
Single atom catalysts: a surface heterocompound perspective
Zongkui
Kou
 a,
Wenjie
Zang
a,
Peikui
Wang
b,
Xin
Li
a and
John
Wang
*a
a,
Wenjie
Zang
a,
Peikui
Wang
b,
Xin
Li
a and
John
Wang
*a
aDepartment of Materials Science and Engineering, National University of Singapore, 117574 Singapore, Singapore. E-mail: msewangj@nus.edu.sg
bDepartment of Chemistry, University of Sherbrooke, Canada
First published on 3rd March 2020
Abstract
The concept of single atom catalysts (SACs) originated from reducing the amount of noble metals used, by steadily refining the particle size loaded on a substrate surface. It has been rapidly moving to non-noble elements and their compounds in recent years, notably transition metals and even non-metals. They are of heterogeneous types, where the active species are refined to atomic dispersion scales on the surfaces/sub-surfaces of the solid support. The catalytic performance is governed by both the type and population of accessible active sites, and their bond and coordination environment, largely as a result of the interactions with the substrate surface. Unlike the internal structure within a crystalline solid, there is a large spatial variation in the bond and coordination environment of different atoms on the solid surface across different length scales, and in particular with the unsaturated surface, where there are various defects. They can also be dramatically altered during both the catalyst synthesis and actual catalysis process. In a way, they form a “surface heterocompound”, where the local bonds for each metal atom are of a compound type, while there can be a large variation from one to another. Herein, we will look into the evolution from traditional heterogeneous catalysts to SACs, from the surface heterocompound perspective. Discussion will then be made on the on-going strategies and challenges in manipulating and identifying the local bond and coordination environment on the hetero-surfaces, in an attempt to develop efficient catalysts for the targeted applications, where both synthesis techniques and analytical tools are critically important, and computational studies can provide the key guiding principles. With selected paradigm studies, we will briefly examine the future perspectives for this newly emerging catalysis frontier.
1. Introduction
Various catalyses and catalysts, which are involved in more than 90% of the chemical, manufacturing and pharm processes, are an indispensable part of the large industry chains. Among them, the most widely used catalysts are of heterogeneous types, which involve the active surfaces in solid catalysts, and they can either be bare or act as the substrate of the active components. Among the earliest applied heterogeneous catalysts were those employed in the catalytic production of sulfuric acid and ammonia oxidation,1 where Pt metal particles were chosen as the main solid catalytic components,2 while the examination into catalytic mechanisms largely remained at the phenomenological understanding, before the early 20th century. Subsequently, these solid catalysts were quickly expanded to other metals, such as transition metal particles of Fe, Co and Ni,3 as well as their oxides and non-oxides,4 since these elements with unfilled d-band electron configurations possess high catalytic activities. In 1925, Taylor first put up the active center theory,5 which marked the beginning of heterogeneous catalysts entering into the large commodity-fuelled growth phase, such as the industrial Fischer–Tropsch synthesis6 and selective catalytic hydrogenation.7 Over the past two decades, since the steady emergence of nanoscience and nanotechnology starting in the early 1990s,8 there have been subjective manipulations in the exploration of various catalysts involving single atoms, in attempts to achieve high efficiency, improved selectivity and 100% atom utilization efficiency.9 Indeed, it has been one of the main advances in heterogeneous catalysts in recent years.10,11 With the metallic-type active centers being dispersed at atomic scales, single atom catalysts (SACs) can effectively break the limit of the poor reaction selectivity and low atom utilization efficiency in some of the traditional heterogeneous catalyses, and therefore they have great potential in the rapidly widening applications in energy, chemical, environment and pharm industries. Unlike the homogeneous catalysts, there is no need for post-use separation, although SACs are dispersed at atomic scales.Among the early studies, the terminology of SACs was first put up by Zhang and co-workers, when they investigated the catalytic performance of Pt1 atoms loaded on the surface of FeOx for CO oxidation.12 Since then, many different types of SACs have been synthesized and investigated as models for both developing catalytic theories13 and the exploration of their catalytic performance in targeted applications. According to a recent definition,14 SACs possess three features which are as follows: (i) they contain isolated active atoms exposed on the substrate surface; (ii) they are often dispersed randomly and with a distribution of heterogeneous coordination types, where they are strongly dependent on supports endowing them with the heterogeneous nature; and (iii) single atom-level active sites may not break the limit of scaling relation in the multistep catalysis15 due to the high step-catalytic selectivity, and therefore a combination of different atom sites can help complete the whole catalytic multistep process.
Given the high catalytic activity, reaction selectivity and high atom utilization efficiency successfully demonstrated for a number of SAC systems in the past decade, there have been ever-intensifying efforts in developing the disruptively different catalysts for targeted applications. Among the on-going activities on SACs are to identify the key configurations of bond and coordination for active centers/species, and then devise synthesis conditions to form them, followed by performance evaluation in the targeted applications. In addition to those SAC systems involving a single metal type, several researchers have looked into the metal dimers,16 trimers17 and multimers,18 where the multiple bonds and coordination environments are expected to fix on the same substrate surfaces. Unlike SACs involving one metal type, they can offer more than one type of active sites for “cosplay” synergistic functions, which are especially useful for those catalytic reactions involving complicated multisteps, such as oxygen-involving reactions (ORR and OER), nitrogen reduction reaction (NRR) and CO2 reduction reactions (CO2 RR). In particular, a number of researchers have primarily tried to synthesize atomic-level metal dimers/trimers on the nitrogen-contained carbon or oxide supports.19 These catalysts with multiple and neighboring active centers as well as well-confined coordination among them can apparently add more advantages to SACs, such as a much higher efficiency, excellent reaction selectivity and wider flexibility in devising the most desired multi-coordination environment. With the multiple and synergistic active sites, they can therefore present several distinct features, for example, including: (i) enabling the complementary advantages of different atomic types for the multiple steps of certain catalyses; (ii) maximizing the limited active area of the heterogeneous surface/interface; and (iii) achieving a higher efficiency and selectivity for specific catalytic processes via the purposely devised bond and coordination of atoms.
With the steady downsizing, from small particles, clusters, dots and eventually to single atoms supported on a substrate surface for heterogeneous catalysts, there are apparent changes in the bond and coordination environment for almost each and every atom involved. For a relatively large metal particle, the internal metallic bond dominates, although those atoms on the surface will interact with the substrate support. With the steady refining in the particle size, where the ultimate dispersion consists of single atoms, there is an increasing fraction of atoms exposed on the surfaces, where they have opportunities to interact/react with the substrate surfaces, thus giving rise to bond/interaction with the unsaturated and defective support surface. A single atom cannot possibly stay by itself at the surface, if without any bond/interaction with the substrate surface.9 This is especially so for the transition metals, which are much more reactive than the noble metals, and they have been widely studied as SACs. Such high reactivity can limit the loading in order to avoid the aggregation of isolated single atoms, and gives rise to poor stability of SACs during both synthesis and catalytic applications. Therefore, the local bond and interactions of single atoms with the substrate are also crucial for improving the loading and stability of SACs. Considering the local bonds between a single metal atom and the support surface, and a likely wide variation among the different metal atoms, one will see the formation of a “surface heterocompound”, where the valence states of the metal atoms involved can vary from the lowest oxidation state (metal) to the highest oxidation state. Together with the widely variable positions available on the substrate surface, these metal atoms can be located at the edges, steps, kicks, corners and/or other defective places, depending on the type and surface condition of the substrate support. Therefore, there is a large spectrum in both the type and population for each type of the active sites for a given SAC system. Further considering the variable unfilled d-orbitals involved in certain metals, there is a further addition on the apparent wide distribution in the type and population of active sites for the adsorption and release of intermediates.20 In the following, we will look into the single atom catalysts from the surface heterocompound perspective.
2. Single metal types with varying bond and coordination
From a thermodynamic point of view, single atoms can hardly exist as a stable species by itself, due to the extremely high surface energy, such that a well-confined bond/interaction with the substrate surface is required. On the other hand, there is no such thing as a “perfect” substrate surface in reality, as shown by selected examples in Fig. 1, where the surface is rather different from the solid interior, and there are considerable variations in both sites and likely defects on the unsatisfied surface. For example, there are a range of edges, steps, kicks, corners, surface reconstruction sites, distortions, and defects on the unsaturated solid surface. Even for a supposedly atomic flat 2D material, such as single-layer graphene and MoS2, there can be a large population of defects, either deliberately or accidentally made, on top of the edge and corner locations, depending on the processing conditions that they have experienced. The presence of any dopants, where N in graphene is one of the best known examples for SACs, will generate different sites and functional groups. Therefore, they create opportunities for metal atoms to be in many different bonds and coordination environments. As such, the largely varying bond and coordination environment of those single atoms on the substrate surface presents a huge challenge to rationally identify them, and understand and recognize the catalytic process by each of the specific active entities, which has been experienced in the on-going research frontier of exploring single atom catalysis in recent several years.21,22 For example, our group recently performed both experimental and DFT simulation studies with Cu single atoms anchored on the carbon nitride surface, where there are different types of Cu–N/C bonds. Among them, Cu–N2 is shown to be an active coordination structure for nitrogen reduction reaction (NRR), where several other possible Cu–Nx (Cu–N3 and Cu–N4) types exist for different functions.23 Although only 20 at% Cu–N2 species were obtained in the Cu single atom catalyst formed by our experimental conditions, they raised the nitrogen reduction reaction dramatically. Given the rather random mixture of the metal–Nx coordination types identified, there is an apparent need to tune the processing conditions in order to maximize the fraction of Cu–N2. Indeed, there have been a few studies on the precise tuning of the bond and coordination of single atom catalysts in the past one decade, in attempts to enhance their performance, such as catalytic efficiency, selectivity and durability, for targeted applications.24–26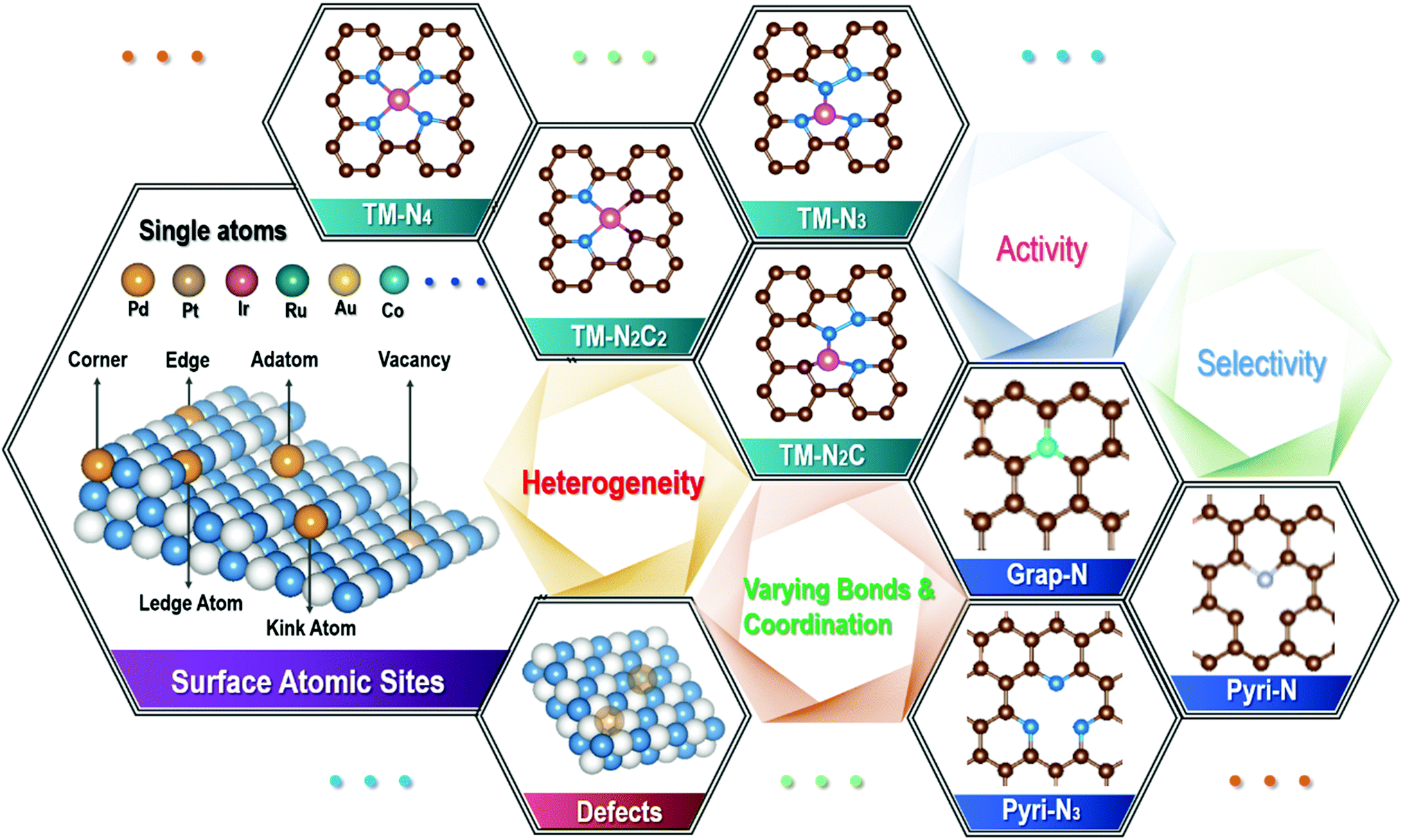 | ||
| Fig. 1 Schematic showing the surface heterocompound perspective of single atom catalysts: there are various sites and defectives for different single atoms, creating different bond and coordination environments for isolated atoms, where transition metal (TM) single atoms anchored on N-doped graphene are a classic example. | ||
To narrow down the distribution of the bond and coordination environments for single atoms, a well-defined substrate surface would be required to achieve a specifically targeted single functionality with a high degree of selectivity. Unfortunately, as has been mentioned above, for a real solid substrate, such as the widely reported metals,27 oxides,28,29 carbides,30 and carbon structures, it hardly exists. Most of these substrate surfaces possess various sites and defects, including those geometrical, valence-varying, dopant/missing sites, strains, distortions, and surface-reconstructions. For example, there exist a range of various defective sites for graphene derived from any solution processing technique, and/or with any hetero-atom doping, such as N-doped graphene (see Fig. 1). Indeed, the majority of N-doped graphene, which have been widely investigated as supports for SACs, are not “perfect”, originating from the synthesis conditions or being purposely engineered (by either chemical or physical means), in order to generate the required bonds with metal atoms,31 leading to a wide variation in the loading sites for metal single atoms on surfaces. Notably, there has been considerable efforts in modulating the N functional groups/species (e.g., graphitic N, pyridinic N and/or pyrrolic N), together with those structural defects (both intrinsic and geometrical types), in N-doped graphene, as a key paradigm to explore the optimum efficiency and selectivity for targeted applications. Even for the supposedly simplest N-doped graphene monolayer, the bond and coordination structures are indeed very complex, where there can be a large number of choices for a transition metal (TM) single atom. These different bond and coordination configurations coexist for different single atoms, giving rise to a huge difficulty in unveiling the authentic active sites among them, and linking the high performance to a specific coordination. Indeed, they undergo changes by different processing conditions, and can also be varied during the actual catalysis process, as transition metal atoms are rather reactive, as has been commonly experienced in OER electrocatalysis.32 Therefore, developing controllable and precision synthesis strategies to produce the designed configuration of bond and coordination of single atoms on a substrate support is a priority. For example, recent efforts have enabled certain single metal atoms to be stabilized by bonding/coordinating with specific single/double vacancy defects,33,34 pyridine-type/pyrrole-type N35–37 and thiophene-type S,38 although other possible bonds/coordination are hardly avoided at the same time.
Although the catalytic activity of most SACs studied so far is largely correlated to the adsorption/desorption of intermediates onto a specific type of metal-based center with an unsaturated d orbital, the overall local bond and coordination environment play a crucial role in controlling the catalytic process. Charge transfer and redistribution between the metal atoms and the surrounding coordinating atoms could well modulate the electronic structure of the metal center to optimize the adsorption properties. Therefore, doping heteroatoms around the single atom sites has been developed to further tune the bond, coordination and electronic structures of the single atom concerned.39 At this stage, theoretical predictions can first be performed to search, identify and rank the most active and stable bond and coordination configuration of the central atoms. For example, theoretical calculations have revealed that Pt single atoms anchored on vacancies and edges of graphene supports are more stable than the corresponding Pt dimers and inhibit the formation of Pt clusters during the water activation process.40 Subsequently, a precise scale-up synthesis can be developed to prepare SACs with the specific bond and coordination configuration under the guidance of thermodynamic principles, where the bond energy of each atom with the substrate shall be smaller than that between single atoms to avoid their reunion.41
3. Surface heterocompounds with metallic dimers, trimers, and multimers
In addition to SACs involving a single metal type, metal dimers, trimers, and multimers have been successfully implanted on support substrates, including metals, metal compounds and carbon-based substrates. These di-/tri-/multi-metal types dispersed at atomic-levels on the substrate surfaces can be either homogeneous or heterogeneous, either orderly or disorderly, in terms of the distribution of the metal atoms concerned. They would be largely determined by the synthesis technique and processing conditions employed to make them. These catalytically active sites in association with di-/tri-/multi metal-types can also exhibit a strong synergistic effect, and some of them have demonstrated superior activities in several catalytic reactions. In addition, most of the multistep catalytic reactions are known to be kinetically sluggish and proceed at rather high overpotential, due to the universal scaling relation, namely, the adsorption energies of intermediates are linearly correlated and cannot be optimized simultaneously on one single active site. Several approaches have been proposed to break the scaling relation by introducing additional active sites,42 although very few positive experimental results have been documented. In this context, constructing multifunctional active sites of di-/tri-/multi metal-types to a maximum exposed level on the substrate surface would be among the best practices. Nevertheless, there have been considerable “deficiency” and/or “misleading”, due to the limitations of currently available characterization techniques. For example, metal–N4 coordination was disclosed as a classic model for TM–N–C SACs, so some research had argued that integrating two different metal atoms into the metal dimer sites could produce TM1TM2–N6 coordination43,44 where the atomic height calculated by software was used to identify and differentiate TM1 from TM2. The research is definitely of interest; however the results need to be further verified. The direct observation, for example by precise and atomic-resolution electron energy loss spectroscopy (EELS), can be a promising choice to show the local atomic distribution one by one. There are few studies showcasing the exact bond and coordination of atomic pairs/ensembles. For example, Wu and Li et al. embedded Fe–Co metal dimers onto the N-doped carbon substrate, and applied the local EEL spectrum to show the existence of Fe–Co diatomic sites in the typical dimers, which exhibited an ORR performance significantly outperforming that of commercial Pt/C.45 Yao et al. used the fast Fourier transformation (FFT) and inverse FFT with noise filtered aiming to directly give the fingerprint of metal dimers.46 Moreover, they made a statistical distribution of 40 pairs and established that most of the metal–metal distances are located between 0.25 and 0.29 nm, which accounts for 32% of them. These results suggest even further variation in the bond and coordination environment in the surface heterocompounds involving di-/tri-/multi metal-types, compared with SACs involving one metal type.Given the high complexity in the surface heterocompounds, computational studies have significantly aided the experimental design in regulating the bond and coordination of one and/or more types of metallic atoms.47,48 They are capable of considering various possibilities on the types of bonds and coordination of metal centers in coping with the increasing variates and uncertainties. Due to the heterogeneous nature, the aggregation among metal atoms on a surface can be efficiently prohibited, based on the thermodynamic principles, leading to a remarkably increasing mass loading of the active metal atoms. In addition, developing effective strategies to tune the bond and coordination can be easily done, by simulation studies, which have been applied to several SAC systems. They have provided some key guiding principles for experimental works.24,49,50
Regulating the designed bond and coordination on the hetero-surfaces is among the main objectives of developing highly efficient and selective catalysis. A well-defined hetero-surface with known active configurations can also provide a model platform to investigate the desperately required structure–activity relationship, by deeply analyzing the local configurations at atomic scales in each of the intermediate catalytic steps. For example, by performing high throughput theoretical calculations, both the efficiency and selectivity of a catalytic heterocompound surface can be established and optimized. In the case of electrocatalysis, it is largely controlled by the strong interactions between the surface-coordinated atoms, rather than a single atom on the surface. In addition, an algorithm in machine-learning has been put up to construct a database able to screen out multi-metal atoms anchored on graphdiyne by analyzing the electron transfer number and direction.51 Nevertheless, such a paradigm is still largely restricted to the theoretical investigations so far but shines light as a pioneering work. Those catalytic surface heterocompounds with the designed bond and coordination present much enhanced performance, which is contributed by the synergistic functions/defects produced in situ by doping of the exotic metal atoms (Fig. 2a) on the surface. Indeed, the diverse catalytic reactions call for different configurations with the surface heterocompounds. In addition to the di-/tri-/multi-metal types, the integration of metal–nonmetal ensembles at molecular levels (Fig. 2b) on a stable support can also lead to a substantial enhancement in catalytic performance, when compared with a single metal type, although they have different coordination structures.19
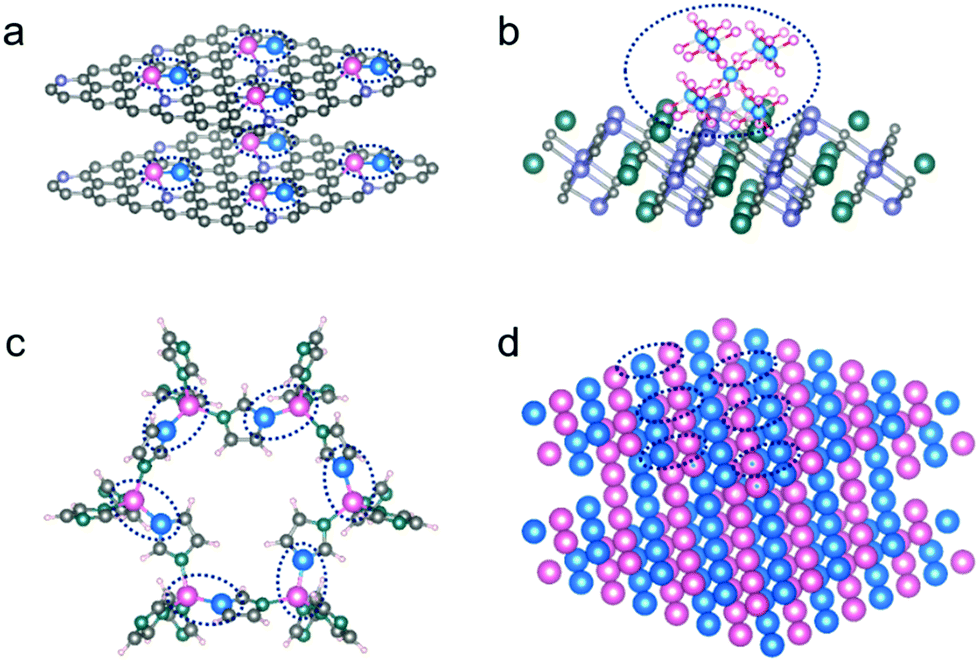 | ||
| Fig. 2 Examples of surface heterocompound catalysts: (a) active metal dimer bonded on a carbon support; (b) active molecule-level multimers bonded on a stable compound; (c) 2D bimetallic organic frameworks with two neighbouring active sites; (d) 2D bimetallene or bimetallic alloy surfaces. The pink and blue atoms represent two different well-confined active sites encircled. | ||
To prepare the di-/tri-/multi-metal types, metal–organic frameworks (MOFs) and certain organometallic complexes are among the useful porous precursor materials that are cross-linked by metal nodes and organic ligands. Some of them possess nearly 100% accessible active sites by themselves, and have controllable multi-metal atomic coordination (Fig. 2c), when used as heterogeneous catalysts. However, their relatively poor stability and low conductivity make them unsuitable for certain catalytic applications, such as those electrocatalyses requiring high conductivity.52 For this reason, there have been considerable efforts to anchor some of them on a strong substrate, e.g., by growing ultrathin MOFs supported on a conductive support, which has been demonstrated widely, especially on carbon-based materials.53 In parallel, there has been considerable success in applying them as a precursor, upon a designed post-synthesis treatment environment, to tune them into the desired chemical bond and coordination environment for isolated metal atoms anchored on the substrate surface. As such, a large number of MOF-derived SAC ensembles have been designed and fabricated.45,54,55 In addition, for example, bimetallene with bimetal alloy and sub-nanometer-thick sheet structure (Fig. 2d) was reported very recently by Guo et al., which was demonstrated with high electrochemical activity and nearly 100% atomic utilization, and thus exhibited an almost exponential enhancement in mass activity, in comparison to single metal sites on the surface.56
4. Case studies of structures and catalytic performance
Given the surface heterocompound perspective, the bond and coordination chemistry of isolated metal atoms with the substrate in SACs enable distinct catalytic activity and selectivity, which have been demonstrated in a large number of them. Some of these atomic ensembles on a well-confined substrate have pioneered certain unique advantages. For example, starting from single-atom Pt1/CeO2, a monolayer multi-atom Pt–O–Pt cluster on the CeO2 surface was devised by applying reductive H2 and oxidized CO + O2 to induce Pt assembly.18 Comparative HAADF-STEM studies of the typical Pt1/CeO2 (Fig. 3a) and Pt–O–Pt/CeO2 (Fig. 3b) catalysts disclosed that the single Pt1 atoms were evolved into a cluster structure with a size of 1 ± 0.1 nm on the CeO2 support. It is worth noting that Pt1/CeO2 possesses a more positive Pt chemical valence (IV, Pt–O) than that in Pt–O–Pt/CeO2 (Fig. 3c). However, Pt atoms in their first-shell coordination are largely bridged by oxygen in Pt–O–Pt/CeO2 (Fig. 3d), which is similar to that of Pt1/CeO2. It is rather challenging to differentiate them from Pt single atoms by EXAFs. A CO adsorption probe was then applied in the FTIR study to identify the Pt–O–Pt ensemble, in which the two CO adsorption bands at 2107 and 2081 cm−1 are assigned to the CO chemisorption of the Pt–O–Pt ensembles (Fig. 3e) at the top and bridge sites, respectively. When applied in a low temperature CO oxidation, the Pt–O–Pt ensemble showed a 100–1000 times activity of Pt1/CeO2 (Fig. 3f). Theoretical calculations further suggested that the bridge oxygen atoms had a synergistic effect on the adjacent Pt atoms. These results indeed demonstrate that Pt1 materials are less competitive than the multiple atoms involving the high coordination environment of Pt, which is a typical surface heterocompound in structure. Several investigations in recent years are also in line with the concept of PGM–O–PGM catalytic units, in breaking the shackles of high-performing catalysis. For example, the Ir–O–Ir and Rh–O–Rh ensembles prepared by Zhao57 and Jeong et al.58 also show more distinguished activities.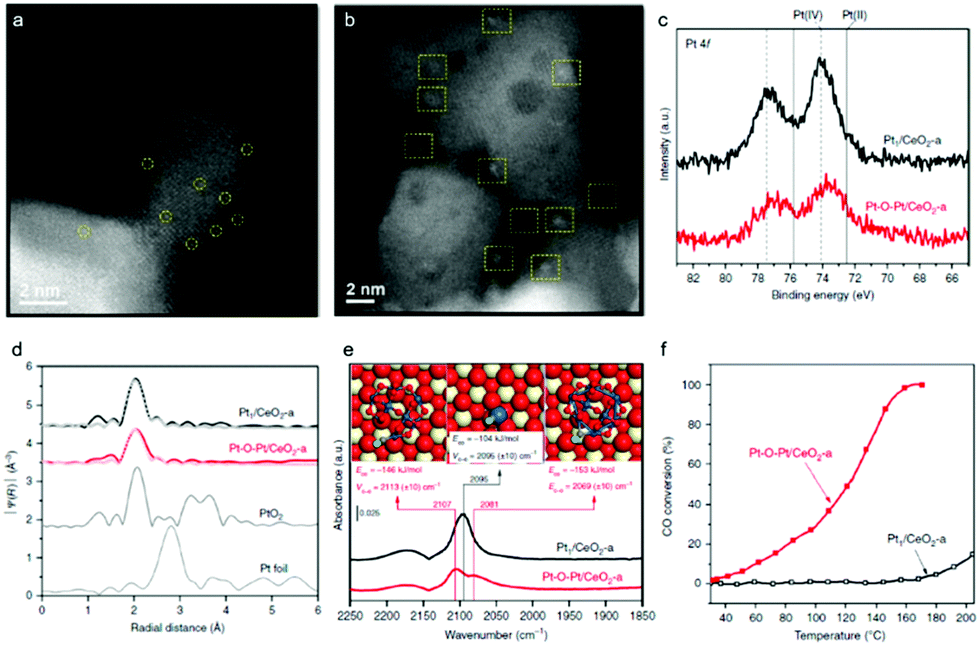 | ||
| Fig. 3 (a and b) HAADF-STEM images of the Pt1/CeO2 and Pt–O–Pt/CeO2 catalysts. Pt1 and Pt–O–Pt ensemble are highlighted by yellow circles and squares, respectively. (c) Pt 4f X-ray photoelectron spectroscopy (XPS) spectra. (d) Fourier transform of extended X-ray absorption fine structure (EXAFS) spectra of the Pt L3 edge. (e) In situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) under the CO oxidation conditions. DFT-predicted CO adsorption modes are inserted. Color legend of atoms: Ce = beige; Pt = blue; C = gray; O (in CeO2 and Pt8O13) = red; O (in CO) = green. (f) CO oxidation light-off performance. Reproduced with permission from ref. 18. | ||
Another example of a study is the dimer/trimer of metal atoms anchored on the support. For example, dual Fe–Fe,59 Fe–Mn,60 and Co–Zn43 sites coordinated with nitrogen-doped carbon have been designed as highly efficient ORR catalysts, although the limitation of current structural characterization is hindering the large-scale rational development. Nevertheless, electron energy-loss spectroscopy (EELS) has been disclosed as a useful analytical tool to precisely identify the dual atom sites. Typically, the atomic-level HAADF-STEM images with EELS mode have been used to observe the atomic coordination among Fe, Co and N.45 According to the Z-contrast among the Fe, Co, C and N atoms, the bright dual-sites are of Fe–Co dimers (Fig. 4a). Clear Fe and Co EELS have further confirmed that these dual-sites are Fe–Co dimers (Fig. 4b). Furthermore, X-ray fine structure analyses have been performed to verify the possible coordinations of the Fe–Co dimers on the carbon support, as shown in Fig. 4c and d. Remarkably, such Fe–Co dimers indeed delivered a higher ORR activity, compared with the single metal counterparts (Fig. 4e). To the best of our knowledge, it is also among the best ORR performance. Theoretical calculations have further demonstrated that both Fe and Co sites in such dimer catalysts are more active than the isolated Fe or Co sites in the single-atom catalysts (Fig. 4f).
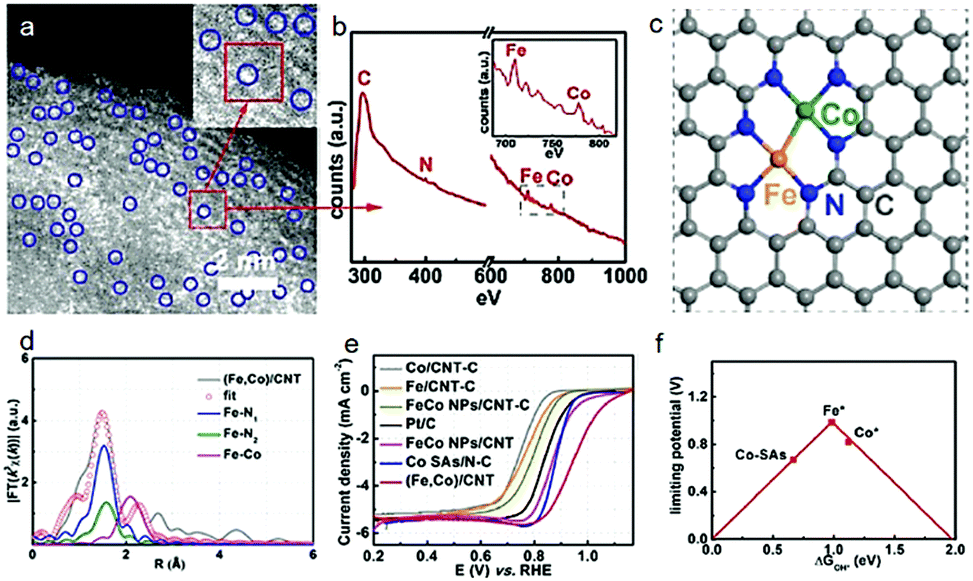 | ||
| Fig. 4 (a) Magnified HAADF-STEM images of the (Fe,Co)/CNT. (b) The EELS was taken on the red rectangle in (a). (c) Proposed model of Fe–Co dual sites. (d) Fe K-edge EXAFS fitting curves of the (Fe,Co)/CNT. (e) LSV curves of samples in O2-saturated 0.1 M KOH at a sweep rate of 10 mV s−1 and a rotation rate of 1600 rpm. (f) Volcano plot of the ORR activity as a function of ΔGOH* where Fe* and Co* represent Fe and Co sites on the OH* anchored (Fe,Co)/CNT, respectively. Reproduced with permission from ref. 45. | ||
Besides the dimer types, a platinum-trimer on the core–shell cobalt–palladium (Co@Pd–Pt) nanocatalyst has been suggested by both theoretical and experimental investigations.17 In the Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra (Fig. 5a), a lower CN (1.95) of Pt–Pt bonds in Co@Pd–Pt than that in the Pd@Pt structure was revealed, demonstrating the formation of Pt3 (possessing an average CN of 2.0 theoretically). Fig. 5b compares the ultraviolet photoelectron spectroscopy (UPS) spectra of the various catalyst samples, in which Co@Pd–Pt showed the lowest work function, rooted from a unique surface effect from the Pt3 decoration. As shown in Fig. 5c, a slightly gradient oxygen adsorption energy (EadsO) can be observed on the surface of the Co@Pd–Pt model. Such an uncommon phenomenon suggests a feasible relocation of the adsorbed oxygen atom between oxygen-involving splitting and reduction sites (site S and V). Moreover, the relocation of Oads to the neighboring sites reduces the possibility of passivation on Pt3 by intermediates. Such relocation undoubtedly demonstrates a great synergistic possibility among the multiple sites. As illustrated in Fig. 5d, Co@Pd–Pt exhibits an initial mass activity which is dozens of times higher than that of the commercial Pt/C, together with good stability.
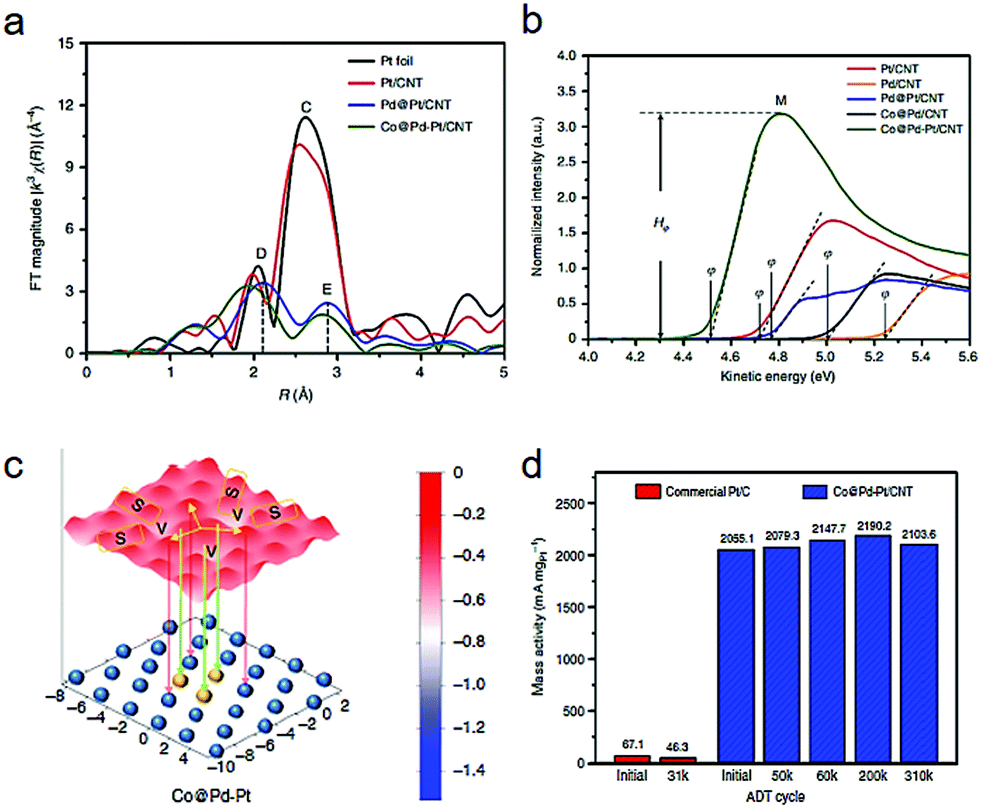 | ||
| Fig. 5 (a) Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS). (b) Ultraviolet photoelectron spectra of Pt-decorated Co–Pd and controls. (c) Contour of oxygen adsorption energy. EadsO in models of Pt-decorated Co–Pd catalyst surface. The atomic structure of the topmost layers is shown in the schematic diagram, in which blue and yellow spheres correspond to Pd and Pt atoms, respectively. Co atoms are not shown in the Pt-decorated Co–Pd (Co@Pd–Pt) model. (d) Comparison of ORR mass activity of commercial Pt/C and Co@Pd–Pt/CNT at selected ADT cycles. Reproduced with permission from ref. 17. | ||
From the surface heterocompound perspective, by devising/achieving the specific bond and coordination structure for the multiple active sites, they not only hammer at decreasing the energy barrier of each of the intermediate steps and dismissing the scaling relations in certain complex catalytic reactions, but also provide diverse possibilities in directing the catalytic reaction routes and thus customizing the selectivity of certain catalytic reactions.
5. Future perspectives
In the rapid evolution of single atom catalysts over the past two decades, by steadily refining the particle size of metal clusters loaded on a substrate support, it has become a rather different regime of studies on the interactions between metal atoms and the substrate surface. Single atoms can hardly exist by themselves; it is their bond and coordination with the substrate surface that have given rise to the high catalytic activity and selectivity, which have been demonstrated in a number of SAC systems. Besides the non-covalent lateral interactions among the active atoms, there can be strong and irreversible interactions with the support surface, which will transform the functional moieties and alter their chemical states. The large variation and possibility in the bond/interactions between these metal atoms and the substrate surface, which by themselves are largely defective and heterogeneous, create the “surface heterocompound”. On one hand, given the highly distributive nature, it is challenging to identify and purposely form the desired bond and coordination environment for a particular catalytic reaction. On the other hand, the surface heterocompound with a large population of different active sites can give rise to a superior catalytic efficiency and selectivity, where some of them can break the limit of scaling relation among catalytic immediate steps.To realize a rational design with the multi-site types and therefore catalytic activities, the surface heterocompound of SACs offers one of the great opportunities to explore for both disruptively enhancing the existing catalysts, where the electrocatalysts have been widely investigated in the past several years, and searching for completely new catalysts that are desperately needed for new chemical processes and/or synthesis of new compounds/medicines and drugs. Considering the structural complexity and the complicated interactions between the central atoms and the substrate surface, Al technologies, deep learning and a big database shall be applied to achieve useful studies with the thermodynamic and physiochemical properties to break the theoretical limit of catalysts and thus achieve the ultrahigh performance for targeted applications.
The currently available morphological and structural analysis tools for SACs and surface heterocompounds include high-resolution image/spectrometry techniques, such as HAADF-STEM, EELS and XAS. However, they only provide an average coordination of different active sites, rather than the local coordination of one particular single site. Moreover, these defective sites often play very important roles in the catalytic reactions, but XAS can only give an average coordination structure, including the edge/corner and planar sites. In addition, the likely deviation of XAS usually involves 20%, which is also not sensitive enough to the multi-sites, especially for metal atoms with similar coordination chemistry. Hence, more powerful analytical tools, such as direct detection EELS or STM with a purposely modified tip, which can be used to acquire the structural information of each active site in high accuracy and at high energy edges, should be employed. Precise spectrometry characterization techniques, such as electron paramagnetic resonance (EPR), nuclear magnetic resonance (NMR), and others, should be developed to probe all the possible bonds and coordination environments, as well as their distributions. In addition, various high resolution in situ and operando techniques would be required to understand the dynamic catalytic processes and the heterogeneous nature of SACs.
Besides the key analytical and calculation tools to be able to quantify the bond and coordination environment of active atoms in the surface heterocompounds and their overall structures, efficient and reproducible fabrication processes will be crucial, especially those capable of controlling the exact types of bonds and coordination. Over the past several years, various wet-chemistry and solution-based synthesis techniques have been widely explored, where the precursors are converted into the desired SAC structures by post-annealing. There has been a mix of success and unsatisfactory results. Among the relatively under-studied fabrication methods, both atomic layer deposition (ALD) and chemical vapour deposition (CVD) as the bottom-up techniques can be advantageous. When properly controlled, they can give better tunability in the population and distribution of single atoms on the designed support, as well as the overall structure and therefore performance, in relation to the self-limiting reactions involved. Nevertheless, for any large-scale production, it would be also essential to consider the overall yield and cost, in addition to the catalyst quality and performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of MOE (MOE2018-T2-2-095, Singapore) for funding of research, conducted at the National University of Singapore.Notes and references
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- J. Contour, G. Mouvier, M. Hoogewys and C. Leclere, J. Catal., 1977, 48, 217–228 CrossRef CAS.
- L. Lietti, C. Cristiani, G. Groppi and P. Forzatti, Catal. Today, 2000, 59, 191–204 CrossRef CAS.
- L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha and R. Dziembaj, Appl. Catal., B, 2005, 58, 235–244 CrossRef CAS.
- H. S. Taylor, Proc. R. Soc. A, 1925, 108, 105–111 CAS.
- F. Fischer, Fischer–Tropsch synthesis, US Pat., 1.246, 1930, p. 464 Search PubMed.
- A. S. Richardson, C. A. Knuth and C. H. Milligan, Ind. Eng. Chem., 1925, 17, 80–83 CrossRef CAS.
- T. Kaehler, Clin. Chem., 1994, 40, 1797–1799 CrossRef CAS.
- W. Zang, Z. Kou, S. J. Pennycook and J. Wang, Adv. Energy Mater., 2020, 1903181 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- J. M. Thomas, R. Raja and D. W. Lewis, Angew. Chem., Int. Ed., 2005, 44, 6456–6482 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65 CrossRef CAS.
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef.
- Y. Pan, C. Zhang, Z. Liu, C. Chen and Y. Li, Matter, 2020, 2, 78–110 CrossRef.
- S. Dai, J.-P. Chou, K.-W. Wang, Y.-Y. Hsu, A. Hu, X. Pan and T.-Y. Chen, Nat. Commun., 2019, 10, 440 CrossRef CAS PubMed.
- H. Wang, J.-X. Liu, L. F. Allard, S. Lee, J. Liu, H. Li, J. Wang, J. Wang, S. H. Oh and W. Li, Nat. Commun., 2019, 10, 1–12 CrossRef PubMed.
- Z. W. Chen, L. X. Chen, C. C. Yang and Q. Jiang, J. Mater. Chem. A, 2019, 7, 3492–3515 RSC.
- Y. Sohtome, G. Nakamura, A. Muranaka, D. Hashizume, S. Lectard, T. Tsuchimoto, M. Uchiyama and M. Sodeoka, Nat. Commun., 2017, 8, 14875 CrossRef CAS PubMed.
- Z. Kou, W. Zang, Y. Ma, Z. Pan, S. Mu, X. Gao, B. Tang, M. Xiong, X. Zhao and A. K. Cheetham, Nano Energy, 2020, 67, 104288 CrossRef.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS PubMed.
- W. Zang, T. Yang, H. Zou, S. Xi, H. Zhang, X. Liu, Z. Kou, Y. Du, Y. P. Feng and L. Shen, ACS Catal., 2019, 9, 10166–10173 CrossRef CAS.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo and L. Zheng, Nat. Commun., 2019, 10, 1–11 CrossRef.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu and S.-F. Hung, Nat. Chem., 2019, 11, 222 CrossRef CAS.
- M. Yoo, Y.-S. Yu, H. Ha, S. Lee, J.-S. Choi, S. Oh, E. Kang, H. Choi, H. An and K.-S. Lee, Energy Environ. Sci., 2020 10.1039/C9EE03492G.
- Y. Pang, W. Zang, Z. Kou, L. Zhang, G. Xu, J. Lv, X. Gao, Z. Pan, J. Wang and Y. Wu, Nanoscale, 2020, 12, 4302–4308 RSC.
- Z. Kou, W. Zang, W. Pei, L. Zheng, S. Zhou, S. Zhang, L. Zhang and J. Wang, J. Mater. Chem. A, 2020, 8, 3071–3082 RSC.
- P. Avouris and C. Dimitrakopoulos, Mater. Today, 2012, 15, 86–97 CrossRef CAS.
- Z. Kou, T. Wang, Q. Gu, M. Xiong, L. Zheng, X. Li, Z. Pan, H. Chen, F. Verpoort, A. K. Cheetham, S. Mu and J. Wang, Adv. Energy Mater., 2019, 9, 1803768 CrossRef.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS.
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang and A. Du, Chem, 2018, 4, 285–297 CAS.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- H. Fei, J. Dong, Y. Feng, C. S. Allen, C. Wan, B. Volosskiy, M. Li, Z. Zhao, Y. Wang and H. Sun, Nat. Catal., 2018, 1, 63 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed.
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed.
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Adv. Energy Mater., 2019, 1902844 Search PubMed.
- H. Wei, K. Huang, D. Wang, R. Zhang, B. Ge, J. Ma, B. Wen, S. Zhang, Q. Li and M. Lei, Nat. Commun., 2017, 8, 1490 CrossRef PubMed.
- X. He, Y. Deng, Y. Zhang, Q. He, D. Xiao, M. Peng, Y. Zhao, H. Zhang, R. Luo and T. Gan, Cell Rep. Phys. Sci., 2020, 1, 100004 CrossRef.
- Z.-J. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt, G. Henkelman and J. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi and Z. Jiang, Angew. Chem., 2019, 131, 2648–2652 CrossRef.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang and C. Zhao, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- L. Zhang, J. Fischer, Y. Jia, X. Yan, W. Xu, X. Wang, J. Chen, D. Yang, H. Liu, L. Zhuang, M. Hankel, D. J. Searles, K. Huang, S. Feng, C. L. Brown and X. Yao, J. Am. Chem. Soc., 2018, 140, 10757 CrossRef CAS PubMed.
- T. He, S. K. Matta, G. Will and A. Du, Small Methods, 2019, 3, 1800419 CrossRef.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 9664–9672 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan and Y. Li, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- M. Sun, T. Wu, Y. Xue, A. W. Dougherty, B. Huang, Y. Li and C.-H. Yan, Nano Energy, 2019, 62, 754–763 CrossRef CAS.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 15341 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao and S. Wei, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- W. Zang, A. Sumboja, Y. Ma, H. Zhang, Y. Wu, S. Wu, H. Wu, Z. Liu, C. Guan and J. Wang, ACS Catal., 2018, 8, 8961–8969 CrossRef CAS.
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne and L. Jin, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902–2907 CrossRef CAS PubMed.
- H. Jeong, G. Lee, B.-S. Kim, J. Bae, J. W. Han and H. Lee, J. Am. Chem. Soc., 2018, 140, 9558–9565 CrossRef CAS PubMed.
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long and M. Chen, Chem, 2019, 5, 2865–2878 CAS.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J.-P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |