
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Salalen vs. thiolen: in the ring(-opening of epoxide and cyclic carbonate formation)†
Oliver J.
Driscoll
,
Jack A.
Stewart
,
Paul
McKeown
 and
Matthew D.
Jones
*
and
Matthew D.
Jones
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: mj205@bath.ac.uk
First published on 30th March 2020
Abstract
A range of Fe(III)-salalen and -thiolen–chloride complexes have been prepared and are shown to be active catalysts for the selective coupling of CO2 and cyclohexene oxide (CHO). The first Fe(III)-thiolen–acetate complex is also reported. The effect of the structure of the complex on activity is explored. An epoxide substrate scope is also provided, using the most active catalyst, as well as a study into the effect of co-catalyst equivalents on activity and selectivity.
Introduction
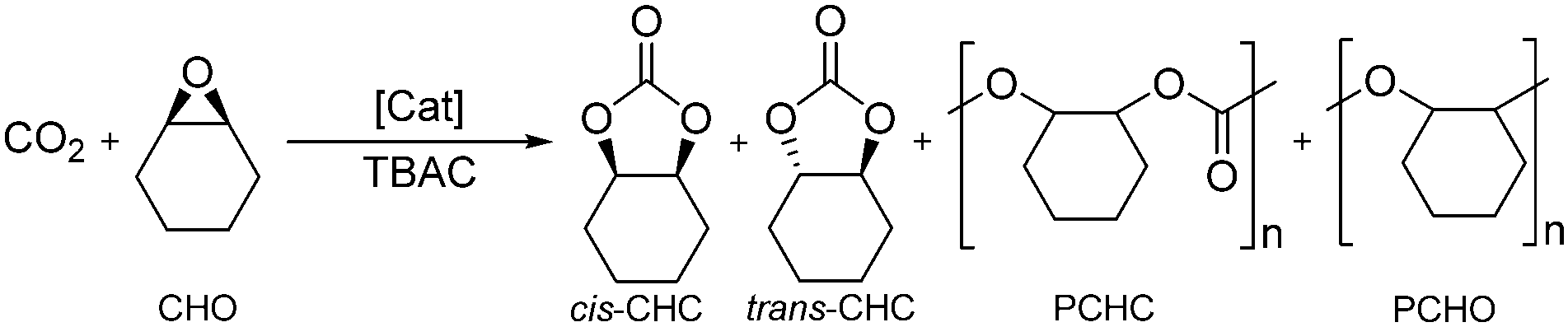
Two contemporary, rapidly growing areas of green chemistry involve the use of sustainable metal complexes, such as iron, for catalysis and catalytic transformations involving CO2, ideally utilising a waste stream as part of a ‘circular economy’ approach.1–12 The coupling of CO2, a renewable, abundant, cheap, non-toxic, ‘waste’ material, with reactive epoxides enables the formation of cyclic organic carbonates (COCs) and/or aliphatic polycarbonates (APCs).3,4,7–9,13–20 COCs are used in a range of applications including: high boiling, polar aprotic solvents; lithium-ion battery electrolytes; plasticisers; anti-foam additives; intermediates in both organic synthesis and industry and as monomers for copolymerisation with cyclic esters.7,8,14,15,21–23 This is a more sustainable method compared to the use of highly toxic phosgene employed in the traditional synthesis.8,14,16,17 Examples for the selective and effective Fe-mediated CO2/epoxide coupling reaction remain less prevalent than complexes containing metals such as Mg, Cr, Co, Zn and Al.15,22–33 This is despite the numerous benefits associated with iron such as high abundance, low toxicity and low commercial and industrial cost.1,2,5Della Monica et al. recently reviewed a variety of ligand classes complexed to iron such as salalen {ONNO} and phenoxy-thioether {OSSO}.7 Lamberti and co-workers reported the first example of a Fe(III)-salalen–chloride complex, together with Fe-salen and -salan complexes, for CO2/epoxide coupling.28 More recently, we reported examples of air-stable Fe(III)–acetate complexes with salalen, salan and salen ligands for the selective coupling of CO2 with a series of epoxides, predominantly cyclohexene oxide (CHO).34cis-Cyclohexene carbonate (cis-CHC) was formed as the exclusive product under mild, solvent-free conditions (80 °C, 10 bar CO2, 0.08 mol% metal loading). This is rare in the literature due to the bicyclic ring strain of CHC and the challenging nature of the internal CHO epoxide which imparts steric hindrance and regioselectivity issues associated with the possibility of four different products.
Della Monica, Capacchione and co-workers have reported a variety of mononuclear and dinuclear Fe(II/III)–bis(thioether)-phenolate {OSSO} complexes with two hemilabile sulfur donor atoms.7,21,35,36 Indeed, the application of the mononuclear Fe(III)–bis(thioether)-diphenolate complexes with an onium salt co-catalyst achieved very high TOFs and selectivity under mild conditions at 1 bar CO2 for a large range of internal and terminal epoxide substrates.21 For example, the conversion of propylene oxide (PO) to propylene carbonate (PC) achieved a TOF of 290 h−1 at 35 °C and 1 bar CO2. However, the cis-CHC product was not observed with CHO and a TBAC co-catalyst, instead polycyclohexene carbonate (PCHC) was selectively formed.21
We recently reported the synthesis of a range of Fe(III)-salalen–chloride {ONNO} and Fe(III)-thiolen–chloride {ONSO} complexes and their application to the isoselective ring-opening polymerisation (ROP) of rac-lactide.37,38 In particular, there are scarce examples of the ‘thiolen’, imine-thiobis(phenolate), family of ligands,39,40 and they have not been applied to CO2/epoxide coupling despite the similarities to Della Monica's bis(thioether)–phenolate {OSSO} complexes.
Herein, this work represents the application of previously synthesised Fe(III)-salalen–chloride and Fe(III)-thiolen–chloride complexes (Scheme 1) to the selective coupling of CO2 and epoxides. Furthermore, a new Fe(III)-thiolen–acetate complex is reported, applied and fully characterised through High-Resolution Mass-Spectrometry (HR-MS) and single crystal XRD. The most effective catalyst was tested with a range of epoxide substrates and co-catalyst concentrations. Modification of the ligand structure allows for comparison between donor atoms (N/S) as well as sterics and electronics.
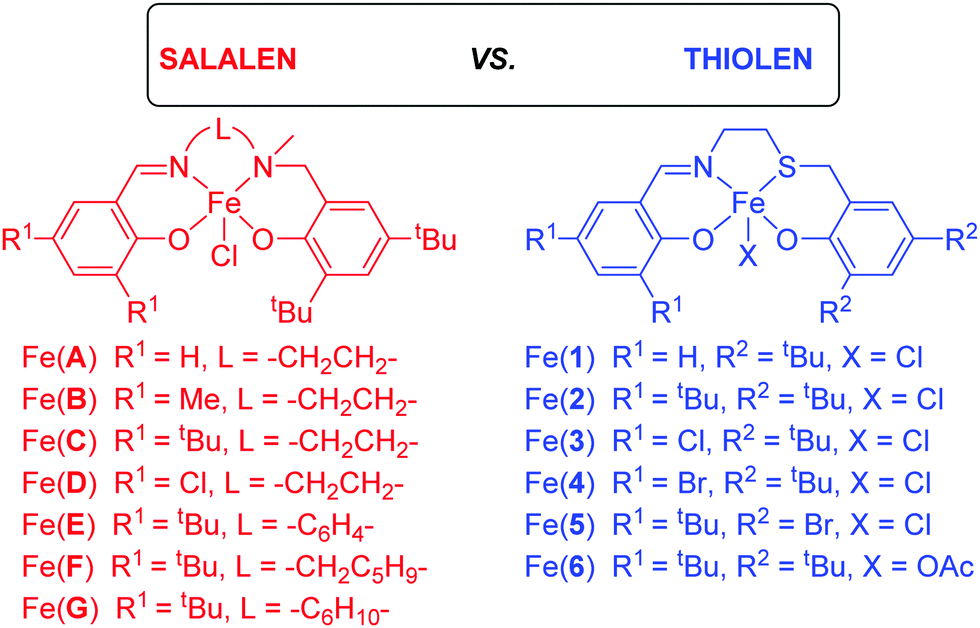 | ||
| Scheme 1 Salalen [Fe(A–G)], and thiolen [Fe(1–6)] complexes used in this study. | ||
Results and discussion
The Fe(III) chloride complexes bearing salalen [Fe(A–G)] and thiolen [Fe(1–5)] ligands39–44 have been previously synthesised (Scheme 1),37,38 and were screened for CO2/epoxide coupling with distilled CHO. Additionally, one Fe(III)-thiolen–acetate complex [Fe(6)] (Fig. 1) has been synthesised with the identity of the ML+ ion confirmed by ESI-MS. The solid-state structure of Fe(6) displays a distorted pseudo-trigonal bipyramidal geometry (τ5 = 0.63) with an equatorial site occupied by an acetate auxiliary group (Fig. 1). Although this geometry leans closer to a square-based pyramid compared with Fe(2) (τ5 = 0.78), bearing a chloride auxiliary group,38,45 it is similar to the salalen complex, Fe(C), and the acetate auxiliary group analogue {Fe(C), τ5 = 0.66 and ‘Fe(C)OAc’, τ5 = 0.65}.34,37 We have previously reported a carbonato-bridged dimer arising from the recrystallisation of Fe(2) in air and suggested that it could represent an intermediate in halide free CO2/epoxide coupling as reported by Muller and co-workers.38,46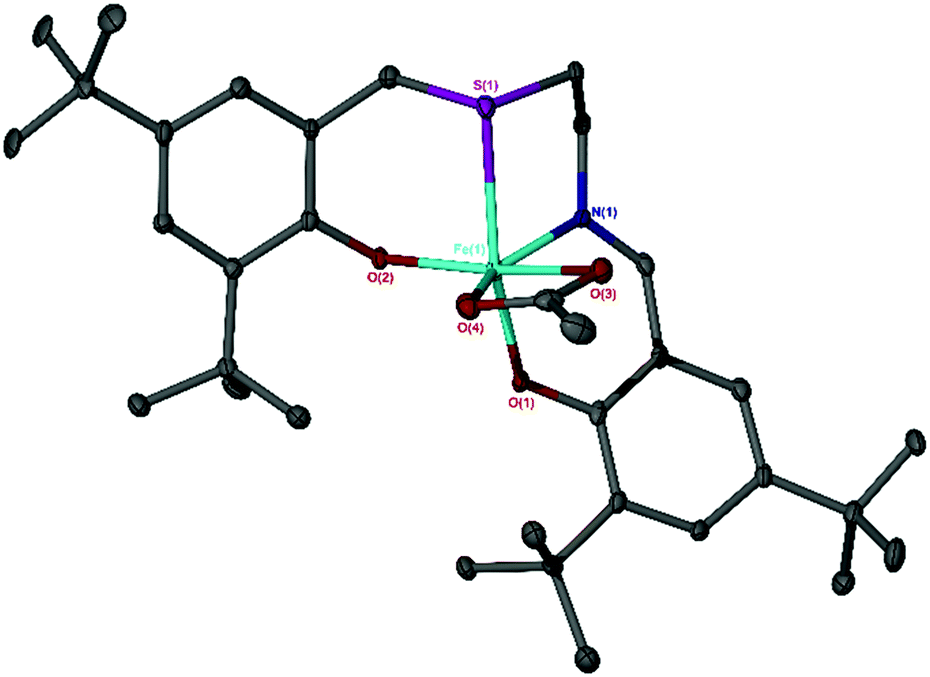 | ||
| Fig. 1 Solid state structure of Fe(6), ellipsoids shown at 30% probability level. Selected bond lengths (Å) and angles (°): Fe–O(1) = 1.887(3), Fe–O(2) = 1.873(3), Fe–N(1) = 2.077(4), Fe–S(1) = 2.5904(17); O(1)–Fe–N(1) = 87.64(14), O(2)–Fe–N(1) = 109.78(14), N(1)–Fe–S(1) = 81.07(12), O(1)–Fe–S(1) = 168.43(10), O(2)–Fe–C(acetate) = 130.40(17). | ||
The reactions were performed, using previously reported conditions,34,47 at a low catalyst loading of 0.08 mol% and 0.64 mol% tetrabutylammonium chloride (TBAC) co-catalyst loading in solvent-free conditions at 80 °C and 10 bar CO2 for 24 h (Table 1). The reaction mixtures changed colour from purple to red over the course of the reaction; this has previously been attributed to the formation of μ-oxo-bridged Fe(III) species.22,34
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Conv.a/% | Selectivity for cis-CHCa/% |
cis-CHC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans-CHC:PCHC:PCHO ratioa/% :trans-CHC:PCHC:PCHO ratioa/% |
TOFb/h−1 |
| Conditions: [Fe] catalyst (0.08 mol%, 1 eq.), TBAC (0.64 mol%, 8 eq.), CHO (5.0 mL), 10 bar CO2, 80 °C, 24 h.a Determined via1H NMR spectroscopy using the methine resonances of cis-CHC (δ = 4.66 ppm), trans-CHC (δ = 3.99 ppm) and PCHO (δ = 3.35 ppm).b TOF = [(Conv. %/100) × (100/0.08 mol%)]/24 h = [(Conv./100) × 1250]/24.c 120 °C, 6 h. | |||||
| 1 | FeCl3 | 44 | 83 | 83:10:16 |
23 |
| 2 | None | 43 | 83 | 83:0:0:17 |
22 |
| 3 | Fe(A) | 53 | >99 | >99:0:0:0 |
28 |
| 4 | Fe(B) | 47 | >99 | >99:0:0:0 |
28 |
| 5 | Fe(C) | 41 | >99 | >99:0:0:0 |
21 |
| 6 | Fe(D) | 44 | >99 | >99:0:0:0 |
23 |
| 7 | Fe(E) | 46 | >99 | >99:0:0:0 |
24 |
| 8 | Fe(F) | 48 | >99 | >99:0:0:0 |
25 |
| 9 | Fe(G) | 51 | >99 | >99:0:0:0 |
27 |
| 10 | Fe(1) | 47 | >99 | >99:0:0:0 |
24 |
| 11 | Fe(2) | 43 | >99 | >99:0:0:0 |
22 |
| 12 | Fe(3) | 54 | >99 | >99:0:0:0 |
28 |
| 13 | Fe(4) | 51 | 90 | 90:1:0:9 |
27 |
| 14 | Fe(5) | 60 | >99 | >99:0:0:0 |
31 |
| 15c | Fe(5) | 75 | 94 | 94:1:0:5 |
156 |
| 16 | Fe(6) | 44 | >99 | >99:0:0:0 |
23 |
Aliquots of the crude product mixtures were taken and analysed using 1H NMR spectroscopy to determine the conversion, product selectivity and TOF values through integration of the methine proton resonance signals for cis-cyclohexene carbonate (cis-CHC), trans-cyclohexene carbonate (trans-CHC), polycyclohexene carbonate (PCHC) and polycyclohexene oxide (PCHO) respectively relative to unreacted CHO. At 80 °C, the strained bicyclic cis-CHC product was formed exclusively for all complexes, with the exception of Fe(4), which is rare in the literature.
A range of techniques were applied to corroborate the findings from 1H NMR spectroscopy. ESI-MS of the crude product mixtures was used to confirm the presence of cyclic CHC. Although ESI-MS is unable to differentiate between the cis- and trans-CHC, 1H NMR spectroscopy identifies the methine resonance signals with cis-CHC appearing at δ 4.66 ppm and trans-CHC at δ 3.99 ppm, in agreement with that previously reported.23 The characteristic methine resonance associated with PCHC is reported to appear at δ 4.65 ppm which is very close to the desired cis-CHC product.23 To confirm the identity of the signal and that no polymer (PCHC) was formed, GPC analysis was carried out which showed no evidence of polymer formation. Therefore, 1H NMR spectroscopy, ESI-MS and GPC analysis confirmed that cis-CHC was formed as the exclusive product.
Control reactions were carried out and activity was observed using the FeCl3 synthetic metal precursor with TBAC co-catalyst (Table 1, entry 1). Although cis-CHC remained the major product, two additional products were present and identified as trans-CHC, the thermodynamic product, and PCHO, formed via polymerisation of the epoxide without CO2 insertion. This PCHO side-product was also produced when TBAC co-catalyst was used without any [Fe] catalyst (Table 1, entry 2). No reaction was observed when [Fe] catalyst was used without co-catalyst; the TBAC being required to open the epoxide and initiate the catalytic cycle (Table 2, entry 1).
| Entry | Eq. of TBAC | Conv.a/% | Selectivity for cis-CHCa/% |
cis-CHC:trans-CHC:PCHC:PCHO ratioa/% |
TOFb/h−1 |
|---|---|---|---|---|---|
| Conditions: [Fe] catalyst (0.08 mol%, 1 eq.), TBAC, CHO (5.0 mL), 10 bar CO2, 80 °C, 24 h.a Determined via1H NMR spectroscopy using the methine resonances of cis-CHC (δ 4.66 ppm), trans-CHC (δ 3.99 ppm) and PCHO (δ 3.35 ppm).b TOF = [(Conv. %/100) × (100/0.08 mol%)]/24 h = [(Conv./100) × 1250]/24. | |||||
| 1 | 0 | 8 | 0 | 0:0:0:>99 |
4 |
| 2 | 2 | 8 | 79 | 79:8:0:13 |
4 |
| 3 | 4 | 42 | 94 | 94:4:0:2 |
22 |
| 4 | 8 | 60 | >99 | >99:0:0:0 |
31 |
The increase of steric bulk at R1, for both salalen and thiolen complexes, decreased conversion {Fe(A), R1 = H, 53% vs. Fe(B), R1 = Me, 47% vs. Fe(C), R1 = tBu, 41% and Fe(1), R1 = H, 47% vs. Fe(2), R1 = tBu, 44% respectively} but maintained product selectivity at >99% for the cis-CHC. Thiolen complexes were generally more active than their salalen counterparts with the exception of the unsubstituted analogues {Fe(A), R1 = H, 53% vs. Fe(1), R1 = H, 47%}. This was proposed to be due to the softer sulfur donor atom being more labile allowing for more space around the Fe(III) centre for the binding of epoxide, enhancing the reactivity. Similar observations were made for the comparisons of Fe(III)-salalen and -thiolen complexes when applied to the ROP of lactide.37,38 Thiolen complex Fe(6), bearing tBu substituents and an acetate auxiliary ligand, displayed similar activity to the analogous chloride complex {Fe(2), X = Cl, 43% vs. Fe(6), X = OAc, 44%}. This is in contrast to the substituted Fe(III)-salalen–chloride/acetate complexes previously investigated, where the acetate complex was more active.34
For the salalen complexes, modifying the ethylene backbone by installing a planar phenyl ring increased activity {Fe(C), L = –CH2CH2–, 41% vs. Fe(E), L = –C6H4–, 46%} and this is further emphasised when using a more rigid aminopiperidine backbone {Fe(F), R1 = –CH2C5H9–, 48%}. Moving from the ethylene backbone to a sterically hindered cyclohexane ring had minimal effect on activity {Fe(A), R1 = H, L = –CH2CH2–, 53% vs. Fe(G), R1 = –C6H10–, 51%}. The trends observed with the modification of backbone structure are in agreement with the Fe(III)–acetate complexes previously reported.34
As expected, installing chloro-functionality at R1, thus increasing the Lewis acidity of the Fe(III) centre, increased conversion; the thiolen complex was moderately more active than the salalen {Fe(D), R1 = tBu, 44% vs. Fe(3), R1 = Cl, 54%}. The use of halide substitution was further explored using the Fe(III)-thiolen complexes. Modifying the R1 position from a chloride to a heavier bromide slightly decreased activity, presumably due to a decrease in Lewis acidity at the Fe(III) centre {Fe(3), R1 = Cl, 54% vs. Fe(4), R1 = Br, 51%}. Additionally, this was accompanied by a decrease in product selectivity to 90%. Transferring the bromide to the thio-phenolate moiety (R2) increased activity while maintaining the cis-CHC as the exclusive product {Fe(5), R2 = Cl, 60%}. Indeed, this complex was found to be the most active in this study with a TOF value of 31 h−1; a TOF value similar to that reported by Lamberti using Fe(III)-salalen, -salen, -salan–chloride complexes at 100 °C, 20 bar CO2 for 22 h using CHO.28 This value was increased to 156 h−1 by increasing the temperature to 120 °C and using a shorter reaction time of 6 hours (Table 1, entry 15). However, the formation of PCHO and trans-CHC decreased cis-CHC selectivity to 94%.
The effect of using different equivalents of TBAC co-catalyst was explored using the most active catalyst, Fe(5) (Table 2). As mentioned previously, no reaction was observed with 0 eq. of TBAC (Table 2, entry 1). Increasing TBAC concentration improved the activity and also had a positive effect on selectivity; a mixture of products was observed using 2 and 4 eq. of TBAC (Table 2, entries 2 and 3). Increasing the TBAC loading to 8 eq. formed cis-CHC as the only product >99% and gave the highest TOF value at 80 °C (31 h−1) (Table 2, entry 4). This underlines the importance of employing an elevated concentration of co-catalyst when targeting CHCs with this reaction. The formation of cis-CHC selectively has previously been reported and discussed in literature; the presence of co-catalyst and particularly excess co-catalyst favours the formation of carbonate anions after CO2 insertion. Therefore intramolecular ring-closing nucleophilic substitution, forming the cis-CHC product, is strongly favoured and potential competitive binding of further CHO and polymerisation is disfavoured (although PCHC is not observed in this study even when 0 eq. of TBAC was employed).23,47
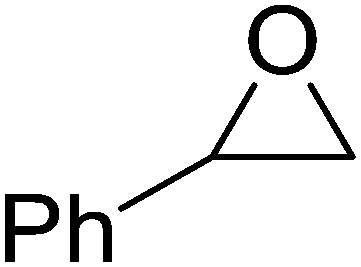
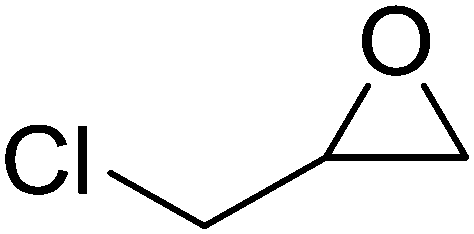
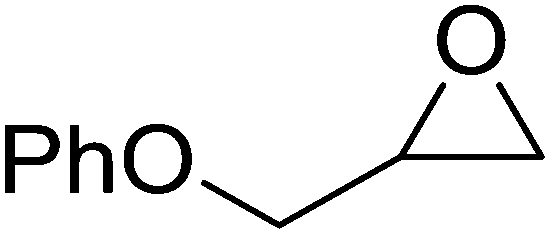
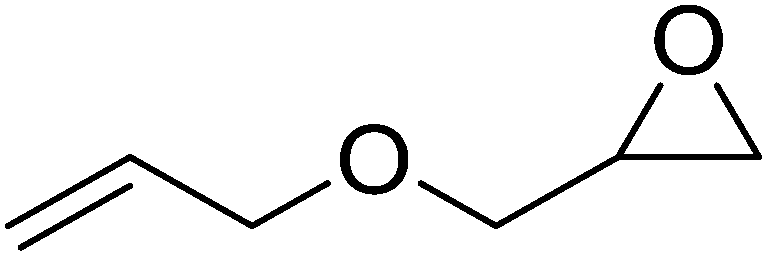
High functional group tolerance was observed when Fe(5) and TBAC co-catalyst were applied to a substrate scope of commercially available, terminal epoxides (Table 3). As with the CHO reactions, identification of the product and determination of selectivity was achieved through a combination of 1H NMR, GPC and ESI-MS. In all cases, the epoxide was selectively converted to the corresponding cyclic carbonate with moderate to high conversions. As expected, the least sterically hindered epoxide, propylene oxide (PO), was significantly more reactive than CHO (PO, TOF = 42 h−1, CHO, TOF = 31 h−1). Styrene oxide (SO), bearing a bulky phenyl group, was closer in reactivity to CHO, presumably as a result of similar steric profiles (TOF = 40 h−1). The addition of electron withdrawing groups increased reactivity, with the exception of epichlorohydrin (ECH) (TOF = 38 h−1). Allylglycidyl ether (AGE) gave the highest conversion in 24 h (93%); however, due to the solidification of phenoxymethyl ethylene carbonate, the reaction time with phenylglycidyl ether (PGE) was reduced to 18 h resulting in a higher TOF value (TOF = 64 h−1).
| Entry | Epoxide | Conv.a/% | Product selectivitya/% | TOFb/h−1 |
|---|---|---|---|---|
| Conditions: [Fe] catalyst (0.08 mol%, 1 eq.), TBAC (0.64 mol%, 8 eq.), CHO (5.0 mL), 10 bar CO2, 80 °C, 24 h.a Determined via1H NMR spectroscopy.b TOF = [(Conv.%/100) × (100/0.08 mol%)]/24 h = [(Conv./100) × 1250]/24.c Reduced reaction time of 18 h. | ||||
| 1 |
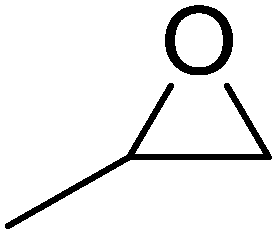
|
81 | >99 | 42 |
| 2 |
|
76 | >99 | 40 |
| 3 |
|
73 | >99 | 38 |
| 4c |
|
92 | >99 | 64 |
| 5 |
|
93 | >99 | 48 |
Conclusions
Fe(III) chloride complexes bearing a range of salalen and thiolen ligands were prepared and applied to the coupling of CO2 and CHO. Additionally, the first Fe(III)-thiolen–acetate was reported, fully characterised and also shown to be active for CO2/epoxide coupling. With the exception of Fe(4), all catalysts gave cis-CHC as the exclusive product at 80 °C. Thiolen complexes tended to be more active than their direct salalen counterparts and structure–activity-relationships were explored within the ligand classes. The most active catalyst, Fe(5), gave a TOF of 31 h−1 at mild conditions that could be increased to 156 h−1 when the temperature was raised to 120 °C. A substrate scope of terminal epoxides demonstrated high functional group tolerance for Fe(5) and the importance of using 8 eq. of TBAC co-catalyst was highlighted.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the University of Bath and the EPRSC (EP/L016443/1 and EP/P016405/1) for funding. We also thank Isabel Thomlinson for help with the graphical abstract. MC2 are also acknowledged for the use of their analytical facilities.References
- Iron Catalysis, ed. B. Plietker, Springer, Berlin, 2011, vol. 33 Search PubMed.
- M. Albrecht, R. Bedford and B. Plietker, Organometallics, 2014, 33, 5619–5621 CrossRef CAS.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163–4170 RSC.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- C. Darcel and J.-B. Sortais, Isr. J. Chem., 2017, 57, 1069 CrossRef CAS.
- F. Della Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2019, 361, 265–282 CrossRef CAS.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- A. J. Hunt, E. H. K. Sin, R. Marriott and J. H. Clark, ChemSusChem, 2010, 3, 306–322 CrossRef CAS PubMed.
- J. A. Martens, A. Bogaerts, N. De Kimpe, P. A. Jacobs, G. B. Marin, K. Rabaey, M. Saeys and S. Verhelst, ChemSusChem, 2017, 10, 1039–1055 CrossRef CAS PubMed.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 1–56 CrossRef PubMed.
- V. Besse, F. Camara, C. Voirin, R. Auvergne, S. Caillol and B. Boutevin, Polym. Chem., 2013, 4, 4545–4561 RSC.
- H. Zhang, H. Liu and J. Yue, Chem. Rev., 2014, 114, 883–898 CrossRef CAS PubMed.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. De Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef CAS.
- K. A. Andrea, T. R. Brown, J. N. Murphy, D. Jagota, D. McKearney, C. M. Kozak and F. M. Kerton, Inorg. Chem., 2018, 57, 13494–13504 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- M. A. Fuchs, T. A. Zevaco, E. Ember, O. Walter, I. Held and E. Dinjus, Dalton Trans., 2013, 42, 5322–5329 RSC.
- E. Fazekas, G. S. Nichol, M. P. Shaver and J. A. Garden, Dalton Trans., 2018, 47, 13106–13112 RSC.
- G. Bresciani, M. Bortoluzzi, F. Marchetti and G. Pampaloni, ChemSusChem, 2018, 11, 2737–2743 CrossRef CAS PubMed.
- D. Alhashmialameer, J. Collins, K. Hattenhauer and F. M. Kerton, Catal. Sci. Technol., 2016, 6, 5364–5373 RSC.
- M. Cozzolino, V. Leo, C. Tedesco, M. Mazzeo and M. Lamberti, Dalton Trans., 2018, 47, 13229–13238 RSC.
- F. Chen, N. Liu and B. Dai, ACS Sustainable Chem. Eng., 2017, 5, 9065–9075 CrossRef CAS.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- M. Taherimehr, J. P. C. C. Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS PubMed.
- C. J. Whiteoak, B. Gjoka, E. Martin, M. Mart, E. C. Escudero-ada, C. Zonta, G. Licini and A. W. Kleij, Inorg. Chem., 2012, 51, 10639–10649 CrossRef CAS PubMed.
- X. Sheng, L. Qiao, Y. Qin, X. Wang and F. Wang, Polyhedron, 2014, 74, 129–133 CrossRef CAS.
- O. J. Driscoll, C. H. Hafford-Tear, P. Mckeown, J. A. Stewart, G. Kociok-Köhn, M. F. Mahon and M. D. Jones, Dalton Trans., 2019, 48, 15049–15058 RSC.
- F. Della Monica, M. Leone, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, Mol. Catal., 2018, 460, 46–52 CrossRef CAS.
- F. Della Monica, A. Buonerba, V. Paradiso, S. Milione, A. Grassi and C. Capacchione, Adv. Synth. Catal., 2019, 361, 283–288 CrossRef CAS.
- O. J. Driscoll, C. K. C. Leung, M. F. Mahon, P. McKeown and M. D. Jones, Eur. J. Inorg. Chem., 2018, 5129–5135 CrossRef CAS.
- J. A. Stewart, P. Mckeown, O. J. Driscoll, M. F. Mahon, B. D. Ward and M. D. Jones, Macromolecules, 2019, 52, 5977–5984 CrossRef CAS.
- A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS.
- A. Stopper, K. Press, J. Okuda, I. Goldberg and M. Kol, Inorg. Chem., 2014, 53, 9140–9150 CrossRef CAS PubMed.
- E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469 RSC.
- E. L. Whitelaw, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 10004–10006 RSC.
- S. M. Kirk, G. Kociok-Köhn and M. D. Jones, Organometallics, 2016, 35, 3837–3843 CrossRef CAS.
- P. McKeown, M. G. Davidson, J. P. Lowe, M. F. Mahon, L. H. Thomas, T. J. Woodman and M. D. Jones, Dalton Trans., 2016, 45, 5374–5387 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- J. A. Castro-Osma, M. North, W. K. Offermans, W. Leitner and T. E. Müller, ChemSusChem, 2016, 9, 791–794 CrossRef CAS PubMed.
- M. Cozzolino, T. Rosen, I. Goldberg, M. Mazzeo and M. Lamberti, ChemSusChem, 2017, 10, 1217–1223 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1980459. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0nj00725k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |