
Implementation of ferroelectric materials in photocatalytic and photoelectrochemical water splitting
Yi
Li
a,
Jun
Li
 b,
Weiguang
Yang
*a and
Xudong
Wang
*b
b,
Weiguang
Yang
*a and
Xudong
Wang
*b
aDepartment of Electronic Information Materials, School of Materials Science and Engineering, Shanghai University, Shanghai 200444, China. E-mail: wgyang@shu.edu.cn
bDepartment of Materials Science and Engineering, University of Wisconsin-Madison, Madison, WI 53706, USA. E-mail: xudong.wang@wisc.edu
First published on 18th June 2020
Abstract
As a promising technology for sustainable hydrogen generation, photocatalytic (PC) and photoelectrochemical (PEC) water splitting have gathered immense attention over a half-century. While many review articles have covered extensive research achievements and technology innovations in water splitting, this article focuses on illustrating how the ferroelectric polarization influences charge separation and transportation in photocatalyst heterostructures during PC and PEC water splitting. This article first discusses the fundamentals of PC and PEC water splitting and how these electrochemical processes interact with the ferroelectric polarization-induced interfacial band bending, known as piezotronics. A few representative ferroelectric material-based heterogeneous photocatalyst systems are then discussed in detail to illustrate the effects of polarization, space charge region, and free charge concentration, which are critical factors determining the ferroelectric influences. Finally, a forward looking statement is provided to point out the research challenges and opportunities in this promising interdisciplinary research field between ferroelectrics and electrochemistry for clean energy applications.
1. Introduction
Confronted with a population boom and technological progression, it is becoming critical for the human community to rely more and more on renewable energy in order to improve the air quality and to mitigate the greenhouse effect for our vulnerable earth. Our environment has been dramatically influenced by human activities. For example, the temperature of the earth has increased at a staggering rate caused by the emissions of greenhouse gas from a huge number of cars that consume fossil fuels. Environmental friendly transportations such as hydrogen fuel cell vehicles (HFCVs) are gradually being manufactured to ensure the sustainable development of human society. HFCVs mainly depend on electricity which is derived from the electrochemical oxidation of hydrogen.1 Apart from being carbon-free and non-toxic, hydrogen has the highest energy-to-mass ratio of any chemical.2 The use of HFCVs to replace gasoline internal combustion engine vehicles can bring many benefits and reduce the reliance on diminishing fossil fuels.3–6 Therefore, extensive research has been allocated to the discovery of reliable methods for hydrogen production.Hydrogen can typically be produced by three energy resources, e.g. fossil fuels, nuclear energy, and renewable energies.7 At present, the majority of hydrogen is produced from fossil fuels via steam reforming and gasification. This technology is commercialized with a reasonably low cost and high production rate. However, it still causes the emission of carbon dioxide. Another approach is to use nuclear reactors to provide high temperature for hydrogen generation through thermochemical water splitting. Nevertheless, most of the existing nuclear power plants are not able to provide sufficiently high temperature.8 Nowadays, producing hydrogen from renewable energy resources has been generally considered as a promising strategy that is sustainable and environmental friendly. A typical route is based on photocatalytic (PC) or photoelectrochemical (PEC) processes by directly harvesting energy from solar illumination, which is also called artificial photosynthesis.
PC or PEC water splitting is potentially an effective way to generate hydrogen with good sustainability and minimal pollution.9–11 Meanwhile, it still faces many challenges, such as low efficiency and high capital cost. Since the first report of titanium dioxide (TiO2) for water splitting by Fujishima and coworkers,12 metal oxides, particularly TiO2, have intrigued great interests owing to their high stability and superior electrochemical properties.13–15 Nonetheless, these oxides typically have limited visible light absorption due to their wide bandgap. Besides, the recombination of photogenerated electrons and holes associated with large interior and surface defects also largely reduces the energy conversion efficiency.16–21 So far, many strategies have been applied to oxide materials to improve the charge separation and transport properties in order to move the overall efficiency close to the theoretical level.22–24 Typical strategies include surface functionalization, building heterojunctions, and tuning defects.25–27 Distinguished from all these chemical and structural modifications, using ferroelectric polarization to modulate the interfacial electronic structure recently stood out as a unique strategy to control electrochemical processes.28–30 Known as a unique application direction of piezotronics, it offers a new opportunity to boost the water splitting efficiencies beyond other conventional approaches. In this article, we will review the most current implementations of ferroelectric materials in both PC and PEC water splitting processes. The mechanism of how the internal electric field from ferroelectric materials is used to enhance water splitting performance will be discussed in detail. Representative application examples of ferroelectric materials in water splitting will be compared and analyzed. Finally, the prospective outlook of this promising subfield of water splitting will be presented.
2. Photocatalytic and photoelectrochemical water splitting
Solar water splitting is a process that converts solar energy into chemical energy, which mimics the photosynthesis by green plants.31,32 From the thermodynamic perspective, splitting of water into H2 and O2 is an uphill reaction that requires a minimum Gibbs free energy of 237 kJ mol−1 to overcome the energy barrier.33 In general, the water splitting process involves two half-reactions, e.g. the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode. Depending on whether an external bias is used, solar water splitting can be divided into PC and PEC categories. In a PC system, photocatalytic semiconductors with appropriate bandgap are applied in direct contact with the electrolyte. As schematically shown in Fig. 1a, under the illumination of incident photons with an energy higher that the bandgap, electrons at the valence band (VB) are excited to the conduction band (CB), leaving holes at the VB. The photo-generated electron–hole pairs are separated driven by the built-in potential and migrate toward the corresponding OER and HER sites to produce oxygen and hydrogen, respectively.34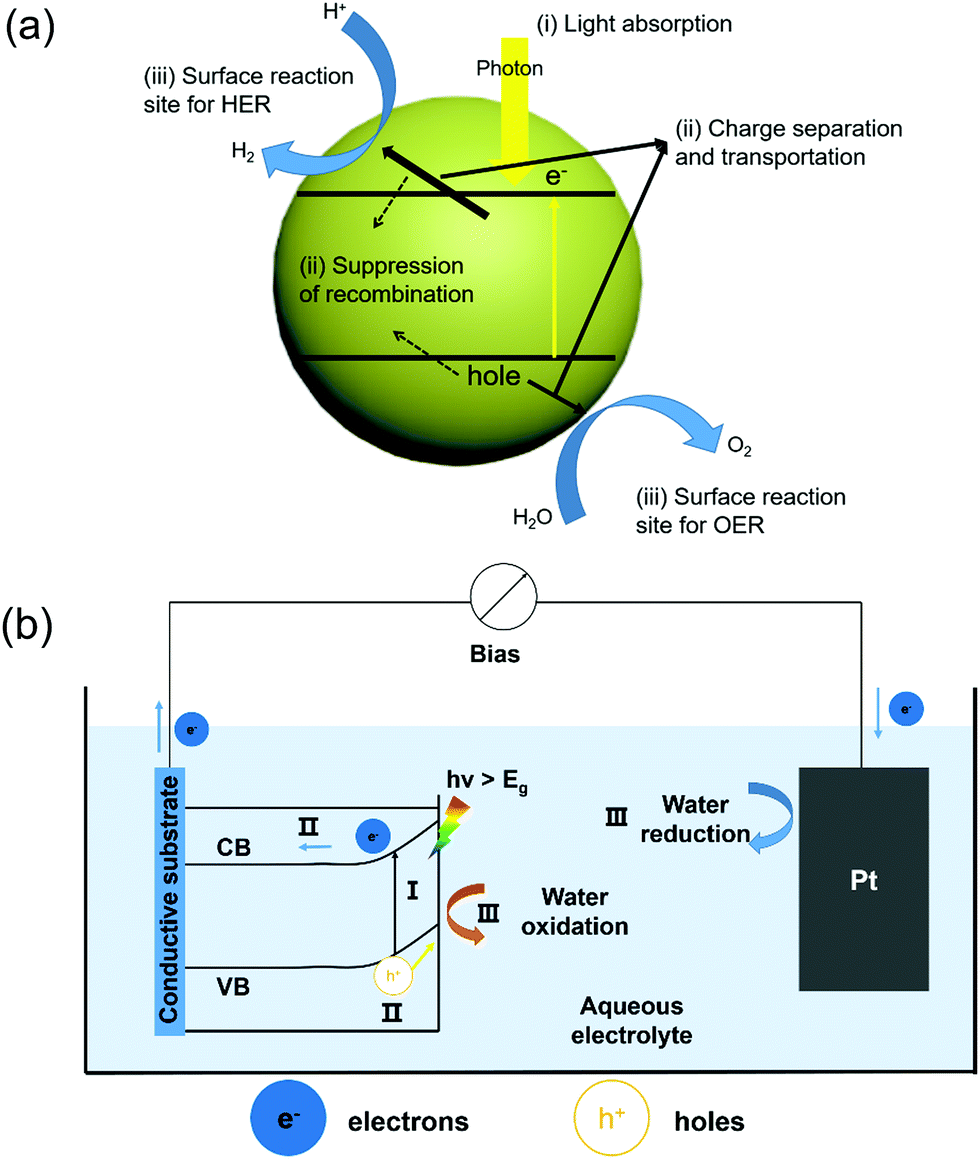 | ||
| Fig. 1 (a) Schematic of the main process of PC water splitting in a simple photocatalytic semiconductor; (b) diagram of a typical PEC water splitting system with an n-type semiconductor as the photoanode and Pt as the counter electrode, both immersed in an aqueous electrolyte. In the picture, process I refers to photon absorption and the resulting excitation of an electron from the VB to CB, leaving a hole at the VB. Process II refers to the separation and transportation of photogenerated electron and hole pairs. Process III refers to the surface redox reactions, where water oxidation occurs at the semiconductor surface and the reduction reaction occurs at the Pt surface. | ||
This system has a simple configuration and a good potential for scaling up. The materials usually present a nanoscale structure in order to increase the specific surface area as well as the reaction sites. However, this compact system also brings two disadvantages. First, there is a high tendency for the photogenerated electrons and holes to recombine instead of drifting to the surface reaction sites.35 Second, the products (H2 and O2) are spatially confined to the vicinity of the material surface, which could easily lead to back reactions. These challenges placed a significant hurdle for improving the overall efficiency near to the theoretical level.36–41
Compared to a PC system, a PEC system has the cathode and anode bridged between an external power source that provides additional bias toward HER and/or OER.42 As illustrated in Fig. 1b, a n-type semiconductor is applied as the photoanode for the OER and connected to the Pt counter electrode for the HER via an external bias. The semiconductor photoanode absorbs sun light and produces electron–hole pairs. Driven by the built-in electric field at the semiconductor–electrolyte interface and the external bias, these electron–hole pairs separate, where holes go across the depletion layer to the semiconductor surface for the OER, and electrons move through the external circuit toward the counter electrode for the HER. In such a system, the OER and HER occur at different locations, so that the oxygen and hydrogen could be effectively separated and collected.
Although both PC and PEC systems have been studied over decades, their efficiencies are still far below the theoretical values. There exist several key challenges in the photocatalyst design and implementation.43,44 The first key requirement for solar water splitting is the suitable bandgap position in correlation with the electrolyte redox energy levels. To satisfy the thermodynamics of water oxidation and reduction, the CB must be more negative than the proton reduction potential of H+ to H2 (0 V vs. NHE at pH = 0); while the VB must be more positive than the oxidation potential of H2O to O2 (1.23 V vs. NHE at pH = 0). Thus, the theoretical minimum bandgap for water splitting is 1.23 eV, corresponding to a wavelength of ∼1100 nm.
Therefore, it is important to select a semiconductor with an appropriate band gap in order to achieve a high light utilization efficiency. However, considering the unavoidable thermal dynamic energy losses and kinetic overpotential, a minimum band-gap of 1.8 eV is typically required to sustain the overall water splitting, equivalent to light of a wavelength of approximately 700 nm. The upper limit of the bandgap energy is 3.2 eV which corresponds to the sharp intensity drop in the solar spectrum below 390 nm. Therefore, semiconductors with a bandgap energy from 1.8 eV to 3.2 eV are appropriate in order to achieve desired water splitting efficiency.45 Considering the thermodynamic band position demands, a bandgap of 2.0–2.2 eV is favorable for achieving excellent solar light utilization.46 The bandgap alignment of typical visible-light active semiconductors is shown in Fig. 2. From this chart, many ferroelectric oxide materials fit in this category, such as SrTiO3, PbTiO3, BaTiO3, and BiFeO3. They readily incorporate ferroelectric polarization with photon excitation for PC or PEC water oxidation, reduction or overall water splitting.
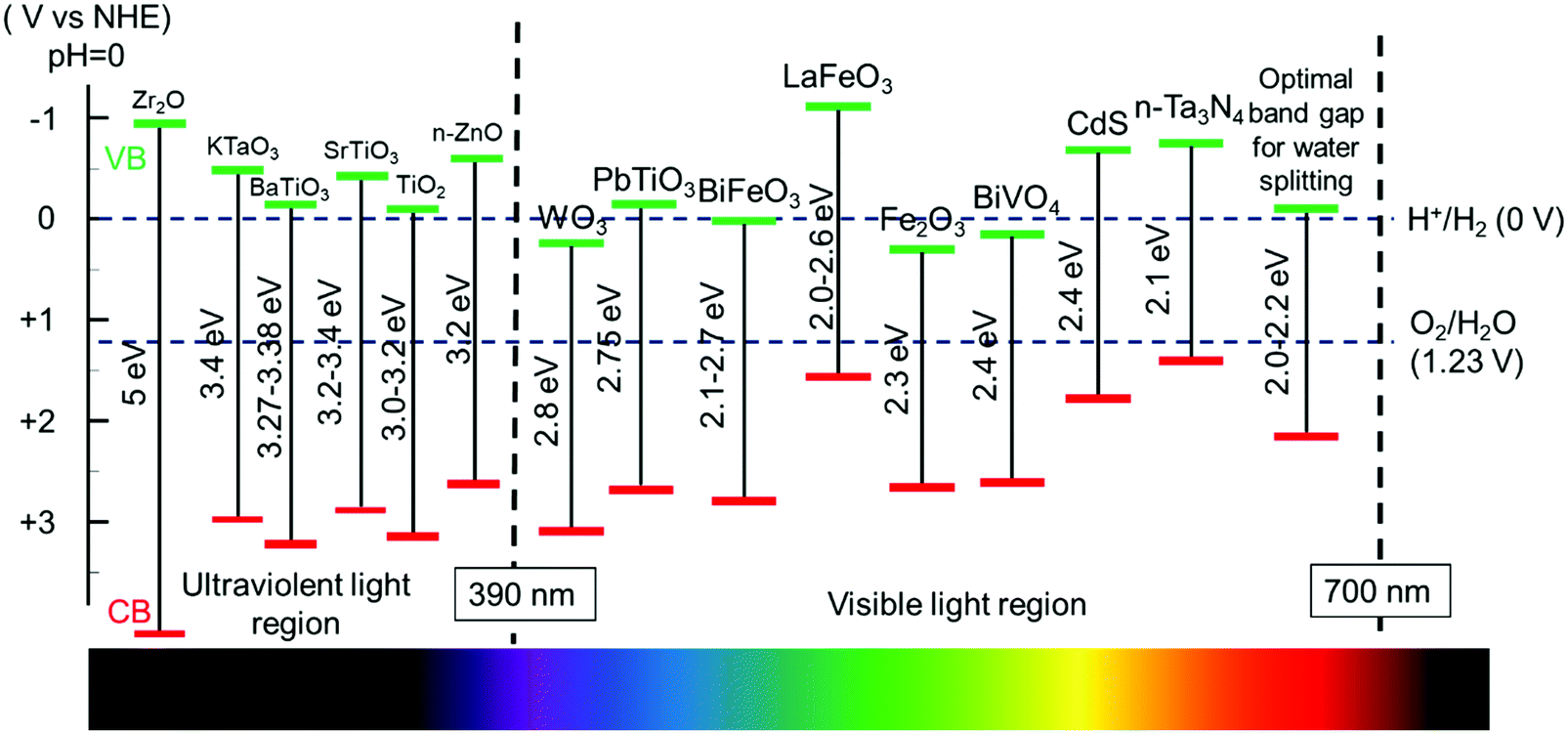 | ||
| Fig. 2 Band edge positions of different semiconductors and ferroelectric materials used in water splitting and their corresponding light absorption regions. | ||
Electron and hole separation and transportation is another critical issue for the PC and PEC system to reach a high overall efficiency. Electron–hole recombination could significantly reduce the efficiency of converting the absorbed photons into redox reactions. The electron–hole recombination is related to many intrinsic and extrinsic factors, such as surface or interface sites, internal defects and dislocations, depletion region, built-in electric field, external bias, drifting velocity, and mean free path. In general, large driving force and high crystal quality are favorable for achieving high charge separation efficiency.
Currently, the widely studied strategies to improve the charge separation and transportation in PEC and PC processes are mainly from three perspectives, e.g. morphology optimization, heterostructuring, and surface functionalization.47–50 For example, nanoscale morphology design was able to shorten the charge diffusion distance and improve the surface reaction sites.51–54 Tuning the band structure by doping or hetero-band structure coupling was found useful to improve light absorption and photo-responses.55–62 Surface decoration is also often used to minimize the surface trapping effect and lower the charge transport energy barrier.63 Nevertheless, these approaches are typically associated with physical and/or chemical alteration of the materials. Undesired coupling among the structure and properties often leads to performance improvements far below expectations.
For materials suffering from rapid recombination of charge carriers and occurrence of back reactions, tuning the internal electric field is considered the most effective strategy to mitigate the losses from recombination and back reaction. The internal electric fields in a water splitting system appear at the dissimilar interfaces, such as between the semiconductor and electrolyte, between metal and semiconductor, and at a p–n junction. Currently, four approaches have been studied to modulate the internal electric field in order to improve electron–hole separation and transportation by introducing ferroelectric polarization, a polar surface, p–n junctions, or polymorphs. As illustrated in Fig. 3,64 ferroelectric materials provide an additional internal electric field to drive charge transport through the semiconductor or across the surface. Polar surface terminations produce an additional field at the semiconductor–electrolyte interface to enhance the photochemical activity.65 A p–n junction could be built to improve the charge separation within the space charge region.66 A polymorph junction consists of two different polymorphs with the same composition, where different electronic properties at the interface can create an internal electric field and facilitate the separation of charge carriers.67–69 A similar electronic property difference within the same composition could also be achieved by gradient doping,70,71 where the internal electric field is induced by varying the space-resolved doping concentration without evolving different phases. The high structuring continuity in gradient doping may offer better charge transport behavior compared to polymorph junctions, but it can be challenging to maintain the doping concentration gradient particularly within small domains and over a long time. Among them, ferroelectric modulation is distinguished by its potential to achieve very high internal potential and good flexibility of forming junctions. It has therefore received a rapid increase of research attention, and become a popular new strategy for improving water splitting efficiency.
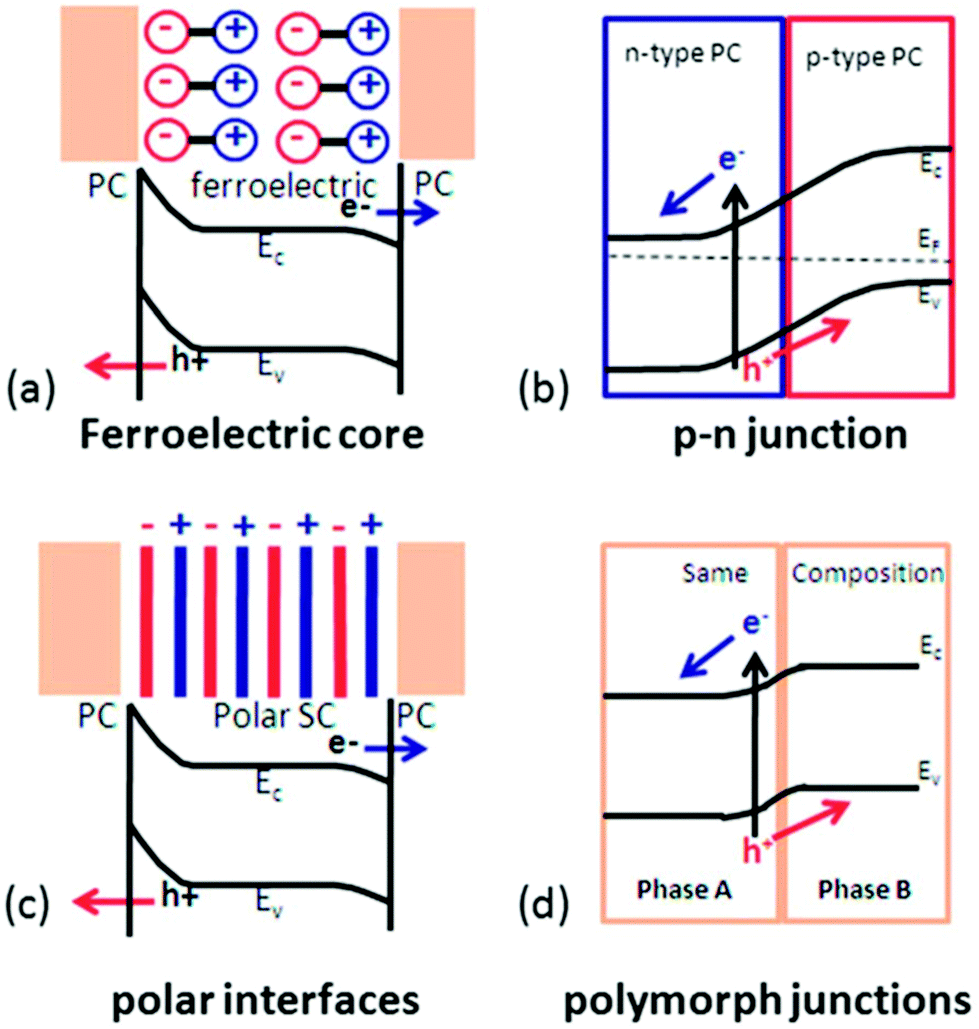 | ||
| Fig. 3 Separation of photogenerated charge carriers enhanced by four different internal electric fields: (a) ferroelectric polarization; (b) p–n junctions; (c) polar surface; and (d) polymorph junctions. (PC: photocatalytic active materials; SC: semiconductor). Adapted from ref. 64. Copyright 2014 Royal Society of Chemistry. | ||
3. Ferroelectric enhancement in water splitting
Ferroelectric materials refer to a category of materials that exhibit spontaneous electric polarization. Ferroelectricity was first reported by Joseph Valasek during his research on the dielectric properties of special salt rock.72 To date, more than 700 ferroelectric materials have been discovered and the majority of them are complex oxides.73 These materials have been widely used in many functional devices such as sensors, dielectric capacitors, non-volatile memories, actuators and energy harvesters.74–78 According to their chemical composition and crystal structure, ferroelectric materials can be divided into three major categories, including inorganic oxides; inorganic non-oxides; and organic materials.79,80 Today, the most widely studied ferroelectric materials are inorganic oxides, particularly the perovskite-based materials due to their strong ferroelectricity and wide range of applications. The definition of perovskite-based materials originates from the same crystalline unit cell as CaTiO3(CTO). The molecular composition of perovskite materials can be described as ABX3, where A and B represent the metal ions (A is usually larger than B), and X is the oxygen element. The crystal structure is shown in Fig. 4(a), and B and X form BX6 octahedra where B lies in the center and X locates at the vertex of the octahedra respectively. The BX6 octahedra link each other forming a network structure in the three-dimensional space by sharing the same X corners.81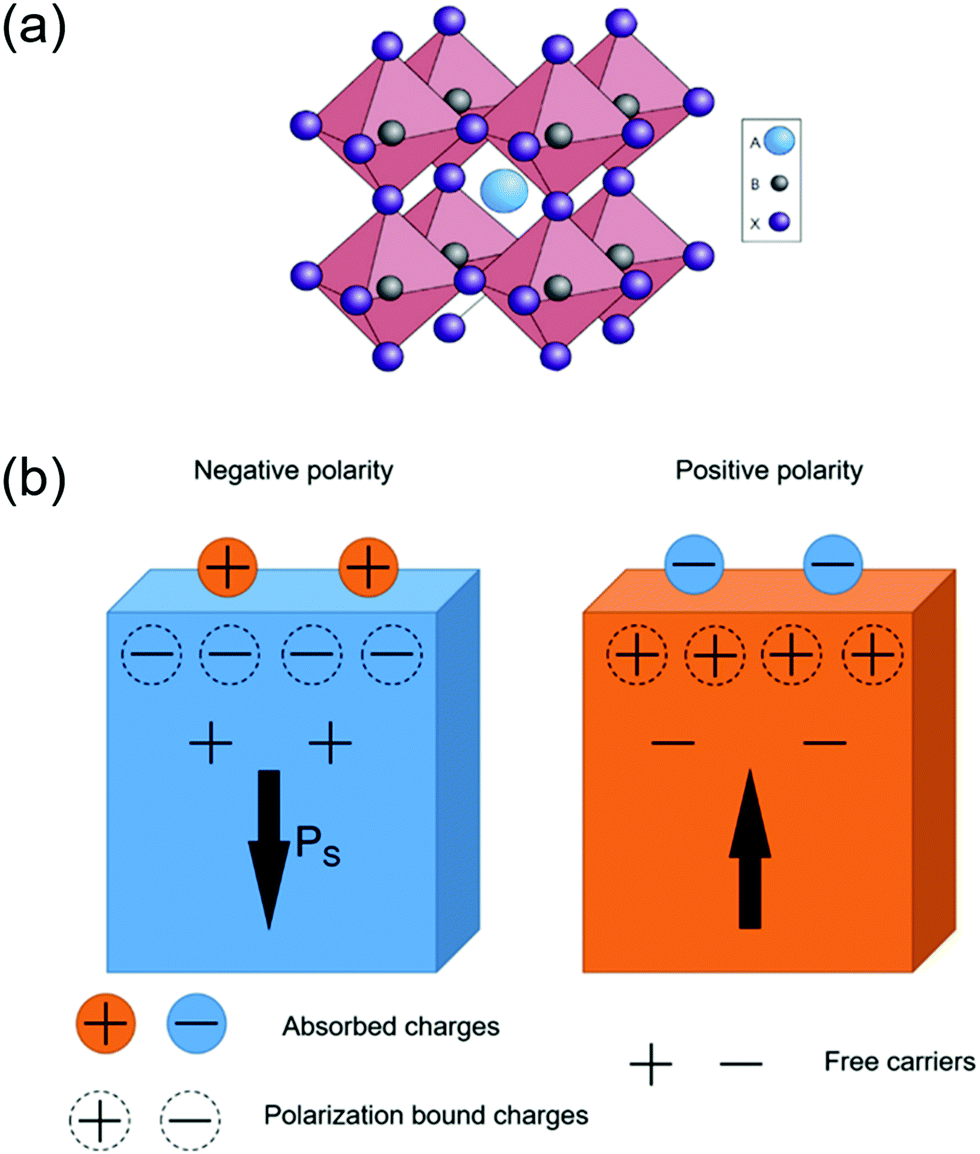 | ||
| Fig. 4 (a) The extended network structure of cubic perovskite linking through the corner-shared octahedra. Reprinted with permission from ref. 81. Copyright 2014 Nature Publishing Groups. (b) A schematic diagram of charge absorption near the polar surface. The direction of the arrow is parallel with the spontaneous polarization at the surface. Modified with permission from ref. 86. Copyright IOP Science. | ||
Ferroelectric materials possess a polar unit cell due to the distorted dimensional structure inside the bulk.82 This polar unit cell can generate a spontaneous and permanent electric polarization, where the direction is switchable by an external electric field.83–85 Deformation caused by cationic substitution can further tune the ferroelectric properties of the perovskite oxides. A ferroelectric phase can transform into a non-ferroelectric phase when the temperature exceeds a particular value. This phase transformation temperature is called the Curie temperature (TC).84 Above TC, the crystal structure of ABO3 perovskites transforms to a cubic structure with minimal polarization, which is defined as a paraelectric phase.86
The spontaneous polarization situation of ferroelectric materials is a high energy state. Therefore, this polarization is often neutralized by the flow of free charge carriers and defects within the crystal (internal screening) or by inducing electrons/holes or charged molecules/ions from the surrounding medium (external screening). The surface electronic structure, as well as the related energy band near the surface, can be modified by the internal and external screening fields. As shown in Fig. 4b, the distribution of absorbed charges is determined by the orientation of spontaneous polarization within the crystal.87,88
Photocatalytic water splitting typically relies on the internal electric field to achieve photogenerated charge separation. The spontaneous electric polarization of ferroelectrics could offer a strong internal electric field toward the photocatalytic water splitting process. Depending on the direction of ferroelectric polarization, it can enhance the separation of charge carriers and/or facilitate the water oxidation and reduction processes.89Fig. 5a illustrates how the electric field facilitates the charge separation inside a ferroelectric semiconductor. The photogenerated electron–hole pairs are forced to move towards opposite surfaces by the internal polarization.90 Specifically, C+ domain refers to the region where the spontaneous polarization points from bulk to surface and attracts free electrons; while C− domain possesses reverse dipoles from surface to bulk and induces the accumulation of holes.91 Thereby, owing to the accumulation of electrons at the C+ surface and holes at the C− surface, bands near opposite surfaces are bended downward and upward, respectively. This additional surface band bending resulting from internal polarization promotes the transportation of charge carriers to absorbed species.92 The variation of surface absorbed intermediates (hydroxyl radicals and atomic hydrogen) further determines outer surface dipoles contributing external screening.93 As such, separation of photogenerated electron–hole pairs could be significantly enhanced. In addition, the HER and OER would occur on the opposite sites determined by the internal polarization, which effectively suppresses back reaction to form water.
 | ||
| Fig. 5 (a) Schematic diagram of PC water splitting by internal and external screening due to the polarization in ferroelectric materials. (b) Schematic diagram of PEC water splitting by internal and external screening due to the polarization in perovskite-based materials. | ||
Ferroelectricity also shows strong influences in tuning the PEC performance, as ferroelectric materials are able to couple with other photoelectrode materials providing substantial impacts to the internal electrical field. A TiO2/ferroelectric perovskite heterostructure is exemplified here to illustrate the role served by a ferroelectric material in a representative PEC system. As depicted in Fig. 5b, the ferroelectric material generates an internal electric field. Analogous to the mechanisms in PC water splitting, the existence of C− and C+ domains induces upward and downward band bending near opposite surfaces, respectively. At the TiO2/perovskite interface, the negative polarization inside ferroelectrics is neutralized by holes extracted from TiO2; while the absorption of charged hydroxyl radicals from alkaline solution balances positive polarity at the ferroelectric/electrolyte interface. As a result, external screening at the TiO2/ferroelectric interface provides an additional driving force to attract holes from TiO2 into the ferroelectric layer, effectively suppressing the recombination inside TiO2. The holes injected into the ferroelectric layer will further move across the ferroelectric/electrolyte interface, participating in the OER process. Similarly, if the polarization direction is pointing away from the TiO2/ferroelectric interface, it will attract electrons from TiO2 and facilitate the HER process if an appropriate ferroelectric/electrolyte interface is created. In general, based on the direction of ferroelectric polarization and electronic band position, a ferroelectric layer can serve as an efficient pump to extract the holes(electrons) out of adjacent photocatalytic semiconductors, and facilitate the OER or HER accordingly.
4. Implementations of ferroelectric materials in water splitting
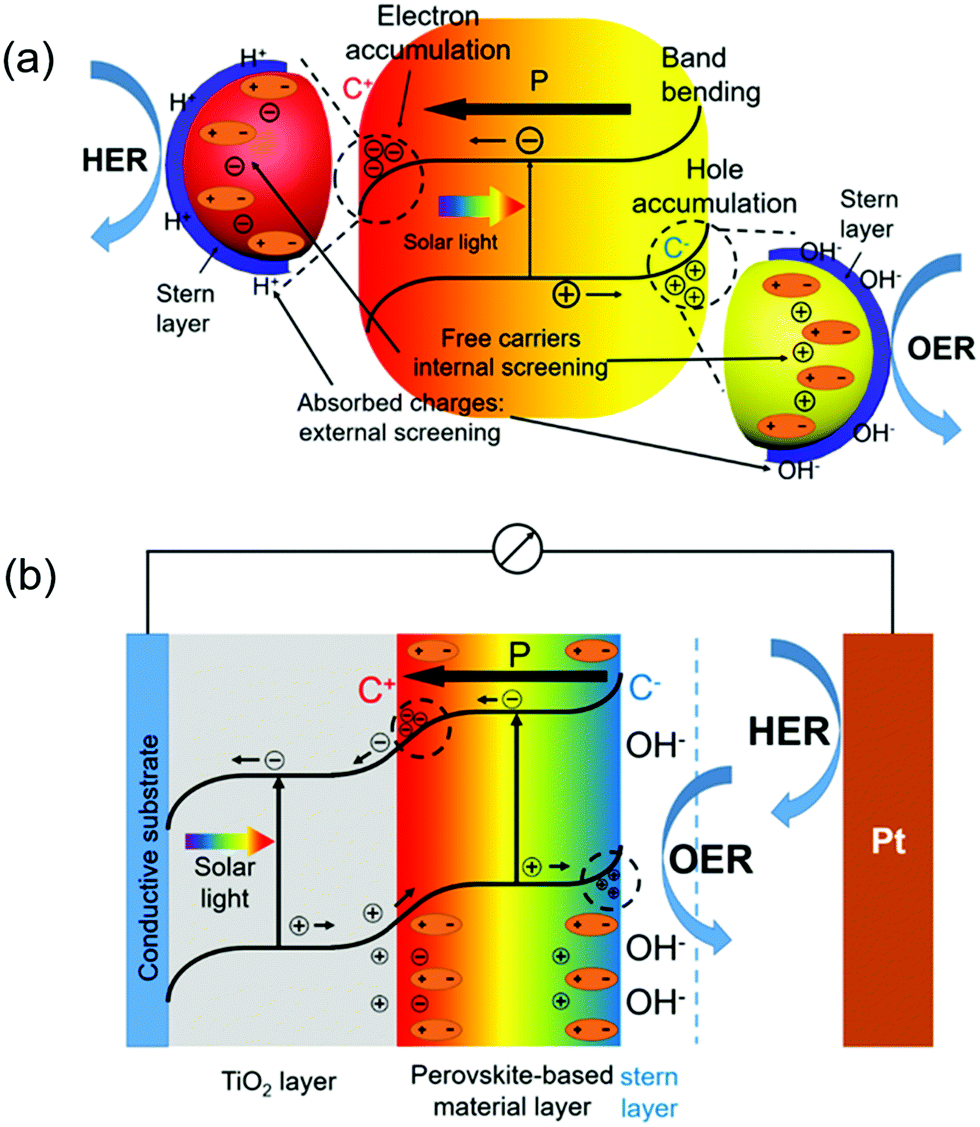
As discussed above, ferroelectric materials offer a promising solution to address the charge separation and transport challenges by introducing an additional strong electric field into the system. Over the last few years, different strategies of introducing various ferroelectrics to PC or PEC systems have been reported.94–100 In this section, the influence of ferroelectric materials on PC/PEC water splitting will be viewed and discussed.Owing to its strong ferroelectric polarization, BaTiO3(BTO) has been applied in conjunction with high-performance photocatalytic materials to improve the overall PEC efficiency. In a representative example, TiO2/BTO core/shell nanowires (NWs) were developed by a two-step hydrothermal process (Fig. 6a).101 A thin layer of BTO epitaxially covered on the TiO2 NW core (Fig. 6b), and the BTO thickness varied from 5 to 40 nm as controlled by temperature (150–210 °C). Ferroelectricity of the tetragonal BTO shell was evidenced by a polarization–electric field (PE) hysteresis loop. Fig. 6c compares the photocurrent density-potential (Jph–V) curves of pristine TiO2 NWs and TiO2/BTO NWs. The TiO2/BTO core/shell NWs yielded markedly higher Jph of 1.30 mA cm−2 at 1.23 V vs. RHE, corresponding to 67% improvements over pristine TiO2 NWs (0.78 mA cm−2 at 1.23 V vs. RHE) in a 1 M NaOH solution under AM 1.5 G illumination. Nevertheless, TiO2/BTO NWs with thick BTO shells had barely any improvements on the performance. This was primarily because the high-resistance of thicker BTO impeded the tunneling of photogenerated holes from the TiO2 core to the electrolyte. To further understand the ferroelectric impacts, potential distribution over the heterostructure was calculated. As revealed in Fig. 6d, the positive polarization in BTO induced upward band bending in TiO2. This additional band bending facilitated the separation of photogenerated electron–hole pairs. In contrast, negative polarization led to downward band bending in TiO2, which was not favorable for charge separation. The calculation was validated by the PEC test of differently poled samples. A positively poled TiO2/BTO NW photoanode enhanced Jph compared to the unpoled sample, whereas Jph and the fill factor were significantly impaired in negatively poled samples.
 | ||
| Fig. 6 TiO2/BTO core/shell NW photoanode for PEC water splitting. (a) Scanning electron microscope (SEM) images of TiO2/BTO nanowires (synthesized at 150 °C) from different views: (i) top-view and (ii) cross-section (15° titling). Scale bars are 500 nm (for i) and 1 μm (for ii) respectively. (b) Representative transmission electron microscope (TEM) image for TiO2/BTO nanowires (synthesized at 150 °C). (c) Photocurrent density–potential (J–V) curves of TiO2 NWs and TiO2/BTO NWs (150–210 °C) measured in 1 M NaOH under 1.5 G illumination. (d) Potential distribution of TiO2/5 nm BTO/NaOH heterojunction, where the spontaneous polarization in the BTO shell induced an upward band bending of the TiO2 core and facilitated the charge separation and transportation inside TiO2. (e) J–V curves of the as-prepared (red), positively poled (blue) and negatively poled (magenta) TiO2/5 nm BTO NWs. Reprinted with permission from ref. 101. Copyright 2015 ACS. | ||
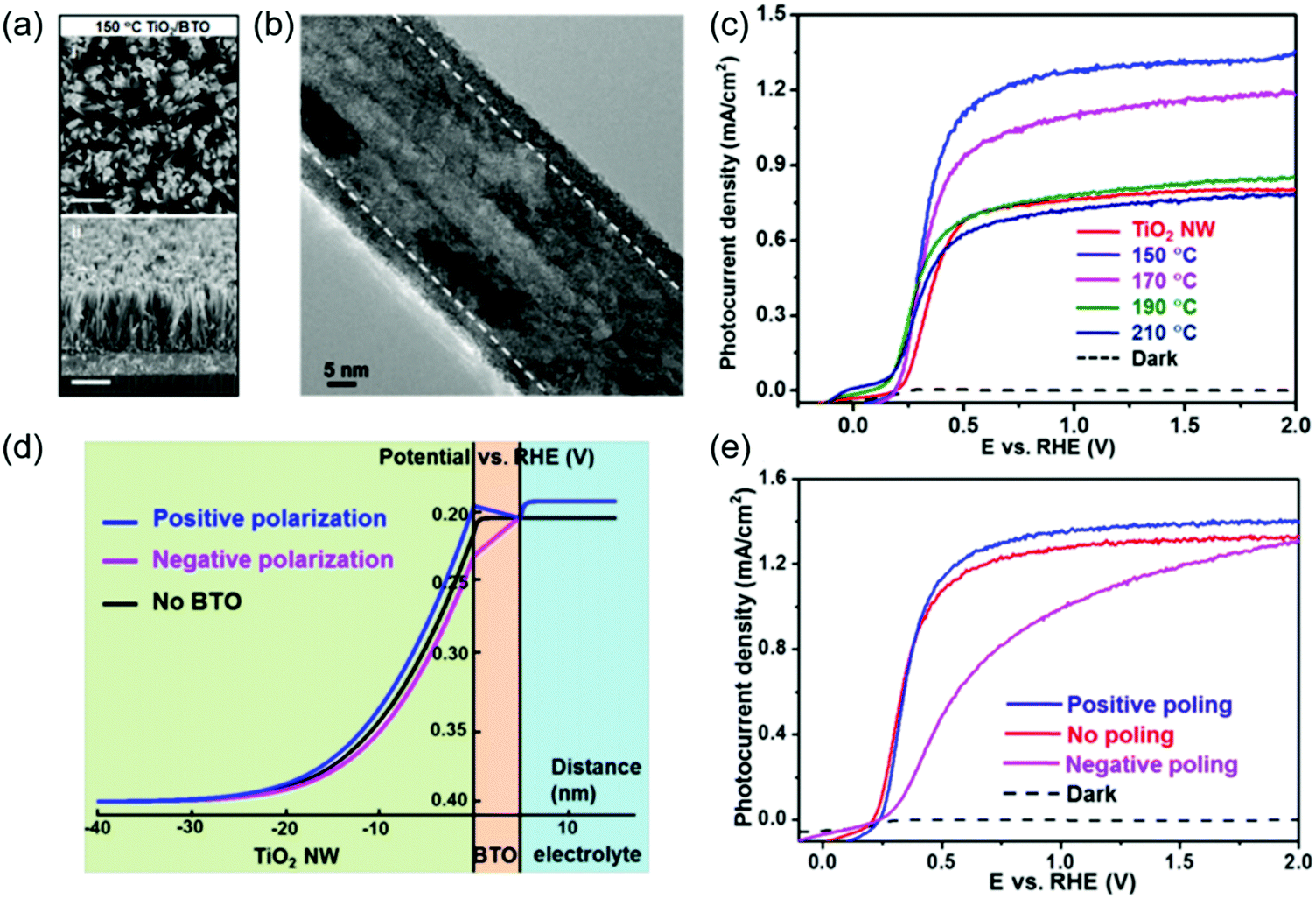
From the TiO2/BTO system, while the enhancement of the ferroelectricity was obvious, the high insulating property of BTO clearly impeded the charge transport as the BTO layer grew thicker. From a similar NW system, Wu et al. showed that changing BTO to another more conductive ferroelectric material SrTiO3 (STO) could simultaneously improve the charge separation and hole transportation.102 TiO2/STO core/shell NWs were obtained by hydrothermally converting the TiO2 NW surface to STO (Fig. 7a). High-resolution TEM evidenced a thin layer of STO shell surrounding the single-crystalline TiO2 core with various thicknesses determined by reaction time (Fig. 7b). Without poling, the as-prepared core/shell NWs exhibited obvious ferroelectricity due to the favorable crystal orientation between the TiO2 core and STO shell. The TiO2/STO heterostructure was applied as photoanodes in the PEC test (1 M NaOH under AM 1.5 G illumination). As shown in Fig. 7c, the TiO2-10 nm STO yielded the highest Jph (1.43 mA cm−2 at 1.23 V vs. RHE), corresponding to 83% improvement compared to that of pristine TiO2 (0.78 mA cm−2). This enhancement was attributed to the higher yield of the photogenerated surface-reaching holes (ηseparation). Jph–V curves of sulfite oxidation verified the highest ηseparation of TiO2–10 nm STO among all samples, which reached 87.7% at 1.23 V vs. RHE, corresponding to 79.3% improvement against pristine TiO2 NWs. The 10 nm optimal STO coating compared to the 5 nm optimal BTO coating suggested that better charge transport from STO offered higher tolerance to the ferroelectric thickness.
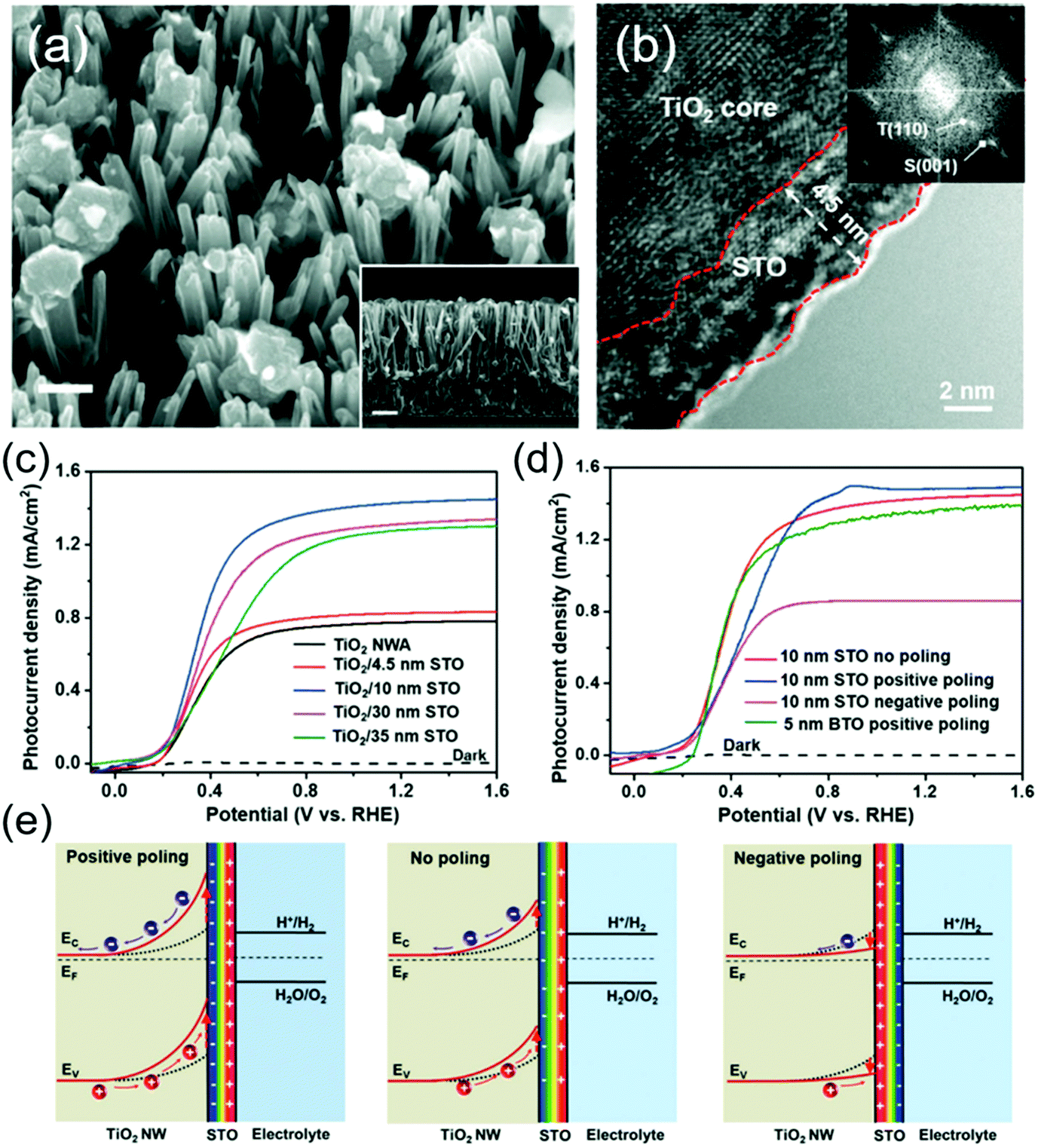 | ||
| Fig. 7 TiO2/STO core/shell NW photoanode for PEC water splitting. (a) SEM image of TiO2/STO core/shell NWs. The insert is a cross sectional SEM image. (b) HRTEM image of the core/shell NW along the edge showing a distinct crystalline 4.5 nm STO shell. The inset is the corresponding FFT image. (c) J–V curves of pristine TiO2 NW arrays and TiO2/STO core/shell NW arrays with different shell thicknesses measured in 1 M NaOH solution under AM 1.5 G illumination. (d) J–V curves of the as-prepared (red), positively poled (blue), and negatively poled (magenta) materials; and (e) schematic electronic band diagram of the TiO2/STO interface with positive poling, no poling and negative poling conditions. Reprinted from ref. 102. Copyright 2017 Wiley. | ||
Similar to the BTO system, PEC performance of this heterostructure can be further improved by positive poling (Fig. 7d). However, the amplitude of improvement was relatively small. Whereas, negative poling significantly reduced the Jph to almost half of that from the unpoled sample. This asymmetric switching behavior was attributed to the spontaneous ferroelectric polarization in the STO shell which was mostly aligned in the favorable direction during the synthesis. Numerical band structure calculation revealed that under the positive polarization, negative ferroelectric charge will present at the TiO2–STO interface and amplify the band bending of TiO2, which eventually enhances the charge separation and photocurrent density (Fig. 7e). In contrast, negative poling will switch the polarization direction and reduce the depletion width of TiO2, resulting in a substantial decrease of Jph.
Leading by these two pioneering and intriguing discoveries, more studies have been implemented to seek the best combination of ferroelectricity, charge transportation and catalytic property in ferroelectric materials and heterostructures. BiFeO3 (BFO) is a promising multiferroic material with a decent remnant ferroelectric polarization (∼90 μC cm−2).103 It has been considered as a novel platform to incorporate multiple functionalities together, including photocatalysis given its favorable bandgap. The ferroelectric polarization influence from BFO was investigated on a TiO2-based photoanode for PEC water splitting.104 Under visible light, the BFO/TiO2 thin film photoanode with a thickness of 50 nm exhibited a maximum Jph of 11.25 mA cm−2 at 1.5 V vs. SCE (in 1 M NaOH under a 300 W Xe lamp with a visible light filter), significantly higher than that achieved by pristine TiO2 (0.33 mA cm−2 at 1.5 V vs. SCE). As shown in Fig. 8a, when the BFO film was positively poled, the Jph jumped to 28.75 mA cm−2 at 1.5 V vs. SCE under the same situation. A similar amount of Jph drop was detected when the BTO film was poled negatively.
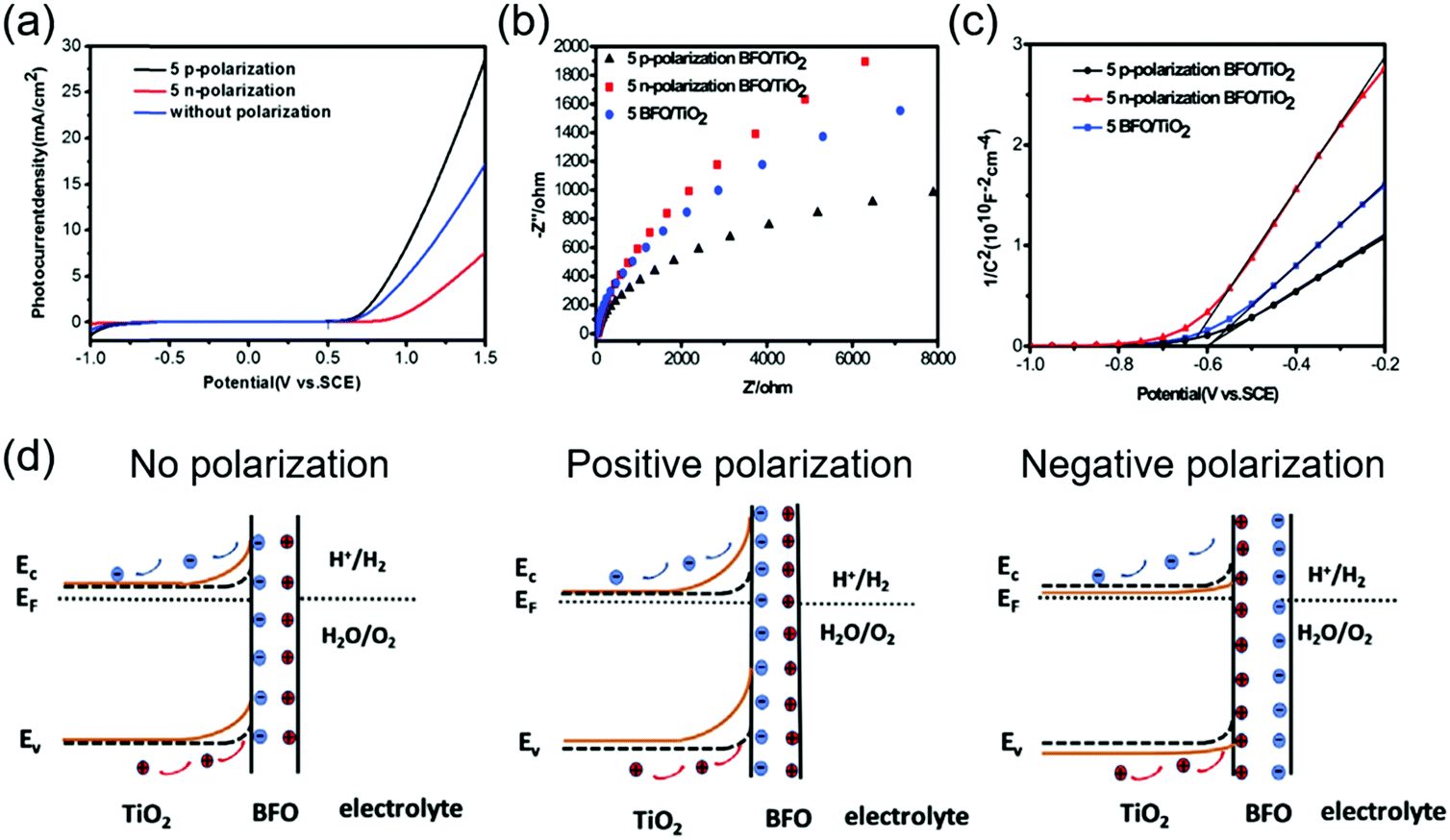 | ||
| Fig. 8 BFO/TiO2 photoanode for PEC water splitting. (a) Jph–V curves of positively and negatively poled and unpoled BFO/TiO2 under AM 1.5 G. (b) Nyquist plots and (c) Mott–Schottky plots of positively and negatively poled and unpoled BFO/TiO2 under visible light. (d) Schematic band diagram of the BFO/TiO2 photoanode interfacing with an electrolyte without poling (left), and with positive (middle) and negative (right) poling, respectively. Reprinted from ref. 104. Copyright 2019 Elsevier. | ||
Electrochemical Impedance Spectroscopy (EIS) revealed that the ferroelectric polarization had a strong influence on charge transfer at the electrolyte interface, as shown in Fig. 8b. The impedance arc of the positively poled BFO/TiO2 film was much smaller compared to those of the pristine film. The negatively poled ones exhibited the highest charge transfer impedance at the electrolyte interface due to the unfavorable polarization direction. The Mott–Schottky plots shown in Fig. 8c revealed that the slope of the positively poled BFO film was decreased, suggesting an increase of carrier density in TiO2. A very similar ferroelectric polarization-regulated band structure change was presented to explain the PEC performance change (Fig. 8d). Here, it was argued that the upward band bending due to favorable ferroelectric polarization would increase the carrier density (holes) in TiO2. The enlarged band bending at the BFO/TiO2 interface and the extended space charge region (hole accumulation) could benefit the transportation and separation of electrons and holes at the interface. Obviously, the negative polarization would bring the opposite effect toward the interface and jeopardized the PEC performance.
When the ferroelectric material has a certain amount of free charge carrier, the space charge region could present at the ferroelectric/electrolyte interface and play an important role in regulating photogenerated charge separation and transportation. In a Ag/Nb-doped STO nanoporous film, additional free charges were introduced to the STO films, therefore the STO film itself could serve as a photosensitizer to produce electron–hole pairs.105 In this system, the ferroelectric Ag/Nb-doped STO could still be poled to introduce additional polarization influences. Fig. 9a–c shows the Jph–V curves of the unpoled, positively poled and negatively poled samples, respectively. The unpoled sample yielded a Jph of 93 μA cm−2 at 1 V vs. Ag/AgCl and an onset potential (Von) at −0.74 V vs. Ag/AgCl. The positively poled sample gave a Jph of 130 μA cm−2 at 1 V vs. Ag/AgCl with a decrease of 20 mV in Von; while Jph of the negatively poled samples was reduced to 40 μA cm−2 at 1 V vs. Ag/AgCl with a 20 mV increase in Von. The corresponding band diagrams are schematically shown in Fig. 9d to explain the possible mechanism. Due to the presence of a sufficient amount of free charge carriers in STO as a result of doping, the electronic band at the middle of STO could become mostly flat. The energy level bends upward at the STO/electrolyte interface, leading to the formation of a space charge region (Wsc). When the STO was poled positively, the width of Wsc at the STO/electrolyte interface would be enlarged. This enhanced potential gradient could drive more holes toward the interface and thus facilitate the interfacial charge transfer. In addition, extended Wsc indicated that there was more volume in STO where the photogenerated electron–hole pairs could be effectively separated by the built-in potential. Similarly, negative polarization would provide opposite effects to the interfacial band structure, which narrows Wsc, reduces the interfacial charge density, and hinders electron–hole separation, leading to poor PEC performance.
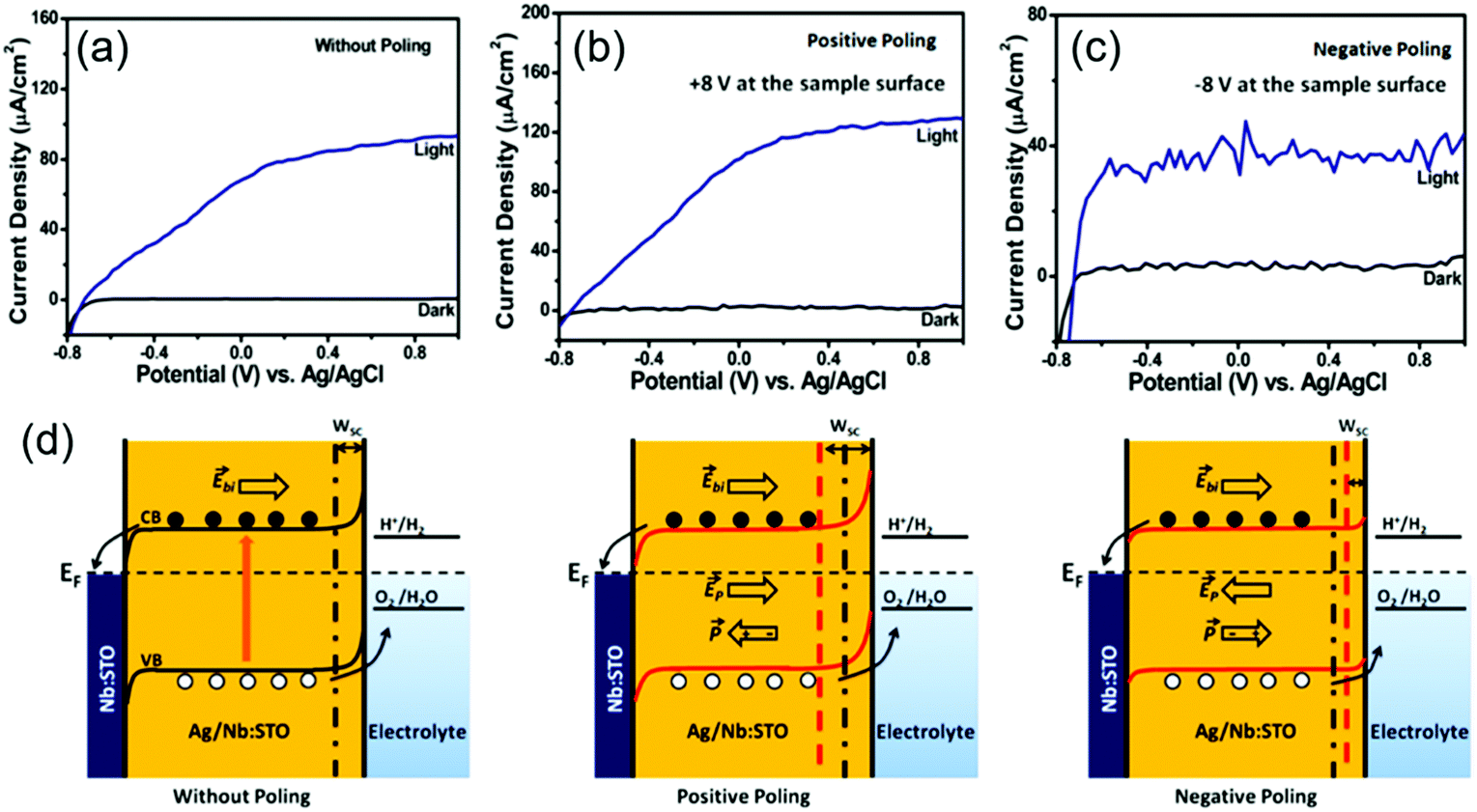 | ||
| Fig. 9 Ag/Nb-doped STO nanoporous film for PEV water splitting. J–V curves of (a) unpoled, (b) positively poled, and (c) negatively poled Ag/Nb-doped STO thin film photoanodes under ∼100 mW cm−2 illumination in 0.5 M NaOH solution. (d) Schematic band diagrams of ferroelectric Ag/Nb-doped STO photoanode interfacing with electrolyte without poling (left), and with positive (middle) and negative (right) poling, respectively. Reprinted with permission from ref. 105. Copyright 2019 ACS. | ||
Doping insulating ferroelectrics could be a general strategy to allow direct application of perovskites as photocatalysts by introducing free charge carriers to the crystals. Insulating BTO, which was primarily used in conjunction with other photocatalysts (e.g. TiO2), could be directly applied for PEC photoanodes when doped with Ce.106 Ce doping could introduce oxygen vacancies and free electrons in BTO, and thus shift the valence and conduction band edges and increase the photoconductivity. Under the same mechanism, positive poling of Ce-doped BTO could induce favorable band bending at the ferroelectric/electrolyte interface, and thus enhanced the PEC performance.
PC systems that do not have an external bias to facilitate charge separation and transportation, are even more sensitive to the internal built-in potential to mitigate the charge recombination challenge. Therefore, implementation of ferroelectric potential might show profound impacts on the overall solar-to-chemical energy conversion efficiency. The enhanced photocatalytic performance introduced by ferroelectric materials has been initially explored in multiple photosensitized electrochemical processes, such as the degradation of organic pollutants and hydrogen evolution.107 However, due to the PC reactions occurring on the catalyst particle surfaces, it is much more challenging to quantify the ferroelectric enhancement compared to PEC systems. As a result, ferroelectric-modulated PC water splitting was not as broadly reported as PEC. Table 1 summarizes a few representative examples of PC water splitting performance assisted by ferroelectric materials. In a Y3+ modified SrBi2Ta2O9 powder system,108 the Y3+ substitutions at the Sr2+ site would induce the formation of Sr vacancies, resulting in lattice rotation and defect charge neutrality. The increase of substitution content (x) was found able to enhance the ferroelectric polarity. The powders were tested for PC water splitting using aqueous CH3OH/H2O solution as a sacrificial electron donor and AgNO3 as a sacrificial electron acceptor, where the sample with x = 0.1 exhibited the highest H2 evolution rate (Fig. 10a). The authors correlated the PC water splitting enhancement with the spontaneous ferroelectric polarization in the powders. As illustrated in Fig. 10b, when immersed in the electrolyte, the spontaneous ferroelectric polarization would be compensated by free charge screening both internally and externally. The accumulated charges at the interface could induce the downward and upward band bending at two oppositely charged surfaces, respectively, resulting in extension of the space charge regions. As a consequence, the separation of photoexcited charge carriers and transporting them toward the oxidation/reduction surface sites would be enhanced.89
| Materials | Conditions | PC hydrogen evolution | Performance comparison | Physical propertiesa | Ref. |
|---|---|---|---|---|---|
| a P r: remnant polarization; Eg: band gap energy. | |||||
| Y3+ modified SrBi2Ta2O9 | 350 W Hg lamp under UV irradiation | 3.95 mmol h−1 | 1.7 times higher than that of pristine SrBi2Ta2O9 | P r 7.03 μC cm−2 | 108 |
| BiOIO3/g-C3N4 Z-scheme heterostructure | 500 W xenon lamp | 56.4 μmol h−1 | 3.5 times higher than that of g-C3N4 (15.8 μmol h−1) | BiOIO3Eg 3.0 eV | 109 |
| Al/BaTiO3/Ti heterostructure | AM 1.5 G, 300 W Xe lamp | 657 μmol (h cm2)−1 | 327 μmol (h cm2)−1 without polarization | 110 | |
| P-Doped g-C3N4/polyaniline/BaTiO3 | 350 W Xe light | 602 μmol (g h)−1 | 2.1 times higher than that of g-C3N4/polyaniline | E g 2.51 eV | 111 |
| Pt-g-C3N4/KNbO3 | 300 W Xe arc lamp | 1019.38 μmol (g h)−1 | 14 times higher than that of Pt-g-C3N4 73.43 μmol (g h)−1 | 112 | |
| (Ba,Ca)TiO3–Ba(Sn,Ti)O3 cermaics | 500 W Xe lamp (λ > 420 nm) | 450 μmol g−1 | E g 3.11 eV | 113 | |
| P r 2.5–5 μC cm−2 | |||||
| g-C3N4/BiFeO3 heterojunction | 125 W Hg lamp as UV source | 160.75 μmol (g h)−1 | 18 μmol (g h)−1 from pristine g-C3N4 | E g 2.32–2.8 eV | 114 |
| Au/BiFeO3 nanosheets | 250 W Xe-arc lamp (λ > 420 nm) | 1.05 mmol h−1 | E g 2.1 eV | 115 | |
| SrTiO3–TiO2 hollow multi-shelled structures | 300 W Xe lamp | 10.6 μmol h−1 | 116 | ||
| g-C3N4 coated SrTiO3 | 250 W iron doped metal halide UV-Vis lamp (λ > 420 nm) | 440 μmol (g h)−1 | 326 μmol (g h)−1 from pristine g-C3N4 | E g 2.92 eV | 117 |
| Corona-poled KxNa1−xNbO3 | 450 W high-pressure mercury lamp under UV-light | 4.7 mmol (g h)−1 | 7 times increase after polarization | P r 14.7 μC cm−2 | 118 |
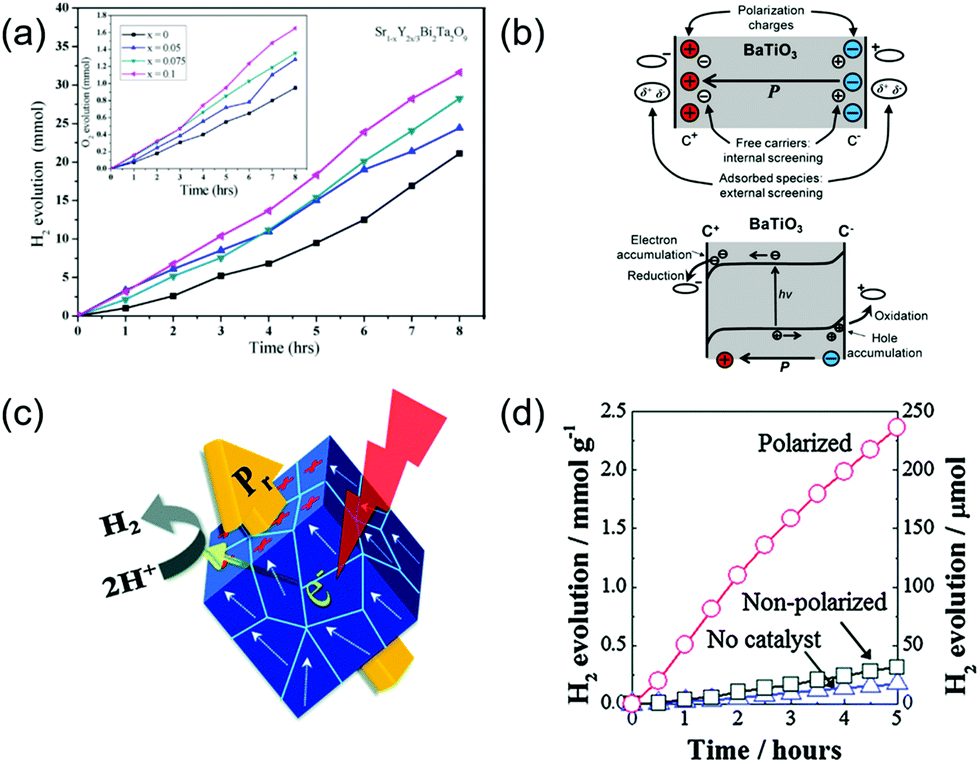 | ||
| Fig. 10 Ferroelectric-enhanced photocatalytic water splitting. (a) The amount of H2/O2 evolution using CH3OH/AgNO3 scavengers for the 0.2 g of Sr1−xY2x/3Bi2Ta2O9 (x = 0–0.1) catalyst under a 350 W Hg lamp at room temperature at normal air pressure in a closed circulation system. Reprinted with permission from ref. 108. Copyright 2019 Elsevier. (b) Schematic illustration of a ferroelectric material. Reprinted with permission from ref. 89. Copyright 2013 ACS. (c) Schematic diagram of a corona-poling system, and (d) amount of H2 evolution for polarized and non-polarized Na0.5K0.5NbO3 powder under UV light illumination. Reprinted with permission from ref. 118. Copyright 2014 RSC. | ||
Nevertheless, powders are likely to show multiple ferroelectric domains, which could cancel out the net polarization in an individual particle and jeopardize the ferroelectric enhancement. This might be the reason that the improvement of hydrogen generation was not significant as shown in Fig. 10a. Park et al. presented a potential solution to this challenge by pre-poling the powders before the PC application.118 In this work, a ferroelectric KxNa1−xNbO3 (NKN) solid-solution acted as the photocatalyst for PC water splitting (Fig. 10c). The as-synthesized ferroelectric NKN powders were first poled by corona-poling to align the dipole within individual particles. The poled powders were then applied to deionized water containing 10 vol% methanol as a hole scavenger under ultraviolent (UV) irradiation. The PC activities of hydrogen evolution are shown in Fig. 10d. A significant enhancement of PC performance was obtained from the poled NKN powders. After polarization, the H2 evolution rate of NKN powders reached 4.7 mmol g−1 h−1, which was more than 7 times higher than that of the non-polarized samples (∼63 μmol g−1 h−1). It was also worthwhile noting that the non-polarized NKN powders only exhibited a slightly higher hydrogen evolution rate. This further confirmed that random ferroelectric polarization had minimal impacts on the PC activity. Many other reports also demonstrated that pre-poling the ferroelectric powers could be an effective solution to enhance the ferroelectric influences to PC performance.119–121
In general, the above discussions clearly revealed that the ferroelectric polarization could have a profound impact on the interfacial electronic band structure and thus tune the PC/PEC performance. This principle applies to all ferroelectric materials that can provide spontaneous polarization. Insulating ferroelectric materials should be integrated with semiconductor photocatalysts to facilitate the charge generation and transport. When the ferroelectric material is semiconducting, it may be used directly as the photocatalyst, but the performance is usually lower than those hybrid systems. Table 2 summarizes the Jph of various ferroelectric material-based PEC photoanodes with different polarization directions, together with bandgap and ferroelectric remnant polarization (Pr) information available. The general positive influences of the ferroelectric polarization could be clearly observed from the list.
| Samples | Photocurrent density | Potential | Illumination | Solution | Band gap (eV) | Remnant polarization (μC cm−2) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|
| Positive polarization | Without polarization | Negative polarization | |||||||
| Two-step hydrothermal synthesized TiO2/SrTiO3 core–shell nanowires | ∼1.48 | 1.43 | 0.8–0.9 | mA cm−2 at 1.23 V vs. RHE | AM 1.5G | 1 M NaOH | 0.05 | 102 | |
| Two-step hydrothermal synthesized TiO2/BaTiO3 core–shell nanowires | ∼1.37 | 1.3 | ∼1.1 | mA cm−2 at 1.23 V vs. RHE | AM 1.5G | 1 M NaOH | 0.045 | 101 | |
| Plused laser deposited Ag/Nb-doped SrTiO3 nanoporous film | 0.13 | 0.93 | 0.04 | mA cm−2 at 1.0 V vs. Ag/AgCl | 100 mW cm−2 | 0.5 M NaOH | 105 | ||
| Sintered Ce-doped BaTiO3 nanoassemblies | 0.25 | 0.2 | mA cm−2 at 1.2 V vs. Pt | 120 mW cm−2 | 1 mM NaOH | 2.73 | 1.76 | 106 | |
| Magnetron sputtering and chemical bath facricated CdS/Au/Ti/Pb(Mg1/3Nb2/3)0.7Ti0.3O3 single crystal | 1.7 | ∼0.085 | mA cm−2vs. Ag/AgCl | AM 1.5 G | 0.25 M Na2SO4 and 0.35 M Na2SO3 | CdS (2.2) | 122 | ||
| Spin-coated and pre-annealed BiFeO3/TiO2 film | 19 | ∼11.02 | ∼4.96 | mA cm−2 at 1.5 V vs. SCE | dark | 1 M NaOH | BFO (2.12) | 104 | |
| Sol–gel fabricated BiFeO3/Sn:TiO2 nanorod | 1.76 | 1.4–1.5 | 1–1.1 | mA cm−2 at 1.23 V vs. RHE | AM 1.5 G | 1 M NaOH | 2.84 | 123 | |
| Laser deposited (111)pc BiFeO3 thin film | 0.08 | 0.001 | mA cm−2 at 0 V vs. Ag/AgCl | AM 1.5 G | 0.5 M Na2SO4 | 2.57 | 110 | 124 | |
| Hydrothermal synthesized KNbO3 nanosheets | 0.82 | 0.53 | μA cm−2 at 0 V vs. Ag/AgCl | AM 1.5 G | 0.5 M Na2SO4 | 3.02 | 125 | ||
| Hydrothermal grown single-crystalline KNbO3 nanowire arrays | 11.5 | 0.7 | μA cm−2 at 0 V vs. Ag/AgCl | AM 1.5 G | 1 M Na2SO4 | 3.28 | 90 | 126 | |
| Sol–gel synthesized Pb(Zr0.2Ti0.8)O3 thin film with Au nanoparticle arrays | 16.7 | 2.4 | μA cm−2 at 0 V vs. Ag/AgCl | 100 mW cm−2 | 0.1 M Na2SO4 | PZT (3.6) | PZT (∼17) | 127 | |
| Sol–gel fabricated polycrystalline BiFeO3 film with Ag nanoparticles | 50 | −45 | μA cm−2 at 0 V vs. Ag/AgCl | 150 mW cm−2 | 0.1 M Na2SO4 | <2.46 | 128 | ||
| Spin-coated BiFeO3 film | 10 | 0 | μA cm−2 at 0 V vs. Ag/AgCl | 100 mW cm−2 | 0.1 M KCl | 2.14 | 1–2 | 129 | |
5. Summary and prospects
Leveraging the spontaneous polarization in a ferroelectric material to regulate the interface electronic band structure, also known as piezotronics, is a promising strategy to manipulate the electrochemical performance in heterogeneous photocatalysis. In this review article, we overviewed how ferroelectric polarization can be implemented to facilitate solar water splitting processes, including both PC and PEC systems. In particular, a positive polarization may broaden the width of the space charge region and increase band bending, which is beneficial for photogenerated charge separation and transportation, and thus improves the solar water splitting efficiency. In contrast, a negative polarization may narrow the space charge region and deteriorate band bending, and thus suppress charge separation and transportation, resulting in lower water splitting efficiency. This general principle may apply to both PC and PEC processes. In this article, we summarized a few representative examples to show how the ferroelectric polarization can be integrated in a heterogeneous photoelectrode. We showed that due to the most insulating electronic property, the ferroelectric material was typically combined together with semiconducting catalytic materials, where the polarization could penetrate through the interface and influence the band structure in the semiconductor material. It is also possible to improve the conductivity of the ferroelectric material, such as by doping to allow better charge transport through the ferroelectric layer, and thus facilitate the overall photocurrent density. Compared to PEC systems, a PC system is relatively simpler but it may require more effort to align the dipole to provide a valuable interface band structure control.In general, initial studies of the ferroelectric influences were very intriguing. Integrating ferroelectric polarization could obviously provide an additional and maybe significant tuning factor to the interfacial energetics beyond what can be achieved by chemical or structural modifications. However, one should also be aware that PC and PEC regulated by ferroelectric polarization is still at the early stage. There are several critical challenges toward practical applications, particularly from the materials science perspectives.
First, the ferroelectric modulation of the interfacial electronic band structure imposes high requirements on the interfaces. As the interfacial band substructure is extremely sensitive to the electronic energy level and free charge carrier concentration, small variations in compositions, atomic structures and defect states at the interface may induce large changes to the built-in potential and depletion region distribution, and thus may significantly jeopardize the impacts of ferroelectric polarization. Therefore, in order to obtain repeatable and reproducible enhancements from ferroelectric modulation, it is critical to build a clean and sharp interface between the ferroelectric and semiconductor materials with defined crystal facets. While epitaxial is always preferred to achieve such a high-quality interface, it may not be generally applicable to ferroelectric-modulated PEC/PC systems, as these two types of materials may not have compatible lattices. Current advancements in heterogeneous materials synthesis, such as remote epitaxy might offer unique advantages in preparing ideal ferroelectric-semiconductor heterostructures with maximized enhancement as the growth by-passes the requirement of lattice matching substrates.130–132
Second, the intrinsically low electronic transport property of ferroelectric materials limits the broad application of this strategy in many photocatalytic processes. Ferroelectric materials are mostly desired to be insulating with a relatively large bandgap (>3 eV) in order to preserve a high built-in polarization. This feature however is in contradiction to the requirements of a high-performance photocatalyst. One big challenge is how to keep a high ferroelectric polarization at the interface while still maintaining a good charge transport property to bring the overall energy conversion efficiency to a theoretically high level. In addition, for photon-driven reactions (e.g. the PEC and PC processes), a smaller bandgap (<2 eV) is also desired to reach efficient absorption of the solar spectrum. However, the small bandgap-associated high free charge concentration and high photocurrent are detrimental to the internal ferroelectric polarization. Indirect solutions, such as introducing an atomic layer of conducting graphene with ferroelectrics to facilitate charge transportation, have shown some promises.133 However, the ultimate solution will still depend on material innovations of novel ferroelectric materials with strong built-in polarization, low carrier concentration, and fast charge mobility, which would allow rapid charge transfer through the ferroelectric materials and across interfaces without impairing the strong built-in potential. In this regard, traditional ferroelectric materials may not be able to satisfy all the requirements simultaneously. The emergence of novel 2D materials that show strong built-in polarization as well as good charge transport properties may be of great potential for future exploitation.
Third, most ferroelectric materials are not good catalytic materials. Their stable and low-energy polar surfaces are usually not sufficiently catalytically active. Direct implementation of ferroelectrics as the catalytic surfaces usually leads to low performances, particularly in PC systems. Loading noble metals such as Pt and Pd, or other electrocatalysts such as Ni(OH)2 could significantly enhance the surface activity. However, it is essential to create a stable bonding between the ferroelectric and catalytic materials, to sustain a long-term high-performance ferroelectric-modulated catalytic activity. New chemical or physical approaches are needed to introduce nanoscale catalytic materials to the ferroelectric polar surfaces with strong chemical bonding and high selectivity. Combining polarization-facilitated electrochemical deposition might be a potential solution that naturally combines the desired ferroelectricity with targeted catalysis.
In general, addressing all these critical challenges would rely on new material designs, discoveries, and synthesis and characterization capabilities to move the heterogeneous ferroelectric-catalysis systems to the next horizon. New materials science and engineering understandings and breakthroughs, together with suitable chemical engineering catalyst design, would eventually bring this promising concept into a practical solar hydrogen fuel generation technology with an unprecedented energy conversion efficiency.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation under grant number DMR-1709025 and the National Key R&D Program of China (No. 2018YFB1502501-05).References
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS.
- L. Schlapbach, Nature, 2009, 460, 809 CrossRef CAS PubMed.
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS.
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc., A, 2010, 368, 3329–3342 CrossRef PubMed.
- S. Stephens-Romero, M. Carreras-Sospedra, J. Brouwer, D. Dabdub and S. Samuelsen, Environ. Sci. Technol., 2009, 43, 9022–9029 CrossRef CAS PubMed.
- S. Stephens-Romero and G. S. Samuelsen, Int. J. Hydrogen Energy, 2009, 34, 628–641 CrossRef CAS.
- C. Acar and I. Dincer, Int. J. Hydrogen Energy, 2014, 39, 1–12 CrossRef CAS.
- G. H. Marcus and A. E. Levin, Phys. Today, 2002, 55, 54–60 CrossRef CAS.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- F. A. Grant, Rev. Mod. Phys., 1959, 31, 646–674 CrossRef CAS.
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 RSC.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- M. Ahmed and I. Dincer, Int. J. Hydrogen Energy, 2019, 44, 2474–2507 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2245 CrossRef CAS PubMed.
- S. Licht, B. Wang, S. Mukerji, T. Soga, M. Umeno and H. Tributsch, J. Phys. Chem. B, 2000, 104, 8920–8924 CrossRef CAS.
- J. Akikusa and S. U. M. Khan, Int. J. Hydrogen Energy, 2002, 27, 863–870 CrossRef CAS.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- S. Chen, S. S. Thind and A. Chen, Electrochem. Commun., 2016, 63, 10–17 CrossRef CAS.
- X. Wang, Z. Li, J. Shi and Y. Yu, Chem. Rev., 2014, 114, 9346–9384 CrossRef CAS.
- Y. Yu, Z. Zhang, X. Yin, A. Kvit, Q. Liao, Z. Kang, X. Yan, Y. Zhang and X. Wang, Nat. Energy, 2017, 2, 17045 CrossRef CAS.
- F. Cao, J. Xiong, F. Wu, Q. Liu, Z. Shi, Y. Yu, X. Wang and L. Li, ACS Appl. Mater. Interfaces, 2016, 8, 12239–12245 CrossRef CAS PubMed.
- X. Wang, G. S. Rohrer and H. Li, MRS Bull., 2018, 43, 946–951 CrossRef.
- M. B. Starr and X. Wang, Nano Energy, 2015, 14, 296–311 CrossRef CAS.
- J. Shi, M. B. Starr and X. Wang, Adv. Mater., 2012, 24, 4683–4691 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- P. D. Yang, Abstr. Pap. Am. Chem. Soc., 2014, 248, 1 Search PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- W. J. Albery and P. N. Bartlett, J. Electrochem. Soc., 1984, 131, 315–325 CrossRef CAS.
- M. D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- R. M. Navarro Yerga, M. C. Álvarez Galván, F. del Valle, J. A. Villoria de la Mano and J. L. G. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef CAS PubMed.
- U. A. Joshi, A. Palasyuk, D. Arney and P. A. Maggard, J. Phys. Chem. Lett., 2010, 1, 2719–2726 CrossRef CAS.
- K. Rajeshwar, J. Appl. Electrochem., 2007, 37, 765–787 CrossRef CAS.
- T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2007, 111, 5259–5275 CrossRef CAS.
- D. A. Tryk, A. Fujishima and K. Honda, Electrochim. Acta, 2000, 45, 2363–2376 CrossRef CAS.
- R. Morrison, Electrochemistry at Semiconductor and Oxidised Metal Surfaces, 1980 Search PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- A. B. Murphy, P. R. F. Barnes, L. K. Randeniya, I. C. Plumb, I. E. Grey, M. D. Horne and J. A. Glasscock, Int. J. Hydrogen Energy, 2006, 31, 1999–2017 CrossRef CAS.
- Z. S. Li, W. J. Luo, M. L. Zhang, J. Y. Feng and Z. G. Zou, Energy Environ. Sci., 2013, 6, 347–370 RSC.
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed.
- W. Ou, J. Pan, Y. Liu, S. Li, H. Li, W. Zhao, J. Wang, C. Song, Y. Zheng and C. Li, J. Energy Chem., 2020, 43, 188–194 CrossRef.
- W.-Z. Xiao, L. Xu, Q.-Y. Rong, X.-Y. Dai, C.-P. Cheng and L.-L. Wang, Appl. Surf. Sci., 2020, 504, 144425 CrossRef.
- S. Banerjee, S. K. Mohapatra and M. Misra, Chem. Commun., 2009, 7137–7139, 10.1039/B912549C.
- Y. Lin, S. Zhou, X. Liu, S. Sheehan and D. Wang, J. Am. Chem. Soc., 2009, 131, 2772–2773 CrossRef CAS.
- R. Liu, Y. Lin, L.-Y. Chou, S. W. Sheehan, W. He, F. Zhang, H. J. M. Hou and D. Wang, Angew. Chem., Int. Ed., 2011, 50, 499–502 CrossRef CAS.
- I. S. Cho, Z. Chen, A. J. Forman, D. R. Kim, P. M. Rao, T. F. Jaramillo and X. Zheng, Nano Lett., 2011, 11, 4978–4984 CrossRef CAS PubMed.
- M. I. Litter, Appl. Catal., B, 1999, 23, 89–114 CrossRef CAS.
- A.-W. Xu, Y. Gao and H.-Q. Liu, J. Catal., 2002, 207, 151–157 CrossRef CAS.
- R. Dholam, N. Patel, M. Adami and A. Miotello, Int. J. Hydrogen Energy, 2009, 34, 5337–5346 CrossRef CAS.
- L. Han, S. J. Dong and E. K. Wang, Adv. Mater., 2016, 28, 9266–9291 CrossRef CAS PubMed.
- L. Z. Ma, K. Zhang, S. Wang, L. N. Gao, Y. F. Sun, Q. Y. Liu, J. X. Guo and X. Zhang, Appl. Surf. Sci., 2019, 489, 815–823 CrossRef CAS.
- Y. J. Wang, Q. S. Wang, X. Y. Zhan, F. M. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326–8339 RSC.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- Y. Li, W. Yang, C. Wang, Z. Li, J. Lai, L. Wang and L. Huang, ACS Appl. Energy Mater., 2019, 2, 8229–8235 CrossRef CAS.
- P. Wen, F. Su, H. Li, Y. Sun, Z. Liang, W. Liang, J. Zhang, W. Qin, S. M. Geyer, Y. Qiu and L. Jiang, Chem. Eng. J., 2020, 385, 123878 CrossRef.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- J. L. Giocondi and G. S. Rohrer, J. Am. Ceram. Soc., 2003, 86, 1182–1189 CrossRef CAS.
- J. J. Loferski, J. Appl. Phys., 1956, 27, 777–784 CrossRef CAS.
- X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089–13092 CrossRef CAS PubMed.
- J. Pan, G. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS PubMed.
- R. Hengerer, L. Kavan, P. Krtil and M. Grätzel, J. Electrochem. Soc., 2000, 147, 1467–1472 CrossRef CAS.
- F. Wang, W. Septina, A. Chemseddine, F. F. Abdi, D. Friedrich, P. Bogdanoff, R. van de Krol, S. D. Tilley and S. P. Berglund, J. Am. Chem. Soc., 2017, 139, 15094–15103 CrossRef CAS PubMed.
- D. Chen and Z. Liu, ChemSusChem, 2018, 11, 3438–3448 CrossRef CAS PubMed.
- J. Valasek, Phys. Rev., 1921, 17, 475–481 CrossRef CAS.
- J. F. Scott, Science, 2007, 315, 954–959 CrossRef CAS PubMed.
- G. H. Haertling, J. Am. Ceram. Soc., 1999, 82, 797–818 CrossRef CAS.
- V. Garcia and M. Bibes, Nature, 2012, 483, 279–280 CrossRef CAS PubMed.
- A. M. Ionescu, Nat. Nanotechnol., 2012, 7, 83–85 CrossRef CAS PubMed.
- S. Xu, B. J. Hansen and Z. L. Wang, Nat. Commun., 2010, 1, 93 CrossRef PubMed.
- R. W. Whatmore, Ferroelectrics, 1991, 118, 241–259 CrossRef CAS.
- P. Paufler, Cryst. Res. Technol., 1988, 23, 1360 CrossRef.
- T. Mitsui, in Springer Handbook of Condensed Matter and Materials Data, ed. W. Martienssen and H. Warlimont, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, pp. 903–938 DOI:10.1007/3-540-30437-1_13.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506 CrossRef CAS.
- K. C. Kao, in Dielectric Phenomena in Solids, ed. K. C. Kao, Academic Press, San Diego, 2004, pp. 213–282 DOI:10.1016/B978-012396561-5/50014-1.
- H. T. Yi, T. Choi, S. G. Choi, Y. S. Oh and S.-W. Cheong, Adv. Mater., 2011, 23, 3403–3407 CrossRef CAS PubMed.
- C. H. Ahn, K. M. Rabe and J.-M. Triscone, Science, 2004, 303, 488–491 CrossRef CAS PubMed.
- M. A. Khan, U. S. Bhansali and H. N. Alshareef, Org. Electron., 2011, 12, 2225–2229 CrossRef CAS.
- M. E. Lines, A. M. Glass and G. Burns, Phys. Today, 1978, 31, 56–58 CrossRef.
- W. C. Yang, B. J. Rodriguez, A. Gruverman and R. J. Nemanich, J. Phys.: Condens. Matter, 2005, 17, S1415–S1426 CrossRef CAS.
- D. Tiwari and S. Dunn, J. Mater. Sci., 2009, 44, 5063–5079 CrossRef CAS.
- Y. Cui, J. Briscoe and S. Dunn, Chem. Mater., 2013, 25, 4215–4223 CrossRef CAS.
- D. von der Linde and A. M. Glass, Appl. Phys., 1975, 8, 85–100 CAS.
- S. Dunn, C. P. Shaw, Z. Huang and R. W. Whatmore, Nanotechnology, 2002, 13, 456–459 CrossRef CAS.
- O. Ambacher, J. Smart, J. R. Shealy, N. G. Weimann, K. Chu, M. Murphy, W. J. Schaff, L. F. Eastman, R. Dimitrov, L. Wittmer, M. Stutzmann, W. Rieger and J. Hilsenbeck, J. Appl. Phys., 1999, 85, 3222–3233 CrossRef CAS.
- W.-C. Yang, B. J. Rodriguez, A. Gruverman and R. J. Nemanich, Appl. Phys. Lett., 2004, 85, 2316–2318 CrossRef CAS.
- T. Sakthivel, G. Venugopal, A. Durairaj, S. Vasanthkumar and X. Huang, J. Ind. Eng. Chem., 2019, 72, 18–30 CrossRef CAS.
- S. Kim, N. T. Nguyen and C. W. Bark, Appl. Sci., 2018, 8, 1526 CrossRef.
- M. Wang, B. Wang, F. Huang and Z. Lin, Angew. Chem., Int. Ed., 2019, 58, 7526–7536 CrossRef CAS PubMed.
- P. Lopez-Varo, L. Bertoluzzi, J. Bisquert, M. Alexe, M. Coll, J. S. Huang, J. A. Jimenez-Tejada, T. Kirchartz, R. Nechache, F. Rosei and Y. B. Yuan, Phys. Rep., 2016, 653, 1–40 CrossRef CAS.
- S. Y. Jeong, J. Song and S. Lee, Appl. Sci., 2018, 8, 1388 CrossRef.
- F. Chen, H. Huang, L. Guo, Y. Zhang and T. Ma, Angew. Chem., Int. Ed., 2019, 58, 10061–10073 CrossRef CAS PubMed.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 RSC.
- W. Yang, Y. Yu, M. B. Starr, X. Yin, Z. Li, A. Kvit, S. Wang, P. Zhao and X. Wang, Nano Lett., 2015, 15, 7574–7580 CrossRef CAS PubMed.
- F. Wu, Y. H. Yu, H. Yang, L. N. German, Z. Q. Li, J. G. Chen, W. G. Yang, L. Huang, W. M. Shi, L. J. Wang and X. D. Wang, Adv. Mater., 2017, 29, 7 Search PubMed.
- W. Ji, K. Yao, Y.-F. Lim, Y. C. Liang and A. Suwardi, Appl. Phys. Lett., 2013, 103, 062901 CrossRef.
- X. Wu, H. Li, X. Wang, L. Jiang, J. Xi, G. Du and Z. Ji, J. Alloys Compd., 2019, 783, 643–651 CrossRef CAS.
- S. Singh, A. L. Sangle, T. Wu, N. Khare and J. L. MacManus-Driscoll, ACS Appl. Mater. Interfaces, 2019, 11, 45683–45691 CrossRef CAS PubMed.
- P. Senthilkumar, D. A. Jency, T. Kavinkumar, D. Dhayanithi, S. Dhanuskodi, M. Umadevi, S. Manivannan, N. V. Giridharan, V. Thiagarajan, M. Sriramkumar and K. Jothivenkatachalam, ACS Sustainable Chem. Eng., 2019, 7, 12032–12043 CAS.
- Q. Jia, A. Iwase and A. Kudo, Chem. Sci., 2014, 5, 1513–1519 RSC.
- V. Senthil and S. Panigrahi, Int. J. Hydrogen Energy, 2019, 44, 18058–18071 CrossRef CAS.
- Y. Gong, X. Quan, H. Yu, S. Chen and H. Zhao, Appl. Catal., B, 2018, 237, 947–956 CrossRef CAS.
- L. Guo, C. Zhong, J. Cao, Y. Hao, M. Lei, K. Bi, Q. Sun and Z. L. Wang, Nano Energy, 2019, 62, 513–520 CrossRef CAS.
- Q. Li, Y. Xia, K. Wei, X. Ding, S. Dong, X. Jiao and D. Chen, New J. Chem., 2019, 43, 6753–6764 RSC.
- D. Xu, L. Li, T. Xia, W. Fan, F. Wang, H. Bai and W. Shi, Int. J. Hydrogen Energy, 2018, 43, 16566–16572 CrossRef CAS.
- K. S. Srikanth, M. K. Hooda, H. Singh, V. P. Singh and R. Vaish, Mater. Sci. Semicond. Process., 2018, 79, 153–160 CrossRef CAS.
- H. Sepahvand and S. Sharifnia, Int. J. Hydrogen Energy, 2019, 44, 23658–23668 CrossRef CAS.
- S. Bera, S. Ghosh, S. Shyamal, C. Bhattacharya and R. N. Basu, Sol. Energy Mater. Sol. Cells, 2019, 194, 195–206 CrossRef CAS.
- Y. Wei, J. Wang, R. Yu, J. Wan and D. Wang, Angew. Chem., Int. Ed., 2019, 58, 1422–1426 CrossRef CAS PubMed.
- X. Xu, G. Liu, C. Randorn and J. T. S. Irvine, Int. J. Hydrogen Energy, 2011, 36, 13501–13507 CrossRef CAS.
- S. Park, C. W. Lee, M.-G. Kang, S. Kim, H. J. Kim, J. E. Kwon, S. Y. Park, C.-Y. Kang, K. S. Hong and K. T. Nam, Phys. Chem. Chem. Phys., 2014, 16, 10408–10413 RSC.
- M. Sharma, A. Halder and R. Vaish, Mater. Res. Bull., 2020, 122, 110647 CrossRef CAS.
- L. Qifeng, M. Jingjun, M. Sharma and R. Vaish, J. Am. Ceram. Soc., 2019, 102, 5807–5817 CrossRef CAS.
- W. Gu, W. Zhang, L. Zhu, W. Zou, H. Liu, Z. Fu and Y. Lu, Mater. Lett., 2019, 241, 115–118 CrossRef CAS.
- Z. Song, B. Hong, X. Zhu, F. Zhang, S. Li, J. Ding, X. Jiang, J. Bao, C. Gao and S. Sun, Appl. Catal., B, 2018, 238, 248–254 CrossRef CAS.
- J. Huang, Y. Wang, X. Liu, Y. Li, X. Hu, B. He, Z. Shu, Z. Li and Y. Zhao, Nano Energy, 2019, 59, 33–40 CrossRef CAS.
- J. Song, T. L. Kim, J. Lee, S. Y. Cho, J. Cha, S. Y. Jeong, H. An, W. S. Kim, Y.-S. Jung, J. Park, G. Y. Jung, D.-Y. Kim, J. Y. Jo, S. D. Bu, H. W. Jang and S. Lee, Nano Res., 2018, 11, 642–655 CrossRef CAS.
- D. Yu, Z. Liu, J. Zhang, S. Li, Z. Zhao, L. Zhu, W. Liu, Y. Lin, H. Liu and Z. Zhang, Nano Energy, 2019, 58, 695–705 CrossRef CAS.
- S. Li, J. Zhang, B.-P. Zhang, W. Huang, C. Harnagea, R. Nechache, L. Zhu, S. Zhang, Y.-H. Lin, L. Ni, Y.-H. Sang, H. Liu and F. Rosei, Nano Energy, 2017, 35, 92–100 CrossRef CAS.
- Z. Wang, D. Cao, L. Wen, R. Xu, M. Obergfell, Y. Mi, Z. Zhan, N. Nasori, J. Demsar and Y. Lei, Nat. Commun., 2016, 7, 10348 CrossRef CAS PubMed.
- Q. Liu, Y. Zhou, L. You, J. Wang, M. Shen and L. Fang, Appl. Phys. Lett., 2016, 108, 022902 CrossRef.
- D. Cao, Z. Wang, Nasori, L. Wen, Y. Mi and Y. Lei, Angew. Chem., Int. Ed., 2014, 53, 11027–11031 CrossRef CAS PubMed.
- Y. Kim, S. S. Cruz, K. Lee, B. O. Alawode, C. Choi, Y. Song, J. M. Johnson, C. Heidelberger, W. Kong, S. Choi, K. Qiao, I. Almansouri, E. A. Fitzgerald, J. Kong, A. M. Kolpak, J. Hwang and J. Kim, Nature, 2017, 544, 340–343 CrossRef CAS PubMed.
- J. Jiang, X. Sun, X. Chen, B. Wang, Z. Chen, Y. Hu, Y. Guo, L. Zhang, Y. Ma, L. Gao, F. Zheng, L. Jin, M. Chen, Z. Ma, Y. Zhou, N. P. Padture, K. Beach, H. Terrones, Y. Shi, D. Gall, T.-M. Lu, E. Wertz, J. Feng and J. Shi, Nat. Commun., 2019, 10, 4145 CrossRef PubMed.
- H. Kum, D. Lee, W. Kong, H. Kim, Y. Park, Y. Kim, Y. Baek, S.-H. Bae, K. Lee and J. Kim, Nat. Electron., 2019, 2, 439–450 CrossRef CAS.
- X. Chen, L. German, J. Bong, Y. Yu, M. Starr, Y. Qin, Z. Ma and X. Wang, Nano Energy, 2018, 48, 377–382 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |