
Ultrafast assembly of swordlike Cu3(1,3,5-benzenetricarboxylate)n metal–organic framework crystals with exposed active metal sites†
Heba
Ahmed
a,
Xinci
Yang
a,
Yemima
Ehrnst
a,
Ninweh N.
Jeorje
 a,
Susan
Marqus
a,
Peter C.
Sherrell
ab,
Ahmed
El Ghazaly
c,
Johanna
Rosen
c,
Amgad R.
Rezk
*a and
Leslie Y.
Yeo
*a
a,
Susan
Marqus
a,
Peter C.
Sherrell
ab,
Ahmed
El Ghazaly
c,
Johanna
Rosen
c,
Amgad R.
Rezk
*a and
Leslie Y.
Yeo
*a
aMicro/Nanophysics Research Laboratory, School of Engineering, RMIT University, Melbourne, VIC 3000, Australia. E-mail: leslie.yeo@rmit.edu.au; amgad.rezk@rmit.edu.au
bDepartment of Chemical Engineering, The University of Melbourne, Parkville, VIC 3010, Australia
cDepartment of Physics, Chemistry & Biology (IFM), Linköping University, Linköping, SE-581 83, Sweden
First published on 14th April 2020
Abstract
Owing to their large surface area and high uptake capacity, metal–organic frameworks (MOFs) have attracted considerable attention as potential materials for gas storage, energy conversion, and electrocatalysis. Various strategies have recently been proposed to manipulate the MOF surface chemistry to facilitate exposure of the embedded metal centers at the crystal surface to allow more effective binding of target molecules to these active sites. Nevertheless, such strategies remain complex, often requiring strict control over the synthesis conditions to avoid blocking pore access, reduction in crystal quality, or even collapse of the entire crystal structure. In this work, we exploit the hydrodynamics and capillary resonance associated with acoustically-driven dynamically spreading and nebulizing thin films as a new method for ultrafast synthesis of swordlike Cu3(1,3,5-benzenetricarboxylate)n (Cu–BTC) MOFs with unique monoclinic crystal structures (P21/n) distinct to that obtained via conventional bulk solvothermal synthesis, with ‘swordlike’ morphologies whose lengths far exceed their thicknesses. Through pulse modulation and taking advantage of the rapid solvent evaporation associated with the high nebulisation rates, we are also able to control the thicknesses of these large aspect ratio (width and length with respect to the thickness) crystals by arresting their vertical growth, which, in turn, allows exposure of the metal active sites at the crystal surface. An upshot of such active site exposure on the crystal surface is the concomitant enhancement in the conductivity of the MOF, evident from the improvement in its current density by two orders of magnitude.
New conceptsA new acoustomicrofluidic method for the synthesis of metal–organic frameworks (MOFs) is reported. Besides being extremely rapid (milliseconds), this unconventional method yields unique large aspect ratio (≈30–50 nm thin, 10 μm wide and 100 μm long) swordlike morphologies that have yet to be reported, with a monoclinic crystal structure (P21/n) that is remarkably distinct to the regular 3D cubic crystal structures of HKUST-1 obtained through conventional bulk solvothermal synthesis. More interestingly, we show that the metal ions, which constitute the crystal's active sites, are exposed along the surface of the structure rather than being embedded internally within it. This has important implications in overcoming a significant bottleneck of MOFs particularly for electrocatalytic applications wherein the metal nodes are usually buried deep within the 3D structure, therefore limiting their access as binding sites and resulting in its poor conductivity. In addition to characterising this new MOF crystal structure and revealing the high density of active sites on its surface, we also elucidate the unique thin film hydrodynamics that gives rise to its peculiar one-dimensional swordlike morphology, as well as demonstrating the possibility for tuning the crystal dimensions. |
1 Introduction
Metal–organic frameworks (MOFs) are a class of crystalline materials that display extraordinary porosity and tremendous potential for physical, chemical and geometrical tunability given the large number of permutations that are possible between the metal and organic linker combinations that make up its composition.1,2 While the metal nodes of a MOF usually constitute the chemically and catalytically active sites that typically confer it with specific functionality for various applications, they are usually embedded internally within the three-dimensional (3D) crystal coordination network structure. It is therefore often necessary to incorporate pre- or post-synthesis steps to overcome the constraints associated with poor accessibility to these metal sites—particularly the diffusion transport limitation through the pore network—in order to enhance the MOFs’ inherently low conductivity3,4 and to improve their performance in applications such as gas storage,5,6 energy conversion,7–9 molecular separation10–12 and heterogeneous catalysis.13–15 For example, recent chemical modification strategies have proposed circumventing the innate burial of active sites either by constructing MOFs on conductive supports such as nickel–iron meshes to enhance its overall conductivity,16–18 or by combining the MOF with other materials such as molybdenum disulfide (MoS2) that possess active metallic edges.19 Such chemical modification protocols can however be complex, either requiring sufficient care to prevent pore blockage, compromising the overall crystal quality, or even resulting in collapse of the entire crystal structure itself.20–22An alternative strategy to facilitate easier and more rapid access to the metal sites, on the other hand, involves their exposure along the crystal surface. This is achieved, for example, by fabricating large aspect ratio (i.e., long and thin) structures through the addition of inhibitors such as small molecule coordination modulators23–25 or biologically-derived macromolecules26 to limit growth in the crystal thickness, although these are often at the expense of the MOF quality and are often difficult to remove post-synthesis.27,28 Two-dimensional (2D) MOFs with even larger aspect ratios—often referred to as MOF nanosheets (MONs), grown through layer-by-layer self assembly or chemical vapor deposition on a functional support followed by subsequent delamination, have also been synthesized.29–31 While these 2D structures succeed in revealing the active sites along the crystal surface, and, in doing so, endowing the crystals with superior performance compared to their 3D counterparts, their low production yield coupled with the laborious nature and complexity of these bottom-up synthesis approaches render them impractical and costly for large scale production.32,33
Recently, we have shown the ability to grow highly-oriented free-standing MOF crystals through a microfluidic centrifugation technique driven by surface acoustic waves (SAWs),34 which are MHz-order nanometer amplitude electromechanical Rayleigh waves that are generated, and which propagate under confinement, along the surface of a piezoelectric substrate.35–38 In that case, the SAW-driven recirculatory flow facilitated convective transport of the solute (MOF precursor) molecules to the contact line of a sessile droplet containing the precursor solution, where they crystallize as the droplet evaporated under the influence of molecular dipole polarization effects (that arise as a consequence of the SAW evanescent electric field that is a result of the SAW electromechanical coupling) to give rise to long-range supramolecular ordering.34
In this work, we instead carry out the synthesis of MOFs under a distinct SAW-driven flow phenomenon involving the formation, spreading and subsequent nebulisation of a thin acoustowetting film.39,40 In contrast to the azimuthal rotational microcentrifugation flow in ref. 34, the dynamic flow in the current setup (see Fig. 1) associated with the spreading of the thin film, is unidirectional. Moreover, the thin film, with typical dimensions that are tens of microns in height and several hundred of microns in length, resembles a slender body (aspect ratio ≫1) unlike the droplet in the preceding work whose radial dimension is comparable to its height (aspect ratio ≈1). A further distinction of the current work is the nebulization that occurs off the surface of the thin film, which is absent from the synthesis of the MOFs with the SAW microcentrifugation; a consequence of the nebulization is the markedly higher evaporation rate that significantly hastens the MOF crystallization process in the thin film such that the entire synthesis process is extremely rapid—occurring over just several milliseconds, as opposed to the microcentrifugation-driven synthesis which required several minutes. Given the 10 MHz frequency employed, we also note that the nature of the acoustic wave generated on the acoustowetting device is no longer a pure SAW, but a hybrid resonant acoustic (HYDRA) configuration that constitutes a unique surface and bulk wave combination (i.e., a surface reflected bulk wave; SRBW),41 although for the intended purpose of this work, the acoustowetting flow and the nebulization it generates is no different than that phenomenologically produced by the SAW, albeit with much greater efficiency. Both continuous and pulse modulation operation, the latter at different pulse durations (or frequencies, which are typically kHz order), are explored and will be shown to facilitate tunability of the crystal thickness, and, as a consequence, the promotion of the active metal sites to the crystal surfaces.
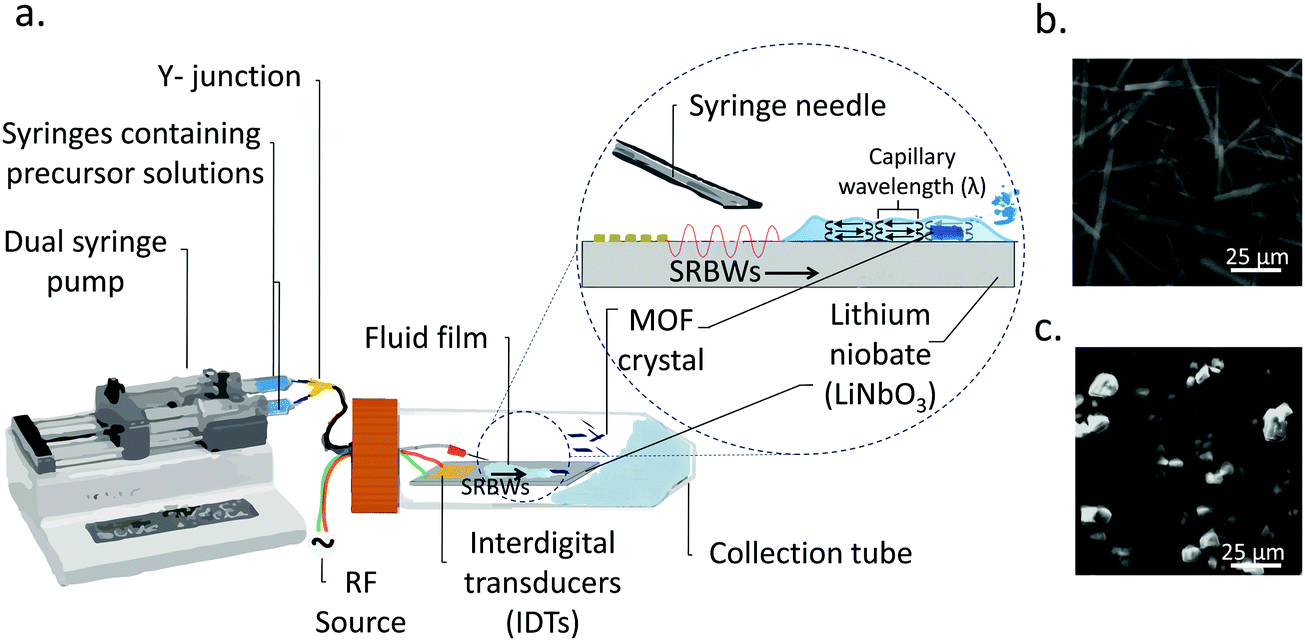 | ||
| Fig. 1 (a) Schematic illustration of the acoustic device for the synthesis of swordlike Cu–BTC MOFs. Upon excitation of the acoustic wave (i.e., the SRBW) on the device, a thin fluid film comprising the MOF precursor solutions is drawn out from the syringe and spreads along the surface of the device. Vertical and lateral dimension confinement of the diffusion kinetics of the precursors inside the thin film, and thus of the crystals that are produced, are imposed by ‘convective mixing cells’ defined by the film thickness and its resonant capillary wavelength, which is equivalent to the SRBW wavelength, respectively. Moreover, the crystallization is accelerated due to the fast evaporation rate that arises as a consequence of the nebulization of the film, which, in turn, further arrests the unidirectional growth of the MOF crystals along the vertical (thickness) direction. (b) and (c) SEM images comparing (b) the swordlike Cu–BTC crystals that form under the acoustic templating to (c) the typical cubic morphology of HKUST-1 crystals formed under conventional bulk solvothermal methods in the absence of the acoustic forcing (control). | ||
More specifically, this novel acoustic-driven templating technique allows us to exploit the combination of dimensional confinement afforded by the spreading hydrodynamics of the thin acoustowetting film40 and the capillary wave resonance imposed on its free surface by the substrate vibration, i.e., the SAW,42 in concert with the fast evaporation dynamics associated with the subsequent nebulization of the film43 to arrest further crystal growth in the thickness dimension. This results in MOFs—in this case, Cu3(1,3,5-benzenetricarboxylate)n (Cu–BTC)—with distinctive and uncommon monoclinic (P21/n) crystal structures with swordlike morphologies whose length far exceeds their vertical dimensions (i.e., their thicknesses). More importantly, we reveal that the large aspect ratio of these swordlike morphologies facilitates exposure of its inherently hidden metal active sites.
2 Materials and methods
2.1 HYDRA device
10 MHz HYDRA devices41 were fabricated by patterning 40 interdigital transducer (IDT) finger pairs comprising 10 and 400 nm thick chromium and aluminium films with 3.9 mm aperture widths on 500 mm thick 128° Y–X double-sided polished single-crystal piezoelectric lithium niobate (LiNbO3) substrates (University Wafer Inc., South Boston, MA, USA) using UV lithography. The acoustic waves, i.e., the SRBWs, were excited by applying a sinusoidal electrical input at the resonant frequency (f = 10 MHz) to the IDTs with a signal generator (SML01; Rhode & Schwarz, North Ryde, NSW, Australia) and amplifier (LYZ-22+, Mini Circuits, Brooklyn, NY, USA). f is related to the SRBW wavelength λ by f = c/λ, wherein c is the speed at which the acoustic wave propagates in LiNbO3; λ is set by the pitch of the IDT fingers (specifically, four times the finger width and gap).2.2 MOF synthesis
0.420 g 1,3,5-benzenetricarboxylic acid (C6H3(CO2H)3 (BTC); Sigma Aldrich Pty. Ltd., Castle-Hill, NSW, Australia) and 0.875 g copper(II) nitrate hemi(pentahydrate) (Cu(NO3)2·2.5H2O; Sigma Aldrich Pty. Ltd., Castle-Hill, NSW, Australia) were each dissolved in 12 ml of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (vol/vol) ethanol (Sigma Aldrich Pty. Ltd., Castle-Hill, NSW, Australia) and MilliQ® water (18.2 MΩ cm, Merck Millipore, Bayswater, VIC, Australia), and vortex spun for 10 min at 500 rpm until dissolved. As schematically depicted in Fig. 1, 10 ml of each of these precursor solutions were then delivered using a dual-syringe pump at a flow rate of 0.3 ml min−1via a 24 gauge syringe needle that is mounted so the fluid it dispenses flows out into the middle of the device. Upon turning on the acoustic excitation, a thin film comprising both precursor solutions is drawn from the needle, which subsequently spreads along the surface of the substrate and is nebulized.
:1 (vol/vol) ethanol (Sigma Aldrich Pty. Ltd., Castle-Hill, NSW, Australia) and MilliQ® water (18.2 MΩ cm, Merck Millipore, Bayswater, VIC, Australia), and vortex spun for 10 min at 500 rpm until dissolved. As schematically depicted in Fig. 1, 10 ml of each of these precursor solutions were then delivered using a dual-syringe pump at a flow rate of 0.3 ml min−1via a 24 gauge syringe needle that is mounted so the fluid it dispenses flows out into the middle of the device. Upon turning on the acoustic excitation, a thin film comprising both precursor solutions is drawn from the needle, which subsequently spreads along the surface of the substrate and is nebulized.
All experiments were carried out at a fixed voltage input of 26 Vrms, either under continuous wave excitation or pulsed modulation (50% duty cycle) with different pulse durations (0.1 ms, 100 ms and 400 ms). The MOFs were observed to nucleate and hence crystallize within the film, following which they were ejected along with the nebulized aerosols and subsequently collected in 50 ml centrifuge tubes. The resulting precipitate containing the crystals was thrice centrifuged for 10 min at 3000 rpm, and subsequently washed in 60 ml of a 1:1 (vol/vol) ethanol/MilliQ® water solution. After the final wash, the precipitated crystals were suspended in 20 ml MilliQ® water unless drying was required. A control sample of the same precursor concentrations was also delivered at the same flow rate via the same syringe pump to the surface of the device although in the absence of the acoustic excitation. The solution was then left to evaporate to allow crystallization to occur, through which a blue crystalline powder was obtained. The powder was then thrice washed in 60 ml of a 1:1 (vol/vol) ethanol/water solution, and reconstituted in 20 ml MilliQ® water for further characterization.
2.3 Materials characterization
The collected MOF crystals were deposited on atomic force microscopy (AFM) mica discs (10 mm diameter, Ted Pella Inc., Redding, CA, USA) glued onto an AFM metal disc (15 mm diameter, Ted Pella Inc., Redding, CA, USA) by drop casting 10 μl of the sample and leaving it to air dry for 10 min, following which they were examined under contact mode AFM (Multimode 8 with PeakForce Tunnelling (TUNA™) module; Bruker Corp., Santa Barbara, CA, USA). The AFM images that were obtained were first flattened at a z-threshold of 0.5 nm, from which the layer thickness and lateral size distributions across 512 bins were acquired by analyzing at least 200 crystals using the supplied software (NanoScope, v6.13; Bruker Corp., Santa Barbara, CA, USA).Scanning electron microscopy (SEM) imaging (Philips XL30; FEI, Hillsboro, OR, USA) was employed to characterize the morphology of the MOF crystals. Briefly, the crystals were deposited on a silicon wafer above which a 5 nm gold layer was sputtered for 60 s. Imaging was subsequently carried out at 10 kV. The size of the MOF crystals was determined through visual inspection of approximately 100 crystals from the SEM digital images using ImageJ (v1.34, National Institutes of Health, Bethesda, MD, USA).
A high-speed video camera (FASTCAM SA-X, Photron USA Inc., San Diego, CA, USA), operated at a maximum rate of 10000 frames s−1 and equipped with a long distance microscopic lens (K2-SC; Infinity Photo-Optical, Centennial, CO, USA) was used to visualize the spreading of the film and the capillary waves that are generated on its free surface.
The structure of the crystals that were synthesized were determined using powder X-ray diffraction ((XRD) D8 Advance; Bruker Pty. Ltd, Preston, VIC, Australia) under Cu Kα radiation at 40 mA and 40 kV (λ = 1.54 Å) with a scan rate of 2° min−1, step size of 0.02°, and a 2θ range of 6° to 90°. Single crystal diffraction data for structure resolution were collected using a diffractometer (Apex Duo; Bruker Pty. Ltd, Preston, VIC, Australia) using monochromated Mo Kα radiation (λ = 0.71069 Å) and a CCD camera (Apex II; Bruker Pty. Ltd, Preston, VIC, Australia) as an area detector. Structure solution and Rietveld refinement was performed using the SHELXTL package (Bruker Pty. Ltd, Preston, VIC, Australia),44 and matched against crystal structure files obtained from the Cambridge Crystallographic Data Center (CCDC data card number 987920; Cambridge, UK).31 The calculated XRD patterns were obtained using the Mercury Crystal Visualisation Software package (v3.8; Cambridge Crystallographic Data Center, Cambridge, UK).45
Fourier Transform Infrared (FTIR) spectroscopy analysis of the samples at room temperature was carried out using a spectrophotometer (Spectrum One; PerkinElmer Inc., Waltham, MA, USA) by placing a 10 μl suspension of the crystals on a diamond substrate, from which transmittance measurements were conducted in the wavenumber range between 500 and 4000 cm−1. The thermal properties of the crystals, on the other hand, were analyzed through thermogravimetric analysis ((TGA) Pyrus 1; PerkinElmer Inc., Waltham, MA, USA). Specifically, 7.5 mg of the crystals were placed in an aluminium pan held at 50 °C for 5 min prior to being heated at a rate of 10 °C min−1 under N2 from 35 °C to 800 °C.
The conductivity enhancement of the synthesized MOFs can be inferred from the current–voltage relationship of 1 cm2 hand-pressed films comprising the optimised (shown below to correspond to a pulse duration of 0.1 ms) Cu–BTC swordlike crystals and the conventional bulk 3D crystals prepared via solvothermal synthesis (control). The crystal mass in both films was the same (0.1 mg). Measurements were conducted using a 2-electrode probe station (C-2-RF, EverBeing International Corp., Hsinchu, Taiwan) together with an electrometer and source meter unit (Model 4200; Keithley Instruments Inc., Cleveland, OH, USA) through the application of a 1 V voltage bias.
3 Results and discussion
Once the liquid containing the Cu–BTC precursors, namely, copper(II) nitrate hemi(pentahydrate) (Cu(NO3)2·2.5H2O) and 1,3,5-benzenetricarboxylic acid (C6H3(CO2H)3), from the dual syringe pump contacts the surface of the HYDRA device, the SRBW, which is launched from the interdigitated transducer (IDT) electrode and propagates along the surface of the substrate, draws the liquid into a film such that it spreads along the surface of the device (Fig. 1a). As can be seen from the images captured via high speed video in Fig. 2 (left column), the dimension of the thin film that is formed is somewhat dependent on the pulse duration, and has a strong influence on the intensity (i.e., amplitude) and resonant frequency (and hence the wavelength) of the standing capillary waves imposed on its free surface as a consequence of the underlying substrate vibration.46 This, in turn, has a direct effect on the corresponding thickness of the MOF crystals that are generated within the film, as observed in the AFM profiles in Fig. 2a–d for a fixed input power to the device, thus alluding to the role of the film hydrodynamics and capillary wave resonant dynamics in the MOF production and hence its potential to be tuned; given the typical millisecond order residence time of the film on the device as it spreads prior to being nebulized, we note that the crystallization of the MOFs occurs rather rapidly.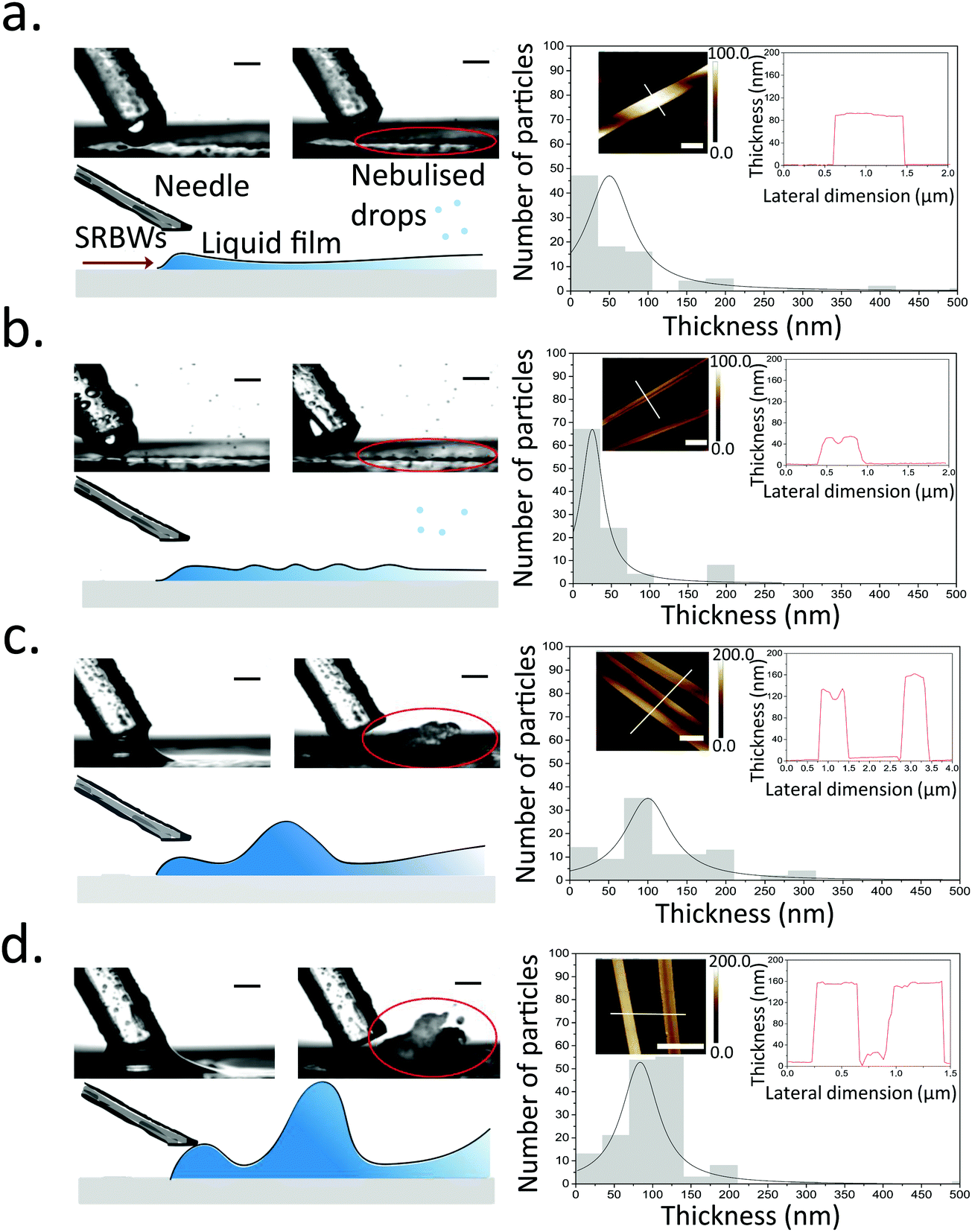 | ||
| Fig. 2 High speed video footage (left column, top) and schematic depiction of the acoustowetting film hydrodynamics (left column, bottom), and, corresponding thicknesses (obtained from analysis of the AFM images) of the swordlike crystals formed in the film under (a) continuous exposure to SRBW, and, under pulsed modulation with pulse durations of (b) 0.1, (c) 100, and, (d) 400 ms. The scale bars in the high speed camera images denote lengths of 500 mm, whereas those in the AFM scans represent a length of 1 μm. The red circled regions in the high speed video images highlight the differences in the film thicknesses and resonant capillary wavelengths for the different cases, which, in turn, has an effect on the thicknesses of the crystals that are produced. | ||
In general, all of the Cu–BTC MOFs that were produced under this acoustic templating technique possessed elongated swordlike morphologies (see, for example, the representative images in Fig. 1b) compared to the regular 3D cubic crystals that are formed in the absence of the acoustic excitation through conventional bulk solvothermal synthesis (Fig. 1c). In particular, the lattice motifs can be seen to preferentially grow along the lateral in-plane direction to form the crystals. While their average widths, approximately 7 μm ± 2 μm, and lengths, around 70 ± 25 μm, do not vary considerably, the thicknesses of the swordlike structures can be seen from Fig. 2a–d to decrease as the pulse duration is increased from continuous operation (limit of zero pulse width) where the average thickness is approximately 50 ± 10 nm (Fig. 2a) to an optimum of around 30 ± 15 nm at a pulse duration of 0.1 ms (Fig. 2b); this optimum pulse duration is associated with the case where the rate at which the liquid is delivered to the substrate roughly matches the rate at which it leaves the substrate via nebulization such that the residence time of the film on the substrate is minimized. With longer pulse durations above this minimum film residence time, the average thickness is however seen to increase (Fig. 2c and d).
Viewed together with the effect of the pulse duration on the film hydrodynamics and capillary wave resonance in Fig. 2, a possible underlying mechanism responsible for the different MOF crystal thicknesses can then be construed. It is likely that the height and the resonant capillary wavelength (approximately 100 μm, which is roughly the same as the SRBW wavelength) of the acoustowetting film46–48 imposes both vertical and lateral confinement to set up compartmentalized ‘cells’ within which convective mixing of the precursors occurs in a manner akin, though considerably distinct in origin, to Rayleigh-Bénard cells arising due to thermal convection49 (parenthetically, we note from our previous work in which the liquid comprised a polymer solution that these ‘cells’ are the sites where regular hexagonal closed-packed polymer spot patterns are formed on the substrate when the solvent, in which the polymer was dissolved, is completely nebulized39). The MOF crystals that form under the precursor mixing within these cells are then restricted by their dimensions, thus resulting in their large aspect ratio (both in terms of width and length, which are micron order, compared to their relatively thin nm-order thicknesses) swordlike morphologies. The subsequent nebulization of the film that ensues drives fast evaporation of the solvent, which additionally reinforces the resultant crystal geometry by further arresting crystal growth along the vertical (thickness) dimension. As such, when the pulse duration exceeds the minimum film residence time, liquid buildup occurs since more liquid is supplied to the substrate than can be nebulized, thus reducing the intensity of the capillary wave resonance (at least its harmonic response associated with the 100 μm wavelength), consistent with our previous observations where the harmonic excitation of the capillary waves becomes progressively weaker as the film height is increased, being subsumed by a larger low frequency (kHz order) broadband response.42,46,50
XRD data of the crystals synthesized under all of the conditions tested reveal primitive monoclinic crystal structures with a P21/n space-group and corresponding cell parameters a = 6.7590 Å, b = 18.8403 Å, c = 8.5206 Å, α = γ = 90°, β = 92.406°, and chemical composition C9H10O9Cu, with coordinated termination of the exposed Cu moieties (Fig. 3a). Interestingly, we observe a relative reduction in the in-plane (i.e., the (020) plane) intensity in comparison to an increase in the out-of-plane (i.e., the (121), (13![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and (140) planes) intensity, confirming the increase in crystal thickness at 100 ms and 400 ms pulse durations (Fig. 4). In contrast, the control sample that was produced using conventional bulk solvothermal synthesis yielded the regular 3D cubic morphology with face-centered (Fm3m) space group and cell parameters a = b = c = 26.343 Å, α = β = γ = 90° typically reported in the literature (Fig. 3b), commonly known as HKUST-1.
) and (140) planes) intensity, confirming the increase in crystal thickness at 100 ms and 400 ms pulse durations (Fig. 4). In contrast, the control sample that was produced using conventional bulk solvothermal synthesis yielded the regular 3D cubic morphology with face-centered (Fm3m) space group and cell parameters a = b = c = 26.343 Å, α = β = γ = 90° typically reported in the literature (Fig. 3b), commonly known as HKUST-1.
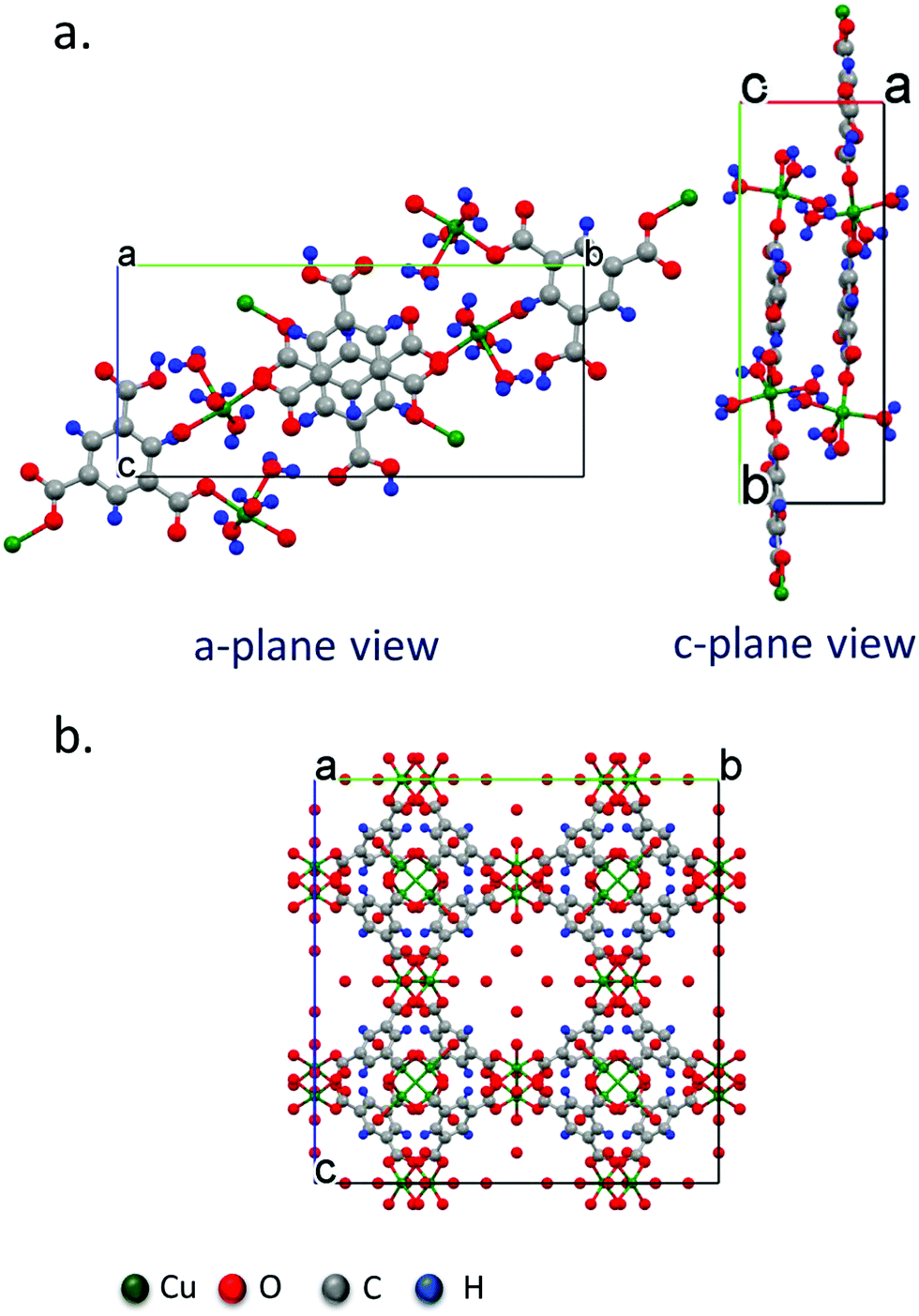 | ||
| Fig. 3 Crystal structure models obtained from single crystal diffraction data of (a) the P21/n monoclinic Cu–BTC MOF crystal with the swordlike morphology shown in Fig. 2 synthesized by the acoustic templating technique reported in this work, as viewed from the (100) and (111) crystal planes, and, (b) the conventional Fm3m cubic morphology of the HKUST-1 crystals synthesized using the bulk solvothermal technique (control). | ||
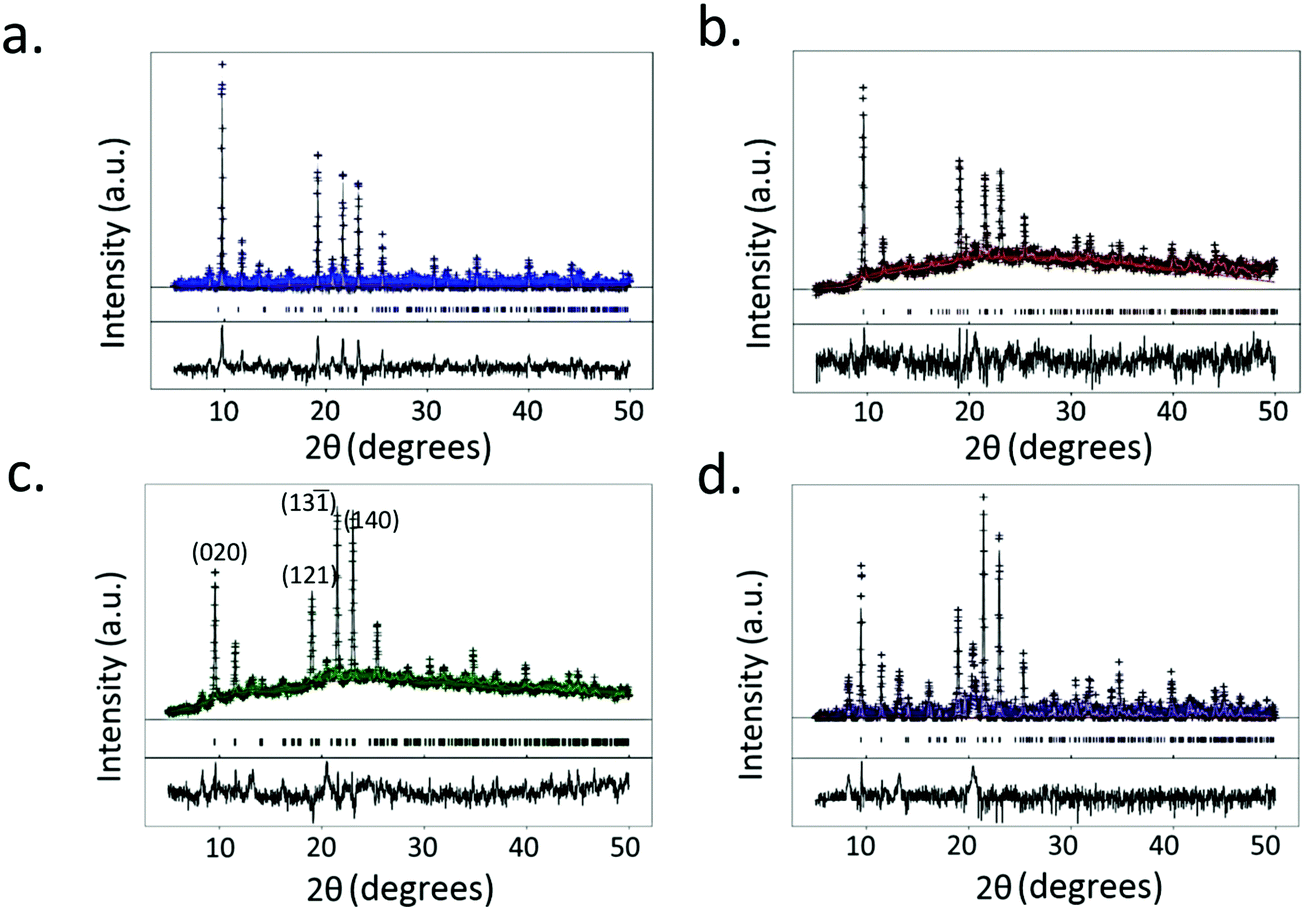 | ||
| Fig. 4 Single crystal X-ray diffraction analysis showing Rietveld refinement of the crystals synthesized under (a) continuous (blue) acoustic excitation, and, (b) 0.1 ms (red), (c) 100 ms (green), and (d) 400 ms (purple) pulse durations, showing monoclinic P21/n crystal structures. The decrease in the relative intensity of the (020) crystal plane in comparison to the increase in intensities of the (121), (13) and (140) crystal planes indicate an increase in crystal thickness for those obtained under the 100 ms and 400 ms pulse durations. | ||
To confirm its structure, TGA of the crystals obtained from the acoustic templating technique (Supplementary Fig. 1, ESI†) showed an initial weight loss occurring at 75 °C, which can be attributed to the release of two out of the three Cu2+ coordinated H2O molecules. The loss of the third coordinated H2O molecule within the structure is associated with a further weight loss observed at 205 °C. The total weight loss observed for all three coordinated H2O molecules is 14.1%. The 51.1% weight loss observed at the onset of 270 °C corresponds to the loss of the BTC ligand. The remaining mass can be attributed to Cu (calculated to be 15.1%) and the degraded sample, with an observed mass of 27.5% at 800 °C. These weight losses correspond to a 3:1:1 ratio between the H2O molecules, Cu, and BTC, respectively. These results strongly agree with the crystal structure of the network. The FTIR spectra for the synthesized crystals (Supplementary Fig. 2, ESI†) further verified the coordination of H2O molecules in the crystal structure as seen by its characteristic broad peak at 3600–3500 cm−1. The appearance of the peaks associated with the asymmetric stretching of the carboxylate groups in the Cu–BTC molecules at 1508–1623 cm−1, the symmetric stretching of the COO–Cu carboxylate groups at 1384 and 1405 cm−1, and the various bands attributed to the out-of-plane vibration of the BTC ligand over 1300–600 cm−1 provides further confirmation of the structure; we note, however, that the infrared (IR) modes associated with Cu below 600 cm−1 are out of the detectable range for the system employed.
A consequence of the increased surface area to volume ratio of the swordlike MOFs and the corresponding exposure of the Cu nodes along their surface (Supplementary Fig. 3, ESI†) is the accompanying enhancement in its conductivity. The current–voltage curves in Supplementary Fig. 4 (ESI†) indicate an approximate 100-fold improvement in the current density (125 μA cm−2 at a low overpotential η10 of 120 mV) of the as-synthesized compressed films comprising the optimized (0.1 ms pulse duration) swordlike Cu–BTC crystals compared to the value (0.002 μA cm−2 at an overpotential η10 of 2 V) obtained for films made from bulk 3D cubic HKUST-1 crystals synthesized using the bulk solvothermal method, consistent with the 50- to 70-fold increase in the surface area to volume ratio (Supplementary Table 1, ESI†). Given that this two order-of-magnitude improvement was achieved with neat MOFs (i.e., without the inclusion of any additives or conductive binders), we anticipate such surface active site exposure via synthesis of large aspect ratio swordlike crystals to complement other strategies for enhancing MOF conductivity3,4 in order to obtain even larger improvements than those already reported.
4 Conclusion
In summary, we have demonstrated a unique approach for the synthesis of large aspect ratio MOFs with exposed active catalytic sites by arresting their growth along the vertical axis through rapid confinement afforded by thin resonant acoustowetting films. These high aspect (length or width respect to thickness) ratio swordlike crystals, whose thicknesses can be controlled through the duration associated with the pulse modulation of the acoustic excitation, possess a structure distinct from that typically obtained using conventional bulk solvothermal synthesis. Given the simplicity and speed (millisecond order) of the process, and the low costs of the acoustic devices, we anticipate that the current production rate of 4 g h−1 per device, which is equivalent to a space-time yield σP of approximately 36000 kg m−3 day−1, can easily be upscaled through massive device parallelization for high throughput production of unique one-dimensional MOF architectures that offer significant advantages over other methods, particularly for applications involving heterogeneous catalysis.51
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. R. R. and L. Y. Y. are grateful for funding from the Australian Research Council (ARC) through Discovery Project DP180102110. J. R. acknowledges funding from the Swedish Foundation for Strategic Research (SSF).References
- H. Furukawa, K. E. Cordova, M. OKeeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- B. Chen, M. Eddaoudi, S. T. Hyde, M. O’Keeffe and O. M. Yaghi, Science, 2001, 291, 1021–1023 CrossRef CAS PubMed.
- S. K. Bhardwaj, N. Bhardwaj, R. Kaur, J. Mehta, A. L. Sharma, K.-H. Kim and A. Deep, J. Mater. Chem. A, 2018, 6, 14992–15009 RSC.
- M. Ko, L. Mendecki and K. A. Mirica, Chem. Commun., 2018, 54, 7873–7891 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- K. Gedrich, I. Senkovska, N. Klein, U. Stoeck, A. Henschel, M. R. Lohe, I. A. Baburin, U. Mueller and S. Kaskel, Angew. Chem., Int. Ed., 2010, 49, 8489–8492 CrossRef CAS PubMed.
- H. B. Wu and X. W. D. Lou, Sci. Adv., 2017, 3, 9252 CrossRef PubMed.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- C.-D. Wu and M. Zhao, Adv. Mater., 2017, 29, 1605446 CrossRef PubMed.
- F. Luo, C. Yan, L. Dang, R. Krishna, W. Zhou, H. Wu, X. Dong, Y. Han, T.-L. Hu, M. O’Keeffe, L. Wang, M. Luo, R.-B. Lin and B. Chen, J. Am. Chem. Soc., 2016, 138, 5678–5684 CrossRef CAS PubMed.
- X. Zhao, X. Bu, T. Wu, S.-T. Zheng, L. Wang and P. Feng, Nat. Commun., 2013, 4, 2344 CrossRef PubMed.
- P.-Z. Li, X.-J. Wang, S. Y. Tan, C. Y. Ang, H. Chen, J. Liu, R. Zou and Y. Zhao, Angew. Chem., Int. Ed., 2015, 54, 12748–12752 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley III, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste, G. W. Peterson, R. Q. Snurr, C. J. Cramer, J. T. Hupp and O. K. Farha, Nat. Mater., 2015, 14, 512 CrossRef CAS PubMed.
- A. Huang and J. Caro, Angew. Chem., Int. Ed., 2011, 50, 4979–4982 CrossRef CAS PubMed.
- C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS PubMed.
- X. Xu, F. Nosheen and X. Wang, Chem. Mater., 2016, 28, 6313–6320 CrossRef CAS.
- A. W. Peters, Z. Li, O. K. Farha and J. T. Hupp, ACS Appl. Mater. Interfaces, 2016, 8, 20675–20681 CrossRef CAS PubMed.
- Y. Shu, Y. Yan, J. Chen, Q. Xu, H. Pang and X. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 22342–22349 CrossRef CAS PubMed.
- W. Ren, H. Zhang, C. Guan and C. Cheng, Adv. Funct. Mater., 2017, 27, 1702116 CrossRef.
- Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4635–4637 CrossRef CAS PubMed.
- K. K. Tanabe, Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2008, 130, 8508–8517 CrossRef CAS PubMed.
- C. D. Fast, J. Woods, J. Lentchner and T. A. Makal, Dalton Trans., 2019, 48, 14696–14704 RSC.
- H. Guo, Y. Zhu, S. Qiu, J. A. Lercher and H. Zhang, Adv. Mater., 2010, 22, 4190–4192 CrossRef CAS PubMed.
- S. Wang, W. Morris, Y. Liu, C. M. McGuirk, Y. Zhou, J. T. Hupp, O. K. Farha and C. A. Mirkin, Angew. Chem., Int. Ed., 2015, 54, 14738–14742 CrossRef CAS PubMed.
- F. Vermoortele, M. Vandichel, B. Van de Voorde, R. Ameloot, M. Waroquier, V. Van Speybroeck and D. E. De Vos, Angew. Chem., Int. Ed., 2012, 51, 4887–4890 CrossRef CAS PubMed.
- P. Serra-Crespo, E. Gobechiya, E. V. Ramos-Fernandez, J. Juan-Alcañiz, A. Martinez-Joaristi, E. Stavitski, C. E. A. Kirschhock, J. A. Martens, F. Kapteijn and J. Gascon, Langmuir, 2012, 28, 12916–12922 CrossRef CAS PubMed.
- K. Bodnár, E. Fegyver, M. Nagy and R. Mészáros, Langmuir, 2016, 32, 1259–1268 CrossRef PubMed.
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC.
- F. Cao, M. Zhao, Y. Yu, B. Chen, Y. Huang, J. Yang, X. Cao, Q. Lu, X. Zhang, Z. Zhang, C. Tan and H. Zhang, J. Am. Chem. Soc., 2016, 138, 6924–6927 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban and W. Yang, Angew. Chem., Int. Ed., 2017, 56, 9757–9761 CrossRef CAS PubMed.
- A. Kojtari, P. J. Carroll and H.-F. Ji, CrystEngComm, 2014, 16, 2885–2888 RSC.
- Z. Kang, L. Fan and D. Sun, J. Mater. Chem. A, 2017, 5, 10073–10091 RSC.
- M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu, H. Xu, Y. Zhao and H. Zhang, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed.
- H. Ahmed, A. R. Rezk, J. J. Richardson, L. K. Macreadie, R. Babarao, E. L. H. Mayes, L. Lee and L. Y. Yeo, Nat. Commun., 2019, 10, 2282 CrossRef PubMed.
- J. Friend and L. Y. Yeo, Rev. Mod. Phys., 2011, 83, 647–704 CrossRef.
- L. Y. Yeo and J. R. Friend, Annu. Rev. Fluid Mech., 2014, 46, 379–406 CrossRef.
- J. Shi, X. Mao, D. Ahmed, A. Colletti and T. J. Huang, Lab Chip, 2008, 8, 221–223 RSC.
- G. Destgeer and H. J. Sung, Lab Chip, 2015, 15, 2722–2738 RSC.
- M. Alvarez, J. R. Friend and L. Y. Yeo, Langmuir, 2008, 24, 10629–10632 CrossRef CAS PubMed.
- A. R. Rezk, O. Manor, J. R. Friend and L. Y. Yeo, Nat. Commun., 2012, 3, 1167 CrossRef PubMed.
- A. R. Rezk, J. K. Tan and L. Y. Yeo, Adv. Mater., 2016, 28, 1970–1975 CrossRef CAS PubMed.
- J. Blamey, L. Y. Yeo and J. R. Friend, Langmuir, 2013, 29, 3835–3845 CrossRef CAS PubMed.
- H. Ahmed, L. Lee, C. Darmanin and L. Y. Yeo, Adv. Mater., 2018, 30, 1602040 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- A. Qi, L. Y. Yeo and J. R. Friend, Phys. Fluids, 2008, 20, 074103 CrossRef.
- M. Tan, J. Friend, O. Matar and L. Yeo, Phys. Fluids, 2010, 22, 112112 CrossRef.
- D. Collins, O. Manor, A. Winkler, H. Schmidt, J. Friend and L. Yeo, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2012, 86, 056312 CrossRef PubMed.
- B. I. Shraiman, Phys. Rev. A: At., Mol., Opt. Phys., 1987, 36, 261–267 CrossRef PubMed.
- H. Li, J. R. Friend and L. Y. Yeo, Phys. Rev. Lett., 2008, 101, 084502 CrossRef PubMed.
- L. Yang, G. L. Ruess and M. A. Carreon, Catal. Sci. Technol., 2015, 5, 2777–2782 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00171f |
| This journal is © The Royal Society of Chemistry 2020 |