
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemical synthesis, luminescent and magnetic properties of lanthanide benzene-1,4-dicarboxylate coordination polymers (Ln0.5Gd0.5)2 (1,4-BDC)3(H2O)4; Ln = Sm, Eu, Tb†
Tarek
Alammar
 *a,
Ihor Z.
Hlova
b,
Shalabh
Gupta
*b,
Anis
Biswas
b,
Tao
Ma
b,
Lin
Zhou
b,
Viktor
Balema
b,
Vitalij K.
Pecharsky
ab and
Anja-Verena
Mudring
*abc
*a,
Ihor Z.
Hlova
b,
Shalabh
Gupta
*b,
Anis
Biswas
b,
Tao
Ma
b,
Lin
Zhou
b,
Viktor
Balema
b,
Vitalij K.
Pecharsky
ab and
Anja-Verena
Mudring
*abc
aDepartment of Materials Science and Engineering, Iowa State University, Ames, IA 50011-2300, USA. E-mail: tarek.alammar@rub.de
bAmes Laboratory, Iowa State University, Ames, IA 500011-3020, USA. E-mail: shalabhg@ameslab.gov
cDepartment of Materials and Environmental Chemistry, Stockholm University, Svante Arrhenius väg 16 C, 106 91 Stockholm, Sweden. E-mail: anja-verena.mudring@mmk.su.se
First published on 12th November 2019
Abstract
Mechanochemical reactions of benzene-1,4-dicarboxylate (BDC2−) and lanthanide carbonates, Ln2(CO3)3·xH2O (Ln = Sm, Eu, Gd, Tb) yield phase pure lanthanide coordination polymers, (Ln0.5Gd0.5)2(1,4-BDC)3(H2O)4 with Ln = Sm, Eu, Tb, which are isostructural with Tb2(1,4-BDC)3(H2O)4 as confirmed by powder X-ray diffraction and vibrational spectroscopy. Upon excitation with UV light all three compounds display strong emissions, characteristic for the respective optically active lanthanide ion, namely, red for Eu3+, green for Tb3+ and orange-red for Sm3+. In case of the Tb3+-containing compound, the energy difference between the triplet energy level of benzene-1,4-dicarboxylate ligand (BDC2−) allows for the most efficient BDC2−–Tb3+ energy transfer. As a consequence, an intense green luminescence with rather long lifetime (0.81 ms) and high quantum yield (22%) is observed after allowed excitation of the BDC2− ligand. The compounds are paramagnetic with no onset of long range magnetic ordering down to liquid He temperatures.
Introduction
The synthesis of luminescent metal–organic frameworks (MOFs) with high fluorescence intensities, lifetimes and quantum efficiencies continues to be a challenging task.1 However, such materials are of interest for a number of applications such as organic light emitting diodes,2 fluorescent lighting,3,4 luminescent probes for medical imaging,5 luminescence thermometry,6 chemical sensors,7 and molecular magnetorefrigeration8 to name a few. The best performing luminescent MOF materials rely on the emission of trivalent lanthanide ions, Ln3+, which arise from intraconfigurational 4f–4f-transitions. This is advantageous as it leads to high color purity. At the same time, the transitions are forbidden, leading to low absorption coefficients and, thus, direct excitation of the Ln3+ ions is very inefficient.9 However, employing a suitable linker system that can participate in luminescence sensitization can improve the materials through efficient light absorption by the linker and subsequent electronic energy transfer to the emissive Ln3+ ion. Often this sensitization process is referred to as the “antenna effect”.10A number of factors influence the efficiency of ligand-to-metal energy transfer such as the intersystem crossing efficiency of the sensitizer, the location of the ligand triplet state through which sensitization occurs with respect to the emitting state of the lanthanide ion, the spectral overlap, potential charge transfer states and the donor–acceptor distance.11 Achieving high efficiency of ligand-to-metal energy transfer, hence, requires careful selection or design of suitable organic linkers including appropriate configuration and energy levels and coordination modes for the respective Ln3+ ion to support an efficient luminescence sensitization. A variety of luminescent MOFs have been prepared using these guidelines.9 During these studies it has been realized that the luminescent properties such as the lifetime and quantum yield are influenced by the employed synthetic protocols, especially when non-conventional approaches like mechanochemical reactions are used.12–14 Experience tells that mechanochemical synthesis leads to materials with unusual morphologies, small particle sizes and low crystallinity, and hence affects the emission and quantum yields which are sensitive to structural defects and impurity doping.15 Recently, MIL-78 type frameworks were synthesized by our group by using a simple, solvent-free mechanochemical approach.16,17 Advantages of this method include, but are not limited to the high conversion rates, simplicity of the experimental set up, exclusion of solvents and reasonably good scalability.18–20 Therefore, here we have adopted the mechanochemical approach for the preparation of metal–organic polymer structures, (Ln0.5Gd0.5)2(BDC)3(H2O)4 with Ln = Sm, Eu and Tb, containing benzene-1,4-dicarboxylate (1,4-BDC) as a linker, and investigate the optical and magnetic properties of the as-synthesized compounds.
Results and discussion
The solid-state mechanochemical reactions between Ln2(CO3)3·xH2O and BDC (benzene-1,4-dicarboxylate) are typically complete after 2 h of milling with a 100% yield. The completion of reactions is judged by the absence of characteristic Bragg peaks from either of the precursors or any other crystalline intermediates (Fig. 1). This is a clear advantage over the classical hydrothermal synthesis where lower yield, such as 67% for the Tb compound have been reported.21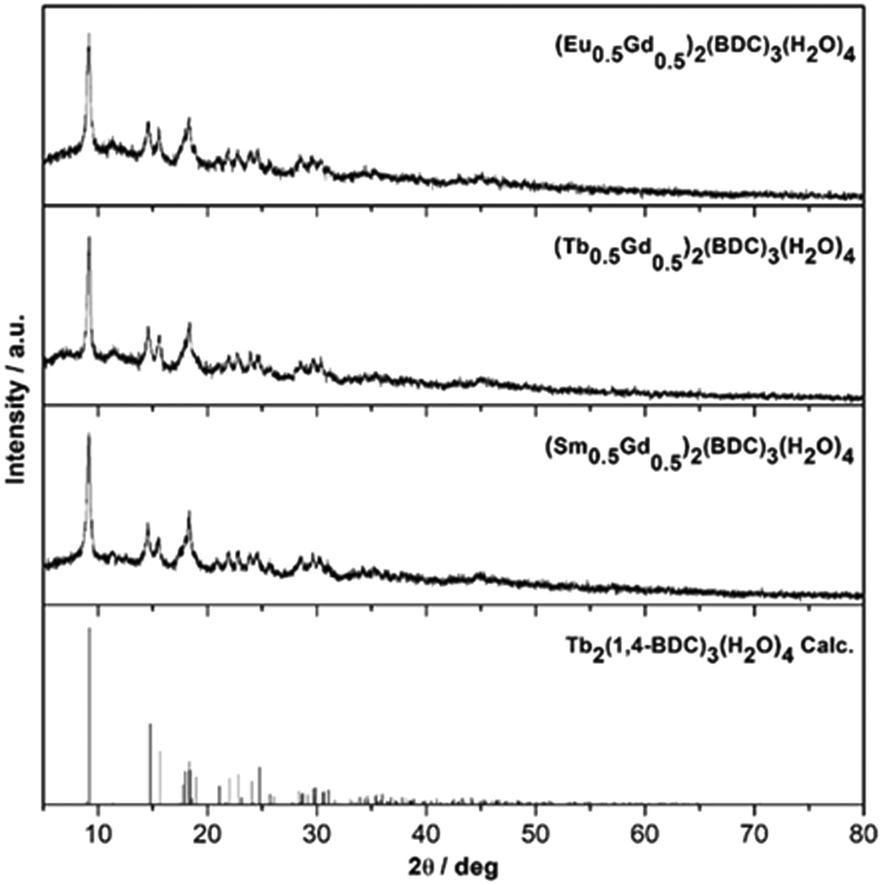 | ||
| Fig. 1 Powder XRD patterns of mechanochemically prepared (Ln0.5Gd0.5)2(1,4-BDC)3(H2O)4, Ln = Eu, Tb, Sm and the calculated pattern of Tb2(1,4-BDC)3(H2O)4 [ref. 21] (top to bottom). | ||
Rietveld refinement using the FULLPROF program of the XRD patterns confirm that, the as-synthesized MOFs are isostructural with Tb2(1,4-BDC)3(H2O)4 (S1 in ESI†).21 In all cases, satisfactory structural refinement was obtained based on the reported crystal structure for the Tb2(1,4-BDC)3(H2O)4 yielding an overall refined composition of (Ln0.5Gd0.5)2(BDC)3(H2O)4. The number of water molecules in the lattice was independently confirmed by TGA as described below.
The refined cell volumes range from 603.31(9) Å3 for (Tb0.5Gd0.5)2(BDC)3(H2O)4 to 607.0(1) Å3 for (Sm0.5Gd0.5)2(BDC)3(H2O)4 and the refined lattice constants are in excellent agreement with the changing ionic radius of the incorporated Ln3+ ions (ionic radii: Tb3+ = 92.3 pm, Eu3+ = 94.7 pm, Sm3+ = 95.8 pm) (Table 1).22
| a, b, c/Å | α, β, γ/° | V/Å3 | |
|---|---|---|---|
| (Sm0.5Gd0.5)2(BDC)3(H2O)4 | 6.2145(9) | 101.93(1) | 607.0(1) |
| 10.098(2) | 91.15(1) | ||
| 10.112(1) | 101.57(1) | ||
| (Eu0.5Gd0.5)2(BDC)3(H2O)4 | 6.195(1) | 102.05(1) | 605.8(2) |
| 10.090(2) | 91.35(1) | ||
| 10.131(1) | 101.34(1) | ||
| (Tb0.5Gd0.5)2(BDC)3(H2O)4 | 6.1936(4) | 102.114(7) | 603.31(9) |
| 10.078(1) | 91.180(8) | ||
| 10.1076(8) | 101.460(7) | ||
| (Tb)2(BDC)3(H2O)4 (See, ref. 21. Data from single crystal XRD) | 6.2142(2) | 102.247(2) | 596.63(3) |
| 10.0694(1) | 91.118(1) | ||
| 10.0956(3) | 101.518(2) |
TG-DSC analyses of the as-prepared (Ln0.5Gd0.5)2(BDC)3(H2O)4 MOFs display a two-step weight loss between room temperature and 1000 °C (Fig. 2). The first loss occurs at 100–215 °C. The weight losses are 8.4% (∼4.1 eq. H2O per f.u.) for Eu-, 8.7% (∼4.2 eq. H2O per f.u.) for Tb-, and 8% (∼3.9 eq. H2O per f.u.) for the Sm-containing MOF, which can be assigned to the dehydration of the as-synthesized samples. For all three samples, the second weight loss is observed at ∼500 °C, which is caused by the thermal decomposition of (Ln0.5Gd0.5)2(BDC)3, resulting in the formation of the respective lanthanide oxides.23 This is in line with the results of previously reported thermal analysis of Tb2(BDC)3(H2O)4 and Er2(BDC)3(H2O)4.21,24 No signals corresponding to the thermal events for benzene-1,4-dicarboxylic acid and/or carbonates can be seen, indicating complete consumption of the starting materials during the mechanochemical syntheses.
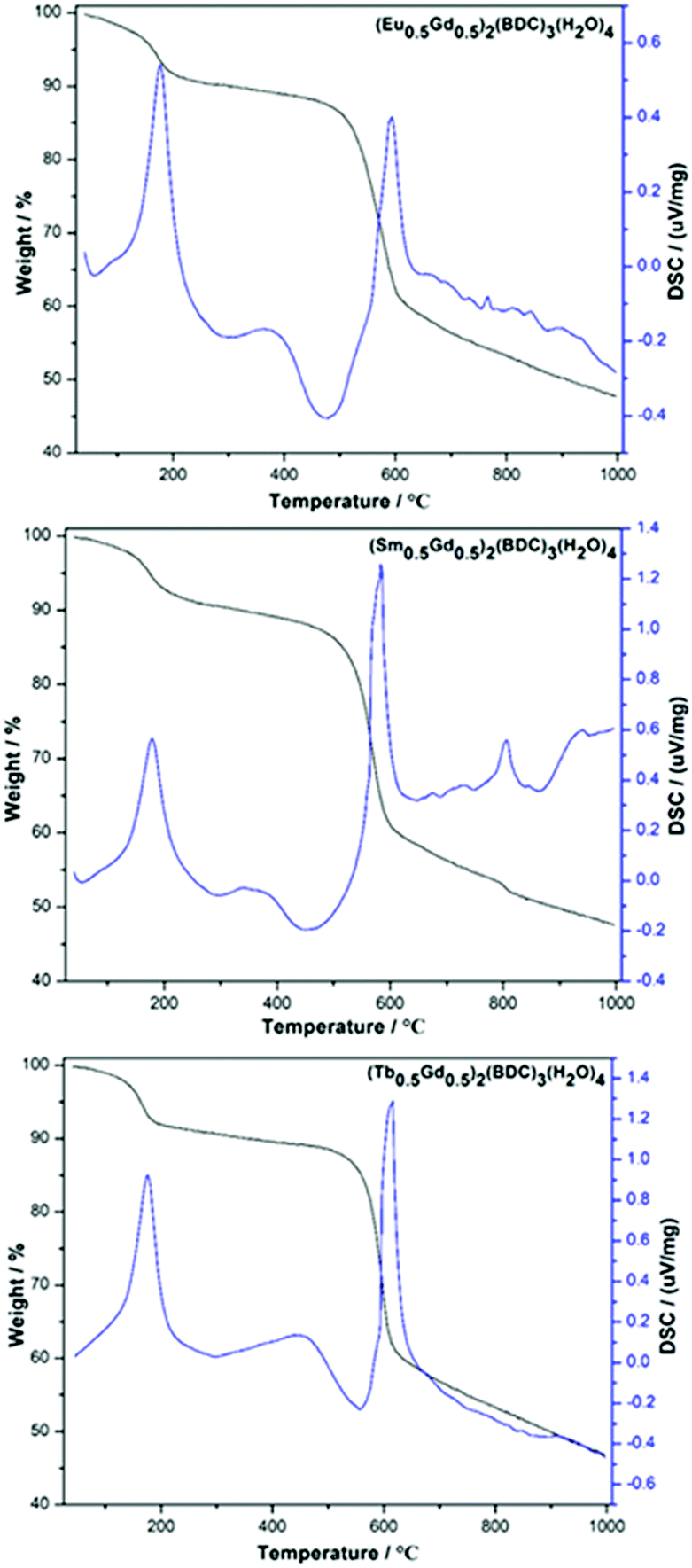 | ||
| Fig. 2 TG-DSC curves of mechanochemically synthesized (Ln0.5Gd0.5)2(BDC)3(H2O)4, Ln = Eu, Sm, Tb (top to bottom). | ||
X-ray photoelectron spectroscopy (XPS) analyses were performed to examine the surface elemental composition and the oxidation state of each element in the mechanochemically synthesized (Ln0.5Gd0.5)2(BDC)3(H2O)4. The XPS survey spectra for the different (Ln0.5Gd0.5)2(BDC)3(H2O)4 materials are shown in Fig. 3, where all peaks corresponding to the characteristic electronic transitions of Sm, Eu, Gd, Tb, O and C are visible. Furthermore, XPS core level spectra of Eu-3d, Tb-3d, and Sm-3d regions are illustrated in Fig. 3. The XPS spectrum of (Eu0.5Gd0.5)2(BDC)3(H2O)4 features two prominent peaks with binding energies of 1164.9 eV (Eu3+ 3d3/2) and 1135.1 eV (Eu3+ 3d5/2) together with three small satellites that occur at 1155.7 eV (Eu2+ 3d3/2), 1143.8 eV (mult), and 1125 eV (Eu2+ 3d5/2). The appearance of Eu2+ is related to the reduction of Eu3+ to Eu2+ as a result of the charge compensation procedure during profiling under the experimental XPS conditions which is not unusual to notice for Eu(III) compounds.25,26 The XPS spectrum of the core levels in the Tb-3d region of (Tb0.5Gd0.5)2(BDC)3(H2O)4 displays two peaks at 1242.8 eV (Tb3+ 3d5/2) and 1277 eV (Tb3+ 3d3/2). Moreover, a satellite peak at 1250.8 eV was detected, which is ascribed to the existence of Tb4+ traces owing to oxidation of Tb3+ to Tb4+ during the measurement, which has been observed for other Tb3+ compounds under similar measurement conditions before.27 The XPS spectrum of (Sm0.5Gd0.5)2(BDC)3(H2O)4 shows two peaks centered at 1083.5 eV and 1111 eV. These peaks originate from Sm3+ 3d5/2 and Sm3+ 3d3/2 levels.
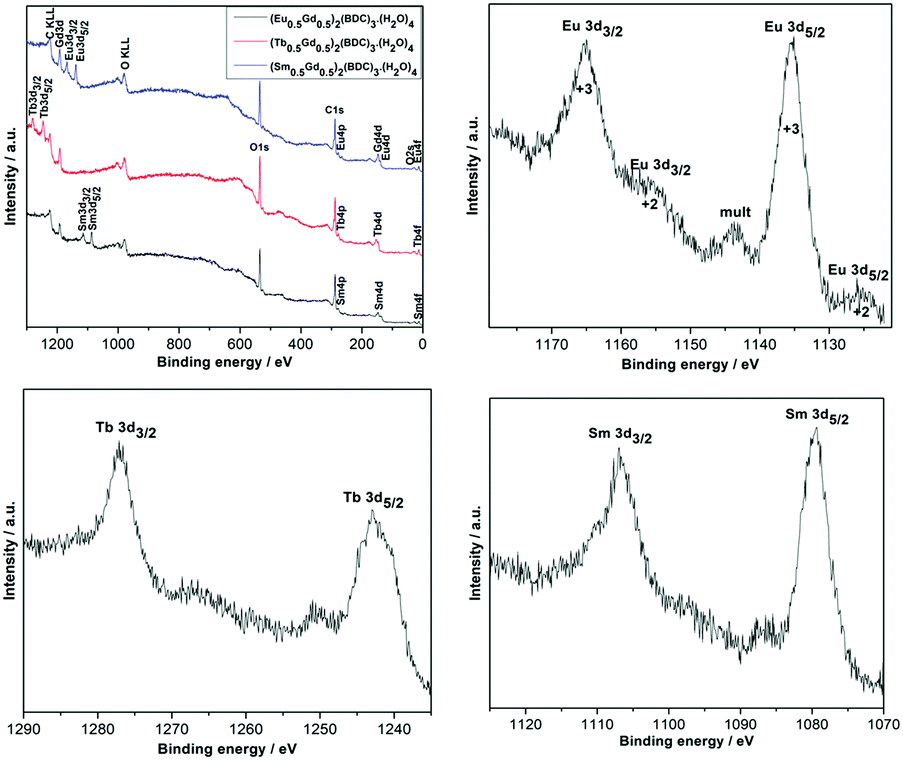 | ||
| Fig. 3 X-ray photoelectron spectrum survey and narrow scans of Sm-3d, Eu-3d and Tb-3d regions for the (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Sm, Eu and Tb. | ||
Electron microscopy reveals that the as-synthesized samples are composed of irregularly shaped and mostly aggregated particles (Fig. 4). A few particles ranging between 50–100 nm were selected for elemental mapping using energy dispersive spectroscopy (EDS) in order to ascertain the homogeneity of metal atoms in the heteronuclear complex compounds. Results are shown in Fig. 4 and Table 2. Element specific (Ln and Gd) images obtained from the same area are shown in two central columns and their combined color-composites are on the extreme right. The images clearly show a homogeneous distribution of metal atoms down to a few nanometers length scale, and shows no sign of clustering or agglomeration of metal centres. These results demonstrate that mechanochemical methods can be effectively utilized to synthesize high quality luminescent MOFs where atomic level control, both chemistry and concentration, of emission centres are necessary to tailor and optimize quantum yields. As expected, the atomic ratios of Gd and respective lanthanide ions are close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
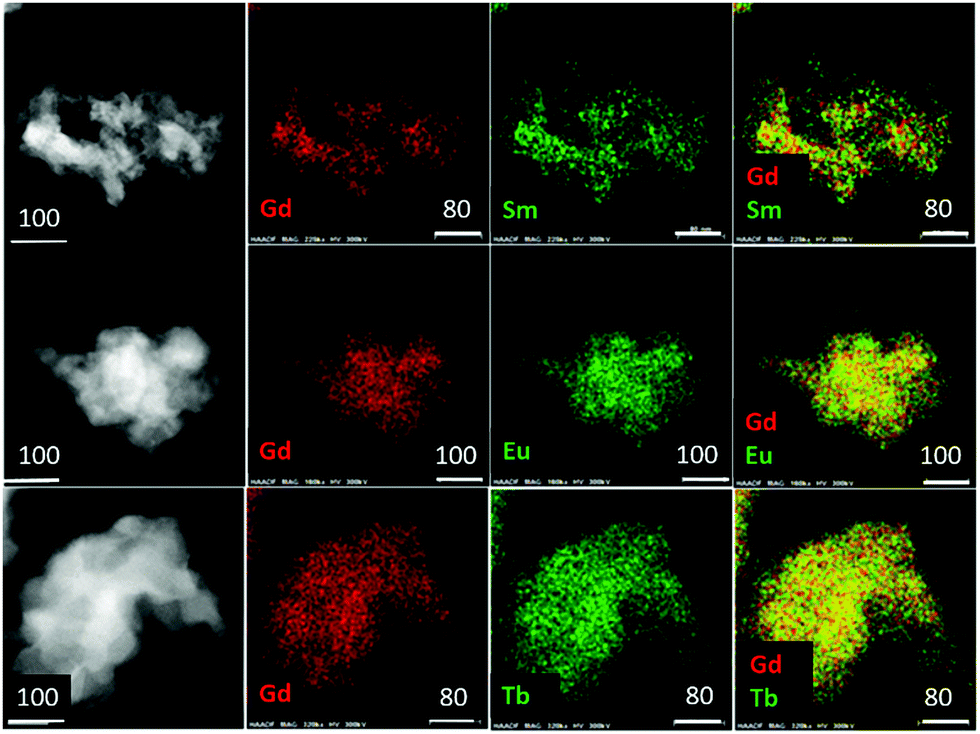 | ||
| Fig. 4 High-angle-annular-dark-field scanning transmission electron microscopy images (extreme left) and corresponding EDS elemental mapping of the (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Sm, Eu and Tb (top to bottom). White bars indicate dimensions in μm. | ||
| Element | Wt% | At% | Error% |
|---|---|---|---|
| Gd/Sm | 55.18/44.82 | 54.07/45.93 | 5.58/4.54 |
| Gd/Eu | 52.79/47.21 | 51.93/48.07 | 5.33/4.77 |
| Gd/Tb | 51.19/48.81 | 50.92/49.08 | 5.15/4.91 |
Fig. 5 shows infrared (IR) spectra of (Ln0.5Gd0.5)2(BDC)3(H2O)4 along with that of benzene-1,4-dicarboxylic acid for reference. Notably, the IR spectra of the three (Ln0.5Gd0.5)2(BDC)3(H2O)4 samples are practically indistinguishable and confirm the formation of isostructural compounds. The absence of bands characteristic of the free acid i.e. the band at 1672 cm−1 which originates from the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of COOH groups, the very broad O–H stretching band in the region between 2200 and 3200 cm−1, and a band at around 1282 cm−1 which belongs to the bending vibration of carboxylic acid (δO–H), as well as the band at about 920 cm−1 attributable to OH out of plane bending in the IR spectra of (Ln0.5Gd0.5)2(BDC)3(H2O)4 prove the complete deprotonation of COOH groups and the coordination of COO− to Ln3+. Also, the signals corresponding to the starting metal carbonates (νCO between 1500 and 300 cm−1)28–30 cannot be observed in the spectra of the final products. The bands in the range between 1501 and 1588 cm−1 and at 1386 cm−1 can be ascribed to asymmetric (νas) and symmetric (νs) stretching vibrations of –COO−, respectively. The broad band centered at 3450 cm−1 corresponds to OH stretch of structural water molecule.31
O stretching band of COOH groups, the very broad O–H stretching band in the region between 2200 and 3200 cm−1, and a band at around 1282 cm−1 which belongs to the bending vibration of carboxylic acid (δO–H), as well as the band at about 920 cm−1 attributable to OH out of plane bending in the IR spectra of (Ln0.5Gd0.5)2(BDC)3(H2O)4 prove the complete deprotonation of COOH groups and the coordination of COO− to Ln3+. Also, the signals corresponding to the starting metal carbonates (νCO between 1500 and 300 cm−1)28–30 cannot be observed in the spectra of the final products. The bands in the range between 1501 and 1588 cm−1 and at 1386 cm−1 can be ascribed to asymmetric (νas) and symmetric (νs) stretching vibrations of –COO−, respectively. The broad band centered at 3450 cm−1 corresponds to OH stretch of structural water molecule.31
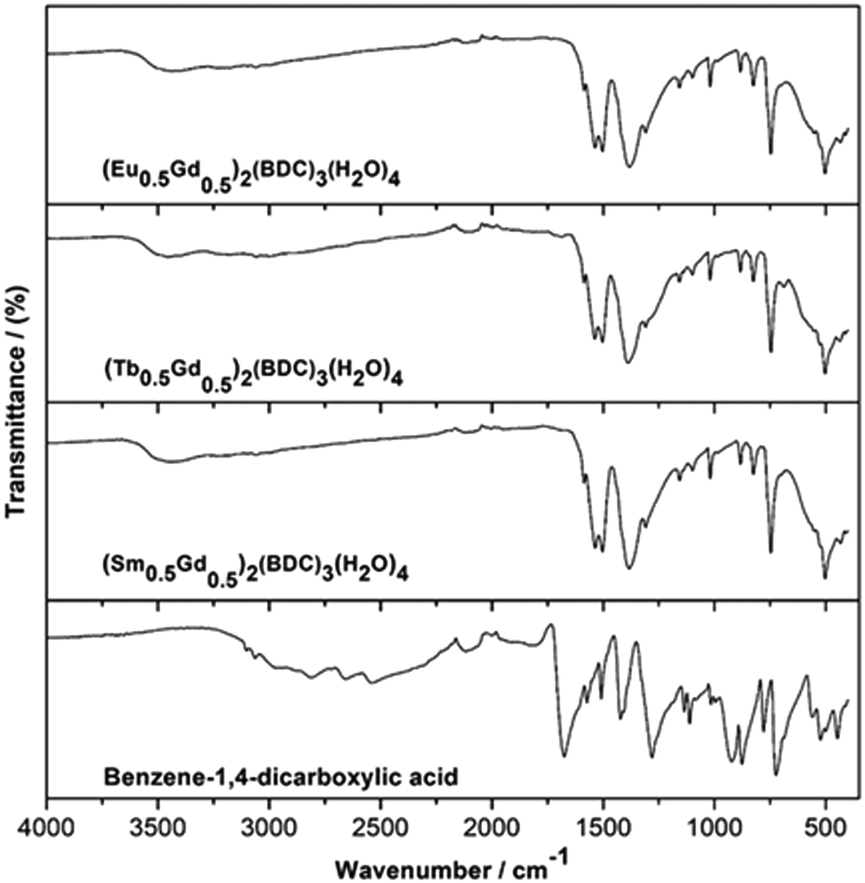 | ||
| Fig. 5 IR spectra of as-prepared (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Sm, Eu and Tb and benzene-1,4-dicarboxylic acid, for comparison. | ||
The UV-Vis absorption spectra of benzene-1,4-dicarboxylic acid and as-synthesized (Ln0.5Gd0.5)2(BDC)3(H2O)4 complexes (see ESI,† Fig. S2) were measured in the solid-state at room temperature. The absorption spectra of (Ln0.5Gd0.5)2(BDC)3(H2O)4 and Ln2(BDC)3(H2O)4 are clearly dominated by features similar to those of benzene-1,4-dicarboxylic acid with bands occurring at 250 and 315 nm attributable to ligand centered and π–π* and n–π* transitions, respectively.32 However, a bathochromic (red-) shift can be noticed between the transitions of the free acid and the coordination polymer, especially for the n–π* transitions, which underpins that BDC2− is coordinating to the Ln3+ ions in the corresponding complexes.
The luminescent properties of the compounds were investigated at room temperature. Excitation spectra of (Ln0.5Gd0.5)2(BDC)3(H2O)4 monitored at 616, 545, and 645 nm corresponding to the characteristically intense 4G5/2–6H9/2, 5D0–7F2 and 5D4–7F5, transitions for Sm3+, Eu3+ and Tb3+ are shown in Fig. 6. All recorded excitation spectra are dominated by a high intensity broad band between 240 nm to 323 nm corresponding to the π–π* and n–π* transitions of the ligand similar to the characteristic absorption spectra of the corresponding samples (ESI,† Fig. S2). In the excitation spectrum of (Sm0.5Gd0.5)2(BDC)3(H2O)4 bands, which belong to the intraconfigurational 4f–4f transitions of Sm3+ can be detected, albeit with much weaker relative intensity. The most intense band centered at 405 nm originates from the 6H5/2–4K11/2 transition of the Sm3+ ion. For (Eu0.5Gd0.5)2(BDC)3(H2O)4, in addition to the broad absorption band, several low intensity narrow bands between 350 nm and 475 nm can be observed which can be assigned to the characteristic intraconfigurational 4f–4f transitions of Eu3+.33 The most intense bands are observed for the 7F0–5L6 (394 nm) and 7F0–5D2 transitions (465 nm). The excitation spectrum of (Tb0.5Gd0.5)2(BDC)3(H2O)4 is clearly dominated by ligand-centered transitions and the characteristic transition for Tb3+ are barely visible. At 487 nm the 7F6–5D5 transition can be weakly seen. Below 380 nm the transitions from 7F6 to the 5L and 5G manifolds are barely visible. The comparatively weak intensities of the intraconfigurational f–f transitions of the Ln3+ ions compared to the ligand-centered transitions in the excitation spectra indicates that BDC2− can transfer energy to the Ln3+ ions as the indirect excitation of the Ln3+ ions through ligand is more effective than the direct excitation of Ln3+ ions, hence, BDC2− appears to be suitable as a sensitizer (antenna) for Ln3+. A schematic energy level diagram showing intramolecular energy transfer processes is presented in Fig. S3 (ESI†).
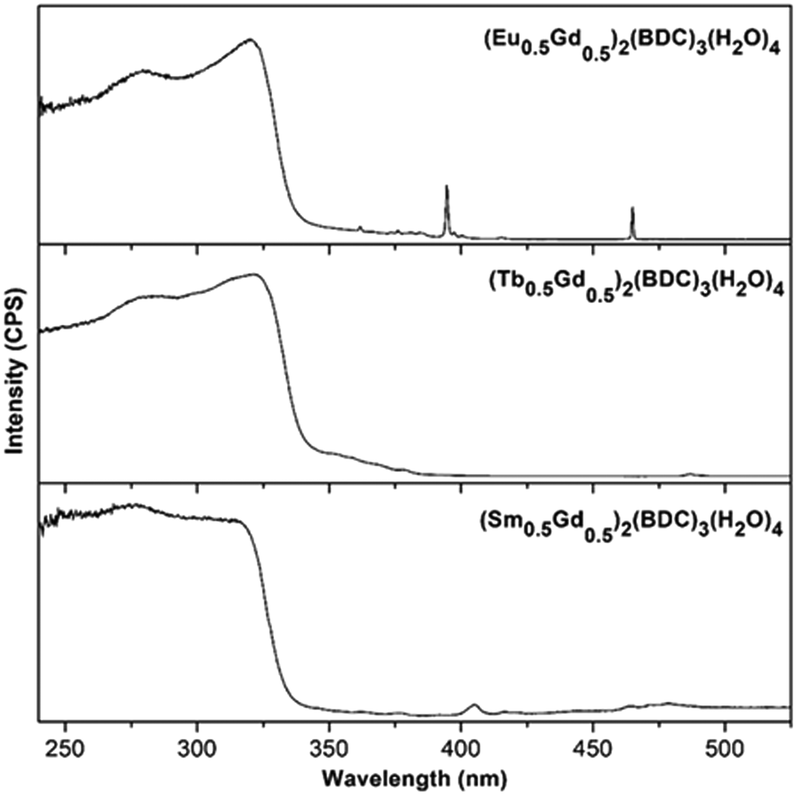 | ||
| Fig. 6 Excitation spectra of (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Eu, Tb, Sm in the solid state at room temperature monitored at 616, 545, and 645 nm, respectively. | ||
The emission spectrum of (Sm0.5Gd0.5)2(BDC)3(H2O)4 reveals the characteristic bands for the Sm3+ ion at 562, 598, 645 and 709 nm corresponding to the 4G5/2–6HJ transitions (J = 5/2, 7/2, 9/2, 11/2) when excited at 315 nm (Fig. 7). The 4G5/2–6H7/2 and 4G5/2–6H9/2 transitions are most prominent bands and they are responsible for the orange-red colour of emission. The hypersensitive electric dipole 4G5/2–6H9/2 transition is sensitive to the symmetry of coordination environment, while the magnetic dipole 4G5/2–6H5/2 transition is insensitive to the coordination environment of the Sm3+ ion. It is clearly seen from Fig. 7 that the 4G5/2–6H9/2 transition is more intense than the 4G5/2–6H5/2 transition. This is in agreement with the fact that the Sm3+ ions are located at a non-centrosymmetric site.34
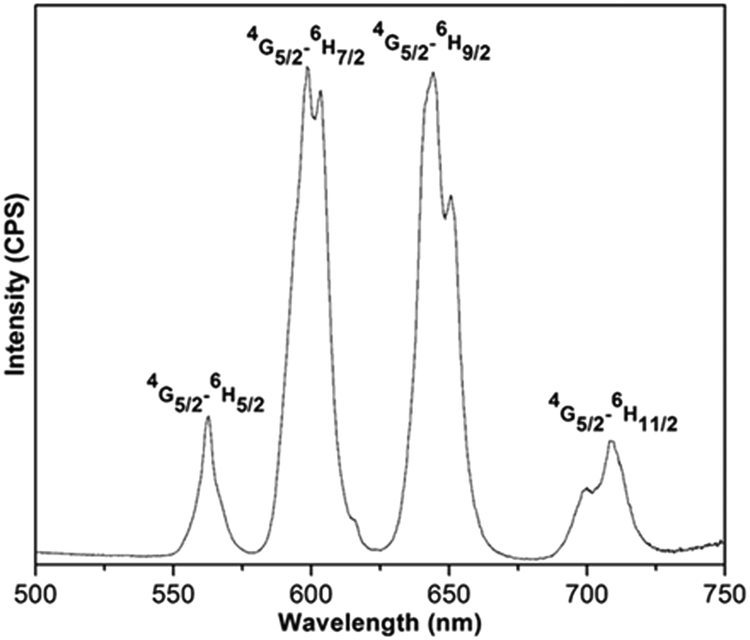 | ||
| Fig. 7 The emission spectrum of (Sm0.5Gd0.5)2(BDC)3(H2O)4 in the solid state at room temperature. | ||
Upon excitation at 320 nm, the (Eu0.5Gd0.5)2(BDC)3(H2O)4 emission spectrum reveals the characteristic Eu3+ luminescence originating from the 5D0–7FJ (J = 0, 1, 2, 3, 4) transitions (Fig. 8). Based on the selection rules of the electric dipole transition, the existence of a single 5D0–7F0 transition at 580 nm informs that the Eu3+ ions occupy a site without inversion symmetry, in agreement with the crystal structure. The weak band at 590 nm (5D0–7F1) belongs to a magnetic dipole transition, which is independent of the coordination and local environment of the Eu3+ ion. The relative emission intensity of 5D0–7F2 transition is much higher than the 5D0–7F1 transition confirming the presence of low symmetry without centre of inversion around the Eu3+ ions. Furthermore, the hypersensitive electric dipole 5D0–7F2 transition centered on 616 nm is the strongest band. This band is dependent on the local environment around Eu3+ ions and is responsible for the red emission. The asymmetry ratio (I) of (5D0–7F2) to (5D0–7F1) was estimated to be 5.08 for (Eu0.5Gd0.5)2(BDC)3(H2O)4. No emission band from ligand centered transition could be observed in the emission spectra of either of the Eu3+ based complex indicating an efficient operative intramolecular energy transfer from the BDC2− ligand to Eu3+ ions.
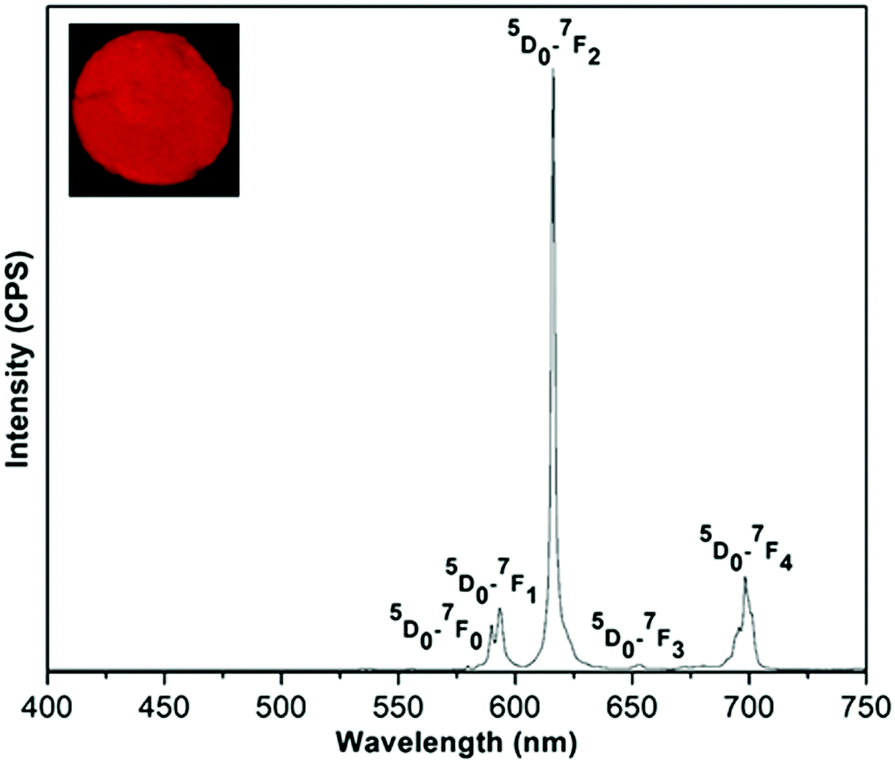 | ||
| Fig. 8 The emission spectrum of (Eu0.5Gd0.5)2(BDC)3(H2O)4 in the solid state at room temperature under excitation with 320 nm. The inset shows luminescence of the sample under UV light (255 nm). | ||
For (Tb0.5Gd0.5)2(BDC)3(H2O)4, under excitation at 323 nm, the typical narrow emission bands of Tb3+ ions arising from 5D4–7FJ (J = 0–6) transitions at 488 (5D4–7F6), 545 (5D4–7F5), 587 (5D4–7F4), 623 (5D4–7F3), 651 (5D4–7F2), 669 (5D4–7F1), and 681 nm (5D4–7F0) can be seen (Fig. 9).33 The hypersensitive 5D4–7F5 transition is the most intense band and gives a rise to the green emission for both Tb-based MOFs. The splitting observed in this band confirms the low site symmetry around Tb3+ ions.
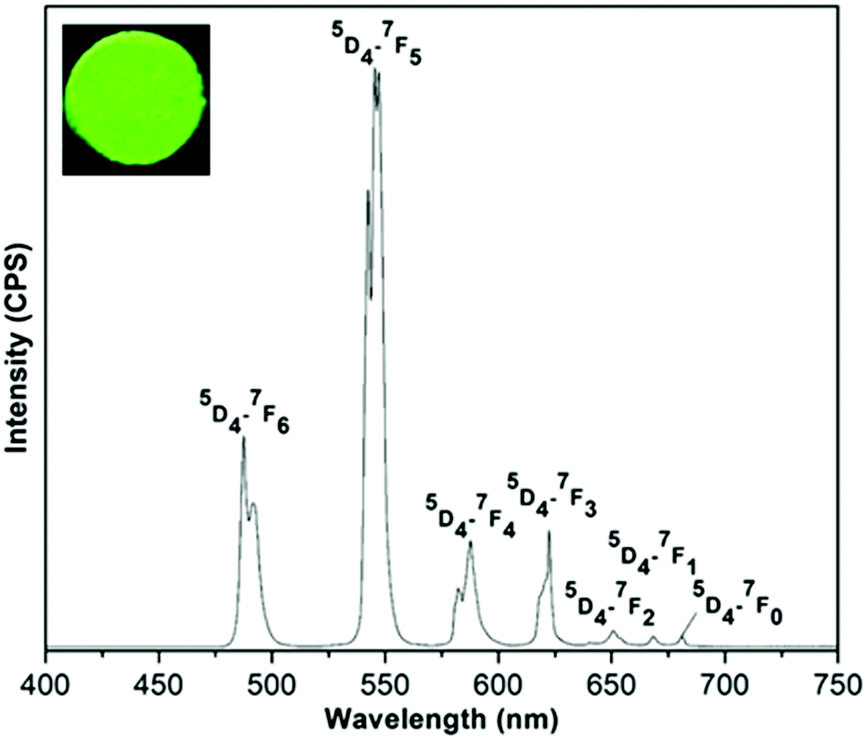 | ||
| Fig. 9 The emission spectra of (Tb0.5Gd0.5)2(BDC)3(H2O)4 in the solid state at room temperature under excitation at 323 nm. The inset shows luminescence of the sample under UV light (255 nm). | ||
The CIE chromaticity coordinates (x, y) of the mechanochemically synthesized (Ln0.5Gd0.5)2(BDC)3(H2O)4 samples calculated from corresponding emission spectra are listed in Table 3. As shown in Fig. 10, the colour coordinates of compounds are located in the orange-red for (Sm0.5Gd0.5)2(BDC)3(H2O)4, the red region for (Eu0.5Gd0.5)2(BDC)3(H2O)4 and in the green for (Tb0.5Gd0.5)2(BDC)3(H2O)4. These values are very close to the reported data by the National Television System committee (NTSC) for primary red (0.630, 0.340) and primary green (0.310, 0.595) indicating that these materials would be of interest in commercial applications.35
| Sample | CIE coordinates (x, y) | Lifetime τ (ms) | Quantum yield (%) |
|---|---|---|---|
| (Sm0.5Gd0.5)2(BDC)3(H2O)4 | 0.539, 0.341 | 0.062, 0.006 | 0.1 |
| (Eu0.5Gd0.5)2(BDC)3(H2O)4 | 0.659, 0.336 | 0.287 | 2.4 |
| (Tb0.5Gd0.5)2(BDC)3(H2O)4 | 0.322, 0.596 | 0.811 | 22 |
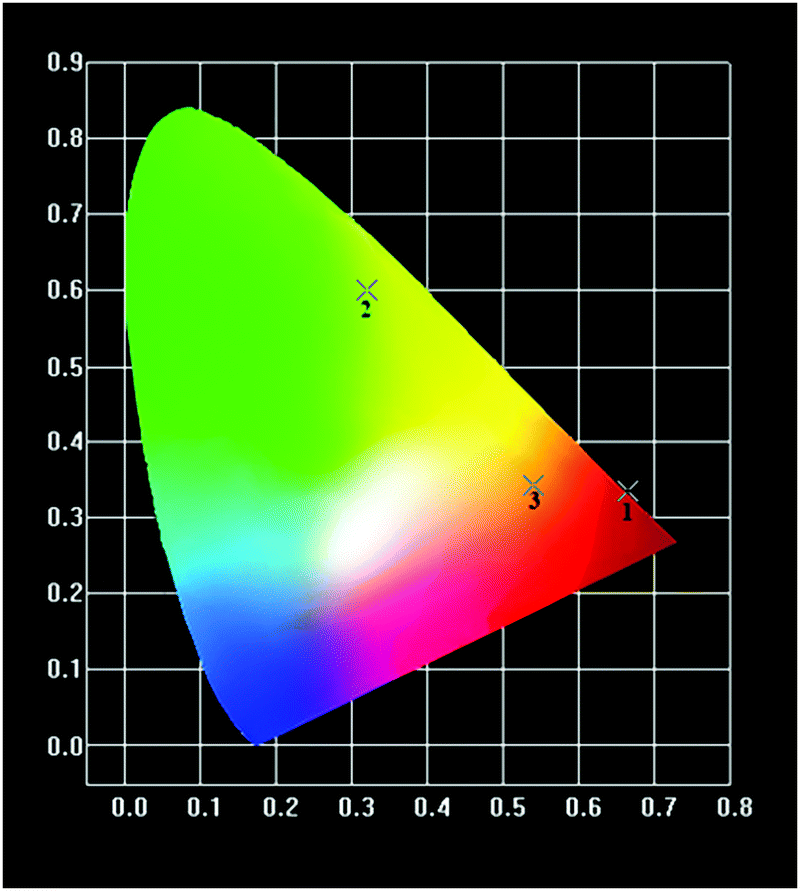 | ||
| Fig. 10 1931 CIE chromaticity diagram of (Sm0.5Gd0.5)2(BDC)3(H2O)4 (3), (Eu0.5Gd0.5)2(BDC)3(H2O)4 (1) and (Tb0.5Gd0.5)2(BDC)3(H2O)4 (2). | ||
The room temperature luminescence lifetimes of (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Sm, Eu, Tb, are measured by monitoring the 4G5/2–6H7/2 transition for Sm3+, 5D0–7F2 for Eu3+ and 5D4–7F5 for Tb3+. The decay curves and fit curves are depicted in Fig. 11 and the data are listed in Table 3. The decay curve for (Sm0.5Gd0.5)2(BDC)3(H2O)4 had to be fitted by a double-exponential function:
| I = I0 + A1exp(−t/τ1) + A2exp(−t/τ2), | (1) |
| I = I0 + A1exp(−t/τ). | (2) |
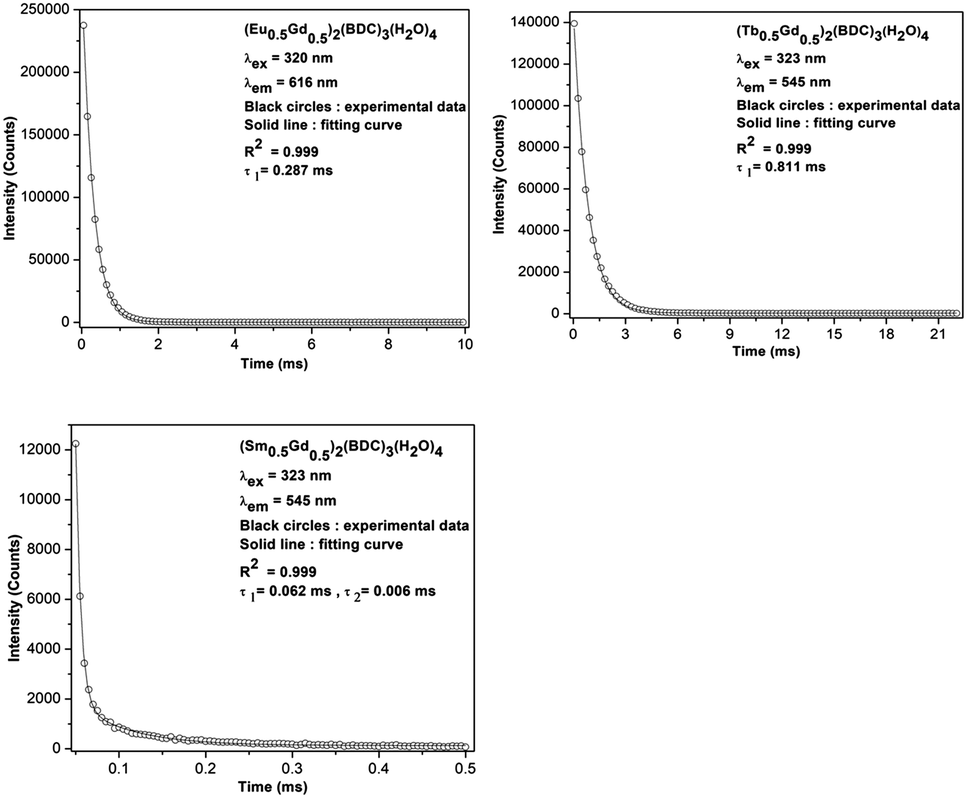 | ||
| Fig. 11 Room-temperature luminescence decay curves of (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Sm, Eu, Tb. | ||
On the basis of eqn (1) and (2), and the decay curves presented in Fig. 11, the lifetime values are calculated as τ1 = 0.062 ms and τ2 = 0.006 ms for (Sm0.5Gd0.5)2(BDC)3(H2O)4, τ = 0.28 ms for (Eu0.5Gd0.5)2(BDC)3(H2O)4 and τ = 0.81 ms for (Tb0.5Gd0.5)2(BDC)3(H2O)4 and Tb2(BDC)3(H2O)4 (Table 3). It is noteworthy to mention that the single exponential behaviour of decay curve of (Ln0.5Gd0.5)2(BDC)3(H2O)4, Ln = Eu, Tb reveals the existence of homogenous chemical environment around the Eu3+ and Tb3+ ions. In contrast, the decay pattern of (Sm0.5Gd0.5)2(BDC)3(H2O)4 shows aside from the expected decay of ions in bulk Sm3+ (Sm0.5Gd0.5)2(BDC)3(H2O)4 a small contribution of a second, fast decaying species.
The emission quantum yields of the mechanochemically prepared (Ln0.5Gd0.5)2(BDC)3(H2O)4 samples were measured in the solid-state by using integrating sphere at room temperature under the excitation wavelength of 323 nm. The recorded values of quantum yields are around 2.4% for (Eu0.5Gd0.5)2(BDC)3(H2O)4, 22% for (Tb0.5Gd0.5)2(BDC)3(H2O)4, and 0.1% for (Sm0.5Gd0.5)2(BDC)3(H2O)4 (Table 3).
The energy transfer mechanism comprises harvesting the light by the ligand then transfer the energy to the excited state of Ln3+. Latva et al. reported that the energy difference between the triplet state of ligand and emissive level of Ln3+ ions ΔE (T1–5DJ) for optimal energy transfer must be around 2500–4000 cm−1 for Eu3+ and 2500–4500 cm−1 for Tb3+.36 The triplet state energy value for the BDC2− ligand was determined by Zhang et al. to be 23641 cm−1.37 This value is located above the emitted levels of 5D0 of Eu3+ (17300 cm−1), 5D4 of Tb3+ (20400 cm−1) and 4G5/2 of Sm3+ (18021 cm−1) with energy differences of ΔE being 6341 cm−1 for (Eu0.5Gd0.5)2(BDC)3(H2O)4, 3241 cm−1 for (Tb0.5Gd0.5)2(BDC)3(H2O)4 and 5620 cm−1 for (Sm0.5Gd0.5)2(BDC)3(H2O)4. Therefore, the longer lifetime and higher quantum yield of (Tb0.5Gd0.5)2(BDC)3(H2O)4 compared to (Eu0.5Gd0.5)2(BDC)3(H2O)4, and (Sm0.5Gd0.5)2(BDC)3(H2O)4 can be explained on the basis of the respective ΔE, which is in the ideal energy difference range for an effective energy transfer only for the Tb-containing compound. In the case of the Sm3+ based compound, the short lifetime and the nearly complete quenching of emission can be attributed to the large ΔE (T1–5G5/2) value as well as the small gaps between the emitting 5G5/2 level and final the 6HJ levels (about 7500 cm−1), where nonradiative deactivation is favoured.
Fig. 12 shows the temperature dependent dc-magnetic susceptibility for all the three samples measured in two different fields: 100 Oe and 1 kOe using two protocols namely ZFC, in which samples were cooled down to 5 K in zero magnetic field and FC in which samples were cooled down to 5 K in the presence of magnetic field. Magnetic data were recorded during heating cycles for each protocol. The dc-susceptibility was calculated as χ = M/H. For all the samples, χ(T) increases continuously with decreasing temperature and shows the same trend irrespective of measurement protocols and magnetic field which indicate absence of any thermomagnetic irreversibility typically seen in materials with long-range magnetic ordering.38 Similar M vs. T behavior was also seen previously in the mechanochemically synthesized lanthanide MOFs with MIL-78 structure.17 The paramagnetic behavior of the samples were further verified by linear χ−1vs. T curves passing through T = 0 K in accordance with Curie–Weiss law (see insets in Fig. 12a–c). The effective magnetic moments, μeff for the samples as mentioned in insets were calculated from the slope of χ−1vs. T plots. For (Ln0.5Gd0.5)-BDC MOF with Ln = Sm and Tb, the experimental μeff (8.37 and 12.28 μB per f.u. respectively) are very close to the theoretical values (7.99 and 12.56 μB per f.u.), considering 3+ oxidation states for Ln atoms. However, for Ln = Eu, the value is slightly higher than the theoretical (10.06 vs. 7.94 μB per f.u.). This is not surprising because even for Eu3+ the experimentally obtained μeff is often reported to be much higher compared to its theoretical value of 0 μB.39 A comparison of experimental and theoretical values of μeff for four relevant lanthanide atoms are shown in Fig. S4 in ESI.†
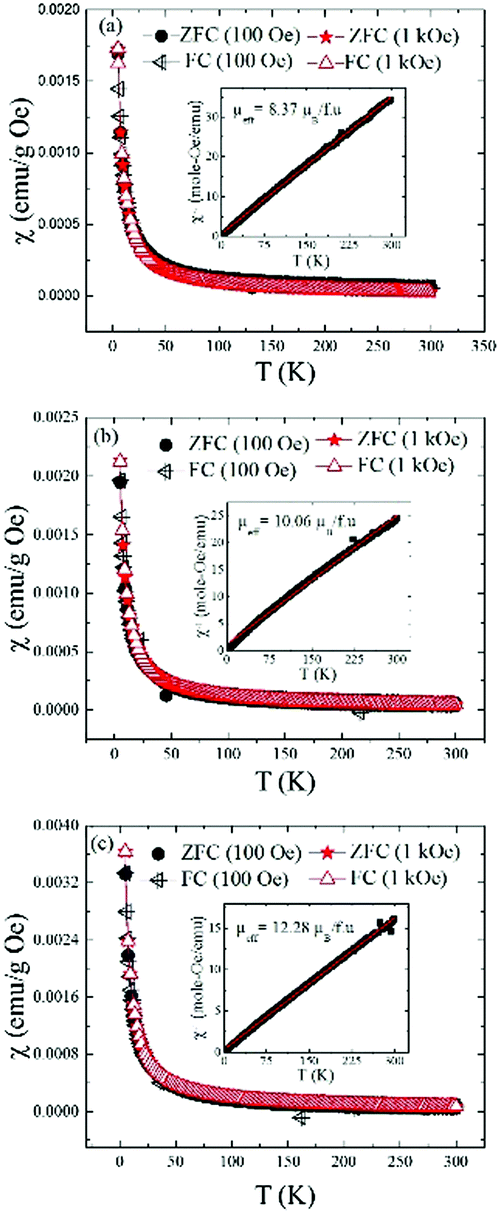 | ||
| Fig. 12 Temperature dependent dc magnetic susceptibility plots for as-synthesized (Ln0.5Gd0.5)-BDC MOFs. The data shown were collected in the zero-field cooled (ZFC) and field cooled (FC) condition on heating under the applied magnetic field of 100 Oe and 1 kOe for Ln = (a) Sm, (b) Eu and (c) Tb respectively. The insets show the inverse susceptibility plots (black symbols) and the Curie–Weiss fits (red). | ||
Conclusions
In summary, three new coordination polymers have been successfully synthesized from lanthanide carbonates and benzene-1,4-dicarboxylate by solvent-free mechanochemistry. Powder X-ray diffraction measurements confirmed that the compounds are isostructural with Tb2(1,4-BDC)3(H2O)4. The luminescent properties of (Ln0.5Gd0.5)2(BDC)3(H2O)4 Ln = Eu, Sm, Tb were investigated in the solid-state at room temperature. The absence of any emission from ligand in the emission spectra demonstrates the utility of BDC2− ligand as a good sensitizer. This can be explained from the position of the ligand T-level with respect to the emitting Ln3+ ions levels. The observed lifetimes range from 0.811 ms for (Tb0.5Gd0.5)2(BDC)3(H2O)4 and 0.287 ms for (Eu0.5Gd0.5)2(BDC)3(H2O)4 to 0.006 ms for (Sm0.5Gd0.5)2(BDC)3(H2O)4 and the quantum yields vary from 22% for (Tb0.5Gd0.5)2(BDC)3(H2O)4 to 0.1% for (Sm0.5Gd0.5)2(BDC)3(H2O)4 to. The superior luminescent properties of (Tb0.5Gd0.5)2(BDC)3(H2O)4 complex can be ascribed to the suitable energy gap between the lowest triplet energy level of BDC2− ligand with the emissive level of Tb3+ which allows for the most efficient ligand-to-metal energy transfer. No long-range magnetic ordering is observed in any of the as-synthesized (Ln0.5Gd0.5)2(BDC)3(H2O)4 MOFs. The results presented here demonstrate that solvent-free mechanochemical synthesis is well suited for synthesis of not only single metal MOFs but also for the synthesis of solid-solutions of two or perhaps more than two lanthanide elements. This opens up an opportunity to tailor the composition and precisely control the dopant concentrations in the luminescent materials that is highly critical for their optimal performance.Experimental
Materials
Precursors Eu2(CO3)3·4H2O, Gd2(CO3)3·4H2O, Sm2(CO3)3·4H2O, Tb2(CO3)3·6H2O, and benzene-1,4-dicarboxylic acid (terephthalic acid, C6H4(COOH)2) were purchased from Alfa Aesar and were used without further purification.Synthesis of (Ln0.5Gd0.5)2(1,4-BDC)3(H2O)4
(Ln0.5Gd0.5)-BDC MOFs were prepared by mechanochemical reaction of mixtures of benzene-1,4-dicarboxylic acid with the respective rare-earth carbonate hydrates Ln2(CO3)3·xH2O (Ln = Sm, Eu, Tb) and Gd2(CO3)3·4H2O taken in 3:1:0 molar ratio for Ln2(BDC)3(H2O)4 and 3:0.5:0.5 molar ratio for (Ln0.5Gd0.5)2(BDC)3(H2O)4. In a typical synthesis, a total of 1 g of the stoichiometric mixture was placed into a ∼65 ml hardened steel container together with two 12.7 mm and four 6.35 mm grinding stainless steel balls (ball-to-sample ratio close to 18:1) and ball-milled in a SPEX 8000M shaker mill for up to 2 h. All milling experiments were performed under argon atmosphere.
Characterization
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Ames Laboratory is operated for the U.S. Department of Energy (DOE) by Iowa State University of Science and Technology under contract No. DE-AC02-07CH11358. Research on mechanochemical synthesis, structural and magnetic characterization was supported by the Division of Materials Sciences and Engineering of Basic Energy Sciences Program of the Office of Science of the U.S. Department of Energy. Research on optical properties was supported by the National Science Foundation, NSF, grant CHE-1465071. AVM acknowledges support from the Swedish Royal Academy of Science through the Göran Gustafsson prize in Chemistry and support through Vetenskapsrådet (research grant 2016-05405).Notes and references
- J. C. G. Bünzli, Coord. Chem. Rev., 2015, 293, 19–47 CrossRef.
- M. A. Katkova and M. N. Bochkarev, Dalton Trans., 2010, 39, 6599–6612 RSC.
- L. D. Carlos, R. A. Ferreira, V. de Zea Bermudez, B. Julian-Lopez and P. Escribano, Chem. Soc. Rev., 2011, 40, 536–549 RSC.
- Ó. Guzmán-Méndez, F. González, S. Bernès, M. Flores-Álamo, J. Ordóñez-Hernández, H. García-Ortega, J. Guerrero, W. Qian, N. Aliaga-Alcalde and L. Gasque, Inorg. Chem., 2018, 57, 908–911 CrossRef PubMed.
- S. Faulkner, S. J. Pope and B. P. Burton-Pye, Appl. Spectrosc. Rev., 2005, 40, 1–31 CrossRef CAS.
- Z. Wang, D. Ananias, A. Carné-Sánchez, C. D. Brites, I. Imaz, D. Maspoch, J. Rocha and L. D. Carlos, Adv. Funct. Mater., 2015, 25, 2824–2830 CrossRef CAS.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- (a) S.-J. Liu, S.-D. Han, J.-P. Zhao, J. Xu and X.-H. Bu, Coord. Chem. Rev., 2019, 394, 39–52 CrossRef CAS; (b) S.-J. Liu, C. Cao, C.-C. Xie, T.-F. Zheng, X.-L. Tong, J.-S. Liao, J.-L. Chen, H.-R. Wen, Z. Chang and X.-H. Bu, Dalton Trans., 2016, 45, 9209–9215 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2011, 112, 1126–1162 CrossRef PubMed.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- E. G. Moore, A. P. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2011, 112, 933–969 CrossRef PubMed.
- K. D. Harris, Nat. Chem., 2013, 5, 12–14 CrossRef CAS PubMed.
- W. Liu, K. Zhu, S. J. Teat, B. J. Deibert, W. Yuan and J. Li, J. Mater. Chem. C, 2017, 5, 5962–5969 RSC.
- D. Yuan, G. S. Yi and G. M. Chow, J. Mater. Res., 2009, 24, 2042–2050 CrossRef.
- N. K. Singh, M. Hardi and V. P. Balema, Chem. Commun., 2013, 49, 972–974 RSC.
- N. K. Singh, S. Gupta, V. K. Pecharsky and V. P. Balema, J. Alloys Compd., 2017, 696, 118–122 CrossRef CAS.
- M. Klimakow, P. Klobes, A. F. Thünemann, K. Rademann and F. Emmerling, Chem. Mater., 2008, 47, 5216–5221 Search PubMed.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- P. Zhang and S. Dai, J. Mater. Chem. A, 2017, 5, 16118–16127 RSC.
- T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651–1657 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- L.-N. Jin, Q. Liu, Y. Lu and W.-Y. Sun, CrystEngComm, 2012, 14, 3515–3520 RSC.
- L. Pan, N. Zheng, Y. Wu, S. Han, R. Yang, X. Huang and J. Li, Inorg. Chem., 2001, 40, 828–830 CrossRef CAS.
- R. Vercaemst, D. Poelman, L. Fiermans, R. L. Van Meishaeghe, W. H. Laflere and F. Cardon, J. Electron Spectrosc. Relat. Phenom., 1995, 74, 45–56 CrossRef CAS.
- S. Kumar, R. Prakash, R. J. Choudhary and D. M. Phase, Mater. Res. Bull., 2015, 70, 392–396 CrossRef CAS.
- W. Cartas, R. Rai, A. Sathe, A. Schaefer and J. F. Weaver, J. Phys. Chem. C, 2014, 118, 20916–20926 CrossRef CAS.
- L. Song and M. Rongjun, J. Cryst. Growth, 1996, 169, 190–192 CrossRef.
- B. Vallina, J. D. Rodriguez-Blanco, A. P. Brown, J. A. Blanco and L. G. Benning, J. Nanopart. Res., 2013, 15, 1438 CrossRef.
- A. Sungur and M. Kizilyalli, J. Less-Common Met., 1983, 93, 419–423 CrossRef CAS.
- R. J. Batrice, A. K. Adcock, P. M. Cantos, J. A. Bertke and K. E. Knope, Cryst. Growth Des., 2017, 17, 4603–4612 CrossRef CAS.
- A. Gilbert and J. Baggott, Essentials of Molecular Photochemistry, CRC Press, Boca Raton, FL, 1991, pp. 87–89 Search PubMed.
- G. H. Dieke and H. M. Crosswhite, Appl. Opt., 1963, 2, 675–686 CrossRef CAS.
- V. Haquin, M. Etienne, C. Daiguebonne, S. Freslon, G. Calvez, K. Bernot, L. Le Pollès, S. E. Ashbrook, M. R. Mitchell, J.-C. Bünzli, S. V. Eliseeva and O. Guillou, Eur. J. Inorg. Chem., 2013, 3464–3476 CrossRef CAS.
- L. De Marsh, TV Display Phosphors/Primaries – Some History, SMPTE J., 1993, 1095–1098 Search PubMed.
- M. Latva, H. Takalo, V. Mukkala, C. Matachescu, J. Rodriguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- H.-J. Zhang, R.-Q. Fan, P. Wang, X.-M. Wang, W. Chen, X.-B. Zheng, K. Li and Y.-L. Yang, J. Inorg. Organomet. Polym. Mater., 2014, 24, 624–632 CrossRef CAS.
- F.-S. Guo, Y.-C. Chen, J.-L. Liu, J.-D. Leng, Z.-S. Meng, P. Vrabel, M. Orendac and M.-L. Tong, Chem. Commun., 2012, 48, 12219–12221 RSC.
- R. L. Carlin, Magnetochemistry, Springer, NY, 1986, ch. 9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nj02583a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 |