
A new visible light triggered Arrhenius photobase and its photo-induced reactions†
Joonyoung F.
Joung‡
,
Jeeun
Lee‡
,
Joungin
Hwang
,
Kihang
Choi
* and
Sungnam
Park
 *
*
Department of Chemistry and Research Institute for Natural Science, Korea University, Seoul, 02841, Korea. E-mail: kchoi@korea.ac.kr; spark8@korea.ac.kr
First published on 18th December 2019
Abstract
Arrhenius photobases are of potential use for excited state hydroxide ion dissociation (ESHID), photo-induced pOH jump experiments, and base-catalyzed reactions. However, previously studied Arrhenius photobases have to be excited by UV light and undergo ESHID reactions only in protic solvents. These characteristics have become a disadvantage to their application in many fields of research. In this work, we have designed and synthesized a new Arrhenius photobase (NO2-Acr-OH), the ESHID reaction of which is readily triggered by visible light excitation. In contrast to previously studied Arrhenius photobases, NO2-Acr-OH undergoes ESHID reactions in protic solvents as well as in polar aprotic solvents. Solvent-dependent photo-induced reactions of NO2-Acr-OH are comprehensively studied by time-resolved fluorescence spectroscopy. Molecular designs for visible light triggered acridinol-based Arrhenius photobases with a large ΔpKb value are proposed.
I. Introduction
A sudden change in the pH of a solution can be made optically by using various photoacids which release protons upon electronic excitation because their pKa value is significantly decreased in their electronic excited state.1–16 As a counterpart of photoacids, quinolines,17,18 aminoanthraquinone,19 3-styrylpyridine,20 xanthone,21 curcumin,22 and Schiff bases23,24 have been reported as Brønsted-Lowry photobases which accept protons upon electronic excitation because their pKa value is increased in their electronic excited state. Based on their interesting photochemical properties, the photoacids and Brønsted-Lowry photobases have been extensively used for a model system to investigate the excited-state proton transfer reaction, proton solvation, ionic effect on the proton transfer reaction, and pH jump induced protein folding.1–14,17–24As opposed to the Brønsted-Lowry photobases, Arrhenius photobases are capable of releasing hydroxide ions (OH−) directly upon electronic excitation. Triphenylmethanol derivatives,25–28 9-phenyl-xanthen-9-ol derivatives,29,30 9-phenyl-acridin-9-ol derivatives,31,32 and fluoren-9-ol33,34 have been studied as Arrhenius photobases. The excited state hydroxide ion dissociation (ESHID) reactions of several Arrhenius photobases have been studied in water and acetonitrile (ACN) mixtures.30 In addition, Arrhenius photobases have been studied for colorimetric and fluorometric pH indicators in aqueous and alcohol solutions because their optical properties are sensitive to pH.29 Furthermore, light-driven pOH jump experiments have been carried out by using Arrhenius photobases.25 As such, Arrhenius photobases are of potential use for various fundamental studies and applications. However, for previously reported Arrhenius photobases, their ESHID reactions have been shown to be triggered by UV light excitation and are found to occur only in protic solvents. UV light excitation may hinder the potential use of currently available Arrhenius photobases (λmaxAbs < ∼310 nm) for applications in materials sciences because highly energetic UV photons can cause degradation of materials under study. Furthermore, biological applications of existing Arrhenius photobases may be limited due to their spectral interferences with amino acids such as tryptophan, tyrosine, and phenylalanine which have strong absorption at 260–310 nm.
In this work, we have designed and synthesized a new Arrhenius photobase, the photoreactions of which can be readily triggered by visible light excitation. Photo-induced reactions of the newly designed Arrhenius photobase NO2-Acr-OH shown in Fig. 1 are studied in various solvents in great detail.
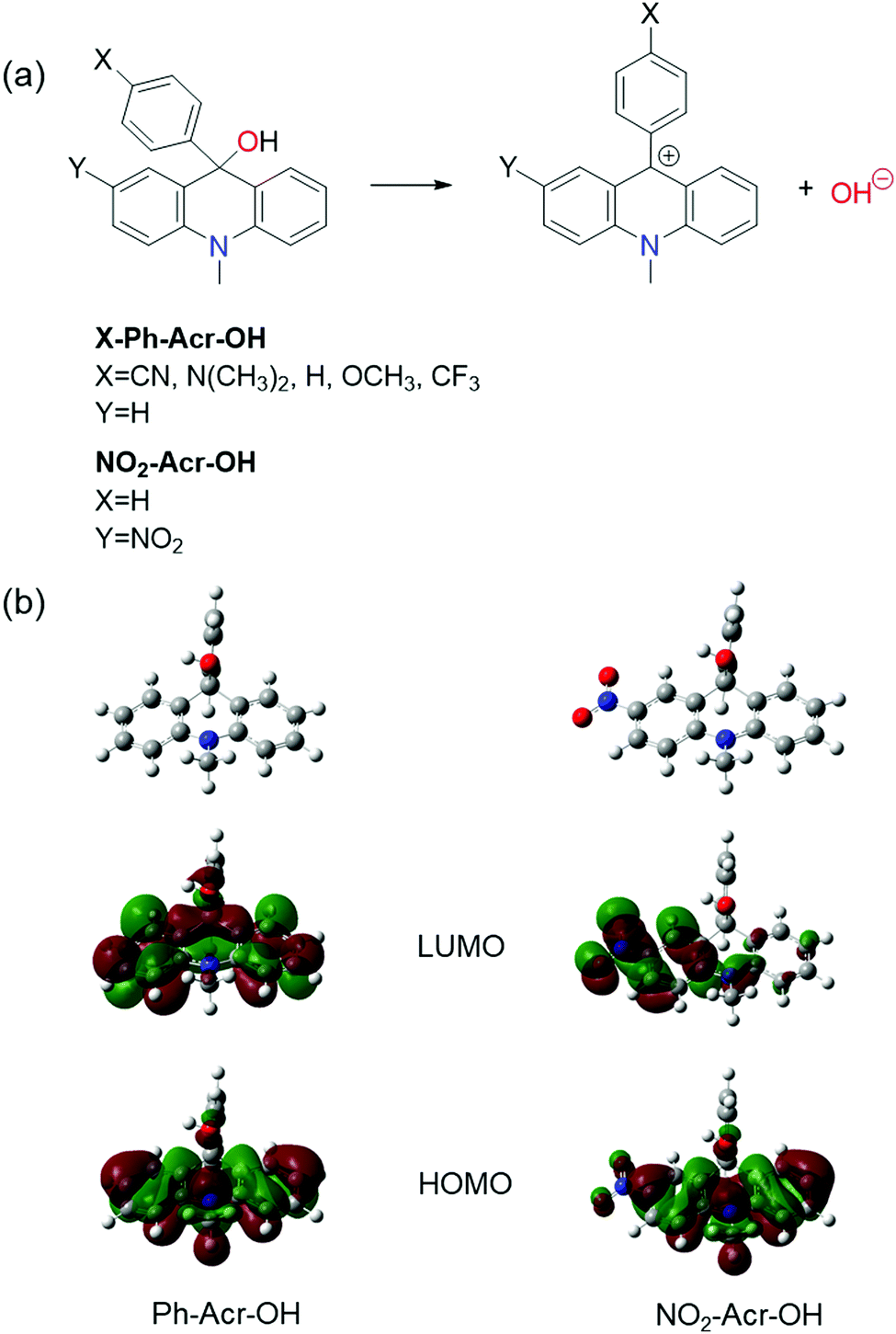 | ||
| Fig. 1 (a) Molecular structures of acridinol-based photobases and their ESHID reactions. (b) Frontier orbitals of Ph-Acr-OH and NO2-Acr-OH. NO2-Acr-OH exhibits more charge transfer (CT) character than Ph-Acr-OH. | ||
II. Results and discussion
II-A. Designing an Arrhenius photobase with visible light absorption
Acridinol-based photobases (X-Ph-Acr-OH, X = CN, CF3, H, OCH3, N(CH3)2 and Y = H) shown in Fig. 1 have been studied as Arrhenius photobases, the ESHID reactions of which can occur by absorption of UV light at ∼310 nm.32 The functional groups (X = CN, CF3, OCH3, and N(CH3)2) are introduced at the para position (X) of the 9-phenyl group in X-Ph-Acr-OH. DFT calculations indicate that the 9-phenyl group in X-Ph-Acr-OH is perpendicular to the acridine backbone as shown in Fig. 1(b) and it does not influence the conjugation of the acridine backbone significantly. Accordingly, the absorption spectra of acridinol derivatives (X-Ph-Acr-OH) are found to be not much different. However, the absorption and emission spectra of X-Ph-Acr+ are significantly influenced by the functional groups at the X position because the 9-phenyl group is placed in the same plane with the acridine backbone in X-Ph-Acr+ forming an extended conjugated structure. Based on the relationship between the optical properties and the molecular structures of acridinol-based photobases, we designed a new visible light triggered photobase by introducing a nitro group at the Y position of the acridine backbone shown in Fig. 1 and extending the conjugation of the acridine backbone. This newly designed Arrhenius photobase (NO2-Acr-OH) in various solvents is found to be readily excited by ∼400 nm light and undergoes ESHID reaction and intersystem crossing, which will be discussed below in great detail.II-B. Photophysical properties of NO2-Acr-OH in water
Fig. 2(a) displays the UV-visible absorption spectra of NO2-Acr-OH and NO2-Acr+ in water, which are significantly red-shifted due to the nitro group which increases the conjugation length of the acridine backbone. In addition, the nitro group is found to destabilize NO2-Acr+ because of its electron withdrawing ability which leads to a significant increase in the pKb value of NO2-Acr-OH. The pKb value of NO2-Acr-OH in water was theoretically estimated to be 7.69 by using eqn (S3) (ESI†) and DFT calculations, and was experimentally determined to be 7.63 at 25 °C as described in the ESI† (Fig. S1–S5).35–41 Obviously, the pKb value of NO2-Acr-OH is larger than that of X-Ph-Acr-OH shown in Fig. 1(a) which is in the range of 2.3 to 3.3.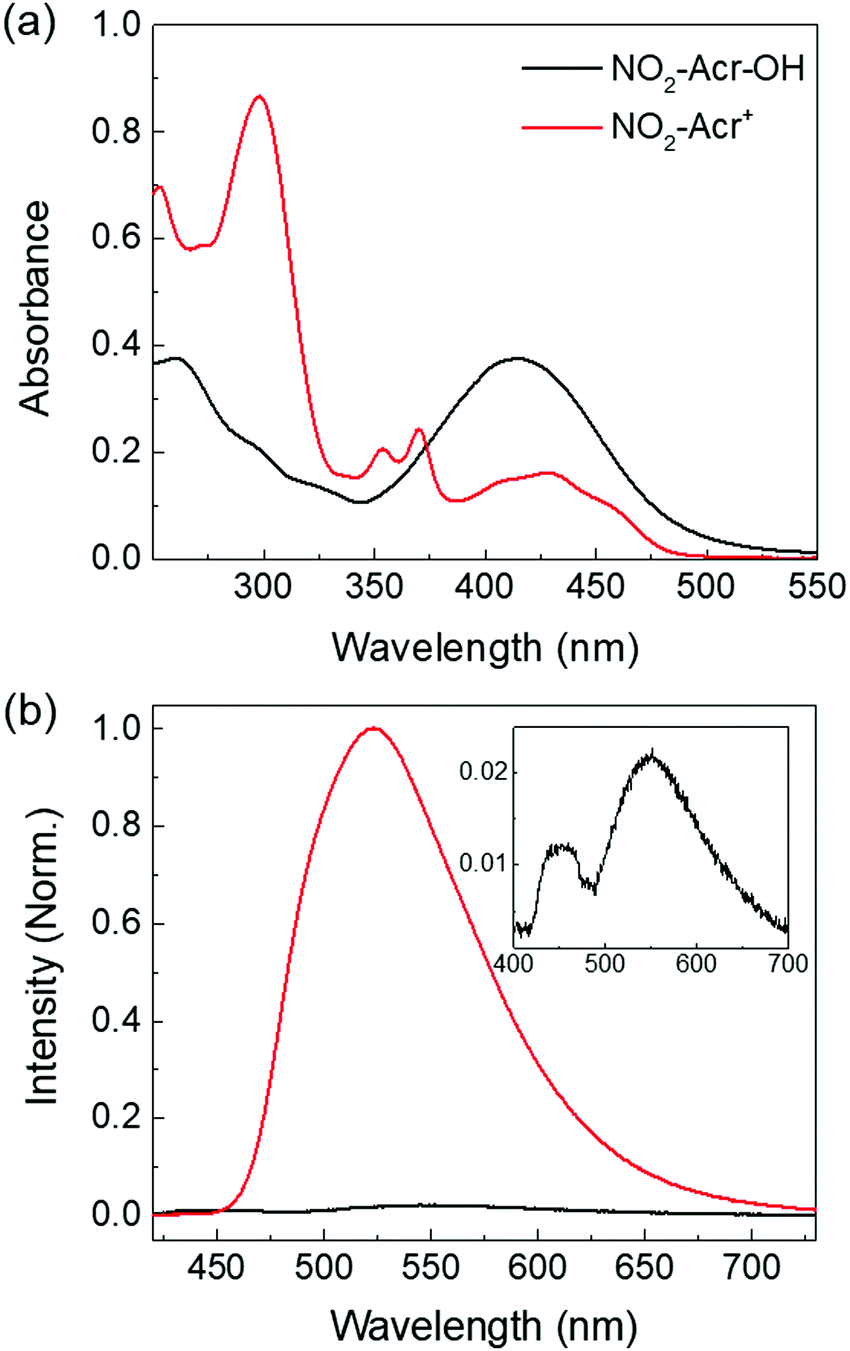 | ||
| Fig. 2 (a) UV-visible absorption spectra of NO2-Acr-OH and NO2-Acr+ in water (38 μM). (b) PL spectra of NO2-Acr-OH (black) and NO2-Acr+ (red) in water (λex = 375 nm) and their relative intensities are compared. The inset shows the emission spectrum of NO2-Acr-OH on a different intensity scale. | ||
Fig. 2(b) shows the photoluminescence (PL) spectra of NO2-Acr-OH and NO2-Acr+ in water. The PL quantum yields of NO2-Acr-OH and NO2-Acr+ in water are determined to be ΦPLQY = 0.003 and 0.010, respectively (Fig. S6, ESI†).42 The PL of NO2-Acr-OH in water is negligibly weak. Two peaks are observed in the PL spectrum of NO2-Acr-OH in water (see the inset of Fig. 2(b)). The PL peaks at ∼450 nm and ∼550 nm are found to result from photo-generated NO2-Acr+ and NO2-Acr-OH, respectively, as will be discussed in the next section. Because of such negligible fluorescence intensity, the photo-induced reaction of NO2-Acr-OH in water is unable to be studied by time-resolved fluorescence experiments.
II-C. Photo-induced reaction of NO2-Acr-OH in organic solvents
The UV-visible absorption and emission spectra of NO2-Acr-OH are measured in various solvents (dimethyl sulfoxide (DMSO), acetonitrile (ACN), methanol, ethanol, tetrahydrofuran (THF), and toluene) as shown in Fig. 3. The absorption peaks of NO2-Acr-OH are near 400 nm and are gradually red-shifted as the solvent polarity is increased (solvatochromic shift). The absorption spectra of NO2-Acr+ in organic solvents, which is prepared by the reaction of NO2-Acr-OH with sulfuric acid (NO2-Acr-OH + H+ → NO2-Acr+ + H2O), are displayed in Fig. 3(b). The ΔpKb (=pKb − pKb*) values of NO2-Acr-OH are estimated to be 2.9, 4.5, 4.4, and 4.7 in DMSO, ACN, methanol, and ethanol, respectively, by using the Förster cycle method.14 The emission spectra of NO2-Acr-OH (Fig. 3(c)) are solvent-dependent, which results from different photo-induced processes occurring in various solvents as will be presented in the next section. | ||
| Fig. 3 (a) UV-visible absorption spectra of NO2-Acr-OH in various solvents. (b) UV-visible absorption spectra of NO2-Acr+ in various solvents. (c) PL spectra of NO2-Acr-OH in various solvents (λex = 375 nm). | ||
To study the photo-induced reaction of NO2-Acr-OH in different solvents, we carried out time-resolved fluorescence (TRF) experiments by using the time-correlated single photon counting (TCSPC) method. Fig. 4 shows the TRF signals measured with NO2-Acr-OH in different solvents following excitation by 375 nm pulses. Note that the TRF signals measured at ∼400 nm are found to be readily quenched by molecular oxygen (O2) as shown in Fig. S7 (ESI†) and are attributed to the phosphorescence of NO2-Acr-OH. This feature has been commonly observed with previously studied Arrhenius photobases.30–34 However, the TRF signals measured at ∼600 nm, which are not quenched by O2 as shown in Fig. S8 (ESI†), result from the fluorescence of NO2-Acr-OH. Accordingly, to analyze the TRF signals of NO2-Acr-OH, the ESHID reaction and intersystem crossing (ISC) process are included in the kinetic model presented in Fig. 4(a).
 | ||
| Fig. 4 (a) Kinetic model for analyzing the TRF signals of NO2-Acr-OH in various solvents. TRF signals measured with NO2-Acr-OH in (b) DMSO, (c) ACN, (d) methanol, (e) ethanol, (f) THF, and (g) toluene (λex = 375 nm). The spectral components and time-dependent population decay of NO2-Acr-OH* (blue), NO2-Acr+* (red), and 3[NO2-Acr-OH*] (green), which are extracted from the global fitting analysis, are presented in the right and bottom panels, respectively. | ||
In the kinetic model shown in Fig. 4(a), kROH, kR+, and ktriplet are the rate constants for the radiative decay of NO2-Acr-OH*, NO2-Acr+*, and 3[NO2-Acr-OH*], respectively. The rate constants for ESHID and reverse ESHID reactions are kOH and krev, respectively, and kISC is the ISC rate constant. By using the global fitting analysis based on the kinetic model,9,43–45 the TRF signals of NO2-Acr-OH were fully analyzed and all the rate constants were extracted as summarized in Table 1. The individual contributions of NO2-Acr-OH*, NO2-Acr+*, and 3[NO2-Acr-OH*] to each TRF signal exhibit distinct spectral and temporal features as shown in Fig. 4.
| k OH (ns−1) | k rev (ns−1) | k ISC (ns−1) | k ROH (ns−1) | k R+ (ns−1) | k triplet (ns−1) | Φ OH | |
|---|---|---|---|---|---|---|---|
| a k triplet is determined by TRF experiments with NO2-Acr-OH in degassed solvents. b Φ OH = kOH/(kOH + kISC + kROH). | |||||||
| DMSO | 5.03 | 0.18 | 0.93 | 0.099 | 0.651 | 0.015 | 0.83 |
| ACN | 4.50 | 0.27 | 3.71 | 0.103 | 0.428 | 0.013 | 0.54 |
| Methanol | 10.63 | 0.05 | 3.40 | 0.247 | 0.542 | 0.012 | 0.74 |
| Ethanol | 8.19 | 0.05 | 4.53 | 0.167 | 0.534 | 0.016 | 0.64 |
| THF | — | — | 1.00 | 0.078 | — | 0.009 | — |
| Toluene | — | — | 3.20 | 0.091 | — | 0.012 | — |
The global fitting analysis of TRF signals reveals a few interesting and important features. First, the ESHID reaction of NO2-Acr-OH does not occur in weakly polar solvents (THF and toluene). In this case, the produced NO2-Acr+ may not be stabilized in THF (ε = 7.52) and toluene (ε = 2.38) with small dielectric constants (ε).37 Second, the ESHID reaction and the ISC process occur competitively in polar solvents but the ESHID reaction is relatively faster than the ISC process (kOH > kISC). Third, the ESHID reaction of NO2-Acr-OH is much faster in polar protic solvents (methanol and ethanol) than in polar aprotic solvents (ACN and DMSO). This implies that the hydroxide ion dissociation reaction of NO2-Acr-OH should occur via acid-catalytic processes. Lastly, the pKb* values of NO2-Acr-OH in various solvents were estimated by the rate constants, e.g., pKb* = −log10(kOH/krev), by assuming that the ESHID equilibrium was rapidly achieved in the electronic excited state. Subsequently, the pKb values were determined by using the Förster cycle method and the pKb* values (Table 2). The OH− dissociation rate (kOH) (Table 1) is shown to become larger as the pKb* value gets smaller, which has been explained by the free-energy-reactivity relationship in the literature.30,32 The quantum yield of OH− production indicates that the OH− dissociation in excited state is more favored than the ISC process in polar solvents.
| DMSO | ACN | Methanol | Ethanol | |
|---|---|---|---|---|
| pKb | 1.5 | 3.3 | 2.1 | 2.5 |
| pKb* | −1.4 | −1.2 | −2.3 | −2.2 |
Before this section is closed, it should be noted that acridinol-based photobases (X-Ph-Acr-OH, X = CN, CF3, H, CH3O, and (CH3)2N) in various solvents have been studied by Glusac and co-workers.31,32 They have found that X-Ph-Acr-OH in S1 state undergoes ESHID reaction and ISC process in alcohol but not in polar aprotic solvents (e.g., ACN). In addition, they have claimed that the ground state X-Ph-Acr+ is directly formed as a result of the ESHID reaction of X-Ph-Acr-OH. In contrast, our new NO2-Acr-OH is found to undergo ESHID reaction in both polar aprotic and protic solvents producing fluorescent NO2-Acr+. The nitro group introduced to the acridine backbone makes significant changes in the chemical and photophysical properties of acridinol-based Arrhenius photobases in terms of the absorption spectrum, pKb value, and solvent-dependent ESHID reaction.
III. Concluding remarks
A new Arrhenius photobase, NO2-Acr-OH, which was readily excited by visible light, was designed and synthesized and its photo-induced reactions were comprehensively studied in various solvents by using steady-state fluorescence and TRF experiments combined with DFT calculations. Unlike previously studied Arrhenius photobases, the ESHID reaction of NO2-Acr-OH was found to occur not only in protic solvents but also in polar aprotic solvents. The ESHID reaction was dependent on solvent polarity. The ESHID reaction was relatively fast in highly polar solvents and protic solvents but did not proceed in weakly polar solvents. In addition, NO2-Acr-OH was found to undergo ISC process in various solvents but the ISC process of NO2-Acr-OH was not significantly solvent-dependent. Note that NO2-Acr-OH in water exhibits weak fluorescence (ΦPLQY = 0.003) and undergoes ESHID reactions producing NO2-Acr+ whose fluorescence is negligibly weak as well. Therefore, NO2-Acr-OH is nearly a non-fluorescent Arrhenius photobase in aqueous solutions and can be used in pOH jump experiments minimizing its fluorescence interference with molecular systems under study.Visible light triggered photobases with large ΔpKb values are ideal for studying ESHID reactions in various molecular systems. NO2-Acr-OH absorbs visible light but exhibits a relatively small ΔpKb value. In the case of Ph-Acr-OH, functionalizing the para position of the 9-phenyl group is shown to increase the ΔpKb value while functionalizing the acridine backbone induces a redshift in the absorption of photobases. Accordingly, we will be able to develop various visible light triggered Arrhenius photobases with large ΔpKb values by introducing different functional groups at the para position of the 9-phenyl group and in the acridine backbone.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was supported by grants from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2019R1H1A2079968 and 2019R1A6A1A11044070 for S. P. and No. 20100020209 for K. C.).References
- E. Pines and D. Huppert, J. Phys. Chem., 1983, 87, 4471–4478 CrossRef CAS.
- S. Abbruzzetti, E. Crema, L. Masino, A. Vecli, C. Viappiani, J. R. Small, L. J. Libertini and E. W. Small, Biophys. J., 2000, 78, 405–415 CrossRef CAS PubMed.
- M. Rini, B. Z. Magnes, E. Pines and E. T. Nibbering, Science, 2003, 301, 349–352 CrossRef CAS PubMed.
- M. Rini, D. Pines, B. Z. Magnes, E. Pines and E. T. Nibbering, J. Chem. Phys., 2004, 121, 9593–9610 CrossRef CAS PubMed.
- N. Agmon, J. Phys. Chem. A, 2005, 109, 13–35 CrossRef CAS PubMed.
- D. B. Spry and M. D. Fayer, J. Chem. Phys., 2008, 128, 084508 CrossRef CAS PubMed.
- Y.-S. Lee, O.-H. Kwon, H. J. Park, J. Franz and D.-J. Jang, J. Photochem. Photobiol., A, 2008, 194, 105–109 CrossRef CAS.
- M. J. Cox, B. J. Siwick and H. J. Bakker, ChemPhysChem, 2009, 10, 236–244 CrossRef CAS PubMed.
- J. F. Joung, S. Kim and S. Park, J. Phys. Chem. B, 2015, 119, 15509–15515 CrossRef CAS PubMed.
- R. Simkovitch, S. Shomer, R. Gepshtein and D. Huppert, J. Phys. Chem. B, 2015, 119, 2253–2262 CrossRef CAS PubMed.
- S. P. Laptenok, J. Conyard, P. C. Page, Y. Chan, M. You, S. R. Jaffrey and S. R. Meech, Chem. Sci., 2016, 7, 5747–5752 RSC.
- W. Liu, Y. Wang, L. Tang, B. G. Oscar, L. Zhu and C. Fang, Chem. Sci., 2016, 7, 5484–5494 RSC.
- J. F. Joung, S. Kim and S. Park, Phys. Chem. Chem. Phys., 2017, 19, 25509–25517 RSC.
- J. F. Joung, S. Kim and S. Park, J. Phys. Chem. B, 2018, 122, 5087–5093 CrossRef CAS PubMed.
- A. A. Awasthi and P. K. Singh, ChemPhysChem, 2018, 19, 198–207 CrossRef CAS PubMed.
- O. Gajst, L. Pinto da Silva, J. C. G. Esteves da Silva and D. Huppert, J. Phys. Chem. A, 2019, 123, 48–58 CrossRef CAS PubMed.
- N. Munitz, Y. Avital, D. Pines, E. T. J. Nibbering and E. Pines, Isr. J. Chem., 2009, 49, 261–272 CrossRef CAS.
- E. W. Driscoll, J. R. Hunt and J. M. Dawlaty, J. Phys. Chem. A, 2017, 121, 7099–7107 CrossRef CAS PubMed.
- T. Yatsuhashi and H. Inoue, J. Phys. Chem. A, 1997, 101, 8166–8173 CrossRef CAS.
- G. Favaro, U. Mazzucato and F. Masetti, J. Phys. Chem., 1973, 77, 601–604 CrossRef CAS.
- B. S. Vogt and S. G. Schulman, Chem. Phys. Lett., 1983, 97, 450–453 CrossRef CAS.
- K. Akulov, R. Simkovitch, Y. Erez, R. Gepshtein, T. Schwartz and D. Huppert, J. Phys. Chem. A, 2014, 118, 2470–2479 CrossRef CAS PubMed.
- A. Jimenez-Sanchez and R. Santillan, Analyst, 2016, 141, 4108–4120 RSC.
- W. Sheng, M. Nairat, P. D. Pawlaczyk, E. Mroczka, B. Farris, E. Pines, J. H. Geiger, B. Borhan and M. Dantus, Angew. Chem., Int. Ed., 2018, 57, 14742–14746 CrossRef CAS PubMed.
- M. Irie, J. Am. Chem. Soc., 1983, 105, 2078–2079 CrossRef CAS.
- S. Abbruzzetti, M. Carcelli, P. Pelagatti, D. Rogolino and C. Viappiani, Chem. Phys. Lett., 2001, 344, 387–394 CrossRef CAS.
- L. Zhou, H. Yang and P. Wang, J. Org. Chem., 2011, 76, 5873–5881 CrossRef CAS PubMed.
- X. Ding and P. Wang, J. Org. Chem., 2018, 83, 10736–10742 CrossRef CAS PubMed.
- E. E. Nekongo, P. Bagchi, C. J. Fahrni and V. V. Popik, Org. Biomol. Chem., 2012, 10, 9214–9218 RSC.
- Y. Xie, H. L. Luk, X. Yang and K. D. Glusac, J. Phys. Chem. B, 2015, 119, 2498–2506 CrossRef CAS PubMed.
- D. Zhou, R. Khatmullin, J. Walpita, N. A. Miller, H. L. Luk, S. Vyas, C. M. Hadad and K. D. Glusac, J. Am. Chem. Soc., 2012, 134, 11301–11303 CrossRef CAS PubMed.
- Y. Xie, S. Ilic, S. Skaro, V. Maslak and K. D. Glusac, J. Phys. Chem. A, 2017, 121, 448–457 CrossRef CAS.
- E. Gaillard, M. A. Fox and P. Wan, J. Am. Chem. Soc., 1989, 111, 2180–2186 CrossRef CAS.
- R. A. McClelland, N. Mathivanan and S. Steenken, J. Am. Chem. Soc., 1990, 112, 4857–4861 CrossRef CAS.
- J. Kielland, J. Am. Chem. Soc., 1937, 59, 1675–1678 CrossRef CAS.
- S. Park, C. Kim, M. H. Kim, I.-J. Lee and K. Kim, J. Chem. Soc., Faraday Trans., 1998, 94, 1421–1425 RSC.
- D. R. Lide, CRC Handbook of chemistry and physics, CRC Press, Boca Raton, Florida, 83rd edn, 2002 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. Matsui, A. Oshiyama and Y. Shigeta, Chem. Phys. Lett., 2011, 502, 248–252 CrossRef CAS.
- T. Matsui, T. Baba, K. Kamiya and Y. Shigeta, Phys. Chem. Chem. Phys., 2012, 14, 4181–4187 RSC.
- T. Matsui, Y. Shigeta and K. Morihashi, J. Chem. Theory Comput., 2017, 13, 4791–4803 CrossRef CAS PubMed.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
- J. F. Joung, J. Baek, Y. Kim, S. Lee, M. H. Kim, J. Yoon and S. Park, Phys. Chem. Chem. Phys., 2016, 18, 23096–23104 RSC.
- J. Baek, J. F. Joung, S. Lee, H. Rhee, M. H. Kim, S. Park and J. Yoon, J. Phys. Chem. Lett., 2016, 7, 259–265 CrossRef CAS PubMed.
- K. Y. Oang, C. Yang, S. Muniyappan, J. Kim and H. Ihee, Struct. Dyn., 2017, 4, 044013 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nj05404a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |