESI†) and the calculated charge of 0.091 e transferred from the γ-PC surface to the NO2 molecule indicated the strong acceptor ability of the molecule. The charge transfer from γ-PC to NO2 occurred through orbital hybridization, which simultaneously induced a magnetic moment of about 0.99 μB in γ-PC. A more detailed explanation of the acceptor nature of the NO2 molecule on γ-PC is presented in the ESI† (Fig. S3 and Table S1). Similarly, NO2 has been found to be a strong acceptor for InSe9 and MoS2.34 The value of the work function of γ-PC remained almost unchanged after the adsorption of NO2, which indicated that the transfer of electrons to the vacuum level was not hindered.
The LDOS and band structure, shown in Fig. 4c and d, respectively, indicate the spin splitting of the 6a1 orbital of NO2 due to its adsorption on γ-PC. Particularly, the 6a1 spin-up state is ∼0.60 eV below the Fermi level, while the 6a1 spin-down state is ∼0.50 eV above the Fermi level. The 4b1 and 1a2 orbitals also exhibit spin splitting and coincide with the valence states of γ-PC. Such strong orbital mixing accounts for the charge transfer between NO2 and γ-PC.
For applications in gas sensors and detectors, the recovery time of sensing materials is an important parameter.28,32 Therefore, the recovery time of γ-PC was also calculated. It was found that at the room temperature of 300 K, the recovery times for NH3, NO, and NO2 on γ-PC were about 0.4 × 10−9, 2.3 × 10−9, and 2.8 × 10−3 s, respectively. This fast recovery time of γ-PC suggests that it is a promising reversible material for room-temperature gas sensors.
Methods
The simulations were based on the density functional theory (DFT), as implemented in the Vienna ab initio simulation package (VASP).37 The exchange correlation interaction was described by the generalized gradient approximation (GGA) with the Perdew, Burke and Ernzerhof (PBE) functional.38 The hybrid Heyd–Scuseria–Ernzerhof (HSE06) functional was employed for the test calculations of the electronic structure of the γ-PC unit cell.39 To obtain a better description of the long-range van der Waals interactions between the surface and gas molecules, the van der Waals-corrected functional with Becke88 optimization was used. The Brillouin zone sampling for k-points was based on a 6 × 6 × 1 Monkhorst–Pack grid. The atomic positions were relaxed until the force on each atom was smaller than 0.01 eV Å−1. The simulated system of a 5 × 3 × 1 supercell was created with the vacuum space in the z-direction larger than 15 Å to avoid interactions with the neighbouring super cells. The kinetic energy cut-off for the plane wave expansion was set to 400 eV. The adsorption energy of gas molecules on the γ-PC surface was calculated as follows:| Ea = EPC/mol − EPC − Emol, | (1) |
Bader analysis40 was conducted to estimate the charge flow between the γ-PC surface and gas molecules. The work function was calculated as the difference between the electrostatic potentials of an electron at points remote from the surface and the Fermi level.
According to the conventional transition state theory, the recovery time32,41 (τ) can be expressed as follows:
| τ = ν−1e(−Ea/KBT), | (2) |
Conclusions
The calculated low adsorption energies of the NH3, NO, and NO2 molecules on the γ-PC surface suggested strong interactions between γ-PC and these molecules. In all the considered cases, the molecules were physiosorbed on γ-PC, which suggested that they may easily desorb from its surface. Due to this, γ-PC possessed a short recovery time. In addition, the adsorption abilities of NH3, NO, and NO2 on γ-PC were superior to those on InSe but lower than those on MoS2. The physisorption of NO and NO2 led to a remarkable change in the band structure of γ-PC. Hence, the presence of these molecules on its surface could be detected. Moreover, the physisorption of NH3 and NO induced significant modulations in the work function of γ-PC.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges CSC – IT Center for Science, Finland, for computational resources and the financial support provided by the Academy of Finland (grant No. 311934).Notes and references
- S. A. Wells, A. Henning, J. T. Gish, V. K. Sangwan, L. J. Lauhon and M. C. Hersam, Nano Lett., 2018, 18, 7876 CrossRef CAS PubMed.
- Q. Li, Q. Zhou, L. Shi, Q. Chen and J. Wang, J. Mater. Chem. A, 2019, 7, 4291 RSC.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS PubMed.
- L. Kou, T. Frauenheim and C. Chen, J. Phys. Chem. Lett., 2014, 5, 2675 CrossRef CAS PubMed.
- S. Zhao, J. Xue and W. Kang, Chem. Phys. Lett., 2014, 595, 35 CrossRef.
- T. Järvinen, G. S. Lorite, J. Peräntie, G. Toth, S. Saarakkala, V. K. Virtanen and K. Kordas, Nanotechnology, 2019, 30, 40550 CrossRef PubMed.
- D. Ma, W. Ju, Y. Tang and Y. Chen, Appl. Surf. Sci., 2017, 426, 244 CrossRef CAS.
- S. Yang, C. Jiang and S. Wei, Appl. Phys. Rev., 2017, 4, 021304 Search PubMed.
- Y. Cai, G. Zhang and Y. W. Zhang, J. Phys. Chem. C, 2017, 121, 10182 CrossRef CAS.
- A. A. Kistanov, Y. Cai, K. Zhou, S. V. Dmitriev and Y. W. Zhang, J. Mater. Chem. C, 2018, 6, 518 RSC.
- Y. Cai, Q. Ke, G. Zhang and Y. W. Zhang, J. Phys. Chem. C, 2015, 119, 3102 CrossRef CAS.
- J. Park, J. Mun, J. S. Shin and S. W. Kang, R. Soc. Open Sci., 2018, 5, 181462 CrossRef CAS PubMed.
- M. Schleicher and M. Fyta, ACS Appl. Electron. Mater., 2020, 2, 74 CrossRef CAS.
- F. Safaria, M. Moradinasab, M. Fathipour and H. Kosina, Appl. Surf. Sci., 2019, 464, 153 CrossRef.
- A. N. Abbas, B. Liu, L. Chen, Y. Ma, S. Cong, N. Aroonyadet, M. Köpf, T. Nilges and C. Zhou, ACS Nano, 2015, 9, 5618 CrossRef CAS PubMed.
- Gaganpreet, Appl. Surf. Sci., 2020, 507, 144967 CrossRef CAS.
- S. Kuriakose, T. Ahmed, S. Balendhran, V. Bansal, S. Sriram, M. Bhaskaran and S. Walia, 2D Mater., 2018, 5, 032001 CrossRef.
- D. R. Kripalani, Y. Cai, M. Xue and K. Zhou, Phys. Rev. B, 2019, 100, 224107 CrossRef CAS.
- B. Liu and K. Zhou, Prog. Mater. Sci., 2019, 100, 99 CrossRef CAS.
- A. A. Kistanov, S. K. Khadiullin, K. Zhou, S. V. Dmitriev and E. A. Korznikova, J. Mater. Chem. C, 2019, 7, 9195 RSC.
- J. Jin, Y. Zheng, S. Huang, P. Sun, N. Srikanth, L. B. Kong, Q. Yan and K. Zhou, J. Mater. Chem. A, 2019, 7, 783 RSC.
- J. Guan, D. Liu, Z. Zhu and D. Tománek, Nano Lett., 2016, 16, 3247 CrossRef CAS PubMed.
- A. Furlana, G. K. Gueorguiev, Z. Czigány, V. Darakchieva, S. Braun, M. R. Correia, H. Högberg and L. Hultman, Thin Solid Films, 2013, 548, 247 CrossRef.
- G. Wang, R. Pandey and S. P. Karna, Nanoscale, 2016, 8, 8819 RSC.
- W. C. Tan, Y. Cai, R. J. Ng, L. Huang, X. Feng, G. Zhang, Y. W. Zhang, C. A. Nijhuis, X. Liu and K. W. Ang, Adv. Mater., 2017, 29, 1700503 CrossRef PubMed.
- K. P. Katin, V. S. Prudkovskiy and M. M. Maslov, Phys. Lett. A, 2017, 381, 2686 CrossRef CAS.
- K. P. Katin, V. S. Prudkovskiy and M. M. Maslov, Physica E, 2016, 81, 1 CrossRef CAS.
- D. Ma, W. Ju, T. Li, G. Yang, C. He, B. Ma, Y. Tang, Z. Lu and Z. Yang, Appl. Surf. Sci., 2016, 371, 180 CrossRef CAS.
- B. Cho, et al. , Sci. Rep., 2015, 5, 8052 CrossRef CAS PubMed.
- S. Posysaev and M. Alatalo, ACS Omega, 2019, 4, 4023 CrossRef CAS PubMed.
- J. Z. Ou, W. Ge, B. Carey, T. Daeneke, A. Rotbart, W. Shan, Y. Wang, Z. Fu, A. F. Chrimes, W. Wlodarski, S. P. Russo, Y. X. Li and K. Kalantarzadeh, ACS Nano, 2015, 9, 10313 CrossRef CAS PubMed.
- D. Ma, J. Zhang, X. Lia, C. Heb, Z. Lu, Z. Luc, Z. Yang and Y. Wang, Sens. Actuators, B, 2018, 266, 664 CrossRef CAS.
- W. Zhang, J. Yin, P. Zhang, X. Tang and Y. Ding, J. Mater. Chem. A, 2018, 6, 12029 RSC.
- L. Kou, A. Du, C. Chen and T. Frauenheim, Nanoscale, 2014, 21, 5156 RSC.
- A. Abbasi and J. J. Sardroodi, Appl. Surf. Sci., 2019, 469, 781 CrossRef CAS.
- Q. Yue, Z. Shao, S. Chang and J. Li, Nanoscale Res. Lett., 2013, 8, 425 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules – A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- B. Roondhea and P. K. Jha, J. Mater. Chem. B, 2018, 6, 6796 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nj01612h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |