
Endosome/lysosome-detained supramolecular nanogels as an efflux retarder and autophagy inhibitor for repeated photodynamic therapy of multidrug-resistant cancer†
Xiaodong
Zhang‡
,
Xiaokai
Chen‡
,
Yuxin
Guo
,
Hao-Ran
Jia
,
Yao-Wen
Jiang
and
Fu-Gen
Wu
 *
*
State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, China. E-mail: wufg@seu.edu.cn
First published on 22nd October 2019
Abstract
The presence of drug efflux pumps and endo/lysosomal entrapment phenomena in multidrug-resistant cancer cells leads to insufficient and off-target accumulation of anticancer drugs in the cells, which severely reduces the drugs’ therapeutic efficacies. Here, we prepare a novel type of photosensitizer (PS)-loaded supramolecular nanogel, which can utilize the endo/lysosomal entrapment for enhanced photodynamic therapy (PDT) of multidrug-resistant cancer. The PS-loaded nanogels can elude the drug efflux pumps, and be markedly internalized by drug-resistant cancer cells through the endocytic pathway. With their pH-sensitive properties, the internalized nanogels can aggregate in the acidic endosomes/lysosomes, thus retarding their exocytosis from the cells. Moreover, the lysosomes of the nanogel-treated cells are severely damaged after irradiation, which inhibits the protective autophagy and improves the photodynamic therapeutic performance of the nanogels. Besides, the in vivo experiments show that the nanogels significantly prolong the tumor retention of the PSs, thus enabling multiple PDT treatments after a single drug injection.
New conceptsWhen small molecule drugs and nanoparticulate drugs enter cancer cells, they are usually trapped in endosomes and/or lysosomes. The endo/lysosomal entrapment will significantly weaken the therapeutic efficacies of these drugs, because it hinders their transportation to other target organelles. Hence, previous efforts have been mainly devoted to realizing endo/lysosomal escape of drugs to enhance their anticancer efficacies. On the other hand, since the drug efflux pumps of multidrug-resistant cancer cells can rapidly pump small molecule drugs out of the cells, but not nanodrugs, utilizing nanoparticles as drug carriers is a practical way to overcome the drug uptake problem. However, the internalized small nanoparticles will still be quickly excreted from the cells through exocytosis, while large ones with slow exocytosis have poor passive tumor targeting ability, thus posing a “size paradox” problem for nanomedicines. To address the above-mentioned issues, we propose a strategy based on pH-sensitive and size-changeable nanogels, and such a strategy does not aim to avoid the endo/lysosomal entrapment problem but to utilize this entrapment phenomenon for retarding the efflux of nanodrugs from drug-resistant cancer cells. It is demonstrated that the nanogels can simultaneously realize increased drug uptake, reduced drug efflux, and inhibited autophagy for enhanced cancer therapy. |
Cancer is one of the leading causes of mortality for humans.1 Multidrug resistance (MDR), which can be found in over 90% of patients with metastatic cancers, is the main reason causing the failure of cancer chemotherapy.2,3 As a non-invasive cancer treatment modality, photodynamic therapy (PDT) involves the administration of photosensitizers (PSs) followed by the generation of toxic reactive oxygen species (ROS) under light irradiation.4–9 It is found that PDT can overcome cancer MDR when combined with chemotherapy,10–14 and even directly kill drug-resistant cancer cells.15,16
Nevertheless, there are still some limitations of PDT in fighting against cancer MDR: (1) Singlet oxygen (1O2), the main form of ROS generated by PSs, has a very short lifetime (half-life: 0.03–0.18 ms) and a limited diffusion distance (radius: <20 nm).17–19 To achieve satisfactory PDT outcomes, PSs should be accumulated in the cells. However, owing to the presence of ATP-binding cassette (ABC) transporters (e.g., P-glycoprotein, P-gp) in multidrug-resistant cancer cells,20–22 PSs, similar to other small molecule drugs (300–2000 Da), will be rapidly pumped out of the cells.23–25 To address this issue, various nanoparticles have been developed as robust drug carriers to help the small molecule drugs to evade the ABC transporters.26–28 However, the internalized small nanoparticles may still be quickly excreted from the cells through exocytosis,29 while the large ones with slow exocytosis have poor passive tumor targeting ability in vivo.30,31 Therefore, it is highly desirable to design size-changeable nanocarriers that are small in the blood but become large inside the cells. (2) Protective autophagy often takes place after sub-lethal PDT, leading to the weakened photodynamic therapeutic efficiency.32 Hence, lysosome-targeted PDT is considered as a potential way of circumventing autophagic protection through the damage of lysosomes.33 (3) Since rapid PS clearance from the tumor leads to ineffective PDT treatment, the long-term tumor accumulation of PSs should be achieved for repeated PDT after a single drug injection to improve the phototherapeutic performance of the PSs. Thus, it is ideal to integrate size changeability, lysosomal targeting property, and long-term tumor retention capability into one nanosystem.
On the other hand, it has been reported that small molecule drugs and nanoparticulate drugs are usually trapped in the endosomes and/or lysosomes of cancer cells after cellular internalization.34,35 The endo/lysosomal entrapment will significantly weaken the therapeutic efficacies of these drugs,36 because it hinders the transportation of the drugs to other target organelles (such as mitochondria and cell nuclei).37 To this end, several strategies have been proposed to realize the endo/lysosomal escape of anticancer drugs to enhance their therapeutic efficacies.38,39 However, few studies have utilized this endo/lysosomal entrapment phenomenon for improving the therapeutic performance of the drugs.
We have recently reported the fabrication of ultrabright organosilica nanodots (OSiNDs) that can aggregate in the acidic lysosomes for long-term lysosome imaging.40 Inspired by that work, we here prepared pH-sensitive nanogels via the supramolecular self-assembly of OSiNDs, the biocompatible copolymer methoxy-poly(ethylene glycol)113-block-poly(L-glutamic acid sodium salt)200 (abbreviated as PEG-PLE), and the PS called tetraphenylporphinesulfonate (TPPS) (Scheme 1). The TPPS-loaded nanogels could elude the drug efflux pumps and realize massive drug influx in multidrug-resistant cancer cells. Moreover, it was found that the small nanogels entered the cells through endocytosis, and then aggregated in the acidic endosomes/lysosomes to form larger particles, thus down-regulating their exocytosis from the cells. Besides, co-treatment of the nanogels and laser irradiation severely damaged the lysosomes, thereby inhibiting the protective autophagy and improving the PDT outcome. In addition, the in vivo experiments showed that the nanogels prolonged the retention of the PSs in a drug-resistant tumor, which was beneficial for the operation of repeated PDT with a single drug injection.
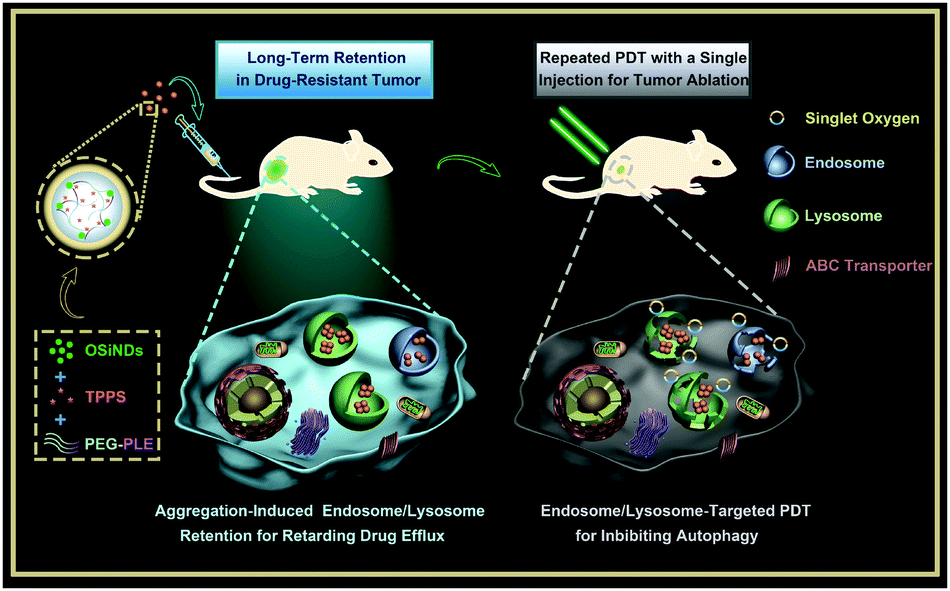 | ||
| Scheme 1 Schematic illustrating the preparation of the nanogels and their application for multiple PDT treatments of drug-resistant cancer. | ||
Ultrabright green fluorescent OSiNDs were synthesized by the hydrothermal treatment of rose bengal and 3-[2-(2-aminoethylamino)ethylamino]propyl-trimethoxysilane, which had an average diameter of ∼2.1 nm (Fig. S1, ESI†). After purification, the OSiND aqueous suspension was added into the solution containing PEG-PLE and TPPS (weight ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to obtain the TPPS-loaded nanogels. The size of the nanogels was tunable through changing the weight ratios of the three components. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images showed that the OSiNDs/PEG-PLE/TPPS weight ratios of 5:1:1, 8:1:1, and 10:1:1 resulted in the formation of nanogels with distinct sizes of 40 (termed SiPT40), 75 (termed SiPT75), and 185 nm (termed SiPT185), respectively (Fig. S2a–i, ESI†). The hydrodynamic (HD) sizes of the three nanogels revealed by dynamic light scattering (DLS) also indicated that the size of the nanogels increased with increasing OSiND contents. Besides, as measured by DLS, the low polydispersity index (PDI) values (<0.1) indicated that all the prepared nanogels had excellent monodispersity in aqueous solutions (Fig. S2j–l (ESI†) and Table 1).
:1) to obtain the TPPS-loaded nanogels. The size of the nanogels was tunable through changing the weight ratios of the three components. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images showed that the OSiNDs/PEG-PLE/TPPS weight ratios of 5:1:1, 8:1:1, and 10:1:1 resulted in the formation of nanogels with distinct sizes of 40 (termed SiPT40), 75 (termed SiPT75), and 185 nm (termed SiPT185), respectively (Fig. S2a–i, ESI†). The hydrodynamic (HD) sizes of the three nanogels revealed by dynamic light scattering (DLS) also indicated that the size of the nanogels increased with increasing OSiND contents. Besides, as measured by DLS, the low polydispersity index (PDI) values (<0.1) indicated that all the prepared nanogels had excellent monodispersity in aqueous solutions (Fig. S2j–l (ESI†) and Table 1).
| SiPT40 | SiPT75 | SiPT185 | |
|---|---|---|---|
| Diameter (nm) | 38.9 ± 9.8 | 75.5 ± 19.8 | 185.5 ± 46.9 |
| HD size (nm) | 60.8 ± 21.2 | 118.7 ± 39.9 | 212.2 ± 71.3 |
| ξ potential (mV) | −19.2 ± 5.8 | −15.6 ± 4.9 | −9.7 ± 4.4 |
| PDI | 0.095 | 0.084 | 0.082 |
| EE (%) | 46.3 ± 4.2 | 94.9 ± 2.3 | 98.6 ± 0.8 |
| LE (%) | 7.2 ± 0.6 | 9.5 ± 0.2 | 8.3 ± 0.1 |
Next, we compared the drug (TPPS) encapsulation efficiencies (EEs) of the three nanogels. For SiPT75 and SiPT185, their EEs were calculated to be ∼94.9% and ∼98.6%, respectively, which were much higher than that of SiPT40 (∼46.3%) (Table 1). The low EE of SiPT40 may stem from its smaller proportion of amine-rich OSiNDs that have strong interaction with negatively charged TPPS. Then the drug (TPPS) loading efficiencies (LEs) of the three nanogels were also calculated. Owing to its low OSiND proportion and high EE, SiPT75 had an LE of 9.5%, which was higher than those of SiPT40 (∼7.2%) and SiPT185 (∼8.3%). Furthermore, the TPPS release profiles of the three nanogels were recorded. The mixture of OSiNDs and TPPS could form aggregates in several seconds (as shown in Fig. S3, ESI†). Meanwhile, TPPS and PEG-PLE were assembled into nanofibers and other nanostructures (Fig. S4, ESI†). The above results indicated that TPPS had strong interaction with OSiNDs and PEG-PLE. As a result, after 48 h, only ∼25.2%, ∼15.1%, and ∼10.1% of the TPPS molecules were released from SiPT40, SiPT75, and SiPT185 in PBS (pH = 7.4), respectively (Fig. S5, ESI†). The low release rates of TPPS from the nanogels ensured the effectiveness and safety of the nanogels for further PDT.
Afterwards, the pH-sensitivity of the nanogels was evaluated. For SiPT75, the TEM image in Fig. 1a showed that the nanogels were well dispersed in the neutral medium (pH = 7.4). Interestingly, small aggregates composed of two or three nanoparticles were formed in a weakly acidic environment (pH = 6.0) (Fig. 1b), and the aggregates further clustered in a more acidic medium (pH = 4.5) (Fig. 1c). Similar results were observed in the TEM images of SiPT40 and SiPT185 (Fig. S6 and S7, ESI†). DLS results further proved that all three nanogels especially SiPT75 and SiPT185 could cluster in the pH range of 4.5–6.0 (Fig. 1d), suggesting that these nanogels can also gather in acidic organelles such as endosomes (pH 5.0–6.0) and lysosomes (pH 4.5–5.0). Then, the time- and concentration-dependent HD sizes of SiPT75 in solutions with different pH values were measured. As shown in Fig. S8a (ESI†), when the nanogels were dispersed in acidic solutions (pH = 4.5 and 6.0), the HD sizes of SiPT75 (TPPS concentration: 10 μg mL−1) increased rapidly at the beginning and then remained relatively constant until 24 h. Besides, it was found that when the nanogels were dispersed in acidic solutions (pH = 4.5 and 6.0) for 24 h, their HD sizes were larger than those of the nanogels dispersed in neutral solutions (pH = 7.4) even at a low TPPS concentration of 1 μg mL−1 (Fig. S8b, ESI†), indicating that the nanogels could easily form aggregates in acidic solutions. As shown in Table 1, the nanogels were negatively charged in the neutral environment (pH = 7.4), possibly due to the presence of negatively charged TPPS and PEG-PLE in the nanogels. Thus, in the acidic environment, the nanogels will be less charged and prone to aggregate. Besides, it has been demonstrated that OSiNDs can form aggregates in acidic solutions.40 Both of the above reasons may explain the low acidity-induced aggregation property of the nanogels.
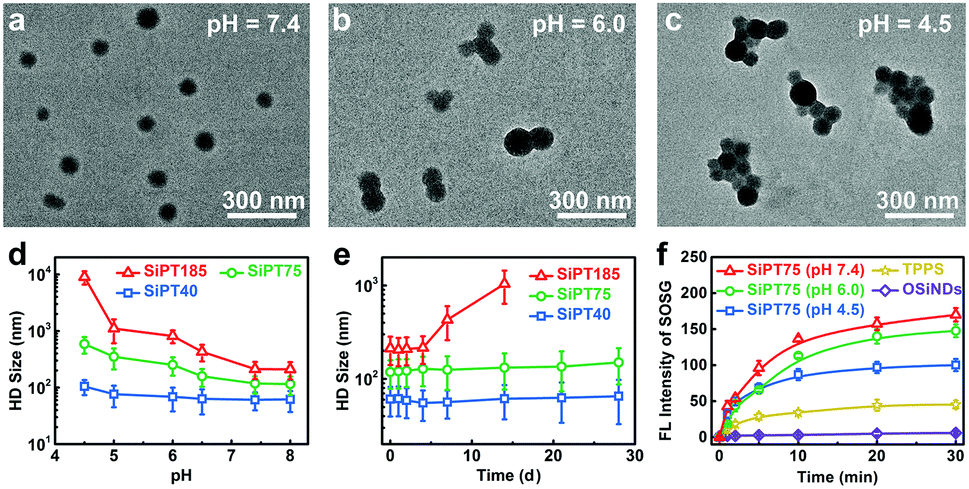 | ||
| Fig. 1 (a–c) TEM images of SiPT75 dispersed in various solutions with different pH values. (d) Effect of pH values on the HD sizes of SiPT40, SiPT75, and SiPT185. (e) Time-dependent changes of the HD sizes of SiPT40, SiPT75, and SiPT185 within 28 d. (f) Fluorescence (FL) intensities of SOSG showing the 1O2 generation of OSiNDs (pH = 7.4), TPPS (pH = 7.4), SiPT75 (dispersed in the solution with a pH value of 7.4 for 1 h), SiPT75 (dispersed in the solution with a pH value of 6.0 for 1 h), or SiPT75 (dispersed in the solution with a pH value of 4.5 for 1 h) after irradiation by a 532 nm laser (20 mW cm−2) for different time periods. | ||
Then, the stability of the nanogels in neutral solutions was evaluated. No apparent changes in the HD sizes of SiPT40 and SiPT75 dispersed in PBS solutions (pH = 7.4) were detected within 4 weeks when the TPPS concentration in the nanogels was at 10 μg mL−1 (Fig. 1e). Moreover, the HD size of SiPT75 remained relatively constant within 7 d in PBS, normal silane and serum-containing normal silane even when the TPPS concentration in the nanogels was up to 100 μg mL−1 (Fig. S9, ESI†), indicating their excellent colloidal stability in neutral solutions. For SiPT185, its HD size was notably increased in a week, possibly due to its larger size and less negatively charged surface (Table 1).
Owing to its good pH-responsibility and aqueous stability, SiPT75 was selected for further characterization. To elucidate its formation mechanism, three inhibitors including NaCl, urea, and Triton X-100 that can destroy the electrostatic interaction, hydrogen bonds, and hydrophobic interaction during the formation of the nanogels, respectively, were separately added to the mixtures of the three raw materials (OSiNDs, PEG-PLE, and TPPS) of the nanogels. The results in Fig. S10 (ESI†) revealed that all three inhibitors could prevent the formation of the nanogels, indicating that the above-mentioned three interactions played indispensible roles in the formation of the nanogels. Under irradiation at 488 and 552 nm, SiPT75 could emit strong fluorescence with the corresponding emission peaks residing at 525 and 648 nm (Fig. S11, ESI†), which were from OSiNDs and TPPS, respectively. Besides, the 1O2 generation abilities of free TPPS and SiPT75 were compared under 532 nm laser irradiation. Compared with free TPPS, SiPT75 could generate more 1O2 under both neutral and acidic conditions when they were dispersed in the solutions for 1 h, as measured by a singlet oxygen sensor green (SOSG) kit (Fig. 1f) and electron paramagnetic resonance spectroscopy (Fig. S12, ESI†). Because the HD sizes of SiPT75 remained relatively constant during the period of 2–24 h (Fig. S8a, ESI†), 24 h was also selected as another time point to evaluate the 1O2 generation ability of the nanogels. As shown in Fig. S13 (ESI†) and Fig. 1f, although the 1O2 generation abilities of the nanogels in acidic solutions (pH = 4.5 and 6.0) at 24 h were weaker than those at 1 h, they were still stronger than that of free TPPS. Similar results have been reported in several other studies, in which PSs loaded in nanogels or hydrogels can generate more 1O2 than those dispersed in aqueous solutions.41–43 This is possibly attributed to the decreased self-quenching effect of the PSs in nanogels or hydrogels.
We then evaluated the cellular internalization abilities of free TPPS and nanogels using cis-dichlorodiammineplatinum(II) (DDP)-resistant A549 lung cancer cells (termed A549/DDP cells). The confocal imaging results (Fig. 2a) showed that free TPPS molecules were hard to enter the A549/DDP cells even after incubation for 24 h, possibly due to the presence of ABC transporters on the plasma membranes of the cells. In sharp contrast, the TPPS-encapsulated nanogels exhibited massive time-dependent cellular influx and could efficiently target lysosomes (Fig. 2a and b). Owing to their higher OSiND proportions that led to less negatively charged surfaces, SiPT75 and SiPT185 had higher cellular uptake than SiPT40, which was proved by the flow cytometric results (Fig. 2c). To investigate the endocytic pathway of the nanogels, 4 °C treatment, amiloride, chlorpromazine (CPZ), and genistein were used to inhibit the energy-dependent, macropinocytosis-, clathrin-, and caveolae-mediated pathways, respectively.44 4 °C treatment, CPZ, and genistein remarkably reduced the endocytosis of SiPT75 (Fig. 2d), indicating that the nanogels were internalized mainly through energy-dependent, clathrin- and caveolae-mediated endocytosis.
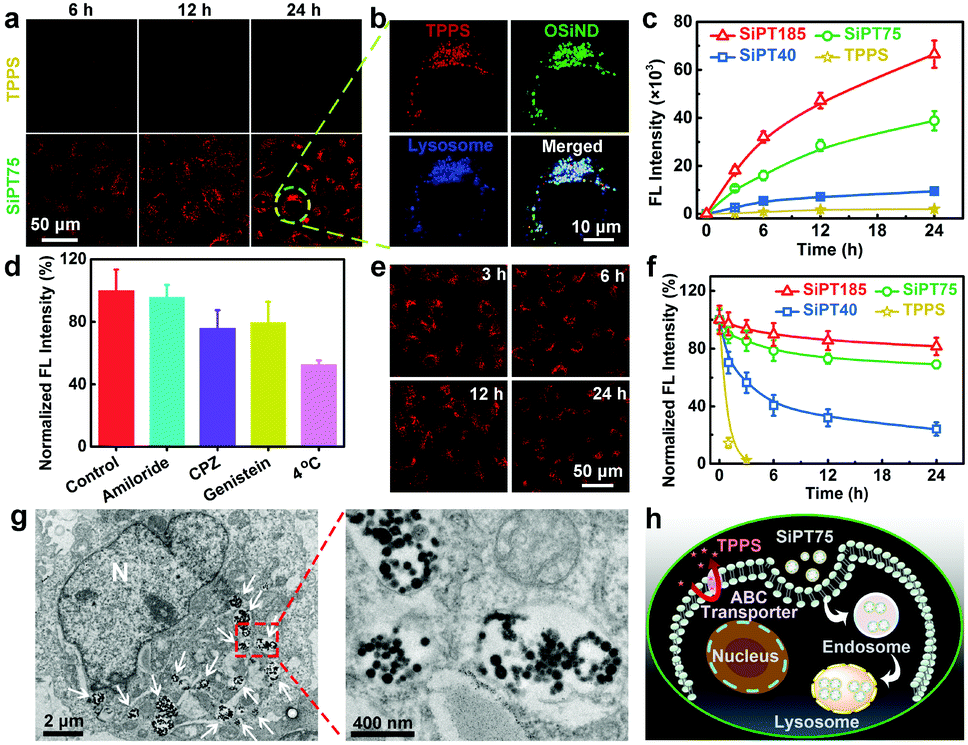 | ||
| Fig. 2 (a) Confocal fluorescence images of A549/DDP cells exposed to TPPS or SiPT75 for various time periods (TPPS concentration: 10 μg mL−1). (b) Confocal fluorescence images showing the co-localization results of TPPS, OSiNDs, and lysosomes in an SiPT75-treated A549/DDP cell. (c) Flow cytometric results of A549/DDP cells after incubation with different drugs for various time periods. (d) Flow cytometric results of A549/DDP cells treated without inhibitors (control) and with amiloride, CPZ, genistein, or 4 °C incubation, followed by the addition of SiPT75. (e) Confocal fluorescence images of A549/DDP cells after treatment with SiPT75 for 24 h and incubation in drug-free culture medium for different time periods. (f) Flow cytometric results showing the anti-efflux performance of different drugs in A549/DDP cells. (g) Bio-TEM images of an A549/ DDP cell treated with SiPT75 for 24 h. “N” presents the nucleus, and the white arrows point to lysosomes or endosomes. (h) Schematic illustrating the anti-efflux mechanism of SiPT75. The concentrations of free TPPS and TPPS in the nanogels were fixed at 10 μg mL−1 in all the above experiments. | ||
Next, we studied the exocytosis of free TPPS and the nanogels from the drug-treated A549/DDP cells. As shown in the confocal fluorescence images in Fig. 2e, SiPT75 was slowly excreted from the cells as time increased. Furthermore, the anti-efflux abilities of various drugs were compared. Free TPPS was fast excreted from the cells with an efflux half-life of ∼0.65 h, while SiPT40 had a significantly prolonged efflux half-life of ∼4.3 h (Fig. 2f). Moreover, even after 24 h, only 19% of SiPT185 and 31% of SiPT75 were excreted from the cells, respectively. We also compared the exocytosis rate of SiPT75 as a function of the nanogel concentration. As shown in Fig. S14 (ESI†), the exocytosis rate of the nanogels decreased with increasing nanogel concentrations in the range of 0.5–10 μg mL−1 (based on TPPS). In addition, it was found that the size of the nanogel aggregates in the acidic solutions (pH = 4.5 and 6.0) increased with increasing concentrations (Fig. S8b, ESI†). Collectively, we proposed that the anti-efflux ability of the nanogels was possibly due to their aggregation in the acidic endosomes/lysosomes to form larger particles, as proved by the biological TEM (Bio-TEM) images of SiPT75-treated cells (Fig. 2g) and the conventional TEM image of the lysosomes extracted from the SiPT75-treated cells (Fig. S15, ESI†). To further verify our hypothesis, we used NaHCO3-treated A549/DDP cells as a control. Bio-TEM imaging results showed that after NaHCO3 treatment, only some small nanogel aggregates were observed in the cells (Fig. S16, ESI†). Moreover, the exocytosis rate in the NaHCO3-treated cells was much higher than that in the cells without NaHCO3 treatment (Fig. S17, ESI†). Taken together, the nanogels can be efficiently endocytosed by the drug-resistant cancer cells even in the presence of ABC transporters on the plasma membranes, and then target and accumulate in the acidic endosomes/lysosomes. The aggregation of the nanogels in the acidic organelles can realize long-term endosomal/lysosomal retention and prevent the efflux of the drugs (Fig. 2h).
Encouraged by the significantly improved endocytosis and anti-efflux ability of the nanogels in drug-resistant cancer cells, we further evaluated their in vitro photodynamic therapeutic efficiencies. Methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay results indicated that free TPPS and the three nanogels had negligible dark toxicity to A549/DDP cells (Fig. 3a). After irradiation (532 nm, 20 mW cm−2, 10 min), free TPPS showed low photodynamic anticancer efficiency even when its concentration was up to 10 μg mL−1, which was mainly due to its poor uptake by the drug-resistant cells. In contrast, the nanogels showed excellent anticancer performance after irradiation, and their anticancer efficacies increased with increasing sizes. For example, SiPT185 with a TPPS concentration of 2 μg mL−1 or SiPT75 with a TPPS concentration of 5 μg mL−1 killed almost all the cells, while SiPT40 with a TPPS concentration of 10 μg mL−1 only killed 80% of the cells. Compared with the A549/DDP cells without NaHCO3 treatment (Fig. 3a), the cells after NaHCO3 pretreatment were more difficult to be killed by SiPT75 after laser irradiation (Fig. S18, ESI†). Thus, the decreased exocytosis was the reason why PDT was enhanced.
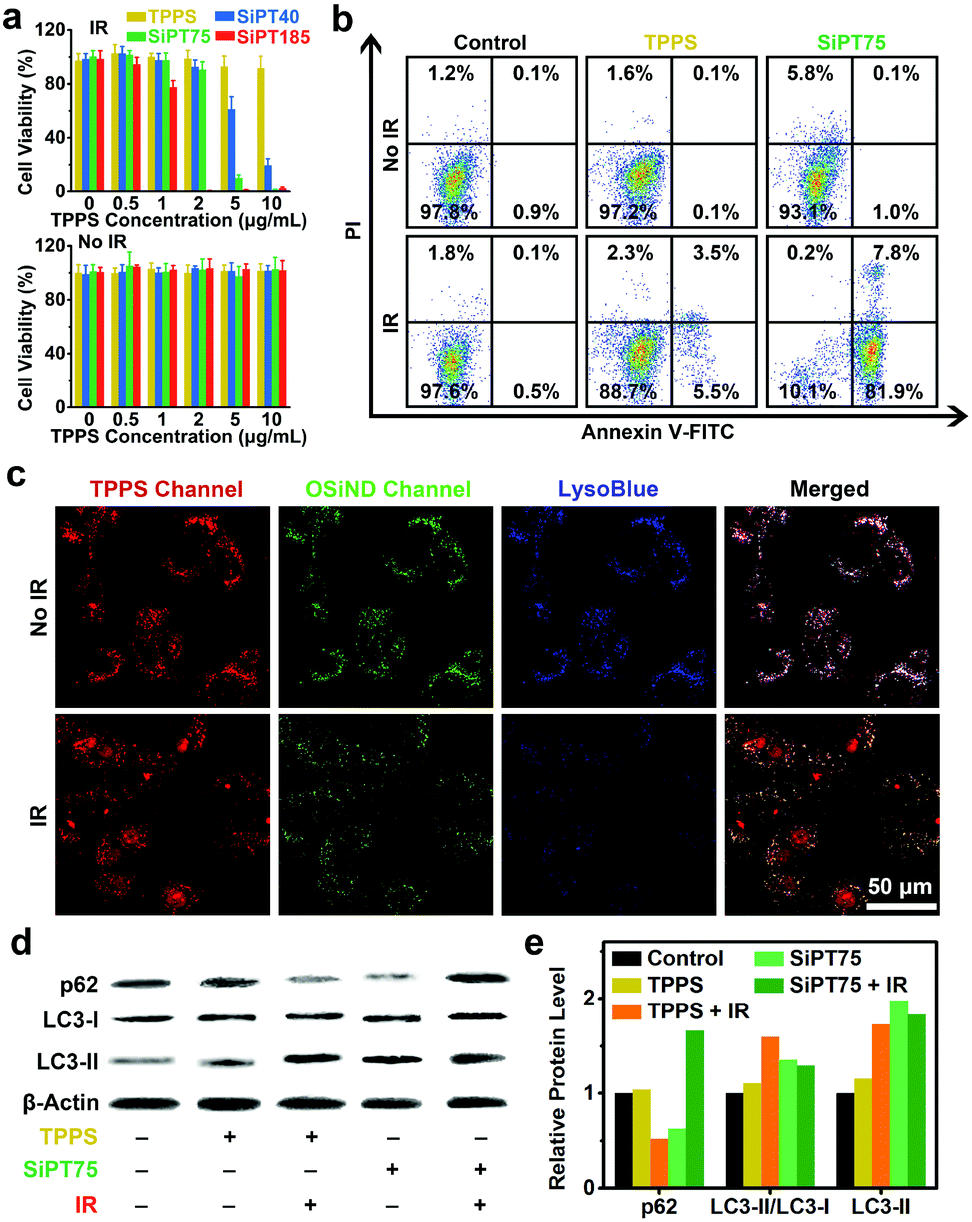 | ||
| Fig. 3 (a) Viabilities of the A549/DDP cells that received various treatments as indicated. “IR” indicates irradiation (532 nm laser, 20 mW cm−2, 10 min). (b) Apoptosis results (measured by flow cytometry) of A549/DDP cells after various treatments as indicated. “IR” indicates irradiation (532 nm laser, 20 mW cm−2, 10 min). (c) Confocal fluorescence images of the A549/DDP cells treated with SiPT75 for 24 h and irradiated by a 532 nm laser (20 mW cm−2, 10 min) or not. Before imaging, the cells were stained by LysoBlue to visualize lysosomes. (d) Image and (e) corresponding statistical histogram of the Western blot results. Before analyses, the cells were treated with free TPPS or SiPT75 for 24 h and irradiated by a 532 nm laser (20 mW cm−2, 10 min) or not. The cells without treatments were set as the control groups. β-Actin serves as a loading control. The concentrations of free TPPS and TPPS in the nanogels were fixed at 10 μg mL−1 in all the above experiments. | ||
To further confirm the good anticancer performance of the nanogels under irradiation, we compared the photodynamic anticancer capability of SiPT75 with that of free TPPS using the apoptosis assay. The apoptosis rate of SiPT75-treated A549/DDP cells sharply increased from 1.1% to 89.7% after irradiation, while only a slight increase from 0.2% to 9.0% was observed in the TPPS-treated cells after irradiation (Fig. 3b). More ROS were generated in the SiPT75-treated cells than in the free TPPS-treated cells after irradiation (Fig. S19, ESI†).
Because SiPT75 was mainly distributed in the endosomes/lysosomes of the cells, we presumed that the lysosomes in the SiPT75-treated cells were damaged after laser irradiation. To verify this hypothesis, the lysosomes were stained with the blue-fluorescent lysosomal dye LysoBlue. Different from the good imaging performance in the non-irradiated cells, LysoBlue could hardly stain the lysosomes of the cells co-treated with SiPT75 and laser irradiation (Fig. 3c), indicating the severe damage of the lysosomes in the cells. In addition, it has been reported that lysosome plays an important role in regulating autophagy,45 and the damage of lysosomes may inhibit cell autophagy.33,46 To study the effect of lysosomal damage on autophagy, the expression levels of autophagy-related proteins (including LC3-I, LC3-II, and p62) in the cells after various treatments were evaluated via Western blot analysis. The conversion of the soluble form of LC3 (LC3-I) to its membrane-bound form (LC3-II) and the degradation of p62 are both important markers of autophagy.47 As shown in Fig. 3d and e, the cells after the combined treatment with TPPS and laser irradiation had a lower p62 expression level and a higher LC3-II/LC3-I ratio compared with the untreated and TPPS-treated cells. The above results indicated that the combination of TPPS and laser irradiation could promote autophagy, similar to the conclusion drawn by previous studies that the autophagy level of the cancer cells increases after PDT.48,49 In sharp contrast, the combination of SiPT75 and laser irradiation could increase the expression of both p62 and LC3-II, suggesting that the fusion of autophagosomes with lysosomes was blocked and the autophagy was inhibited due to the lysosomal damage after PDT treatment.
Considering its suitable size for passive tumor targeting, good stability, and excellent in vitro anticancer activity, SiPT75 was selected as a representative for in vivo imaging experiments. A549/DDP tumor-bearing nude mice were intravenously injected with free TPPS or SiPT75 with the same TPPS concentration, and the fluorescence signals of TPPS were monitored by an in vivo fluorescence imaging system to evaluate the biodistribution of the drugs. The results showed that free TPPS had low tumor accumulation during the observation time periods (Fig. 4a). In contrast, strong fluorescence signals were observed in the tumor region of SiPT75-treated mice at 12 h postinjection, and the fluorescence intensity of the tumor region at 24 h postinjection was much higher than those of the major organs, demonstrating the good tumor targeting ability of SiPT75 (Fig. 4b). As time went by, the fluorescence intensity further increased and reached the maximum at ∼36 h postinjection (Fig. 4c). Moreover, evident fluorescence signals could still be observed for SiPT75 even at 72 h postinjection, indicating that SiPT75 exhibited prolonged tumor retention compared with free TPPS. When the tumor was injected with NaHCO3 to weaken its acidity, some fluorescence signals of the nanogels were observed in the tumor region at 1 d postinjection, but the signals were significantly decreased at 3 d postinjection (Fig. S20, ESI†). Furthermore, the Bio-TEM imaging experiments for the tumor tissue slices were conducted. As shown in Fig. 4d, after intravenous injection of SiPT75, some nanogels were observed in the tumor at 3 d postinjection, and they formed aggregates in the acidic tumor region. In contrast, the nanogels were hardly observed in the tumor region of the mouse after intravenous injection of SiPT75 and intratumoral injection of NaHCO3 (Fig. S21, ESI†). The results indicated that the long-term tumor retention ability of the nanogels might be attributed to their low acidity-induced aggregation properties. Besides, it was found that the average size of the nanogels in the tumor region (∼37 nm, Fig. S22, ESI†) was smaller than that of the nanogels dispersed in a neutral environment (∼76 nm, Fig. S2h, ESI†), indicating that the nanogels degraded in vivo, possibly due to the biodegradation of PEG-PLE. Although the nanogels could realize long-term tumor retention, the fluorescence signals of SiPT75 could be hardly detected in the major organs of the mice on the 10th day postinjection (Fig. S23, ESI†).
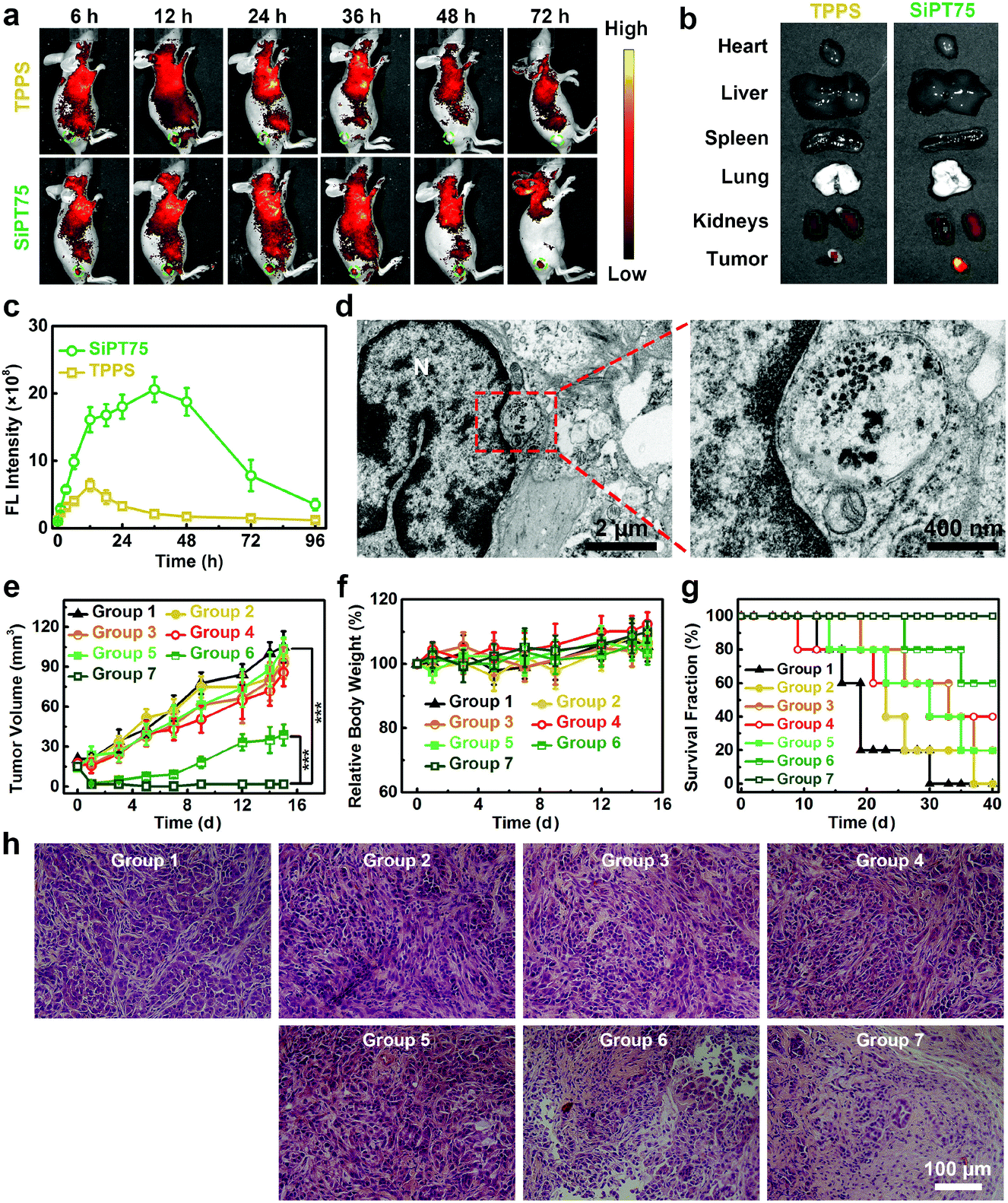 | ||
| Fig. 4 (a) Time-dependent in vivo fluorescence images of A549/DDP tumor-bearing mice after intravenous injection of TPPS solution or SiPT75 suspension (200 μL, TPPS concentration: 1 mg mL−1). (b) Ex vivo fluorescence images of major organs and tumor tissues excised from the mice at 24 h post the intravenous injection of TPPS solution or SiPT75 suspension (200 μL, TPPS concentration: 1 mg mL−1). (c) Time-dependent fluorescence intensities of TPPS in the tumor tissues of the mice intravenously injected with TPPS solution or SiPT75 suspension (200 μL, TPPS concentration: 1 mg mL−1). (d) Bio-TEM images of the tumor cell slice from the tumor-bearing mouse sacrificed on the 3rd day after intravenous injection of SiPT75 suspension (200 μL, TPPS concentration: 1 mg mL−1). (e) Tumor growth curves, (f) relative body weight changes, (g) survival fractions, and (h) H&E-stained tumor slices from the mice in various groups (n = 5). ***P < 0.001. Group 1: PBS, group 2: TPPS, group 3: TPPS + IR for once, group 4: TPPS + IR for twice, group 5: SiPT75, group 6: SiPT75 + IR for once, group 7: SiPT75 + IR for twice. “IR” indicates irradiation (532 nm laser, 20 mW cm−2, 20 min). The mice were intravenously injected with 200 μL of free TPPS solution or SiPT75 suspension with the same TPPS concentration of 1 mg mL−1. | ||
Motivated by its long-term tumor accumulation ability, we then tested if SiPT75 can be used for repeated PDT under laser irradiation with a single injection of the drug. To this end, A549/DDP tumor-bearing mice were randomly divided into the following 7 groups: group 1: PBS, group 2: TPPS, group 3: TPPS + IR for once, group 4: TPPS + IR for twice, group 5: SiPT75, group 6: SiPT75 + IR for once, group 7: SiPT75 + IR for twice. The mice in the groups 1, 2, and 5 were placed in the dark. The mice in groups 3, 4, 6, and 7 were irradiated by a 532 nm laser (20 mW cm−2, 20 min) at 1 d postinjection, and the mice in groups 4 and 7 had a second PDT treatment at 2 d postinjection. The tumor volumes, body weights, and survival fractions of the mice were monitored (Fig. 4e–g). The results showed that both free TPPS and SiPT75 had negligible inhibitory effect on the tumor growth in the dark. After irradiation, SiPT75 showed much higher PDT efficiency than free TPPS. Nevertheless, the tumors of the SiPT75-treated mice with one PDT treatment only showed delayed growth and could not be completely ablated. In contrast, the tumors of SiPT75-treated mice with two PDT treatments were completely ablated and no tumor regrowth was observed during the observation period. Hematoxylin and eosin (H&E)-stained histopathology images further demonstrated the excellent anticancer performance of SiPT75 after irradiation twice (Fig. 4h). To evaluate the biocompatibility of the nanogels, hemanalyses and H&E staining of major organs from healthy mice receiving the administration of PBS or SiPT75 were conducted. No apparent toxicities were observed (Fig. S24, ESI†), indicating the excellent biosafety of the nanogels.
Conclusions
In summary, utilizing the endo/lysosomal entrapment phenomenon in the cells, we have demonstrated that the as-prepared size-changeable supramolecular nanogels can be an efflux retarder and autophagy inhibitor for enhanced PDT of multidrug-resistant cancer. On one hand, the nanogels could elude the drug efflux pumps on the plasma membrane, target the endosomes/lysosomes, and form large aggregates in these acidic organelles, thus simultaneously realizing enhanced drug influx and reduced drug efflux in drug-resistant cancer cells. On the other hand, PDT treatment could damage the lysosomes of the nanogel-treated cells, leading to the inhibition of protective autophagy. Hence, the photodynamic therapeutic performance of the nanogels was dramatically improved. Furthermore, the nanogels could prolong the tumor accumulation of the PSs for realizing multiple PDT with a single drug injection. We hope that the developed strategy can provide a new method for overcoming cancer MDR, and can be expanded to other cancer remedies such as chemotherapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21673037) and Scientific Research Foundation of the Graduate School of Southeast University (YBJJ1777).Notes and references
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef.
- D. B. Longley and P. G. Johnston, J. Pathol., 2005, 205, 275–292 CrossRef CAS.
- X. F. Chen, X. M. Yao, L. Chen and X. S. Chen, Macromol. Biosci., 2015, 15, 1563–1570 CrossRef CAS.
- M. Abbas, Q. L. Zou, S. K. Li and X. H. Yan, Adv. Mater., 2017, 29, 1605021 CrossRef.
- H. J. Zhu, Y. Fang, Q. Q. Miao, X. Y. Qi, D. Ding, P. Chen and K. Y. Pu, ACS Nano, 2017, 11, 8998–9009 CrossRef CAS.
- S. K. Li, Q. L. Zou, Y. X. Li, C. Q. Yuan, R. R. Xing and X. H. Yan, J. Am. Chem. Soc., 2018, 140, 10794–10802 CrossRef CAS.
- H. J. Zhu, J. C. Li, X. Y. Qi, P. Chen and K. Y. Pu, Nano Lett., 2018, 18, 586–594 CrossRef CAS.
- Z. J. Yang, Q. Chen, J. W. Chen, Z. L. Dong, R. Zhang, J. J. Liu and Z. Liu, Small, 2018, 14, 1803262 CrossRef.
- X. D. Zhang, X. K. Chen, H. Y. Wang, H. R. Jia and F. G. Wu, Adv. Therap., 2019, 2, 1800140 CrossRef.
- L. Y. Zeng, Y. W. Pan, Y. Tian, X. Wang, W. Z. Ren, S. J. Wang, G. M. Lu and A. G. Wu, Biomaterials, 2015, 57, 93–106 CrossRef CAS.
- Y. Y. Yuan, C. J. Zhang and B. Liu, Chem. Commun., 2015, 51, 8626–8629 RSC.
- C. B. He, D. M. Liu and W. B. Lin, ACS Nano, 2015, 9, 991–1003 CrossRef CAS.
- X. Wei, L. Q. Liu, X. Guo, Y. Wang, J. Y. Zhao and S. B. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 17672–17684 CrossRef CAS.
- G. Q. Wei., Y. Wang, X. H. Huang, H. B. Hou and S. B. Zhou, Small Methods, 2018, 2, 1700358 CrossRef.
- K. D. Lu, C. B. He and W. B. Lin, J. Am. Chem. Soc., 2014, 136, 16712–16715 CrossRef CAS.
- H. Li, C. Liu, Y. P. Zeng, Y. H. Hao, J. W. Huang, Z. Y. Yang and R. Li, ACS Appl. Mater. Interfaces, 2016, 8, 31510–31523 CrossRef CAS PubMed.
- L. Cheng, C. Wang, L. Z. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 10869–10939 CrossRef CAS.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS.
- X. D. Zhang, L. Y. Xia, X. K. Chen, Z. Chen and F. G. Wu, Sci. China Mater., 2017, 60, 487–503 CrossRef CAS.
- X. Wei, Y. Wang, X. Xiong, X. Guo, L. Zhang, X. B. Zhang and S. B. Zhou, Adv. Funct. Mater., 2016, 26, 8266–8280 CrossRef CAS.
- K. Han, J. Y. Zhu, H. Z. Jia, S. B. Wang, S. Y. Li, X. Z. Zhang and H. Y. Han, ACS Appl. Mater. Interfaces, 2016, 8, 25060–25068 CrossRef CAS.
- Z. Y. Ma, L. Wu, K. Han and H. Y. Han, Nanoscale Horiz., 2019, 4, 1124–1131 RSC.
- L. P. Qiu, T. Chen, I. Ocsoy, E. Yasun, C. C. Wu, G. X. Zhu, M. Y. You, D. Han, J. H. Jiang, R. Q. Yu and W. H. Tan, Nano Lett., 2015, 15, 457–463 CrossRef CAS.
- C. Zhang, L. H. Liu, W. X. Qiu, Y. H. Zhang, W. Song, L. Zhang, S. B. Wang and X. Z. Zhang, Small, 2018, 14, 1703321 CrossRef.
- X. K. Chen, X. D. Zhang, Y. X. Guo, Y. X. Zhu, X. Y. Liu, Z. Chen and F. G. Wu, Adv. Funct. Mater., 2019, 29, 1807772 CrossRef.
- M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- G. Lin, P. Mi, C. C. Chu, J. Zhang and G. Liu, Adv. Sci., 2016, 3, 1600134 CrossRef.
- Y. Ma, Z. H. Wang, Y. X. Ma, Z. H. Han, M. Zhang, H. Y. Chen and Y. Q. Gu, Angew. Chem., Int. Ed., 2018, 57, 5389–5393 CrossRef CAS.
- B. D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef CAS.
- C. B. He, Y. P. Hu, L. C. Yin, C. Tang and C. H. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS.
- E. A. Sykes, J. Chen, G. Zheng and W. C. W. Chan, ACS Nano, 2014, 8, 5696–5706 CrossRef CAS.
- X. Duan, B. Chen, Y. N. Cui, L. Zhou, C. K. Wu, Z. L. Yang, Y. Wen, X. Y. Miao, Q. L. Li, L. Xiong and J. He, Apoptosis, 2018, 23, 587–606 CrossRef.
- B. Q. Spring, I. Rizvi, N. Xu and T. Hasan, Photochem. Photobiol. Sci., 2015, 14, 1476–1491 RSC.
- H. Y. Nam, S. M. Kwon, H. Chung, S. Y. Lee, S. H. Kwon, H. Jeon, Y. Kim, J. H. Park, J. Kim, S. Her, Y. K. Oh, I. C. Kwon, K. Kim and S. Y. Jeong, J. Controlled Release, 2009, 135, 259–267 CrossRef CAS.
- R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Chem. Soc. Rev., 2012, 41, 2539–2544 RSC.
- K. Dong, Z. Z. Wang, Y. Zhang, J. S. Ren and X. G. Qu, ACS Appl. Mater. Interfaces, 2018, 10, 31998–32005 CrossRef CAS.
- M. H. Sui, W. W. Liu and Y. Q. Shen, J. Controlled Release, 2011, 155, 227–236 CrossRef CAS.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, J. Controlled Release, 2011, 151, 220–228 CrossRef CAS.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263–272 CrossRef CAS.
- X. K. Chen, X. D. Zhang, L. Y. Xia, H. Y. Wang, Z. Chen and F. G. Wu, Nano Lett., 2018, 18, 1159–1167 CrossRef CAS.
- J. Y. Kim, W. I. Choi, M. Kim and G. Tae, J. Controlled Release, 2013, 171, 113–121 CrossRef CAS.
- L. Y. Xia, X. D. Zhang, M. Cao, Z. Chen and F. G. Wu, Biomacromolecules, 2017, 18, 3073–3081 CrossRef CAS PubMed.
- S. A. Lim, H. Park, J. M. Lee and E. S. Lee, Polym. Adv. Technol., 2018, 29, 2766–2773 CrossRef CAS.
- X. K. Chen, X. D. Zhang, H. Y. Wang, Z. Chen and F. G. Wu, Langmuir, 2016, 32, 10126–10135 CrossRef CAS.
- G. Kroemer and M. Jäättelä, Nat. Rev. Cancer, 2005, 5, 886–897 CrossRef CAS PubMed.
- S. S. Wan, L. Zhang and X. Z. Zhang, ACS Cent. Sci., 2019, 5, 327–340 CrossRef CAS PubMed.
- P. D. Jiang and N. Mizushima, Methods, 2015, 75, 13–18 CrossRef CAS PubMed.
- J. J. Reiners, P. Agostinis, K. Berg, N. L. Oleinick and D. H. Kessel, Autophagy, 2010, 6, 7–18 CrossRef CAS PubMed.
- I. Coupienne, G. Fettweis and J. Piette, Lasers Surg. Med., 2011, 43, 557–564 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00643e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |