
Ultrathin boron nanosheets as an emerging two-dimensional photoluminescence material for bioimaging†
Dingtao
Ma‡
a,
Jinlai
Zhao‡
ac,
Jianlei
Xie‡
b,
Feng
Zhang
b,
Rui
Wang
b,
Leiming
Wu
 a,
Weiyuan
Liang
b,
Delong
Li
b,
Yanqi
Ge
b,
Jianqing
Li
a,
Yupeng
Zhang
*b and
Han
Zhang
*b
a,
Weiyuan
Liang
b,
Delong
Li
b,
Yanqi
Ge
b,
Jianqing
Li
a,
Yupeng
Zhang
*b and
Han
Zhang
*b
aFaculty of Information Technology, Macau University of Science and Technology, Taipa, Macau SAR 999078, P. R. China
bCollaborative Innovation Center for Optoelectronic Science and Technology and Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, Shenzhen University, Shenzhen 518060, P. R. China. E-mail: hzhang@szu.edu.cn; ypzhang@szu.edu.cn
cCollege of Materials Science and Engineering, Shenzhen Key Laboratory of Polymer Science and Technology, Guangdong Research Center for Interfacial Engineering of Functional Materials, Shenzhen 518060, P. R. China
First published on 5th February 2020
Abstract
Two-dimensional (2D) metal-free sheets with atomic thickness have been highly considered as promising candidates for fluorescent probes, due to their intriguing characteristics. In this work, 2D ultrathin boron nanosheets (B NSs) with a surface defect nanolayer can be effectively prepared by modified liquid phase exfoliation. The as-prepared ultrathin B NSs show blue fluorescence characteristics even with a quantum yield efficiency of up to 10.6%. Such luminescent behavior originates from the quantum confinement effect and the existence of a surface defect layer. In light of the advantages of being environmentally friendly, having high photostability and good biocompatibility, for the first time we have shown that ultrathin B NSs can be used as an emerging fluorescent probe for application in cellular bioimaging. It is believed that this work will open new avenues for ultrathin B NSs in biomedical fields, and it will also inspire the development of other elemental 2D nanomaterials.
New conceptsRecently, two-dimensional (2D) elemental nanosheets have attracted great attention in biomedical fields, and have also been considered as promising candidates for fluorescent probes. Among them, the pursuit of fluorescent probes with characteristics of good biocompatibility, low cytotoxicity, and good photostability has always been a research hotspot. In this work, we report the efficient fabrication of blue luminescent ultrathin B nanosheets using a modified liquid-phase exfoliation method. Such luminescence behavior mainly results from the quantum confinement effect and the existence of a surface defect layer. As an emerging luminescent material, such 2D B nanosheets were found to have a high quantum yield efficiency of 10.6%. In addition, we demonstrated that such a material exhibits several ideal properties of being environmentally friendly, high photostability, and good biocompatibility. Furthermore, for the first time, such 2D B nanosheets were successfully applied to cellular bioimaging. It is anticipated that this first report of an emerging luminescent material of ultrathin B nanosheets will have far-ranging impact and will open up the applications of B nanosheet materials in biomedical fields. |
Introduction
Bioimaging is a crucial biotechnology process aimed at the development of novel methods to image specific molecular pathways in living cells or in the human body.1–3 In order to non-invasively investigate the biological processes in vivo at the cellular and molecular levels, there has been a continual search for new materials that can be engineered for bioimaging purposes in the nanotechnology field. In general, good biocompatibility, low cytotoxicity, and photostability are the most important factors that determine the fluorescent probes applied for bioimaging. Among the developed candidates, traditional organic dyes and polymers have a relatively low stability, whilst the high price and unsatisfactory cytotoxicity greatly limit the practical applications of rare-earth metals and noble metal systems.4,5 In contrast, two-dimensional (2D) heavy-metal free nanosheets/quantum dots (QDs) such as graphene,6–8 boron nitride,9–12 and g-C3N413,14 have attracted research attention in biomedical applications, due to their fascinating physicochemical characteristics, as well as their low cost, environmental friendliness, and high stability.As an emerging member of this category, borophene, namely ultrathin boron nanosheets (B NSs) with only atomic thickness, exhibits many fascinating properties, such as Dirac fermions, in-plane anisotropy and superconductivity.15,16 In addition, the electron-deficient characteristics of the multi-center-two-electron bonds of boron lead to a complicated in-plane atomic bonding manner, thereby rendering borophene to have structural polymorphism and a tunable band structure.15–18 However, only a few studies concerning its photoluminescent properties have been reported up to now. Previously, Tai et al.19 synthesized a novel 2D monolayer γ-boron film, and the as-prepared sample was demonstrated to have excellent photoluminescence characteristics, due to strong quantum-confinement effects. Therefore, more in-depth research is still required in order to discover the practical applications of the materials. In addition, considering the abundant reserves, cost effectiveness and environmental friendliness of boron, the development of 2D ultrathin B NSs as an emerging fluorescent probe for bioimaging can be greatly expected, and should urgently be demonstrated.20–22
In order to achieve this target, the effective preparation of ultrathin B NSs is the first priority. For example, Mannix et al.23 and Feng et al.24 have reported the experimental synthesis of 2D borophene by molecular beam epitaxy technology on a Ag(111) substrate, and two structural polymorphs of borophene, i.e. rectangular and rhombohedral phases, were successfully observed. However, it should be pointed out that such preparative technologies usually require harsh operating conditions. In addition, the obtained yield is very low, and the additional transfer step leads to increased time consumption and a high cost, thereby hindering its practical applications. In contrast to the vapor-based methods, liquid-phase exfoliation (LPE) has recently been considered to be an effective strategy for fabricating 2D nanomaterials, and this combines the advantages of a facile operation, a high yield, low cost, and it being environmentally friendly.25–27 With this in mind, it also raises the question as to whether LPE can be applied for preparing 2D ultrathin B NSs derived from the corresponding bulk boron powder. Although several previous studies have suggested that B NSs can be obtained through a probe sonication treatment, it is still difficult for them to obtain B NSs with an average thickness down to a few nanometers, due to its complex and diverse bulk structure.28–33 In this way, the development of a green strategy for fabricating ultrathin B NSs is extremely necessary and remains a challenge.
Herein, we put forward a modified LPE technique combining probe and water bath sonication methods, in order to realize the effective fabrication of ultrathin B NSs. The as-obtained ultrathin B NSs retained good structural integrity, with an average thickness of down to 1.5 nm. In addition, the ultrathin B NSs exhibit strong blue fluorescence characteristics with a quantum yield efficiency up to 10.6%. Due to the good biocompatibility and high photostability, the practical application of ultrathin B NSs as a cellular imaging probe was first demonstrated by labeling HeLa and Huh-7 cells. Briefly, this work is expected to not only provide a facile solution for synthesizing ultrathin B NSs, but also to extend the use of this emerging multi-functional material in biomedical applications.
Results and discussion
As illustrated in Fig. 1a, bulk B powder can be effectively exfoliated to obtain ultrathin 2D B NSs via a modified liquid-phase method, which combines probe sonication and subsequent water bath sonication. Notably, unlike the isotropic characteristic of water bath sonication, the probe sonication performs a unique anisotropically processing effect. In detail, during probe sonication, the probe-induced ultrasonic waves produce oriented bubbles in the vertical direction, then the B block distributed around the probe is preferentially cut into slices by the ultrasonic wave in the vertical direction. Through this procedure, the 3D B block can be effectively transformed to 2D B slices with large lateral sizes and thicknesses. Subsequently, along with the water bath sonication treatment, the size and thickness of the 2D B slices can be greatly reduced and exfoliated to obtain B nanosheets. Since the transfer direction of ultrasonic waves in this process is isotropic, the uniform size and thickness of the sheet-like product can be guaranteed. Finally, the ultrathin B NSs with uniform size can be easily obtained through centrifugation treatment with speed control.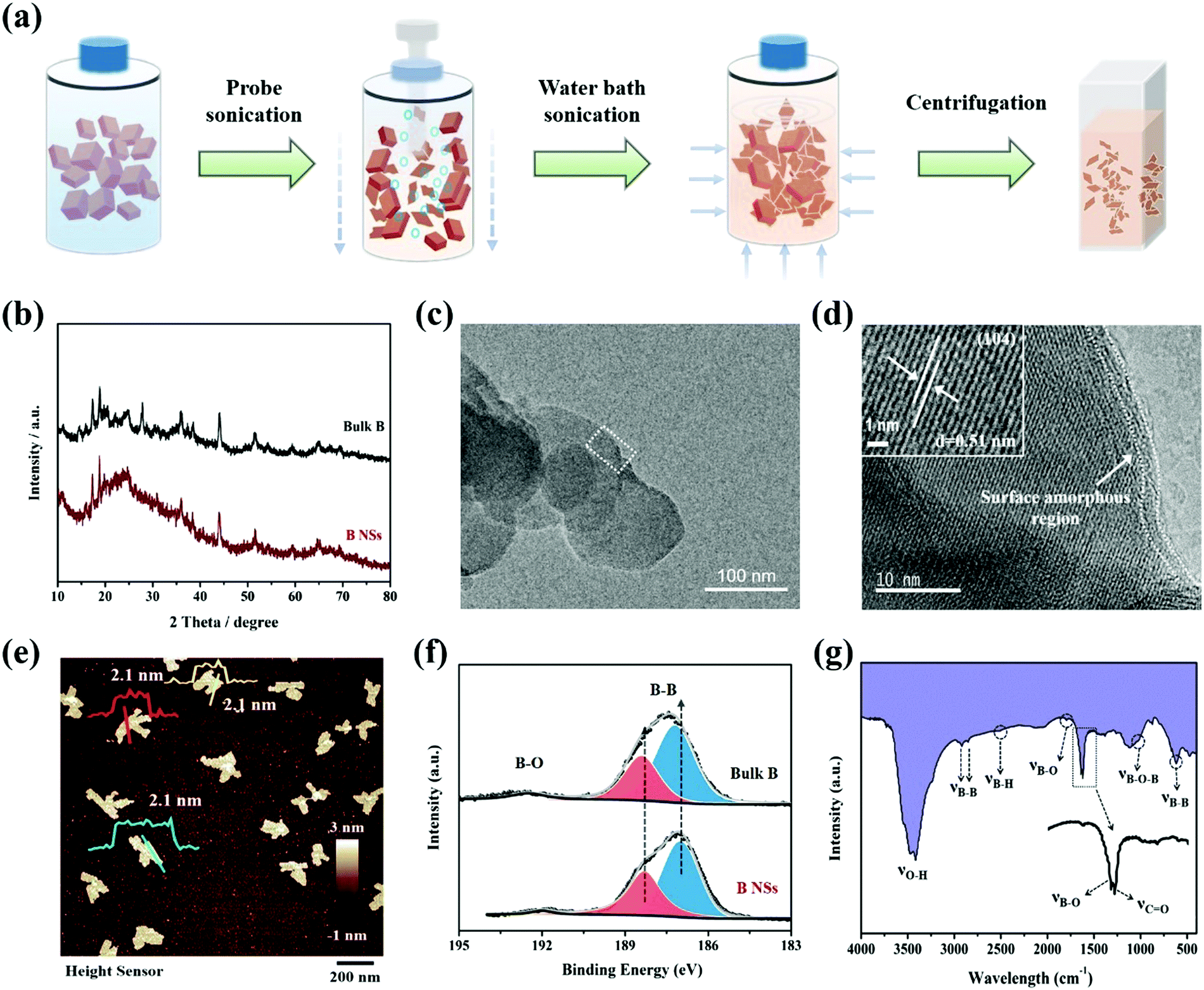 | ||
| Fig. 1 (a) The illustration of the 2D ultrathin B NSs prepared by a modified liquid-phase exfoliation process. (b) The XRD patterns of bulk B and ultrathin B NSs. (c) The TEM and (d) corresponding HRTEM images of ultrathin B NSs. (e) The AFM image of B NSs. (f) The XPS spectra for bulk B and ultrathin B NSs in the B 1s region. (g) The FTIR spectrum of ultrathin B NSs. | ||
Fig. 1b shows the X-ray diffraction (XRD) patterns of the bulk B and exfoliated B NSs. Apart from the weakening and broadening of several diffraction peaks that probably originate from the formation of structural defects during exfoliation, the two samples exhibited similar (003), (110), (104), (113), and (217) planes, which can be indexed to β-rhombohedral boron (JCPDS no. 11-0618).28,29,33,34 Compared with bulk B (Fig. S1, ESI†), Fig. S2 (ESI†) clearly shows that the exfoliated product exhibits small size dimensions whilst maintaining its 2D lamellar structure. In addition, the corresponding elemental mapping images also demonstrate the uniform distribution of the boron element. Moreover, the weak signal of the oxygen element could possibly correspond to air contamination and/or the surface oxygen-containing functional groups, and this will be further discussed herein. The TEM and corresponding HRTEM images of B NSs are respectively displayed in Fig. 1c and d. It is clear that the high crystalline nature of B NSs can be confirmed, and a clear lattice fringe spacing of ∼0.51 nm is in good agreement with the (104) plane of boron. However, an amorphous layer on the edge of the B NSs can be also detected, which can be ascribed to the surface defects formed during the exfoliating process. The Raman spectra was obtained and is represented in Fig. S3 (ESI†). Specifically, the bulk B and ultrathin B NSs exhibited similar multiple Raman peaks, and the peaks located at 436 cm−1, 636.5 cm−1, 801.8 cm−1, 1052 cm−1, and 1153 cm−1 correspond to the B13g, A1g, A2g, B11g, and A1g vibration modes, respectively, and this is consistent with the previous result of the β-type boron structure.35 In addition, an AFM test was conducted to characterize the thickness of the B NSs. As shown in Fig. 1e and Fig. S4 (ESI†), the average thickness was calculated to be 2.1 nm, with a predominant size distribution of 82 nm (Fig. S5, ESI†).
In order to understand the influence of exfoliation on the formation of surface defects of ultrathin B NSs, the surface chemical compositions of the bulk B and the as-prepared ultrathin B NSs were characterized by X-ray photoelectron spectroscopy (XPS). For their full spectrums, no obvious change was detected apart from a small amount of N species in the ultrathin B NSs, which comes from surface contamination (Fig. S6, ESI†). As can be seen in the high-resolution B 1s XPS region (Fig. 1f), two obvious peaks for both the bulk B and ultrathin B NSs are shown. Specifically, the weak peak located at high binding energy corresponds to B–O bonding, whilst the strong peak at low binding energy is attributed to B–B bonding. Here, the peaks of B–B and B–O bonding can be fit using the asymmetric and symmetric Gaussian–Lorentzian functions, respectively. Then, three peaks resolved at 187.3, 188.4, and 190.4 eV for bulk B, and 187, 188.2, and 190 eV for the ultrathin B NSs could be obtained, respectively. Notably, the B–B bonding of ultrathin B NSs shows a blue-shift of approximately 0.3 eV relative to that of bulk B noted above, and this is consistent with previous works.29,33 However, the presence of B–O bonding can be assigned to the interaction between the ultrathin B NSs with oxygen-containing functional groups, coupled with the observations in the HRTEM. On the other hand, Fourier transform infrared (FTIR) spectroscopy was also employed to reveal the structure of the ultrathin B NSs. As shown in Fig. 1g, several stretching vibrations including O–H at 3450 cm−1, B–B at 2920, 2840 and 617 cm−1, B–H at 2500 cm−1, B–O at 1780 and 1642 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1640 cm−1, and B–O–B at around 1020 cm−1 can be detected.36 Among them, the B–O and B–O–B vibrations are induced by the edge oxidization, and this can compensate for the electron deficiency of B NSs during exfoliation. In combination with the XPS analysis, the existence of oxygen-containing functional groups modified at the edges of the ultrathin B NSs after exfoliation can be confirmed.
O at 1640 cm−1, and B–O–B at around 1020 cm−1 can be detected.36 Among them, the B–O and B–O–B vibrations are induced by the edge oxidization, and this can compensate for the electron deficiency of B NSs during exfoliation. In combination with the XPS analysis, the existence of oxygen-containing functional groups modified at the edges of the ultrathin B NSs after exfoliation can be confirmed.
The optical properties of ultrathin B NSs were investigated by UV-vis absorption spectroscopy and fluorescence spectroscopy. Fig. 2a shows that the UV-vis absorption spectrum of the ultrathin B NSs exhibited two absorption bands (black line). Among them, the broad peak located at 240–300 nm (red area) is related to the formation of defects on the edges of the ultrathin B NSs, while the other absorption band at 350–400 nm (blue area), in particular at 356, 375, and 395 nm, can be assigned to a π electron transition at the edges of the oxygen-containing ultrathin B NSs, due to its high interaction with the solvent (IPA). A blue luminescence emission can be observed by the excitation of ultrathin B NSs by a 365 nm wavelength of light, shown in the inset. The fluorescence emission spectroscopy results show that the maximum emission occurs at 408 and 433 nm with a shoulder peak, and this may indicate the presence of multi active luminescence centers. Such a phenomenon is also similar with that of the boron nitride system that has been reported previously.9–11Fig. 2b shows that the ultrathin B NSs exhibit a similar absorption feature at a wide range of wavelengths from 200 nm to 800 nm at different concentrations. In addition, Fig. 2c reveals that the absorbance of the dispersed solution under different concentrations exhibits a linear relationship at 370 nm, and this is indicative of the good dispersion of the ultrathin B NSs in IPA. Moreover, the absorption coefficient (α) of 1964 L g−1 m−1 can also be calculated according to the Lambert–Beer law. Fig. S7 (ESI†) shows the comparison of the fluorescence properties of the bulk B and ultrathin B NSs, as well as the pure IPA solvent. Except for the peak at 415 that corresponds to the IPA solvent, several weak emission peaks located at 408, 439, 454, 468, 537, 574, and 626 nm were clearly detected for bulk B. However, unlike in the bulk form, two strong emission peaks with a shoulder peak were observed in the range of 400–550 nm for the ultrathin B NSs. Interestingly, unlike the excitation-dependent fluorescence of IPA (Fig. S8, ESI†), the ultrathin B NSs exhibited excitation-independent fluorescence (Fig. 2d). In particular, the emission intensity reached a maximum when excited at a wavelength of 370 nm (Fig. S9, ESI†). In addition, no other obvious emission peaks can be detected when the excitation wavelength was extended from 500 to 800 nm (Fig. S10, ESI†). As shown in Fig. S11 (ESI†), the fluorescence intensity of the ultrathin B NSs was extremely stable even after irradiation with a UV lamp for 100 min. To further evaluate the photostability of the B NSs, we compared it with a known dye, rhodamine B, under irradiation with a specific power of 300 W (Fig. S12, ESI†). The corresponding results indicated that the B NSs exhibited a superior stability with a retention of 94.1% after 50 min, which is much higher than that of 58.4% for rhodamine B. Consequently, together with the characterizations and the discussions above, the possible structure of the luminescent ultrathin B NSs is given in Fig. 2e. On the one hand, strong quantum confinement effects might be induced in the exfoliated B NSs due to their ultrathin thickness. On the other hand, the formation of a defects layer modified with chemical functional groups on the edges of the ultrathin B NSs could produce a π electron transition. In this way, we propose that such a fluorescence mechanism of ultrathin B NSs will originate from the synergistic effect of the quantum confinement effect and surface defects, although the exact mechanism responsible for the luminescence still needs to be elucidated.37–41
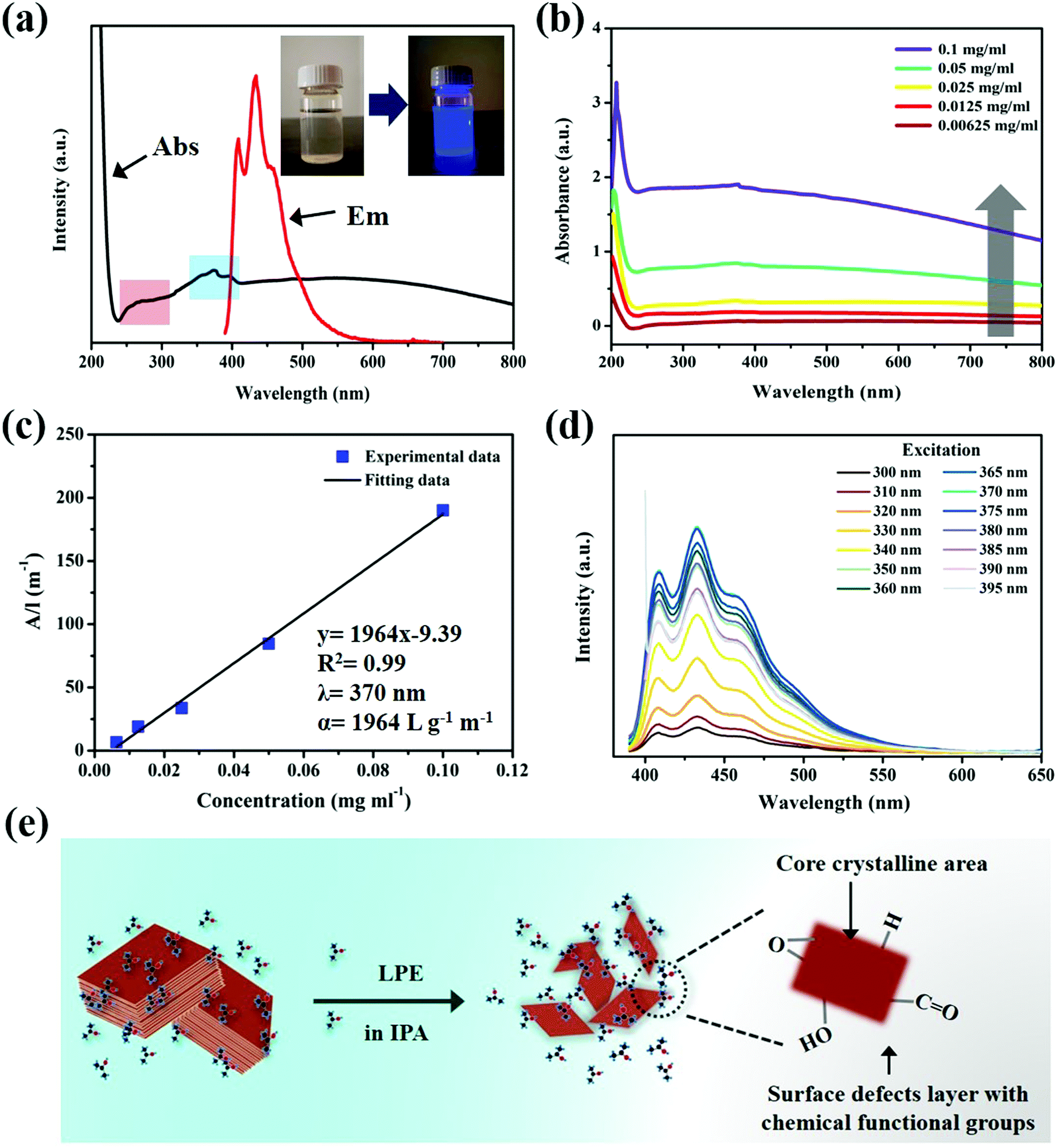 | ||
| Fig. 2 (a) UV-vis absorption and fluorescence emission spectra (the inset photograph is the ultrathin B NSs/IPA dispersion before and after irradiation with a 365 nm UV lamp). (b) UV-vis spectra for ultrathin B NSs/IPA dispersion at various concentrations of 0.1, 0.05, 0.025, 0.0125, and 0.00625 mg ml−1. (c) Absorbance at 370 nm plotted against concentration, showing a nearly linear manner. (d) Fluorescence spectra of the ultrathin B NSs/IPA dispersion under excitation at various wavelengths. (e) The possible structure of the fluorescent ultrathin B NSs. | ||
As well as the influence of the excitation wavelength on the fluorescence, the case of the dispersion concentration is also discussed. Fig. 3a shows the fluorescence spectra of the ultrathin B NSs at different concentrations of 0.2, 0.1, 0.05, 0.025, 0.0125, 6.25 × 10−3, and 3.125 × 10−3 mg ml−1. In particular, the moderate concentration (0.025 mg ml−1) exhibited the highest emission intensity, while an extremely high concentration or an extremely low concentration decreased the corresponding intensity, especially when the values were larger than 0.1 mg ml−1. Moreover, the quantum yield (QY) of the ultrathin B NSs was also measured by the Quantaurus-QY system. As shown in the inset of Fig. 3b, a clear Tyndall effect was observed for all the samples with different concentrations, and the corresponding QY values maximally reached 10.6% at a concentration of 6.25 × 10−3 mg ml−1. Such a value is higher than that of the Au nanoclusters, MoS2 QDs, WS2, and of Ti3C2 MXenes QDs, and is even comparable with that of graphene QDs (see Table S1, ESI†). Under irradiation with a 365 nm UV lamp, the samples at lower concentrations of 0.025 and 6.25 × 10−3 mg ml−1 exhibited strong luminescence, compared with that at higher concentration, and this is consistent with the above results (Fig. 3c). As shown in Fig. S13 (ESI†), such a phenomenon may be attributed to the follow reason, which is that the ultrathin B NSs can guarantee a state of uniform dispersion when at a low concentration. However, such a balance would be broken with an increase in concentration, since the ultrathin B NSs would inevitably agglomerate, leading to an increase in size and thickness of the B NSs, as well as a reduction in the surface defects area. In this case, the strong luminescence characteristic would be weakened.
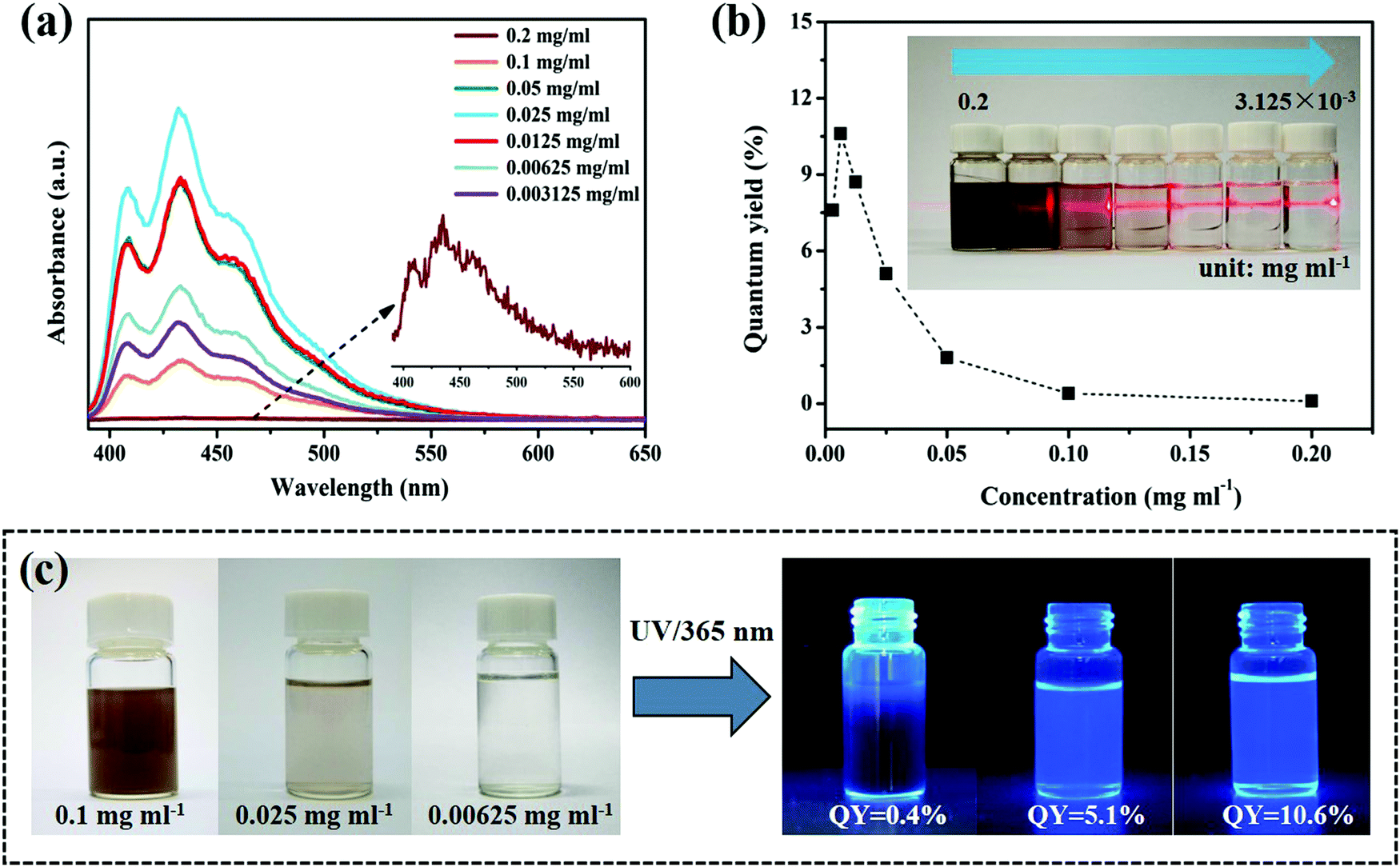 | ||
| Fig. 3 (a) Fluorescence spectra of the ultrathin B NSs with different concentrations excited at 370 nm. (b) The relationship between the concentration of ultrathin B NSs dispersion and the corresponding quantum yield. (c) The fluorescence behavior of ultrathin B NSs dispersion with a concentration of 0.1, 0.025, 6.25 × 10−3 and 3.125 × 10−3 mg ml−1, respectively, under irradiation with a 365 nm UV lamp. | ||
Femtosecond transient absorption (TA) spectroscopy was applied to investigate the ultrafast exciton dynamics of the ultrathin B NSs, in which the photons with a high energy of 3.65 eV (340 nm) were used to excite the electrons from the ground state to the high energy state. The absorption difference of the probe light versus the pump–probe delay time and probe wavelength were calculated according to ΔA = Aex − A00, where Aex and A00 represent the absorption of the probe light with and without the pump light, respectively. The excitation and cooling processes of the photo-induced carriers could be obtained, and this could reveal the photo-physical properties of the free carriers,42 the excitons,43 and of the plasmons.44 A distinct phenomenon of a photoinduced bleach (PIB) signal at short wavelength (380–500 nm) and a photoinduced absorption (PIA) signal at long wavelength (500–640 nm) in the TA spectrum was clearly observed, as shown in Fig. 4a. Generally, the negative PIB signal corresponded to the band filling induced ground state bleaching or stimulated emission, while the positive PIA signal corresponded to the absorbance of excited states and new photoproducts.45 Based on these principles, the PIB signal here corresponded to stimulated emissions due to the observation of fluorescence in the ultrathin B NSs.
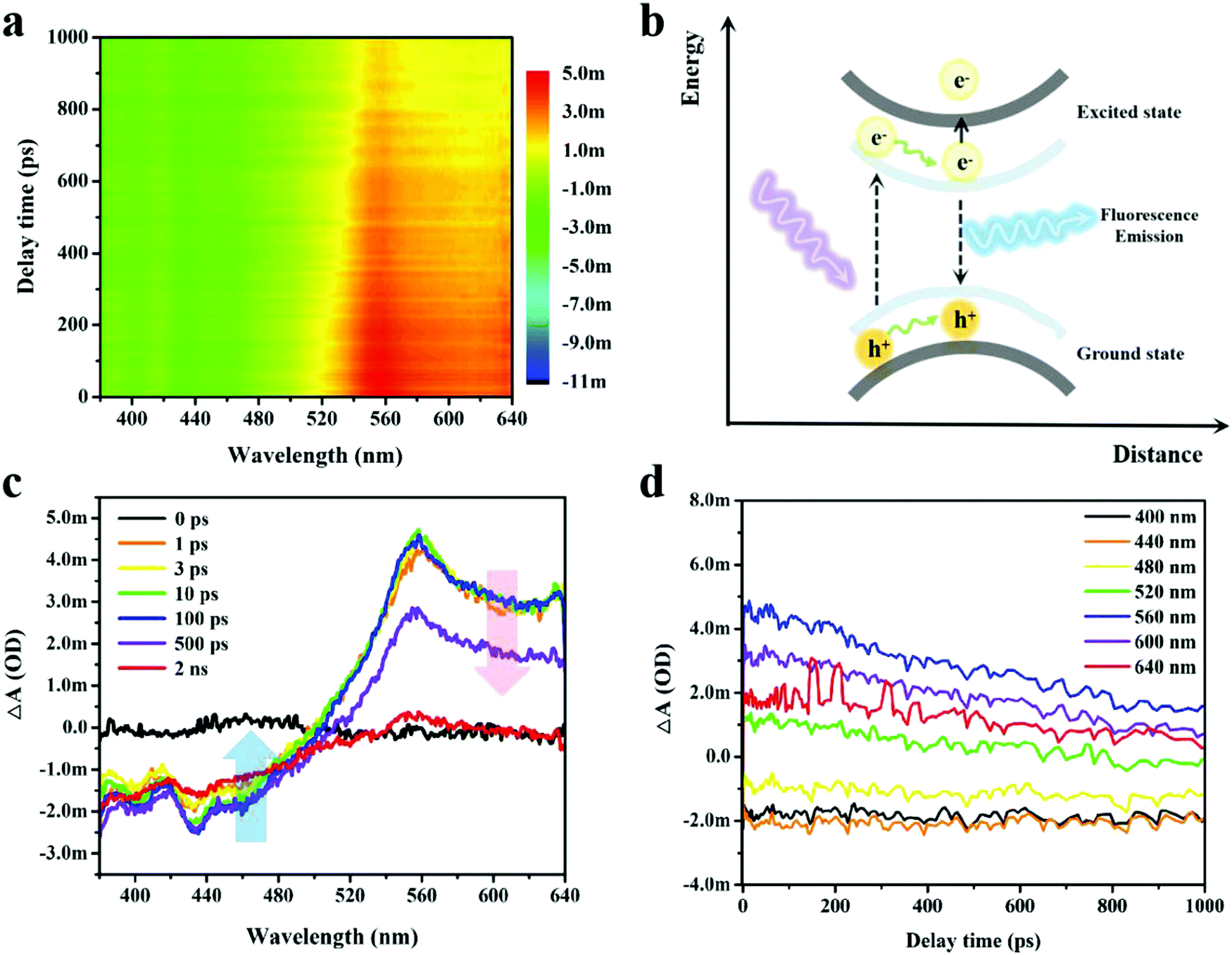 | ||
| Fig. 4 Transient absorption signals of the ultrathin B NSs measured by femtosecond transient optical absorption spectroscopy. (a) Temporally and spectrally resolved transient absorption signals. (b) Schematic diagram of the carrier transfer process in ultrathin B NSs. (c) Transient absorption spectra at various delay times. (d) The kinetic curves at various wavelengths. | ||
The electron–hole pairs underwent separation by the excitation of the tested sample under irradiation of pump light. Then, the electrons were able to transit from the ground state to the excited state and this could be followed by other subsequent possible activities (Fig. 4b). On the one hand, the excited electrons relaxed to their ground state with light emission, while on the other hand, a second transition of the excited electrons to high energy levels may have occurred. Fig. 4c shows the representative TA spectra measured at different probe delay times ranging from 0 to 2 ns. Notably, the lifetime of the PIB signal was beyond the range of the optical delay time (2 ns) for our setup, indicating the effective separation of the electron–hole pairs with subsequent strong fluorescence emission, and this was also consistent with our analysis above. Contrastingly, the PIA signal exhibited a shorter lifetime as the ΔA decayed to zero within 2 ns. In addition, the long-lived trend of PIB and the rapid cooling of PIA were further verified by the comparison of carrier dynamic curves selected at a certain wavelength (Fig. 4d). To better decompose and analyze the TA spectrum, singular-value decomposition and global lifetime time strategies were employed (Fig. S14, ESI†),46 whilst three principal time constants of τ1 = 63.6 ps, τ2 = 337.8 ps and τ3 = 1465 ps were obtained, and the deposited signals complied with aforementioned TA results.
In light of the above-mentioned favorable luminescence characteristics, ultrathin B NSs hence demonstrated great potential for application as fluorescent probes. The inherent cytotoxicity of the ultrathin B NSs was evaluated using a CCK-8 viability assay with the cervical cancer cell line HeLa and the hepatocarcinoma cell line Huh-7, and the corresponding viabilities of these two cells were measured after incubating them with the ultrathin B NSs for 24 h. Both of them showed high cell viability for incubation with ultrathin B NSs even with a high concentration of 400 μg ml−1 (Fig. 5). In particular, compared to Huh-7 cells, HeLa cells exhibited a higher survival rate of more than 90%. Thus, such a low cytotoxicity also confirms the excellent biocompatibility and low toxicity of the ultrathin B NSs, making them definitely suitable for bioimaging applications.
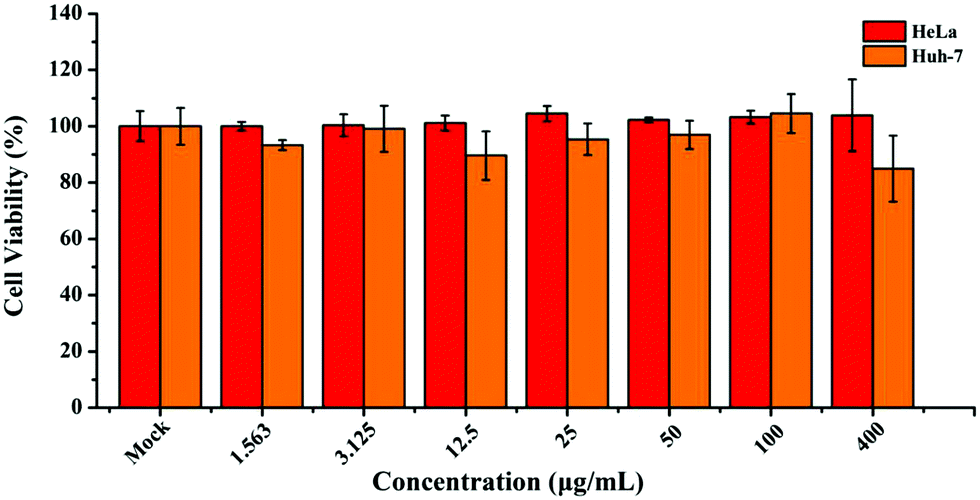 | ||
| Fig. 5 Cell viability of the HeLa and Huh-7 cells after incubation with different concentrations of ultrathin B NSs for 24 h. | ||
Benefiting from high quantum yield and high photostability, as well as from high biocompatibility and low toxicity, ultrathin B NSs were further applied for cellular imaging. After incubation with ultrathin B NSs for 4 h, bright-field images of HeLa cells and Huh-7 cells are shown in Fig. 6a and d, respectively. The observations in their corresponding overlay images (Fig. 6b and e) indicate that the ultrathin B NSs were mainly distributed in the cytoplasm of the cells. In order to prove this, we further adopted a marker dye of the lysosome to detect the precise location of the ultrathin B NSs in HeLa cells (Fig. S15, ESI†). It was clear that most of the ultrathin B NSs co-localized well with the Lyso-Tracker marker, thus demonstrating it in the HeLa cells through the endocytosis pathway.47 In addition, the confocal images (Fig. 6c and f) show a bright blue light emission upon excitation at 405 nm, while no apparent signal was detected for the control cells (Fig. S16, ESI†). Therefore, our results demonstrated the ultrathin B NSs to be a promising biolabeling material, which can be further utilized for biomedical applications in future research.
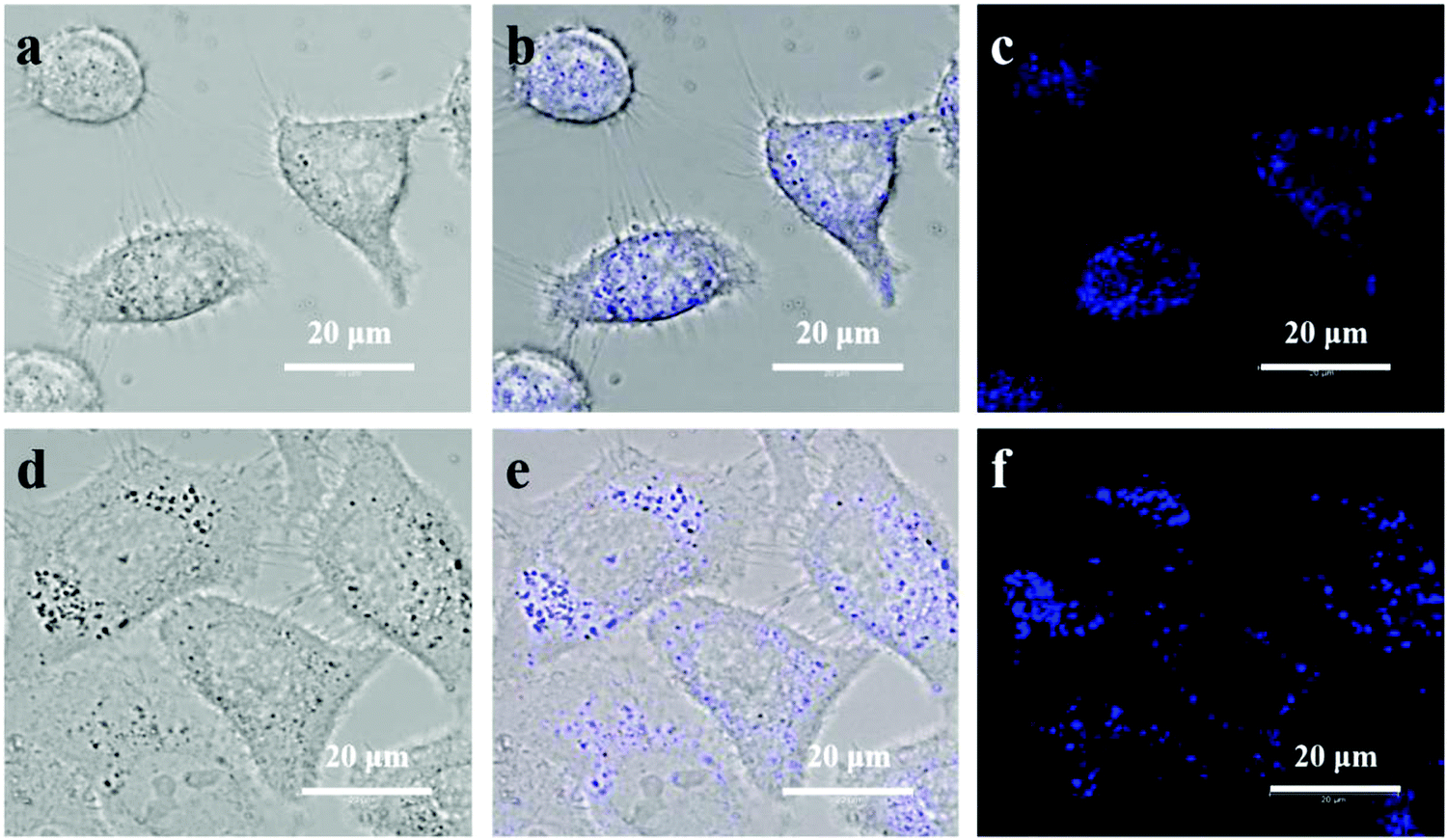 | ||
| Fig. 6 The bright field images with corresponding overlay images of the (a and b) HeLa cells, and (d and e) Huh-7 cells. Confocal laser scanning microscopy images of the (c) HeLa cells and (f) Huh-7 cells, incubated with the ultrathin B NSs for about 4 h. | ||
Conclusions
In summary, we developed a facile, high-output method for fabricating ultrathin B nanosheets with an average thickness of ∼2.1 nm and a lateral size of ∼82 nm. The systematic characterizations demonstrated that the as-exfoliated ultrathin B nanosheets maintained good structural integrity, whilst forming a surface defect layer at the edge connected with chemical functional groups. Due to the synergistic effect of the quantum confinement effect and surface defects, the ultrathin B nanosheets exhibited strong fluorescence with excitation-independent characteristics, as well as many striking luminescence features including a high quantum yield (as high as 10.6%) and high photostability. Furthermore, we also demonstrated that such ultrathin B nanosheets have high biocompatibility and low toxicity. Based on these premises, ultrathin B nanosheets as an emerging fluorescent probe applied for cellular bioimaging was successfully revealed here. It is anticipated that this work will not only provide a high-efficient strategy to prepare ultrathin B nanosheets, but it will also broaden the practical application of ultrathin B nanosheets, thus stimulating the rapid development of other elemental 2D materials.The statement of author contributions
D. Ma and J. Zhao conceived the idea, designed all the experiments, and wrote the manuscript. D. Ma, J. Zhao, and R. Wang performed the measurements. J. Xie and W. Liang carried out the bioimaging experiment. F. Zhang, L. Wu, D. Li, and Y. Ge analyzed the experimental data. J. Li, Y. Zhang and H. Zhang revised the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is supported by the State Key Research Development Program of China (Grant No. 2019YFB2203503), National Natural Science Fund (Grant No. 61875138, U1801254, and 61961136001), the Science and Technology Planning Project of Guangdong Province (Grant No. 2016B050501005), the Science and Technology Innovation Commission of Shenzhen (Grant No. KQTD2015032416270385), the Student Innovation Development Foundation of Shenzhen University (PIDFP-ZR2017002), and the Science and Technology Development Fund (Grant No. 007/2017/A1), Macao SAR, China.References
- W. Tao, N. Kong, X. Ji, Y. Zhang, A. Sharma, J. Ouyang, B. Qi, J. Wang, N. Xie, C. Kang, H. Zhang, O. Farokhzad and J. Kim, Chem. Soc. Rev., 2019, 48, 2891 RSC.
- J. Du, Z. Gu, L. Yan, Y. Yong, X. Yi, X. Zhang, J. Liu, R. Wu, C. Ge, C. Chen and Y. Zhao, Adv. Mater., 2017, 29, 1701268 CrossRef PubMed.
- T. Xue, W. Liang, Y. Li, Y. Sun, Y. Xiang, Y. Zhang, Z. Dai, Y. Duo, L. Wu, K. Qi, B. Shivananju, L. Zhang, X. Cui, H. Zhang and Q. Bao, Nat. Commun., 2019, 10, 28 CrossRef CAS.
- S. He, J. Song, J. Qu and Z. Cheng, Chem. Soc. Rev., 2018, 47, 4258 RSC.
- R. Chandan, J. Schiffman and R. Balakrishna, Sens. Actuators, B, 2018, 258, 1191 CrossRef.
- D. Wang, J. Chen and L. Dai, Part. Part. Syst. Charact., 2015, 32, 515 CrossRef CAS.
- J. Ren, F. Weber, F. Weigert, Y. Wang, S. Choudhury, J. Xiao, I. Lauermann, U. Genger, A. Bande and T. Petit, Nanoscale, 2019, 11, 2056 RSC.
- M. An, J. Wu, P. Wang and Y. Fang, Appl. Phys. Lett., 2019, 114, 022102 CrossRef.
- S. Angizi, A. Hatamie, H. Ghanbari and A. Simchi, ACS Appl. Mater. Interfaces, 2018, 10, 28819 CrossRef CAS PubMed.
- H. Li, R. Tay, S. Tsang, X. Zhen and E. Teo, Small, 2015, 11, 6491 CrossRef CAS PubMed.
- L. Lin, Y. Xu, S. Zhang, I. Ross, A. Ong and D. Allwood, Small, 2014, 10, 60 CrossRef CAS PubMed.
- P. Kumbhakar, A. Kole, C. Tiwary, S. Biswas, S. Vinod, J. Tijerina, U. Chatterjee and P. Ajayan, Adv. Opt. Mater., 2015, 3, 828 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- M. Zeng, Y. Xiao, J. Liu, K. Yang and L. Fu, Chem. Rev., 2018, 13, 6236 CrossRef PubMed.
- A. Mannix, Z. Zhang, N. Guisinger, B. Yakobson and M. Hersam, Nat. Nanotechnol., 2018, 13, 444 CrossRef CAS PubMed.
- X. Liu, Z. Zhang, L. Wang, B. Yakobson and M. Hersam, Nat. Mater., 2018, 17, 783 CrossRef CAS PubMed.
- G. Qin, A. Du and Q. Sun, Phys. Chem. Chem. Phys., 2018, 20, 16216 RSC.
- N. Gao, X. Wu, X. Jiang, Y. Bai and J. Zhao, FlatChem, 2018, 7, 48 CrossRef CAS.
- G. Tai, T. Hu, Y. Zhou, X. Wang, J. Kong, T. Zeng, Y. You and Q. Wang, Angew. Chem., Int. Ed., 2015, 54, 15473 CrossRef CAS PubMed.
- X. Liu, L. Wang, S. Li, M. S. Rahn, B. I. Yakobson and M. C. Hersam, Nat. Commun., 2019, 10, 1642 CrossRef PubMed.
- Z. Zhang, E. S. Penev and B. I. Yakobson, Nat. Chem., 2016, 8, 525 CrossRef CAS PubMed.
- R. Wu, I. Drozdov, S. Eltinge, P. Zahl, S. Beigi, I. Bozovic and A. Gozar, Nat. Nanotechnol., 2019, 14, 44 CrossRef CAS PubMed.
- A. Mannix, X. Zhou, B. Kiraly, J. Wood, D. Alducin, B. Myers, X. Liu, B. Fisher, U. Santiago, J. Guest, M. Yacaman, A. Ponce, A. Oganov, M. Hersam and N. Guisinger, Science, 2015, 350, 1513 CrossRef CAS PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563 CrossRef CAS PubMed.
- V. Nicolosi, M. Chhowalla, M. Kanatzidis, M. Strano and J. Coleman, Science, 2013, 340, 1226419 CrossRef.
- L. Niu, J. Coleman, H. Zhang, H. Shin, M. Chhowalla and Z. Zheng, Small, 2016, 12, 272 CrossRef CAS PubMed.
- W. Zhang, Y. Hu, L. Ma, G. Zhu, P. Zhao, X. Xue, R. Chen, S. Yang, J. Ma, J. Liu and Z. Jin, Nano Energy, 2018, 53, 808 CrossRef CAS.
- Q. Guo, K. Wu, Z. Shao, E. Basore, P. Jiang and J. Qiu, Adv. Opt. Mater., 2019, 1900322 CrossRef.
- H. Li, L. Jing, W. Liu, J. Lin, R. Tay, S. Tsang and E. Teo, ACS Nano, 2018, 12, 1262 CrossRef CAS PubMed.
- H. Zhong, K. Huang, G. Yu and S. Yuan, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 98, 054104 CrossRef CAS.
- Z. Wang, T. Lu, H. Wang, Y. Feng and J. Zheng, Front. Phys., 2019, 14, 33403 CrossRef.
- W. Li, L. Kong, C. Chen, J. Gou, S. Sheng, W. Zhang, H. Li, L. Chen, P. Cheng and K. Wu, Sci. Bull., 2018, 63, 282 CrossRef CAS.
- X. Ji, N. Kong, J. Wang, W. Li, Y. Xiao, S. Gan, Y. Zhang, Y. Li, X. Song, Q. Xiong, S. Shi, Z. Li, W. Tao, H. Zhang, L. Mei and J. Shi, Adv. Mater., 2018, 1803031 CrossRef PubMed.
- P. Ranjan, T. Sahu, R. Bhushan, S. Yamijala, D. Late, P. Kumar and A. Vinu, Adv. Mater., 2019, 1900353 CrossRef PubMed.
- S. Sheng, J. Wu, X. Cong, Q. Zhong, W. Li, W. Hu, J. Gou, P. Cheng, P. Tan, L. Chen and K. Wu, ACS Nano, 2019, 13, 4133 CrossRef CAS PubMed.
- F. Ghezzi, L. Laguardia, R. Caniello, A. Canton, S. D. Bello, B. Rais and M. Anderle, Appl. Surf. Sci., 2015, 354, 408 CrossRef CAS.
- Q. Xue, H. Zhang, M. Zhu, Z. Pei, H. Li, Z. Wang, Y. Huang, Y. Huang, Q. Deng, J. Zhou, S. Du, Q. Huang and C. Zhi, Adv. Mater., 2017, 1604847 CrossRef PubMed.
- X. Zhang, H. Wang, H. Wang, Q. Zhang, J. Xie, Y. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438 CrossRef CAS.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356 CrossRef CAS PubMed.
- S. Zou, C. Liu, R. Li, F. Jiang, X. Chen, Y. Liu and M. Hong, Adv. Mater., 2019, 1900606 CrossRef PubMed.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1603443 CrossRef PubMed.
- J. He, D. He, Y. Wang, Q. Cui, M. Bellus, H. Chiu and H. Zhao, ACS Nano, 2015, 9, 6436 CrossRef CAS PubMed.
- L. Wibmer, S. Lages, T. Unruh and D. Guldi, Adv. Mater., 2018, 30, 1706702 CrossRef.
- R. Sato, S. Ishii, T. Nagao, M. Naito and Y. Takeda, ACS Photonics, 2018, 5, 3452 CrossRef CAS.
- C. Ruckebusch, M. Sliwa, P. Pernot, A. Juan and R. Tauler, J. Photochem. Photobiol., C, 2012, 13, 1 CrossRef CAS.
- F. Zhang, K. Chen, X. Jiang, Y. Wang, Y. Ge, L. Wu, S. Xu, Q. Bao and H. Zhang, J. Mater. Chem. C, 2018, 6, 8977 RSC.
- W. Tao, X. Zhu, X. Yu, X. Zeng, Q. Xiao, X. Zhang, X. Ji, X. Wang, J. Shi, H. Zhang and L. Mei, Adv. Mater., 2017, 29, 1603276 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00698b |
| ‡ D. Ma, J. Zhao and J. Xie contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |