
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVinylimidazole coordination modes to Pt and Au metal centers†
Sirpa
Jääskeläinen
 *,
Igor O.
Koshevoy
,
Sari
Suvanto
,
Tiina
Ryhänen
and
Pipsa
Hirva
*,
Igor O.
Koshevoy
,
Sari
Suvanto
,
Tiina
Ryhänen
and
Pipsa
Hirva
Department of Chemistry, University of Eastern Finland, P. O. Box 111, Fi-80101 Joensuu, Finland. E-mail: sirpa.jaaskelainen@uef.fi
First published on 3rd July 2020
Abstract
The coordination modes of 1-vinylimidazole to platinum and gold were studied. Complexes [PtCl3(Hvinylimidazole)]·H2O (1), [Au(vinylimidazole)2]+[AuBr2]− (2), [Hvinylimidazole]+[AuCl4]− (3), and [Hvinylimidazole]+[AuBr4]− (4) were prepared and structurally characterized. Compound 1 is the first structurally characterized transition metal complex containing a protonated vinylimidazole, which is coordinated through the vinyl group in the side-on position. In compound 2, the neutral ligands coordinate through the imidazole nitrogens to the reduced gold(I) center and the charge balancing counter anion [Au(I)Br2]− has a short Au–Au contact with the cationic part. In 3 and 4, the acidic reaction conditions lead to the protonation of the imidazole nitrogen and an ion pair with tetrahalogenide gold(III) is obtained. The tendency to the different crystallized products is attributed to the combination of the metal and the halogen properties with the reaction conditions. Computational chemistry was used to explain the preference of the vinyl coordination type, as well as in the interpretation of the spectroscopic details and the nature of the intra- and intermolecular interactions present in the solid state.
Introduction
1-Vinylimidazole or N-vinylimidazole is a two nitrogen containing, five-membered, aromatic compound with the vinyl group at one of the nitrogen atoms. Imidazoles, in general, are important in biomolecules,1–4 pharmaceutical applications5–10 and in industrial utilization as corrosion inhibitors.11 Vinylimidazole is particularly known for its homo and co-polymers. The monomer is easily polymerized through the vinyl group by exploiting UV-irradiation, heat or activator agents. Furthermore, vinylimidazole monomers can be quaternized to ionic liquids and polymerized through the side chains.12,13 The active imidazole nitrogens in these polyionic hydrogels effectively bind metal atoms. Due to the mechanical stability, strength and recoverability of the materials, diverse applications in polymer anchored metal catalysts,14 drug delivery,15 and water purification16,17 are produced.In the context of transition metal complexes, the most probable coordination sites in 1-vinylimidazole are the imidazole nitrogen and the vinyl group. The substituent at the heteroatom blocks up the possibility for tautomerism typical of the original imidazole. The non-coordinated nitrogen atom is slightly basic and protonation of this site is possible. The high tendency to coordination to metal centers and formation of molecular complexes, as in the case of polymeric structures, is a source of diverse applications such as pharmacologically active species.18,19
A survey of the CCDC database and literature reveals that exclusively nitrogen bound ligands are present in transition metal vinyl imidazole complexes. A quite large selection of complexes for lighter transition metals is published. However, going towards the heavier metals, the number of synthesized and characterized complexes is reduced. In group ten in the periodic table, Ni has several vinylimidazole derivatives.20–22 For palladium, tetranuclear planar structures with one to four ligands have been reported,23,24 while only the octahedral [PtCl2(vinylimidazole)4]Cl2 is crystallographically characterized for platinum.25
Among the coinage metals, copper has several derivatives of this ligand. The most common coordination type is octahedral, but also tetrahedral complexes are known. Typically, these have two or four vinylimidazole ligands.26–37 The catena compounds, where Cu centers with 1-vinylimidazole ligands and ReCl6-units are connected through chloro bridges, have shown magnetic properties.38 Surprisingly, no compounds of Ag or Au have been reported. In group twelve, Zn29,38–40 and Cd33,41 form MOF complexes with auxiliary linking ligands, and the vinylimidazoles coordinate monodentately.
Ruthenium has a unique cluster derivative [HRu3(CO)10(vinylimidazole)], where the deprotonated ligand acts as a bidentate bridging C,N-ligand. The imidazole carbon between the nitrogen atoms is utilized in coordination, but the vinyl group remains intact.42
A similar type of ligand, 4-vinylpyridine typically coordinates through the nitrogen atom, as in the complexes of Cu,43 Pt44,45 and Zn.46 The second isomer, 2-vinylpyridine, has been the subject of a relatively limited number of studies on the synthesis, structure and reactivity, which have involved metals Ru,47–52 Os,50,53–57 Co,58 Rh,59,60 Ir,61 Pd,62 Pt63,64 and Au.65,66 2-vinylpyridine can form five-membered cyclometalated complexes with the high versatility of applications in the fields of biological, catalytic and luminescence properties.64
In this work we concentrated on the reactions of vinylimidazole with late transition metals, Pt and Au, to clarify the relative reactivity of the imidazole group or the vinyl group towards the metal ions. The experimental crystal structures of the products were analyzed via X-ray diffraction, NMR, IR and Raman spectroscopy, and computational DFT methods.
Results and discussion
Reactions and structural studies
The reaction of 1-vinylimidazole with K2PtCl4 in water solution led to the formation of a white product, which by spectroscopic measurements and elemental analysis was interpreted as the square planar [PtCl2(vinylimidazole)2] with nitrogen coordinated ligands. This is in good agreement with the earlier reported reactions in water, ethanol or acetone.67On the other hand, the reaction of 1-vinylimidazole with K2PtCl4 in acidic medium generated a novel vinylimidazole derivative type [PtCl3(Hvinylimidazole)]·H2O (1), where the partially protonated ligand is bound to metal through the vinyl group in side-on geometry (Fig. 1a). The Pt(II) center has further three chloro ligands in the perpendicular plane against the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of 1.395(2) Å. The Pt–C(6) bond is 2.135(2) Å and Pt–C(7) is slightly shorter 2.109(2) Å, showing a strong side-on coordination type of the vinyl group. The sum of the van der Waals radii of Pt and C is 3.45 Å. The Pt–Cl bonds are 2.2972(5) and 2.2922(5) Å in the cis-position and 2.3056(5) Å in the trans-position to the vinylimidazole.
C bond of 1.395(2) Å. The Pt–C(6) bond is 2.135(2) Å and Pt–C(7) is slightly shorter 2.109(2) Å, showing a strong side-on coordination type of the vinyl group. The sum of the van der Waals radii of Pt and C is 3.45 Å. The Pt–Cl bonds are 2.2972(5) and 2.2922(5) Å in the cis-position and 2.3056(5) Å in the trans-position to the vinylimidazole.
 | ||
| Fig. 1 Structure of the [PtCl3(Hvinylimidazole)]·H2O monomer (a) and packing (b). Selected bond lengths: Pt–C(6) 2.135(2), Pt–C(7) 2.109(2), Pt–Cl(1) 2.3056(5), Pt–Cl(2) 2.2972(5), Pt–Cl(3) 2.2922(5), and C(6)–C(7) 1.395(2) Å. Thermal ellipsoids are shown at the 50% probability level. | ||
Reaction of vinylimidazole with AuBr3 in organic aprotic THF solvent led to the formation of an ion pair [Au(vinylimidazole)2]+[AuBr2]− (2) (Fig. 2). In the cation, two neutral ligands are attached to the Au(I) center via imidazole nitrogens, the vinyl groups remaining unreacted. Anionic [Au(I)Br2]− acts as the counter ion. Therefore, reduction of the metal with partial oxidation of the bromines has taken place during coordination.
 | ||
| Fig. 2 Structure of [Au(vinylimidazole)2]+[AuBr2]− (2). Selected bond lengths and angles: Au(1)–Au(2) 3.1200(5), Au(2)–Br(1) 2.3826(9), Au(2)–Br(2) 2.3843(9), Au(1)–N(3) 2.017(5), Au(1)–N(3A) 2.018(5), C(6)–C(7) 1.306(10), C(6A)–C(7A) 1.300(10) Å, N(3)–Au(1)–N(3A) 176.1(2)°, N(3)–Au(1)–Au(2) 91.0(2)°, N(3A)–Au(1)–Au(2) 90.8(2)°. Thermal ellipsoids are shown at the 50% probability level. | ||
The coordination at both gold atoms is almost ideal T-shape with Au–Au–N bond angles of 90.8(2)° and 91.0(2)°. The Au–N distances are 2.018(5) and 2.017(5) Å. The CC bond lengths in vinyl groups are considerably shorter, 1.306(10) and 1.300(10) Å, than in 1. The Au–Au distance is 3.1200(5) Å, which is slightly shorter than the sum of the van der Waals radii of two gold atoms (3.32 Å). The cationic part is planar and the Br–Au–Br moiety lies perpendicular to this plane.
The ion pairs form a chain structure in the solid state via hydrogen bonds (Fig. 3). The bromine forms an intramolecular hydrogen bond of 3.0687(8) Å to H(2). The bromine atoms have also several short contacts of 2.8839(7)–3.0437(8) Å to hydrogens of neighboring ion pairs.
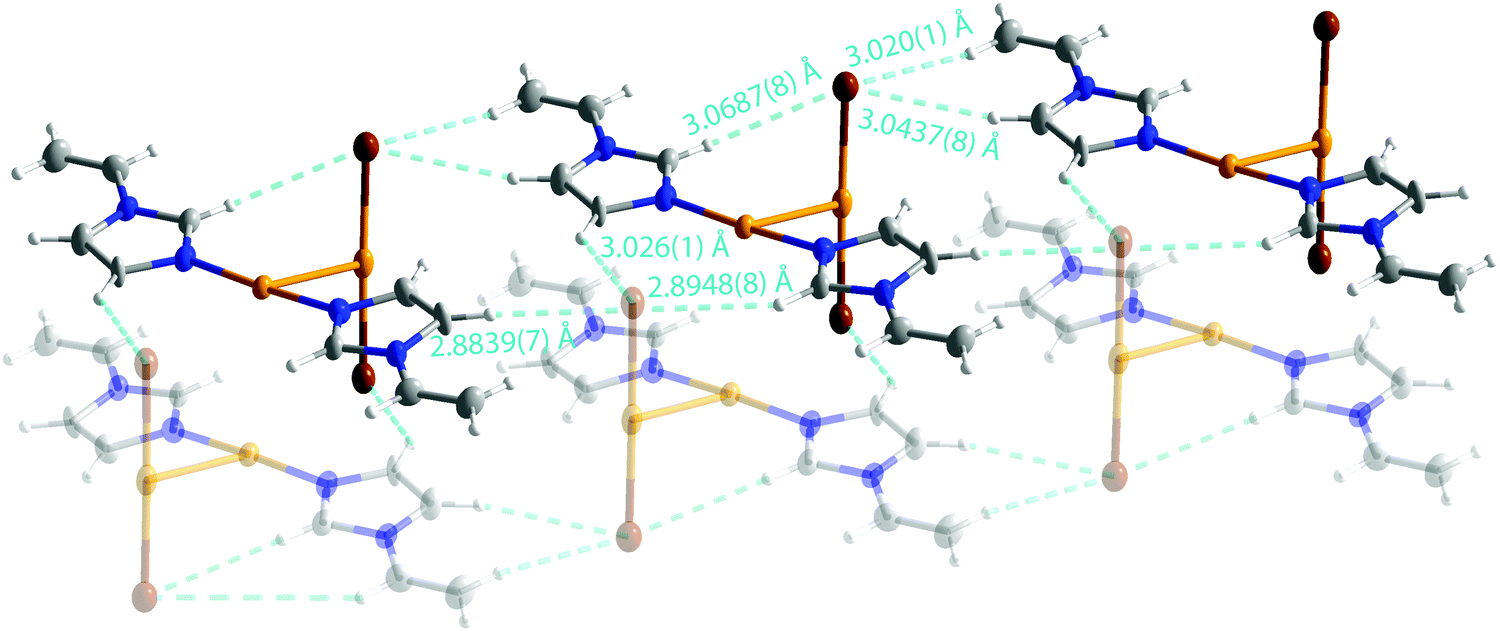 | ||
| Fig. 3 Hydrogen bonds in 2. | ||
The nature of the hydrogen bonding network was further studied computationally by analyzing the charge density of an extended model by Quantum Theory of Atoms in Molecules (QTAIM). The model with four adjacent ion pairs was cut directly from the experimental crystal structure (see Fig. S1 in the ESI†) and analyzed using the DFT wavefunction. The results for the properties of the electron density are shown in Table 1.
| BCP# | Type | ρ (e Å−3) | |V|/G | E INT (kJ mol−1) |
|---|---|---|---|---|
| 1 | Au(1)–N(3) | 0.898 | 1.37 | −278 |
| 1′ | Au(1)–N(3A) | 0.899 | 1.38 | −275 |
| 2 | C(6)C(7) |
2.398 | 3.73 | −777 |
| 2′ | C(6A)C(7A) |
2.408 | 3.73 | −784 |
| 3 | Au(1)⋯Au(2) | 0.175 | 1.12 | −26 |
| 4 | Au(2)⋯H(4) | 0.048 | 0.80 | −5 |
| 4′ | Au(2)⋯H(2) | 0.057 | 0.82 | −6 |
| 5 | Br(1)⋯H(4) | 0.044 | 0.74 | −4 |
| 5′ | Br(1)⋯H(2) | 0.054 | 0.73 | −5 |
| 6 | Au(1)⋯Au(1′) | 0.105 | 1.03 | −13 |
| 7 | Au(1)⋯Br(1′) | 0.073 | 0.89 | −7 |
| 8 | Br(2)⋯H(5) | 0.040 | 0.75 | −3 |
| 9 | Br(2)⋯H(7) | 0.042 | 0.76 | −4 |
| 10 | (π–π)vinyl | 0.023 | 0.82 | −2 |
| 11 | (π–π)imidazole | 0.042 | 0.80 | −4 |
The [Au(vinylimidazole)2]+[AuBr2]− ion pairs show a large number of relatively weak intermolecular hydrogen bonds to the adjacent ion pairs, explaining the packing of the ions into rows. The properties of the electron density, such as small ρ, the ratio between potential energy density and kinetic energy density |V|/G < 1, and interaction energy EINT < 10 kJ mol−1, all point out to typical noncovalent intermolecular hydrogen bonds. In addition, the aurophilic Au(1)⋯Au(2) interactions at BCP 3 exhibit slightly larger strength with EINT of −26 kJ mol−1. Notably, the aurophilic interactions to the neighboring cations have comparable strength (EINT = −13 kJ mol−1 at BCP 6), enhancing the self-assembly of the ion pairs. Furthermore, π–π interactions form between vinyl groups and the imidazole rings of the adjacent ion pairs, explaining the torsional behavior of the N-ligands as they stack together.
In the reaction of the ligand with either NaAuCl4 or HAuCl4 in water solution containing HCl, protonation of the ligand took place giving compound [Hvinylimidazole]+[AuCl4]− (3) (Fig. 4) with a disordered structure. Thus, the protonated ligand does not coordinate to the metal like in the case of platinum. This type of ion pair was earlier known for instance for pyridine.68 In the solid state, a network supported by hydrogen bonds H(3)–Cl(1) of 2.4256(5) Å and H(7B)–Cl(4) of 2.9059(5) and 2.9139(5) Å was obtained (Fig. 4).
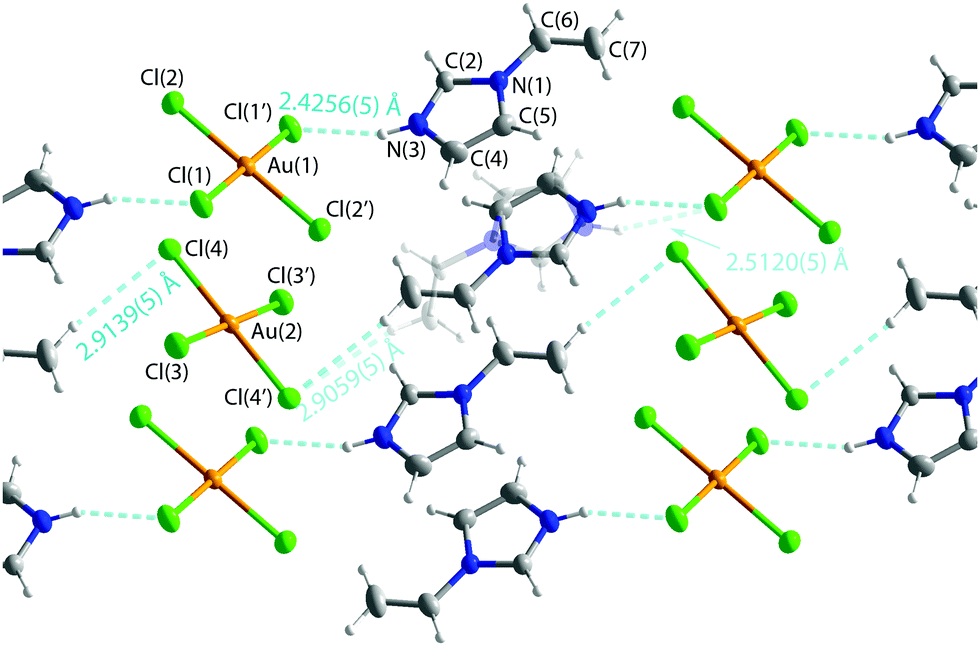 | ||
| Fig. 4 Structure of [Hvinylimidazole]+[AuCl4]− (3). Selected bond length: C(6)–C(7) 1.302(3), C(6A)–C(7A) 1.26(2) Au–Cl 2.2738(5)–2.2798(5) Å. The structure of [Hvinylimidazole]+ [AuBr4]− (4) is similar, but in this case with no disorder and with C(6)–C(7) 1.291(4) and Au–Br distances of 2.4227(2)–2.4286(2) Å. Thermal ellipsoids are shown at the 50% probability level. | ||
The synthesis of [Hvinylimidazole]+[AuBr4]− (4) from AuBr3 in acidic HBr solution proceeded analogously to that of 3. The crystal structure and spectroscopic data showed a similar structure type to 3, but in this case with no disorder.
IR and FT-Raman spectroscopy
The appearance of the ν(N–H) vibration at 3281, 3254 and 3256 cm−1 supports the protonation of the imidazole nitrogen in compounds 1, 3 and 4. The interpretation of the spectra was verified via computational simulation at the DFT level of theory on optimized molecular models. The experimental and simulated spectra are shown in Fig. S6–S8 (ESI†).Vinylic ν(CC) in the free ligand lies at 1630 cm−1 (computational value at 1626 cm−1). In 1 it is clearly shifted to a lower frequency of 1437 cm−1 (simulated 1441 cm−1), which is attributed to π-bonded CC-ligands like ethylene and related ligands.69 In a σ-bonded ligand, the shift is typically to higher frequencies. In 2, 3 and 4, ν(CC) appears at 1640 cm−1 (sim. 1632), so the shift is not remarkable.
The Raman spectra were measured for complexes 1 and 2 in the solid state and for vinylimidazole in the liquid form, and also simulated at the DFT level of theory using single molecular models to facilitate interpretation. The computational values are given in Table 2, and an example of the interpretation of the most important signals for compound 1 is given in Fig. S4 and S5 (ESI†).
| Vibration | 1 | 2 | L | HL |
|---|---|---|---|---|
| n.r. = not resolved. | ||||
| ν(N–H) | 3468 (3281) | — | — | 3431 |
| ν(C–H)im,H(2) | 3141 (3152) | 3111 (3113) | 3106 (3108) | 3102 |
| ν(C–H)vinyl | 3009 (3026) | 2968 (n.r.) | 3014 (3044) | 3040 |
| ν(C–H2)vinyl,s | 2967 (2951) | 2994 (n.r.) | 2993 (2999) | 2997 |
|
ν(CC)vinyl |
1441 (1408) | 1632 (1645) | 1625 (1653) | 1627 |
| s(C–H2)vinyl | 1315 (1322) | 1370 (n.r.) | 1375 (1367) | 1331 |
| ν(M–C)vinyl,as | 544 (503) | — | — | — |
| ν(M–C)vinyl,s | 476 (456) | — | — | — |
| ν(M–N) | — | 239 (n.r.) | — | — |
The full interpretation of the solid state Raman spectra was possible only for the platinum complex 1, which yielded the best quality experimental spectrum. Fig. S4 and S5 (ESI†) compare the experimental and simulated signals, which were further interpreted by animation of the vibrations. Even though the wavenumbers do not exactly match because all signals in the simulation do not scale similarly, the general appearance of the spectra is similar enough to allow interpretation (Table 2).
The coordination mode of 1 and 2 can most easily be seen in the stretching vibration of the CC double bond of the vinyl group, which is strongly shifted to smaller wavenumbers in the platinum complex 1, but shows a similar value to that in the free vinylimidazoles for the gold complex 2. The same, but less clear trend is seen in the ν(C–H) values of the CH2 protons of the vinyl group, as well as in the scissor vibration of the CH2 protons. Furthermore, clear Pt–C(vinyl) signals could be obtained in the spectrum of 1, verifying the side-on coordination of the metal.
The poor quality of the experimental Raman spectrum of compound 2, resulting from the degradation of the crystals during the measurement, did not allow all the signals to be resolved, most importantly the Au–N stretching frequency. However, indirect evidence on the coordination mode could be obtained by the absence of interaction with the vinyl group, since ν(CC)vinyl and ν(C–H2)vinyl were not shifted compared to the free 1-vinylimidazole. Additionally, there was no N–H stretching signal, which verified the coordination of the ligand in the non-protonated form.
It has been observed in the previous computational Raman studies of platinum complexes that the effect of temperature and isotopes of chlorine complicate the situation and some contradiction among the assignments exists.70,71
NMR spectroscopy
The 1H NMR spectra of all compounds were measured in d6-DMSO solvent and interpreted by comparison with the spectrum of the free ligand. The spectra are presented in Fig. S2 (ESI†).Compound 1 shows two sets of sharp peaks with a very similar pattern to the free ligand shifted to slightly larger chemical shifts. One possibility to the two sets of signals is the existence of another isomer with a non-protonated and possibly N-coordinated ligand. This suggestion is supported by the observation that the imidazole protons (2, 4, and 5) in isomer b show the same chemical shifts as in the spectrum of 2, while the peaks of the crystallized product appear at the same location as in 3. The protonation of the vinylimidazole ligand was computationally found to change the charge distribution especially in the aromatic ring of the free ligand, which could explain the difference in the chemical shift values.
The spectrum of 2, as in the case of 1, has also a pattern of two signal sets shifted toward higher chemical shifts. In this case, the small difference in chemical shifts suggests quite similar structures for the isomers, indicating the presence of two compounds in solution, probably due to the rotation around the Au(1)–Au(2), Au(1)–N(3/3A), and N(1/1A)–C(6/6A) bonds. The major compound has reasonably sharp signals. However, the minor compound shows broad signals, which can be assumed to rise from hydrogen bonding of the protons to the near-by quadrupole bromines. Since the rotation in solution can be rather free around single bonds, in the minor isomer the protons can have short contact with bromines. The elemental analysis indicates the presence of one pure product. The NMR spectra of 3 and 4 are similar with one set of peaks describing one pure product without the possibility of fluxionality.
Overall, the most notable shift of the signals is observed at the imidazolic H(2) proton signal in those structures, where protonation of the ligand has taken place. This shift can be interpreted to have originated from the protonation of the imidazole nitrogen N3, which was found to modify considerably the calculated charge distribution of the whole imidazole ring and especially the charge of C(2) and H(2).
Both proton coupled and proton decoupled 13C NMR spectra of the vinylimidazole ligand in the basic form as well as HCl treated vinylimidazole to simulate the protonation were measured (Fig. S3, ESI†), but only the proton decoupled spectra of the products (1–4) were obtained. Comparison of the experimental and the calculated72–74 spectra is given in Table S1 (ESI†). The spectra show the remaining vinylimidazole assembly in all cases. In complexes 1, 3 and 4, containing the protonated ligands, the signal of C(6) is slightly shifted to lower values as in the case of the protonated free ligand. Otherwise, the structure of the spectrum remains practically unchanged. The presence of different types of metals and paramagnetic species as well as the solvent effect has earlier shown to cause peculiarities in the spectrum appearance of 1-vinylimidazole and even the relative positions of the signals can change.75,76
Stability of the coordination modes
In order to find explanation for the preferred coordination mode in the Pt or Au vinylimidazoles, we performed computational optimization for models mimicking both N-coordination (A) of the deprotonated ligand and the side-on coordination (B) to the vinyl group of the protonated ligand. Since the HOMO–LUMO energy gap can give information on the relative stability of the models, the highest occupied and the lowest unoccupied orbitals along with the corresponding energies of the small molecular models are presented in Fig. 5.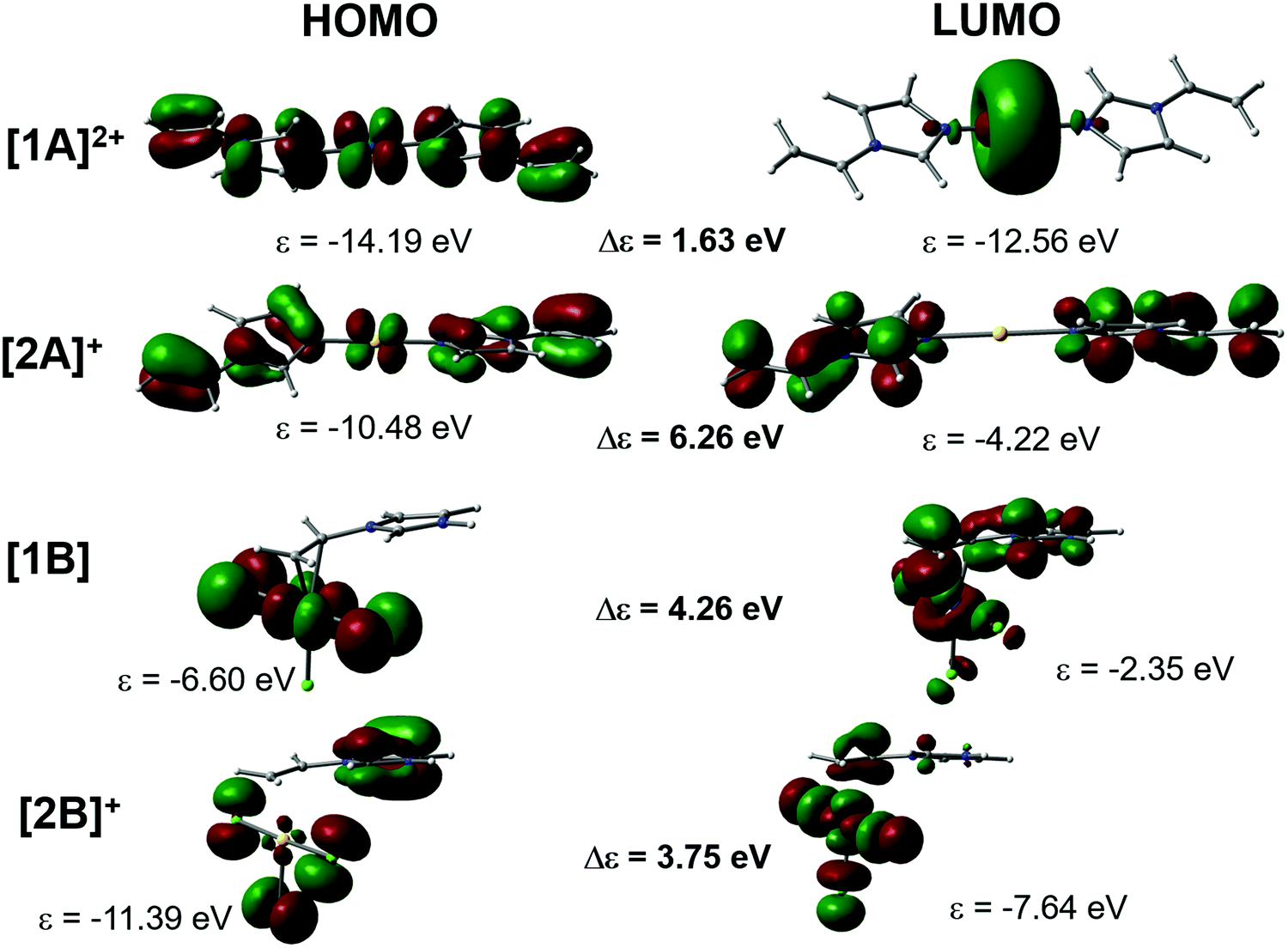 | ||
| Fig. 5 The appearance of the HOMO and LUMO in the N-coordinated cationic models [1A]2+ (Pt) and [2A]+ (Au), and the C,C-coordinated model [1B] (Pt) and [2B]+ (Au). | ||
There is a clear difference in the frontier molecular orbitals of the N-coordinated cations. With platinum, the strong involvement of the metal d orbitals in both the HOMO and LUMO stabilizes their energy, leading to a small energy gap. In contrast, the LUMO of the gold cation is expanded over the two ligands without contribution from the metal center, which destabilizes strongly the LUMO energy and leads to a large energy gap and hence to very stable coordination. The opposite is true in the CC coordination mode B, where the LUMO of the Au complex concentrates over the MX orbitals, again stabilizing LUMO energy, whereas in the Pt complex the LUMO is expanded mostly on the ligand π orbitals. According to the frontier orbital energies, platinum would prefer coordination to the vinyl group and gold would prefer N-coordination of the 1-vinylimidazole, provided that the Au(III) center is reduced to Au(I). It should be noted that we also tested the side-on coordination with Au(I), and even though the stability of the product, Δε = 4.58 eV, was slightly larger than for Au(III), the preferred coordination mode would still be A. Experimentally, we tested also the reaction HAuCl4 + vinylimidazole in THF solution. The crystals formed showed again the structure of 3. The 1H NMR spectrum of the reaction mixture verified this, but two minor products were present, thus other coordination modes are possible for gold.
Experimental
Materials and methods
Commercially available reagents AuBr3 (99%, Alfa Aesar), HAuCl4·3H2O (Au 49.5%, Alfa Aesar), NaAuCl4·2H2O (99%, Sigma Aldrich), K2PtCl4 (99,9% Alfa Aesar) and 1-vinylimidazole (99%, Alfa Aesar) were used without purification. The organic solvents were dried using molecular sieves. Infrared spectra were measured from KBr pellets using a Shimadzu IRAffinity-1 Fourier transform infrared spectrophotometer in the range of 4000–400 cm−1. The elemental analysis was performed on a varioMICRO V1.7. The 1H NMR spectra were recorded on a Bruker Avance 400 MHz or Bruker AMX 400 MHz spectrometer. Raman spectra were recorded on a Renishaw inVia Raman Microscope with 514 nm excitation laser wavelength. Raman shifts ranging from 3200–100 cm−1 were collected.Crystal structure determination
The crystals of 1–4 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 150 K. The X-ray diffraction data were collected on a Bruker Kappa Apex II diffractometer using Mo Kα radiation (λ = 0.71073 Å). The APEX277 program package was used for cell refinements and data reductions. The structures were solved by direct methods using the SHELXS-201878 program with the WinGX79 graphical user interface. A numerical absorption correction (SADABS)80 was applied to all data. Structural refinements were carried out using SHELXL-2018.78The crystallization solvent in 2 was heavily disordered and could not be resolved unambiguously. The contribution of the missing solvent to the calculated structure factors was taken into account by using a SQUEEZE routine of PLATON.81 The missing solvent was not taken into account in the unit cell content.
The N–H hydrogen atoms in 1, 2 and 4 and O–H water hydrogen atoms in 1 were located from the difference Fourier map and constrained to ride on their parent atoms, with Uiso = 1.2–1.5Ueq (parent atom). All other hydrogen atoms were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95 Å, N–H = 0.88 Å and Uiso = 1.2Ueq (parent atom). The crystallographic details are summarized in Table 3.
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| a R 1 = Σ||Fo| − |Fc||/Σ|Fo|; wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. | ||||
| Empirical formula | C5H9Cl3N2O Pt | C10H12Br2Au2N4 | C5H7Cl4AuN2 | C5H7Br4AuN2 |
| f w (g mol−1) | 414.58 | 741.99 | 433.89 | 611.73 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
| a (Å) | 6.6036(13) | 7.3655(13) | 10.5561(4) | 7.8576(3) |
| b (Å) | 12.158(2) | 9.5071(16) | 8.2051(3) | 8.7084(3) |
| c (Å) | 12.513(2) | 14.007(2) | 12.2809(5) | 9.5630(3) |
| a (°) | 90 | 71.099(3) | 90 | 110.7660(10) |
| β (°) | 95.214(4) | 84.673(4) | 93.8790(10) | 92.5480(10) |
| γ (°) | 90 | 68.279(4) | 90 | 102.5670(10) |
| Z | 4 | 2 | 4 | 2 |
| ρ calc (Mg m−3) | 2.752 | 2.860 | 2.716 | 3.433 |
| μ(Mo Kα) (mm−1) | 14.780 | 21.634 | 14.818 | 25.884 |
| F(000) | 760 | 656 | 792 | 540 |
| θ range for data collection (°) | 3.351 to 40.255 | 3.269 to 28.998 | 2.463 to 29.994 | 2.742 to 32.203 |
| Refl. collected | 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 058 058 |
17834 |
14680 |
11784 |
| Independent refl. | 6022 [R(int) = 0.0229] | 4535 [R(int) = 0.0348] | 3092 [R(int) = 0.0186] | 4085 [R(int) = 0.0205] |
| GOOF on F2 | 1.037 | 1.086 | 1.057 | 1.048 |
| Final R indices [I > 2σ(I)]a | R 1 = 0.0176, wR2 = 0.0372 | R 1 = 0.0323, wR2 = 0.0757 | R 1 = 0.0148, wR2 = 0.0318 | R 1 = 0.0158, wR2 = 0.0354 |
| R indices (all data) | R 1 = 0.0221, wR2 = 0.0383 | R 1 = 0.0373, wR2 = 0.0777 | R 1 = 0.0230, wR2 = 0.0349 | R 1 = 0.0195, wR2 = 0.0365 |
| Largest diff. peak and hole (e Å−3) | 0.729 and −1.214 | 2.992 and −1.442 | 0.466 and −0.812 | 0.608 and −1.187 |
Computational details
All calculations were performed by applying the Gaussian 09 software package.82 The optimized geometry and simulated scaled infrared and Raman spectra of all the complexes were obtained by the PBE0 functional83 with the 6-311++G(d,p) basis set for non-metal atoms and the Def2-TZVPPD basis set84 for Pt and Au atoms.To obtain the electronic properties of the solid state structure of 2, we performed topological charge density analysis with the QTAIM (Quantum Theory of Atoms in Molecules)85 method, which allowed us to access the nature of the bonding via calculating different properties of the electron density at the bond critical points (BCPs). The analysis was done with the AIMALL program86 using the wavefunction obtained from the single point DFT calculations of an extended model cut from the crystal structure.
Synthesis of complexes 1–4
C, vinyl) 1437 cm−1. 1H NMR (400 MHz, d6-DMSO, 298 K, δ): 9.37 (H2, s), 8.19 (H5, s), 7.82 (H4, s), 7.69 (H6, m), 5.98 (H7, d) and 5.42 (H7, d) ppm. For isomer b: 8.63 (H2, s), 7.92 (H5, s), 7.33 (H4, s), 7.30 (H6, m), 5.75 (H7, d) and 5.15 (C7, d) ppm. 13C NMR (400 MHz, d6-DMSO, 298 K, δ): 138.0 (C2), 128.9 (C4), 120.8 (C6), 118.6 (C5), 108.5 (C7) ppm.
C, vinyl) 1640 cm−1. 1H NMR (400 MHz, d6-DMSO, 298 K, δ): 8.76 (H2, s), 8.01 (H5, s), 7.41 (H4, s), 7.18 (H6, m), 5.73 (H7, d) and 5.16 (H7, d) ppm. For minor isomer: 8.65 (H2, br), 7.92 (H5, br), 7.30 (H4, br), 7.11 (H6, m), 5.66 (H7, br) and 5.09 (H7, br) ppm. 13C NMR (400 MHz, d6-DMSO, 298 K, δ): 139.9 (C2), 130.0 (C4), 129.2 (C6), 118.4 (C5), 106.3 (C7) ppm.
From NaAuCl4. NaAuCl4·2H2O (56.1 mg, 0.141 mmol) was dissolved in 5 M HCl (5 ml) and one drop of vinylimidazole was added. Yield: 34.5 mg, 56%. Anal. calcd for AuCl4N2C5H7: C, 13.84; H, 1.63; N, 6.46. Found: C, 14.04; H, 1.60; N, 6.35%. Mp 141–142 °C. IR (KBr): ν(N–H) 3254, ν(CC, vinyl) 1640 cm−1. 1H NMR (400 MHz, d6-DMSO, 298 K, δ): 9.36 (H2, s), 8.18 (H5, s), 7.81 (H4, s), 7.31 (H6, m), 5.97 (H7, d) and 5.41 (H7, d) ppm. 13C NMR (400 MHz, d6-DMSO, 298 K, δ): 134.8 (C2), 128.9 (C4), 120.9 (C6), 118.6 (C5), 108.6 (C7) ppm.
C, vinyl) 1640 cm−1. 1H NMR (400 MHz, d6-DMSO, 298 K, δ): 9.10 (H2, s, 1H), 8.23 (H5, s), 7.81 (H4, s), 7.34 (H6, m), 5.99 (H7, d) and 5.41 (H7, d) ppm. 13C NMR (400 MHz, d6-DMSO, 298 K, δ): 134.9 (C2), 128.9 (C4), 120.9 (C6), 118.7 (C5), 108.7 (C7) ppm.
Conclusions
1-Vinylimidazole has different coordination modes towards platinum and gold. Reaction conditions were found to have a major effect on the obtained crystalline products, which was attributed to the protonation/deprotonation of the 1-vinylimidazole free nitrogen in acidic/basic medium. Under the acidic conditions, the first complex of this ligand with the vinyl group coordinated to platinum metal was structurally characterized. On the other hand, the protonated ligand did not directly coordinate to gold, but formed halide salts with [AuX4]− as the counter anion. Under the basic conditions, the gold complex was observed as an ion pair with cation [AuL2]+ and anion [AuBr2]− connected via aurophilic interactions.According to the DFT calculations, the reduction of the Au(III) to Au(I) center leads to the formation of a very stable cationic N-coordinated complex with 1-vinylimidazole. For platinum, the calculations predict that the side-on coordination of the vinyl group is energetically more favorable than N-coordination, which explains the structure of complex 1. The stability of the molecular models was fully consistent with the experimentally obtained structures, which were verified via X-ray diffraction and IR, Raman, and NMR spectroscopy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge grants of computer capacity from the Finnish Grid and Cloud Infrastructure (persistent identifier urn:nbn:fi:research-infras-2016072533).References
- M. Satterfield and J. S. Brodbelt, Inorg. Chem., 2001, 40, 5393–5400 CrossRef CAS PubMed.
- Z.-M. Wang, H.-K. Lin, S.-R. Zhu, T.-F. Liu and Y.-T. Chen, J. Inorg. Biochem., 2002, 89, 97–106 CrossRef CAS.
- M. A. Masood and D. J. Hodgson, Inorg. Chem., 1993, 32, 4839–4844 CrossRef CAS.
- I. Erden, N. Demirhan and U. Avciata, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2007, 36, 559–562 CrossRef.
- T. M. Handyside, J. C. Lockhart, M. B. McDonnell and P. V. S. Rao, J. Chem. Soc., Dalton Trans., 1982, 2331–2336 RSC.
- U. Sandbhor, P. Kulkarni, S. Padhye, G. Kundu, G. Mackenzie and R. Pritchard, Bioorg. Med. Chem. Lett., 2004, 14, 2877–2882 CrossRef CAS PubMed.
- A. R. Katritzky and C. W. Rees, Comprehensive heterocyclic chemistry: the structure, reactions, synthesis, and uses of heterocyclic compounds, Pergamon Press, 1984 Search PubMed.
- M. R. Grimmett, Imidazole and benzimidazole synthesis, Academic Press, 1997 Search PubMed.
- A. F. Pozharskiĭ, A. T. Soldatenkov and A. R. Katritzky, Heterocycles in life and society: an introduction to heterocyclic chemistry and biochemistry and the role of heterocycles in science, technology, medicine, and agriculture, Wiley, 1997 Search PubMed.
- E. G. Brown, Ring nitrogen and key biomolecules: the biochemistry of N-heterocycles, Kluwer Academic, 1998 Search PubMed.
- F. Zhou, H. Wang and Q. Dai, IOP Conf. Series: Earth and Environmental Science, 2018, 153, 052001 CrossRef.
- M. D. Green and T. E. Long, Polym. Rev., 2009, 49(4), 291–314 CrossRef CAS.
- S. Santanakrishnan and R. A. Huttchinson, Macromol. Chem. Phys., 2015, 214, 1140–1146 CrossRef.
- A. Pourjavadi, N. Safaie, S. H. Hosseini and C. Bennett, Appl. Organomet. Chem., 2015, 29(9), 601–607 CrossRef CAS.
- B. Singh and A. Kumar, Int. J. Biol. Macromol., 2018, 120(Part B), 1369–1378 CrossRef CAS PubMed.
- Y. Ren, J. Zhang, J. Guo, F. Chen and F. Yan, Macromol. Rapid Commun., 2017, 38(14), 1700151(1–7) CrossRef PubMed.
- M. Takafuji, S. Ide, H. Ihara and Z. Xu, Chem. Mater., 2004, 16(10), 1977–1983 CrossRef CAS.
- G. Aliev, Y. Li, V. N. Chubarev, S. A. Lebedeva, L. N. Parshina, B. A. Trofimov, S. S. Sologova, A. Makhmutova, M. F. Avila-Rodriguez, S. G. Klochkov, P. A. Galenko-Yaroshevsky and V. V. Tarasov, Int. J. Mol. Sci., 2019, 20, 2104 CrossRef CAS PubMed.
- L. N. Parshina, L. A. Grishchenko, M. Ya. Khilko, N. K. Gusarova and B. A. Trofimov, Dokl. Chem., 2016, 471, 360 CrossRef CAS.
- A. V. Artem’ev, N. K. Gusarova, I. Y. Bagryanskaya, Y. V. Gatilov and A. O. Sutyrina, Polyhedron, 2016, 111, 79–85 CrossRef.
- S. J. Pang, J. Su and Q. Lin, Acta Crystallogr., 2007, E63(7), m2369 Search PubMed.
- K. S. Pedersen, M. Sigrist, M. A. Soerensen, A. L. Barra, T. Weyhermueller, S. Piligkos, C. A. Thuesen, H. Mutka, H. Weihe, R. Clérac and J. Bendix, Angew. Chem., 2014, 53(5), 1351–1354 CrossRef CAS PubMed.
- K. Kurdziel and T. Glowiak, J. Coord. Chem., 2002, 55(3), 327–334 CrossRef CAS.
- H. Lv, L. Zhu, Y. Q. Tang and J. M. Lu, Appl. Organomet. Chem., 2014, 28(1), 27–31 CrossRef CAS.
- E. S. Domnina, V. N. Voropaev, G. G. Skvortsova, M. V. Sigalov, T. K. Voropaeva and G. S. Muraveiskaya, Koord. Khim., 1983, 9, 1101 Search PubMed.
- W. H. Li, H. N. Chen, G. H. Li and F. Q. Liu, Acta Crystallogr., 2007, E63(2), m519–m520 Search PubMed.
- F. Q. Liu, R. X. Li, S. X. Li, C. Q. Li and G. Y. Liu, Acta Crystallogr., 2007, E63(9), m2437 Search PubMed.
- I. Sterkhova, V. Smirnov, A. Artemev and B. Trofimov, CSD Commun., 2014, 1031424 Search PubMed.
- Y. P. Tong, G. T. Luo, J. Zhen, Y. Shen and H. R. Liu, Cryst. Growth Des., 2013, 13(2), 446–454 CrossRef CAS.
- F.-Q. Liu, R.-X. Li, S.-X. Li, L.-S. Sun and G.-Y. Liu, Acta Crystallogr., 2007, E63, m2441 Search PubMed.
- T. Li and Z. Xing, Z. Kristallogr. – New Cryst. Struct., 2019, 234, 363–635 CAS.
- J. Zhao, Acta Crystallogr., 2008, E64, m1321 Search PubMed.
- H. Yilmaz, S. Gördük and Ö. Andaç, Inorg. Chim. Acta, 2017, 469, 154–163 CrossRef.
- F. Sen, R. Sahin, Ö. Andaç and M. Tas, Acta Crystallogr., 2012, E68, m1045 Search PubMed.
- S. Hamamci Alisir, N. Dege and R. Tapramaz, Acta Crystallogr., 2019, C75, 388 Search PubMed.
- A. A. Kashaev, E. A. Zel'bst, M. P. Demidov, M. P. Yu, L. Frolov, N. N. Chipanina, E. S. Domnina and G. G. Skvortsova, Koord. Khim., 1978, 4, 785 CAS.
- Z. Zhang, Y. Zhang, Z. Li, N. Jiao, L. Liu and S. Zhang, Eur. J. Inorg. Chem., 2018, 981 CrossRef CAS.
- A. H. Pedersen, M. Julve, J. Martínez-Lillo, J. Cano and E. K. Brechin, Dalton Trans., 2017, 46, 16025 RSC.
- H. Yilmaz, Ö. Andaç and S. Gördük, Polyhedron, 2017, 133, 16–23 CrossRef CAS.
- R. X. Li, S. X. Li, Q. Y. Wu, G. Y. Liu and F. Q. Liu, Acta Crystallogr., 2007, E63(12), m2874 Search PubMed.
- G. Y. Liu, H. N. Chen, F. Q. Liu, S. X. Li, R. X. Li and S. Y. Huang, Chin. J. Inorg. Chem., 2007, 23(6), 1085–1088 CAS.
- R. Agarwala, K. A. Azam, R. Dilshad, S. E. Kabir, R. Miah, M. Shahiduzzaman, K. I. Hardcastle, E. Rosenberg, M. B. Hursthouse and K. M. A. Malik, J. Organomet. Chem., 1995, 492(2), 135–144 CrossRef CAS.
- R. Yang, Y. Sun, D. Zhao and L. Guo, J. Coord. Chem., 2003, 56(13), 1169–1177 CrossRef CAS.
- M. Kubiak and J. Kuduk-Jaworska, Acta Crystallogr., 1986, C42, 1703 CAS.
- M. P. Shaver, C. M. Vogels, A. I. Wallbank, T. L. Hennigar, K. Biradha, M. J. Zaworotko and S. A. Westcott, Can. J. Chem., 2000, 78, 568 CrossRef CAS.
- C. Hu, J. Huster and U. Englert, Z. Kristallogr., 2003, 218(11), 761–765 CAS.
- K. Hiraki, N. Ochi, Y. Sasada, H. Hayashida, Y. Fuchita and S. Yamanaka, J. Chem. Soc., Dalton Trans., 1985, 873–877 RSC.
- G. Jia, D. W. Meek and J. C. Gallucci, Organometallics, 1990, 9, 2549–2555 CrossRef CAS.
- J. N. Coalter III, W. E. Streib and K. G. Caulton, Inorg. Chem., 2000, 39, 3749–3756 CrossRef PubMed.
- M. L. Buil, M. A. Esteruelas, E. Goni, M. Oliván and E. Oñate, Organometallics, 2006, 25, 3076–3083 CrossRef CAS.
- L. Zhang, L. Dang, T. B. Wen, H. H.-Y. Sung, I. D. Williams, Z. Lin and G. Jia, Organometallics, 2007, 26, 2849–2860 CrossRef CAS.
- K. A. Azam, D. W. Bennett, M. R. Hassan, D. T. Haworth, G. Hogarth, S. E. Kabir, S. V. Lindeman, L. Salassa, S. R. Simi and T. A. Siddiquee, Organometallics, 2008, 27, 5163–5166 CrossRef CAS.
- K. Burgess, H. D. Holden, B. F. G. Johnson, J. Lewis, M. B. Hursthouse, N. P. Walker, A. J. Deeming, P. J. Manning and R. Peters, J. Chem. Soc., Dalton Trans., 1985, 85–90 RSC.
- W.-Y. Wong and W.-T. Wong, J. Organomet. Chem., 1996, 513, 27–29 CrossRef CAS.
- P. Barrio, M. A. Esteruelas and E. Oñate, Organometallics, 2004, 23, 3627–3639 CrossRef CAS.
- B. Eguillor, M. A. Esteruelas, M. Oliván and E. Oñate, Organometallics, 2005, 24, 1428–1438 CrossRef CAS.
- M. A. Esteruelas, F. J. Fernández-Alvarez, M. Oliván and E. Oñate, J. Am. Chem. Soc., 2006, 128, 4596–4597 CrossRef CAS PubMed.
- H.-F. Klein, S. Camadanli, R. Beck, D. Leukel and U. Flörke, Angew. Chem., Int. Ed., 2005, 44, 975–977 CrossRef CAS PubMed.
- R. J. Foot and B. T. Heaton, J. Chem. Soc., Dalton Trans., 1979, 295–298 RSC.
- J. Müller, C. Hirsch and K. Ha, Z. Anorg. Allg. Chem., 2003, 629, 2180–2185 CrossRef.
- J. Navarro, E. Sola, M. Martín, I. T. Dobrinovitch, F. J. Lahoz and L. A. Oro, Organometallics, 2004, 23, 1908–1917 CrossRef CAS.
- G. R. Newkome, K. J. Theriot, B. K. Cheskin, D. W. Evans and G. R. Baker, Organometallics, 1990, 9, 1375–1379 CrossRef CAS.
- A. Zucca, L. Maidich, V. Carta, G. L. Petretto, S. Stoccoro, M. A. Cinellu, M. I. Pilo and G. J. Clarkson, Eur. J. Inorg. Chem., 2014, 2278 CrossRef CAS.
- M. Niazi and H. R. Shashavari, J. Organomet. Chem., 2016, 803, 82–91 CrossRef CAS.
- P. K. Monaghan and R. J. Puddephatt, Inorg. Chim. Acta, 1975, 15, 231–234 CrossRef CAS.
- M. A. Cinellu, F. Cocco, G. Minghetti, S. Stoccoro, A. Zucca and M. Manassero, J. Organomet. Chem., 2009, 694, 2949–2955 CrossRef CAS.
- L. N. Parshina and B. A. Trofimov, Russ. Chem. Bull. Int. Ed., 2011, 60, 601–614 CrossRef CAS.
- H. N. Adams and J. Strähle, Z. Anorg. Allg. Chem., 1982, 485, 65 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, Inc., USA, Hoboken, New Jersey, 6th edn, 2009 Search PubMed.
- N. Bélanger-Desmarais, M. Schütz and C. Reber, J. Phys. Chem. A, 2019, 123, 5574–5579 CrossRef PubMed.
- D. Michalska and R. Wysokinski, J. Phys. Chem. Lett., 2005, 403, 211–217 CrossRef CAS.
- D. Banfi and L. Patiny, Resurrecting and processing NMR spectra on-line, Chimia, 2008, 62(4), 280–281 CrossRef CAS , http://www.nmrdb.org.
- A. M. Castillo, L. Patiny and J. Wist, J. Magn. Reson., 2011, 209, 123–130 CrossRef CAS.
- C. Steinbeck, S. Krause and S. Kuhn, J. Chem. Inf. Comput. Sci., 2003, 43(6), 1733–1739 CrossRef CAS PubMed.
- V. K. Voronov, Int. J. Exp. Spectroscopic Tech., 2018, 3, 019 Search PubMed.
- V. K. Voronov, I. A. Ushakov and L. V. Baikalova, Russ. Chem. Bull., Int. Ed., 2005, 54, 1473–1476 CrossRef CAS.
- APEX2 – Software Suite for Crystallographic Programs, Bruker AXS, Inc., Madison, WI, USA, 2010 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., 2015, C71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, SADABS-2008/1 – Bruker AXS Area Detector Scaling and Absorption Correction, Bruker AXS, Madison, Wisconsin, USA, 2008 Search PubMed.
- A. L. Spek, Acta Crystallogr., 2015, C71, 9–18 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox and D. Gaussian, Gaussian 09, Revision C.01, Inc., Wallingford CT, 2009 Search PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- T. A. Keith, AIMAll (Version 12.06.03), TK Gristmill Software, Overland Park, KS, USA, 2003, http://aim.tkgristmill.com Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 1–4, figures of experimental and calculated Raman spectra of 1, as well as bond paths and bond critical points of the extended model of 2. The crystallographic data for compounds [PtCl3(Hvinylimidazole)]·H2O (1), [Au(vinylimidazole)2]+[AuBr2]− (2), [Hvinylimidazole]+[AuCl4]− (3), and [Hvinylimidazole]+[AuBr4]− (4) have been deposited. CCDC 1984394–1984397. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nj00845a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |