
Reducing end modification on cellulose nanocrystals: strategy, characterization, applications and challenges†
Han
Tao
a,
Nathalie
Lavoine
 b,
Feng
Jiang
c,
Juntao
Tang
d and
Ning
Lin
*a
b,
Feng
Jiang
c,
Juntao
Tang
d and
Ning
Lin
*a
aSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan 430070, P. R. China. E-mail: ninglin.whut@gmail.com; ninglin@whut.edu.cn; Fax: +86-27-87152611; Tel: +86-27-87152611
bDepartment of Forest Biomaterials, College of Natural Resources, North Carolina State University, Campus Box 8005, Raleigh NC 27695-8005, USA
cSustainable Functional Biomaterials Laboratory, Department of Wood Science, University of British Columbia, Vancouver, BC V6T1Z4, Canada
dCollege of Chemistry and Chemical Engineering, Central South University, Changsha, Hunan 410083, P. R. China
First published on 31st January 2020
Abstract
Different from traditional chemical surface modification, localized modification of the reducing end groups of cellulose nanocrystals (CNCs), i.e. the active aldehyde groups, provides new opportunities for diverse functional applications of this renewable nanomaterial without altering its surface chemistry and properties. Numerous reviews have deeply discussed the surface modification of the hydroxyl groups of CNCs, but no critical comment has been reported on the reducing end modification approach. This review is a comprehensive summary on the modification of the CNC reducing end, presenting the reaction mechanisms and conditions, discussing the different chemical modification strategies and characterization techniques, potential applications and future challenges in this field. In addition, the comparison between surface and end modification strategies of CNCs will highlight the potential of reducing end-functionalized CNCs to be used in various applications as an alternative to traditional surface-modified CNCs, or as additional functional nanoparticles for the design of advanced functional materials.
 Nathalie Lavoine | Dr Nathalie Lavoine is an Assistant Professor in the Department of Forest Biomaterials, at North Carolina State University (Raleigh, NC USA). She received her PhD in 2013 from the Laboratory of Pulp and Paper Science and Graphic Arts in Grenoble (France) under the direction of Dr Julien Bras and Dr Isabelle Desloges. Her specific research interests are in the area of renewable nanotechnology, especially cellulose nanomaterial (CNM) production and characterization, advanced functional materials, active and intelligent bio-based packaging, controlled release systems and sustainable materials processing. |
 Feng Jiang | Dr Feng Jiang is an Assistant Professor in the Department of Wood Science at the University of British Columbia, and a Tier II Canada Research Chair in Sustainable Functional Biomaterials. He obtained his PhD in Macromolecular Science and Engineering from the Department of Sustainable Biomaterials at Virginia Tech in 2011. His current research interests include isolation and modification of lignocellulosic nanomaterials, assembly of nanocellulose into hierarchical structures, biomimetic design, additive manufacturing, and advanced materials for energy and environmental applications. He has published over 50 peer-reviewed articles in high impact journals, and is an emerging young researcher in the bio-based nanomaterials field. |
 Ning Lin | Dr Ning Lin is an Assistant Professor at the School of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology. He received his PhD in 2014 from the International School of Paper, Print Media and Biomaterials (Pagora) at the University Grenoble Alpes in France and continued his one-year post-doctoral research. He has published over 40 peer-reviewed articles in high impact journals, and his research interests include chemical modification, design and development of nanopolysaccharides (nanocellulose, nanochitin, and starch nanocrystals) and their functional applications. |
1. Introduction
Extracted from native cellulose fibers via treatments such as strong acid hydrolysis, enzymatic degradation, oxidation, and hydrothermal hydrolysis, cellulose nanocrystals (CNCs) are highly crystalline and rigid nanoparticles with a rod-like or needle-like morphology.1–3 The treatment step aims at primarily degrading the amorphous and para-crystalline regions of cellulose fibers, leaving the crystalline regions available for further purification.4,5 Besides numerous advantages of renewability, biodegradability, biocompatibility, low density, and nontoxicity inherited from natural cellulose, CNCs also possess some unique properties, including high aspect ratio, rod-like morphology, liquid crystalline behavior with a chiral nematic phase, high mechanical properties, and versatile surface chemistry.6 Specifically, the dimensions of CNCs vary from 100 nm to 1000 nm in length and 5–20 nm in diameter with aspect ratios of 10–120, depending on the cellulosic sources and hydrolysis approaches.7 The chiral nematic structures of surface-charged or surface-modified CNCs originate from their rod-like morphology and repulsive interactions. These CNCs can self-assemble into an ordered alignment and form a liquid crystal phase in suspensions.8,9 As a result of the high-crystallinity of CNCs, these nanoparticles exhibit an impressive specific modulus of 60–120 GPa cm3 g−1, which is approximately 4–5 times higher than the specific modulus of steel.10 In comparison with inorganic nanoparticles, the abundant available active groups of CNCs (surface hydroxyl and reducing end aldehyde groups) make them accessible for a variety of chemical modification, aiming to tune the physicochemical properties of CNCs for diverse applications. Over the past twenty years, CNCs have received tremendous interest from academia and industry. As summarized in Table S1 (ESI†) of the review articles in the last three years, academic studies on the topic of CNCs mostly focused on the preparation and optimization, characterization techniques for property analysis, surface modification and functionalization strategies for designing high-performance tailored materials with diverse applications such as optical-tunable materials,11–14 biomedicines,15–18 biosensing materials,19,20 catalysis,21 aerogels and foams,22–25 mechanically enhanced nanocomposites,26–28 adsorbing materials29–31 and energy-storage devices.32–35 On a larger scale, pilot and commercial CNC production facilities and plants have been built in e.g. Canada, the USA, Sweden and Israel.36In CNC with the cellulose I allomorph, the cellulose chains are parallelly packed into crystallites, leaving the reducing end with hemiacetal groups at one end and the non-reducing end with pendant hydroxyl groups at the other. Previous studies on CNC modification mainly focused on the chemical modification of the surface hydroxyl groups,37,38 but ignored the chemical modification of the reducing end, i.e. chemical modification of the aldehyde groups, because of the relatively low amount of reactive sites. However, as a regio-selective and local reaction site, the reactive reducing end can provide direct functionalization of CNCs on one end to introduce some unique features. The research interest in chemical modification of the CNC reducing end has intensified over the past five years, particularly in chemical reactions and potential applications of functionalized CNCs. As shown in Fig. 1, earlier studies investigated the characteristic of the reducing end to serve as evidence of the parallel-chain structure in native cellulose (cellulose I) and parallel-up packing in cellulose Iα and Iβ.39–42 The first report referring to the chemical modification of the CNC reducing end groups was reported in 2003 and describes complicated chemical reactions to graft aromatic molecules and polymer chains.43 A later study reported a precise control over the self-assembly of reducing end-modified CNCs on a gold (Au) surface by regio-selective functionalization.44 Decorating the CNC reducing end with fluorescent molecules paved the way for CNC functionalization located at the terminal rather than at the surface.45 Surfactant-like amphiphilic nanoparticles were created by introducing hydrophobic molecule chains at the reducing end of CNCs, while preserving the hydrophilic surface hydroxyl groups.46 The reducing end-modified CNCs were further used in reactively-compounding composites based on a thiol–ene click reaction between the CNC reducing end and rubber matrix, as a strategy to improve the interfacial compatibility and architecture of the surface hydrogen-bonding percolating network.47,48 In addition, reducing end-modified CNCs showed unique advantages in the field of nanomedicine, as a potential nanodevice for theranostics in bone tumors or nuclear imaging probes.49,50
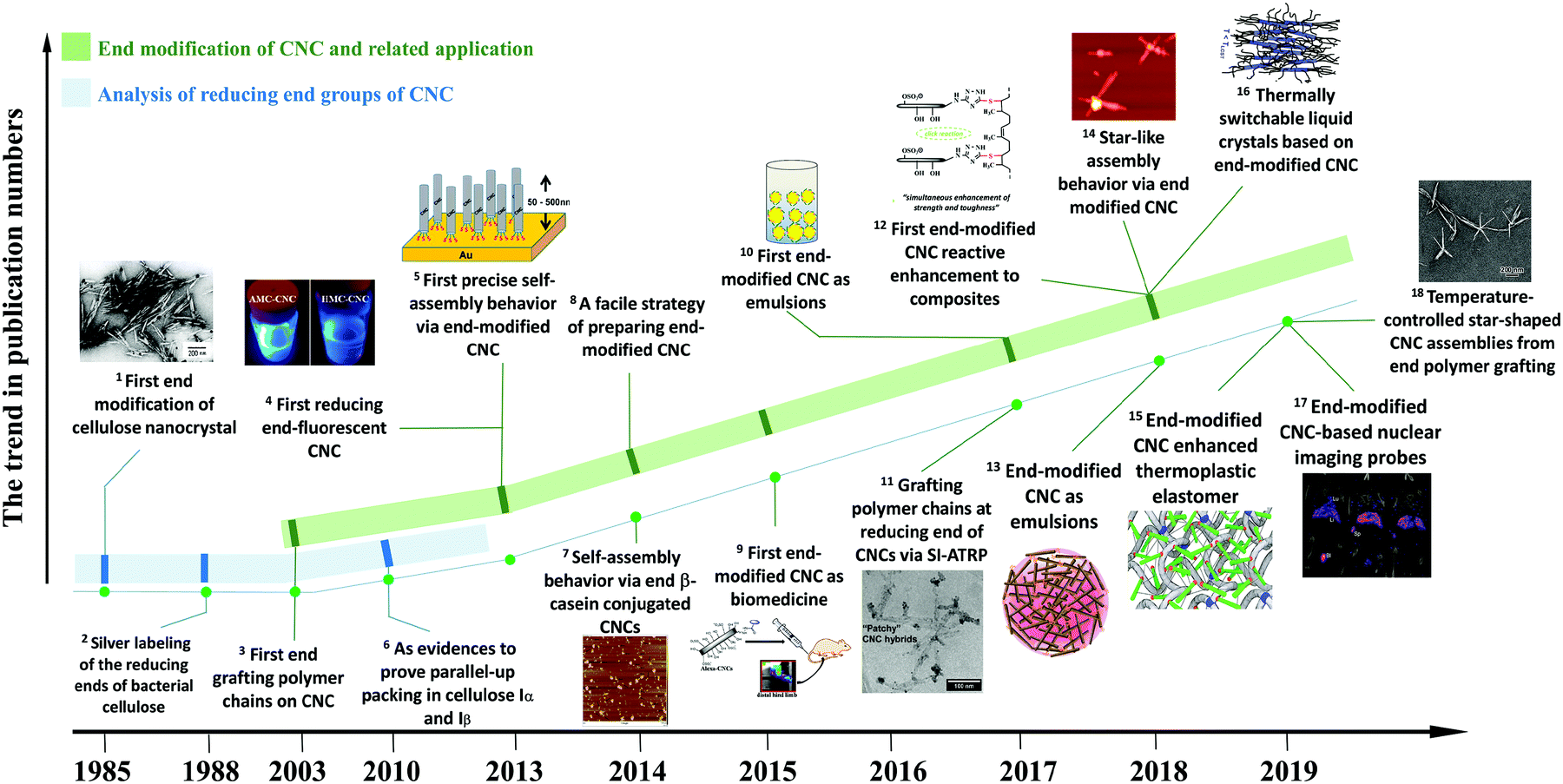 | ||
| Fig. 1 A roadmap on the studies of reducing end modification of CNCs. Related publications in the panels: 1 – [ref. 39]. (Reproduced with permission, Copyright 1984, John Wiley and Sons.) 5 – [ref. 44]. (Reproduced with permission, Copyright 2013, American Chemical Society.) 7 – [ref. 52]. (Reproduced with permission, Copyright 2013, Springer.) 9 – [ref. 49]. (Reproduced with permission, Copyright 2015, American Chemical Society.) 10 – [ref. 54], 11 – [ref. 55]. (Reproduced with permission, Copyright 2017, American Chemical Society.) 4 – [ref. 45], 12 – [ref. 47], 13 – [ref. 46], 14 – [ref. 56]. (Reproduced with permission, Copyright 2018, American Chemical Society.) 16 – [ref. 57] (Reproduced with permission, Copyright 2018, Wiley-VCH.) 15 – [ref. 48], 17 – [ref. 50]. 18 – [ref. 57]. (Reproduced with permission, Copyright 2019, American Chemical Society.) 2 – [ref. 40], 3 – [ref. 43], 6 – [ref. 42], 8 – [ref. 53]. | ||
2. Origin and nature of the reducing end groups of CNCs
The biosynthesis of cellulose is a very complex process. From the view of molecular structure, the C1 aldehyde group in a glucose molecule reacts spontaneously with the C5 hydroxyl groups via a nucleophilic addition reaction, forming a D-glucopyranose unit with the C1 aldehyde group being converted to a hemiacetal group.58 Subsequently, the β-1,4-D-glucopyranose chain is synthesized by the polymerization of D-glucopyranose in the presence of enzymes, where the C1 anomeric carbon of the glucopyranose is attacked by the C4 hydroxyl of another free glucopyranose unit. In fact, this process is a nucleophilic substitution reaction from a hemiacetal to an acetal group with the initial carbonyl oxygen leaving as a water molecule.59 Therefore, an individual cellulose chain will be synthesized by the successive enzymatic polymerization of glucose, as a high molecular weight homopolymer of β-1,4-linked anhydro-D-glucose units (Fig. 2A). Each glucopyranose unit on cellulose chains contains three highly-reactive hydroxyl groups, which are responsible for the intrinsic properties of cellulose such as hydrophilicity, chirality, and crystallinity, among others.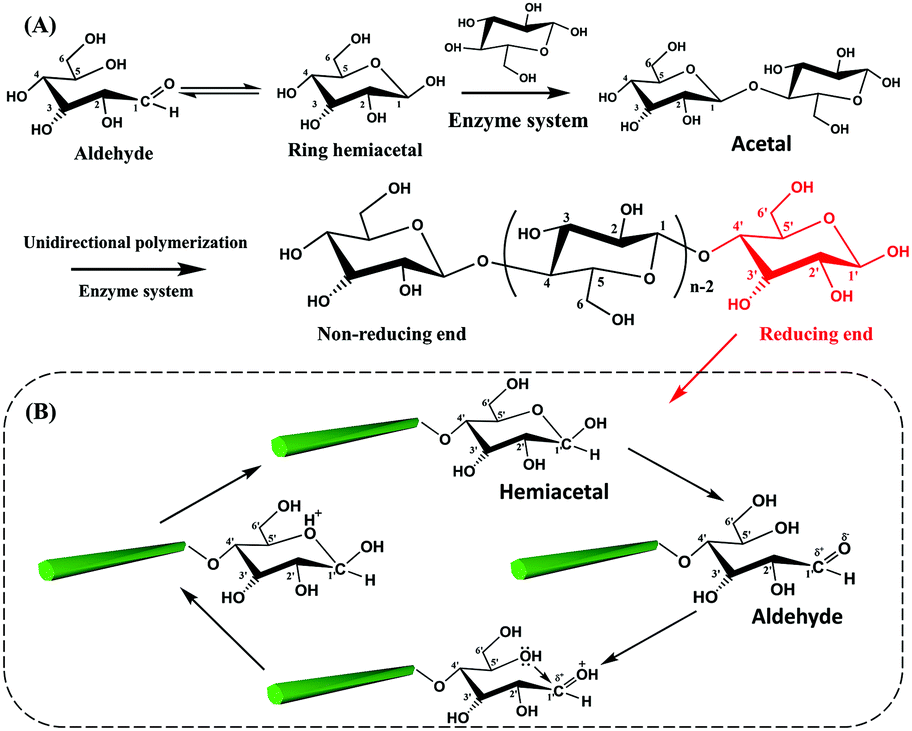 | ||
| Fig. 2 Polymerization of glucopyranose units for a cellulose chain (A) and the reversible equilibrium between hemiacetal and aldehyde groups at the reducing end of CNCs (B). | ||
As mentioned before, the two ends of a cellulose chain are different in chemical structure, with the anomeric carbon involved in a glycosidic linkage on one end and the anomeric carbon atom on the other end being free. As shown in Fig. 2B, this cyclic hemiacetal group is in an equilibrium in which a small proportion is an aldehyde state with reducing properties at this end of the cellulose chain. This end is referred to as the reducing end because it has the ability to reduce Cu(II) ions into Cu(I) ions in Fehling's solution.2 Changes in external conditions, such as pH, will affect the reversible equilibrium of the CNC reducing end. Under acidic conditions, the presence of protons drives the shift of the equilibrium to the direction of aldehyde groups.60 In particular, the excessive amount of water in the CNC suspension is expected to promote the generation of aldehyde groups at the reducing end.59 It is worth noting that the peeling reaction may take place under alkaline conditions and result in the spilt off of reducing end groups from the cellulose chains accompanied by the destruction of the crystalline structure.61 Consequently, appropriate reaction conditions are very important for the reducing end modification on CNCs.
3. Strategies of reducing end modification and comparison between surface and end modification on CNCs
3.1 Strategies and mechanisms of reducing end modification on CNCs
The reducing end modification of CNCs is also known as topochemical, regio-selective and local modification. As introduced before, a dynamic reversible conversion takes place between the aldehyde and the cyclic hemiacetal at the reducing end of CNCs. Therefore, such specific-site modification can be achieved by aldehyde-selective reactions under controlled conditions. As shown in Fig. 3, two strategies are commonly implemented for CNC reducing end modification, viz. the direct aldehyde-selective reaction and the indirect carboxyl-selective reaction involving aldimine condensation and carboxyamine condensation respectively.47 In the case of the aldimine condensation, the active aldehyde groups at the reducing end of CNCs are randomly attacked by the nucleophilic primary amino group of the modified molecules, which results in the aldimine linkage accomplished with the formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond by the removal of one molecule of by-product (H2O). In some cases, the addition of a reducing agent can convert the unstable CN bond to a C–N bond, offering a more stable covalent linkage between the reducing end of the CNCs and the modified agents. The indirect end modification with carboxyamine condensation is a two-step reaction, occurring under mild oxidizing conditions that converts the terminal aldehyde groups of CNCs to carboxyl groups for subsequent end modification steps, enriching the available species for reducing end modification of CNCs. The selected oxidant in the process of mild pre-oxidization should ensure the conversion of terminal aldehyde groups to carboxyl groups alone, while preserving the surface hydroxyl groups intact (or unmodified). Hence, the use of strong oxidant such as (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)-mediated oxidation is avoided in the reducing end modification of CNCs, and instead, mild sodium chlorite (NaClO2) oxidation can be used for the oxidation. The second step of the carboxyamine condensation for the reducing end modification of CNCs is the grafting reaction between the end-carboxylated CNCs and the amine-bearing compounds in the presence of the catalyst N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS). Typically, the covalent reaction between carboxyl and amine groups is induced by the EDC catalyst to form an O-acylisourea active intermediate, which then transformed as the active ester product under NHS catalysis. The reducing end modification of CNCs is achieved by the removal of one molecule of NHS under nucleophilic attack of the primary amino group of the reactant. Owing to the EDC-NHS catalytic effect, the reaction conditions of carboxyamine condensation are supposed to be milder than those of the aldimine condensation (i.e. use of lower reaction temperature and duration). However, more reactive steps are included in the indirect carboxyamine condensation, which may result in the decrease of grafting efficiency in comparison with the direct aldimine condensation approach.
N bond by the removal of one molecule of by-product (H2O). In some cases, the addition of a reducing agent can convert the unstable CN bond to a C–N bond, offering a more stable covalent linkage between the reducing end of the CNCs and the modified agents. The indirect end modification with carboxyamine condensation is a two-step reaction, occurring under mild oxidizing conditions that converts the terminal aldehyde groups of CNCs to carboxyl groups for subsequent end modification steps, enriching the available species for reducing end modification of CNCs. The selected oxidant in the process of mild pre-oxidization should ensure the conversion of terminal aldehyde groups to carboxyl groups alone, while preserving the surface hydroxyl groups intact (or unmodified). Hence, the use of strong oxidant such as (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO)-mediated oxidation is avoided in the reducing end modification of CNCs, and instead, mild sodium chlorite (NaClO2) oxidation can be used for the oxidation. The second step of the carboxyamine condensation for the reducing end modification of CNCs is the grafting reaction between the end-carboxylated CNCs and the amine-bearing compounds in the presence of the catalyst N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide (EDC) and N-hydroxysuccinimide (NHS). Typically, the covalent reaction between carboxyl and amine groups is induced by the EDC catalyst to form an O-acylisourea active intermediate, which then transformed as the active ester product under NHS catalysis. The reducing end modification of CNCs is achieved by the removal of one molecule of NHS under nucleophilic attack of the primary amino group of the reactant. Owing to the EDC-NHS catalytic effect, the reaction conditions of carboxyamine condensation are supposed to be milder than those of the aldimine condensation (i.e. use of lower reaction temperature and duration). However, more reactive steps are included in the indirect carboxyamine condensation, which may result in the decrease of grafting efficiency in comparison with the direct aldimine condensation approach.
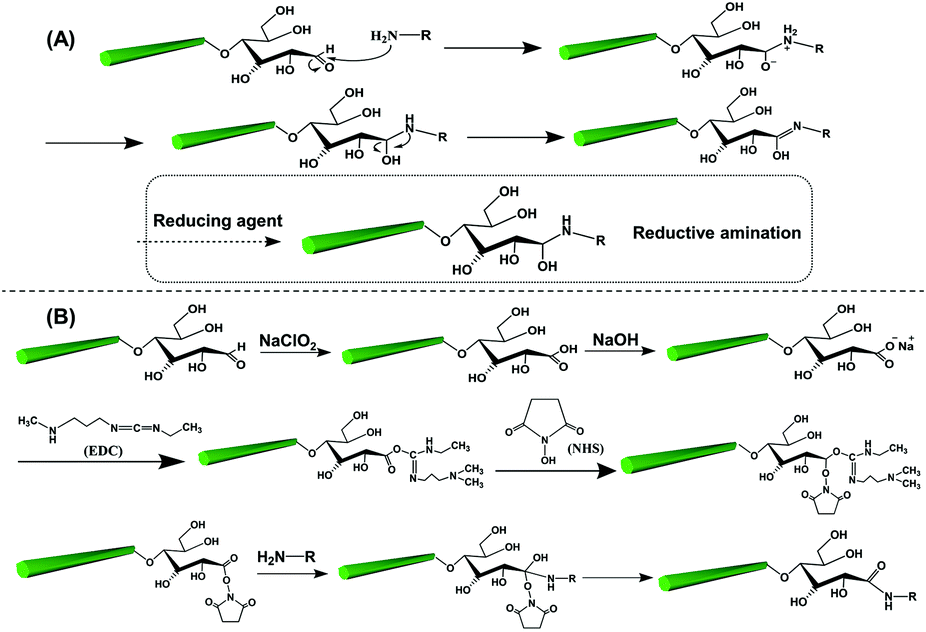 | ||
| Fig. 3 The two common strategies and respective mechanisms for the reducing end modification of CNCs: (A) the aldimine condensation and (B) the carboxyamine condensation. | ||
3.2 Comparison between the reducing end and surface modification on CNCs
Conventional surface modification of CNCs can include covalent attachment of functional groups (derivatization), molecules, polymers and/or nanoparticles on the surface of the CNCs, which provides various properties and functions for the development of CNCs with high-value added applications. The reducing end modification of CNCs is different from the conventional surface modification approaches in terms of both reactive species, namely, the group types and quantity, and reaction sites.The determined amounts of available aldehyde groups and surface hydroxyl groups of CNCs are summarized in Table 1. The amount of total hydroxyl groups on the CNC surface can vary greatly with the cellulosic sources and crystal dimensions, ranging from 0.955 to 2.646 mmol g−1 (Table 1). Note that the content of sulfate esters on the CNC surface derived from sulfuric acid hydrolysis is commonly reported within the range of 0.08–0.35 mmol g−1.68,69 The amount of terminal aldehyde groups of CNCs can be determined via direct and indirect measurements, with the reported values ranging from 0.018 to 0.0738 mmol g−1 (Table 1).
| Active group | Cellulose source | CNC dimension | Content of group (mmol g−1) | Measurement techniques | Ref. | ||
|---|---|---|---|---|---|---|---|
| Length (nm) | Width (nm) | Height (nm) | |||||
| Aldehyde group | Whatman filter paper | n/a | n/a | n/a | 0.035–0.045 | n/a | 53 |
| Cotton linter | 100 | — | — | 0.0548–0.0738 | Formazan method | 43 | |
| Wood pulp | 106 ± 39 | 11 ± 2.5 | 0.018 | Colorimetric assays | 51 | ||
| CelluForce product | n/a | n/a | n/a | 0.0245 | Hydroxylamine hydrochloride | 46 | |
| n/a | n/a | n/a | 0.0311 | Potentiometric titration | |||
| Cotton linters | 150 | 22 | 6 | 0.0249 | Fehling test | 57 | |
| Hydroxyl group | Cotton | 169.9 ± 86.8 | 23.0 ± 8.9 | 5.2 ± 1.9 | 0.955–2.646 | Theoretical calculation | 65 |
| Whatman filter paper | 131 ± 10 | 13 ± 1 | 1.57–1.82 | Theoretical calculation | 68 | ||
The direct measurement of terminal aldehyde groups of CNCs involves a chemical reaction which results in the consumption of reactants or generation of by-products. The Fehling test is a common method to measure the aldehyde content of CNCs and entails a reduction of Cu(II) to Cu(I) ions. Aldehyde content of cotton cellulose nanocrystals was determined as 0.0249 mmol g−1via this method.57 The Fehling solution consists of two equal volumes of solutions A and B. Normally, solution A is CuSO4 solution at a concentration of 0.05 g mL−1, while solution B is prepared by mixing an equal volume of 0.2 g mL−1 potassium sodium tartrate tetrahydrate (KNaC4H4O6·4H2O) and 0.1 g mL−1 NaOH solution. The CNC suspension is heated to 65 °C under gentle stirring and then titrated dropwise with the Fehling solution using a micropipette until the red precipitate disappears.70 The formazan method is another direct method to determine the amount of aldehyde groups. This technique is a colorimetric method using 2,3,5-triphenyltetrazolium chloride as the reagent. Reported values of aldehyde group contents on untreated and ultrasonically-treated CNCs were 0.0548 and 0.0738 mmol g−1, respectively.43 Recently, Zoppe et al. reported the quantification of aldehyde groups of CNCs by the colorimetric method with the addition of bicinchoninic acid assay to form a purple-colored complex. A low value of 0.018 mmol g−1 for the amount of terminal aldehyde groups of CNCs was obtained via this method.51 The hydroxylamine hydrochloride method was also reported for estimating the amount of terminal aldehyde groups, and in this case a value of 0.0245 mmol g−1 was obtained.46 Specifically, the reaction between hydroxylamine hydrochloride and terminal aldehyde groups of CNCs produces oxime and releases equivalent HCl for each formyl residue. The amount of produced HCl can be measured by potentiometric titration, corresponding to the amount of aldehyde groups at the CNC reducing end. Indirect methods to determine the aldehyde contents are based on the formed carboxyl groups, which are generated at the reducing end by oxidation of aldehyde groups or introduction of the additional species carrying carboxyl groups. The aldehyde groups at the reducing end of CNC are primarily oxidized to carboxyl groups by NaClO2, followed by conductometric titration using NaOH. The reported value of the aldehyde group content was 0.0331 mmol g−1 for the reducing end of CNC.46 Another indirect method to estimate the amount of formed carboxyl groups of CNCs is the addition of a cationic dye, namely, methylene blue, to promote its selective adsorption on the acidic groups, which can then be followed by UV spectroscopy at a wavelength of 664 nm. In this case, the reported value of the formed carboxyl groups at the reducing end of CNC was 0.031 mmol g−1.52
Because of the relatively low content of reducing end aldehyde groups, the measurements mentioned before have some limitations and therefore reported a wide range of data. When using the formazan method, the prepared cellulose nanocrystals were completely dissolved in the dimethylacetamide/LiCl solution,71 resulting in the destruction of cellulose crystalline regions. Hence, the content of aldehyde groups estimated by the formazan method is those at the reducing end of individual cellulose chains but not the available groups at the reducing end of crystalline CNCs. Therefore, the data from the formazan method are generally higher than those from other measurements. With regards to the hydroxylamine hydrochloride method, the content of aldehyde groups at the reducing end of CNCs was indirectly determined by the amount of resultant HCl, which may be sensitively affected by the ambient conditions during the potentiometric titration experiment. The colorimetric assays on the measurement of reducing end aldehyde groups is based on the generation of colored products by the reduction of Cu(II) ions to Cu(I) ions and complexation with chromogenic agents, which may be more accurate when performing the measurement of UV absorbance for the products.
Based on the reported results, the amount of surface hydroxyl groups is approximately 50–150 times higher than that of terminal aldehyde groups of CNCs. A recent study used a crude model of crystal with a square cross-section to calculate the numbers of anhydrous glucose units at the surface and reducing end of cotton CNCs of 200 nm length and 20 nm width, suggesting that the number of surface hydroxyl groups is 122 times higher than that of terminal aldehyde groups.45 In terms of quantity of available groups for chemical modification, the content of aldehyde groups at the reducing end of CNCs is far less than that of the surface hydroxyl groups even when excluding the existing sulfate ester groups. Other than the different chemical reactivity and amounts of aldehyde and hydroxyl groups, steric hindrance also plays a critical role in the modification efficiency. The hydroxyl groups are located at the surface of nanocrystals with a higher specific surface area and therefore are more accessible to chemical modification than the aldehyde groups at the reducing end with limited accessibility. The intrinsic self-aggregation tendency of the cellulose nanoparticles can indeed shield the reactive sites and active groups from surface and reducing end modification. Therefore, a diluted suspension of individual CNCs should be prepared from a homogeneous and stable dispersion of nanocrystals before performing any surface or reducing end modification in order to obtain the higher end modification efficiency.
In comparison with the surface hydroxyl-selective modification, the reducing end modification of CNCs alters the CNC chemistry to a relatively lower degree, and can only occur at one terminal of the nanocrystals, thus resulting in small and even negligible changes of the properties of reducing end-modified CNCs. The surface modification of CNCs can, for example, alter the hydrophilic nature of CNCs or their surface charge chemistry from e.g. anionic or neutral to cationic charge, while the reducing end modification would result in target functionalization of the terminal group with the preservation of the bulk CNC properties. In the case of precisely regulating the self-assembly behavior of CNCs, end-to-end, star-like, and hairbrush-like nanocrystals can be designed with the introduction of tailored molecules by reducing end modification.44,52,56,57 Such regio-selective and local modification can endow CNCs with structural design of grafted functional molecules, keeping intact the cellulose surface chemistry, as for example reported with the design of cilia-mimetic materials by end modification of thiol groups absorbed at an Au surface.44
4. Preparation of reducing end-modified CNCs
Various functional small molecules and polymer chains with amino-group moieties have been grafted onto the reducing end of CNCs. Typical functional molecules for reducing end modification of CNCs include fluorescent molecules, such as 7-hydrazino-4-methylcoumarin, 7-amino-4-methylcoumarin45 and Alexa Fluor 633 hydrazide.49 The approaches of “grafting onto” and “grafting from” are carried out for the end grafting of polymers on CNCs. The method of grafting onto is a peptide coupling reaction relying on the chemical reaction between the terminal aldehyde or carboxyl groups of CNCs and the amino groups of targeted polymers, such as poly(ethylene glycol)43 and polystyrene;46 while the approach of grafting from includes the introduction of initiator molecules first at the reducing end of CNCs and then inducing the chain growth for polymeric grafting of poly(N-isopropylacrylamide), poly[2-(methacryloyloxy) ethyltrimethylammonium chloride], and poly(sodium 4-styrenesulfonate) by atom transfer radical polymerization (ATRP) and polyacrylamide by free radical polymerization (FRP).43,51,55For both surface and reducing end modification of CNCs, one critical prerequisite is to keep the original morphology and crystal structure of cellulose intact, without any polymorphic conversion or damage of crystalline integrity.72 Therefore, the reactive conditions of reducing end modification are supposed to be controlled to avoid any destruction of the CNC structure. The reaction conditions such as pre- and post-treatments, pH, solvent polarity and reactant addition will significantly affect the efficiency of the reducing end modification of CNCs. Diverse cases of reducing end modification on CNCs are classified based on the properties of grafted species such as additional active groups, functional molecules and polymeric chains, aiming to compare the different procedures, influential factors and designability.
4.1 Reducing end grafting of CNCs with additional active groups
The aldimine condensation is a straightforward method to introduce additional active groups at the reducing end of CNCs by direct coupling of the aldehyde groups with molecules bearing both reactive amino groups and targeted active groups. Common chemical reaction pathways are illustrated in Fig. 4 and include the introduction of additional carboxyl groups (–COOH), thiol groups (–SH), amino groups (–NH2), and azide groups (–N3) to the reducing end of CNCs. It is worth noting that a recent study recommended the use of hydrazine derivatives such as hydrazinium hydroxide (HH) for direct coupling with the reducing end aldehyde groups of CNCs, as more stable conjugates of aldehydes and ketones can be formed during aldimine condensation.49 To achieve a stable hydrazone linkage, hydrazine-terminated molecules, carrying the targeted active group, should be directly coupled with the CNC aldehyde groups. The active groups can thus be immobilized at the CNC reducing end and serve as “anchor groups” to be further bonded to other targeted molecules or react with other components. Certain studies have exploited this chemical modification as a mean to activate the reducing end of CNCs. For instance, the introduced –SH and –N3 groups can be used in thiol–ene and azide–alkyne click reactions to link the CNC reducing end to unsaturated polymers and alkynyl-functionalized β-casein.47,48,52 Furthermore, the reducing end modification of CNCs can be enriched with the introduced –NH2 and –COOH groups via additional amino-selective and carboxyl-selective reactions. For example, a primary amino group introduced at the reducing end of CNCs can react with 2-bromoisobutanoic acid N-hydroxysuccinimide ester (NHS-BiB) to introduce a macromolecular initiator at the active terminal sites.55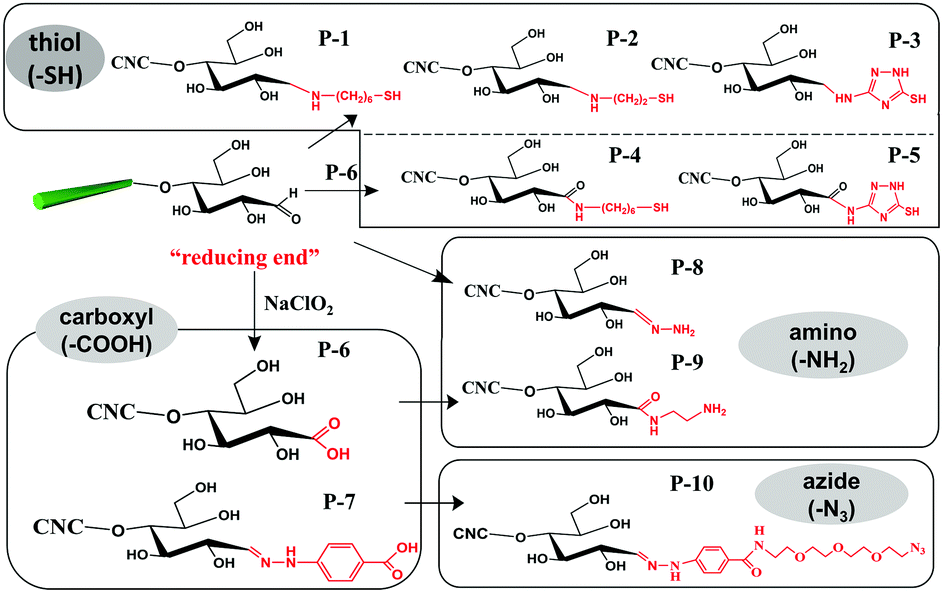 | ||
| Fig. 4 Reducing end modification of CNCs with the introduction of active groups (–COOH, –SH, –NH2, –N3). | ||
The reaction and purification conditions for introducing active groups at the reducing end of CNCs are summarized in Table 2. With regard to both aldimine and carboxyamine condensation reactions, the reaction durations and temperatures are relatively mild. Due to the catalysis effect of NHS and EDC, the duration of the carboxyamine condensation is shorter than that of the aldimine condensation. The reducing end modification of CNCs is generally carried out in water, as most of the modifying reactants are water-soluble, and CNCs can form a stable aqueous suspension that promotes the grafting efficiency. In addition, the presence of water can also promote the generation of aldehyde groups from the terminal hemiacetal groups according to the equilibrium mechanism mentioned before.59 With the purpose of eliminating the stereo-hindrance effect during the reducing end modification, the concentration of the CNC aqueous suspension is generally controlled at diluted levels, and ranges from 2 to 33 mg mL−1 in the previously mentioned studies.
| Grafted groups | Products | Pre-treatments | pH | CNC concentration (mg mL−1) | Solvent | Reducing agent | Duration and temperature | Post-treatments | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a r.t. = room temperature. b MES = 2-(N-morpholino)-ethanesulfonic acid buffer. | |||||||||
| –SH | P-1 | Sodium acetate buffer | 5.0 | 10 | Water | Cn, Ac, or Pc | 70 °C, 72 h | Addition of HCl to remove excess reducing agent; dialysis against water by the addition of KCl | 44 |
| P-1 | Sodium phosphate buffer | 7.0 | |||||||
| P-1 | Sodium carbonate buffer | 9.2 | |||||||
| P-2, P-3 | Degas by bubbling nitrogen; reaction in sodium carbonate buffer | 9.2 | 10 | Water | Ac | 47 and 48 | |||
| P-4 | Degas by bubbling nitrogen and pH adjustment to 7.0; addition of NHS, EDC and pH adjustment to 6.5, then addition of KCl; pH adjustment to 9.2 | 9.2 | 2 | Water | r.t.,a 2.5 h | Dialysis against water | 53 | ||
| P-5 | 10 | Water | 70 °C, 72 h | Dialysis against water | 47 | ||||
| –COOH | P-6 | Acetic acid | 3.5 | 10 | Water | r.t.,a 20 h | Dialysis against water | 39, 47, 51, 53 and 55–57 | |
| P-7 | Suspension in calcium acetate solution, borate buffer with sonication | 9.0 | 33 | Water | 35 °C, 72 h | Washing with borate buffer, calcium acetate and tetrahydrofuran; then dialysis for 3 days | 52 | ||
| 56.6 | Water | 35 °C, 48 h | Centrifugation in calcium acetate solution and washing with tetrahydrofuran | 43 | |||||
| –NH2 | P-8 | Suspension in calcium acetate solution, borate buffer with sonication | 9.0 | 6.265 | Water | 35 °C, 72 h | Dialysis against calcium acetate solution and then centrifugation with calcium acetate solution and tetrahydrofuran | 43 and 54 | |
| P-9 | Sodium phosphate buffers | 6.5 | 1 | Water | r.t.,a 4 h | Dialysis against water | 55 | ||
| –N3 | P-10 | Sonication | 4.0 | 8.3 | MESb buffer | r.t.,a 24 h | Dialysis against brine 1 day and water 3 days | 52 | |
Most of the reducing end modifications are performed under weak alkaline conditions (at pH of 9.2), which is beyond the isoelectric pH of amine to promote its deprotonation during the modification.53 Alkaline reaction conditions are selected mainly for three reasons: (1) the CN bond formed by aldimine condensation is stable in alkaline solution and could be destroyed under suitable acidic conditions;73 (2) the protonated amine groups generated under acidic conditions can be adsorbed onto the CNC surface via electrostatic attraction, therefore limiting their availability for the reducing end modification; and (3) the deprotonation of amino groups under alkaline conditions can result in a higher efficiency for end modification. Weak alkaline conditions can thus be achieved by either extended period of activation using calcium acetate solution followed by transfer to a buffer solution or by directly adjusting the pH of the suspensions.
A pioneering study involving the reducing end modification of CNCs was reported in 1984 by Hieta et al., even though the authors did not propose the concept of cellulose nanocrystals and reducing end modification. An oxidization reaction was performed at the reducing end of Valonia CNCs (Fig. 4, P-6) using chlorite oxidation (sodium chlorite, pH 3.5, room temperature, 20 h) to convert the aldehyde groups to carboxyl groups followed by the adsorption of silver nanoparticles, thus providing direct evidence of the parallel-chain structure in cellulose I.39 In a follow-up study, the silver proteinate-nucleated modification was performed on bacterial cellulose, which was the first case of introducing functional molecules at the reducing end of cellulose.40 The thiosemicarbazide-silver proteinate was grafted at the reducing end of cellulose and provided a nucleus for silver deposition. The silver ammonia solution was then converted to silver nanoparticles for observation using an electron microscope. This technique is an effective staining method to prove the presence of active reducing end of cellulose.
In the preparation of end-thiolated CNCs by aldimine condensation using NH2-R-SH as active groups (Fig. 4, P-1), the effects of different reducing agents [NaBH3CN(Cn), sodium cyanoborohydride, C6H10BNaO6(Ac), sodium triacetoxyborohydride and C6H7N·BH3(Pc), 2-picoline–borane complex] and pH conditions (5.0, 7.0 or 9.2) on the end grafting efficiency were discussed.44 The addition of reducing agents can convert the CN to C–N linkage between the grafted molecules and CNC reducing end for an optimized end grafting efficiency obtained at pH 9.2, which was attributed to the deprotonation effect of the amino groups on the grafted molecules under weak alkaline conditions.74 The post-treatment in this protocol included the addition of hydrochloric acid to neutralize the excess of reducing agent. A similar method was applied for the reducing end modification by carboxyamine condensation with a pre-oxidization treatment to convert the end aldehyde groups to carboxyl groups (Fig. 4, P-4).53 In this case, the reducing end grafting efficiency of CNCs was reported to be ca. 60%. Changes in ionic strength of the system, in both pre- and post-treatments, were reported to affect the efficiency of the reducing end modification of CNCs. The addition of KCl (1 mol L−1) in the starting suspension was shown to prevent electrostatic interactions between the positively charged protonated modifier and negatively charged sulfate ester groups along the surface of CNCs, and to thus remarkably increase the efficiency of amide bond formation at pH 9.2. As post-treatment, the addition of KCl (2 mol L−1) was reported to significantly remove the physisorbed molecules from the CNC surface. Recently, cyclic molecule 3-amino-1,2,4-triazole-5-thiol was explored to be grafted at the reducing end of CNCs using both modification strategies, i.e. the direct aldimine and the indirect carboxyamine condensation strategies (Fig. 4, P-3, P-5).47 The reducing end-modified CNCs exhibited an improved re-dispersibility in aqueous suspension, which was attributed to the synergistic effect between the spatial stability of the grafted triazole molecules and the electrostatic repulsion of the preserved surface sulfate groups. In a later study, a cysteamine hydrochloride bearing active thiol group was grafted at the reducing end of CNCs (Fig. 4, P-2).48 An inert environment is required for this reaction in order to inhibit any oxidization of the grafted thiol groups.
Introduction of active amino groups at the reducing end of CNCs can be achieved by covalent coupling using hydrazinium hydroxide to form a stable hydrazone linkage (Fig. 4, P-8). In this procedure, a calcium acetate solution was added to CNC aqueous suspension under continuous stirring for 16 hours at room temperature as pre-treatment. The suspension was then transferred to a borate buffer (pH 9.0) for further reaction.54 The introduction of Ca2+ was reported to restrain the peeling-off effect of cellulose chains, particularly well-known under alkaline conditions.75 The reducing end grafting of azide groups on CNCs can be achieved by converting terminal aldehyde to carboxyl groups by reaction with 4-hydrazinobenzoic (Fig. 4, P-7), and then carbodiimide-mediated grafting with an amine terminated azide, namely, 11-azido-3,6,9-trioxaundecan-1-amine (Fig. 4, P-10). Hydrochloric acid-hydrolyzed CNCs (i.e. CNCs produced using HCl instead of H2SO4, thus without negative half sulfate ester groups) were used for this reducing end modification, and therefore successive sonication was performed between each step to avoid the aggregation of nanocrystals.52
4.2 Reducing end grafting of CNCs with functional molecules
With the purpose of developing new functionalities on cellulose, some functional molecules were grafted at the reducing end of CNCs. These functional molecules were attached at the reducing end of CNCs by reaction with pre-immobilized active groups, as introduced before. Generally, reported functional species for reducing end modification of CNCs include fluorescent molecules, radiolabeled molecules, bio-active molecules (biotin derivatives, β-casein) and hydrophobic alkyl chains, as shown in Fig. 5. The reaction and purification conditions for introducing functional molecules at the reducing end of CNCs are summarized in Table 3.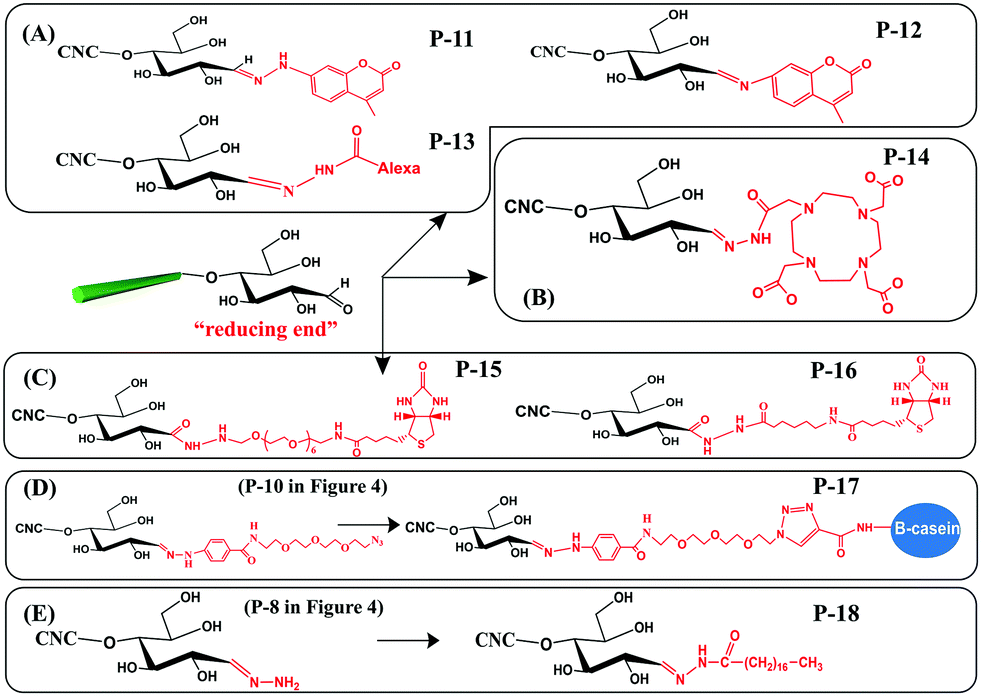 | ||
| Fig. 5 Reducing end modification of CNCs with grafting of functional molecules: (A) fluorescent molecules, (B) radiolabeled molecules, (C) biotin derivatives, (D) β-casein, and (E) C18 alkyl chain. | ||
| Functional species | Products | Pre-treatments | pH | CNC concentration (mg mL−1) | Solvent | Duration and temperature | Post-treatments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Fluorescent molecule | P-11, P-12 | — | n/a | n/a | Water | 25 °C, 24 h in the dark | Soxhlet extraction with acetone and methanol; dialysis against water | 45 |
| P-13 | Bis(triethylammonium) salt, acetic acid | n/a | n/a | Water | 25 °C, 24 h in the dark | Dialysis against water | 49 | |
| Radiolabeled molecules | P-14 | Sonication | n/a | 5 | Dimethyl sulfoxide | 4 days, 50–60 °C | Centrifugation and washing with dimethyl sulfoxide 4 times; dialysis against ultrapure water for 4 days | 51 |
| Biotin derivatives | P-15, P-16 | Degas by bubbling nitrogen; addition of NHS and EDC; pH adjustment to 6.5; addition of KCl; sonication; pH adjustment to 9.2 | 9.2 | 8 | Water | r.t., 12 h | Centrifugation and dialysis against water | 56 |
| β-Casein | P-17 | Sonication | 8.5 | 2 | Water | r.t., 24 h | Dialysis against EDTA for 1 day and against water for 3 days | 52 |
| C18 alkyl chain | P-18 | — | n/a | 6.265 | Polytetramethylene | 0 °C, 1 h and then at r.t. for 40 h | Centrifugation and washing with THF | 54 |
Fluorescent molecules were introduced at the reducing end of CNCs by attaching hydrazine- or amino-substituted fluorophores [7-hydrazino-4-methylcoumarin (HMC) and 7-amino-4-methylcoumarin (AMC)] to the terminal aldehyde groups, producing end fluorescent nanocrystals (Fig. 5A, P-11, P-12). It is worth noting that the purification of the modified product by successive Soxhlet extraction with acetone and methanol is especially important to remove free and physically adsorbed fluorophores.45 The two fluorescent modified CNCs exhibited absorbance peaks at 325 nm and 350 nm together with emission peaks at 452 nm and 440 nm corresponding to the grafted HMC and AMC, respectively. The formation of hydrazone bonds in the HMC-modified CNCs shifted both the absorption and emission maxima to longer wavelengths.76 Another study reported the reducing end grafting of the Alexa Fluor 633 fluorophore onto CNCs via the reaction of hydrazo and aldehyde groups (Fig. 5A, P-13) as a fluorescent nanocarrier for theranostics in living biological matrices.49 The UV-vis spectrum of the Alexa Fluor 633 end-labeled CNCs displayed characteristic signals at 560 nm and 630 nm of the fluorescent molecules, suggesting that the end-grafting was successful, while the negatively charged CNC surface with sulfate ester groups was preserved intact.
The introduction of a radiolabeling function was achieved by chemical grafting of a macrocyclic radiometal chelator (namely, DOTA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) onto the reducing end of CNCs by aldimine condensation (Fig. 5B, P-14).50 The end-modified DOTA-CNCs were further radiolabeled with indium-111 chloride (111InCl3) to generate stable 111In-DOTA-CNC nanoparticles, with a radiolabeling yield of 7 to 18% and preservation of the surface properties of the CNCs. The reducing end radiolabeling of CNCs was carried out in a neutral or alkaline medium to avoid any degradation of the formed hydrazone bonds between the DOTA chelator and the CNC terminal aldehyde groups under acidic conditions.
Several bioactive species were also successfully grafted onto the reducing end of CNCs, including two different lengths of biotin derivatives, namely biotin-amidohexanoic acid hydrazide (HC-Biot) and biotin-poly(ethylene glycol)-NH2 (PEG-Biot) (Fig. 5C, P-15, P-16).56 In another study, the alkynyl-functionalized β-casein was grafted onto the reducing end of CNCs after pre-treatment with active azide groups by a click reaction (Fig. 5D, P-17).52 Because one β-casein molecule possesses 11 reactive amino groups, more individual CNCs were reported to be attached on the β-casein by the end click reaction, thus forming micelles of arranged mushroom-like aggregates.
To enhance the emulsifying efficiency of an oil-in-water Pickering emulsion, a hydrophobic C18 alkyl chain was grafted onto the reducing end of CNCs by successive hydrazone and amidation reactions (Fig. 5E, P-18). Tetrahydrofuran (THF) was used as a solvent to completely dissolve the stearic acid and catalysts (EDC, NHS). After introduction of the amino-terminated CNC (Fig. 4, P-8) suspension in THF, the reaction was performed at 0 °C for 1 h and then for 40 h at room temperature in order to obtain the C18 alkyl chain end-modified CNCs. Ascribed to the formation of unstable CN linkages between C18 chains and the reducing end of CNCs, the grafted alkyl chains could be cleaved by adjusting pH, which endowed the end-modified CNCs with a pH-triggered de-emulsification property.54
4.3 Reducing end grafting of CNCs with polymeric chains
Two main approaches of “grafting onto” and “grafting from” can be performed at the reducing end of CNCs, namely (i) the direct coupling of polymers with active reducing end groups of CNCs and (ii) the polymerization of monomers by initiators grafted at the reducing end of CNCs. The grafting densities of polymeric chains at the reducing end of CNCs are expected to be lower as compared to those of small molecules due to more severe steric hindrance of the grafted macromolecules. Therefore, in order to reduce the interference and shielding effect of the reactive reducing end groups of neighbor CNCs, the grafting of polymeric chains at the reducing end of CNCs is generally carried out using very dilute CNC suspension (2 mg mL−1). When using organic solvent as the reaction medium, solvent exchange is commonly performed for obtaining stable and homogeneous CNC suspension prior to reducing end modification with grafting polymeric chains. Moreover, most polymer chain grafting modifications on CNCs are performed under an inert gas to avoid interference by atmospheric oxygen. Diverse polar and non-polar polymers have been grafted at the end of CNCs by direct coupling or living radical polymerization. The reaction conditions of reported studies on grafting polymers at the reducing end of CNCs are summarized in Table 4.| Reaction strategies | Products | Pre-treatments | CNC concentration (mg mL−1) | Solvent | Duration and temperature | Post-treatments | Ref. |
|---|---|---|---|---|---|---|---|
| Grafting onto | P-19 | — | 125 | Tetrahydrofuran | r.t., 16 h | Centrifugation and washing with tetrahydrofuran, calcium acetate solution; treatment with tetrahydrofuran for 1 h, and finally suspended in water | 43 |
| P-20 | |||||||
| P-21 | Freeze-dried CNC was re-dispersed in the solution of NMMO·H2O at 80 °C for 30 min | 10 | Water | 4 °C, 24 h | The suspension was filtered to remove the catalyst and dialysis against the water. | 57 | |
| P-22 | |||||||
| P-23 | Sonication | 10 | N,N-Dimethyl formamide | r.t., 1 day | Centrifugation and washing with tetrahydrofuran and acetone; redispersed in water and dialysis against water | 46 | |
| r.t. or 70 °C, 3 days | |||||||
| r.t., 7 days | |||||||
| Grafting from | P-24 | Freeze-dried CNC was re-dispersed in calcium acetate solution and then transferred into borate buffer | 250 | Water | r.t., 16 h | Centrifugation with calcium acetate solution and tetrahydrofuran | 43 |
| P-26 | Freeze-dried product of P-8 was re-dispersed in calcium acetate solution and then transferred into borate buffer | 300 | Water | 25 °C, 72 h | |||
| P-25 | Degassed in vacuum | 30 | Water | 60 °C, 48 h | Successive centrifugation and washing with water and ethanol; and finally suspended in water | ||
| P-27 | 25 | Water | 70 °C, 48 h | ||||
| P-28 | Sonicated in Na2HPO4 solution | 10 | Water/dimethyl sulfoxide | r.t., 4 h | Dialysis against water | 55 | |
| P-29 | Subjected to 3–4 pump/N2 purge cycles | 2 | Water/methanol | r.t., 2 h | Dialysis against water until neutral pH | ||
| P-30 | |||||||
| P-31 |
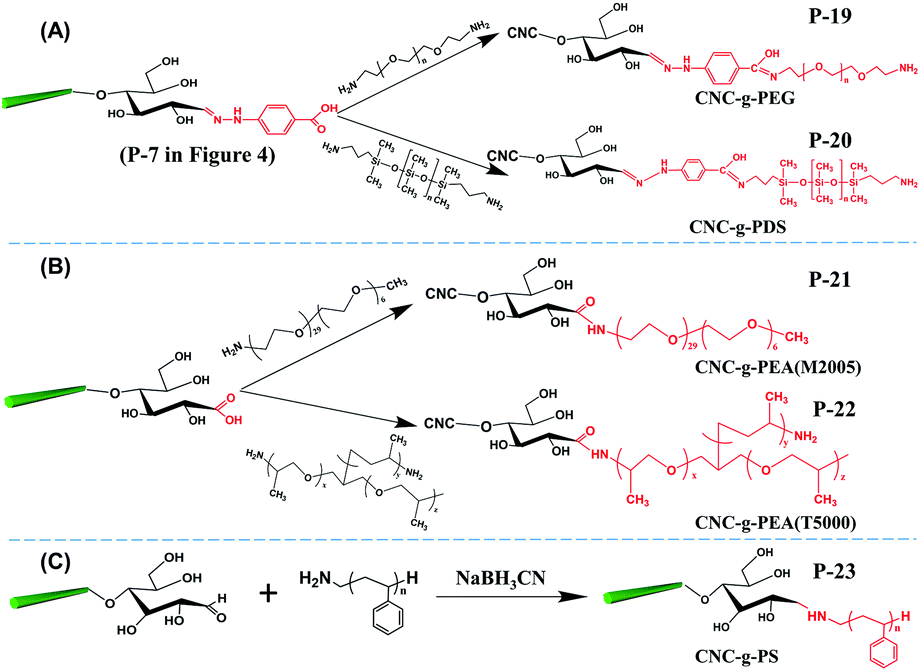 | ||
| Fig. 6 Covalent attachment of polymeric chains at the reducing end of CNCs following the “grafting onto” approach: (A) PEG, poly(ethylene glycol); PDS, poly(dimethyl siloxane); (B) PEA, polyetheramines; and (C) PS, polystyrene. | ||
With the preservation of hydrophilic surface hydroxyl groups on CNCs, hydrophobic polystyrene (PS) chains were grafted onto the reducing end of CNCs to create amphiphilic nanoparticles with the end topo-hydrophobic region (Fig. 6C, P-23).46 This study proposed a one-step end-modification strategy to graft PS onto the reducing end of CNCs by reductive amination. The optimal conditions for this reaction was 70 °C for 3 d, with a maximum reducing end aldehyde group grafting efficiency of 72.24% as estimated by the hydroxylamine hydrochloride method.
 | ||
| Fig. 7 Covalent attachment of polymeric chains at the reducing end of CNCs following the “grafting from” approach: (A) PAM, polyacrylamide; (B) PMETAC, poly[2-(methacryloyloxy)ethyltrimethyl ammonium chloride]; PNIPAM, poly(N-isopropyl acrylamide); and PSS, poly(sodium 4-styrenesulfonate). | ||
A recent study reported the grafting of several hydrophobic polymers at the reducing end of CNCs via ATRP, creating asymmetric CNC grafted polymers. The aldehyde groups of the CNC reducing end were first pre-oxidized and modified with ethylenediamine for attaching the initiator 2-bromoisobutanoic acid N-hydroxysuccinimide ester (NHS-BiB) via a stable hydrazone linkage.55 This CNC macro-bromine-initiator was further grafted with N-isopropylacrylamide (NIPAM), [2-(methacryloyloxy)ethyl]-trimethylammonium chloride (METAC) and sodium 4-vinylbenzenesulfonate (4-SS) to produce CNC-g-PMETAC, CNC-g-PNIPAM and CNC-g-PSS, respectively (Fig. 7B).
Despite the advantages of the “grafting from” approach (e.g. living and controllable polymerization), several limitations still restrict its practical application at the reducing end modification of CNCs. Most “grafting from” protocols for reducing end modification of CNCs involve three or four steps of reactions including carboxylation, carboxyamine condensation for the formation of hydrazone bonds, immobilization of the initiator and monomer polymerization. This number of steps remarkably reduces the overall grafting efficiency. For instance, ca. 50% of the terminal aldehyde groups only can react during the activation step (likewise for the conversion to carboxyl groups), which subsequently lowers the content of additional active groups (e.g., only 36% for carboxyl groups) that can participate in the next reaction.49 Another limitation of the “grafting from” approach comes from the uncontrollable polymerization on the surface of CNCs. For instance, partial polymerization of the PMETAC chains was observed by cryo-TEM along the surface of CNCs (CNC-g-PMETAC (P-30)) because of the potential reaction between NHS active esters and CNC surface hydroxyl groups during the reducing end modification.55 This unexpected polymerization on the surface of CNCs may cause interference to the end modification and meanwhile introduce impurities into the modified product.
The covalent grafting of the thermoresponsive polymer PNIPAM was reported on both the reducing end and surface of CNCs following a grafting from approach.51 As shown in Fig. 8, the surface carboxylated CNCs were directly used as the starting material. The content of carboxyl groups on the surface of CNCs was reported as 0.026 mmol g−1 as determined by conductometric titration and the terminal aldehyde groups were determined to be 0.0182 mmol g−1 as quantified by the colorimetric method. The reducing end aldehyde groups were oxidized to carboxyl groups, followed by coupling with an excess of ethylenediamine to form stable acylhydrazone bonds with the introduction of active amino groups on both end and surface of CNCs. Similarly, a macro-bromine-initiator attached to the initiating sites on both the end and surface of nanocrystals was further used to initiate the polymerization of N-isopropylacrylamide monomers on both the reducing end and surface of CNCs.51 The amount of grafted PNIPAM was estimated by thermogravimetric analysis to be 18 wt% and 37 wt% for the reducing end- and surface-modified products, respectively. The higher surface grafting density is due to the larger surface area and availability of CNCs than at the reducing end, which can provide more space for the reaction to occur and limit steric hindrance. In view of the different grafting densities of PNIPAM chains on the surface and end of CNCs, the modified product (P-32) was designated as a “patchy” nanoparticle.
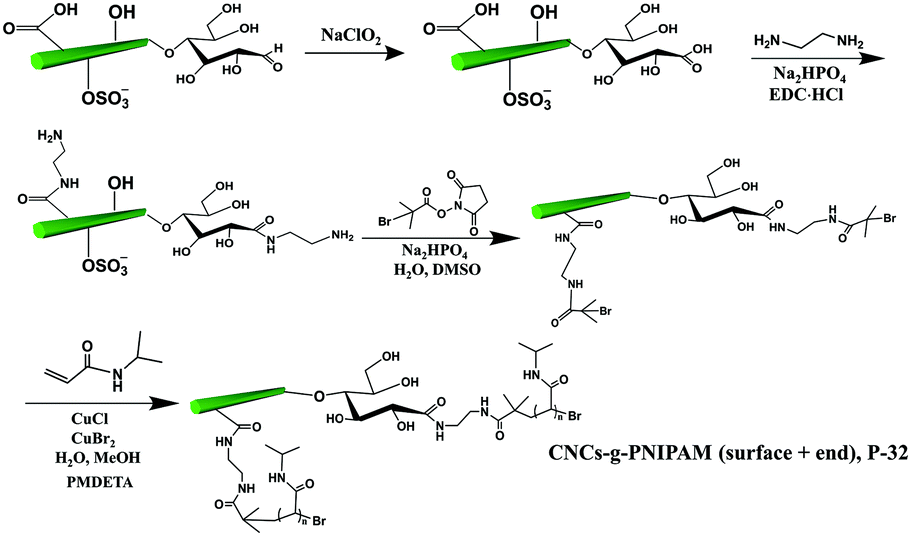 | ||
| Fig. 8 Grafting of PNIPAM on both the surface and the reducing end of CNCs (CNC-g-PNIPAM (surface + end)). | ||
In order to purify the end-modified products, appropriate post-treatments are performed during the reducing end modifications according to different properties of modifying reactants, as summarized in the previous tables. In the case of small molecules grafting at the reducing end of CNCs, most of the reactants are water-soluble, and therefore repeated centrifugation, washing against water and then dialysis in water for 3–5 days are the general purification treatments. In the case of the grafting polymer chains at the reducing end of CNCs, purification is more complicated because of the organic medium. The end-modified products often suffered successive washing against various organic solvents to remove free polymer chains and were then suspended in water.46,55 Several purification treatments are reported with the purpose of removing the physically absorbed reactants. For instance, hydrochloric acid was added into the reaction mixture in order to neutralize the excess reducing agent, followed by the addition of KCl to remove the reactants adsorbed on the CNC surface.44,47,48 Successive Soxhlet extraction with acetone and methanol was also reported to significantly remove the free and physically absorbed reagents.45
5. Characterization of reducing end modification on CNCs
The characterization of the reducing end modification of CNCs is challenging, because of the relatively scarce reaction sites of terminal groups and therefore low grafting density. Limited by the precision of instruments, some mature techniques used for characterizing surface modified CNCs cannot be directly applied for characterizing reducing end modification. In order to get precise characterization of reducing end-modified CNCs, sufficient purification after modification is necessary to remove any free or adsorbed species from the end modified CNC surface, for instance, by Soxhlet extraction, repeated washing or successive precipitation centrifugation. Typically, several characterization techniques are synergistically used to prove the success of the reducing end modification of CNCs.5.1 Characterization tracing the additional elements of grafted species
The use of elemental analysis to characterize the grafting density and efficiency of the reducing end modification is limited by both the precision of the instrument and the possible interference of surface reactions. Typically, the sensitivity of the elemental analysis instrument is in the range of 0.01–0.001%,77–79 leading to unreliable or even erroneous results when the grafting efficiency is low. As previously described, reducing end modification of CNCs is commonly conducted along with surface modification, and therefore cannot be differentiated by elemental analysis. For instance, the N content of CNC-g-PNIPAM (P-30) was estimated to be 7.54% by EA, which is slightly lower than the 8.66% content of surface-grafted PNIPAM CNCs.55 Other than the previously mentioned extra N content in the polymeric chains, this extraordinarily high N content of reducing end-modified CNCs could also be explained by the presence of unavoidable surface functionalization along the principal nanocrystal axis (surface) during the complicated three-step reducing end modification treatments. In conclusion, the amount of N element detected by EA should only be considered as evidence for the successful reducing end modification of CNCs, and not as quantitative information related to the grafting density and efficiency.
| Binding energies of sulfur | Binding energies of nitrogen | |||
|---|---|---|---|---|
| –OSO3H | –SH, –S–S–, –S–metal | Quaternary ammonium N+ | –N(H)CO | –NH2 |
| 169 eV | 165–162 eV | 402 eV | 400 eV | 399 eV |
5.2 Techniques for characterization of functional groups at the reducing end of CNCs
N) from reducing end-modified CNCs (Table 7). During the amidation condensation strategy, imine bond (CN) can be generated at the reducing end modification of CNCs, exhibiting a characteristic stretching vibration peak located at 1544 cm−1 in the FTIR spectrum of the modified CNCs (P-11, P-12).45 In another study, a small shift of stretching vibration for the formed CN was reported at 1560 cm−1 in the FTIR spectrum of end-modified CNC-C18 product (P-18).54 The CN bond is unstable in acidic treatment, which may be destroyed completely at a pH value below 6.5, as indicated by the disappearance of the absorption peak at 1560 cm−1 in the spectrum of the CNC-C18 product after acid treatment.54,73 The addition of a reducing agent can convert the CN bond from end modification of CNCs to a more stable C–N bond. Theoretically, the stretching vibration of primary amine C–N bond is located at 1230–1030 cm−1 in the FTIR spectrum. However, no study reported this C–N peak for end-modified CNCs after chemical reduction, which may be due to a low grafting density and therefore, weak IR signals. In addition, the strong stretching vibrations of CNCs (C–O) are also located in the same wavenumber range and can thus overlap with the C–N bond signal.
| Samples | Assignment | Wavenumber (cm−1) |
|---|---|---|
| Pristine CNCs | Stretching vibration of O–H bonds with primary and secondary hydroxyl groups | 3000–3700 |
| Stretching vibration of C–H bond | 2900 | |
| In-plane bending vibration of primary and secondary hydroxyl groups | 1315, 1335, 1430 and 1470 | |
| Antisymmetric stretching vibration of the C–O–C glycosidic bond | 1160 | |
| Vibrations of C–O bonds | 1110, 1060 and 1035 | |
| Out-of-plane torsional vibrations of the hydrogen bonded O–H bonds | 665 and 705 | |
| Adsorbed moisture | 1650 | |
| Reducing end-modified CNCs | Primary C–N | 1230–1030 |
| CN |
1560–1544 | |
| CO from carboxylic acid |
1720–1730 | |
| Primary amide (OC–N) |
1680–1655 | |
| Primary amide (C–N–H) | 1550–1530 | |
After carboxyamine condensation, the newly formed amide bond between the carboxyl groups and amino groups is the primary target in the FTIR spectrum of reducing end modified CNCs. As shown in Table 7, the stretching vibration of the carbonyl from carboxylic acid appears at around 1760 cm−1, while the stretching vibrations of primary amide are located at 1680–1655 cm−1 and 1550–1530 cm−1 for the OC–N and C–N–H bonds, respectively. The stretching and bending bands of the primary amide of PNIPAM in CNC-g-PNIPAM (P-32) appeared at 1650 cm−1 and 1540 cm−1 respectively,51,84 which provided direct evidence of successful reducing end modification of CNCs by PNIPAM.
5.3 Morphological observation and end adsorption of metallic nanoparticles
The indirect characterization method can also provide clues for the end modification of CNCs, viz. morphological observation with the assistance of reducing end adsorption of metallic nanoparticles. The introduced metallic nanoparticles at the reducing end-modified CNCs can be observed using a transmission electron microscope (TEM), resulting from the high interaction potential with the electron beam.84 By taking advantage of high affinity between specific chemical groups at the reducing end of CNCs and metallic nanoparticles, this method can be considered an indirect characterization technique to identify the reducing end modification on CNCs. The first study that reported the observation of silver nanoparticles deposited at the reducing end of carboxyl-modified CNCs by TEM was published in 1984. The aim of this work was to originally prove the parallel arrangement of the cellulose I chains.39 Inspired by this study, silver nanoparticles were synthesized and directly adsorbed at the reducing end of CNCs with carboxyl/thiol groups by reduction of silver nitrate with sodium borohydride.44,47,48,53 The protocol includes premixing of silver nitrate solution with a diluted end-modified CNC suspension by magnetic stirring at room temperature and for 30 min, followed by the generation of silver nanoparticles with the addition of sodium borohydride in solution. Fig. 9 shows that Ag nanoparticles can be selectively adsorbed at the reducing end of CNCs with diverse functional groups. Interestingly, silver nanoparticles generally do not adsorb on unmodified CNCs or randomly distribute along the surface of only CNCs.54 While this method can thus really help in quantifying the grafting density and yield of the reducing end modification, it is yet difficult to make a distinction between adsorbed groups of different affinities such as –SH and –COOH. This technique can provide inaccurate information on end-thiolated CNCs prepared by carboxyamine condensation. To avoid any interference of larger-scale nanocrystals, the use of slightly dilute negative staining solution is also recommended for the preparation of Ag nanoparticle-decorated CNCs.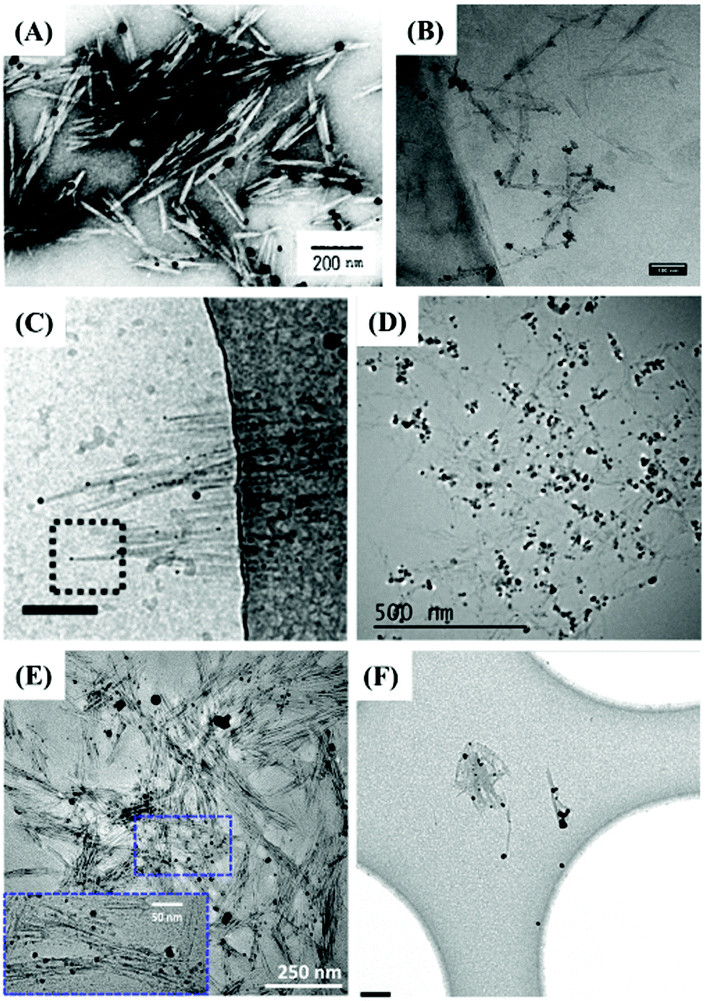 | ||
| Fig. 9 TEM images of reducing end-modified CNCs with the end-capped Ag nanoparticles: (A) CNC-COOH (P-6). (Reproduced with permission,43 Copyright 1984, John Wiley and Sons.) (B) CNC-g-PMETAC (P-29). (Reproduced with permission,55 Copyright 2018, American Chemical Society.) (C) CNC-NH(CH2)6SH (P-1). (Reproduced with permission,44 Copyright 2013, American Chemical Society.) (D) CNC-triazole-SH (P-2). (Reproduced with permission,47 Copyright 2018, American Chemical Society.) (E) CNC-NH(CH2)2-SH (P-3). (Reproduced with permission,48 Copyright 2019, American Chemical Society.) (F) CNC-NH(CH2)6-SH (P-4). (Reproduced with permission,53 Copyright 2014, Springer.) | ||
Atomic force microscopy (AFM) is another technique to indirectly confirm successful reducing end modification of CNCs. Although direct observation of grafted molecules or even longer polymeric chains by AFM is impossible, the reducing end functionalized CNCs can show clear differences in self-assembly into distinguishable supramolecular structure. The mushroom-like supramolecular morphology of reducing end-modified CNCs with β-casein52 was, for instance, clearly observed by AFM imaging. Details on the assembly of reducing end modified CNCs will be discussed in the last section of this review article.
Besides the morphological observation of reducing end-modified CNCs, quartz crystal microbalance with dissipation (QCM-D) was also used to analyze the adsorption behavior of end modified CNCs onto substrates of different chemical affinities, to prove the successful chemical end modification. With adsorption of end-modified CNCs-SH on a gold-coated QCM-D crystal, the oscillation frequency of the QCM-D crystals was reduced proportionally to the amount of adsorbed mass.44 At the same CNC concentration, the decrease in oscillation frequency was reported to be 9–10 times higher for CNC-SH than for pristine CNCs, suggesting that a higher amount of CNC-SH was adsorbed on the surface of the gold crystal. The measured adsorption between the end-modified –SH of the CNCs and the surface of the QCM-D gold crystal can also be imaged and quantified as direct evidence of the presence of the –SH reducing end. Characterization of reducing end modified CNCs with biotin molecules was also successfully achieved by QCM-D, which demonstrated the selective binding of end-modified nanocrystals to the surface of streptavidin.56
6. Applications of reducing end-modified CNCs
The component's property is the kernel factor of the constituted material's application, whereas the material's application is the external expression of the originated component's property. Similar to conventional CNCs or surface modified CNCs, reducing end-modified CNCs can also be found applicable in varied occasions where CNCs are generally applied, such as nanocomposites, fluorescent labeling, and stabilization of Pickering emulsions (Fig. 10). The preservation of both the physical and surface chemistry properties of end-modified CNCs makes it possible to use these nanoparticles in a similar manner to the unmodified and surface modified ones. Reducing end modified CNCs, however, also have unique application areas, such as the regulation of self-assembly owing to the regio-selective chemical modification at the reducing end of the particles. Both the preserved surface chemistry and additional regio-selective reducing end functionalities are expected to provide more interesting and diverse performance to the particles, paving the way for their use in a broader spectrum of applications.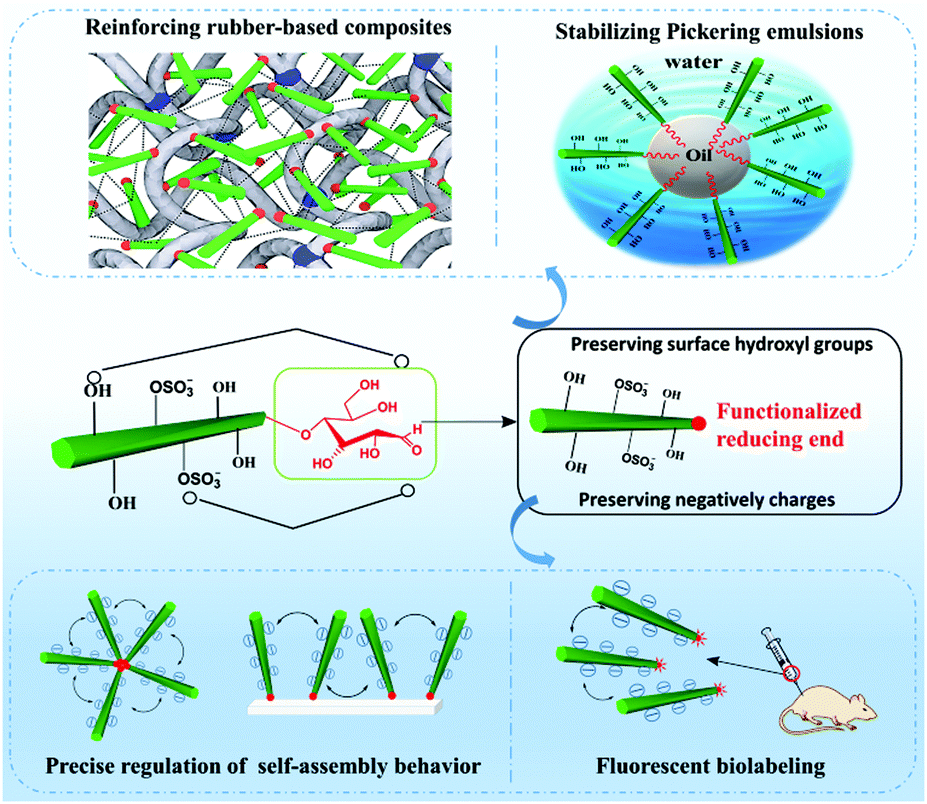 | ||
| Fig. 10 Several reported applications of reducing end-modified CNCs. | ||
6.1 Reducing end modified CNC reinforced rubber-based nanocomposites
Aiming to enhance the interfacial compatibility between hydrophilic CNC and hydrophobic matrices, traditional surface modification of CNCs is carried out at the expense of consuming or shielding the surface hydroxyl groups, which will disrupt OH-bond driving forces in the construction of a rigid percolation network for improvement of nanocomposite properties. How to obtain a good dispersion of CNCs in the composites with strong CNC-polymer interfacial adhesion while maintaining the percolating network among CNCs is an important issue in this field. Based on the reducing end modification of CNCs, the reactive molecule was only introduced at the end of the nanocrystal without consuming the polar surface functional groups. For example, end-thiolated CNCs (P-3) were produced by reductive amination and further combined with a natural rubber matrix to form a covalent linkage between the CNCs and the matrix by a UV-induced thiol–ene click reaction.47 The strong interfacial adhesion between the nanocrystals and rubber was achieved by reactive compounding, and meanwhile the surface hydroxyl groups of nanocrystals were preserved for the construction of the percolating network. The obtained nanocomposites showed strength and ductility improvement, with respective increase by 160, 468, and 8% of tensile strength, Young's modulus and elongation at break, respectively. The reason behind the remarkable enhancement of the composites’ mechanical performance was attributed to the creation of a stable percolation network within the composites at a critical percolation threshold of 7.5 wt%. Besides the observed mechanical enhancement of the end-modified CNC/rubber composites, the reducing end modification effectively promoted the re-dispersion of CNCs with reducing end grafted triazole molecules in aqueous suspension.47 The modified CNCs could thus be dried by either freeze-drying or oven drying and used directly as fillers in the design of high performance nanocomposites. The improved redispersion and stability of the end-modified CNCs in aqueous medium resulted from the synergistic action of both electrostatic and special repulsions provided by the half sulfate ester groups and the end-grafted cyclic molecules, respectively. Another study investigated the influence of reducing end-modified CNCs with reactive thiol groups (P-2) as fillers in thermoplastic elastomers [styrene–butadiene–styrene copolymer (SBS)].48 In addition to the formation of a percolating hydrogen bonding network by the end-modified CNCs, the SBS rubber was crosslinked in the bulk to form a double-network structure, resulting in a simultaneous increase of the strength (239%), modulus (411%), work of fracture (330%), and elongation at break (7%) of the composites.6.2 Reducing end fluorescently labeled CNCs in biomedical applications
The introduction of fluorescent molecules at the reducing end of CNCs with preserved surface chemistry broadens their application in the biomedical field. The signal detected from fluorescently labeled end-modified CNCs can be used to track and record the biodistribution, accumulation and clearance in cells or organs. Due to extremely low tropism for the conventional drug carriers toward anatomical targets in bone diseases, end fluorescently labeled CNCs (P-13) were incorporated into healthy mice.49 The preservation of negatively-charged sulfate esters on the surface of CNCs during the reducing end modification was reported to be a prerequisite for the delivery of nanocrystals to the cells and bone matrix. The fluorescent molecule, namely, Alexa Fluor 633, was immobilized at the reducing end of CNCs to provide an easily detectable signal for tracking the nanocrystals in the body of healthy immunocompetent mice, thus avoiding the biological elution of the dyes. The end-modified CNCs displayed a slow but evident internalization in cells without compromising their survival, and more importantly, exhibited a peculiar and transient tropism to the limb bones because of the strong interaction between the negative surface charges and the Ca2+ deposited in the bone.Recently, radiolabeled molecular imaging probes were prepared by conjugating DOTA-hydrazide at the reducing end of the CNCs (CNC(–CHO)-g-DOTA) (P-14) and DOTA-amine on their surface (CNC(–OH)-g-DOTA).50 The 111In-DOTA-CNC nanoparticles were further radiolabeled by single photon emission computed tomography (SPECT) imaging for evaluating their behavior in biological systems in vitro and in vivo. The influence of the preserved surface charge of unmodified and modified CNCs on their dispersion, cell interactions and plasma protein adsorption was also discussed. More 111In was reported to be chelated on the product CNC(–OH)-g-DOTA than on CNC(–CHO)-g-DOTA resulting from a lower amount of conjugated DOTA chelators at the reducing end of nanocrystals as well as the degradation of the hydrazone bond under acidic conditions for radiolabeling with 111In. However, both modified nanocrystals were observed in ex vivo biodistribution in the liver, lung, and spleen. As shown in Fig. 11, attributed to the reduction of surface charged density by the surface chemical modification, the transient accumulation of the CNC(–OH)-g-DOTA in the lung of mice was observed at early time points after systemic administration. This study recommended to introduce DOTA molecules at the end of CNCs as a promising approach for their free motion in the organs and biolabeling application with the preservation of the surface chemistry.
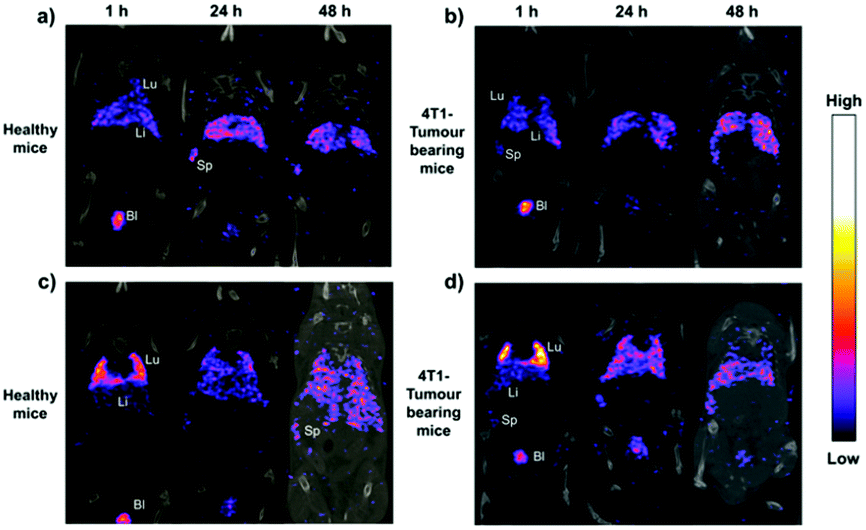 | ||
| Fig. 11 Representative coronal SPECT/CT fusion images after administration of (a and b) CNC(–CHO)-g-DOTA and (c and d) CNC(–OH)-g-DOTA in healthy and 4T1 tumor-bearing animal models at 1, 24, and 48 h. Lu: lung, Li: liver, Sp: spleen, and Bl: bladder. (Reproduced with permission,50 Copyright 2019, American Chemical Society.) | ||
6.3 Reducing end-modified CNCs as stabilizers of Pickering
The superior emulsifying performance of CNCs is due to their rod-like shape and the high aspect ratio.86–88 The introduction of hydrophobic segments (such as polymeric chains) at the reducing end of CNCs with the preservation of surface hydrophilic hydroxyl groups is expected to endow CNCs with an amphiphilic feature, therefore imparting the ability to adsorb at the oil–water interface of Pickering emulsions. Surfactant-like nanoparticles with end-tethered polystyrene chains (CNC-g-PS (P-23)) were prepared as stabilizing particles for Pickering emulsions, and were able to stabilize both toluene and hexadecane-in-water emulsions.46 The Pickering emulsions showed improved stability against coalescence and a low creaming rate over 4 months (Fig. 12) with the added reducing-end modified CNCs, and for different concentrations (0.3, 0.5, 1.0 wt%). Because of the lower polarity of hexadecane than toluene, the hexadecane-in-water emulsions stabilized by the CNC-g-PS exhibited better stability and much lower creaming rate compared with the toluene-in-water emulsions. The critical concentration of CNC-g-PS for emulsion stabilization was reported to be 0.3 wt% to ensure stability of the toluene-in-water emulsion against coalescence. For concentrations of CNC-g-PS higher than 0.5 wt%, a complete coverage of the oil–water interface by the nanocrystals was achieved with unchanged droplet size. With higher molecular weight of end-grafted PS (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 vs. 5000 g mol−1), the reducing end-grafting efficiency was decreased and led therefore to an adverse effect for the stability of oil–water interface.
000 vs. 5000 g mol−1), the reducing end-grafting efficiency was decreased and led therefore to an adverse effect for the stability of oil–water interface.
 | ||
| Fig. 12 Optical micrographs of toluene-in-water emulsions stabilized by CNC-g-PS at room temperature for CNC concentrations of (A) 0.3 wt%, (B) 0.5 wt%, and (C) 1.0 wt%; (D) optical micrographs of hexadecane-in-water emulsions stabilized by 0.3 wt% CNC-g-PS at room temperature. (Reproduced with permission,46 Copyright 2018, American Chemical Society.) | ||
Another study reported the utilization of end modified CNCs with a grafted C18 alkyl chain (P-18) as an emulsifier for oil–water Pickering emulsions.52 In comparison with pristine CNCs, highly-stable n-hexane-in-water Pickering emulsions with an oil/water volume ratio (1:1) was successfully stabilized by the C18 end-modified CNCs with concentrations ranging from 0.05 to 0.3 wt%. The end grafted alkyl chain contributed to the wetting behavior and strong adsorption of the hydrophilic CNCs at the oil–water interface, thus exhibiting good emulsifying effect over 4 months.
6.4 Assembly manipulation of reducing end-modified CNCs
The most attractive and unique application of reducing end modified CNCs may be the precise manipulation of their self-assembly behavior to produce structures like end-to-end, star-like, and hairbrush-like aggregates with potential applications in functional materials. A pioneer work on this topic was performed with end-thiolated CNCs (P-1) immobilized on a gold surface from one end owing to the high affinity between the introduced thiol groups and the gold surface.44 The reducing end-modified CNCs were able to swing on the gold surface because of their orientation and mobility, which resulted in the formation of cilia-like functional hairy surfaces. Moreover, the lateral distance between the two end-attached CNCs could be regulated by varying the intensity of electrostatic interactions of the preserved surface sulfate groups.The introduction of reactive molecules at the reducing end of CNCs makes it possible to tune the self-assembly of the rod-like nanocrystals into desired morphologies through carefully designed interactions by either covalent linkage or physical adsorption. Mushroom-like nanoparticles can be obtained through the assembly of reducing end-modified CNCs with grafted micelle-formed by a model protein (β-casein),52 as shown in Fig. 13A. To graft the β-casein at the reducing end of CNCs, reactive azide groups were first introduced at the reducing end of CNCs, then reacted with the acetylene-functionalized β-casein by an azide–alkyne click reaction. Several end-modified CNCs were observed to be conjugated on the surface of β-casein to form micelles. The biorecognition between streptavidin (SAV) and biotin molecules was also used to tune and control the assembly of reducing end-modified CNCs. A unique star-like assembly was designed by noncovalent adsorption of SAV on the end-modified CNCs grafted with biotin molecules.56 In the study, SAV was separately added to the suspension of two end-modified CNCs namely, CNC-g-PEG-Biot (P-15) and CNC-g-HC-Biot (P-16) of different molecular weights (long-chain PEG and short-chain HC). The end-grafted 6-carbon aliphatic chain (HC) at the reducing end of CNCs was reported to be too short to promote the complexation of end-modified nanocrystals with SAV. In contrast, the mixture of CNC-g-PEG-Biot and SAV could result in star-like aggregates, because of the reduced steric hindrance by the long length of PEG chains with seven repeating units, thus enabling the binding between the end-grafted biotin molecules with SAV (Fig. 13B).
 | ||
| Fig. 13 (A) AFM height image (10 × 10 μm) of end-modified CNC-g-β-casein nanoparticles assembled into mushroom-like structures as highlighted by the arrows. (Reproduced with permission,52 Copyright 2013, Springer.) (B) AFM topographical image of end-modified CNC-g-PEG-Biot with the addition of streptavidin. (Reproduced with permission,56 Copyright 2018, American Chemical Society.) (C) TEM images of negatively-stained CNC-g-PEA (M2005) at 40 °C and (D) self-assembly model of end-modified CNCs as the swollen aggregate in suspension at T > LCST. (Reproduced with permission,57 Copyright 2019, American Chemical Society.) AFM images of end-to-end assembled nanofibers induced by (E) a click reaction between end-modified CNCs with grafted alkyne and azide groups and (F) carboxyamine condensation between the terminal carboxyl groups of CNC and the end-modified CNCs with grafted active amino groups. (Reproduced with permission,89 Copyright 2016, American Chemical Society.) | ||
Thermoresponsive polymers grafted at the reducing end of CNCs by the grafting onto approach exhibited thermally reversible aggregation and gelation at ambient temperature higher than the polymers’ LCST, which resulted in the controlled assembly of the end-modified CNCs by the temperature-triggered physical curling and entanglement of the grafted thermoresponsive polymers. Such temperature-dependent assembly behavior of rod-like nanocrystals was also reported with end-modified CNCs with grafted thermoresponsive PEA (P-21 and 22).57 When the temperature of the CNC-g-PEA suspension was increased from 4 °C to 40 °C (i.e. above the LCST of PEA (M2005) and PEA (T5000) of 16 °C and 12 °C, respectively), the grafted polymeric chains at the reducing end of the CNCs gradually collapsed and entangled together into star-shaped aggregates organized by four, five, or six nanocrystals hooked by their ends (Fig. 13C and D). The obtained star-shaped aggregates exhibited a high degree of symmetry due to the electrostatic repulsive forces along the CNCs, arising from the preservation of negatively charged sulfate groups on the surface. While the temperature was cooled below the LCST, the polymer chains would be compatible with water and the repulsive forces among them can lead to disintegration of the assembled star-like structure into individual nanocrystals. The corresponding hydrodynamic diameter (Dh) of the nanoparticles in suspension at 40 °C was estimated to be 3-fold higher than that of the suspension at 4 °C. The end-modified CNCs demonstrated a reversible and reproducible dynamic assembly-disintegration behavior triggered by a temperature change between 4 and 40 °C.
Recently, streamlined hydrolysis and sequential oxidations were used to prepare CNCs with terminal carboxyl groups. The hydrolysis of raw softwood Kraft pulp was performed in a solution of ammonium persulfate followed by sequential periodate and chlorite oxidation steps.89 The produced CNCs contained a content of carboxyl groups of 0.06 mmol g−1. Both active alkyne and azide groups were introduced at the reducing end carboxyl group by reacting with either 1-amino-3-butyne or 3-azido-1-propanamine, respectively. As shown in Fig. 13E, the two types of end-modified CNCs bearing alkyne and azide groups were covalently linked together by an azide–alkyne click reaction to produce an end-to-end assembly of one-dimensional nanofibers of 1 μm length and 6 nm width. Following a similar approach, another end-to-end aggregated nanofiber was also prepared by carboxyamine condensation between the terminal carboxyl groups of the obtained CNCs and the end-modified CNCs with grafted active amino groups (Fig. 13F). The strategy of end modification and covalent linkage between several individual rod-like nanocrystals is a novel assembly method to produce one-dimensional nanofibers with a high aspect ratio.
7. Future challenges and concluding remarks
Extensive studies have focused on the modification of cellulose nanocrystals (CNCs) with the aim of regulating their properties for various potential applications. However, most CNC modification methods emphasize on surface modification because of the availability and accessibility of numerous hydroxyl groups on the surface. Only in the last five years have reducing end modification of CNCs started to receive more attention, and flourishing publications were reported in this field. One of the important features of end modification strategies of CNCs is the specific-site modification at the terminal region with preservation of the surface chemistry and properties of CNCs. Two main approaches, namely, aldimine condensation and carboxyamine condensation, have been exploited to introduce diverse reactive groups, functional molecules and polymeric chains at the end of CNCs, thus providing a reactive and identified reducing end for these rod-like nanoparticles.Some challenges in the reducing end modification of CNCs still remain. (1) The accurate measurement of the effective amount of reducing end aldehyde groups is a prerequisite for any reducing end modification of CNCs. However, results from four different characterization methods showed high discrepancies between the measured values. A more user-friendly and accurate method is needed to quantify the exact amount of available aldehyde groups at the reducing end of CNCs, associated with diverse cellulosic sources and dimensions of nanocrystals. (2) The reducing end modification strategies of CNCs require improved grafting efficiency and particularly milder conditions, fewer preparation steps and lower reaction time. The current protocols include complex procedures (for instance, including repeated addition of reagents) with severe side reactions and long reaction times, which leads to reduced end grafting density. (3) The characterization techniques (especially lack of powerful and direct characterization) for the reducing end modification of CNCs are still insufficient because of the low grafting density and therefore low detectability. The high-precision characterization technique, such as the two-dimensional nuclear magnetic resonance (2D NMR), may be the choice for the analysis of the CNC reducing end modification. (4) The combination of reducing end modification and surface modification of CNCs deserves further investigations to design CNCs with diverse structures and functionalities based on different regio-selective and local modifications. In comparison with the rich and eye-catching surface modification, there are only a few reports on the reducing end modification of CNCs. However, the few yet promising results discussed in this review article highlight the potential of end modification strategies to broaden the utilization of CNCs to novel functional applications requiring end recognition and end adhesion properties.
Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (51603159). The authors also thank for the support of the Fundamental Research Funds for the Central Universities (WUT: 2019III169CG).References
- A. Dufresne, Curr. For. Rep., 2019, 5, 76–89, DOI:10.1007/s40725-019-00088-1.
- A. Dufresne, Nanocellulose: from nature to high performance tailored materials, 2nd edn, Walter de Gruyter GmbH, Berlin/Boston, 2017, DOI:10.1515/9783110480412-001.
- B. Thomas, M. C. Raj, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625, DOI:10.1021/acs.chemrev.7b00627.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994, 10.1039/C0CS00108B.
- D. Klemm, E. D. Cranston, D. Fischer, M. Gama, S. A. Kedzior, D. Kralisch, F. Kramer, T. Kondo, T. Lindström, S. Nietzsche, K. Petzold-Welcke and F. Rauchfuß, Mater. Today, 2018, 21, 720–748, DOI:10.1016/j.mattod.2018.02.001.
- H. Kargarzadeh, J. Huang, N. Lin, I. Ahmad, M. Mariano, A. Dufresne, S. Thomas and A. Gałęski, Prog. Polym. Sci., 2018, 87, 197–227, DOI:10.1016/j.progpolymsci.2018.07.008.
- D. Trache, M. H. Hussin, M. M. Haafiz and V. K. Thakur, Nanoscale, 2017, 9, 1763–1796, 10.1039/c6nr09494e.
- R. M. Parker, G. Guidetti, C. A. Williams, T. Zhao, A. Narkevicius, S. Vignolini and B. Frka-Petesic, Adv. Mater., 2018, 30, 1704477, DOI:10.1002/adma.201704477.
- E. Kontturi, P. Laaksonen, M. B. Linder, A. H. Gröschel, O. J. Rojas and O. Ikkala, Adv. Mater., 2018, 30, 1703779, DOI:10.1002/adma.201703779.
- A. Dufresne, Mater. Today, 2013, 16, 220–227, DOI:10.1016/j.mattod.2013.06.004.
- M. K. Khan, A. Bsoul, K. Walus, W. Y. Hamad and M. J. MacLach-lan, Angew. Chem., Int. Ed., 2016, 54, 4304–4308, DOI:10.1002/anie.201410411.
- A. P. Almeida, J. P. Canejo, S. N. Fernandes, C. Echeverria, P. L. Almeida and M. H. Godinho, Adv. Mater., 2018, 30, 1703655, DOI:10.1002/adma.201703655.
- T. D. Nguyen, J. A. Kelly, W. Y. Hamad and M. J. MacLachlan, Adv. Funct. Mater., 2015, 25, 2175–2181, DOI:10.1002/adfm.201404304.
- S. Agate, M. Joyce, L. Lucia and L. Pal, Carbohydr. Polym., 2018, 198, 249–260, DOI:10.1016/j.carbpol.2018.06.045.
- H. Du, W. Liu, M. Zhang, C. Si, X. Zhang and B. Li, Carbohydr. Polym., 2019, 209, 130–144, DOI:10.1016/j.carbpol.2019.01.020.
- A. B. Seabra, J. S. Bernardes, W. J. Fávaro, A. J. Paula and N. Durán, Carbohydr. Polym., 2018, 181, 514–527, DOI:10.1016/j.carbpol.2017.12.014.
- Q. Wang, J. Sun, Q. Yao, C. Ji, J. Liu and Q. Zhu, Cellulose, 2018, 25, 4275–4301, DOI:10.1007/s10570-018-1888-y.
- D. M. Nascimento, Y. L. Nunes, M. C. Figueirêdo, H. M. de Azeredo, F. A. Aouada, J. P. Feitosa, M. F. Rosa and A. Dufresne, Green Chem., 2018, 20, 2428–2448, 10.1039/c8gc00205c.
- K. M. Chin, S. Sung Ting, H. L. Ong and M. Omar, J. Appl. Polym. Sci., 2018, 135, 46065, DOI:10.1002/app.46065.
- H. Golmohammadi, E. Morales-Narvaez, T. Naghdi and A. Merkoci, Chem. Mater., 2017, 29, 5426–5446, DOI:10.1021/acs.chemmater.7b01170.
- M. S. Islam, L. Chen, J. Sisler and K. C. Tam, J. Mater. Chem. B, 2018, 6, 864–883, 10.1039/c7tb03016a.
- J. Shojaeiarani, D. Bajwa and A. Shirzadifar, Carbohydr. Polym., 2019, 216, 247–259, DOI:10.1016/j.carbpol.2019.04.033.
- S. S. Athukoralalage, R. Balu, N. K. Dutta and N. Roy Choudhury, Polymers, 2019, 11, 898, DOI:10.3390/polym11050898.
- K. J. De France, T. Hoare and E. D. Cranston, Chem. Mater., 2017, 29, 4609–4631, DOI:10.1021/acs.chemmater.7b00531.
- N. Lavoine and L. Bergström, J. Mater. Chem. A, 2017, 5, 16105–16117, 10.1039/c7ta02807e.
- A. Balea, A. Blanco and C. Negro, Polymers, 2019, 11, 518, DOI:10.3390/polym11030518.
- R. Z. Khoo, W. S. Chow and H. Ismail, Cellulose, 2018, 25, 4303–4330, DOI:10.1007/s10570-018-1879-z.
- J. Wang, D. J. Gardner, N. M. Stark, D. W. Bousfield, M. Tajvidi and Z. Cai, ACS Sustainable Chem. Eng., 2017, 6, 49–70, DOI:10.1021/acssuschemeng.7b03523.
- H. Kargarzadeh, M. Mariano, J. Huang, N. Lin, I. Ahmad, A. Dufresne and S. Thomas, Polymer, 2017, 132, 368–393, DOI:10.1016/j.polymer.2017.09.043.
- D. Wang, Cellulose, 2019, 26, 687–701, DOI:10.1007/s10570-018-2143-2.
- N. Grishkewich, N. Mohammed, J. Tang and K. C. Tam, Curr. Opin. Colloid Interface Sci., 2017, 29, 32–45, DOI:10.1016/j.cocis.2017.01.005.
- J. Ru, B. Y. Geng, C. C. Tong, H. Y. Wang, S. C. Wu and H. Z. Liu, Prog. Chem., 2017, 29, 1228–1251, DOI:10.7536/PC170616.
- W. Chen, H. Yu, S. Y. Lee, T. Wei, J. Li and Z. Fan, Chem. Soc. Rev., 2018, 47, 2837–2872, 10.1039/c7cs00790f.
- Z. Wang, P. Tammela, M. Strømme and L. Nyholm, Adv. Energy Mater., 2017, 7, 1700130, DOI:10.1002/aenm.201700130.
- X. Du, Z. Zhang, W. Liu and Y. Deng, Nano Energy, 2017, 35, 299–320, DOI:10.1016/j.nanoen.2017.04.001.
- N. Lin, J. Tang, A. Dufresne and M. K. Tam, Advanced Functional Materials from Nanopolysaccharides, Springer, Germany, 2019, DOI:10.1007/978-981-15-0913-1.
- S. Wohlhauser, G. Delepierre, M. Labet, G. Morandi, W. Thielemans, C. Weder and J. O. Zoppe, Macromolecules, 2018, 51, 6157–6189, DOI:10.1021/acs.macromol.8b00733.
- Y. Habibi, Chem. Soc. Rev., 2014, 43, 1519–1542, 10.1039/c3cs60204d.
- K. Hieta, S. Kuga and M. Usuda, Biopolymers, 1984, 23, 1807–1810, DOI:10.1002/bip.360231002.
- S. Kuga and R. Malcolm Brown Jr, Carbohydr. Res., 1988, 180, 345–350, DOI:10.1016/0008-6215(88)80091-0.
- M. Koyama, W. Helbert, T. Imai, J. Sugiyama and B. Henrissat, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9091–9095, DOI:10.1073/pnas.94.17.9091.
- N. H. Kim, T. Imai, M. Wada and J. Sugiyama, Biomacromolecules, 2006, 7, 274–280, DOI:10.1021/bm0506391.
- E. Sipahi-Sağlam, M. Gelbrich and E. Gruber, Cellulose, 2013, 10, 237–250, DOI:10.1023/A:1025151701985.
- L. R. Arcot, A. Nykänen, J. Seitsonen, L. S. Johansson, J. Campbell, O. J. Rojas, O. Ikkala and J. Laine, Biomacromolecules, 2013, 14, 2807–2813, DOI:10.1021/bm400633r.
- J. L. Huang, C. J. Li and D. G. Gray, ACS Sustainable Chem. Eng., 2013, 1, 1160–1164, DOI:10.1021/sc400074e.
- C. Tang, S. Spinney, Z. Shi, J. Tang, B. Peng, J. Luo and K. C. Tam, Langmuir, 2018, 34, 12897–12905, DOI:10.1021/acs.langmuir.8b02437.
- L. Li, H. Tao, B. Wu, G. Zhu, K. Li and N. Lin, ACS Sustainable Chem. Eng., 2018, 6, 14888, DOI:10.1021/acssuschemeng.8b03407.
- H. Tao, A. Dufresne and N. Lin, Macromolecules, 2019, 52, 5894–5906, DOI:10.1021/acs.macromol.9b01213.
- L. Colombo, L. Zoia, M. B. Violatto, S. Previdi, L. Talamini, L. Sitia, F. Nicotra, M. Orlandi, M. Salmona, C. Recordati, P. Bigini and B. L. Ferla, Biomacromolecules, 2015, 16, 2862–2871, DOI:10.1021/acs.biomac.5b00805.
- S. Imlimthan, S. Otaru, O. Keinänen, A. Correia, K. Lintinen, H. A. Santos, A. J. Airaksinen, M. A. Kostiainen and M. Sarparanta, Biomacromolecules, 2018, 20, 674–683, DOI:10.1021/acs.biomac.8b01313.
- B. Risteen, G. Delepierre, M. Srinivasarao, C. Weder, P. Russo, E. Reichmanis and J. Zoppe, Small, 2018, 14, 1802060, DOI:10.1002/smll.201802060.
- M. A. Karaaslan, G. Gao and J. F. Kadla, Cellulose, 2013, 20, 2655–2665, DOI:10.1007/s10570-013-0065-6.
- L. R. Arcot, M. Lundahl, O. J. Rojas and J. Laine, Cellulose, 2014, 21, 4209–4218, DOI:10.1007/s10570-014-0426-9.
- W. Du, J. Guo, H. Li and Y. Gao, ACS Sustainable Chem. Eng., 2017, 5, 7514–7523, DOI:10.1021/acssuschemeng.7b00375.
- J. O. Zoppe, A. V. M. Dupire, T. G. Ge. Lachat, P. Lemal, L. Rodriguez-Lorenzo, A. Petri-Fink, C. Weder and H. A. Klok, ACS Macro Lett., 2017, 6, 892–897, DOI:10.1021/acsmacrolett.7b00383.
- A. Villares, C. Moreau and B. Cathala, ACS Omega, 2018, 3, 16203–16211, DOI:10.1021/acsomega.8b02559.
- F. Lin, F. Cousin, J. L. Putaux and B. Jean, ACS Macro Lett., 2019, 8, 345–351, DOI:10.1021/acsmacrolett.8b01005.
- E. B. Sanders, A. I. Goldsmith and J. I. Seeman, J. Anal. Appl. Pyrolysis, 2003, 66, 29–50, DOI:10.1016/S0165-2370(02)00104.
- T. Soderberg, Organic Chemistry with a Biological Emphasis, University of Minnesota Morris Digital Well, Morris, 2012 Search PubMed.
- L. T. Fan, M. M. Gharpuray and Y. H. Lee, Cellulose Hydrolysis, Springer Science & Business Media, 2012, vol. 3, DOI:10.1007/9783-642-72575-3.
- C. J. Knill and J. F. Kennedy, Carbohydr. Polym., 2013, 51, 281–300, DOI:10.1016/S0144-8617(02)00183-2.
- S. Eyley and W. Thielemans, Nanoscale, 2014, 6, 7764–7779, 10.1039/C4NR01756K.
- P. J. Wakelyn, N. R. Bertoniere, A. D. French, D. P. Thibodeaux, B. A. Triplett, M.-A. Rousselle, W. R. Goynes, J. V. Edwards, L. Hunter, D. D. McAlister and G. R. Gamble, Handbook of Fiber Chemistry, 2007, pp. 584–628 Search PubMed.
- J. Araki, Soft Matter, 2013, 9, 4125–4141, 10.1039/C3SM27514K.
- N. Lin and A. Dufresne, Nanoscale, 2014, 6, 5384–5393, 10.1039/C3NR06761K.
- J. Gu, J. M. Catchmark, E. Q. Kaiser and D. D. Archibald, Carbohydr. Polym., 2013, 92, 1809–1816, DOI:10.1016/j.carbpol.2012.10.078.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466, DOI:10.1002/anie.201001273.
- T. Abitbol, E. Kloser and D. G. Gray, Cellulose, 2013, 20, 785–794, DOI:10.1007/s10570-013-9871-0.
- T. Abitbol, A. Palermo, J. M. Moran-Mirabal and E. D. Cranston, Biomacromolecules, 2013, 14, 3278–3284, DOI:10.1021/bm400879x.
- K. P. C. Vollhardt and N. E. Schore, Organic Chemistry; Palgrave version: Structure and Function, Macmillan International Higher Education, 2014 Search PubMed.
- O. Szabolcs, Papier, 1961, 15, 41 CAS.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110, 3479–3500, DOI:10.1021/cr900339w.
- Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045–3055, 10.1039/C2PY20290E.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, Amsterdam, Boston, 2008, DOI:10.1016/B978-0-12-370501-3.X0001-X.
- L. R. Van Loon and M. A. Glaus, J. Environ. Polym. Degrad., 1997, 5, 97–109, DOI:10.1007/bf02763593.
- O. Dilek and S. L. Bane, J. Fluoresc., 2011, 21, 347–354, DOI:10.1007/s10895-010-0723-0.
- 2400 Series II CHNS/O User Manual, PerkinElmer, 2011, pp. 1–12.
- Carbon, Hydrogen, and Nitrogen Determination using the PerkinElmer 2400 Series II CHNS/O Analyzer, PerkinElmer, 2011.
- T. Xia, H. Yuwen and N. Lin, Compos. Sci. Technol., 2018, 161, 8–15, DOI:10.1016/j.compscitech.2018.03.038.
- D. Littlejohn and S. J. Chang, Electron Spectrosc., 1995, 71, 47–50, DOI:10.1016/0368-2048(94)02244-5.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083–5086, DOI:10.1021/la960465w.
- K. S. Siow, L. Britcher, S. Kumar and H. J. Griesser, Plasma Processes Polym., 2006, 3, 392–418, DOI:10.1002/ppap.200600021.
- E. J. Foster, R. J. Moon, U. P. Agarwal, M. J. Bortner, J. Bras, S. Camarero-Espinosa, K. J. Chan, M. J. Clift, E. D. Cranston and S. J. Eichhorn, Chem. Soc. Rev., 2018, 47, 2609–2679, 10.1039/C6CS00895J.
- U. D. Hemraz, A. Lu, R. Sunasee and Y. Boluk, J. Colloid Interface Sci., 2014, 430, 157–165, DOI:10.1016/j.jcis.2014.05.011.
- G. Zhu, Z. Chen, B. Wu and N. Lin, Ind. Crops Prod., 2019, 139, 111580, DOI:10.1016/j.indcrop.2019.111580.
- S. Fujisawa, E. Togawa and K. Kuroda, Sci. Technol. Adv. Mater., 2017, 18, 959971, DOI:10.1080/14686996.2017.1401423.
- F. Jiang and Y. L. Hsieh, Biomacromolecules, 2015, 16, 1433–1441, DOI:10.1021/acs.biomac.5b00240.
- J. Tang, M. F. X. Lee, W. Zhang, B. Zhao, R. M. Berry and K. C. Tam, Biomacromolecules, 2014, 15, 3052–3060, DOI:10.1021/bm500663w.
- H. Yang and T. G. van de Ven, Biomacromolecules, 2016, 17, 2240–2247, DOI:10.1021/acs.biomac.6b00480.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 covering review articles on the topic of cellulose nanocrystals in the last three years is included in this file. See DOI: 10.1039/d0nh00016g |
| This journal is © The Royal Society of Chemistry 2020 |