
Molybdenum carbide nanostructures for electrocatalytic polysulfide conversion in lithium–polysulfide batteries†
Yunling
Wu‡
,
Jun
Deng‡
,
Yuan
Zhou
,
Yang
Huang
and
Yanguang
Li
 *
*
Institute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials and Devices, Soochow University, Suzhou 215123, China. E-mail: yanguang@suda.edu.cn
First published on 6th November 2019
Abstract
Introduction of appropriate cathode electrocatalysts in lithium–sulfur or lithium–polysulfide batteries can accelerate the polysulfide interconversion and suppress the shuttle effect. However, improvements are often limited especially under high sulfur loading. Herein, we prepare molybdenum carbide nanostructures and investigate their potential as the cathode electrocatalyst for lithium–polysulfide batteries. The product is prepared by the self-polymerization of dopamine in the presence of Mo7O246− ions, followed by high-temperature carburization. It features ultrasmall α-MoC1−x nanoparticles uniformly dispersed on a hierarchical carbonaceous support. Polysulfide adsorption experiments and electrochemical measurements show that this material has a strong surface affinity toward polysulfides, and can greatly enhance their conversion rate, in particular the Li2S4 ↔ Li2S2/Li2S conversion. When assessed as the cathode electrocatalyst, it enables lithium–polysulfide batteries with large specific capacity (up to 1400 mA h g−1), impressive rate capability (800 mA h g−1 at 3200 mA g−1) and excellent cycling stability even at high sulfur loading.
New conceptsLithium–sulfur batteries generally suffer from the notorious shuttle effect. Conventional wisdom mainly focuses on designing appropriate electrode materials or membranes to physically or chemically constrain polysulfide intermediates. It has not been realized until recently that the introduction of appropriate cathode electrocatalysts can accelerate the interconversion of polysulfides, and suppress the shuttle effect by reducing their concentration built up near the cathode. However, this catalytic effect is usually weak at high sulfur loading. Our study here shows that α-MoC1−x nanoparticles dispersed on a hierarchical carbonaceous support are an excellent cathode electrocatalyst for lithium–polysulfide batteries. This electrocatalyst material not only has a strong adsorption capability toward polysulfides, but can also greatly promote their interconversion, in particular the Li2S4 ↔ Li2S2/Li2S conversion. Excellent battery performance is therefore demonstrated even at high sulfur loading. |
1. Introduction
Lithium-ion batteries (LIBs) are the electrochemical energy storage solution of choice for a large variety of portable electronics.1,2 Despite their great commercial success, they still suffer from insufficient energy density and relatively high costs, and can barely meet the increasing energy demand for electric vehicles (EVs) and smart grid applications.3–5 Among the several potential beyond lithium-ion technologies, lithium–sulfur (Li–S) batteries stand out thanks to their large theoretical energy density and potential low costs.6–8 However, their practical viability is yet to be proven. One of the most severe challenges is the dissolution of soluble polysulfide intermediates in common ether electrolytes and their shuttle between the cathode and anode (known as the shuttle effect), leading to not only active material loss and lithium anode corrosion, but also reduced coulombic efficiency and fast capacity decay.9–11Over the past decade, researchers have been actively searching for possible solutions to suppress the shuttle effect. It is usually approached by designing appropriate electrode materials or membranes to physically or chemically constrain polysulfide intermediates.12–18 Despite these significant advances, these approaches are generally less effective at practically meaningful sulfur loading.8 On the other hand, dissolved polysulfides may facilitate the redox reaction of sulfur at the electrode–electrolyte interface.19 Studies have shown that if the solvation of polysulfides is fully prohibited, the cycling of the cathode would proceed via solid-phase reactions at considerably lower rates.19 As a result, there is an increasing awareness now of the importance of maintaining the necessary solvation of polysulfide intermediates while at the same time suppressing their shuttle effect in the pursuit of high-performance Li–S batteries.20 Very recently, an alternative strategy demonstrated to inhibit the shuttle effect is to expedite the electrochemical conversion of polysulfide intermediates, reduce their concentration built up near the cathode, and consume them before they are able to diffuse away.21,22 The introduction of cathode electrocatalysts would particularly benefit the conversion of lower-order polysulfides (such as Li2S4) to insoluble Li2S/Li2S2, which involves the heterogeneous nucleation of the solid phase and is kinetically sluggish. Up to now, many candidate materials have been investigated as such cathode electrocatalysts for Li–S or Li–polysulfide batteries, including MoS2, WS2, CoSx, TiO2, TiN, WxC and so on.23–34 However, it is still challenging to achieve decent battery performances at high surface loadings (such as 4–6 mg cm−2) that are generally desirable for practical applications.8,9
In this study, we report our attempt to prepare α-MoC1−x nanoparticles and explore them as the cathode electrocatalyst for Li–polysulfide batteries. α-MoC1−x has demonstrated applications in, for example, CO2 hydrogenation catalysis and hydrogen evolution electrocatalysis.35,36 Here, we synthesize ultra-small α-MoC1−x nanoparticles dispersed on a hierarchical carbonaceous support. They markedly accelerate the polysulfide interconversion and enable Li–polysulfide batteries with large specific capacity and impressive cycle lives even at high sulfur loading.
2. Results and discussion
The preparation of MoC1−x nanostructures is schematically illustrated in Fig. 1a. It started with the self-polymerization of dopamine in weakly alkaline solution in the presence of Mo7O246− ions as the Mo precursor (see Experimental in the ESI† for details). The solid product collected from this step has a hierarchical flower-like morphology (Fig. S1, ESI†) and is denoted as Mo/PDA. Our previous study showed that the interaction between dopamine and Mo species could modify the growth behavior of polydopamine (PDA) and ultimately give rise to a hierarchical structure that was unique only to the Mo–PDA composite.37,38 In the second step, Mo/PDA was annealed at 750 °C under Ar for the transformation of the Mo precursor to MoC1−x and the carbonization of the PDA support. The final product is denoted as MoC1−x/C. The purpose of this two-step synthetic method is to take advantage of the strong confinement effect of the support, and to restrict the overgrowth of MoC1−x nanoparticles during the high-temperature carburization.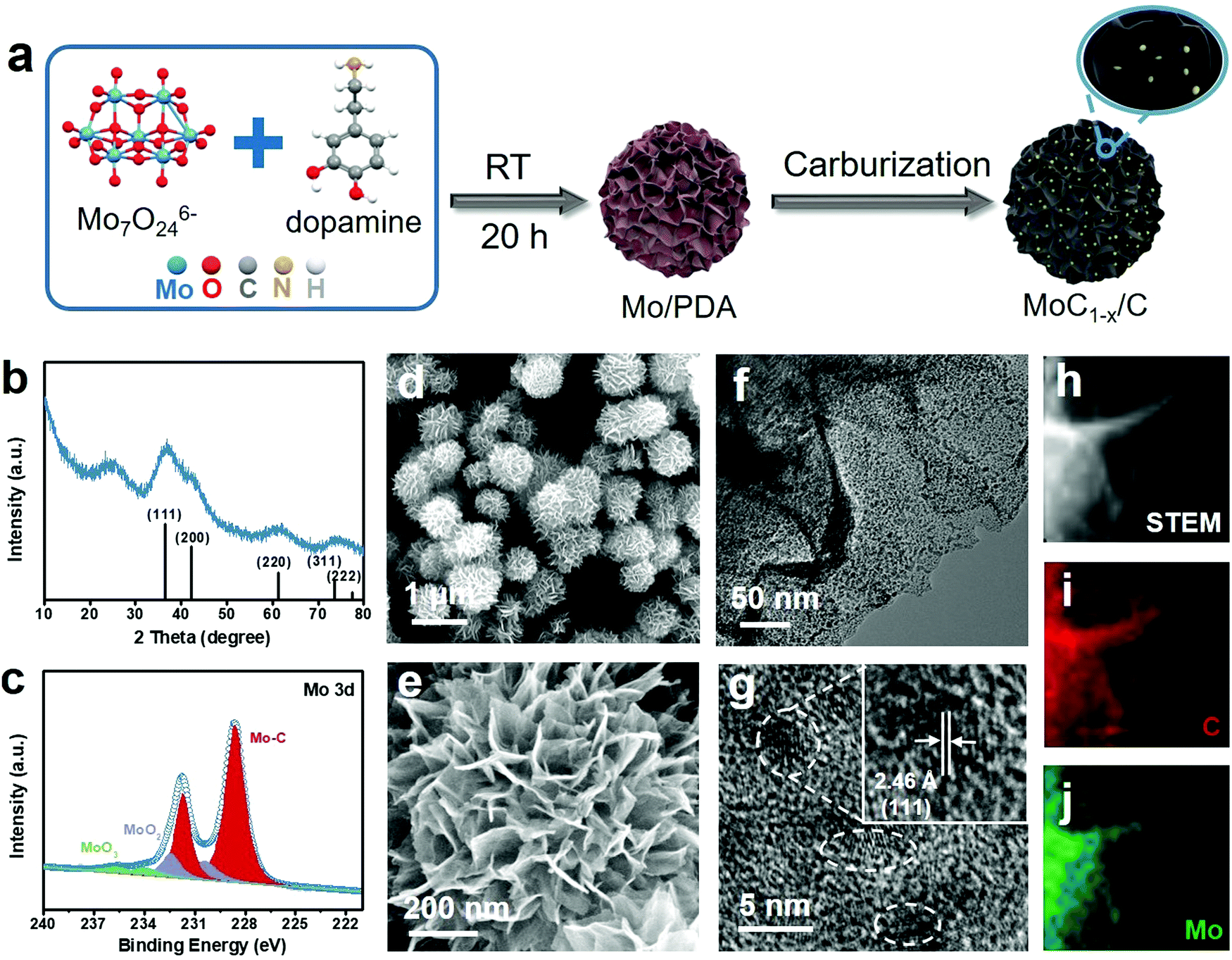 | ||
| Fig. 1 Structural characterization of MoC1−x/C. (a) Schematic synthetic procedure for MoC1−x/C; (b) XRD pattern, (c) Mo 3d XPS spectrum, (d and e) SEM images, (f and g) TEM images and (h–j) STEM image and the corresponding EDS elemental mapping of MoC1−x/C. | ||
The final product was subjected to different spectroscopic characterization studies. The powder X-ray diffraction (XRD) pattern of MoC1−x/C exhibits broad diffraction peaks between 10o and 80° that are assignable to face-centred cubic α-MoC1−x (JCPDS No. 03-065-0280) (Fig. 1b). The broadness of the peaks clearly indicates the ultra-small MoC1−x particle size. The additional signal at ∼25° can be indexed to the (002) diffraction of graphite, evidencing the partially graphitic nature of the support. Moreover, the Mo 3d X-ray photoelectron spectroscopy (XPS) spectrum of MoC1−x/C shows predominant signals from Mo–C with the 3d5/2 and 3d3/2 peaks centered at 228.6 eV and 231.7 eV, respectively, in good agreement with previous observations about molybdenum carbide (Fig. 1c).39,40 Deconvolution of its C 1s XPS spectrum reveals the contributions from the carbide (at 284.2 eV), C–C (at 284.9 eV), C–N (at 285.9 eV), and C–O (at 286.6 eV) (Fig. S2a, ESI†).41,42 Nitrogen mostly in the pyridinic form is also detected from the N 1s XPS spectrum, presumably inherited from the amine functionalities of PDA (Fig. S2b, ESI†).
We further investigated the microstructure of the final product using electron microscopy. Scanning electron microscopy (SEM) images show that MoC1−x/C has a flower-like morphology similar to Mo/PDA (Fig. 1d and e). Each flower is about 0.8–1 μm in diameter, and consists of hundreds of nanosheets radiating out from the center and resembling the petals of the flower. The open space between neighboring nanosheets makes even the deep inside accessible and thus exposes large surface areas for catalytic applications. When examined by transmission electron microscopy (TEM), an individual nanosheet is found to be densely decorated with dark-contrasted nanoparticles over its light carbonaceous background (Fig. 1f). Close observation shows that these nanoparticles are crystalline and ∼3 nm in size (Fig. 1g). They exhibit discernible lattice fringes consistent with the (111) plane of α-MoC1−x. Energy dispersive spectroscopy (EDS) elemental mapping of one nanosheet demonstrates that the spatial distribution of Mo and C signals agrees with the physical location of the nanosheet, evidencing the uniform dispersion of MoC1−x nanoparticles on the support (Fig. 1h–j). It is worth noting that in most conventional syntheses the high annealing temperature required for transition metal carbides usually results in undesirable particle agglomeration.43–45 Such an issue is circumvented here by using PDA as the support to constrain the growth of α-MoC1−x nanoparticles.
Next, MoC1−x/C was assessed as the cathode electrocatalyst for Li–polysulfide batteries. Catalyst powder was loaded onto a carbon cloth current collector at 0.5 mg cm−2 (see Experimental in the ESI† for details). A polysulfide-containing catholyte was prepared by dissolving 0.5 M Li2S6 in dioxolane (DOL)/dimethoxyethane (DME) containing 1 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI). A calculated amount of catholyte was used to wet the cathode, which was then paired with the Li metal anode, added with additional blank electrolyte if necessary, and assembled to standard CR2302 coin cells. It is believed that using soluble Li2S6 instead of elemental sulfur powder as the starting active material may simplify our study about the electrocatalytic effect at the solid–liquid interface. The sulfur loading was calculated by normalizing the sulfur weight in the catholyte with respect to the cathode area, and controlled within 2–6 mg cm−2 throughout our investigation.
Fig. 2a depicts the cyclic voltammetry (CV) profiles of Li–polysulfide batteries using MoC1−x/C as the cathode electrocatalyst at a sulfur loading of 2 mg cm−2 under different scan rates. The negative scans feature two cathodic waves located at ∼2.3 V and ∼2.0 V, corresponding to the reduction of long-chain polysulfides to Li2S4, and subsequently the reduction of Li2S4 to Li2S2/Li2S. Positive scans show two anodic waves centered at ∼2.3 V and ∼2.4 V assignable to the stepwise re-oxidation of Li2S2/Li2S back to Li2S8/Li2S6. The larger polarization of the low-potential redox pair is indicative of the slower conversion kinetics of Li2S4 ↔ Li2S2/Li2S.22,46 Galvanostatic charge and discharge experiments at 0.2 C (or 320 mA g−1, normalized to the sulfur weight) yield consistent results (Fig. 2b). The discharge voltage profile exhibits distinct plateaus at ∼2.3 V and ∼2.1 V with a roughly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 capacity ratio, typical of Li–S or Li–polysulfide batteries.11 Its specific capacity is ∼1400 mA h g−1 at the first discharge. Almost the whole capacity is recovered when recharged. A control experiment in polysulfide-free electrolyte shows that MoC1−x/C itself has a negligible capacity contribution between 1.8 V and 2.6 V (Fig. S3, ESI†).
:3 capacity ratio, typical of Li–S or Li–polysulfide batteries.11 Its specific capacity is ∼1400 mA h g−1 at the first discharge. Almost the whole capacity is recovered when recharged. A control experiment in polysulfide-free electrolyte shows that MoC1−x/C itself has a negligible capacity contribution between 1.8 V and 2.6 V (Fig. S3, ESI†).
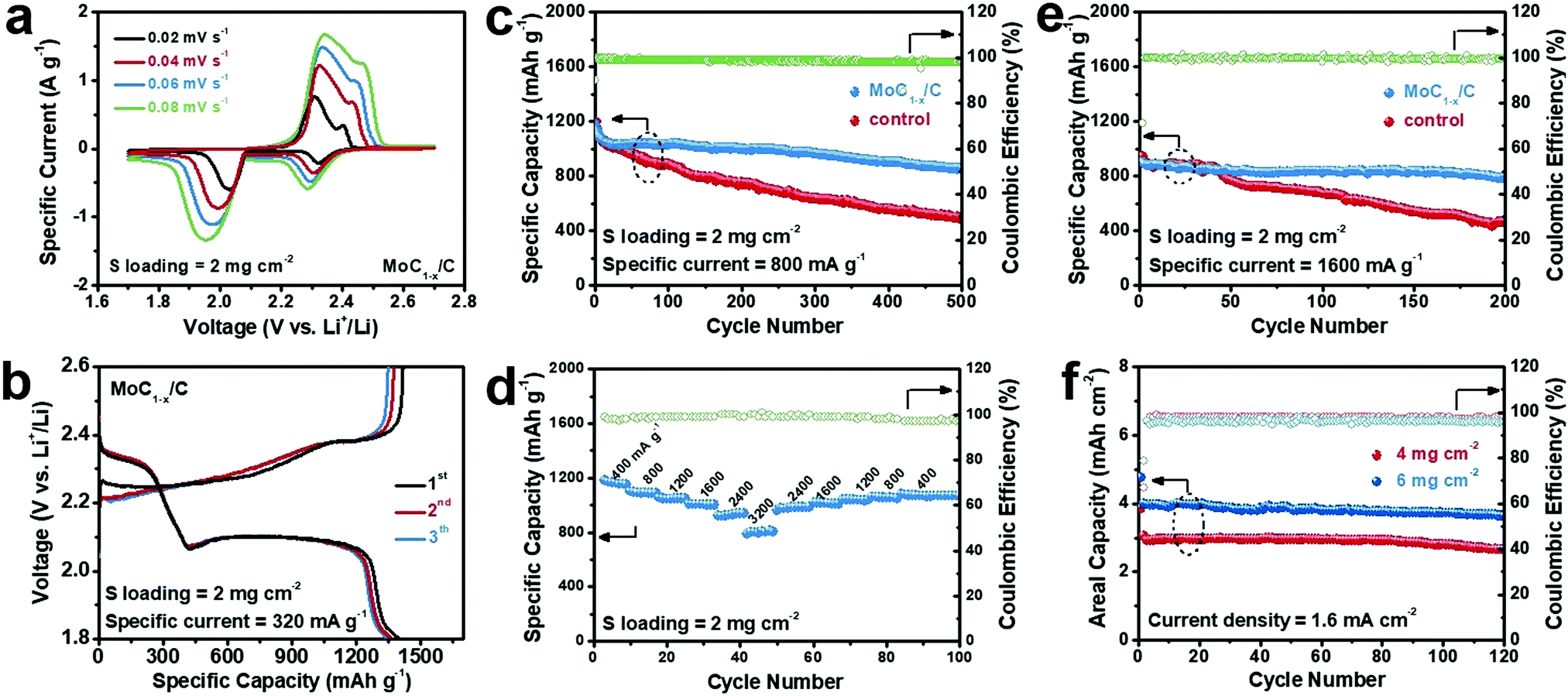 | ||
| Fig. 2 Electrochemical performances of Li–polysulfide batteries. (a) CV curves of Li–polysulfide batteries with MoC1−x/C as the cathode electrocatalyst at various scan rates; (b) galvanostatic charge and discharge curves of Li–polysulfide batteries with MoC1−x/C as the cathode electrocatalyst at 320 mA g−1; (c) cycling stability of Li–polysulfide batteries with MoC1−x/C or carbon black (control) as the cathode electrocatalyst at 800 mA g−1; (d) rate capability of Li–polysulfide batteries with MoC1−x/C as the cathode electrocatalyst; (e) cycling stability of Li–polysulfide batteries with MoC1−x/C or carbon black as the cathode electrocatalyst at 1600 mA g−1; and (f) cycling stability of Li–polysulfide batteries with MoC1−x/C as the working electrocatalyst under a high sulfur loading of 4 or 6 mg cm−2. | ||
The cycling stability of our Li–polysulfide batteries using MoC1−x/C as the cathode electrocatalyst was then evaluated at 800 mA g−1 (Fig. 2c). The battery delivers an initial discharge capacity of 1200 mA h g−1, which decays during the first several cycles and gradually levels off and stabilizes around 1000 mA h g−1 over the next 200–300 cycles. After 500 cycles, ∼860 mA h g−1 was still retained. Its corresponding coulombic efficiency remains at >98% throughout the cycling, evidencing no obvious shuttle effect. For control experiments, we assembled Li–polysulfide batteries using only carbon black covered carbon cloth as the catalyzing cathode under otherwise identical conditions. This battery has similar initial capacity at ∼1200 mA h g−1, but is subjected to faster degradation, and retains only ∼490 mA h g−1 after 500 cycles. The inferior cycling performance is probably caused by the ineffective catalyzing effect of carbon black toward Li2S4 ↔ Li2S2/Li2S, resulting in the surface passivation of the cathode by insoluble Li2S2/Li2S films. Moreover, we also investigated the effect of catalyst loading on the battery performance. As shown in Fig. S4 (ESI†), decreasing the catalyst loading to 0.25 mg cm−2 slightly compromises the cycling stability at 800 mA g−1, while increasing the catalyst loading to 1.0 mg cm−2 doesn’t give rise to an additional performance gain. This leads us to conclude that 0.5 mg cm−2 is the optimal catalyst loading.
Our Li–polysulfide batteries using MoC1−x/C as the cathode electrocatalyst also demonstrate decent rate capability (Fig. 2d). When the current rate is increased stepwise from 400 mA g−1 to 800 mA g−1, 1200 mA g−1, 1600 mA g−1, 2400 mA g−1 and 3200 mA g−1, specific capacities of ∼1100 mA h g−1, ∼1040 mA h g−1, ∼1000 mA h g−1, ∼930 mA h g−1 and ∼800 mA h g−1 are retained, respectively. More importantly, good cycling stability is also observed under 1600 mA g−1, sustaining ∼900 mA h g−1 for 200 cycles (Fig. 2e). This high-rate performance makes our battery highly competitive among all Li–polysulfide batteries.47–50 By contrast, a control experiment shows that batteries using only carbon black as the cathode electrocatalyst rapidly lose more than half of the starting capacity after 200 cycles at 1600 mA g−1.
Another general concern about conventional Li–S or Li–polysulfide batteries is that their capacity and cycling performance quickly deteriorate with increasing sulfur loading. Most previous studies have adopted low sulfur loadings of 1–2 mg cm−2, translating to areal capacities of <2 mA h cm−2—even smaller than those of current lithium-ion batteries. It is believed that a sulfur loading of >5 mg cm−2 is essential to the practical viability of Li–S batteries.8 To this end, in this study, we also assessed the cycling performance of our batteries at a higher sulfur loading of 4 or 6 mg cm−2. When cycled at a current density of 1.6 mA cm−2, they reversibly delivered an areal capacity of 2.6 mA h cm−2 and 3.6 mA h cm−2, respectively, with a negligible capacity loss for over 100 cycles (Fig. 2f). To our best knowledge, such a large and stable areal capacity is superior to those of other Li–polysulfide batteries previously reported as well as many Li–S batteries (Table S1, ESI†).49,51–57 It would be impossible if it was not for MoC1−x/C that we introduce as the cathode electrocatalyst. The energy efficiencies of our Li–polysulfide batteries is estimated to be in the range of 87–91% depending on the sulfur loading and current density (Table S2, ESI†).
In order to decipher the key role that MoC1−x/C plays during the battery cycling, adsorption experiments were carried out to probe its affinity for polysulfide intermediates. It is suggested that stronger surface binding of polysulfide intermediates would better constrain them at the cathode and retard their diffusion.19,58 Here, we soaked MoC1−x/C-loaded carbon cloth and carbon-black-loaded carbon cloth in 2 mL of polysulfide-containing DOL/DME electrolyte. As shown in the ESI,† Fig. S4, the original orange solution becomes completely colorless after aging for 48 h in the presence of MoC1−x/C, whereas the color only partially fades after the same period of aging in the presence of the carbon-black-loaded carbon cloth. This contrast reflects that polysulfides can be more effectively adsorbed onto the surface of MoC1−x/C, which cannot be accounted for by merely the effect of large surface areas, and more likely results from the strong chemical interaction between the carbide surface and polysulfides.28
In addition to being a good polysulfide-adsorber, MoC1−x/C as the cathode electrocatalyst can markedly accelerate the interconversion of polysulfides. To quantitatively evaluate this effect, batteries were subjected to potentiostatic discharges by decreasing the voltage from 2.6 V to 2.2 V or from 2.2 V to 2.0 V, and recording the transient current responses with time.59 The transient currents reflect the reaction kinetics when the electrochemical system is suddenly brought off-equilibrium. The 2.6–2.2 V potentiostatic discharge corresponds to the reduction of a higher-order polysulfide (Li2S8 or Li2S6) to a lower-order polysulfide (Li2S4), and the 2.2–2.0 V potentiostatic discharge corresponds to the reduction of a lower-order polysulfide (Li2S4) to insoluble Li2S2/Li2S. For the former, batteries with MoC1−x/C or carbon black exhibit similar transient current patterns, with the curve smoothly decaying to zero within ∼2000 s (Fig. 3a). It can be rationalized that the reduction of higher-order polysulfides homogeneously takes place in the liquid phase (Fig. 3b) and is probably first-order to the reactant concentration so the curve is asymptotic to zero.28 The very similar shape suggests essentially no difference between the catalytic effects of MoC1−x/C and carbon black for this step of reduction.
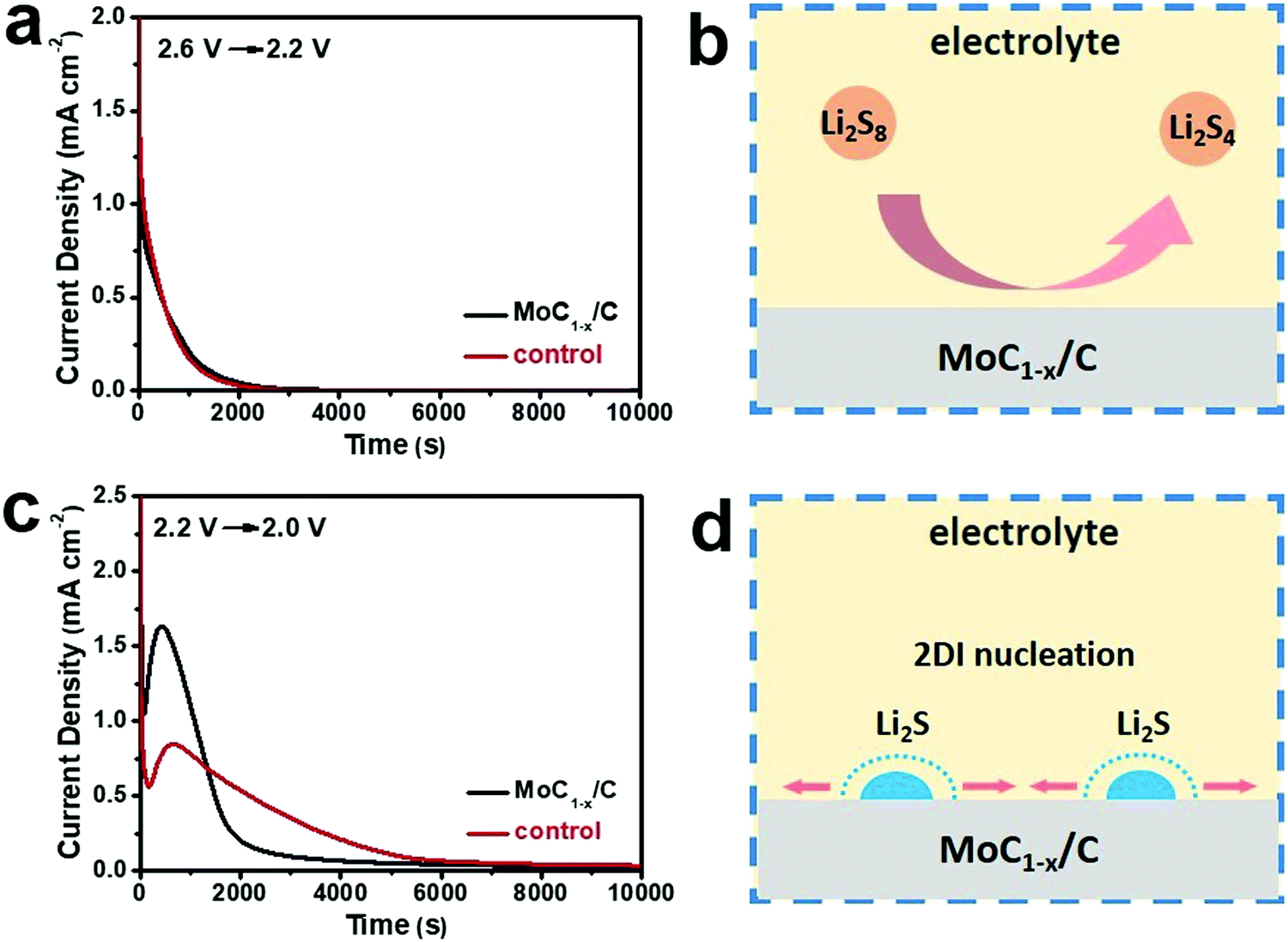 | ||
| Fig. 3 Mechanistic study about the electrocatalytic effect of MoC1−x/C. (a) Transient current curves of Li–polysulfide batteries using MoC1−x/C or carbon black as the cathode electrocatalyst when the voltage is decreased from 2.6 V to 2.2 V; (b) schematic showing that the reduction of higher-order polysulfides homogeneously takes place in the liquid phase; (c) transient current curves of Li–polysulfide batteries when the voltage is decreased from 2.2 V to 2.0 V; and (d) schematic showing the reduction of lower-order polysulfides involving the heterogeneous nucleation and growth of the solid phase following the 2DI model. | ||
By contrast, different transient current patterns are revealed when the battery voltage is decreased from 2.2 V to 2.0 V. Distinct peaks are observed at <1000 s followed by longer decaying tails (Fig. 3c). The reduction of Li2S4 to insoluble Li2S2 or Li2S involves the heterogeneous nucleation and growth of the solid phase, and has to overcome a much higher activation barrier and therefore is kinetically challenging (Fig. 3d).60 The current increase before the peak is caused by the increasing number of Li2S2/Li2S2 nuclei on the electrode surface and/or the growing size of each nucleus. The current decrease after the peak is due to the impingement between neighboring nuclei centers and/or the depletion of active species.59 Interestingly, we find that the transient current curves of Fig. 3c can be well fit with the two-dimensional instantaneous (2DI) nucleation equation (Fig. S5, ESI†).59 For the 2DI model, the reaction rate is inversely proportional to the square of tm, which is the corresponding time at the peak current. From Fig. 3c, we derive that tm is 432 s and 680 s for MoC1−x/C and carbon black, respectively. As a result, the reaction rate on MoC1−x/C is estimated to be ∼2.5 times faster than that on carbon black. The above result unambiguously highlights that MoC1−x/C can significantly catalyze the Li2S4 to Li2S2/Li2S2 conversion, presumably by binding polysulfide intermediates and lowering the necessary activation barrier.
3. Conclusion
In summary, we here reported the preparation of ultra-small α-MoC1−x particles uniformly dispersed on a hierarchical carbonaceous support, and investigated their application as the cathode electrocatalyst for Li–polysulfide batteries. MoC1−x/C not only showed an improved adsorption capability toward polysulfides, but also greatly promoted their interconversion, in particular the Li2S4 ↔ Li2S2/Li2S conversion. Our Li–polysulfide batteries using MoC1−x/C as the cathode electrocatalyst exhibited great discharge capacity (up to 1400 mA h g−1), impressive rate capability (800 mA h g−1 at 3200 mA g−1) and excellent cycling stability at different current rates. A large areal capacity (up to 4 mA h cm−2) was also achieved at a high sulfur loading of 6 mg cm−2. Our study offers a new approach to the rational design and development of high-performance Li–S or Li–polysulfide batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51972219), the Priority Academic Program Development of Jiangsu Higher Education Institutions and the Collaborative Innovation Center of Suzhou Nano Science and Technology.Notes and references
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- A. Eftekhari, ACS Sustainable Chem. Eng., 2019, 7, 5602–5613 CrossRef CAS.
- A. Eftekhari, ACS Sustainable Chem. Eng., 2017, 5, 2799–2816 CrossRef CAS.
- A. Manthiram, S.-H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- S.-H. Chung, C.-H. Chang and A. Manthiram, Adv. Funct. Mater., 2018, 28, 1801188 CrossRef.
- H.-J. Peng, J.-Q. Huang, X.-B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 7, 1700260 CrossRef.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 CrossRef.
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef CAS PubMed.
- H. Wang, W. Zhang, J. Xu and Z. Guo, Adv. Funct. Mater., 2018, 28, 1707520 CrossRef.
- Q. Pang, X. Liang, C. Y. Kwok and L. F. Nazar, Nat. Energy, 2016, 1, 16132 CrossRef CAS.
- X. Chen, T. Hou, K. A. Persson and Q. Zhang, Mater. Today, 2019, 22, 142–158 CrossRef CAS.
- R. Fang, S. Zhao, Z. Sun, D. W. Wang, H. M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef.
- Z. Li, H. B. Wu and X. W. Lou, Energy Environ. Sci., 2016, 9, 3061–3070 RSC.
- M. Liu, X. Qin, Y.-B. He, B. Li and F. Kang, J. Mater. Chem. A, 2017, 5, 5222–5234 RSC.
- H. Yuan, H.-J. Peng, J.-Q. Huang and Q. Zhang, Adv. Mater. Interfaces, 2019, 6, 1802046 CrossRef CAS.
- G. Zhang, H. J. Peng, C. Z. Zhao, X. Chen, L. D. Zhao, P. Li, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16732–16736 CrossRef CAS.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef.
- Y. Song, W. Cai, L. Kong, J. Cai, Q. Zhang and J. Sun, Adv. Energy Mater., 2019 DOI:10.1002/aenm.201901075.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- J. Park, B.-C. Yu, J. S. Park, J. W. Choi, C. Kim, Y.-E. Sung and J. B. Goodenough, Adv. Energy Mater., 2017, 7, 1602567 CrossRef.
- H. Lin, S. Zhang, T. Zhang, S. Cao, H. Ye, Q. Yao, G. W. Zheng and J. Y. Lee, ACS Nano, 2019, 13, 7073–7082 CrossRef CAS PubMed.
- Y. Wang, R. Zhang, J. Chen, H. Wu, S. Lu, K. Wang, H. Li, C. J. Harris, K. Xi, R. V. Kumar and S. Ding, Adv. Energy Mater., 2019, 9, 1900953 CrossRef.
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC.
- Y. Wu, X. Zhu, P. Li, T. Zhang, M. Li, J. Deng, Y. Huang, P. Ding, S. Wang, R. Zhang, J. Lu, G. Lu, Y. Li and Y. Li, Nano Energy, 2019, 59, 636–643 CrossRef CAS.
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed.
- Z. L. Xu, S. Lin, N. Onofrio, L. Zhou, F. Shi, W. Lu, K. Kang, Q. Zhang and S. P. Lau, Nat. Commun., 2018, 9, 4164 CrossRef.
- J. Wu, J. Chen, Y. Huang, K. Feng, J. Deng, W. Huang, Y. Wu, J. Zhong and Y. Li, Sci. Bull., 2019 DOI:10.1016/j.scib.2019.08.016.
- M. Zhao, H. J. Peng, Z. W. Zhang, B. Q. Li, X. Chen, J. Xie, X. Chen, J. Y. Wei, Q. Zhang and J. Q. Huang, Angew. Chem., Int. Ed., 2019, 131, 3819–3823 CrossRef.
- M. Zhao, H. J. Peng, J. Y. Wei, J. Q. Huang, B. Q. Li, H. Yuan and Q. Zhang, Small Methods, 2019 DOI:10.1002/smtd.201900344.
- A. Eftekhari and D.-W. Kim, J. Mater. Chem. A, 2017, 5, 17734–17776 RSC.
- A. M. Alexander and J. S. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- M. Miao, J. Pan, T. He, Y. Yan, B. Y. Xia and X. Wang, Chem. – Eur. J., 2017, 23, 10947–10961 CrossRef CAS.
- J. Chen, Y. Huang, F. Zhao, H. Ye, Y. Wang, J. Zhou, Y. Liu and Y. Li, J. Mater. Chem. A, 2017, 5, 8125–8132 RSC.
- Y. Huang, Q. Gong, X. Song, K. Feng, K. Nie, F. Zhao, Y. Wang, M. Zeng, J. Zhong and Y. Li, ACS Nano, 2016, 10, 11337–11343 CrossRef CAS.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- D. S. Baek, G. Y. Jung, B. Seo, J. C. Kim, H. W. Lee, T. J. Shin, H. Y. Jeong, S. K. Kwak and S. H. Joo, Adv. Funct. Mater., 2019, 29, 1901217 CrossRef.
- D. Wang, T. Liu, J. Wang and Z. Wu, Carbon, 2018, 139, 845–852 CrossRef CAS.
- H. J. Song, M. C. Sung, H. Yoon, B. Ju and D. W. Kim, Adv. Sci., 2019, 6, 1802135 CrossRef PubMed.
- A. M. Nartowski, I. P. Parkin, M. Mackenzie and A. J. Craven, J. Mater. Chem., 2001, 11, 3116–3119 RSC.
- J. P. Kelly, R. Kanakala and O. A. Graeve, J. Am. Ceram. Soc., 2010, 93, 3035–3038 CrossRef CAS.
- C. Giordano and M. Antonietti, Nano Today, 2011, 6, 366–380 CrossRef CAS.
- G. Zhou, Y. Zhao, C. Zu and A. Manthiram, Nano Energy, 2015, 12, 240–249 CrossRef CAS.
- X. Li, X. Pu, S. Han, M. Liu, C. Du, C. Jiang, X. Huang, T. Liu and W. Hu, Nano Energy, 2016, 30, 193–199 CrossRef CAS.
- Y. Cui and Y. Fu, ACS Appl. Mater. Interfaces, 2015, 7, 20369–20376 CrossRef CAS.
- F. Zhou, Z. Li, X. Luo, T. Wu, B. Jiang, L. L. Lu, H. B. Yao, M. Antonietti and S. H. Yu, Nano Lett., 2018, 18, 1035–1043 CrossRef CAS PubMed.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 14627 CrossRef.
- L. Li, L. Chen, S. Mukherjee, J. Gao, H. Sun, Z. Liu, X. Ma, T. Gupta, C. V. Singh, W. Ren, H. M. Cheng and N. Koratkar, Adv. Mater., 2017, 29, 1602734 CrossRef PubMed.
- H. Al Salem, G. Babu, C. V. Rao and L. M. Arava, J. Am. Chem. Soc., 2015, 137, 11542–11545 CrossRef CAS.
- Z. Li, Q. He, X. Xu, Y. Zhao, X. Liu, C. Zhou, D. Ai, L. Xia and L. Mai, Adv. Mater., 2018, 30, e1804089 CrossRef.
- S. H. Chung and A. Manthiram, Adv. Mater., 2014, 26, 1360–1365 CrossRef CAS PubMed.
- J. He, L. Luo, Y. Chen and A. Manthiram, Adv. Mater., 2017, 29, 1702707 CrossRef.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 4325–4329 CrossRef CAS.
- C. Li, Z. Xi, D. Guo, X. Chen and L. Yin, Small, 2018, 14, 1701986 CrossRef.
- Z. Li, Y. Zhou, Y. Wang and Y.-C. Lu, Adv. Energy Mater., 2019, 9, 1802207 CrossRef.
- F. Y. Fan, W. C. Carter and Y. M. Chiang, Adv. Mater., 2015, 27, 5203–5209 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00618d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |