DOI:
10.1039/D0NJ00143K
(Paper)
New J. Chem., 2020,
44, 9896-9902
Enhancing the mechanical and thermal properties of waterborne polyurethane composites with thermoset epoxy resin microspheres
Received
15th January 2020
, Accepted 13th May 2020
First published on 14th May 2020
Abstract
Epoxy resin, amine curing agent (IPDA) and polytetramethylene ether glycol (PTG2000) were used to prepare a new type of highly cross-linked and uniform thermoset epoxy resin microspheres (Ems) through reaction-induced phase separation. Its structure, particle size and thermal properties were characterized. The results showed that the epoxy resin microsphere was an active microsphere containing hydroxyl and amino groups on the surface, with certain monodispersity and a high glass transition temperature (Tg) (178.33 °C). Ems were further successfully used to prepare waterborne polyurethane/epoxy resin microsphere (WPU/Ems) composite films by in situ polymerization. When the composite film contained 4 wt% Ems, the tensile strength increased by 2.5-fold and the elongation-at-break hardly changed compared with pure WPU. Also, the thermal stability of the composite films was significantly improved with the addition of Ems. Fourier transform infrared spectroscopy and scanning electron microscopy results proved that WPU was grafted onto Ems with specific association. Ems were well dispersed in the WPU matrix and acted as chemical cross-linkers. Ems and WPU formed strong interfacial interaction through covalent grafting, which significantly improved the mechanical properties and thermal stability of the prepared composites.
1. Introduction
Homogeneous cross-linked polymer microspheres have long been used as colloidal crystals,1,2 coatings,3–5 drug controlled release microcapsules,6 templates of porous materials,7,8etc. At present, the methods of preparing polymer microspheres include emulsion polymerization, suspension polymerization, seed polymerization and dispersion polymerization. Emulsion polymerization and suspension polymerization are traditional methods to prepare polymer microspheres. However, the diameter of microspheres prepared by the former is relatively small (less than 0.7 μm), and the post-processing of microspheres was complex in operation. The diameter of the microspheres prepared by the latter is relatively large (100–1000 μm), and the monodispersity of the microspheres is difficult to control. Moreover the application of these methods is limited due to utilization of polymethyl methacrylate,9,10 polystyrene11 and polyester.12 Thermosetting polymers such as epoxy resin have better heat resistance and mechanical properties. In addition, epoxy resin is universal and easy to obtain. Therefore, in recent years, how to obtain uniform microspheres through epoxy chemistry has attracted great academic interest.
Thermosetting resin microspheres have a high research value and broad application prospects. Woo et al.13 synthesized micron-sized cross-linked epoxy resin microspheres in a DGEBA/DDS/PMMA curing reaction system by the reaction-induced phase separation method. Wu et al.14 prepared epoxy resin microspheres by mixing an OHT-T (self-made polyester)/epoxy resin/curing agent through the reaction-induced phase separation process with poor repeatability. Microspheres with controllable particle size and excellent monodispersity could be prepared by the reaction-induced phase separation method. However, when preparing Ems with thermoplastic resin as the continuous phase, most of the thermoplastic resins are polar materials with high viscosity and molecular interaction force in the melting state, and it is difficult to elute the Ems in the later stage.
Waterborne polyurethane (WPU) has been widely used in various industrial fields, including coatings, adhesives and inks because it shows a variety of excellent performances,15–19 which meet environmental protection needs. However, since WPU is a linear thermoplastic polymer with hydrophilic groups in the main chain, the mechanical properties (modulus and strength) of WPU are not as good as the traditional solvent-based polyurethanes, which limits its use as a high-performance material.
Nowadays, improving the performance of WPU has attracted great attention, for example, by hybridization with organic compounds such as starch20 or inorganic oxides such as attapulgite,21 clay,22,23 silica,24–26 titanium dioxide,27etc. However, a complex modification process was needed to solve the problem of the accumulation of inorganic oxide particles and the limitation of the doping level. In general, particles with multiple reactive vertex groups act as three-dimensional chemical crosslinkers of relatively high quality. Therefore, Ems may be an ideal structural block due to the versatility of reactive groups.
In this study, a novel type of highly cross-linked and uniform epoxy resin microspheres (Ems) was prepared. The influence of the molar ratio of amine/epoxy on the performance of Ems was investigated. Moreover, WPU/Ems composite films were successfully prepared by in situ polymerization. The thermal properties and mechanical properties of the prepared composites were evaluated.
2. Experimental
2.1 Materials
Bisphenol A epoxy resins (EP, E-51) with a viscosity of 2500 mPa s at 40 °C was supplied by Wuxi A Erzi Chemical Co., Ltd, Wuxi, China. The epoxy value (eq. per 100 g) of EP is 0.48–0.54. Isophorone diamine (IPDA, purity ≥99.7%) was supplied by BASF SE, Germany. Poly(tetramethylene glycol) (PTG, average Mn = 2000), isophorone diisocyanate (IPDI), dibutyltin dilaurate (DBTDL) and ethylenediamine (EDA) were obtained from Aladdin. Poly(propylene glycol) (PPG, Mn = 2000, Haian County National Power Chemical Co., Ltd) was distilled under vacuum at 100 °C for 2 h before use. Dimethylol propionic acid (DMPA, Perstorp) was dried at 80 °C for 24 h in a vacuum oven. Acetone was dried over 4 Å molecular sieves before use.
2.2 Synthesis of Ems
Epoxy resin and PTG were added into a 500 mL round-bottom three-necked flask in turn, and the mixture was stirred at 60 °C until it was dispersed evenly. IPDA was added to mix at high speed until the mixture was transparent and uniform. The mixture was poured into a preheated PTFE mould and cured in an oven. The curing condition was 85 °C/1 h + 150 °C/4 h. After curing, the mixture was washed with acetone to remove PTG. The solid powder obtained consisted of Ems, which are dried in a vacuum oven at 25 °C for 48 hours. The filtrate was rotated and steamed to recover acetone and PTG. All the details concerning the experimental conditions are reported in Table 1.
Table 1 Composition of EP/IPDA/PTG blends for Ems synthesis
| Amine/epoxy (A/E) |
Ep/pbwa |
IPDA/pbw |
PTG/pbw |
|
pbw is parts by weight and amine/epoxy (A/E) is the molar ratio.
|
| 0.4 |
2.0 |
0.69 |
10 |
| 0.7 |
2.0 |
0.56 |
10 |
| 1.0 |
2.0 |
0.46 |
10 |
| 1.3 |
2.0 |
0.30 |
10 |
| 1.6 |
2.0 |
0.17 |
10 |
2.3 Preparation of WPU/Ems composite films
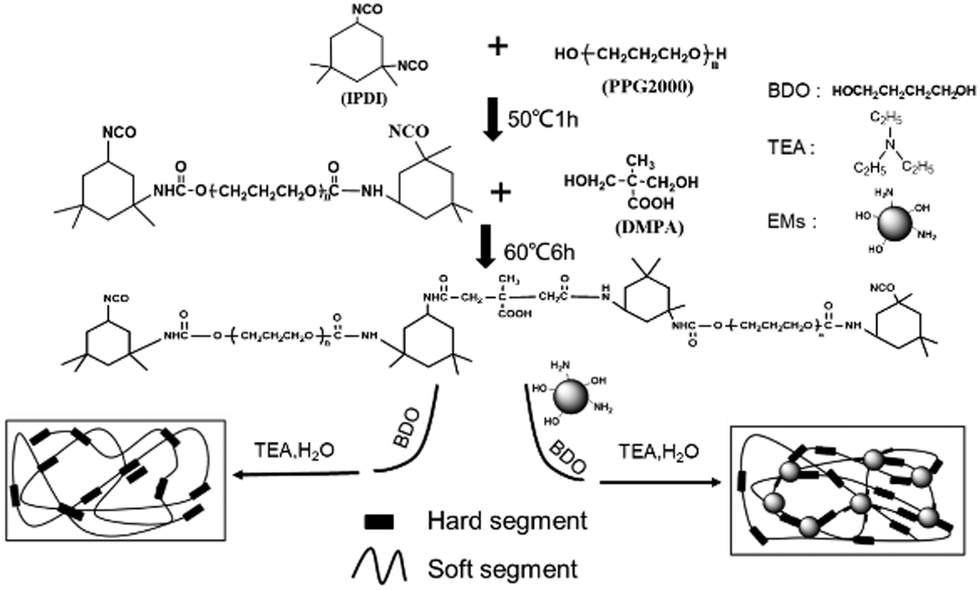
As shown in Scheme 1, the WPU/Ems composite films were prepared as follows. Firstly, IPDI was charged to a 250 mL round-bottom four-necked flask equipped with a mechanical stirrer, thermometer, condenser, and nitrogen inlet/outlet. Subsequently, PPG with drops of DBTDL was added dropwise and reacted at 50 °C for 1–2 hours. The isocyanate (–NCO) content was monitored during the course of the reaction using standard dibutylamine back titration. Upon reaching the theoretical –NCO value, DMPA 1,4-BDO, Ems and some DBTDL droplets were added and allowed to react at 60 °C until the –NCO content reached another theoretical value. After the prepolymer was cooled to room temperature, TEA was introduced into the system as a neutralizing agent and stirred for 1 hour to obtain a WPU/Ems dispersion. The obtained dispersion was transferred to a glass mold, dried at room temperature, and then dried in an oven at 60 °C for 8 hours to obtain a composite film. WPU composite films including 2, 4, 6, and 8 wt% Ems were fabricated through changing the loading of Ems and 1,4-BDO.
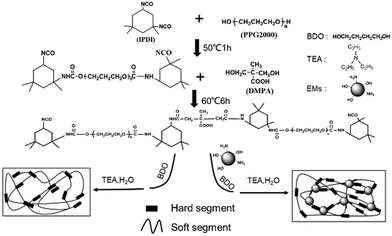 |
| | Scheme 1 Synthetic procedure to prepare WPU and WPU/Ems composites. | |
2.4 Characterization
Optical microscopy (OM).
The reaction-induced phase separation was recorded by optical microscopy (OM) conducted using a microscope equipped with a CCD camera and a hot stage (Axio Imager A2POL, Carl Zeiss AG, Germany).
Scanning electron microscopy (SEM).
The morphologies of Ems, WPU/Ems composites, and fracture surfaces of WPU/Ems composites were observed with scanning electron microscopy (SEM; S-4800, Hitachi Metals Group, Japan). The Ems particle size measurements were done using the open source software ImageJ for each sample; at least 300 particles were measured in order to obtain statistical meaning to the data.
Differential scanning calorimetry (DSC).
The glass transition temperature for Ems was assessed by differential scanning calorimetry using a differential scanning calorimeter (DSC 8000, PerkinElmer, USA). An empty aluminum crucible was used as a reference. The samples were heated at a rate of 10 °C min−1 over the range of 25 to 200 °C in a N2 atmosphere.
ATR-FTIR.
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR; Nicolet 6700, Thermo Fisher Scientific Co., Ltd, USA) analysis of the Ems and WPU/Ems was carried out in the range of 4000–400 cm−1. The test resolution was 2 cm−1, and the scanning time was 16 times.
Mechanical properties.
The mechanical properties of the WPU/Ems composite films, such as tensile strength and elongation at break, were tested by using a universal tensile tester (Instron 5967, USA), which was operated at a rate of 100 mm min−1 at room temperature.
Thermal properties.
Thermogravimetric analysis (TGA) was performed using a TGA/1/1100 SF (Mettler Toledo) instrument. The samples were heated from 20 °C to 800 °C at a heating rate of 10 °C min−1 in a N2 atmosphere (flow rate 20 mL min−1).
3. Results and discussion
3.1 Analysis of the phase separation process for Ems preparation
The reaction-induced phase separation process traced by an optical microscope during the isothermal curing of the blend at 85 °C for A/E = 1.0 is shown in Fig. 1. Phase separation followed the spinodal separation mechanism: the double continuous phase structure of the blend (Fig. 1a) was present at 38 min, which was the initial stage of reaction induced phase separation. As the epoxy resin curing reaction progressed, the molecular weight gradually increased. Under the action of increasing interfacial tension, the continuity of the epoxy resin phase was broken, and an irregular island structure was observed at 51 minutes (Fig. 1b). Prior to the curing gel point of the epoxy resin, the blend system had high flow and the microspheres formed in the epoxy-rich phase dispersed rapidly (Fig. 1c). Over time, after the epoxy resin reached the solidified gel point, the fluidity of the system was reduced and the Ems were finally fixed in the PTG phase with a uniform form.
 |
| | Fig. 1 OM images of the blend system at 85 °C for different curing times: (a) 38 min, (b) 51 min, (c) 64 min, and (d) 89 min. | |
3.2 Characterization of Ems
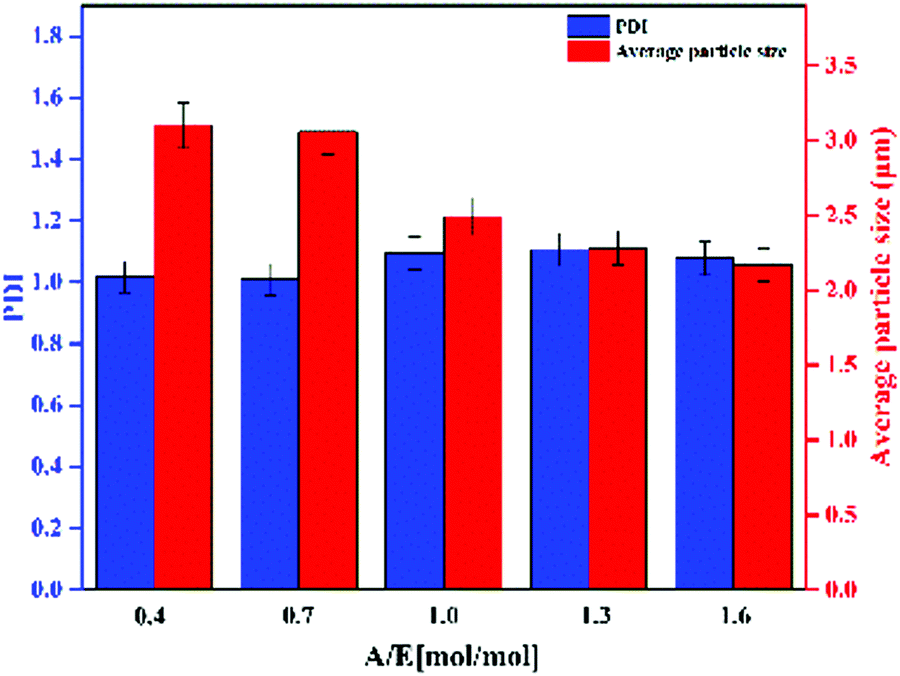
As shown in Fig. 2, Ems appeared to be single particles with a perfect spherical shape, with dimensions on the order of a few microns and with relatively low polydispersity. SEM was used to determine the particle size distribution. PDI and average particle size of Ems are presented in Fig. 3. At the same time, the photograph of Ems (A/E = 1.3) powders washed with acetone is also shown. With an increase of the A/E molar ratio, the average particle size of Ems decreased and the particle size distribution showed a non-uniform change. This phenomenon could be attributed to the epoxy system quickly reaching the gel point. The curing reaction accelerated due to the addition of excessive curing agent, which would promote the reaction-induced phase separation. Then the two-phase structure of the epoxy system and PTG was fixed in the shortest time, which was not conducive for the homogeneous growth of Ems.
 |
| | Fig. 2 SEM images of Ems with different molar ratios of the A/E in the blend and photograph of Ems (A/E = 1.3): (a) 0.4; (b) 0.7; (c) 1.0; (d and d′) 1.3; and (e) 1.6. | |
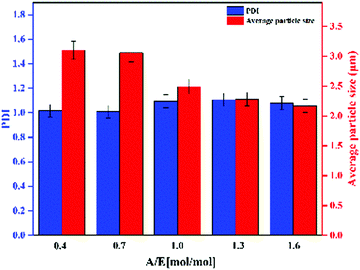 |
| | Fig. 3 PDI and average particle size of Ems. | |
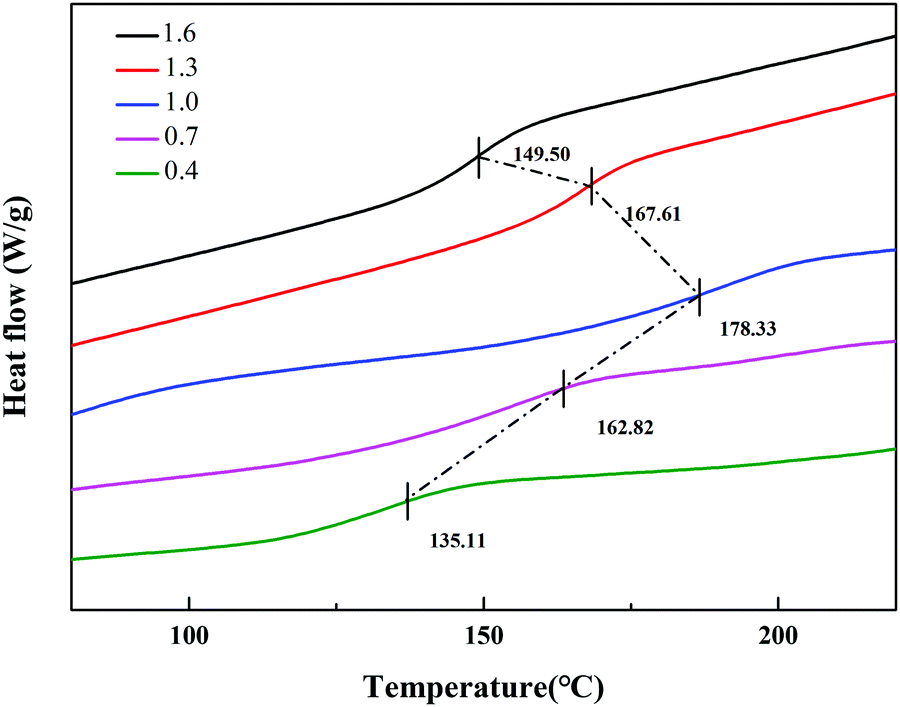
Under the same curing conditions, Fig. 4 shows the glass transition temperature (Tg) with different molar ratios of A/E. When A/E < 1, the density of cross-linking increased with an increase of the curing agent. This corresponded to the decreased free volume of the polymer and the degree of molecular chain activity was restricted. Besides, the average chain length between adjacent cross-linking points became shorter. In conclusion, cross-linking increased the glass transition temperature. When A/E > 1, the lower Tg resulted from an increased amount of curing agent, which formed oligomers with a more linear structure.28 However, these particles have an amino functionalized surface that arose from the residual excess IPDA. They could be further used to graft or covalently attach to an organic substrate.
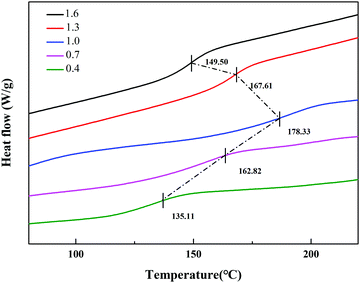 |
| | Fig. 4 DSC curves of different molar ratios of the A/E in the blend. | |
3.3 Grafting of WPU on the surface of Ems
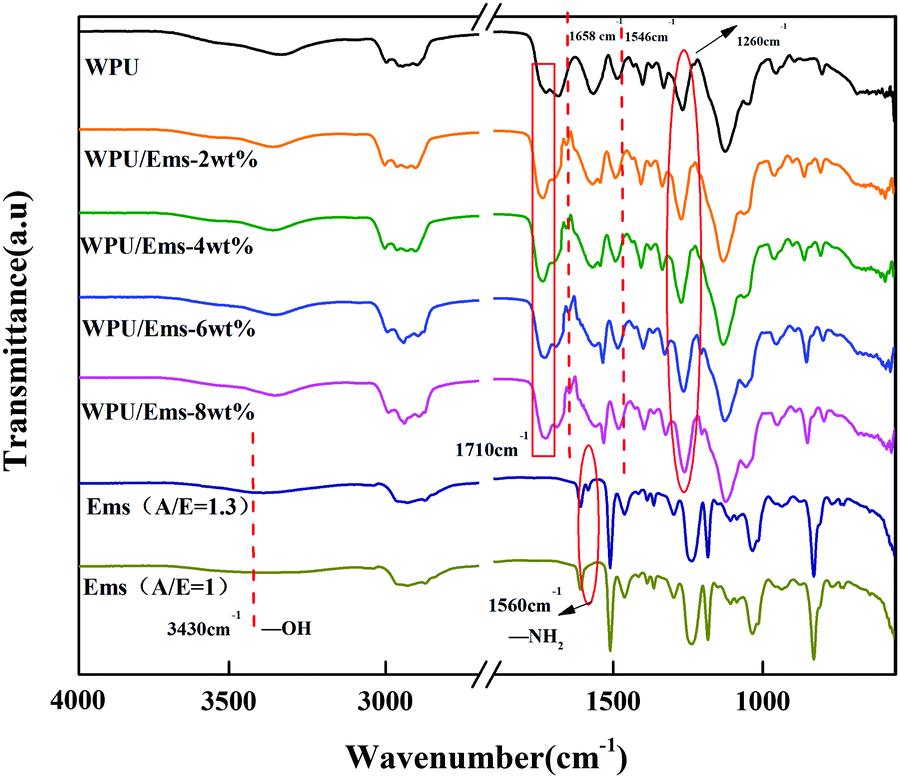
The chemical structures of WPU, Ems (A/E = 1.3) and WPU/Ems composite films were determined by FTIR spectroscopy. The typical FTIR spectra of WPU, Ems (A/E = 1.3) and WPU/Ems composites are presented in Fig. 5. Compared with Ems (A/E = 1), the peaks of Ems (A/E = 1.3) at 3430 cm−1 and 1560 cm−1 showed that the tensile vibration of the hydroxyl group and the tensile vibration of the first-order amino group in the microspheres were caused by ring opening of epoxy and residual excess IPDA, respectively.29 For WPU/Ems composites, the new absorption peak at about 1658 cm−1 was attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and another peak at about 1546 cm−1 was attributed to N–H and C–N.30 At the same time, the peak strength of the C–N peak at 1260 cm−1 and CO peak at 1710 cm−1 increased slightly, which might be caused by the chemical interaction between WPU and Ems. These results indicated that the hydroxyl group and the amino group of Ems and –NCO successfully reacted.
O, and another peak at about 1546 cm−1 was attributed to N–H and C–N.30 At the same time, the peak strength of the C–N peak at 1260 cm−1 and CO peak at 1710 cm−1 increased slightly, which might be caused by the chemical interaction between WPU and Ems. These results indicated that the hydroxyl group and the amino group of Ems and –NCO successfully reacted.
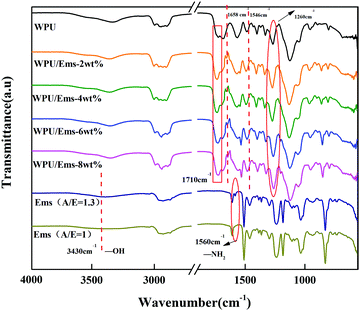 |
| | Fig. 5 FTIR spectra of WPU, Ems, and WPU/Ems composites. | |
The SEM image showed that Ems were spherical in shape, with diameters of approximately 1–3 μm (Fig. 6a). The preparation of WPU/Ems composites was carried out via polymerization of isocyanate on the ends of the WPU prepolymer with hydroxyl groups or primary amine groups on the Ems surface, as presented in Scheme 1. Through a Soxhlet extraction process, the residues (WPU/Ems composites) were obtained and the SEM image is given in Fig. 6b. In general, after the Soxhlet extraction process, WPU chains that are physically adsorbed on Ems would be dissolved in acetone and easily removed, leaving only the WPU covalently linked to Ems. Compared with the original Ems, Ems were uniformly wrapped with WPU, as it could be observed that Ems exhibited irregular spheres on the rough surface. The presence of WPU chains grafted on the Ems surface could be used to explain these phenomena.
 |
| | Fig. 6 SEM images of (a) Ems and (b) the residue obtained from the WPU/Ems composites after the Soxhlet extraction process. | |
3.4 Morphology of the WPU/Ems composite films
The WPU/Ems composite films were brittle in liquid nitrogen and its cross-sectional morphology was observed by field emission scanning electron microscopy to determine the dispersion of Ems in the WPU matrix. As shown in Fig. 7, Ems (white spheres) were irregularly distributed in the WPU matrix and as the Ems content increased, they were evenly distributed throughout the composite and were more compact. When the content of Ems was 8 wt%, they were uniformly dispersed in the WPU, and no agglomeration was observed. However, there was no obvious interface between Ems and the WPU matrix in all SEM images; this further illustrated the strong interaction between Ems and WPU.
 |
| | Fig. 7 SEM images of composite films: (a) WPU/Ems-2 wt%, (b) WPU/Ems-4 wt%, (c) WPU/Ems-6 wt%, and (d) WPU/Ems-8 wt%. | |
3.5 Mechanical properties of WPU/Ems composite films
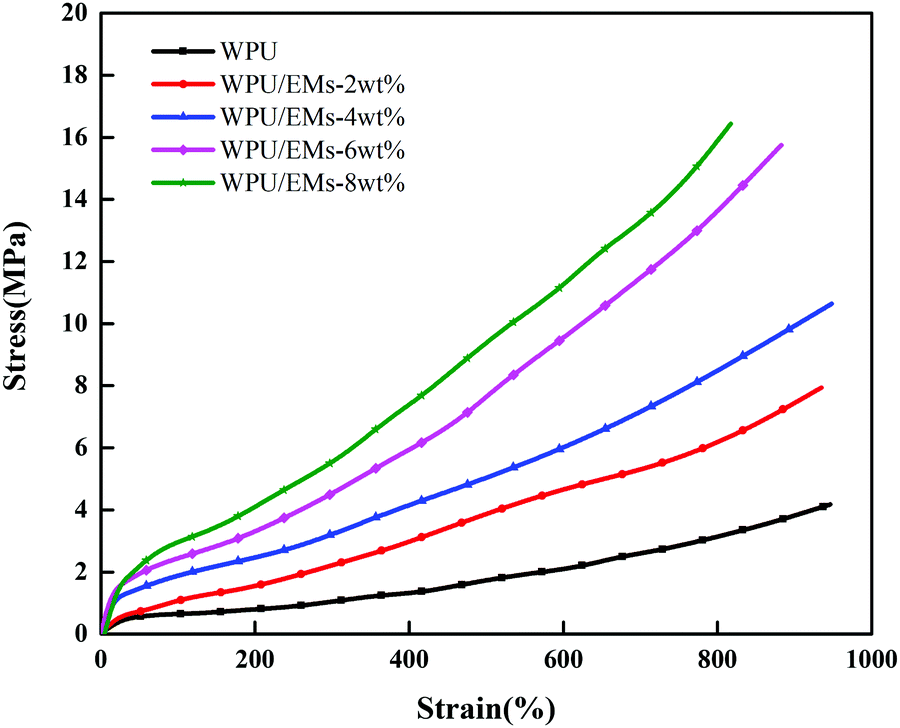
Ems could be used to enhance the mechanical properties of WPU due to their high crosslinking nature. Representative stress–strain curves for WPU and its composite films are shown in Fig. 8. With the addition of Ems, the tensile strength of the WPU films increased to 16.49 MPa. Compared with neat WPU, the tensile strength increased to 4.13 ± 0.42 MPa for WPU/Ems-2 wt% and further to 10.66 ± 0.93 MPa for WPU/Ems-4 wt%, where a more than 2.5-fold improvement had been achieved. Surprisingly, the values of the elongation-at-break for the WPU/Ems composite films had a subtle change in the range of 0–4 wt% Ems content, from 947 ± 42.4% to 952 ± 45.6%. In general, directly mixing thermoset matrices and rigid fillers provides improvements for modulus and tensile stress at the expense of ductility. The result above indicated that a small amount of Ems enhanced the tensile strength while at the same time the elongation at break of WPU films was maintained in this work.
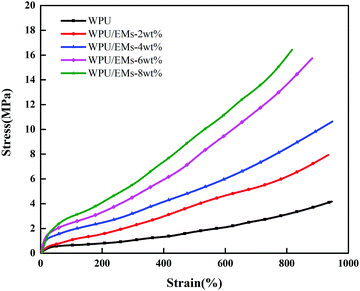 |
| | Fig. 8 Tensile testing curves of WPU and WPU/Ems composite films. | |
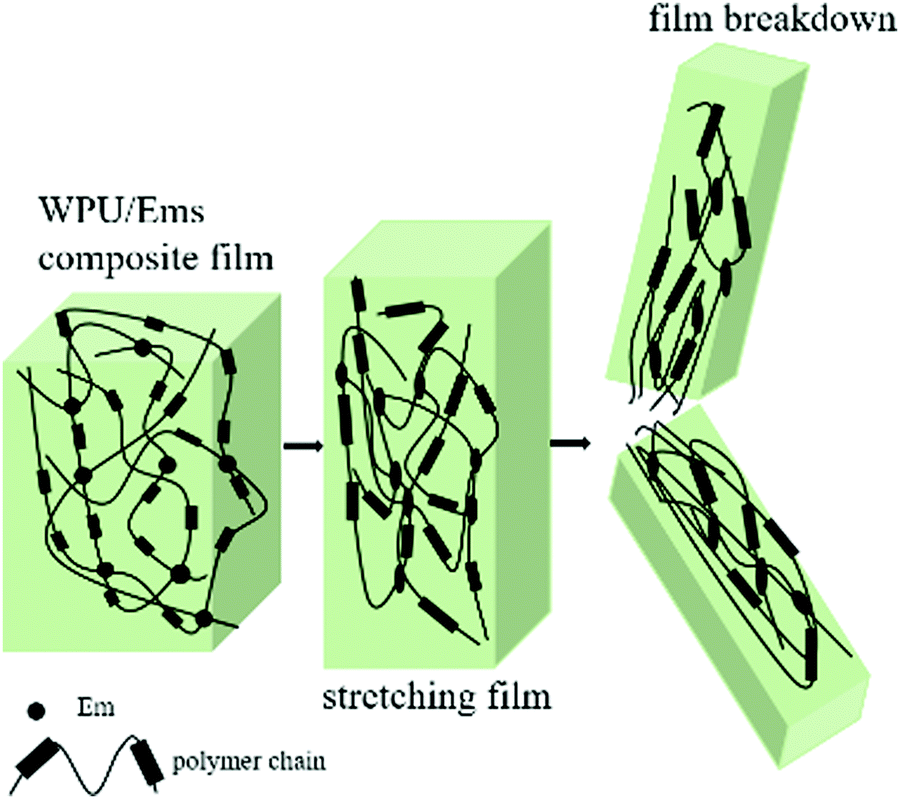
The tensile fracture surfaces of the deformed tensile specimens and the tensile specimen cryo-fractured along the draw direction are depicted in Fig. 9. Compared to a continuous and smooth fractured surface of pure WPU (Fig. 9a), the fractured surface of the WPU/Ems-4 wt% composite film exhibited a relatively coarse and irregular fractured morphology (Fig. 9b and d), indicating changes in the composite film microstructure. As can be seen in Fig. 9c, this morphology was desired for polymer toughening since this deformation of Ems could absorb and dissipate a large amount of energy during tensile tests. This was the important reason why WPU could maintain the elongation at break of the composite film after adding Ems. As shown in Fig. 9b and d, Ems were clearly wrapped with WPU, indicating a strong interaction between Ems and WPU to reinforce the mechanical properties of composite films. The Ems added showed higher intrinsic moduli and acted as cross-linkers, which would improve the cohesion force between hard segments and increase the crosslinking density of the WPU matrix to enhance the stiffness of composite films. Fig. 10 illustrates the main mechanism of enhancing and maintaining the toughness of WPU composite films with Ems. However, with the increase of the Ems content, the tensile strength of composite films significantly enhanced and its elongation at break showed an apparently decreasing trend. These changes could be attributed to a highly cross-linked network structure formed during polymerization, which caused a negative impact on the mechanical properties of the composite films.
 |
| | Fig. 9 SEM images of fracture surfaces of composite films: (a) WPU and (b and d) WPU/Ems-4 wt%, (c) the surface of WPU/Ems-4 wt% of tensile specimen cryo-fractured along the draw direction. | |
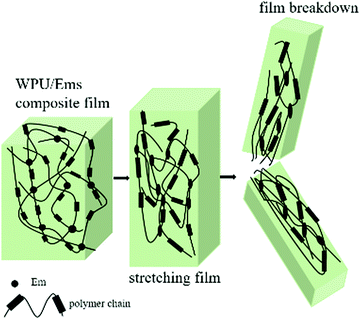 |
| | Fig. 10 Illustration of fracture mechanisms in the WPU/Ems composite film. | |
3.6 Thermal properties of WPU/Ems composite films
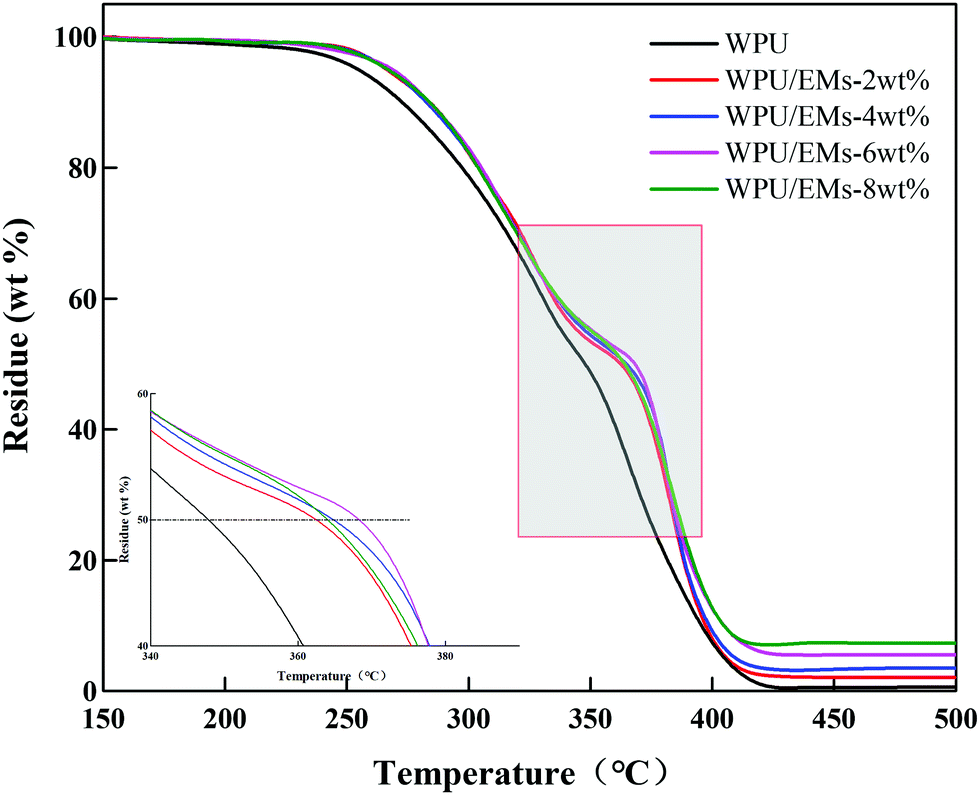
In general, neat WPU has relatively low thermal stability due to the presence of labile urethane bonds in the WPU.31 The TGA of the WPU/Ems composite films is shown in Fig. 11. It could be clearly seen that pure WPU decomposed at around 219 °C, and the thermal degradation of WPU/Ems at 6 wt% Ems was delayed by 18 °C. Moreover, it was interesting to find that adding Ems into WPU resulted in a shift of the Td(1/2) (the half-decomposition temperature) toward a higher temperature (about 20 °C). The increase could be explained by the fact that Ems were polymerized with WPU by in situ polymerization as a cross-linker, and the higher cross-linking density would enhance the thermal stability.32 In addition, the uniform dispersion of Ems in the constrained network structure and the strong interfacial interaction between Ems and the WPU matrix through covalent bonding could be due to the “tortuous path” that delays the escape of thermal decomposition volatiles.33 Obviously, these results further illustrated that the Ems were successfully wrapped with WPU chains.
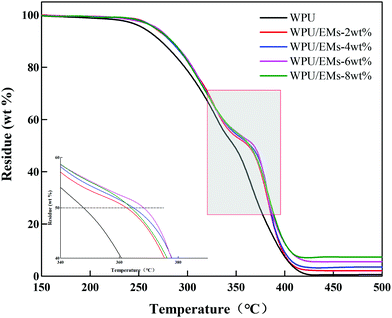 |
| | Fig. 11 TGA curves of WPU and WPU/Ems composite films. | |
4. Conclusions
In this work, highly cross-linked and uniform micro-sized Ems were synthesized through reaction induced phase separation of epoxy resin and IPDA in PTG. The effect of different molar ratios of the amine/epoxy of Ems was investigated. It was found that the molar ratio of the amine/epoxy played an important role in the size and Tg of particles. When the ratio of the amine/epoxy was greater than 1, Ems became active microspheres due to hydroxyl groups and primary amino groups were present on their surface, as indicated by FTIR. Thus, Ems could be adopted as preformed particulate enhancers to improve the stiffness and maintain the toughness of WPU/Ems composites. The partially pre-synthesized WPU chains were successfully grafted onto the Ems by a reaction between hydroxyl and primary amino groups on the surface of Ems with isocyanate (–NCO). Ems were strongly correlated with hard fragments of WPU by covalent attachment which was demonstrated by FTIR and SEM. Therefore, good dispersion and strong interfacial association between Ems and WPU were achieved. Meanwhile, the rigidity of Ems and the absorption of energy by deformation during the stretching process of WPU composites improved the mechanical properties of WPU. Comprehensive consideration of mechanical and thermal properties found that when the addition of Ems was 4 wt%, WPU/Ems composite films showed excellent performance. Overall, this study provided a simple and convenient method for preparing epoxy resin microspheres and a facile way to prepare WPU with good mechanical and thermal properties with the addition of Ems.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 20876070).
References
- S. Yuan, F. Ge, X. Yang and S. Guang, J. Fluoresc., 2016, 26, 2303–2310 CrossRef CAS PubMed.
- K. Manivannan, Y. S. Huang, B. R. Huang, C. F. Huang and J. K. Chen, Polymers, 2016, 8, 428 CrossRef PubMed.
- E.-J. Lee, B. J. Deka and A. K. An, J. Membr. Sci., 2019, 573, 570–578 CrossRef CAS.
- Q. Zhang, Z. Zhu, D. Shen and H. Yang, Colloids Surf., A, 2019, 569, 171–178 CrossRef CAS.
- R. H. Patel and P. M. Kapatel, Int. J. Polym. Anal. Charact., 2018, 24, 1–9 Search PubMed.
- C. Y. Wong, H. Al-Salami and C. R. Dass, Int. J. Pharm., 2018, 537, 223–244 CrossRef CAS PubMed.
- H. Liu, C. Zhou, X. Liu, Y. Xu, S. Geng, Y. Chen, C. Wei and C. Yu, J. Appl. Polym. Sci., 2018, 135, 46127 CrossRef.
- X. Yu, C. Zhang, Y. Ni and S. Zheng, J. Appl. Polym. Sci., 2013, 128, 2829–2839 CrossRef CAS.
- B. Peng, E. van der Wee, A. Imhof and A. van Blaaderen, Langmuir, 2012, 28, 6776–6785 CrossRef CAS PubMed.
- S. Omi, K. Katami, T. Taguchi, K. Kaneko and M. Iso, J. Appl. Polym. Sci., 1995, 57, 1013–1024 CrossRef CAS.
- I. Capek, Adv. Colloid Interface Sci., 2001, 92, 195–233 CrossRef CAS PubMed.
- H. L. Cong, B. Yu, L. L. Gao, B. Yang, F. Gao and H. B. Zhang, RSC Adv., 2018, 8, 2593–2598 RSC.
- H. Kun Hseih and E. M. Woo, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 2591–2598 CrossRef.
- Y. Wu, Y. Luo and J. Han, China Plast., 2008, 30 Search PubMed , in Chinese.
- F. Yu, L. Cao, Z. Meng, N. Lin and X. Y. Liu, Polym. Chem., 2016, 7, 3913–3922 RSC.
- Y. Xiao, X. Fu, Y. Zhang, Z. Liu, L. Jiang and J. Lei, Green Chem., 2016, 18, 412–416 RSC.
- D. K. Chattopadhyay and D. C. Webster, Prog. Polym. Sci., 2009, 34, 1068–1133 CrossRef CAS.
- A. M. Nelson and T. E. Long, Macromol. Chem. Phys., 2014, 215, 2161–2174 CrossRef CAS.
- J. Bullermann, S. Friebel, T. Salthammer and R. Spohnholz, Prog. Org. Coat., 2013, 76, 609–615 CrossRef CAS.
- G. Chen, M. Wei, J. Chen, J. Huang, A. Dufresne and P. R. Chang, Polymer, 2008, 49(7), 1860–1870 CrossRef CAS.
- L. Peng, L. Zhou, Y. Li, F. Pan and S. Zhang, Compos. Sci. Technol., 2011, 71, 1280–1285 CrossRef CAS.
- H. T. Lee and L. H. Lin, Macromolecules, 2006, 39, 6133–6141 CrossRef CAS.
- L. Liao, X. Li, Y. Wang, H. Fu and Y. Li, Ind. Eng. Chem. Res., 2016, 55, 11689–11699 CrossRef CAS.
- D. Sun, X. Miao, K. Zhang, H. Kim and Y. Yuan, J. Colloid Interface Sci., 2011, 361, 483–490 CrossRef CAS PubMed.
- N.-H. Park, J.-W. Lee and K.-D. Suh, J. Appl. Polym. Sci., 2002, 84, 2327–2334 CrossRef CAS.
- L. Zhang, H. Zhang and J. Guo, Ind. Eng. Chem. Res., 2012, 51, 8434–8441 CrossRef CAS.
- D. M. Wu, F. X. Qiu, H. P. Xu and D. Y. Yang, Plast., Rubber Compos., 2013, 40, 449–456 CrossRef.
- V. Ambrogi, S. Cosco, C. Carfagna, G. Cicala and A. Recca, Polym. Eng. Sci., 2006, 46, 1739–1747 CrossRef CAS.
- X. Wu, X. Yang, R. Yu, X.-J. Zhao, Y. Zhang and W. Huang, Polymer, 2018, 143, 145–154 CrossRef CAS.
- X. Yao, X. Qi, Y. He, D. Tan, F. Chen and Q. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 2497–2507 CrossRef CAS PubMed.
- K. K. Jena and K. V. S. N. Raju, Eur. Polym. J., 2007, 43, 1825–1837 CrossRef CAS.
- L. Zhang, H. Zhang and J. Guo, Ind. Eng. Chem. Res., 2012, 51, 8434–8441 CrossRef CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327–331 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a