
Highly uniform Rh nanoparticles supported on boron doped g-C3N4 as a highly efficient and recyclable catalyst for heterogeneous hydroformylation of alkenes†
Yukun
Shi
 *a,
Gang
Ji
a,
Qiqige
Hu
a,
Yang
Lu
a,
Xiaojing
Hu
b,
Baolin
Zhu
b and
Weiping
Huang
b
*a,
Gang
Ji
a,
Qiqige
Hu
a,
Yang
Lu
a,
Xiaojing
Hu
b,
Baolin
Zhu
b and
Weiping
Huang
b
aInstitute of Chemistry for Functionalized Materials, College of Chemistry and Chemical Engineering, Liaoning Normal University, Dalian 116029, China. E-mail: syk312@yeah.net
bCollege of Chemistry, Nankai University, Tianjin 300071, China
First published on 22nd November 2019
Abstract
A series of boron doped g-C3N4 supported rhodium nanoparticle catalysts has been synthesized for the first time and exhibited excellent catalytic activity and easy recycling properties in the hydroformylation of styrene due to the boron doping, which modified the delocalized conjugated π structure of g-C3N4 and increased the adsorption and dispersion of Rh nanoparticles.
Hydroformylation of alkenes, discovered by Roelen as early as 1938, represents one of the most significant and largest catalytic industrial processes for the production of aldehydes and alcohols.1,2 As a 100% atom-economic reaction, nowadays, the production capacity is beyond 12 million tons every year.3–5 More importantly, these aldehydes and alcohols are considered as important versatile intermediates, which can be further transformed into a wide variety of high-performance chemicals, such as detergents, surfactants and plasticizers.6–9
In traditional technology, homogeneous Rh-based complexes are commonly used and widely accepted as catalysts for hydroformylation of alkenes.10–12 However, the application of homogeneous catalytic systems confronts problems of high cost of noble metals and difficulty in the separation of thermally sensitive Rh-based complexes from the low volatilized products.13–15 With the increasing development of green chemistry and sustainable chemistry,16 transition metal nanocatalysts lead the organic synthesis towards a more economical and more sustainable development direction due to their novel electronic, magnetic, optical and catalytic properties.17,18 To date, great progress in the hydroformylation of alkenes catalyzed by rhodium nanoparticle catalysts has been made; nevertheless, Rh nanoparticles alone usually suffer from serious agglomeration, leading to the rapid decay of catalytic activity and the poor durability of the catalyst. To stabilize the nanocatalyst system, a common colloidal method to prepare metal particles involves the reduction of metal ions in the presence of stabilizing or capping materials such as surfactants and polymers.19–22 The important benefit of colloidal nanoparticle synthesis for catalytic applications is the ability to systematically tune particle size and shape, as well as the composition of multielement particles. The stability of Rh nanoparticles can be controlled by the type of stabilizers; however, the strong encapsulation of metal particles within the stabilizing agents results in a loss of catalytic activity due to a capping agent effect.23–26
To stabilize and expose the catalytically “clean” metal nanoparticles, an additional approach is immobilizing metal nanoparticles onto various solid supports, such as dendritic scaffolds, polymers, metal oxides, mesoporous materials and various kinds of carbon.27–30 Among these support materials, graphitic carbon nitride (g-C3N4) has been widely attractive for heterogeneous catalysis due to its unique 2-dimensional (2D) structure, high surface area, facile modification, desirable thermal and chemical stability and facile preparation from abundantly available precursors such as urea and melamine.31–34 In particular, the framework topology of g-C3N4 is in fact a defect-rich, N-bridged “poly (tri-s-triazine ring)”,35,36 which would tend to form π-conjugated planar layers like that of graphite.37 This electronic structure of the π-network would effectively adsorb metal nanoparticles and prevent them from agglomeration on the g-C3N4.38 Furthermore, g-C3N4 contains so-called “nitrogen pots” with abundant melon moieties, which are potential ideal sites for heteroatom doping.37 Among chemical modifications with foreign atoms, boron-doped g-C3N4 has been broadly covered to amplify activity for the targeted reaction.39–41 In particular, the selection of boron source may result in different doping sites and further affect Lewis acidity on the surface of g-C3N4, which can modify the electronic structure and also probably act as specific reaction sites for reactant molecules.42,43 Inspired by these findings in hydroformylation, it is expected that introducing boron in g-C3N4 may be an effective strategy to optimize the catalytic performance of Rh nanoparticles.
In the present study, a boron doped g-C3N4 supported Rh nanoparticle catalyst has been synthesized (cf. ESI,† Scheme S1) and tested in styrene hydroformylation. The g-C3N4, with rich pyridine-like nitrogen, can trap Rh nanoparticles to form potential active sites, while the loading boron can modulate the textural, electronic, and structural properties of g-C3N4 to promote the electron transfer and form a more stable π conjugation system, which strengthens the adsorption and deposition of Rh nanoparticles and alkenes onto the g-C3N4. By considering the importance of boron for deposition of Rh, the deposited Rh nanoparticles on boron doped g-C3N4 could exhibit excellent catalytic activity in the hydroformylation of styrene, as well as easy separation and recyclability.
The structures of the as-prepared pure g-C3N4, 3%B-g-C3N4 and Rh/X%B-g-C3N4 were evaluated by X-ray diffraction (XRD), as shown in the Fig. S1 (cf. ESI†). The XRD patterns confirmed the formation of graphitic stacking C3N4 layers. Two peaks are observed in the XRD patterns for all the samples. The high-intensity peak at around 27.7° is a characteristic of an interlayer stacking of conjugated aromatic systems, which is indexed to the (002) plane of graphitic stacking g-C3N4 corresponding to the average interlayer distance of d = 0.326 nm. The small peak observed at 13.1°, indexed as the (100) plane, is relevant to the in-plane structural packing motif of tri-s-triazine units, with an average distance of d = 0.675 nm. This suggests that the support materials are intact and maintained their graphitic structure after the doping process. For the boron doped samples, the (100) peak shifts slightly to higher angles with increasing content of H3BO3, which should be caused by the lattice contraction from the doping behavior.40 After the anchoring of Rh nanoparticles, there is no obvious diffraction peak related to Rh due to the low loading amount and fine dispersion of Rh nanoparticles on the g-C3N4 support.
The morphologies of pure g-C3N4, 3%B-g-C3N4 and Rh/3%B-g-C3N4 were examined by scanning electron microscopy (cf. ESI,† Fig. S3). The SEM picture of pure g-C3N4 clearly reflects a two-dimensional lamellar structure consisting of small flat sheets with wrinkles and rolling edges. The thickness of the layer is measured to be about 20 nm. Further observation of the SEM picture indicates that there are some pores on the layers’ surface, which is due to the release of a large number of gases such as NH3 and CO2 during thermal condensation of urea. After the doping of boron and deposition of Rh nanoparticles on the g-C3N4 (cf. ESI,† Fig. S3b–d), the platelet-like morphology of g-C3N4 remains intact, which is consistent with the XRD and FTIR results (cf. ESI,† Fig. S1 and S2).
In order to study the promoting effect of boron on the catalytic performance of the Rh/g-C3N4 catalyst, the distributions of the Rh nanoparticle size in Rh/g-C3N4 and Rh/3%B-g-C3N4 were investigated by TEM. As shown in Fig. 1, both Rh/g-C3N4 and Rh/3%B-g-C3N4 samples exhibit a clear lamellar structure of g-C3N4, which means the boron doping does not affect the morphology of g-C3N4. After the introduction of Rh nanoparticles, numerous dark Rh nanoparticles appear on the lamellar g-C3N4. All the Rh nanoparticles are attached to the surface of g-C3N4 strongly.
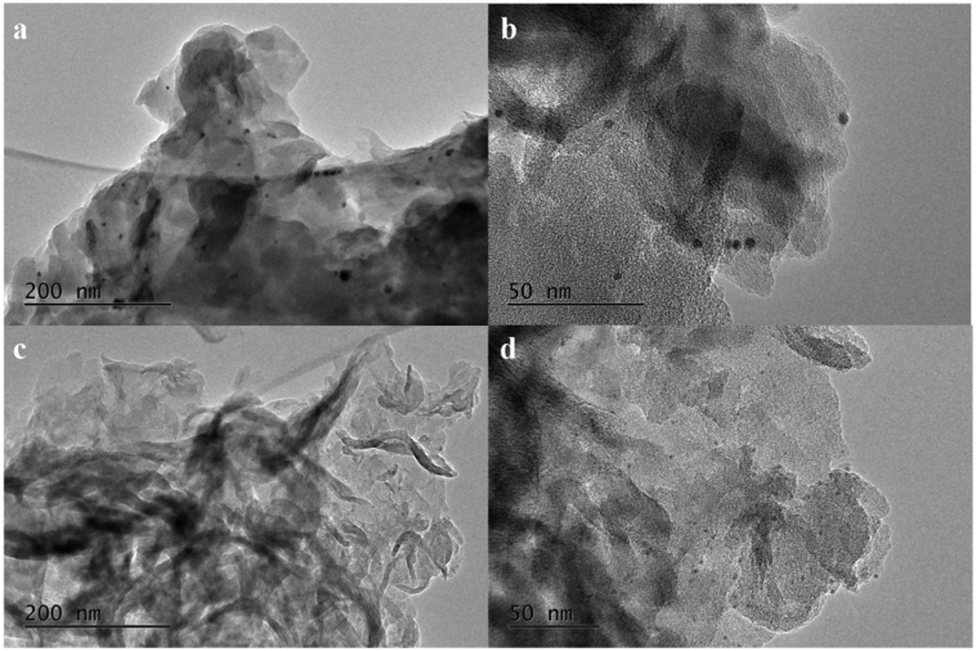 | ||
| Fig. 1 TEM images of Rh/g-C3N4 (a and b) and Rh/3%B-g-C3N4 (c and d). | ||
The TEM micrograph of Rh/g-C3N4 (Fig. 1b) reveals that Rh nanoparticles with an average diameter of 6–7 nm are well dispersed on the surface of g-C3N4. As for Rh/3%B-g-C3N4 (Fig. 1d), the Rh nanoparticles homogeneously disperse on the surface of g-C3N4 and the particle size of the deposited Rh nanoparticles becomes small and evenly-distributed (ca. 1 nm). Thus, these results reveal that highly uniform Rh nanoparticles can be supported successfully on the boron doped g-C3N4. Moreover, the strong interaction between boron and the π bonded planar-layers could induce the formation of small-sized Rh nanoparticles and then stabilize the resulting nanoparticles.
To further probe the composition of samples, X-ray photoelectron spectroscopy (XPS) was carried out to explore the presence of C, N, B and Rh peaks in the sample of Rh/3%B-g-C3N4, as shown in Fig. 2. Fig. 2a shows the high-resolution spectrum of C 1s. The C 1s XPS spectrum reveals two different signals at 284.6 and 287.9 eV, respectively. The peak at 284.8 eV is ascribed to graphitic carbon. The weaker C peak located at 288.7 eV can be assigned to N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups of triazine rings. The N 1s peak in the relevant XPS spectrum can be deconvoluted into three peaks centered at 398.1, 399.8, and 404.2 eV (Fig. 2b), assigned to sp2-bonded N in triazine rings (CN–C), the bridging N atoms in N(–C)3 and terminal NH2, respectively. The B 1s XPS spectrum of Rh/3%B-g-C3N4 in Fig. 2c displays one peak at 191.6 eV, which is higher than 190.1 eV for h-BN and lower than 194 eV for B2O3.44 This indicates that boron inserts in the BCN frame in the form of N–B–N coordination, which implies boron is a substitute for some carbon atoms in the CN framework. XPS analysis of the Rh 3d core level shows the presence of two bands at 307.1 and 311.6 eV which may be assigned to metallic Rh 3d5/2 and Rh 3d3/2, respectively. Another two peaks at 309.1 eV and 314.4 eV are attributed to the Rh3+ 3d5/2 and Rh3+ 3d3/2, respectively. This confirms that the deposition–precipitation method effectively loaded nanosized particles of Rh on the boron doped g-C3N4.
N groups of triazine rings. The N 1s peak in the relevant XPS spectrum can be deconvoluted into three peaks centered at 398.1, 399.8, and 404.2 eV (Fig. 2b), assigned to sp2-bonded N in triazine rings (CN–C), the bridging N atoms in N(–C)3 and terminal NH2, respectively. The B 1s XPS spectrum of Rh/3%B-g-C3N4 in Fig. 2c displays one peak at 191.6 eV, which is higher than 190.1 eV for h-BN and lower than 194 eV for B2O3.44 This indicates that boron inserts in the BCN frame in the form of N–B–N coordination, which implies boron is a substitute for some carbon atoms in the CN framework. XPS analysis of the Rh 3d core level shows the presence of two bands at 307.1 and 311.6 eV which may be assigned to metallic Rh 3d5/2 and Rh 3d3/2, respectively. Another two peaks at 309.1 eV and 314.4 eV are attributed to the Rh3+ 3d5/2 and Rh3+ 3d3/2, respectively. This confirms that the deposition–precipitation method effectively loaded nanosized particles of Rh on the boron doped g-C3N4.
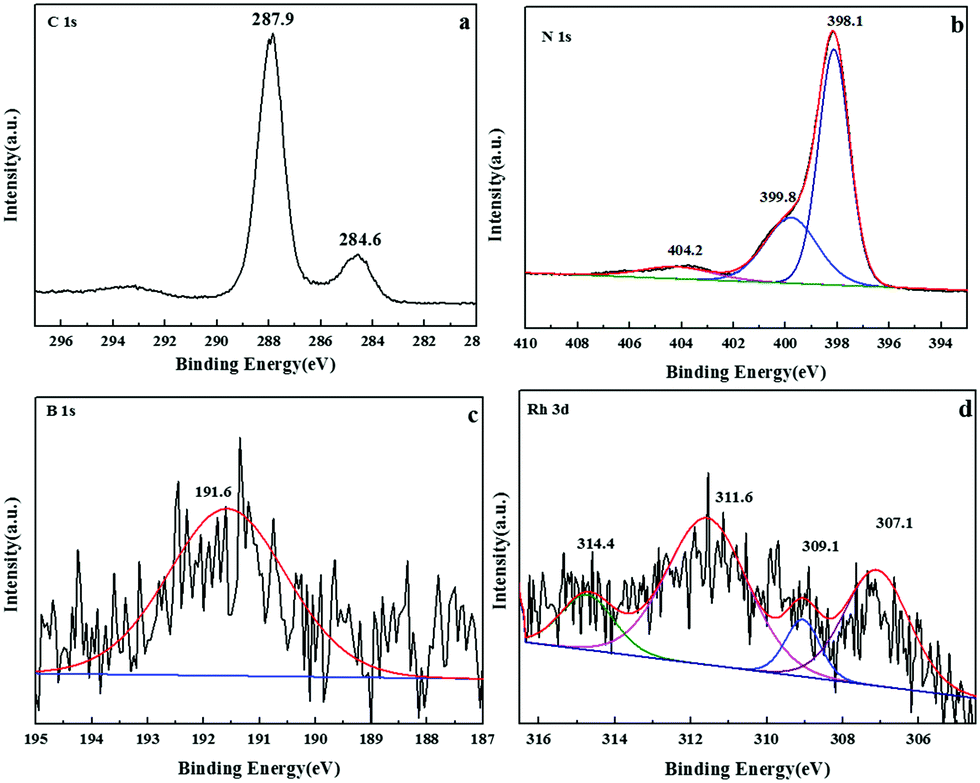 | ||
| Fig. 2 XPS spectra of (a) C 1 s, (b) N 1 s, (c) B 1s and (d) Rh3d of the Rh/3%B-g-C3N4 sample. | ||
In order to further understand the promotion effect of boron addition, the catalytic performances of styrene hydroformylation over the Rh/g-C3N4 and Rh/X%B-g-C3N4 catalysts were evaluated in detail, as shown in Table 1. It can be seen that the Rh/g-C3N4 catalyst exhibits an excellent catalytic activity for the hydroformylation of styrene (entry 1, TOF = 7100 h−1). For the Rh/1%B-g-C3N4 catalyst, the conversion of styrene increases to 79.1% with a higher TOF (entry 2). Encouraged by these results, a series of boron doped g-C3N4 supported Rh catalysts were prepared with increasing boron loading. As shown in entries 2–7, the modification of boron results in higher activity of the catalysts. Doping boron atoms on the surface of g-C3N4 can act as the anchoring sites to support Rh nanoparticles, resulting in the uniform deposition of Rh nanoparticles on the surface of g-C3N4. In addition, the styrene with the delocalized π bonds, as an electron donor, can be adsorbed easily on the boron Lewis acid sites with the formation of electron-deficient aromatic intermediates, resulting in an increase in the catalytic performance of the Rh/X%B-g-C3N4 catalysts. The effect of boron loading in the hydroformylation of styrene catalyzed by Rh/X%B-g-C3N4 catalysts was also evaluated in detail, the conversion of styrene gradually increases until a loading of boron up to 3%, suggesting that the introduction of boron into the catalyst made the catalyst expose many more active sites and that a synergetic effect could be occurring between the Rh species and the boron species. On further increasing the boron loading, the styrene TOF kept unchanged, which may be due to the much higher amount of boron on the surface which cannot disperse and expose more active Rh sites. Thus, the optimum loading of boron in the Rh/X%B-g-C3N4 catalysts is found to be 3% in the tested range. This loading was chosen for further studies.
| Entry | Catalyst | Conversion (%) | TOFb (h−1) | Selectivity (%) | |
|---|---|---|---|---|---|
| Aldehydes | B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Lc :Lc |
||||
|
a Reaction conditions: catalyst: 0.01 g, toluene: 20 mL, styrene: 1.0 mL, reaction time: 3 h, temp.: 100 °C, syngas (CO/H2 = 1): 6.0 MPa.
b TOF = number of moles of product formed/(number of moles of Rh × time).
c B:L = 2-phenylpropanal:3-phenylpropanal.
|
|||||
| 1 | Rh/g-C3N4 | 58.9 | 7100 | 100 | 57:43 |
| 2 | Rh/1%B-g-C3N4 | 79.1 | 9600 | 100 | 58:42 |
| 3 | Rh/2%B-g-C3N4 | 85.2 | 10300 |
100 | 57:43 |
| 4 | Rh/3%B-g-C3N4 | 99.0 | 12000 |
100 | 57:43 |
| 5 | Rh/4%B-g-C3N4 | 99.0 | 12000 |
100 | 56:44 |
| 6 | Rh/5%B-g-C3N4 | 99.0 | 12000 |
100 | 56:44 |
| 7 | Rh/6%B-g-C3N4 | 99.0 | 12000 |
100 | 55:45 |
The lifetime is an important aspect concerning the use of catalysts, and therefore the reusability of the Rh/3%B-g-C3N4 was examined for 7 cycles under the optimum reaction conditions and the results are displayed in Fig. 3. After the reaction, the catalyst was easily separated from the reaction solution and directly reused in the next catalytic cycles. As shown in Fig. 3, the Rh/3%B-g-C3N4 catalyst can be reused at least 7 times with little change in the catalytic performance and selectivity, indicating that the Rh/3%B-g-C3N4 is an efficient and stable catalyst for hydroformylation of alkenes. Namely, the strong interaction between the doped boron and π bonded planar-layers of g-C3N4 may be acting to stabilize the Rh nanoparticles, which can strengthen the adsorption and dispersion of Rh nanoparticles on the surface of g-C3N4.
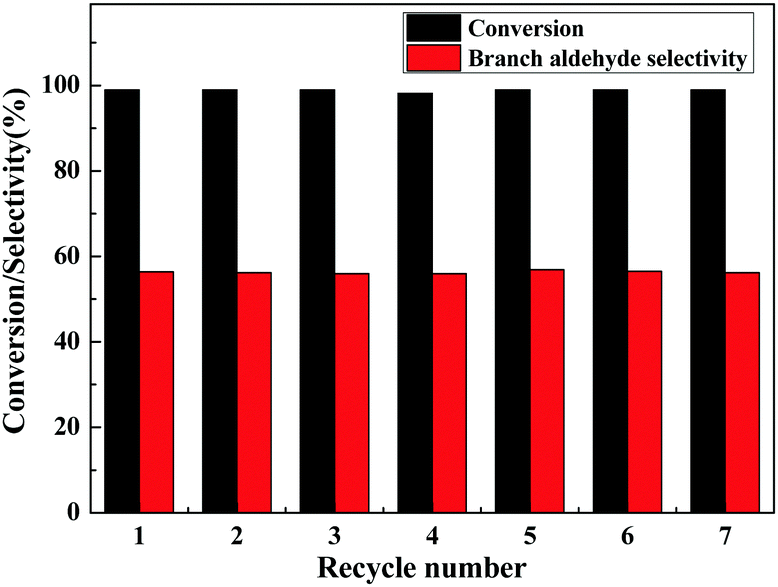 | ||
| Fig. 3 Reusability of the Rh/3%B-g-C3N4 catalyst for the hydroformylation of styrene. Reaction conditions: Rh/3%B-g-C3N4: 0.01 g, toluene: 20 mL, styrene: 1.0 mL, temp.: 100 °C, syngas (CO/H2 = 1): 6.0 MPa. | ||
In this work, a series of boron doped g-C3N4 supported rhodiun nanoparticle catalysts has been prepared and exhibit significantly enhanced activity in the hydroformylation of styrene (the highest TOF = 12000 h−1). The excellent catalytic performances of the Rh/B-g-C3N4 catalysts should be attributed to its unique two-dimensional lamellar structure, which determined its largest exposed Rh surface area, highest nanoparticle dispersion and smallest nanoparticle size. More importantly, the Rh nanoparticles of Rh/B-g-C3N4 dispersed more homogeneously and the Rh particle size (about 1 nm) was more uniform than those of the Rh/g-C3N4. The strong interactions between boron and the π bonded planar-layers of g-C3N4 could induce the formation small-sized Rh nanoparticles and then stabilize the resulting nanoparticles. In addition, Rh/B-g-C3N4 also exhibited excellent catalytic activities for the hydroformylation of various alkenes with high selectivity to aldehydes. More importantly, the Rh/B-g-C3N4 can be easily separated from the products and directly reused in the next cycle without an evident loss in the catalytic activity and selectivity after 7 cycles. In summary, we envision that the research results will constitute a new insight for further applications of boron-doped or boron dominant materials towards heterogeneous hydroformylation catalysis.
Conflicts of interest
There are no conflicts to declare.References
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS.
- R. Franke, D. Selent and A. Borner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed.
- S. A. Jagtap and B. M. Bhanage, Appl. Organomet. Chem., 2018, 32, 4478–4486 CrossRef.
- I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., 2011, 123, 294–298 CrossRef.
- M. Beller, B. Cornils, C. D. Frohning and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17–85 CrossRef CAS.
- S. Fuchs, D. Lichte, T. Jolmes, T. Rösler, G. Meier, H. Strutz, A. Behr and A. J. Vorholt, ChemCatChem, 2018, 10, 4126–4133 CrossRef CAS.
- G. M. Torres, R. Frauenlob, R. Franke and A. Borner, Catal.: Sci. Technol., 2015, 5, 34–54 RSC.
- J. Wildt, A. C. Brezny and C. R. Landis, Organometallics, 2017, 36, 3142–3151 CrossRef CAS.
- C. Y. Li, K. J. Sun, W. L. Wang, L. Yan, X. P. Sun, Y. Q. Wang, K. Xiong, Z. P. Zhan, Z. Jiang and Y. J. Ding, J. Catal., 2017, 353, 123–132 CrossRef CAS.
- M. Haumann and A. Riisager, Chem. Rev., 2008, 108, 1474–1497 CrossRef CAS.
- M. Ahmeda and A. Sakthivel, J. Mol. Catal. A: Chem., 2016, 424, 85–90 CrossRef.
- J. Liu, L. Yan, Y. J. Ding, M. Jiang, W. D. Dong, X. E. Song, T. Liu and H. J. Zhu, Appl. Catal., A, 2015, 492, 127–132 CrossRef CAS.
- Y. P. Zhao, X. M. Zhang, J. Sanjeevi and Q. H. Yang, J. Catal., 2016, 334, 52–59 CrossRef CAS.
- N. C. C. Breckwoldt, N. J. Goosen, H. C. M. Vosloo and P. V. Gryp, React. Chem. Eng., 2019, 4, 695–704 RSC.
- B. C. E. Makhubela, A. Jardine and G. S. Smith, Green Chem., 2012, 14, 338–347 RSC.
- L. B. Wang, W. B. Zhang, S. P. Wang, Z. H. Gao, Z. H. Luo, X. Wang, R. Zeng, A. W. Li, H. L. Li, M. L. Wang, X. S. Zheng, J. F. Zhu, W. H. Zhang, C. Ma, R. Si and J. Zeng, Nat. Commun., 2016, 7, 14036–14043 CrossRef CAS PubMed.
- F. Y. Xiao, Y. W. Qin, N. Wang and D. W. Pan, Appl. Clay Sci., 2018, 166, 74–79 CrossRef CAS.
- F. Huang, Y. C. Deng, Y. L. Chen, X. B. Cai, M. Peng, Z. M. Jia, P. J. Ren, D. Q. Xiao, X. D. Wen, N. Wang, H. Y. Liu and D. Ma, J. Am. Chem. Soc., 2018, 140(41), 13142–13146 CrossRef CAS.
- I. Nakamula, Y. Yamanoi, T. Imaoka, K. Yamamoto and H. Nishihara, Angew. Chem., Int. Ed., 2011, 50, 5830–5833 CrossRef CAS.
- X. R. Xue, Y. Song, Y. C. Xu and Y. H. Wang, New J. Chem., 2018, 42, 6640–6643 RSC.
- W. Alsalahi, W. Tylus and A. M. Trzeciak, ChemCatChem, 2018, 10, 2051–2058 CrossRef CAS.
- Z. Sun, Y. H. Wang, M. M. Niu, H. Q. Yi, J. Y. Jiang and Z. L. Jin, Catal. Commun., 2012, 27, 78–82 CrossRef CAS.
- Y. K. Shi, X. J. Hu, B. L. Zhu, S. R. Wang, S. M. Zhang and W. P. Huang, RSC Adv., 2014, 4, 62215–62222 RSC.
- M. E. Grass, S. H. Joo, Y. W. Zhang and G. A. Somorjai, J. Phys. Chem. C, 2009, 113, 8616–8623 CrossRef CAS.
- Y. Borodko, S. E. Habas, M. Koebel, P. D. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052–23059 CrossRef CAS PubMed.
- Y. Borodko, S. M. Humphrey, T. D. Tilley, H. Frei and G. A. Somorjai, J. Phys. Chem. C, 2007, 111, 6288–6295 CrossRef CAS.
- Z. M. Jia, F. Huang, J. Y. Diao, J. Y. Zhang, J. Wang, D. S. Su and H. Y. Liu, Chem. Commun., 2018, 54, 11168–11171 RSC.
- L. X. Xia, D. Li, J. Long, F. Huang, L. N. Yang, Y. S. Guo, Z. M. Jia, J. P. Xiao and H. Y. Liu, Carbon, 2019, 145, 47–52 CrossRef CAS.
- C. Hou, G. F. Zhao, Y. J. Ji, Z. Q. Niu, D. S. Wang and Y. D. Li, Nano Res., 2014, 79, 1364–1369 CrossRef.
- X. J. Hu, Y. K. Shi, Y. J. Zhang, B. L. Zhu, S. M. Zhang and W. P. Huang, Catal. Commun., 2015, 59, 45–49 CrossRef CAS.
- W. Tian, Q. H. Shen, N. X. Li and J. C. Zhou, RSC Adv., 2016, 6, 25568–25576 RSC.
- W. Liu, M. L. Wang, C. X. Xu and S. F. Chen, Chem. Eng. J., 2012, 209, 386–393 CrossRef CAS.
- Y. J. Fu, C. A. Liu, C. Zhu, H. B. Wang, Y. J. Dou, W. L. Shi, M. W. Shao, H. Huang, Y. Liu and Z. H. Kang, Inorg. Chem. Front., 2018, 5, 1646–1652 RSC.
- Y. S. Fu, T. Huang, B. Q. Jia, J. W. Zhu and X. Wang, Appl. Catal., B, 2017, 202, 430–437 CrossRef CAS.
- Y. Wang, X. C. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- J. D. Hong, X. Y. Xia, Y. S. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006–15012 RSC.
- Q. Liu and J. Y. Zhang, Langmuir, 2013, 29, 3821–3828 CrossRef CAS.
- W. Y. Zhang, H. J. Huang, F. Li, K. M. Deng and X. Wang, J. Mater. Chem. A, 2014, 2, 19084–19094 RSC.
- J. R. Wei, W. L. Shen, J. Zhao, C. W. Zhang, Y. H. Zhou and H. Y. Liu, Catal. Today, 2018, 316, 199–205 CrossRef CAS.
- C. H. Lu, R. Y. Chen, X. Wu, M. F. Fan, Y. H. Liu, Z. G. Le, S. J. Jiang and S. Q. Song, Appl. Surf. Sci., 2016, 360, 1016–1022 CrossRef CAS.
- W. Q. Kong, X. F. Zhang, B. B. Chang, Y. N. Zhou, S. R. Zhang, G. L. He, B. C. Yang and J. J. Li, Electrochim. Acta, 2018, 282, 767–774 CrossRef CAS.
- G. B. Zhou, Y. Pei, Z. Jiang, K. N. Fan, M. H. Qiao, B. Sun and B. N. Zong, J. Catal., 2014, 311, 393–403 CrossRef CAS.
- Y. K. Shi, X. J. Hu, L. Chen, Y. Lu, B. L. Zhu, S. M. Zhang and W. P. Huang, New J. Chem., 2017, 41, 6120–6126 RSC.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization, and supporting figures and tables. See DOI: 10.1039/c9nj05385a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |