
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salicylaldehyde thiosemicarbazone copper complexes: impact of hybridization with estrone on cytotoxicity, solution stability and redox activity†
Tatsiana V.
Petrasheuskaya
 ab,
Márton A.
Kiss
c,
Orsolya
Dömötör
ab,
Tamás
Holczbauer
de,
Nóra V.
May
d,
Gabriella
Spengler
bf,
Annamária
Kincses
f,
Ana
Čipak Gašparović
g,
Éva
Frank
c and
Éva A.
Enyedy
*ab
ab,
Márton A.
Kiss
c,
Orsolya
Dömötör
ab,
Tamás
Holczbauer
de,
Nóra V.
May
d,
Gabriella
Spengler
bf,
Annamária
Kincses
f,
Ana
Čipak Gašparović
g,
Éva
Frank
c and
Éva A.
Enyedy
*ab
aDepartment of Inorganic and Analytical Chemistry, Interdisciplinary Excellence Centre, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: enyedy@chem.u-szeged.hu
bMTA-SZTE Lendület Functional Metal Complexes Research Group, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
cDepartment of Organic Chemistry, University of Szeged, Dóm tér 8, H-6720 Szeged, Hungary
dResearch Centre for Natural Sciences, Magyar tudósok körútja 2, H-1117 Budapest, Hungary
eInstitute of Organic Chemistry, Research Centre for Natural Sciences, 1117 Magyar tudósok körútja 2, Budapest, Hungary
fDepartment of Medical Microbiology and Immunobiology, University of Szeged, Dóm tér 10, H-6720 Szeged, Hungary
gRudjer Boskovic Institute, HR-10000, Zagreb, Croatia
First published on 1st July 2020
Abstract
An estrone–salicylaldehyde thiosemicarbazone hybrid (estrone–TSC) containing integrated domains was designed and synthesized with excellent yield via the condensation reaction of thiosemicarbazide and 2-formyl-estrone under optimized microwave reaction conditions. A structurally related bicyclic derivative (thn-TSC) starting from 5,6,7,8-tetrahydro-1-naphtol (th-1-n) was also prepared in addition to their copper(II) complexes. The ligands have somewhat higher pKa values determined for the deprotonation of the hydroxyl group by UV-visible spectrophotometric and fluorometric titrations than the reference compound salicylaldehyde thiosemicarbazone (STSC), and are neutral at physiological pH. The novel conjugates are more lipophilic and possess higher membrane permeability than STSC based on the n-octanol/water partitioning and the parallel artificial membrane permeability assays, respectively. The isolated [Cu(estrone–TSCH−2)] and [Cu(thn-TSCH−2)] complexes were characterized by ESI-MS, UV-visible and EPR spectroscopy and a detailed solution study was performed to reveal their stoichiometry, stability and reduction by glutathione. The crystal structure of the ligand thn-TSC and its complex [Cu(thn-TSCH−1)Cl] was studied by single crystal X-ray diffraction method. The complexes are fairly stable at pH 7.4, the observed stability order is STSC < thn-TSC < estrone–TSC, and are able to oxidize glutathione readily. The novel ligands thn-TSC and estrone–TSC were found to be only moderately cytotoxic against several human cancer cell lines; however rather low IC50 values were measured in the hormone-responsive MCF-7 breast cancer cell lines (thn-TSC: 3.7 μM, estrone–TSC: 6.4 μM). The copper(II) complexes exhibited high cytotoxicity (IC50 < 0.3–2 μM) and were considerably more cytotoxic than the respective ligands. Low level of reactive oxygen species was measured and a weak GSH depletion was observed for the complexes of thn-TSC and estrone–TSC in SUM159 breast cancer cells, thus their mechanism of action might be related to the induction of oxidative stress.
Introduction
Thiosemicarbazones (TSCs) have been investigated for their versatile pharmacological activity including anticancer properties for many decades.1–3 To date, 3-aminopyridine-2-carbox-aldehyde thiosemicarbazone (Triapine) is the most prominent representative of the compound class of α-N-heterocyclic TSCs has been already tested in more than 30 clinical phase I and II trials4,5 and currently is involved in a Triapine–cisplatin–radiation combination therapy in a phase III trial.6 According to these studies, Triapine has some adverse effects (e.g. methemoglobinemia) and unfavorable pharmacokinetic profile, such as short plasma half-life arising from rapid metabolism and excretion7 which leads to the inefficiency of the active agent against solid tumors.8 Therefore, development of novel TSCs in order to increase the drug efficacy and decrease the side effects is of high research interest. Notably, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC) and 4-(2-pyridinyl)-2-(6,7-dihydro-8(5H)-quinolinylidene)-hydrazide (COTI-2) are currently undergoing phase I evaluation for treatment of advanced solid tumors and gynecologic malignancies, respectively.9,10 The iron containing enzyme ribonucleotide reductase is considered as the main target for Triapine and related TSCs due to their prominent iron binding ability.11 On the other hand, the role of the formation of intracellular copper(II) complexes that can be involved in redox cycling in the presence of reducing agents leading to the production of reactive oxygen species (ROS) is also discussed for certain subclasses of TSCs (mainly in case of N-terminally disubstituted α-N-pyridyl TSCs).12,13Salicylaldehyde TSC (STSC, Chart 1) and its derivatives were also reported to form high stability complexes with transition metal ions in solution.14STSC-based compounds generally exhibit lower cytotoxic activity in human cancer cells in comparison to α-N-pyridyl TSCs, although their copper(II) complexes are often much more cytotoxic than the corresponding ligands most probably due to their redox properties.15–17 The antiproliferative activity and the pharmacokinetic properties of STSC and its copper(II) complexes can be tuned by e.g. the attachment of electron-donating substituents18,19 or via various conjugations.20 The sterane backbone is considered as a potential site-directing molecular unit and its conjugation to bidentate TSCs containing (N,S) donor set was reported by Huang et al. to yield efficient anticancer ligands and metal complexes.21,22 Copper(II) and platinum(II) complexes of sterane–TSC hybrid molecules were developed via the condensation of the carbonyl group of estrone and pregnenolone with thiosemicarbazide.21,22 These steroidal copper(II) complexes exhibited good in vitro cytotoxic activity against Bel-7404 (liver carcinoma) and HeLa (cervical carcinoma) human cancer cell lines.22 Structurally modified cytotoxic and cytostatic steroids are highly relevant as lead compounds for anticancer drug discovery.23–25 A number of steroidal complexes connected to a platinum(II) pharmacophore had also been designed and synthesized in addition to the evaluation of their antiproliferative activities.26–29 Their most important representative is VP-128, which is a cisplatin-type complex conjugated to the clinically approved drug 17β-estradiol.29 This platinum(II) complex showed excellent in vitro efficiency toward hormone-dependent breast cancer cells and higher in vivo antitumor activity against breast cancer xenografts in nude mice compared with cisplatin.29
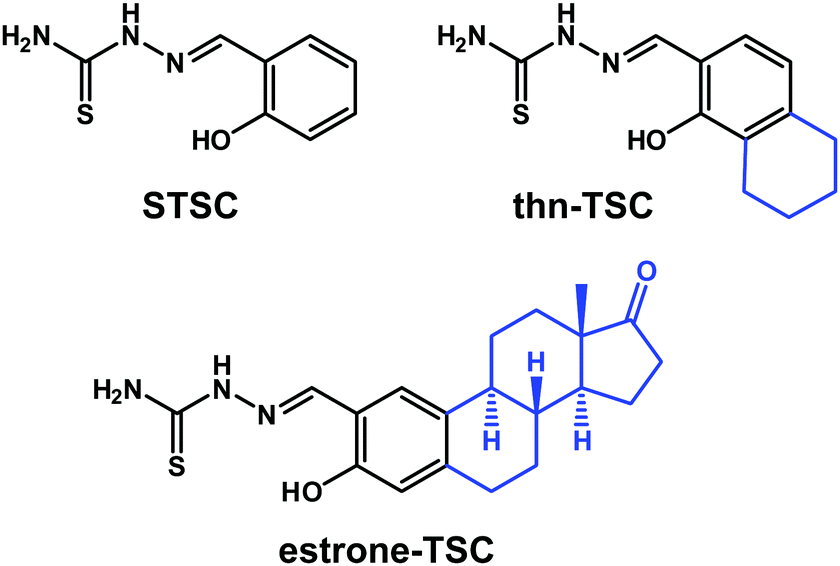 | ||
| Chart 1 Chemical structures of the investigated compounds: salicylaldehyde thiosemicarbazone (STSC), 2-((1-hydroxy-5,6,7,8-tetrahydronaphthalen-2-yl)methylene)hydrazine-1-carbothioamide (thn-TSC) and 2-((3-hydroxy-estra-1,3,5(10)-triene-2-yl)methylene)hydrazine-1-carbothioamide (estrone–TSC) in their neutral forms (HL). | ||
Notably, the primary hormonal activity of the steroidal carrier is undesirable, thus it should be eliminated. Since C-2 aldehyde of estrogens has been demonstrated to display reduced or no estrogenic activity,30 they can be suitable precursors for the synthesis of TSC–estrone hybrid molecules. Moreover, numerous 2-substituted estrone derivatives have been reported to display significant antitumor activity with negligible hormonal effect.24 In this work we developed a tridentate estrone–STSC hybrid ligand (estrone–TSC, Chart 1) and a simpler bicyclic derivative as a structural model (thn-TSC, Chart 1) in addition to their copper(II) complexes. The solution behavior of the ligands as well as the solution stability and redox properties of the complexes were investigated by UV-visible (UV-vis) spectrophotometry. The ligand thn-TSC and its copper(II) complex [Cu(thn-TSCH−1)Cl] could be crystallized and the molecular structures and secondary interactions have been studied by single crystal X-ray diffraction. The anticancer activity of the free ligands and their copper(II) complexes was tested against a series of human cancer cell lines via cytotoxicity assays, furthermore ROS production, catalase activity and L-glutathione (GSH) levels were also monitored.
Results and discussion
Synthesis of thn-TSC and estrone–TSC
STSC is a commercially available compound, while estrone–TSC and thn-TSC were developed in this work using an ortho-formylation reaction of estrone and the related bicyclic compounds (th-1-n, th-2-n) as the first step of the synthesis. The classical formylating procedures of phenol derivatives (e.g. Gattermann–Koch synthesis, Reimer–Tiemann reaction and Vilsmeier–Haack formylation or Duff, Casnati and Casiraghi reactions)31,32 suffer to a greater or lesser extent from serious drawbacks, such as the necessity of strongly toxic reagents, harsh reaction conditions or lack of regiocontrol leading to the aldehydes in low to moderate yields. An exception is the regioselective ortho-formylation of phenols using MgCl2, triethylamine (TEA) and paraformaldehyde (PFA) in refluxing tetrahydrofuran (THF), providing high yields of salicylaldehydes.33 The method has also been applied for the formylation of estrone to afford excellent overall yield of regioisomeric aldehydes in a ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with high preference for the 2-isomer over the 4-isomer.34 In this work the ortho-formylation of estrone was carried out by the method of Hofsløkken and Skattebøl33 (Scheme 1), but both the overall yield and selectivity were found to be lower than that previously reported by Akselsen et al.34 Moreover, the regioisomers (1 and 2) could only be separated by repeated column chromatography due to their similar polarities. Analogous transformation of th-2-n, structurally related to estrone, resulted in an inseparable mixture of regioisomers (3 and 4) in a comparable ratio, although in a higher overall yield. Therefore, compounds 3 and 4 were not used for further transformations. On the other hand, formylation of th-1-n afforded a single product (5) in good yield.
:1, with high preference for the 2-isomer over the 4-isomer.34 In this work the ortho-formylation of estrone was carried out by the method of Hofsløkken and Skattebøl33 (Scheme 1), but both the overall yield and selectivity were found to be lower than that previously reported by Akselsen et al.34 Moreover, the regioisomers (1 and 2) could only be separated by repeated column chromatography due to their similar polarities. Analogous transformation of th-2-n, structurally related to estrone, resulted in an inseparable mixture of regioisomers (3 and 4) in a comparable ratio, although in a higher overall yield. Therefore, compounds 3 and 4 were not used for further transformations. On the other hand, formylation of th-1-n afforded a single product (5) in good yield.
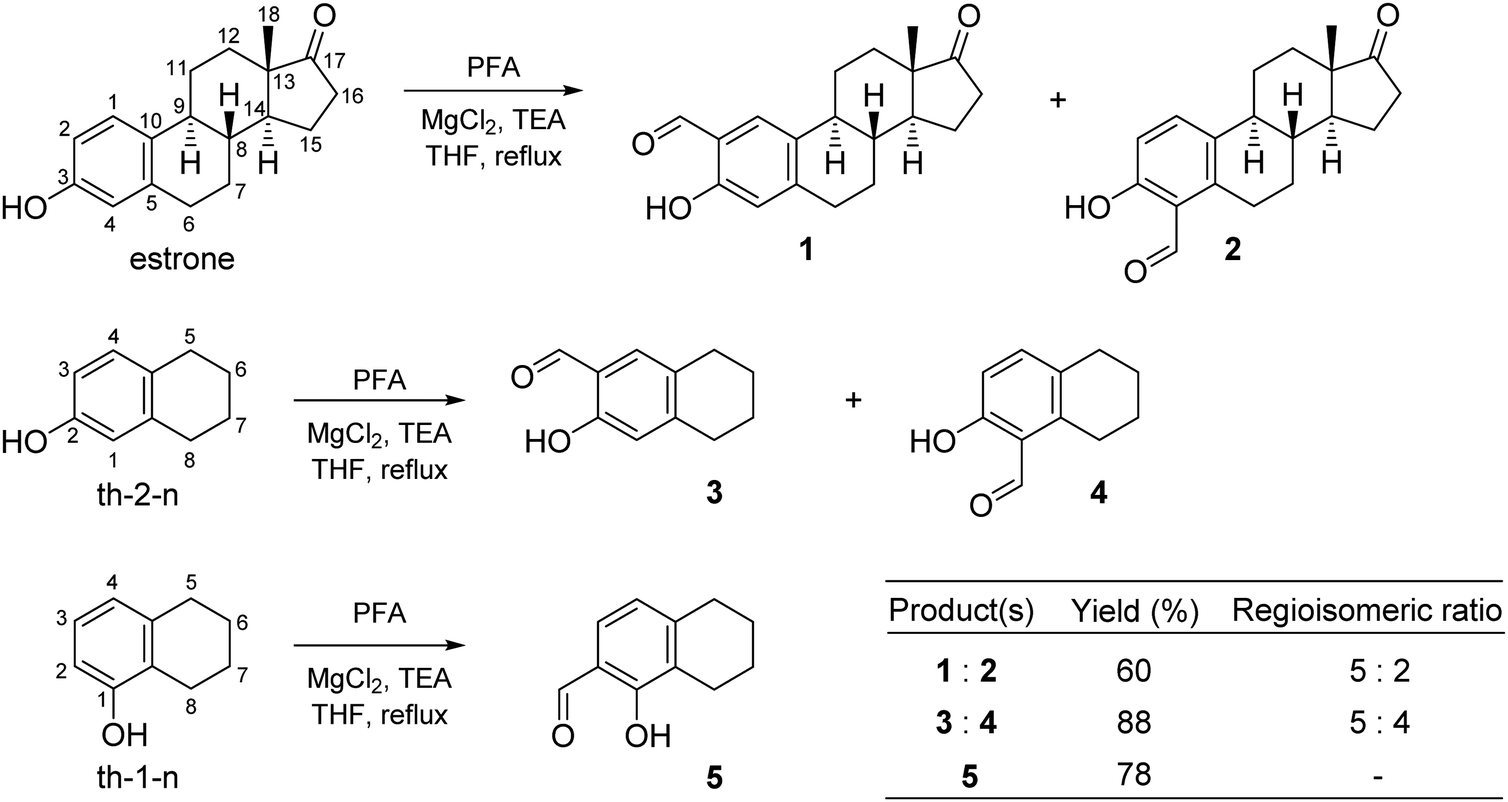 | ||
| Scheme 1 Formylation of estrone and related bicyclic compounds. (Abbreviations: PFA: paraformaldehyde, th-2-n: 5,6,7,8-tetrahydronaphthalen-2-ol, th-1-n: 5,6,7,8-tetrahydronaphthalen-1-ol, TEA: triethylamine, THF: tetrahydrofuran.) | ||
As a continuation, the successfully separated 2-formyl-estrone (1) and 1-hydroxy-5,6,7,8-tetrahydronaphthalene-2-carbaldehyde (5) were subjected to condensation reactions with thiosemicarbazide in ethanol (EtOH) under microwave (MW)-irradiation (Scheme 2) in the presence of a catalytic amount of acetic acid.
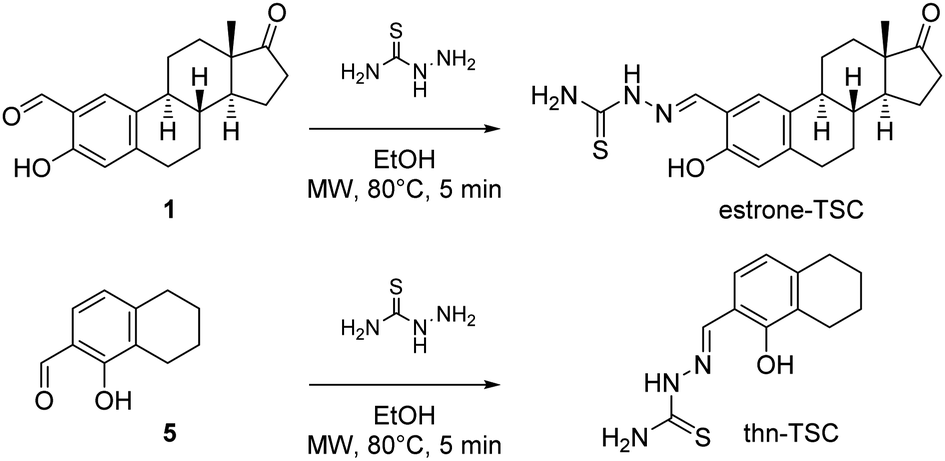 | ||
| Scheme 2 Synthesis of estrone–TSC (yield: 94%) and thn-TSC (yield: 72%). | ||
The transformations occurred at 80 °C within 5 min, and the corresponding thiosemicarbazones (estrone–TSC and thn-TSC, Chart 1) were obtained in good-to-excellent yields after purification by column chromatography. The novel tridentate products were considered to form as (E) configurational isomers in dimethyl sulfoxide (DMSO) and proved to be quite stable due to the extended conjugation. The structures of all synthesized compounds were confirmed by 1H and 13C NMR measurements (see Experimental section). The structure of thn-TSC was also verified by single crystal X-ray crystallography (vide infra).
Solution phase characterization of the salicylaldehyde thiosemicarbazone ligands
The studied compounds have limited water solubility that hindered the use of pH-potentiometry; therefore the proton dissociation processes of estrone–TSC and thn-TSC (Chart 1) were primarily studied by UV-vis spectrophotometry at low ligand concentration (55 μM) in a 30% (v/v) DMSO/H2O solvent mixture. The same medium was used for the reference compound STSC in our former work.14STSC has been characterized by two proton-dissociation processes and the first pKa (8.84) was mainly attributed to the deprotonation of the phenolic OH group, while the much higher pKa (12.57) belongs to the NH group of the thiosemicarbazone moiety.14UV-vis spectra recorded at various pH values for estrone–TSC (Fig. 1a) and thn-TSC (Fig. 1b) show only a single deprotonation process up to pH ∼ 12, and the appearance of well-defined isobestic points (estrone–TSC: 279 and 362 nm, thn-TSC: 272 and 350 nm) demonstrates that only two species (HL, L−) are involved in the chemical equilibrium. This deprotonation step results in the development of a band with an increased λmax (estrone–TSC: 374, thn-TSC: 360 nm) originating from the more extended conjugated π-electron system in the deprotonated form of the ligands, thus it most probably belongs to the hydroxyl group. pKa values were calculated on the basis of deconvolution of recorded UV-vis spectra (Table 1). The thiosemicarbazone-NH group in estrone–TSC and thn-TSC seems to have higher pKa compared to STSC and could not be determined (pKa > 12.5).
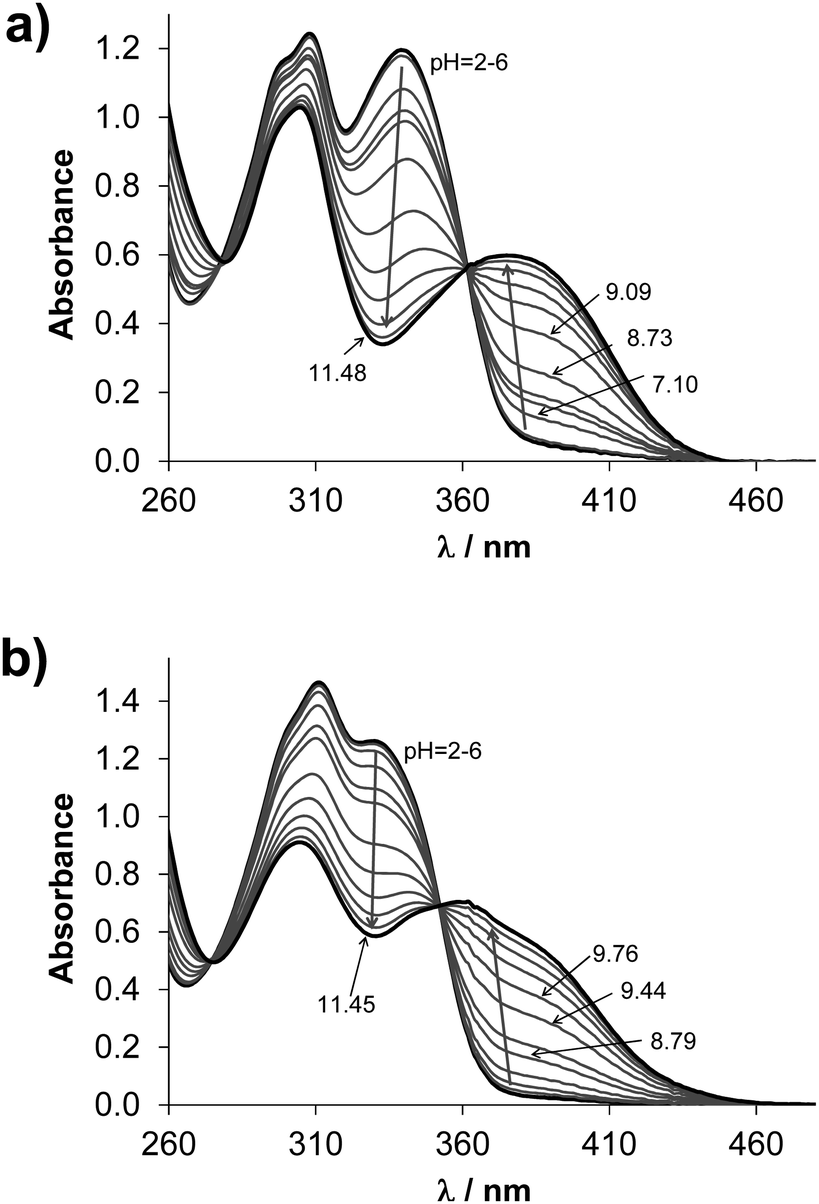 | ||
Fig. 1 UV-vis absorption spectra of estrone–TSC (a) and thn-TSC (b) pH range 2.0–11.5 in 30% (v/v) DMSO/H2O solvent mixture {cL = 55 μM; T = 25.0 °C; I = 0.1 M (KCl); ![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) = 1 cm}. = 1 cm}. | ||
D7.4 (n-octanol/water) and effective passive permeability values (Peff) of compounds studied in addition to logP values predicted by MarvinSketch software35 {T = 25 °C; I = 0.1 M (KCl)}
| Medium | Method | estrone–TSC | thn-TSC | STSC | |
|---|---|---|---|---|---|
|
a Data taken from ref. 14.
b pKa = 8.88 in ref. 14.
c logD7.4 = +1.74 in ref. 36.
|
|||||
| pKa | 30% (v/v) DMSO/H2O | UV-vis | 9.00 ± 0.01 | 9.40 ± 0.01 | 8.84a |
| pKa | H2O | Fluorometry | 8.94 ± 0.01 | 9.15 ± 0.01 | 8.74 ± 0.01b |
| HL%7.4 | 30% (v/v) DMSO/H2O | Calculated | 98 | 99 | 96 |
| HL%7.4 | H2O | Calculated | 97 | 98 | 96 |
| logD7.4 |
n-Octanol/H2O | Partitioning | >+2 | >+1.7 | +1.59 ± 0.05c |
| logP |
— | Predicted | +5.8 | +2.9 | +1.7 |
| P eff (×10−6 cm s−1) | H2O, pH 7.4 | PAMPA | n.d. | 19 ± 6 | 5.7 ± 0.6 |
Similarly to STSC, compounds estrone–TSC and thn-TSC also possess intrinsic fluorescence as the representative 3D spectrum recorded for estrone–TSC shows in Fig. 2a. This technique requires fairly low concentrations, thus fluorometric titrations could be performed in pure aqueous solutions. The emission intensity was found to be sensitive to the pH (see the 3D spectra recorded at different pH values for estrone–TSC in Fig. S1, ESI†), namely a significant increase in the intensity was detected in the basic pH range upon increasing the pH (Fig. 2b). Based on these spectral changes pKa values could be determined in the aqueous solution for the phenolic hydroxyl group as well (Table 1).
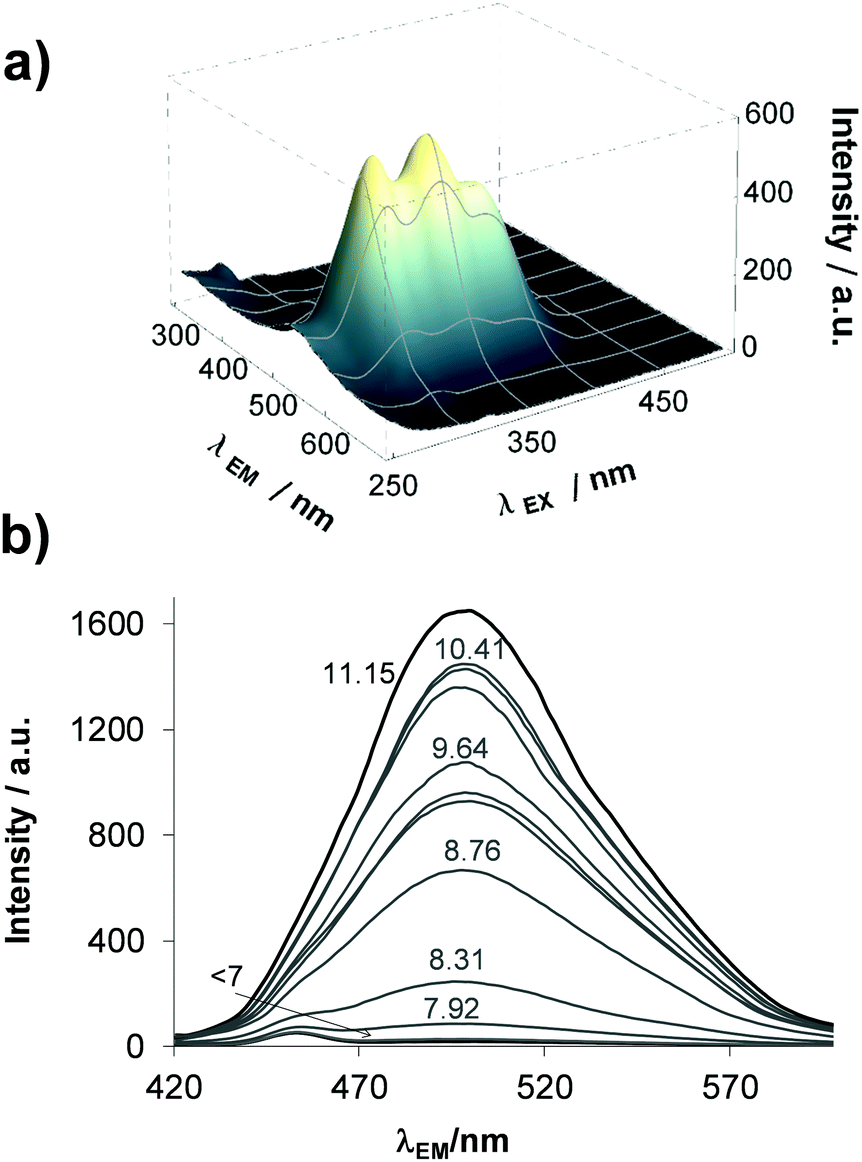 | ||
| Fig. 2 Three dimensional fluorescence spectrum of estrone–TSC at pH 7.4 (a) and its emission spectra recorded at 390 nm excitation wavelength in the pH range 6–12 (b) in aqueous solution {cL = 1 μM; T = 25.0 °C; I = 0.1 M (KCl)}. | ||
All the experimentally obtained and the predicted data reveal the same trend of the pKa values, namely STSC < estrone–TSC < thn-TSC. Most probably the increased pKa values are due to the electron-donating effect of the neighboring cyclohexyl moieties in estrone–TSC and thn-TSC. Notably, the pKa values are higher in the presence of DMSO compared to those obtained in pure water as it is expected for these anionic bases (L−), which are less solvated in the DMSO/H2O mixture making the proton dissociation more difficult. Based on the pKa values, it can be concluded that the studied compounds are found mostly in their neutral form at pH 7.4 in both media (see HL%7.4 data in Table 1).
The lipophilic character and membrane permeability are important drug properties as they strongly affect the passage via biological membranes, since most drugs have to pass through at least one cell membrane in order to reach their targets. As a first step, we attempted to determine the distribution coefficients (D7.4) for the compounds using the shake-flask method in n-octanol/buffered aqueous solution at pH 7.40 (Table 1). The logD7.4 values and the predicted partition coefficients (P, Table 1) represent strong lipophilic character of the compounds; however estrone–TSC and thn-TSC are so lipophilic that only threshold limit for their logD7.4 could be estimated since almost the whole amount of them remained in the n-octanol phase. Then the in vitro cell-free parallel artificial membrane permeability assay (PAMPA) was used to monitor the ability of compounds to penetrate membranes by passive diffusion.
UV-vis spectra recorded for the donor and acceptor phases allowed the calculation of the effective passive permeability coefficients (Peff, Table 1) for thn-TSC and STSC at pH 7.4 (Fig. S2a and c, ESI†), while formation of precipitate in case of estrone–TSC under the experimental setup hindered the determination of its Peff. Notably, data obtained for thn-TSC is merely an estimated value due to the high level of retention (as indicated by the significant difference between the spectra of the original stock solution and the sum of the spectra of the acceptor and donor phases in Fig. S2c, ESI†). Both thn-TSC and STSC exhibit high membrane permeability (Peff ≥ 1.5 × 10−6 cm s−1).
Solution equilibrium of copper(II) complexes of estrone–TSC and thn-TSC
Solution speciation of copper(II) complexes of STSC has already been characterized in details in our previous work by the combined use of pH-potentiometry, UV-vis spectrophotometric titrations and electron paramagnetic resonance (EPR) spectroscopy.14 Measurements performed in 30% DMSO/H2O solvent mixture revealed the formation of mono-ligand complexes exclusively in which the ligand coordinates tridentately via (O−,N,S) donor set. Namely, in the acidic pH range a protonated species is formed with (O−,N,S) donors coordinated, in which the non-coordinating hydrazonic nitrogen is still protonated. The complex, in which the ligand is coordinated in the (O−,N,S−) mode due to the deprotonation of the hydrazonic nitrogen, is found in the pH range 6–9. It is important to note that in the latter complex the negatively charged sulfur atom is coordinated due to the thione-thiol tautomeric equilibrium. Additionally, a mixed hydroxido species with (O−,N,S−)(OH−) coordination mode is present in the basic pH range.14 Based on chemical evidences a similar binding pattern is expected in case of the studied estrone–TSC and thn-TSC complexes. Since estrone–TSC and thn-TSC have even worse water solubility than, STSC UV-vis spectrophotometric titrations were applied to determine the stability constants of the complexes in 30% (v/v) DMSO/H2O medium. UV-vis spectra show characteristic spectral changes upon the variation of the pH (see representative UV-vis spectra of the copper(II)–estrone–TSC system in Fig. S3, ESI†).Overall stability constants (logβ) could be calculated by the deconvolution of the spectra for [CuL]+, [CuLH−1] and [CuLH−2]− species (Table 2) in addition to their individual molar spectra (Fig. 3a). Concentration distribution curves were also computed using the stability constants determined (for the copper(II)–estrone–TSC system see Fig. 3b).
β), pKa of the copper(II) complexes of estrone–TSC, thn-TSC and STSC for comparison determined by UV-vis titrations in 30% (v/v) DMSO/H2O, and calculated pCu values at pH 7.4 using cCu = 50 μM, cligand = 50 μM. logD7.4 and Peff values determined for the complexes. Observed rate constants (kobs) obtained for the redox reaction of the copper(II) complexes (25 μM) with GSH (1.25 mM) at pH 7.4 under anaerobic conditions {T = 25 °C; I = 0.1 M (KCl)}
| estrone–TSC | thn-TSC | STSC | ||
|---|---|---|---|---|
| a Data taken from ref. 14 and STSC is considered as a ligand with two dissociable protons, pKa1 = 8.84; pKa2 = 12.57, unlike estrone–TSC and thn-TSC which have only one measurable pKa value. | ||||
| logβ [CuL]+ |
13.18 ± 0.01 | 13.58 ± 0.01 | logβ [CuLH]+ |
23.03a |
| logβ [CuLH−1] |
9.44 ± 0.01 | 9.35 ± 0.01 | logβ [CuL] |
19.02a |
| logβ [CuLH−2]− |
−1.26 ± 0.04 | −1.15 ± 0.02 | logβ [CuLH−1]− |
8.75a |
| pKa [CuL]+ | 3.74 | 4.23 | pKa [CuLH]+ | 4.01 |
| pKa [CuLH−1] | 10.70 | 10.50 | pKa [CuL] | 10.27 |
| pCu | 11.53 | 11.05 | 8.77 | |
| logD7.4 |
>2 | +1.23 ± 0.2 | +0.97 ± 0.01 | |
| P eff (×10−6 cm s−1) | n.d. | 5.6 ± 0.2 | 1.5 ± 0.4 | |
| k obs (min−1) | 0.13 ± 0.02 | 0.10 ± 0.02 | 0.11 ± 0.02 | |
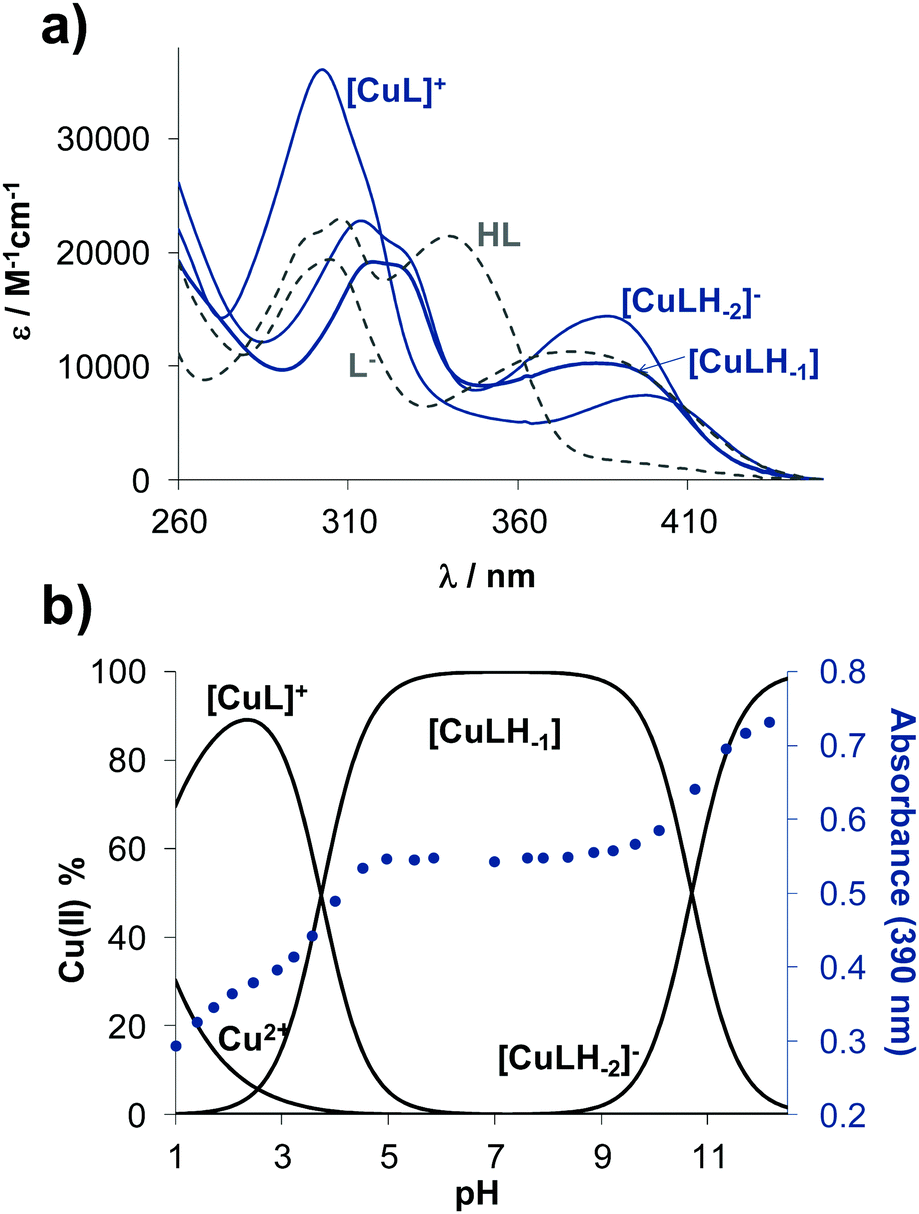 | ||
| Fig. 3 Individual UV-vis absorption spectra of the different complexes (blue lines) and ligand species (grey dashed lines) calculated for the copper(II)–estrone–TSC system in 30% (v/v) DMSO/H2O solvent mixture (a). Concentration distribution curves for the same system plotted together with the absorbance changes at 390 nm (●) {cligand = 50 μM; cCu(II) = 50 μM; T = 25.0 °C; I = 0.1 M (KCl)}. | ||
The protonated neutral form of the ligands estrone–TSC and thn-TSC was formulated as HL due to the single proton dissociation step observed in the studied pH range. Therefore, in the [CuL]+ complex, that is formed in the acidic pH range, the ligand coordinates via replacement of one proton as an (O−,N,S) donor. The replacement of phenolic OH proton by the copper ion and the chelate coordination of (O−,N,S) donor atoms are supported by our solid state results as crystals of the complex of [Cu(thn-TSCH−1)Cl] (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) [CuL]Cl) could be grown and the structure could be studied by single crystal X-ray diffraction (vide infra). Deprotonated [CuLH−1] complex predominates in the pH range 5–9 and most probably is formed by the deprotonation of the hydrazonic nitrogen, thus it contains an (O−,N,S−) donor set. While [CuLH−2]− is a mixed hydroxido species, its more correct formula is [CuLH−1(OH)]. The suggested structures for the complexes formed with thn-TSC are represented in Scheme 3.
[CuL]Cl) could be grown and the structure could be studied by single crystal X-ray diffraction (vide infra). Deprotonated [CuLH−1] complex predominates in the pH range 5–9 and most probably is formed by the deprotonation of the hydrazonic nitrogen, thus it contains an (O−,N,S−) donor set. While [CuLH−2]− is a mixed hydroxido species, its more correct formula is [CuLH−1(OH)]. The suggested structures for the complexes formed with thn-TSC are represented in Scheme 3.
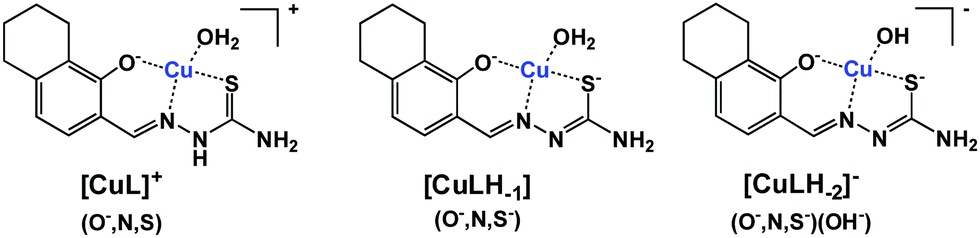 | ||
| Scheme 3 Suggested structures for the copper(II) complexes formed with ligand thn-TSC (HL). | ||
The direct comparison of the logβ values of the complexes formed with estrone–TSC, thn-TSC to those of STSC (Table 2) is not adequate as the ligands have distinct pKa values and were differently formulated (notably for STSC two pKa values were determined14). On the other hand, the pKa values of the complexes regarding the same deprotonation steps are comparable (e.g. pKa [CuL]+ for estrone–TSC, thn-TSCvs. pKa [CuLH]+ for STSC, Table 2). These pKa values show the deprotonation of the hydrazonic-NH moiety in the slightly acidic pH range (pKa ∼ 3.74–4.23) and that of the coordinated water molecule in the basic pH range (pKa ∼ 10.27–10.70). Based on these data it can be concluded that the [CuLH−1] complex of estrone–TSC is present in a wider pH range compared to the case of the other two ligands (Fig. S4, ESI†), suggesting its superior stability. To compare the solution stability of the complexes, thus the copper binding ability of the studied ligands, pCu values were also computed on the basis of the stability constants at pH 7.4 and at 50 μM concentrations of both the ligand and the metal ion (Table 2). pCu is the negative decadic logarithm of the unbound metal ion concentration under a chosen condition and provides a solid basis for comparison of the complex stabilities. The higher pCu value shows higher stability. First of all, the obtained pCu values reflect significantly high stability of these copper(II) complexes at pH 7.4 in all cases (<1% decomposition), and reveal the following copper(II) binding ability of the ligands: STSC < thn-TSC < estrone–TSC.
Synthesis and characterization of copper(II) complexes of estrone–TSC and thn-TSC
Based on our solution equilibrium data it could be concluded that complexes with the (O−,N,S−) coordination mode are formed in a wide pH range including the neutral pH in all cases. This type of complexes of estrone–TSC and thn-TSC could be isolated from a DMSO/water solvent mixture at pH ∼ 7.4. The characterization of the synthesized complexes ([Cu(estrone–TSCH−2)] and [Cu(thn-TSCH−2)] ([CuLH−1]) where the abbreviation of the ligand's name stand for the HL forms) was performed by EPR spectroscopy, ESI-MS and UV-vis spectrophotometry. For comparative purposes the copper(II) complex of STSC was also prepared and characterized by ESI-MS and UV-vis spectrophotometry; however, the synthesis of this complex has been already reported by several authors previously.16,17 The ESI-MS data (collected in Experimental) strongly support the suggested chemical structures of the complexes. In addition, UV-vis spectra recorded in methanol and in n-octanol (Fig. S5 and S6, ESI†) also confirm the complex formation due to the appearance of the well-known S → Cu charge transfer bands at 380, 395 and 406 nm for [Cu(STSCH−2)], [Cu(thn-TSCH−2)] and [Cu(estrone–TSCH−2)], respectively, and the lack of the characteristic bands of the free-ligands.
In order to confirm the coordination modes in the isolated [Cu(thn-TSCH−2)] and [Cu(estrone–TSCH−2)] complexes, EPR spectra were recorded in DMSO at room temperature (Fig. 4) and at 77 K (Fig. S7, ESI†). First of all, no free copper(II) ions were detected. Simulation of the EPR spectra resulted in the isotropic and anisotropic EPR parameters (g and A values in Table 3), which were compared to each other and to the isotropic values of STSC.14 The complexes have similar Hamiltonian EPR parameters with well resolved hyperfine coupling of one nitrogen donor atom, thus similar binding mode is suggested. The somewhat lower g0 value measured for the estrone- and thn-derivatives shows slightly higher ligand field in these complexes compared to that of STSC, in agreement with the electron donating effect of the ring substituents. Comparing the estrone- and thn-derivatives, the only difference is due to the slower rotational motions of the larger estrone-containing ligand, which resulted in an increased line width.
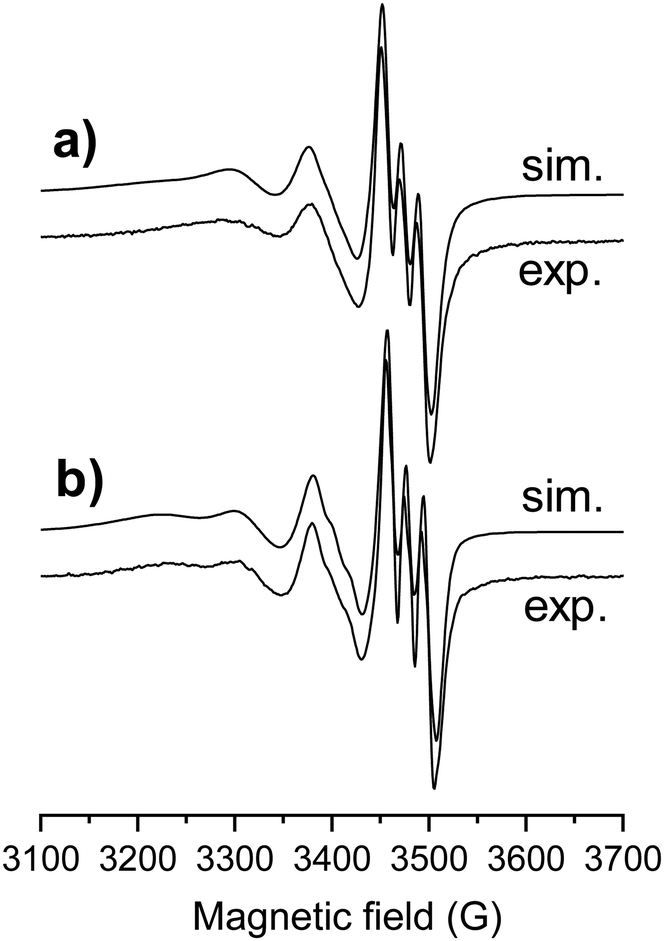 | ||
| Fig. 4 Experimental (exp.) and simulated (sim.) isotropic EPR spectrum for complexes [Cu(estrone–TSCH−2)] (a) and [Cu(thn-TSCH−2)] (b) in DMSO at room temperature. | ||
| estrone–TSC | thn-TSC | STSC | |
|---|---|---|---|
| a Uncertainties (SD) of the last digits are shown in parentheses. b The experimental error were ±0.001 for g, ±1 G for A and aN. c Data taken from ref. 14. | |||
| Isotropic parametersa | |||
| g o | 2.0889(3) | 2.0874(1) | 2.0945 |
| A o (G) | 71.3(1) | 72.8(1) | 73.1 |
| a N (G) | 18.1(1) | 18.1(1) | 17.7 |
| Anisotropic parametersb | |||
| g x /gy/gz | 2.018/2.048/2.182 | 2.026/2.052/2.203 | — |
| A x /Ay/Az (G) | 36.0/23.3/180.8 | 30.8/17.2/174.4 | — |
| a Nx/aNy/aNz (G) | 9.5/16.4/8.5 | 11.6/15.6/13.0 | — |
In frozen solution (77 K) a high extent of dimerization was detected in DMSO, especially for the estrone–TSC derivative, therefore, the solutions were diluted with water to their half concentration (1.5 mM). The recorded spectra revealed the presence of both monomeric and dimeric species (Fig. S7a and b, ESI†), e.g. in case of [Cu(estrone–TSCH−2)], 76% of the complex was still present in the dimeric form (Fig. S7c and d, ESI†). Formation of dimers in case of the copper(II) complex of STSC was also reported in the solid phase based on the structure established by single crystal X-ray diffraction.16,17
Distribution coefficients (logD7.4) and effective passive permeability values (Peff) were also determined for the complexes where the solubility allowed the calculations (Table 2, Fig. S2b, d and S6, ESI†). The complexes were found to be somewhat less lipophilic than the ligands, but their lipophilicity followed the same trend as it was observed for the ligands: STSC < thn-TSC < estrone–TSC. The complexes were characterized by lower Peff values (thus somewhat lower permeability) than the ligands, most probably due to their reduced lipophilic character.
Solid state structures of crystal thn-TSC and [Cu(thn-TSCH−1)Cl]
The crystal structure of thn-TSC (I) and its copper(II) complex [Cu(thn-TSCH−1)Cl] (II) have been determined by single crystal X-ray diffraction. Crystal I crystallized in monoclinic P21/n, while the complex II in the orthorhombic Pbca space group. Crystal data and structure refinement data are collected in Table S1 (ESI†), selected bond length and angles are presented in Table S2 (ESI†). The ORTEP representations of the two compounds are depicted in Fig. 5. In crystal I the conformation of the molecule is stabilized by the O1–H1O⋯N1 intramolecular hydrogen bond. The 5,6,7,8-tetrahydro-1-naphtol ring was found in two disordered positions owing to the two possible ring conformations. The major component was noted with ‘A’ and the occupancy ratio was 68%. The hydrazonic nitrogen N2 is protonated, which is consistent with the shorter bond distances between C11–N1 1.291(3) Å. The molecules are connected to each other via N–H⋯S, C–H⋯S or N–H⋯O hydrogen bonds (Fig. S8 and Table S3, ESI†) turning slightly out of the molecular plane. There is no considerable π⋯π stacking interaction between the molecules.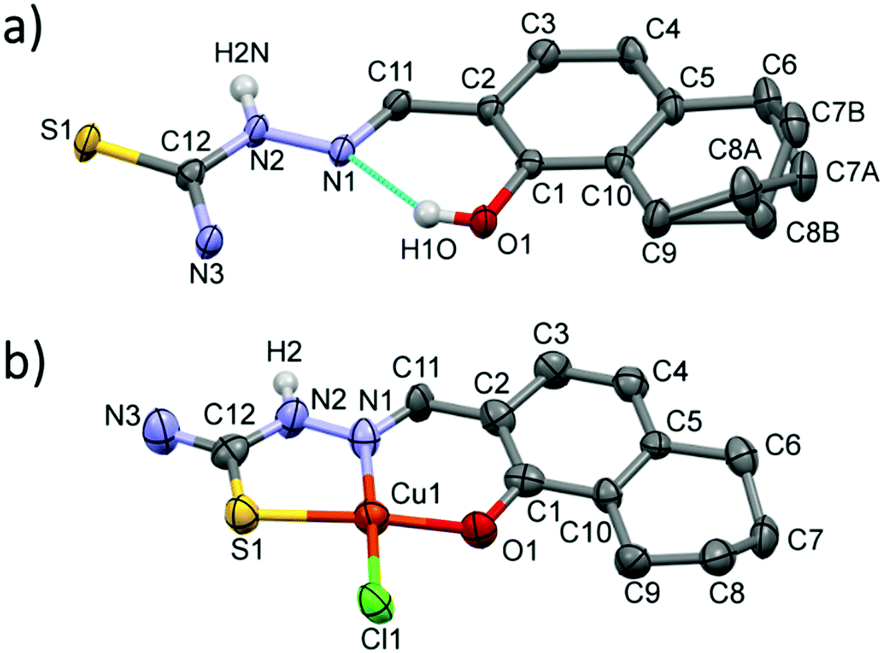 | ||
| Fig. 5 Molecular structure of thn-TSC (I) (a) and [Cu(thn-TSCH−1)Cl] (II) (b) with atom numbering. Displacement parameters are drawn at 50% probability level. Hydrogen atoms on the hydrazinic N2 nitrogen and on O1 atom involved in intramolecular hydrogen bond for crystal I are shown, other hydrogen atoms are omitted for clarity. | ||
In crystal [Cu(thn-TSCH−1)Cl] (II) the ligand coordinates to the copper(II) ion through deprotonated O1 and its N1 and S1 atoms and the fourth position is occupied by a chloride ion in a square planar arrangement. The bond length is shorter for Cu–O and Cu–N (∼1.90 Å) than for Cu–S and Cu–Cl (2.26 Å). The two chelate rings formed by Cu1–S1–C12–N2–N1 and Cu1–N1–C11–C3–C1–O1 atoms are slightly twisted; the angle between the two planes defined by the atoms of the ring is 8.51°. This results in a considerable deviation of S1–Cu1–O1 angle of 170.9(3)° from the ideal 180° of a square planar geometry (Table S2, ESI†). In this crystal the 5,6,7,8-tetrahydro-1-naphtol ring has only one conformation, which is equal to the conformation of the major disorder found in the ligand crystal. The two N–H protons of the amino group are involved in the main H-bond interactions with a neighbouring chloride and an oxygen atom evolving columns in the crystallographic direction ‘a’ (Fig. S9, ESI†). There is no considerable axial coordination to the copper(II) ion and the closest copper–copper distance is 5.63 Å.
Reduction of the copper(II) complexes of TSCs by glutathione and ascorbic acid
The mechanism of action of the copper(II)–TSC complexes is often related to their redox properties, namely their redox reaction with physiological reductants. GSH was reported to be able to reduce copper(II) complexes of α-N-pyridyl TSCs resulting in the formation of copper(I) species that can react with oxygen and its re-oxidation generates intracellular ROS accumulation.12,13,37 Di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT) and DpC were reported to form copper(II) complexes in the lysosomes, which can be involved in ROS production via redox cycling leading to lysosomal membrane permeabilization and then to apoptosis.38 In this work, the direct reaction of the copper(II) complexes of STSC, thn-TSC and estrone–TSC with GSH and ascorbic acid was studied spectrophotometrically. The reaction was monitored in the wavelength range where only the TSC and its complex absorb light in 30% (v/v) DMSO/H2O solvent mixture under strictly anaerobic condition at pH 7.4 using the high excess of the reducing agent (50 equiv.). The reaction with ascorbic acid was very slow resulting in minor spectral changes, which suggests that the studied copper(II) complexes cannot be reduced efficiently by this reducing agent. Contrarily, the stronger reductant, GSH, reduced these complexes. As exemplary shown for estrone–TSC (Fig. 6) and for STSC (Fig. S10a, ESI†), recorded spectra reveal that after mixing the reactants a fast and significant change is observed (comparing spectra at 0 and 0.2 min), most probably as a result of the formation of a mixed ligand complex [Cu(II)LH−1(GSH)] with GSH, as it was reported for other TSCs.37 Then the spectral changes represent the relatively slow release of the ligand, most likely the generated unstable copper(I) complex [Cu(I)LH−1(GSH)] is decomposing, that leads to the formation of the Cu(I)–GSH complex and the free TSC (see spectra in Fig. S11, ESI† for clarity). The redox reaction resulted in the complete reduction of the copper(II) complexes under the applied conditions. Bubbling oxygen into the solution regenerated the [Cu(II)LH−1] complex (Fig. S10b, ESI†). This finding strongly suggests the reversibility of the redox process.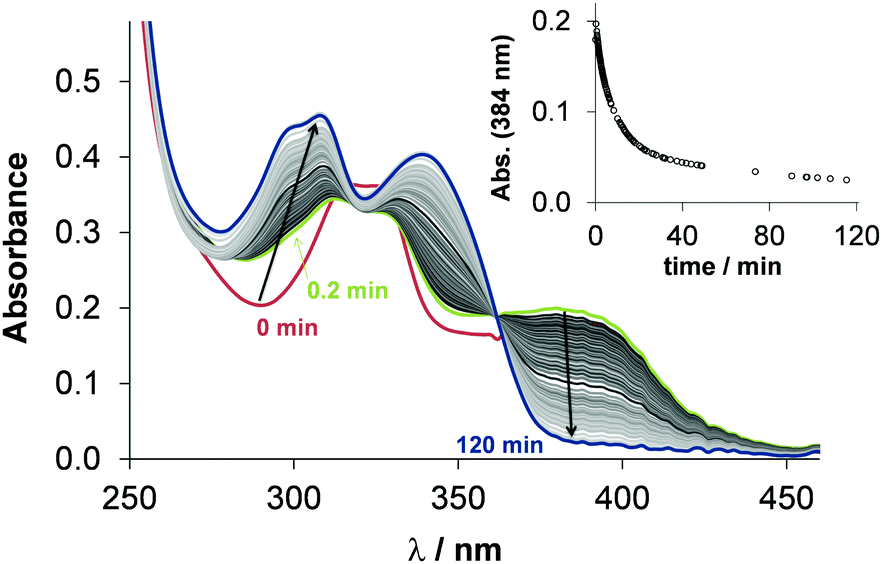 | ||
| Fig. 6 Time dependent changes of the UV-vis spectra of the [Cu(estrone–TSCH−2)] complex (25 μM) in the presence of 50 equiv. GSH (1.25 mM) at pH 7.4 in 30% (v/v) DMSO/H2O under anaerobic conditions and the inserted figure shows the absorbance values at 384 nm plotted against the time {T = 25 °C; I = 0.1 M (KCl)}. | ||
The recorded absorbance–time curves were further analyzed at the λmax of the copper(II) complexes (Fig. 6). However, our interpretation of these kinetic runs is considered only as a semi-quantitative description, it can give information about the differences in the reaction rates. Observed rate constants (kobs) were calculated as the slope of the ln(A/A0) vs. time plots and the calculated values are collected in Table 2. The kobs values are very similar to each other and to the reported value for the copper(II) complex of Triapine (0.11 min−1) obtained under the same condition, but are much higher than those obtained for the complexes of Dp44mT (2.12 × 10−3 min−1) or DpC (2.30 × 10−3 min−1).39
In vitro cytotoxicity and antioxidant properties
The colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay was applied in the doxorubicin-sensitive Colo205, the multidrug resistant Colo320 human colonic adenocarcinoma and the hormone-responsive MCF-7 breast cancer cell lines to monitor the anticancer activity of the TSC ligands and their copper(II) complexes. Notably, the resistance of Colo320 cells is primarily mediated by the overexpression of the transporter P-glycoprotein, which pumps out xenobiotics from the cells. Doxorubicin was applied as a positive control, and CuCl2 was also tested for comparison. In addition, cytotoxicity was measured in normal human embryonal lung fibroblast cells (MRC-5). The determined IC50 values using 72 h incubation time are collected in Table 4. The stock solutions of the compounds were prepared in a 90% (v/v) DMSO/H2O mixture, and in the final samples the DMSO content was always lower than 1%.| IC50 (μM) | Colo205 | Colo320 | MCF-7 | MRC-5 |
|---|---|---|---|---|
| STSC | 65.3 ± 2.2 | 56.6 ± 2.2 | 45.2 ± 3.2 | 72.4 ± 4.8 |
| thn-TSC | 84.3 ± 4.3 | 50.6 ± 1.0 | 3.73 ± 0.83 | 34.0 ± 3.9 |
| estrone–TSC | 20.3 ± 1.4 | 9.85 ± 0.66 | 6.42 ± 0.40 | 22.6 ± 3.5 |
| [Cu(STSCH−2)] | 0.99 ± 0.09 | 0.90 ± 0.05 | 0.31 ± 0.01 | 1.20 ± 0.09 |
| [Cu(thn-TSCH−2)] | 0.61 ± 0.03 | 0.60 ± 0.04 | 0.26 ± 0.02 | 0.48 ± 0.02 |
| [Cu(estrone–TSCH−2)] | 1.99 ± 0.19 | 1.61 ± 0.56 | 0.57 ± 0.03 | 1.59 ± 0.13 |
| CuCl2 | 19.7 ± 0.9 | 20.0 ± 2.3 | 36.6 ± 1.5 | 24.5 ± 2.6 |
| Doxorubicin | 3.28 ± 0.22 | 3.12 ± 0.27 | n.d. | 5.19 ± 0.21 |
STSC was found to be weakly cytotoxic in all tested cell lines, while thn-TSC showed higher activity in the MCF-7 breast cancer cell line. The estrone-conjugate was the most cytotoxic against all cancer cells, and lower IC50 values were obtained in the multidrug resistant Colo320 and MCF-7 cells. Notably, these compounds were found to be moderately toxic in the normal fibroblast cells. Fairly low IC50 values (<0.3–2 μM) were determined for the copper(II) complexes, which were much more cytotoxic than the respective ligands, the copper(II) salt and doxorubicin. Unfortunately, the complexes did not show selectivity against the cancer cells compared to the normal cells.
Since the TSC compounds showed synergism with CuCl2, and the cytotoxic activity of copper(II)–TSC complexes is often associated with generation of reactive oxygen species,12,13,38,39 the compounds were further investigated regarding their intracellular ROS production, catalase activity and their effect on cellular GSH level. These assays were performed in MCF-7 and the triple-negative SUM159 breast cancer cells; therefore cytotoxicity was also measured in these cell lines using 24 h incubation time. In addition IC50 values were determined in the HER2-positive SkBr3 breast cancer and the hepatocellular carcinoma HepG2 cell lines. The in vitro cytotoxicity data in these cells are shown in Table 5, which show a similar activity trend of the tested compounds as it was observed in the Colo205, Colo320, MCF-7 cells using 72 h exposure time; however, the IC50 values are higher due to the shorter incubation period. Notably, estrone–TSC was more cytotoxic against the tested breast cancer cells than the non-steroidal STSC and thn-TSC suggesting that the steroidal carrier might have a role in the cellular uptake, however the copper(II) complexes of the three ligands did not show this difference in the activity. ROS production was measured in MCF-7 and SUM159 cell lines using the ROS sensitive cell permeable dye 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) in the presence and absence of N-acetyl-cysteine (NAC) (Fig. 7 and Table S4, ESI†). Results are expressed as fold change in the emission intensities after exposure to the test compound relative to the solvent control (without the use of NAC). The ligands and their copper(II) complexes showed no or weak ability to produce ROS under the applied conditions (1 μM concentration where the compounds are non-cytotoxic, 60 min incubation time); and the somewhat higher fold change of intensity was observed only in the case of the copper(II) complexes in SUM159 cells and for the complex of thn-TSC and estrone–TSC in MCF-7 cells. Addition of the reducing agent NAC decreased the ROS production in all cases of the compounds tested.
| IC50 (μM) | MCF-7 | SkBr3 | SUM159 | HepG2 |
|---|---|---|---|---|
| STSC | >100 | >50 | >100 | >50 |
| thn-TSC | >100 | >50 | >100 | >50 |
| estrone–TSC | 31.4 ± 0.4 | 19.3 ± 0.6 | 54.8 ± 2.6 | >50 |
| [Cu(STSCH−2)] | 5.77 ± 0.47 | 0.84 ± 0.04 | 2.96 ± 0.41 | 4.34 ± 0.98 |
| [Cu(thn-TSCH−2)] | 3.02 ± 0.04 | 0.89 ± 0.09 | 1.03 ± 0.48 | 3.39 ± 0.75 |
| [Cu(estrone–TSCH−2)] | 11.5 ± 1.1 | 3.28 ± 0.39 | 4.27 ± 0.47 | 6.59 ± 0.16 |
| CuCl2 | >100 | >50 | 73.7 ± 0.5 | >50 |
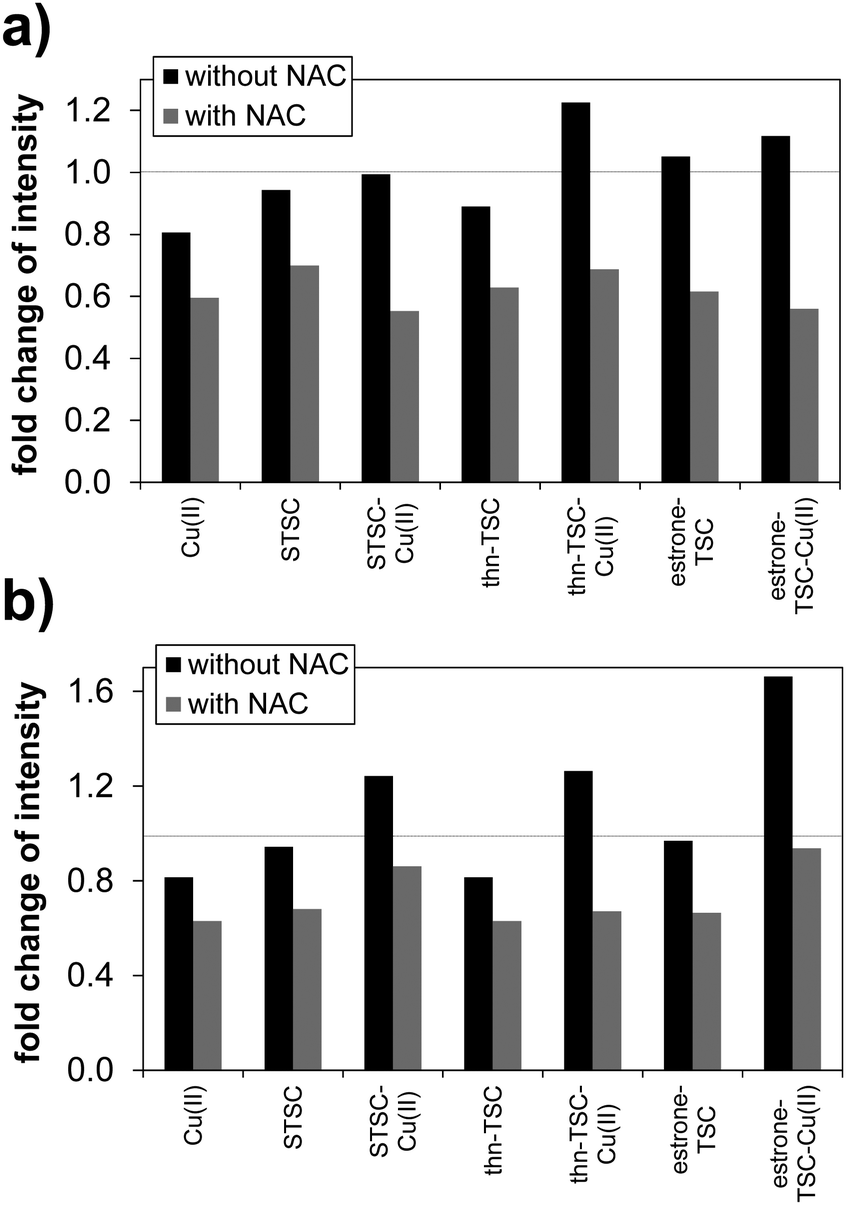 | ||
| Fig. 7 Characterization of intracellular ROS production with DCFH-DA assay in MCF-7 (a) and SUM159 (b) cells. ROS induction by 1 μM compound was evaluated with or without 1 mM NAC. Fluorescence emission intensity was measured following 60 min incubation with the indicated compounds. Fold change in intensity represents the ratio of the measured intensity to that of the solvent control (without NAC). Values show the mean of three experiments (see data in Table S4, ESI†) {λEX = 500 nm; λEM = 529 nm}. | ||
As a next step GSH level was measured in SUM159 cells selected by the higher ROS production in the presence of the copper(II) complexes. GSH level might have importance in activity, as disturbance in GSH homeostasis is often involved in cancer progression.40 Based on the data obtained for the GSH levels (Fig. S12a, ESI†), it can be concluded that the studied compounds decreased the GSH level compared to the solvent control except the copper(II) complex of STSC, although the increment was not significant. Catalase activity was also measured in SUM159 cell lines to monitor the antioxidant status of the cells (results are shown in Fig. S12b, ESI†). Catalase is an antioxidant enzyme that converts H2O2 to H2O and O2; therefore it is able to protect cells against H2O2 stress. The tested compounds showed similar and low catalase activity, and an increased catalase activity was detected only for the copper(II) complex of STSC.
On the whole, the studied copper(II) complexes are highly and more cytotoxic against the tested cancer cell lines than the ligands. They can be reduced by GSH with a similar reaction rate, they showed a slightly elevated intracellular ROS production in MCF-7 and SUM159 cell lines, but their effect on the catalase activity and GSH level was minor.
Conclusions
Novel thiosemicarbazone derivatives were prepared through molecular hybridization of salicylaldehye TSC (STSC) with estrone and th-1-n, respectively, which yielded copper(II) complexes with marked cytotoxic activity in various human cancer cell lines. The introduction of the TSC moiety to the sterane backbone resulting in estrone–TSC was performed via the condensation reaction of 2-formyl-estrone, obtained from estrone by ortho-formylation, and thiosemicarbazide in EtOH under MW-irradiation. A simpler bicyclic derivative (thn-TSC) was also prepared from th-1-n using a similar procedure. Copper(II) complexes of both ligands and of STSC, for comparison, were synthesized and characterized by ESI-MS, UV-vis and EPR spectroscopy. The neutral ligand thn-TSC and the complex of [Cu(thn-TSCH−1)Cl] could be crystallized and the (O−,N,S) coordination of the ligand to the copper ion in a square planar geometry could be confirmed by single crystal X-ray diffraction. Detailed solution equilibrium studies were performed to study the proton dissociation processes of the ligands and their complexation with copper(II) ions by spectrophotometric titrations in a 30% DMSO/H2O solvent mixture due to their limited water solubility. In addition, pKa values of the ligands attributed to the deprotonation of the phenolic OH moiety were determined by fluorometry in pure aqueous solutions. Notably, somewhat higher pKa values were obtained for estrone–TSC and thn-TSC compared with the reference compound STSC in both media. Based on the pKa values, all three compounds are present practically in their neutral forms (almost 100%) at physiological pH. The estrone–TSC and thn-TSC conjugates were found to be more lipophilic, showed higher effective passive permeability coefficients, thus higher membrane permeability based on the PAMPA assay compared to STSC.Formation of three different types of copper(II) complexes was identified in the DMSO/H2O medium according to the spectro-photometric titrations in all cases. Namely, a protonated complex is formed in the acidic pH range with (O−,N,S) donors containing the non-coordinating hydrazonic nitrogen protonated. By the deprotonation of the latter moiety, the ligand coordinates via an (O−,N,S−) donor set and the forming complex predominates in a wide pH range (6–9) including the physiological pH. This coordination mode was confirmed by EPR spectroscopy in the isolated [Cu(thn-TSCH−2)] and [Cu(estrone–TSCH−2)] complexes. With increasing pH a mixed hydroxido species is formed with (O−,N,S−)(OH−) coordination mode. In spite of the similar stoichiometry of the complexes, differences in their solution stability were observed giving the following order: STSC < thn-TSC < estrone–TSC. Interestingly, the isolated neutral complexes were found to be less lipophilic possessing lower (but still high) permeability than the ligands. The copper(II) complexes of estrone–TSC, thn-TSC and STSC could be efficiently reduced with similar reaction rates by the physiological reducing agent GSH under anaerobic conditions accompanied by the release of the ligand, and could be oxidized back by atmospheric oxygen reversibly.
Among the ligands estrone–TSC was the most cytotoxic against the doxorubicin-sensitive Colo205 and the multidrug resistant Colo320 human colonic adenocarcinoma cell lines. Both novel ligands showed low micromolar IC50 values in the hormone-responsive MCF-7 breast cancer cell lines (thn-TSC: 3.7 μM, estrone–TSC: 6.4 μM). The copper(II) complexes of estrone–TSC, thn-TSC and STSC were significantly more cytotoxic than the ligands characterized by 1–2 orders of magnitude lower IC50 values in all tested cancer cell lines (Colo205, Colo320, MCF-7, SkBr3, SUM159 and HepG2), and complex of thn-TSC exhibited the lowest IC50 values. Low intracellular ROS production was observed for the copper(II) complexes in SUM159 cells, but it was higher than obtained for the ligands at 1 μM compound concentration. Meanwhile, somewhat lower catalase activity and intracellular GSH level were found almost in all cases (except the Cu–STSC complex). Based on the results, the role of the redox properties of the copper(II) complexes in the mechanism of action cannot be excluded, however, more biological assays are needed to elucidate the exact mechanism of action.
Experimental section
Chemicals and instruments used for the syntheses and characterization
STSC, CuCl2, KCl, HCl, KOH, NaH2PO4, Na2HPO4, DMSO, EDTA, GSH, ascorbic acid, MTT, NAC, DCFH-DA, 2,2′-dinitro-5,5′-dithiodibenzoic acid (DTNB), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) and doxorubicin were purchased from Sigma-Aldrich in puriss quality. Milli Q water was used for sample preparation. CuCl2 stock solution was made by the dissolution of anhydrous CuCl2 in water and its exact concentration was determined by complexometry through the EDTA complex. The stock solution of STSC, thn-TSC and estrone–TSC was prepared on a weight-in-volume basis dissolved in DMSO.Reagents and materials used for the synthesis were purchased from commercial suppliers (TCI, Tokyo, Japan; Alfa Aesar, Haverhill, MA, USA and Sigma-Aldrich Corporation, St. Louis, MO, USA). All solvents were dried and purified according to standard procedures.
MW-assisted reactions were carried out with a CEM Discover SP instrument (CEM Corporation, Matthews, NC, USA) using a maximum power of 200 W with dynamic control program. Thin layer chromatography was carried out on Kieselgel-G (Si 254 F, Merck KGaA, Darmstadt, Germany) plates (0.25 mm thick). The spots were detected by spraying with phosphomolybdic acid (5%) in aqueous phosphoric acid (50%) or visualized by UV light (254 nm). The products were purified by preparative column chromatography on Merck silica gel 60, 40–63 μm (Merck KGaA, Darmstadt, Germany). Melting points (Mps) were measured on an SRS Optimelt digital device (Stanford Research Systems Inc, Sunnyvale, CA, USA). NMR spectra were recorded at 298 K with a Bruker Avance III HD Ascend 500 Plus instrument. Chemical shifts are reported in ppm (δ scale) and coupling constants (J) in Hz. The 1H resonance signals are indicated as a singlet (s), a broad singlet (bs), a doublet (d), a double doublet (dd), a triplet (t) or a multiplet (m). The 13C NMR spectra are 1H-decoupled. The J-MOD pulse sequence was applied to determine multiplicities.
Synthesis of thn-TSC and estrone–TSC
:70 v/v%) was used for further purification to give 1 as a white solid (255 mg, 43%), Mp 160–162 °C (164–165 °C34); 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 0.92 (s, 3H, 18-H3), 1.33–1.60 (overlapping m, 6H), 1.77 (m, 1H), 1.95 (m, 2H), 2.07 (m, 1H), 2.08 (m, 1H), 2.35 (m, 1H), 2.44 (m, 1H), 2.85 (m, 2H, 6-H2), 6.70 (s, 1H, 4-H), 7.57 (s, 1H, 1-H), 10.14 (s, 1H, CHO), 10.45 (bs, 1H, OH); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 13.4 (C-18), 21.1 (CH2), 25.3 (CH2), 25.6 (CH2), 29.3 (CH2), 31.2 (CH2), 35.3 (CH2), 37.4 (CH), 42.9 (CH), 47.3 (C-13), 49.5 (CH), 116.6 (C-4), 120.2 (C-2), 126.4 (C-1), 131.4 (C-10), 146.6 (C-5), 158.5 (C-3), 192.0 (CHO), 219.4 (C-17).
:4 regioisomeric mixture of 3 and 4 was obtained as a yellowish oil (311 mg, 88%). The two products were assigned based on the NMR spectra recorded for the mixture.
Compound 3. 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 1.69 (overlapping m, 4H, 5-H2 and 6-H2), 2.63 (m, 2H, 4-H2), 2.70 (m, 2H, 7-H2), 6.67 (s, 1H, 8-H), 7.35 (s, 1H, 3-H), 10.13 (s, 1H, CHO); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 22.3 and 22.7 (C-6 and C-7), 27.7 (C-5), 29.4 (C-8), 116.6 (C-1), 120.4 (C-4a), 128.0 (C-3), 129.6 (C-4), 146.6 (C-8a), 158.2 (C-1), 191.7 (CHO).
Compound 4. 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 1.69 (overlapping m, 4H, 4-H2 and 5-H2), 2.64 (m, 2H, 6-H2), 3.06 (t, 2H, J = 6.1 Hz, 3-H2), 6.74 (d, 1H, J = 8.5 Hz, 8-H), 7.24 (d, 1H, J = 8.5 Hz, 7-H), 10.38 (s, 1H, CHO); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 22.0 (C-7), 22.3 (C-6), 25.3 (C-8), 28.5 (C-5), 114.7 (C-3), 118.4 (C-1), 127.9 (C-4a), 138.0 (C-4), 139.7 (C-8a), 160.6 (C-2), 195.3 (CHO).
:20 v/v%) as eluents, 5 was obtained as a colorless oil (276 mg, 78%) (Mp: 29–30 °C41); 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 1.73 (overlapping m, 4H, 6-H2 and 7-H2), 2.58 (t, 2H, J = 6.1 Hz) and 2,75 (t, 2H, J = 5.9 Hz): 6-H2 and 7-H2, 6.80 (d, 1H, J = 8.0 Hz, 4-H), 7.48 (d, 1H, J = 8.0 Hz, 3-H), 9.92 (s, 1H, CHO), 11.23 (bs, 1H, OH); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 21.7 and 21.9 (2C): C-6, C-7 and C-8, 29.8 (C-5), 118.1 (C-2), 120.8 (C-4), 124.9 (C-8a), 129.8 (C-3), 147.1 (C-4a), 158.8 (C-1), 196.7 (CHO).
:90 → 20:80 v/v%), estrone–TSC was obtained as a white solid (175 mg, 94%), Mp >200 °C (decomp.); 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 0.83 (s, 3H, 18-H3), 1.28–1.59 (overlapping m, 6H), 1.77 (m, 1H), 1.93 (m, 2H), 2.07 (m, 1H), 2.14 (m, 1H), 2.43 (m, 1H), 2.56 (m, 1H), 2.77 (m, 2H, 6-H2), 6.56 (s, 1H, 4-H), 7.72 (s, 1H, 1-H), 7.93 (s, 1H, one H of NH2), 8.05 (s, 1H, the other H of NH2), 8.32 (s, 1H, CHN), 9.55 (s, 1H, OH), 11.26 (s, 1H, NH); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 13.6 (C-18), 21.1 (CH2), 25.3 (CH2), 25.9 (CH2), 29.1 (CH2), 31.4 (CH2), 35.4 (CH2), 37.9 (CH), 43.6 (CH), 47.3 (C-13), 49.6 (CH), 115.6 (C-4), 117.8 (C-2), 123.3 (C-1), 130.9 (C-10), 140.0 (C-5), 140.3 (CHN), 154.3 (C-3), 177.3 (CS), 219.7 (C-17).
:80 v/v%), → EtOAc), thn-TSC was obtained as a yellowish solid (90 mg, 72%), Mp 238–240 °C; 1H NMR (DMSO-d6, 500 MHz): δ (ppm) 1.72 (overlapping m, 4H, 6-H2 and 7-H2), 2.59 (t, 2H, J = 6.1 Hz) and 2.68 (t, 2H, J = 5.9 Hz): 6-H2 and 7-H2, 6.35 (bs, 2H, NH2), 6.61 (d, 1H, J = 7.9 Hz, 4-H), 7.10 (d, 1H, J = 7.9 Hz, 3-H), 8.03 (s, 1H, CHN), 10.15 (s, 1H, NH), 10.31 (bs, 1H, OH); 13C NMR (DMSO-d6, 125 MHz): δ (ppm) 22.3 (2C) and 22.6: C-6, C-7 and C-8, 29.3 (C-5), 115.7 (C-2), 120.1 (C-4), 124.2 (C-8a), 126.3 (C-3), 139.6 (C-4a), 142.9 (CHN), 154.1 (CO), 155.9 (C-1).
ESI-MS measurements were performed using a Waters Q-TOF Premier (Micromass MS Technologies, Manchester, UK) mass spectrometer equipped with electrospray ion source. Samples contained 20–50 μM complex dissolved in methanol containing 0.5% (v/v) acetic acid in the case of the estron-TSC complex (see spectra in Fig. S13, ESI†).
UV-vis spectra on an Agilent Carry 8454 spectrophotometer were also recorded for the complexes and ligands in methanol and in n-octanol for the sake of comparison.
EPR spectra for the copper(II) complexes were recorded with a BRUKER EleXsys E500 spectrometer (microwave frequency 9.54 GHz, microwave power 13 mW, modulation amplitude 5 G, modulation frequency 100 kHz). Powder of the copper(II) complexes was dissolved in pure DMSO to obtain 3 mM concentration solutions. The room temperature EPR spectra were recorded in capillaries applying 12 scans. For the frozen solution spectra 0.10 mL samples were diluted with 0.10 mL water and transferred into EPR tubes and the spectra were recorded in Dewar containing liquid nitrogen (77 K). The room temperature spectra were corrected by subtracting the background spectrum of pure DMSO. The spectra were simulated by the “EPR” program42 using the parameters go, Ao copper hyperfine (ICu = 3/2) coupling and four linewidth parameters. The anisotropic EPR spectra were analyzed with the same program. Rhombic g-tensor (gx, gy, gz) and copper hyperfine tensor (ACux, ACuy, ACuz) have been used. The nitrogen superhyperfine structure was taken into account with a rhombic hyperfine tensor (aNx, aNy, aNz) were the xyz directions referred to the g-tensor orientations. Orientation dependent linewidth parameters (α, β, and γ) were used to fit the linewidths through the equation σMI = α + βMI + γMI2, where MI denotes the magnetic quantum number of copper(II) ion. Since a natural CuCl2 was used for the measurements, all spectra were calculated by the summation of spectra 63Cu and 65Cu weighted by their natural abundances. The hyperfine and superhyperfine coupling constants and the relaxation parameters were obtained in field units (Gauss = 10−4 T). The results of the EPR spectroscopic measurements are detailed in the Results.
Characterization of the complexes. Complex [Cu(estrone–TSCH−2)]: yield: 61%; ESI-MS (methanol, positive): m/z 433.0883 [M + 1]+, 433.0891 calcd for C20H24CuN3O2S; λmax in methanol: 318, 328, 392 nm; in n-octanol: 318, 328, 394 nm.
Complex [Cu(thn-TSCH−2)]: yield: 45%; ESI-MS (methanol, positive): m/z 311.0156 [M + 1]+, 311.0154 calcd for C12H14CuN3OS; λmax in methanol: 305, 396 nm; in n-octanol: 318, 328, 396 nm.
Complex [Cu(STSCH−2)]: yield: 38%; ESI-MS (methanol, positive): m/z 256.9682 [M + 1]+, 256.9684 calcd for C8H8CuN3OS; λmax in methanol: 269, 321, 386 (broad band) nm; in n-octanol: 324, 388 (broad band) nm.
Solution equilibrium studies: UV-visible spectrophotometric and fluorometric titrations
An Orion 710A pH-meter equipped with a Metrohm combined electrode (type 6.0234.100) and a Metrohm 665 Dosimat burette were used for the UV-vis and fluorometric titrations at 25.0 ± 0.1 °C. Ionic strength of 0.10 M (KCl) was used in order to keep the activity coefficients constant. The titrations were performed with carbonate-free KOH solution of known concentration (0.10 M) with or without 30% (v/v) DMSO content. The electrode system was calibrated to the pH = −log[H+] scale by means of blank titrations (HCl vs. KOH) according to the method suggested by Irving et al.43 The average water ionization constant (pKw) is 13.76 ± 0.05 in water and 14.52 ± 0.05 in the 30% (v/v) DMSO/H2O mixture. Argon was always passed over the solutions during the titrations.An Agilent Cary 8454 diode array spectrophotometer was used to record the UV-vis spectra at an interval of 200–800 nm. The path length was 1 cm. Spectrophotometric titrations were performed in a 30% (v/v) DMSO/H2O mixture on samples containing the ligands at 50–70 μM concentration, in the pH range from 1.0 to 12.5 in the absence or in the presence of 1 or 0.5 equiv. copper(II) ions. The fluorescence spectra were recorded for TSC ligands on a Hitachi-4500 spectrofluorometer using a 1 cm quartz cell. The fluorometric titrations were performed on samples containing the ligands at 1 μM concentration in the pH range from 5 to 11.5 titrated by KOH solution in pure aqueous solution.
Proton dissociation constants (Ka) of the TSC ligands, the overall stability constants (β) of the copper(II) complexes and the UV-vis and/or emission spectra of the individual species were calculated by the computer program PSEQUAD44 as it was done in our previous works.14,45
Solution kinetic studies on the reduction of the copper(II) complexes
The redox reaction of the copper(II) complexes with GSH and ascorbic acid was studied at 25.0 ± 0.1 °C on a Hewlett Packard 8452A diode array spectrophotometer using a special, tightly closed tandem cuvette (Hellma Tandem Cell, 238-QS). The reactants were separated until the reaction was triggered. Both isolated pockets of the cuvette were completely deoxygenated by bubbling a stream of argon for 10–10 min before mixing the reactants. Spectra were recorded before and then immediately after the mixing, and changes were followed till no further absorbance change was observed. One of the isolated pockets contained the reducing agent (GSH or ascorbic acid), while the other contained the copper(II) complex, and their final concentrations were 1250 μM and 25 μM, respectively after mixing. The pH of all the solutions was adjusted to 7.40 by 100 mM HEPES buffer and an ionic strength of 0.1 M (KCl) was applied. The stock solutions of the reducing agents and the complexes were freshly prepared every day.During the calculations the absorbance (A)–time (t) curves were fitted and analyzed at the λmax of the complex. (A0 − Afinal) × e(−a×t) + Afinal equation was used where A0, Afinal and a parameters were refined and accepted at the minimal value of the weighted sum of squared residuals (difference between the measured and calculated absorbance values) at the given wavelength. Then observed rate constants (kobs) of the redox reaction were obtained from the data points of the simulated absorbance–time curves as the slope of the ln(A/A0) vs. time plots.
Crystallographic structure determination of compounds thn-TSC (I) and [Cu(thn-TSCH−1)Cl] (II)
Single crystals suitable for X-ray diffraction experiment of compound thn-TSC (I) were grown from warmed and then slowly evaporated acetone at room temperature. For the crystallization of its complex thn-TSC (0.027 mmol) was dissolved in methanol (5.4 mL), then 1.11 mL CuCl2 in an aqueous solution (0.027 mmol) was added. The reaction mixture was stirred at 65 °C during 2 h and after filtered, and the solvent was concentrated under reduced pressure to 2 ml. Then the crystal of copper complex [Cu(thn-TSCH−1)Cl] (II) was obtained through slow evaporation from concentrated methanol–water solution at room temperature. Single crystals were mounted on loops and transferred to the goniometer. X-ray diffraction data were collected at 165(5) K on a Rigaku RAXIS-RAPID II diffractometer using Cu-Kα radiation. Numerical absorption correction46 was carried out using the program CrystalClear,47 SHELX48 program package under WinGX49 or Olex250 softwares were used for structure solution and refinement. The structures were solved by direct methods. The models were refined by full-matrix least squares on F2. Refinement of non-hydrogen atoms was carried out with anisotropic temperature factors. For crystal thn-TSC (I) two carbon atoms C7 and C8 was found in disordered positions. The two positions were denoted by ‘A’ and ‘B’ and the occupancy ratio between them was 68% and 32%, respectively. For crystal [Cu(thn-TSCH−1)Cl] (II) small size and platelet shape crystal prevented the collection of high resolution data which resulted in a higher Rint and final R1 and wR2 values. In the refinement process restrains have been used for the ring carbon atoms. In both structures, hydrogen atoms were placed into geometric positions. They were included in structure factor calculations but they were not refined. The isotropic displacement parameters of the hydrogen atoms were approximated from the U(eq) value of the atom they were bonded to. The summary of data collection and refinement parameters are collected in Table S1 (ESI†). Selected bond lengths and angles of compounds were calculated by PLATON software.51 The graphical representation and the edition of CIF files were done by Mercury52 and PublCif53 softwares. The crystallographic data files for the complexes have been deposited with the Cambridge Crystallographic Database as CCDC 1986236 and 1986237.†Lipophilicity and artificial membrane permeability assays
Distribution coefficient (D7.4) values of ligands and the complexes were determined (or attempted to be determined) by the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.40 (20 mM phosphate buffered saline (PBS)) at 25.0 ± 0.2 °C as described previously.54 Additionally, logP values were estimated for the ligands by the means of the MarvinSketch software.35
Parallel artificial membrane permeability assay (PAMPA) was applied for the ligands and complexes with a Corning Gentest pre-coated PAMPA Plate System.55 In summary, the 96-well filter plate was used as the permeation acceptor and the 96-well bottom plate was used as the permeation donor. For simplicity, PBS (containing 4% (v/v) DMSO) was used both as donor and acceptor buffer throughout this study. The initial donor solutions were prepared by diluting DMSO stock solutions (500 μM) in PBS (25-fold dilution). Donor plate was filled with 300 μL of the donor solutions (containing the test compounds). Each well of the filter plate contained 200 μL buffer as acceptor phase. The resulting ‘sandwich’ was protected with parafilm to prevent evaporation and incubated at room temperature at 25 °C for 5 h. Then, solutions from the donor and acceptor wells were transferred to Eppendorf tubes and their UV-vis spectra were recorded to determine the concentration of the components. Peff values were calculated according to the equation reported by Yu et al.56
In vitro cell studies
control)/(ODcellcontrol − ODmediumcontrol)) × 100 values were plotted against the logarithm of compound concentrations. Curves were fitted by GraphPad Prism software57 using the sigmoidal dose–response model (comparing variable and fixed slopes). The IC50 values were obtained from at least 3 independent experiments.
ROS production measurement was performed in MCF-7 and SUM159 cell lines with DCFH-DA. The emission intensity of the fluorescent 2,7-dichlorofluorescein (DCF) was measured at 529 nm using 500 nm excitation wavelength. The human cancer cells were seeded 1 × 104 in 100 μL DMEM/10% fetal bovine serum (FBS) into in 96-well microtiter plates. After allowing cells to adhere for 24 h, 1 mM NAC was added to some wells while 100 μM DCFH-DA was added to all wells. After removing the media with the excess of DCFH-DA, cells were treated with the compounds in 1 μM concentration (where the compounds are not toxic to the cells). The increase in ROS caused by compounds applied was measured at different time points – prior to treatment and 60 min after the treatment.
For the catalase activity and GSH level assays cells were prepared in a same manner. The SUM159 cells were seeded in 6-well plates at density of 5 × 105 cell per well, and were allowed 24 h to attach to the well. Then, cells were treated with 1 μM compound and left for 24 h after which they were harvested, and the dry pellet was stored at −80 °C until analysis. For analyses, cells were lysed in PBS by 4 freeze–thaw cycles, and total protein content was measured by Bradford method.58 Total GSH was measured spectro-photometrically at 450 nm by modified Tietze method based on reduction of DTNB (Ellman's reagent) to 2-nitro-5-thiobenzoate (TNB anion) by GSH.59 Catalase activity was assayed by measuring H2O2 decomposition by catalase in the whole cell lysate by Góth method.60
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by National Research, Development and Innovation Office-NKFIA through projects GINOP-2.3.2-15-2016-00038, FK 124240, PD 128504, FIKP program TUDFO/47138-1/2019-ITM, J. Bolyai Research Scholarship of the Hungarian Academy of Sciences (T. H., N. V. M.), Visegrad Scholarship 51910905 (T. V. P.). This article is also based upon work from COST Action CA17104 supported by COST (European Cooperation in Science and Technology).References
- J. R. Dilworth and R. Hueting, Inorg. Chim. Acta, 2012, 389, 3–15 CrossRef CAS.
- P. Heffeter, V. F. S. Pape, É. A. Enyedy, B. K. Keppler, G. Szakacs and C. R. Kowol, Antioxid. Redox Signaling, 2019, 30, 1062–1082 CrossRef CAS PubMed.
- D. S. Kalinowski, P. Quach and D. R. Richardson, Future Med. Chem., 2009, 1, 1143–1151 CrossRef CAS PubMed.
- A. B. Miah, K. J. Harrington and C. M. Nutting, Eur. J. Clin. Med. Oncol., 2010, 2, 1–6 CAS.
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Antioxid. Redox Signaling, 2013, 18, 973–1006 CrossRef CAS PubMed.
- https://clinicaltrials.gov/ct2/show/NCT02466971, accessed on 26/02/2020.
- C. Nutting, C. Van Herpen, A. Miah, S. Bhide, J.-P. Machiels, J. Buter, C. Kelly, D. De Raucourt and K. Harrington, Ann. Oncol., 2009, 20, 1275–1279 CrossRef CAS PubMed.
- K. Pelivan, W. Miklos, S. van Schoonhoven, G. Koellensperger, L. Gille, W. Berger, P. Heffeter, C. R. Kowol and B. K. Keppler, J. Inorg. Biochem., 2016, 160, 61–69 CrossRef CAS PubMed.
- K. Y. Salim, W. R. Danter, V. S. Maleki and J. Koropatnick, Oncotarget, 2016, 7, 41363–41379 CrossRef PubMed.
- P. J. Jansson, D. S. Kalinowski, D. J. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed.
- J. Shao, B. Zhou, A. J. Di Bilio, L. Zhu, T. Wang, C. Q. J. Shih and Y. Yen, Mol. Cancer Ther., 2006, 5, 586–592 CrossRef CAS PubMed.
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, J. Biol. Inorg. Chem., 2012, 17, 409–423 CrossRef CAS PubMed.
- D. B. Lovejoy, P. J. Jansson, U. T. Brunk, J. Wong, P. Ponka and D. R. Richardson, Cancer Res., 2011, 71, 5871–5880 CrossRef CAS PubMed.
- É. A. Enyedy, É. Zsigó, N. V. Nagy, C. R. Kowol, A. Roller, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2012, 4036–4047 CrossRef.
- D. X. West and A. E. Liberta, Coord. Chem. Rev., 1993, 123, 49–71 CrossRef CAS.
- M. Belicchi Ferrari, S. Capacchi, G. Pelosi, G. Reffo, P. Tarasconi, R. Albertini, S. Pinelli and P. Lunghi, Inorg. Chim. Acta, 1999, 286, 134–141 CrossRef CAS.
- Z. Zhang, Y. Gou, J. Wang, K. Yang, J. Qi, Z. Zhou, S. Liang, H. Liang and F. Yang, Eur. J. Med. Chem., 2016, 121, 399–409 CrossRef CAS PubMed.
- I. Dilović, M. Rubcić, V. Vrdoljak, S. Kraljević Pavelić, M. Kralj, I. Piantanida and M. Cindrić, Bioorg. Med. Chem., 2008, 16, 5189–5198 CrossRef PubMed.
- A. Gulea, D. Poirier, J. Roy, V. Stavila, I. Bulimestru, V. Tapcov, M. Birca and L. Popovschi, J. Enzyme Inhib. Med. Chem., 2008, 23, 806–818 CrossRef CAS PubMed.
- M. N. M. Milunovic, É. A. Enyedy, N. V. Nagy, T. Kiss, R. Trondl, M. A. Jakupec, B. K. Keppler, R. Krachler, G. Novitchi and V. B. Arion, Inorg. Chem., 2012, 51, 9309–9321 CrossRef CAS PubMed.
- Y. Huang, E. Kong, C. Gan, Z. Liu, Q. Lin and J. Cui, Bioinorg. Chem. Appl., 2015, 2015, 742592 Search PubMed.
- Y. Huang, E. Kong, J. Zhan, S. Chen, C. Gan, Z. Liu, L. Pang and J. Cui, Bioinorg. Chem. Appl., 2017, 2017, 4276919 Search PubMed.
- A. Gupta, B. S. Kumar and A. S. Negi, J. Steroid Biochem. Mol. Biol., 2013, 137, 242–270 CrossRef CAS PubMed.
- É. Frank and G. Schneider, J. Steroid Biochem. Mol. Biol., 2013, 137, 301–315 CrossRef PubMed.
- J. A. R. Salvador, J. F. S. Carvalho, M. A. C. Neves, S. M. Silvestre, A. J. Leitao, M. M. C. Silva and M. L. Sa e Melo, Nat. Prod. Rep., 2013, 30, 324–374 RSC.
- M. Kvasnica, L. Rarova, J. Oklestkova, M. Budesinsky and L. Kohout, Bioorg. Med. Chem., 2012, 20, 6969–6978 CrossRef CAS PubMed.
- P. Saha, C. Descôteaux, K. Brasseur, S. Fortin, V. Leblanc, S. Parent, É. Asselin and G. Bérubé, Eur. J. Med. Chem., 2012, 48, 385–390 CrossRef CAS PubMed.
- C. Descôteaux, J. Provencher-Mandeville, I. Mathieu, V. Perron, S. K. Mandal, E. Asselin and G. Bérubé, Bioorg. Med. Chem. Lett., 2003, 13, 3927–3931 CrossRef PubMed.
- C. Van Themsche, S. Parent, V. Leblanc, C. Descôteaux, A.-M. Simard, G. Bérubé and E. Asselin, Endocr.-Relat. Cancer, 2009, 16, 1185–1195 CAS.
- R. H. Peters, W.-R. Chao, B. Sato, K. Shigeno, N. T. Zaveri and M. Tanabe, Steroids, 2003, 68, 97–110 CrossRef CAS PubMed.
- G. A. Olah, L. Ohannesian and M. Arvanaghi, Chem. Rev., 1987, 87, 671–686 CrossRef CAS.
- W. Kantlehner, Eur. J. Org. Chem., 2003, 2530–2546 CrossRef CAS.
- N. U. Hofsløkken and L. Skattebøl, Acta Chem. Scand., 1999, 53, 258–262 CrossRef.
- Ø. W. Akselsen and T. V. Hansen, Tetrahedron, 2011, 67, 7738–7742 CrossRef.
- MarvinSketch (version 16.12.12.0, calculation module developed by ChemAxon), http://www.chemaxon.com/products/marvin/marvinsketch/, 2016 (accessed on 26/02/2020).
- C. R. Kowol, N. V. Nagy, T. Jakusch, A. Roller, P. Heffeter, B. K. Keppler and É. A. Enyedy, J. Inorg. Biochem., 2015, 152, 62–73 CrossRef CAS PubMed.
- A. Santoro, B. Vileno, O. Palacios, M. D. Peris-Díaz, G. Riegel, C. Gaiddon, A. Krężel and P. Faller, Metallomics, 2019, 11, 994–1004 RSC.
- N. A. Seebacher, D. R. Richardson and P. J. Jansson, Cell Death Dis., 2016, 7, e2510 CrossRef CAS PubMed.
- S. Hager, V. F. S. Pape, V. Pósa, B. Montsch, L. Uhlik, G. Szakács, S. Tóth, N. Jabronka, B. K. Keppler, C. R. Kowol, É. A. Enyedy and P. Heffeter, Antioxid. Redox Signaling, 2020 DOI:10.1089/ars.2019.7854.
- N. Traverso, R. Ricciarelli, M. Nitti, B. Marengo, A. L. Furfaro, M. A. Pronzato, U. M. Marinari and C. Domenicotti, Oxid. Med. Cell. Longevity, 2013, 2013, 972913 Search PubMed.
- H. Wynberg and W. S. Johnson, J. Org. Chem., 1959, 24, 1424–1428 CrossRef CAS.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- H. M. Irving, M. G. Miles and L. D. Petit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. L. Leggett, Plenum Press, New York, 1985, pp. 291–353 Search PubMed.
- O. Dömötör, M. A. Kiss, G. T. Gál, N. V. May, G. Spengler, M. Nóvé, A. Čipak Gašparović, É. Frank and É. A. Enyedy, J. Inorg. Biochem., 2020, 202, 110883 CrossRef PubMed.
- T. Higashi, Numerical Absorption Correction, NUMABS, 2002 Search PubMed.
- CrystalClear SM 1.4.0 Rigaku/MSC Inc., 2008.
- SHELXL-2013 Program for Crystal Structure Solution, University of Göttingen, Germany, 2013.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- S. P. Westrip, J. Appl. Crystallogr., 2010, 43, 920–925 CrossRef CAS.
- É. A. Enyedy, D. Hollender and T. Kiss, J. Pharm. Biomed. Anal., 2011, 54, 1073–1081 CrossRef PubMed.
- X. Chen, A. Murawski, K. Patel, C. L. Crespi and P. V. Balimane, Pharm. Res., 2008, 25, 1511–1520 CrossRef CAS PubMed.
- H. Yu, Q. Wang, Y. Sun, M. Shen, H. Li and Y. Duan, PLoS One, 2015, 10, e0116502 CrossRef PubMed.
- GraphPad Software I, GraphPad Prism, GraphPad Software, Inc., 2007, http://www.graphpad.com (accessed on 26/02/2020).
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- F. Tietze, Anal. Biochem., 1969, 27, 502–522 CrossRef CAS PubMed.
- L. Góth, Clin. Chim. Acta, 1991, 196, 143–151 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis, fluorescence, EPR spectral and crystal data. Cellular GSH level, catalase activity and DCFDA assay data. CCDC 1986236 and 1986237. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nj01070g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |