
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA graphene-sandwiched DNA nano-system: regulation of intercalated doxorubicin for cellular localization†
Semonti
Nandi
 ac,
Narendra
Kale
ac,
Ashwini
Patil
ac,
Shashwat
Banerjee
b,
Yuvraj
Patil
*b and
Jayant
Khandare
*cd
ac,
Narendra
Kale
ac,
Ashwini
Patil
ac,
Shashwat
Banerjee
b,
Yuvraj
Patil
*b and
Jayant
Khandare
*cd
aMAEER's Maharashtra Institute of Pharmacy, Kothrud, Pune 411038, India
bMaharashtra Institute of Medical Education and Research Medical College, Talegaon Dabhade, Pune 410507, India. E-mail: yuvrajpatil@mitmimer.com
cSchool of Pharmacy, Dr Vishwanath Karad MIT World Peace University, MIT Campus, S. No. 124, Paud Road, Kothrud, Pune 411038, India. E-mail: jayant.khandare@mippune.edu.in
dSchool of Consciousness, Dr Vishwanath Karad MIT World Peace University, MIT Campus, S. No. 124, Paud Road, Kothrud, Pune 411038, India
First published on 5th October 2020
Abstract
Control of the sub-cellular localization of nanoparticles (NPs) with enhanced drug-loading capacity, employing graphene oxide (GO), iron oxide (Fe3O4) NPs and sandwiched deoxyribonucleic acid (DNA) bearing intercalated anticancer drug doxorubicin (DOX) has been investigated in this work. The nanosystems G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX differentially influence serum protein binding and deliver DOX to lysosomal compartments of cervical cancer (HeLa) cells with enhanced retention. Stern–Volmer plots describing BSA adsorption on the nanosystems demonstrated the quenching constants, Ksv for G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX (0.025 mL μg−1 and 0.0103 mL μg−1 respectively). Nuclear DOX intensity, measured at 24 h, was ∼2.0 fold higher for Fe3O4–DNA–DOX in HeLa cells. Parallelly, the cytosol displayed ∼2.2 fold higher DOX intensity for Fe3O4–DNA–DOX compared to G–DNA–DOX–Fe3O4. Fe3O4–DNA–DOX was more efficacious in the cytotoxic effect than G–DNA–DOX–Fe3O4 (viability of treated cells: 33% and 49% respectively). The DNA:nanosystems demonstrated superior cytotoxicity compared to mole-equivalent free DOX administration. The results implicate DNA:DOX NPs in influencing the cellular uptake mechanism and were critically subject to cellular localization. Furthermore, cell morphology analysis evidenced maximum deformation attributed to free-DOX with 34% increased cell roundness, 63% decreased cell area and ∼1.9 times increased nuclear-to-cytoplasmic (N/C) ratio after 24 h. In the case of Fe3O4–DNA–DOX, the N/C ratio increased 1.2 times and a maximum ∼37% decrease in NSA was noted suggesting involvement of non-canonical cytotoxic pathways. In conclusion, the study makes a case for designing nanosystems with controlled and regulated sub-cellular localization to potentially exploit secondary cytotoxic pathways, in addition to optimized drug-loading for enhanced anticancer efficacy and reduced adverse effects.
Introduction
Complex nanosystems have been used to modify drug-nanocarrier delivery systems to safely deliver drugs to the cells and specific intracellular sites.1,2 The nanosystems including PEG, phosphocholines etc. have paved the way in advancement of delivering drugs clinically. However, their targetability using synergistic and complimentary carrier systems with an actively targeting moiety is highly imperative.3 Such a complex structural framework may impede control over cellular kinetics in regulating the cell organelle targetability, unless specific targeting moieties are deployed.4,5 The incremental complexity may further compromise pharmacokinetics (PK), targeting ability, enhanced efficacy, and potential systemic toxicity.6,7 It is therefore desirable to compound multiple functional roles within limited nanocomponents. Deoxyribonucleic acid (DNA) for instance, has demonstrated roles in conjugation with biomaterial surfaces as well as physical incorporation of drugs such as doxorubicin (DOX) via intercalation within the guanosine and cytosine base pairs.8 It would therefore be intriguing to explore if incorporation of a nucleic acid onto a nanoparticle (NP) would improve its drug loading capacity and display superior cell kinetics. Graphene oxide (GO) has been widely employed in delivering drugs, proteins, probes etc.9,10 The integration of GO with other NPs like iron oxide (Fe3O4) has been evidenced to possess additional physico-chemical advantages including achieving the controlled delivery of anticancer drugs for their distribution, disposition, and localization (positioning) in cell micro-structures.11,12 We have addressed interactions between GO-based biomaterials and cancer cells in the past,13 and also demonstrated that cellular deformations induced by similar multicomponent systems and post chemotherapeutic cycles in vitro implicate activation of acquired drug resistance mechanisms in cells.14 In addition, such nanosystem-architectures may have optimal interactions with serum proteins and influence the protein binding affinities and finally may improve the PK.7,15 Nanocarriers may enhance the drug delivery efficacy by influencing the internalization mechanisms such as receptor mediated endocytosis and may further also modify the cell morphology index and potentiate acquired resistance in repetitive chemocycles.14,16,17Previously we have shown the nuclear DOX-enriching efficiency of modular NPs with or without cell-targeting moieties.14,18,19 DOX is a potent chemotherapy drug which inhibits topoisomerase-II resulting in cell death.20 DOX is known to elicit a direct cytotoxic effect in cancer cells mediated by reactive oxygen species (ROS) accumulation within the cytoplasm. Redox intermediates of DOX metabolism, particularly semiquinone lead to spontaneous free radical formation, in addition to DOX-mediated mitochondrial activity derangements. It is therefore interesting to evaluate the efficacy of DOX-nanocarriers in this context. Tunable sub-cellular localization would be an ideal characteristic for a drug delivery system, in that a drug may be delivered closer to its site of inhibition/activity, thus hastening the drug kinetics, improving the efficacy and potentially reducing the volume of distribution of the drug within the cell, thereby reducing the amount of drug required for dosing at the cellular level.
We assembled NP drug-carrier components, in the nano range, which involved a planar structure of GO sheets, spherical aggregates of Fe3O4 NPs and short double-stranded helical DNA to build multifunctional and efficacious DOX delivery systems. GO has been employed extensively owing to its improved solubility and it also provides a relatively large surface area for covalent conjugation reactions, to introduce reactive functional groups on GO sheets.21
Fe3O4 is a popular material for magnetically driven targeted drug delivery. This property is therefore utilized for the delivery of graphene conjugated DNA–DOX, targeted to cancer cells. Although the magnetic properties of the DOX nanocarrier have not been explored in this work, previous work in this lab has demonstrated the utility of magnetic NPs. Furthermore, Fe3O4 is known to be highly biocompatible with no toxic response in vivo.22,23 In addition to these traits, we have recently shown the self-propulsion property of Fe3O4 based NPs, specifically in the tumor microenvironment. This unique property has been previously shown to aid NP movement into and within the (in vitro) tumor spheroid.18 Fe3O4 is thus envisioned as a valuable tool in the current delivery system. DNA has been emerging as an efficient nanostructure for drug targeting and lowering drug toxicity and it can be engineered to optimize base sequence, shape, size and terminal active groups.24 Interestingly, DNA alone has restricted entry into cells due to its high negative charge which also results in its electrostatic repulsion with GO and the fact that DNA delivery into the nucleus generally entails chaperoned assistance; DNA may also be subject to endonuclease mediated degradation.25 Thus, to regulate the NP-cell kinetics, and localization control at nuclear, lysosomal and the cytoplasmic domains by the GO–DNA complex, DNA can be covalently conjugated onto GO sheets to create a stable nucleic-acid mediated delivery system for DOX.
We hypothesize that the varied multicomponent NPs modulated with bio-chemo-mediated entities including DNA will result in differential drug delivery mediated by an altered cellular internalization mechanism that may influence the nanosystem–drug influx kinetics, sub-cellular localization and ultimately alter the drug efflux kinetics. Further, such systems may affect plasma protein binding resulting in extended circulation time, improve the PK, enhance drug efficiency and ultimately lower the drug dose and frequency. In addition, the nano-carrier systems may by chemically modulated with DNA and DOX which may further influence the regulation and control over their cellular localization. Here, based on our earlier work, we further hypothesized that Fe3O4 as an additional nanocarrier along with graphene and DNA, will alter the cell entry kinetics for DOX. NPs such as Fe3O4 offer a chemo-structure–activity relationship for the delivery of pharmacologically active compounds through altered cell entry mechanisms similar to other entities such as PEGs, dendrimers, and carbon allotropes.3,6 Thus, while cell entry of DNA was inhibited in cells due to its high negative charge (particularly in live cells bearing negative charge on the outer cell membrane), it may additionally impart electrostatic repulsion in conjugation with graphene. The phenomenon necessitated the use of yet another component such as Fe3O4 which is independently known to be highly biocompatible and associated with enhanced cell entry dynamics. All together, the G–Fe3O4–DNA–DOX complex was expected to enhance the cell entry of the DNA system represented here which furthermore, would demonstrate the intercalated DNA–DOX effect on cellular retention kinetics and morphology. As in conventional chemotherapy the DNA–DOX delivery system(s) are expected to achieve incremental PK, targeting ability, enhanced efficacy and finally, reduced systemic toxicity. We have recently reported that such synergetic effects are notable when the nanocarriers deliver chemotherapy drugs, inducing cellular morphological changes which may translate into plausible kinetic perturbations in uptake of anticancer drugs leading to acquired resistance specially in repeated chemotherapy cycles for DOX.14
Thus, the objectives of this work were to: (1) enhance the drug delivery ability using GO and complementary Fe3O4 multicomponent differentiating architectures, to synthesize G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX and (2) achieve controlled cellular localization using the uniqueness of DNA's traits, including intercalation capacity for DOX, (3) evaluate nanosystems for their plasma protein binding ability, (4) study cellular kinetics, cell distribution and disposition at the nucleus, lysosomal region and the cytoplasm using HeLa cells, (5) assess the morphological changes induced in HeLa cells in terms of the nuclear surface area (NSA), cell surface area (CSA), cell roundness and nuclear to cytoplasmic (N/C) ratio, and (6) to compare nanosystem efficacy with the free form of DOX.
We report, multicomponent nanosystems mediated through GO conjugation and DNA–DOX intercalation, further coordinated with Fe3O4 NPs. We characterized G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX for their physicochemical traits such as structure, composition, functional groups, intercalation and conjugation chemistry, size and DOX loading. Adsorption of BSA and its further interaction with the nanosystems were evaluated via protein binding studies using the Stern–Volmer plot. We noted the modified DOX internalization mechanism influenced by the nanosystems. HeLa cells were employed to evaluate the cellular kinetics, drug distribution and nanosystem localization inside specific subcellular compartments. Specific subcellular compartment localization and morphological changes were noted at definite time points and correlated with other regulating parameters. Finally, the toxicity induced by the synthesized nanosystems was compared with free DOX.
Experimental section
Materials
Doxorubicin hydrochloride (DOX), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl), herring sperm DNA, BSA, ethylene diamine and imidazole were obtained from Sigma Aldrich. L-Cysteine hydrochloride monohydrate (Cys), was procured from HiMedia. Graphene (grade 2) was received from Global Nanotech. 4′,6-Diamidino-2-phenylindole (DAPI) was obtained from SRL Pvt. Ltd. 4-Dimethylaminopyridine, 98% (DMAP) was procured from Avra. Water filtered in a Millipore Direct Q-3 system (MilliQ) was used throughout. All other reagents used were of analytical grade.Synthesis of Fe3O4 NPs
Ferrous and ferric chlorides were dissolved in water in molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 0.3 M of Fe ions. The salts were then precipitated using 25% NH4OH solution at 25 °C, maintaining pH 10.0. The precipitate was subsequently heated to 80 °C for 30 minutes. Fe3O4 NPs were collected by magnetic decantation. The particles were washed repeatedly with ethanol and water and finally dried at 70 °C.26
:1 and 0.3 M of Fe ions. The salts were then precipitated using 25% NH4OH solution at 25 °C, maintaining pH 10.0. The precipitate was subsequently heated to 80 °C for 30 minutes. Fe3O4 NPs were collected by magnetic decantation. The particles were washed repeatedly with ethanol and water and finally dried at 70 °C.26
Synthesis of Cys–Fe3O4
5 mg Fe3O4 NPs were dispersed in a mixture of methanol and water at 1:3 (methanol:water) ratio. After 15 min sonication of the dispersion, Cys (4 mg) solution in water (800 μL) was poured into the Fe3O4 NPs suspension. The suspension was re-sonicated for 2 h. The resultant Cys–Fe3O4 complex was collected magnetically which also removed the unreacted Cys. It was then washed with repeated cycles of water, and finally dried under vacuum at room temperature.
Synthesis of GO
Graphene (200 mg) and 5 mL of 98% H2SO4 were constantly stirred for 2 h maintaining the mixture in an ice-bath between 0 and 5 °C. 600 mg KMnO4 was added in small quantities at regular intervals within 1 h, keeping the mixture below 20 °C throughout this reaction step. For the next two days the reaction was stirred at 35 °C. 10 mL water was poured into the above mixture forming a brown coloured solution. The solution was subsequently heated and maintained for 15 min at 98 °C. Another 20 mL of water was poured with continuous stirring. 1 mL H2O2 was finally added changing the solution to light brown in colour. The GO prepared was washed with HCl and water alternately several times and dried using a rotary evaporator.Synthesis of G–DNA–DOX–Fe3O4
5 mg of DNA was reacted for 24 h with 0.5 mg DOX to form a DNA–DOX complex. In a separate reaction, GO (5 mg) was activated for 1 h using EDC·HCl. Then the activated GO and solution of the DNA–DOX complex were mixed and stirred for 48 h. The carboxylic group of GO reacted with an end hydroxyl group of DNA to form an ester bond (molar ratio 1:1). Here a molar excess of DNA was used to react with GO, to ideally form an ester linkage with every –COOH group on GO. Further, the free –OH of DNA was reacted with the COOH of Cys on Fe3O4. Here it was assumed that all DNA added was conjugated to GO, and the free –OH group on DNA was conjugated to Cys–Fe3O4 with the free COOH group of Cys on Fe3O4. Finally the synthesised G–DNA–DOX–Fe3O4 was magnetically separated and washed several times to remove unreacted substances and dried at room temperature.
Synthesis of Fe3O4–DNA–DOX
DNA (5 mg) was activated for 1 h using EDC·HCl in PBS. Then imidazole was added in equivalent moles of phosphate groups of DNA and stirred for 24 h. In the next step, we added ethylene diamine and the reaction continued for 24 h resulting in formation of DNA with terminal NH2 groups. DOX (0.5 mg) reacted with the modified DNA for 24 h in water to obtain DNA–DOX. In a separate reaction, Cys–Fe3O4 was prepared as mentioned above and EDC·HCl activated the COOH groups of Cys. The DNA–DOX complex was added to Cys–Fe3O4 (1:2 molar ratio) and reacted for 48 h. The product was washed and collected using magnetic separation to finally obtain Fe3O4–DNA–DOX.
Characterization of synthesized nanosystems
A JASCO FTIR 4600 system was used for obtaining the Fourier transform infrared (FTIR) absorption spectra. The samples were scanned between 4000 cm−1 and 400 cm−1. Differential light scattering (DLS) using a Nano Partica SZ-100, Horiba was employed for determining the hydrodynamic particle size. FEI Tecnai G2 20-S Twin was used for transmission electron microscopy (TEM) operated at 200 kV. A Cary Eclipse, Varian spectrophotometer recorded the fluorescence emission spectra at different time intervals and concentrations. The excitation wavelength was 480 nm and the emission range was set from 500 nm to 650 nm for detection of DOX.Drug loading
The fluorescence intensity of intercalated DNA–DOX was determined using its calibration curve prepared under the same conditions. The free DOX present in the supernatant was calculated to determine the DOX bound to the nanosystem. Further the amount of DOX bound to the nanosystem was calculated as (total amount of DOX added during the reaction-amount of free unbound DOX in solution). The final synthesised product was dried and weighed to determine the drug loading capacity described as (weight of DOX conjugated to the nanosystem/weight of nanosystem).Protein binding studies using the Stern–Volmer plot
Protein binding of G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX and their components was determined by evaluating the fluorescence spectra after incubating the material with BSA. Stern–Volmer plots were established from the fluorescence signals of BSA and BSA adsorbed NPs. The excitation wavelength (λex) was considered at 280 nm, while the emission (λem) monochromator scanned the wavelength region between 300 and 450 nm. BSA (2 μM) used for the binding analysis, was incubated with the nanosystem components for about 30 min prior to the spectral measurements. Solutions or suspensions of the synthesized nanosystems G–DNA–DOX–Fe3O4, Fe3O4–DNA–DOX and individual components free GO, free DOX, free DNA and Fe3O4 NPs were prepared in PBS of pH 7.4 and added to the BSA solution to yield final concentrations of the conjugates as 2, 4, 6, 8, and 10 μg mL−1.Cell culture
HeLa cells were grown in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic. Cells were grown for 24 h and then G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX were added for cell viability assay and cell kinetics studies.Cellular kinetic studies
The cellular kinetics were determined using HeLa cells, grown in DMEM containing 10% FBS and 1% antibiotic. After 24 h incubation (5000 cells/180 μL), the cells were exposed to 20 μL free DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX. The cells were fixed at 1, 3, 6, 24 and 48 h and the nucleus was stained with DAPI (blue). An Axio Observer.A1, Zeiss, India fluorescence microscope was used for capturing images.Quantification of fluorescence intensity
To confirm the internalisation and accumulation of the nanosystems and free DOX inside the cells, the fluorescence intensity in the cytoplasm, lysosomal compartment and nucleus was quantified separately at 1, 3, 6 and 24 h time points. 50 cells at every time point were analysed using Carl Zeiss Zen proprietary software.Morphological studies on free DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX
The study was performed on HeLa cells, grown in DMEM containing 10% FBS and 1% antibiotic. Post 24 h incubation in a 96-well plate (5000 cells/180 μL), 20 μL GO, free DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX were added into these wells. The wells were fixed at 1, 3, 6, 24 and 48 h. The nucleus was stained with DAPI (blue). An Axio Observer.A1, Zeiss, India fluorescence microscope used for capturing images. Parameters such as the cell surface area (CSA), nuclear surface area (NSA), roundness and nuclear-to-cytoplasmic (N/C) ratio were measured using ImageJ software. Control cells were untreated HeLa cells. The N/C ratio was determined from the following equation:| N/C ratio = NSA/CSA | (1) |
Cytotoxicity studies on free DOX, GO, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX
5000 cells per well were seeded and maintained for 24 h in DMEM media. Free DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX were added in mole equivalent concentration of DOX into the wells. Also, GO was added in the concentration range 3–15 μg mL−1 and incubated with the cells for 48 h. In each well, 20 μL (5 mg mL−1) MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) was added and incubated with the cells for 4 h.The wells were later washed with PBS, carefully leaving the crystals at the bottom of each well. 100 μL of DMSO was added which dissolved the crystals and absorbance of this solution was noted at 570 nm in a Tecan Plate Reader. The DMSO filled well without any cells was taken as the blank reading. Cell viability (%) was computed from:
| (A × 100)/C | (2) |
The concentration of DOX in G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX equivalent to 0.2 μg mL−1 concentration of free DOX, was used to compare and determine the cytotoxicity of these nanosystems.
Statistical analysis
Three replicates of each treatment concentration were performed for each analysis. Values are the mean ± standard deviation of three independent experiments. For morphological analysis, the values are the mean ± standard error of mean. The two groups were compared for analyses using the unpaired Student t-test, with p < 0.05 taken as the value of statistical significance.Results
Synthesis and characterization of G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX (Fig. 1) were carried out through a series of multi-step reactions involving GO, Fe3O4 NPs, Cys, DNA and DOX. As shown in the synthesis scheme, graphene flakes were exposed to strong oxidising agents to form GO sheets with abundant carboxyl groups (Fig. 2a). An acid–base titration method estimated carboxyl groups generated on GO surfaces were determined to be ∼14% w/w of GO. Fe3O4 NPs were synthesised from ferrous and ferric salts using ammonium hydroxide to form a precipitate. The precipitate obtained was then heated and later washed and separated magnetically to obtain Fe3O4 NPs. Cys is an amino acid residue with multifunctional groups that is utilised as a linker for further chemical conjugations. Cys has active terminal groups viz., –COOH, –SH and –NH2.27 Therefore, in the next step, Fe3O4 NPs were conjugated to Cys via coordination chemistry (Fig. 2b) and Fe3O4 NPs interacted with the –SH group of Cys via the coordinate covalent bond. In another reaction, the phosphate group of DNA was selected for modification using a carbodiimide-facilitated reaction. The water-soluble carbodiimide EDC·HCl can react with the phosphate groups of DNA in the presence of imidazole to form a phosphorimidazolide intermediate which can rapidly couple with ethylene diamine to form a stable phosphoramidate linkage. In the next step, DOX was intercalated with the terminally modified DNA. Intercalation of DOX with DNA is known to occur via its anthraquinone ring structures which can fit between the base pairs of DNA.28 The terminal amine group of modified DNA was reacted with the free carboxyl group of Cys in the Cys–Fe3O4 complex to form Fe3O4–DNA–DOX (Fig. 2c). In this reaction Cys–Fe3O4 was capable of conjugating to both terminals of DNA through an amide linkage. Thus, a single DNA–DOX moiety is chemically sandwiched between two Cys–Fe3O4 complexes.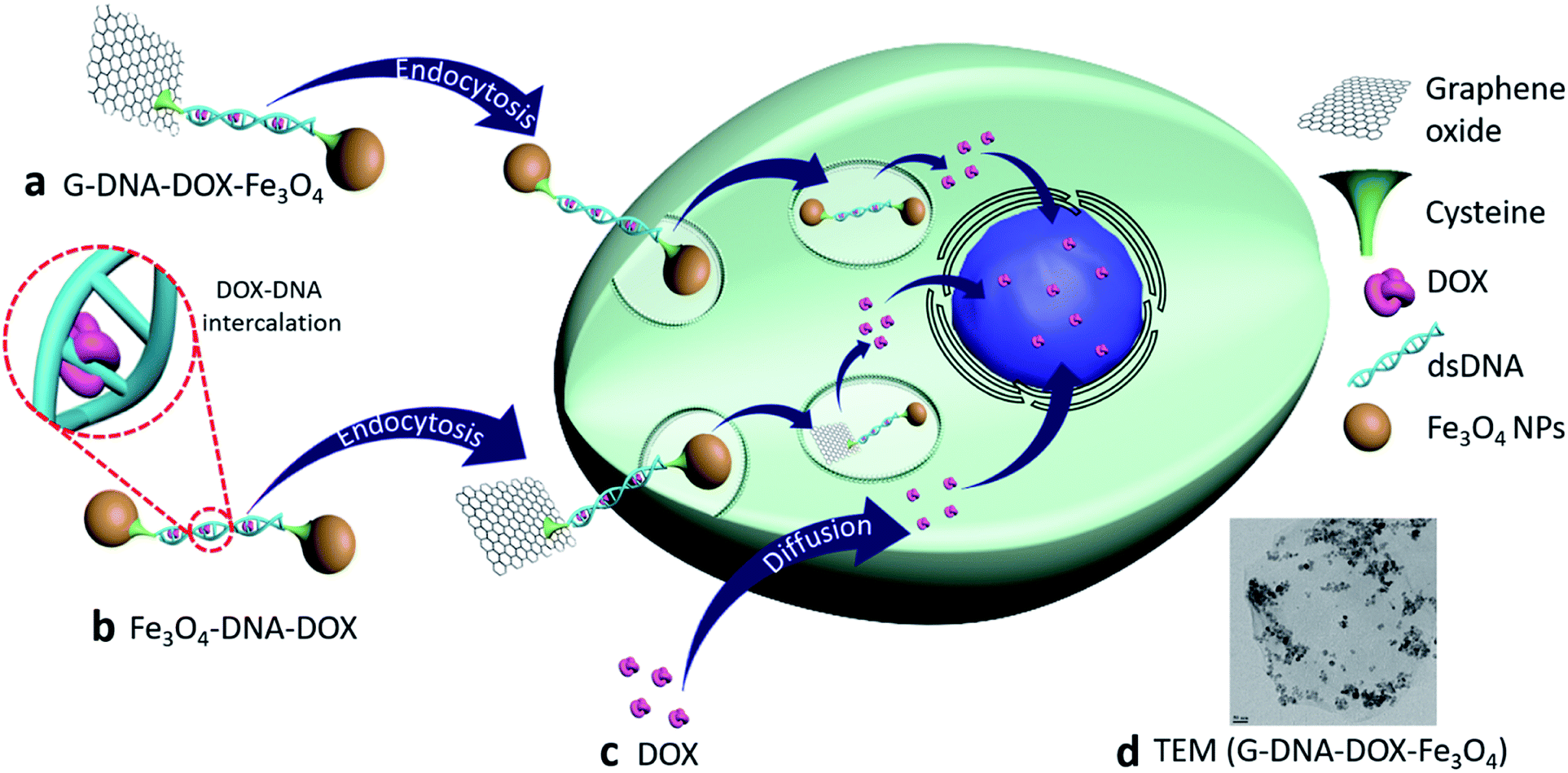 | ||
| Fig. 1 Multicomponent nanosystems composed of GO, Fe3O4 NPs and DOX intercalated to DNA (inset) namely (a), G–DNA–DOX–Fe3O4 and (b) Fe3O4–DNA–DOX were studied in comparison to (c) free DOX, (d) G–DNA–DOX–Fe3O4 was characterised using TEM and showed Fe3O4 NPs on the GO sheet. HeLa cells were exposed to free DOX and the synthesised nanosystems to study the morphological changes and the nanosystem localisation in subcellular compartments by quantifying the DOX intensity in the cells. | ||
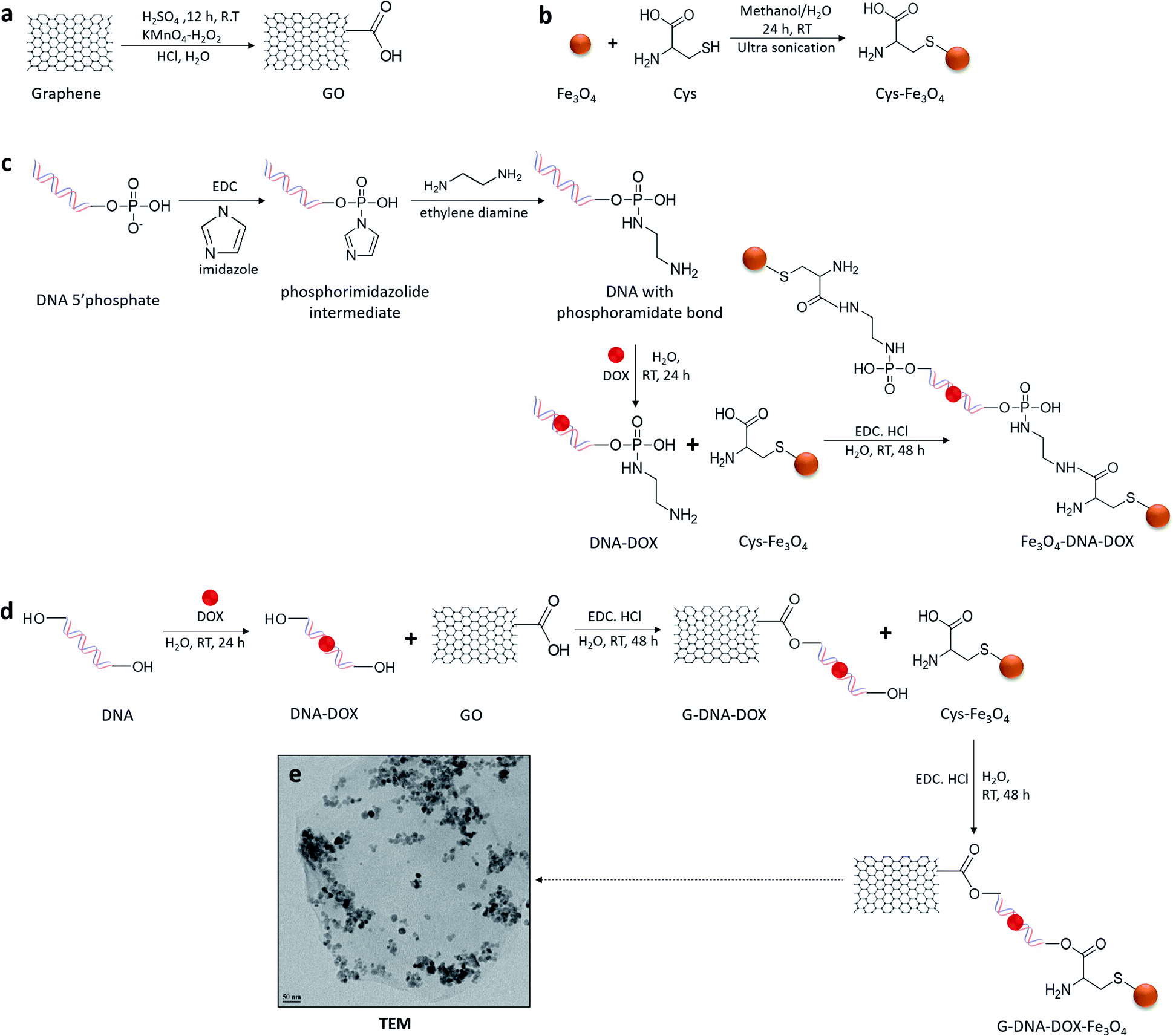 | ||
| Fig. 2 Synthetic scheme for DOX nanosystems. (a) Oxidation in the presence of strong acids resulted in abundant carboxyl groups on the surface of graphene. (b) Synthesis of Cys–Fe3O4 conjugate via formation of the coordinate bond between Fe3O4 NPs and the thiol group of Cys. (c) DNA modified with the terminal NH2 group was intercalated with DOX and further reacted with Cys–Fe3O4 and activated by EDC·HCl to produce Fe3O4–DNA–DOX. (d) DNA was intercalated with DOX and further conjugated to the carboxyl group of GO through an ester linkage, Cys–Fe3O4 was conjugated through the COOH group of Cys, to the hydroxyl group of DNA to form G–DNA–DOX–Fe3O4. (e) TEM image of G–DNA–DOX–Fe3O4. Scale bar represents 50 nm. | ||
In a separate reaction, GO sheets were dispersed into a solution of intercalated DNA–DOX, in the presence of EDC·HCl. This resulted in formation of an ester bond between carboxyl groups of GO and one hydroxyl terminal end of DNA. The other terminal hydroxyl group of DNA was esterified with the carboxyl group of the Cys–Fe3O4 complex to form G–DNA–DOX–Fe3O4 as the final product (Fig. 2d). Here, the DNA–DOX moiety was sandwiched between the GO sheet at one side and the Cys–Fe3O4 complex at the other side.
The size of the G–DNA–DOX–Fe3O4 nanosystem was determined using transmission electron microscopy (TEM) (Fig. 2e). Fe3O4 NPs were mostly anchored to the edges of GO sheets. TEM images showed the size of Fe3O4 NPs between 11 and 23 nm.
G–DNA–DOX–Fe3O4, Fe3O4–DNA–DOX and all intermediate conjugates were assessed by FTIR spectroscopy (Fig. S1†). A broad peak at 3432 cm−1 (O–H) and peaks at 1731 cm−1 (carboxylic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) confirmed oxygen functionalities on GO. Peaks at 614 cm−1, 601 cm−1 and 602 cm−1 observed due to the Fe–O band were present in the spectrum of Cys–Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 respectively. In DNA there were several strong spectral absorption peaks. 1696 cm−1 corresponded to purine and pyrimidine rings. 1219 cm−1 was attributed to DNA deoxyribose sugar. The spectral region between 1200 and 800 cm−1 corresponded to phosphate groups. The peak at 965 cm−1 was due to the O–P–O bending of the backbone of DNA, which was also observed at 983 cm−1 (Fe3O4–DNA–DOX) and 941 cm−1 (G–DNA–DOX–Fe3O4). 1058 cm−1 was due to C–O of deoxyribose in the DNA spectra. A new peak at 1747 cm−1 for G–DNA–DOX–Fe3O4 was due to the formation of an ester bond. A new peak at 1737 cm−1 corresponded to formation of an amide bond in Fe3O4–DNA–DOX. In Cys–Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4, peaks between 1041 and 1407 cm−1 denoted the presence of Cys.
O) confirmed oxygen functionalities on GO. Peaks at 614 cm−1, 601 cm−1 and 602 cm−1 observed due to the Fe–O band were present in the spectrum of Cys–Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 respectively. In DNA there were several strong spectral absorption peaks. 1696 cm−1 corresponded to purine and pyrimidine rings. 1219 cm−1 was attributed to DNA deoxyribose sugar. The spectral region between 1200 and 800 cm−1 corresponded to phosphate groups. The peak at 965 cm−1 was due to the O–P–O bending of the backbone of DNA, which was also observed at 983 cm−1 (Fe3O4–DNA–DOX) and 941 cm−1 (G–DNA–DOX–Fe3O4). 1058 cm−1 was due to C–O of deoxyribose in the DNA spectra. A new peak at 1747 cm−1 for G–DNA–DOX–Fe3O4 was due to the formation of an ester bond. A new peak at 1737 cm−1 corresponded to formation of an amide bond in Fe3O4–DNA–DOX. In Cys–Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4, peaks between 1041 and 1407 cm−1 denoted the presence of Cys.
The hydrodynamic size of Cys–Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 were determined using DLS analysis. The average diameters of Fe3O4 NPs, GO, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 in water were approximately 19.0 ± 4.8 nm, 680.7 ± 10.5 nm, 41.2 ± 11.6 nm, 712.9 ± 14.4 nm respectively (Fig. S2†). The results indicated an increased average diameter of G–DNA–DOX–Fe3O4 as compared to GO, confirming conjugation of Fe3O4 NPs on GO.
The fluorescence studies showed the emission wavelength (λem) of free DOX at 591.10 nm, while the synthesised nanosystems showed a red shift in λem at 596.95 nm for both Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 (Fig. S3†). The data confirmed the presence of DOX in the synthesised nanosystems and the bathochromic shift in the spectra suggested successful intercalation of DOX to DNA.
Drug intercalation and loading in DNA based NPs
The calibration curve of intercalated DNA–DOX was determined to calculate the unbound DNA–DOX in the supernatant (Fig. S4†). The calibration curve was obtained such that DNA:DOX was maintained at a 10:1 ratio with DNA concentrations ranging between 50 and 400 μg mL−1 (and therefore DOX concentrations were between 5 and 40 μg mL−1). Accordingly, the amount of free DOX was determined and the amount of bound DOX was estimated. The drug loading capacity was calculated to be 25 μg mg−1 and 18 μg mg−1 for Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 respectively which corresponded to ∼28% higher DOX loading in Fe3O4–DNA–DOX compared to G–DNA–DOX–Fe3O4.
Protein binding studies using the Stern–Volmer plot
The binding of DNA, DOX, GO, Fe3O4 NPs, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 to BSA were established from the tryptophan fluorescence emission of BSA. The tryptophan residue was selected as the binding component in BSA and the extent of quenching interactions were determined via changes in its fluorescence characteristics. The fluorescence spectra of BSA were recorded at different concentrations ranging from 40 to 250 μg mL−1 at a characteristic emission wavelength of 347.07 nm (λem of tryptophan) (Fig. S5†). We further measured the fluorescence of BSA after incubation for 30 min with DNA, DOX, GO, Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 to estimate the extent of fluorescence quenching of BSA. The fluorescence emission of BSA, excited at 280 nm was inversely proportional to its interaction with the ligands/drug concentration (Fig. 3a–f).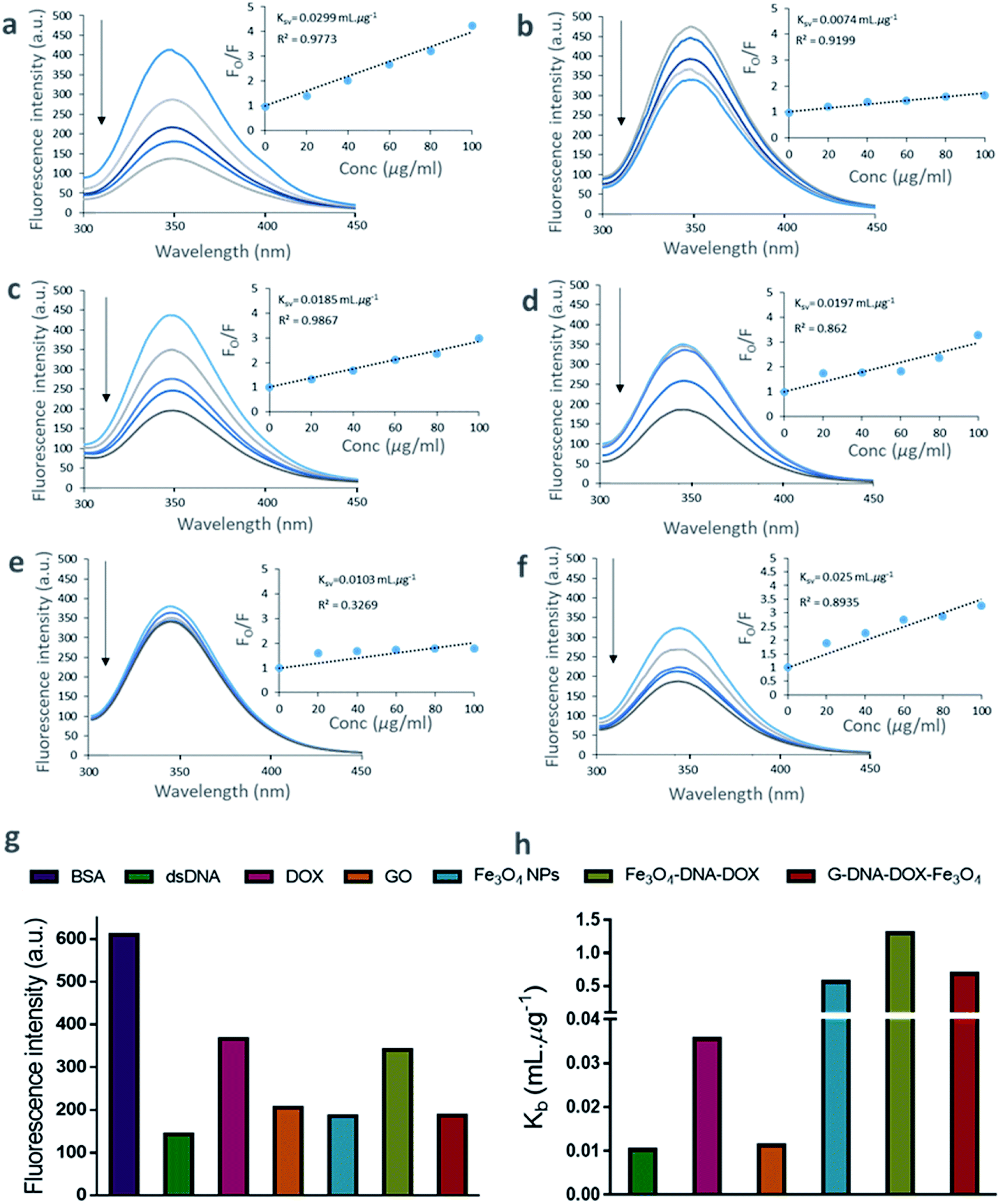 | ||
| Fig. 3 Fluorescence emission spectra and Stern–Volmer plots for BSA–ligand interactions. Representative fluorescence emission spectra of (a) DNA, (b) DOX, (c) GO, (d) Fe3O4 NPs, (e) Fe3O4–DNA–DOX and (f) G–DNA–DOX–Fe3O4. The concentration of BSA was fixed at 2 μM while those of the individual components and synthesized nanosystems were 20, 40, 60, 80 and 100 μg mL−1. The corresponding Stern–Volmer plots for binding of BSA with the various ligands are shown against each fluorescence spectrum. Fo/F is the ratio of the fluorescence intensity of BSA in the absence of ligand to the fluorescence intensity of BSA in the presence of ligand. Graphs comparing the (g) fluorescence quenching intensities of BSA due to interaction with various ligands at 100 μg mL−1, and (h) binding constant (Kb). | ||
A substantial proportion of drug and nanosystem components interacted with BSA contributing to a significant decrease in the detected signal. The decrease in fluorescence intensity with increase in ligand concentration is evident from the quenching profiles corresponding to the ligand concentrations (20–100 μg mL−1). A comparison between the λem intensity of various quenchers (at 100 μg mL−1) and BSA (2 μM) is displayed in Fig. 3g. DNA showed a sharp decrease in fluorescence intensity (∼76.60%) within 30 min. Similarly, Fe3O4, GO and DOX showed ∼69.60%, ∼66.35% and ∼40.01% decreases in signal respectively. ∼44.20% and ∼69.38% decreases in λem intensity were observed for Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 respectively. The observations showed that significant interactions occurred within 30 minutes. The quenching data of BSA–ligand interactions were determined from Stern–Volmer plots. The Ksv was estimated according to the Stern–Volmer equation:29
| Fo/F = 1 + Ksv[Q], | (3) |
| log[(Fo/F)/F] = logKb + nlogQ, | (4) |
The plot of log[(Fo/F)/F] as a function of logQ is shown in Fig. S6.† The values of Kb were obtained from the intercept of the plot.30 Thus, Kb values were 0.0102 mL μg−1 (DNA), 0.0355 mL μg−1 (DOX), 0.0112 mL μg−1 (GO), 0.5619 mL μg−1 (Fe3O4), 1.2999 mL μg−1 (Fe3O4–DNA–DOX), and 0.6830 mL μg−1 (G–DNA–DOX–Fe3O4) (Fig. 3h).
The plot of Fo/F against Q (concentration of ligands in μg mL−1) is linear for DNA, DOX and GO bound to BSA. Fe3O4, Fe3O4–DNA–DOX, and G–DNA–DOX–Fe3O4 bound to BSA showed deviation from linearity using the linear Stern–Volmer equation.
The linearity demonstrated by Fo/F is indicative of concentration dependent binding of the components to BSA, whereas the non-linear binding curves were suggestive of polyphasic binding kinetics. Specifically, as Fig. 2e depicts, Fe3O4–DNA–DOX deviates significantly from the concentration-dependent binding model; Fe3O4–DNA–DOX likely has a poor affinity for BSA leading to extremely limited protein binding. On the other hand, Fe3O4 demonstrates a multi-order protein binding curve, which is reflected in the Fo/F curve for G–DNA–DOX–Fe3O4 as well.
We also noted that fluorescence quenching observed upon addition of the NPs, showed no spectral shift in the fluorescence λem of BSA, within the concentration range studied. This suggested that conformational changes around the tryptophan residue of BSA did not occur due to ligand interactions.
Cellular uptake kinetics and cell organelle localization studies
HeLa cells were incubated with Fe3O4–DNA–DOX, G–DNA–DOX–Fe3O4 and free DOX to compare the efficiency in cellular uptake of DOX from the synthesized nanosystems (Fig. 4a–c). Fluorescence microscopy imaging was conducted at 1 h, 3 h, 6 h, 24 h and 48 h post transfection. The treated HeLa cells were washed prior to imaging in order to eliminate any extracellular NPs adhering to the cell membrane. Under identical imaging conditions, non-treated HeLa cells did not show any apparent observable auto fluorescence. The rate of cellular uptake and DOX localization within the cells were assessed from comparing the relative fluorescence (emission) intensity (RFI) within the nuclear (Fig. 4d) and cytoplasmic compartments (Fig. 4e) at each time point post transfection. Free DOX was found to localize in the nucleus after 24 h, confirmed by the colocalization with the fluorescence signal of DAPI. Moreover, a rapid increase in intensity of free DOX in the nucleus within 1 h (14.66 ± 5.85 RFI) to 24 h (70.29 ± 8.90 RFI) was observed indicating a rapid diffusion of DOX, contrary to the negligible rise in intensity of DOX (upto 4.86 ± 2.55 RFI in 24 h) in the cytoplasm, suggesting that free DOX preferably accumulated in the nucleus.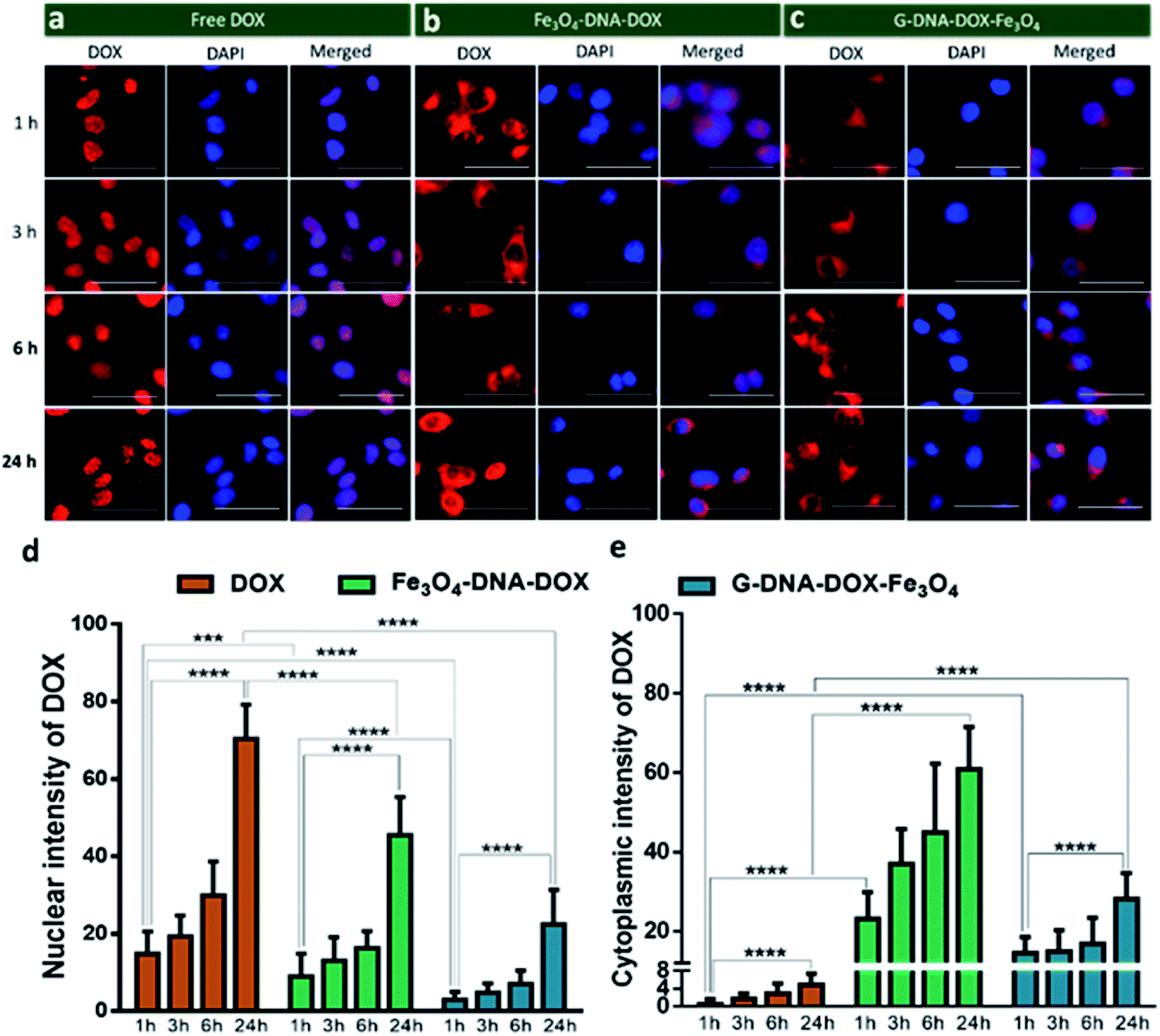 | ||
| Fig. 4 Kinetics of synthesised nanosystems in HeLa cells. Fluorescence images of HeLa cells treated with (a) free DOX, (b) Fe3O4–DNA–DOX and (c) G–DNA–DOX–Fe3O4, and imaged after 1 h, 3 h, 6 h, and 24 h. Scale bar is 50 μm. (d) Nuclear intensity of DOX and (e) cytoplasmic intensity of DOX estimated from fluorescence intensity at each time point. The values were statistically analysed using the unpaired Student t-test, **** represents p ≤ 0.0001 and *** represents p ≤ 0.001. | ||
In the case of Fe3O4–DNA–DOX, the intensity of DOX in the nucleus was significantly low after 24 h (45.42 ± 9.85 RFI) as compared to free DOX (p ≤ 0.0001). Although a steady increase in nuclear intensity of DOX was noted, the cytoplasmic intensity of DOX ranged between 23.18 ± 6.69 RFI (1 h) to 60.91 ± 10.54 RFI (24 h). Similarly G–DNA–DOX–Fe3O4 also had extremely low nuclear intensity upto 6 h (6.97 ± 3.43 RFI) which increased to 22.38 ± 8.93 RFI at 24 h; whereas in the cytoplasm, the intensity was steady between 14.49 ± 4.09 RFI (1 h) to 28.18 ± 6.45 RFI (24 h).
There was ∼64.28% and ∼26.12% increase in DOX intensity from 6 h to 24 h in the nucleus and cytoplasm respectively for Fe3O4–DNA–DOX. Similarly, ∼69.17% and ∼40.21% increase in DOX intensity in the nucleus and cytoplasm respectively was observed in G–DNA–DOX–Fe3O4 from 6 h to 24 h time points. However on comparing the nuclear DOX intensity for both nanosystems at 24 h, there was a ∼2.0 fold higher DOX intensity for Fe3O4–DNA–DOX than for G–DNA–DOX–Fe3O4 whereas a ∼2.2 fold higher DOX intensity in the cytoplasm was observed for Fe3O4–DNA–DOX compared to G–DNA–DOX–Fe3O4. Similarly, comparing the cytoplasm and nuclear intensities after 24 h, DOX primarily accumulated in the nucleus with ∼14 times higher nuclear intensity and negligible localization in the cytoplasm. In the case of Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4, there was ∼1.3 and 1.2 times greater cytoplasmic intensity compared to the nucleus.
In Fig. 5, comparison of the nuclear intensity of DOX at 1 h, 3 h, 6 h and 24 h in free DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX is depicted. RFI was measured using ImageJ software whereby the red fluorescence intensity at various locations within the cell was measured. The background space devoid of the cell was considered to be RFI = 0. On the cell, three areas were selected namely, nucleus, lysosomal region and the cytoplasm. RFI measurements showed that in free DOX exposed cells, RFI was the highest in the nucleus at all time points (∼12 at 1 h, ∼46 at 3 h, ∼107 at 6 h, ∼115 at 24 h) with no fluorescence in the lysosomal region or the cytoplasm.
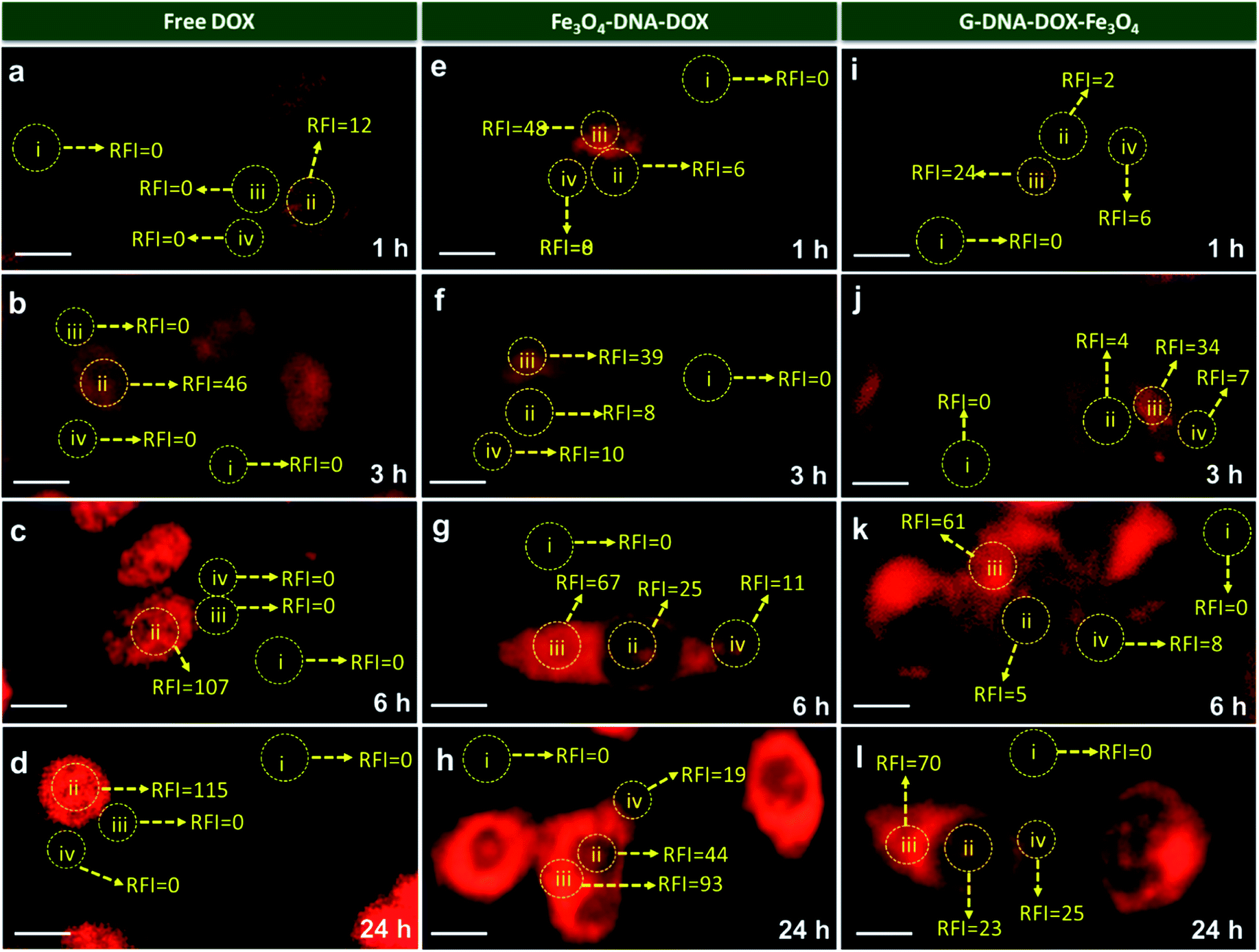 | ||
| Fig. 5 Fluorescence microscope images of HeLa cells imaged at 1 h, 3 h, 6 h and 24 h time points after treatment with (a–d) free DOX, (e–h) Fe3O4–DNA–DOX and (i–l) G–DNA–DOX–Fe3O4. The relative fluorescence intensity (RFI) was measured at four regions i.e. (i) background, (ii) nucleus, (iii) lysosome, and (iv) cytoplasm. Yellow dotted circles indicate the region of interest chosen to measure the RFI. Scale bar is 10 μm. | ||
For cells exposed to Fe3O4–DNA–DOX, nuclear RFI at 1 h was ∼6 which steadily increased to ∼8 at 3 h, ∼25 at 6 h and ∼44 within 24 h depicting a steady rise in concentration of DOX in the nucleus. RFI in the cytoplasm was low throughout (∼19 at 24 h) and there was increased accumulation of Fe3O4–DNA–DOX in the lysosome (∼93 at 24 h). In the case of G–DNA–DOX–Fe3O4, up to 6 h, the nuclear RFI was low at ∼5, with a significantly higher RFI in the lysosome (∼61 at 6 h). At 24 h, RFI in the lysosome was stable around ∼70 with considerable increased RFI detected in the cytoplasm (∼25) and nucleus (∼23). Upon comparison of RFI in the nucleus at 24 h for G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX, nearly two-fold increased RFI was observed for Fe3O4–DNA–DOX and also its cytoplasmic RFI (∼19) was significantly lower than in the nucleus (∼44). RFI values in the case of free DOX have shown maximum intensity in the nucleus at all the time points.
In Table S1,† the statistically significant values of RFI at the nucleus, lysosome and cytoplasm of HeLa cells, over all time points analysed (as shown in Fig. 5), have been listed. The most distinct observation is the elevated levels of DOX in the lysosomal compartment, attributable to the Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4.
The other significant finding is the free drug content in the nucleus which accumulates within 24 h, attributed to free DOX movement, whereas G–DNA–DOX–Fe3O4 is effective in time-dependent restriction of nuclear DOX-loading. As noted, the GO and DNA provide a duality of physical mechanisms which allow selective retention of the payload drug/molecule in a compartment-specific manner. At 48 h, the DOX signal continued to be observable in the lysosomal region (Fig. S8†) for G–DNA–DOX–Fe3O4. Significantly high DOX intensity was noted in the nucleus for Fe3O4–DNA–DOX.
Cell morphological studies
HeLa cells were incubated with GO, free DOX, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 to examine the morphological alterations at each time point (1, 3, 6 and 24 h) and untreated HeLa cells were considered to be the control (Fig. S9†). Statistical differences were compared for morphological changes at 6 h and 24 h for all parameters measured (Fig. 6). CSA of control cells varied from 733.85 ± 22.37 μm2 (at 1 h) to 759.34 ± 42.13 μm2 (at 24 h) and no significant difference in CSA was observed for cells exposed to GO. However, in the case of DOX exposure, there was a drastic reduction in the CSA from 436.84 ± 4.8 μm2 at 1 h to 273.47 ± 10.89 μm2 at 24 h. With Fe3O4–DNA–DOX, a significant decrease in CSA was observed from 6 h (607.46 ± 38.34 μm2) to 24 h (378.17 ± 35.53 μm2). Similarly, for G–DNA–DOX–Fe3O4, a significant change in CSA was observed from 6 h (726.93 ± 23.52 μm2) to 24 h (480.22 ± 45.16 μm2). The highest reduction in CSA was observed for DOX (∼64%), followed by Fe3O4–DNA–DOX (∼50%) and G–DNA–DOX–Fe3O4 (∼36%) when compared to control cells at the end of 24 h.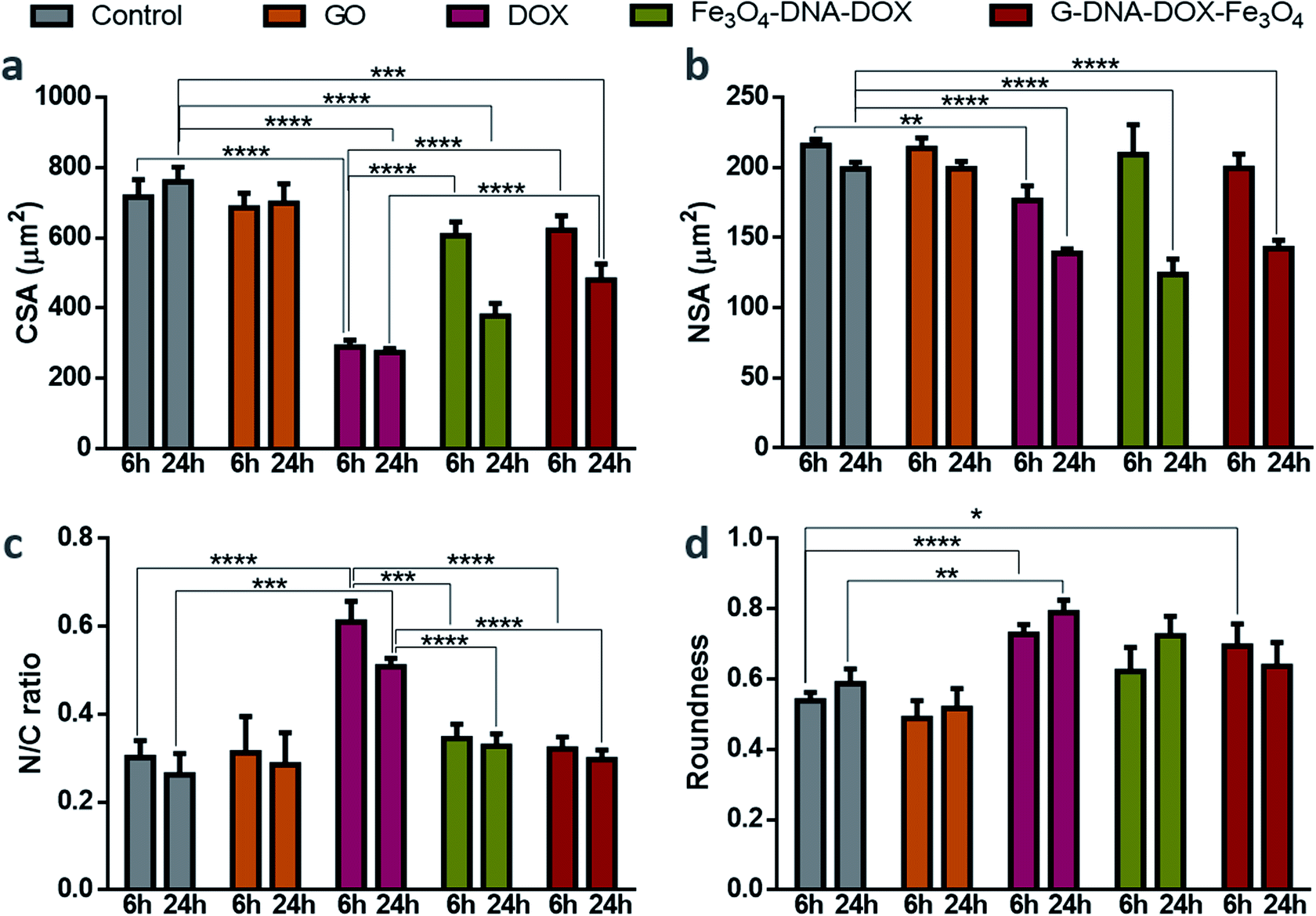 | ||
| Fig. 6 Morphological parameters (a) CSA, (b) NSA, (c) N/C ratio and (d) roundness of HeLa cells compared at 6 and 24 h. The values were statistically analyzed using a two-tailed unpaired t-test, **** represents p ≤ 0.0001, *** represents p ≤ 0.001, ** represents p ≤ 0.01 and * represents p ≤ 0.05. | ||
In the case of NSA, the control HeLa cells and GO maintained NSA around 200 μm2 from 1 h through 24 h. Free DOX treated cells displayed nuclear bulging upto 3 h (from 181.02 ± 6.95 μm2 to 205.53 ± 8.78 μm2) and then gradually decreased to 139.00 ± 2.90 μm2 in 24 h. Fe3O4–DNA–DOX showed enhanced NSA upto 6 h (209.51 ± 21.02 μm2) and then a significant reduction upto 24 h (123.78 ± 11.14 μm2). In the case of G–DNA–DOX–Fe3O4, NSA decreased from 203.60 ± 14.23 μm2 at 1 h to 142.47 ± 5.72 μm2 at 24 h. In comparison to control cells at 24 h, an overall significant decrease in NSA was observed for free DOX, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4.
The N/C ratio of DOX was significantly increased compared to control HeLa cells at 6 h (0.61 ± 0.04 for DOX and 0.30 ± 0.03 for control) and 24 h (0.50 ± 0.2 for DOX and 0.26 ± 0.05 for control). Similarly, a significant difference with DOX was observed for Fe3O4–DNA–DOX (0.33 ± 0.03) and G–DNA–DOX–Fe3O4 (0.30 ± 0.02) at 24 h.
DOX demonstrated a significantly increased cell roundness (0.73 ± 0.03) compared to control (0.54 ± 0.02) at 6 h. Roundness of cells exposed to free DOX (0.79 ± 0.03) and Fe3O4–DNA–DOX (0.72 ± 0.07) increased after 24 h while G–DNA–DOX–Fe3O4 had a slightly lower roundness at 0.64 ± 0.07. Multiple membrane blebs were another morphological feature observed on HeLa cells exposed to G–DNA–DOX–Fe3O4 (Fig. S10†).
At the end of 48 h, the cells exposed to Fe3O4–DNA–DOX (Fig. S8a†) were shrunken with strikingly low NSA and CSA, approaching a roundness value close to 1 (near-spherical), indicating a propensity to detach from the tissue culture substrate.
Cytotoxicity assay
Different concentrations of GO were added to the HeLa cells, incubated for 48 h and then assessed for cytotoxicity using MTT reagent. GO showed a dose dependent decrease in cell viability. The cell viability varied between 93.76 ± 0.11% and 78.50 ± 5.12% as concentration increased from 3 to 15 μg mL−1 (Fig. 7a). Thus, GO was found to have a low toxic effect at the concentration used in HeLa cells. Further, cell viability was evaluated by maintaining equivalent concentration of DOX at 0.2 μg mL−1 for free DOX, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX, under similar experimental conditions (Fig. 7b). Free DOX (0.2 μg mL−1) showed 67.29 ± 7.04% cell viability. Fe3O4–DNA–DOX (11 μg mL−1) showed a significant decrease in cell viability (33.62 ± 3.03%) compared to free DOX (p < 0.01) and G–DNA–DOX–Fe3O4 (p < 0.05) treated cells. G–DNA–DOX–Fe3O4 (8 μg mL−1) also showed significantly decreased cell viability (48.75 ± 8.03%) with p < 0.05 compared to free DOX. Thus, in the case of G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX, the synthesized nanosystems with incorporated DOX enhanced the cytotoxicity compared to free DOX.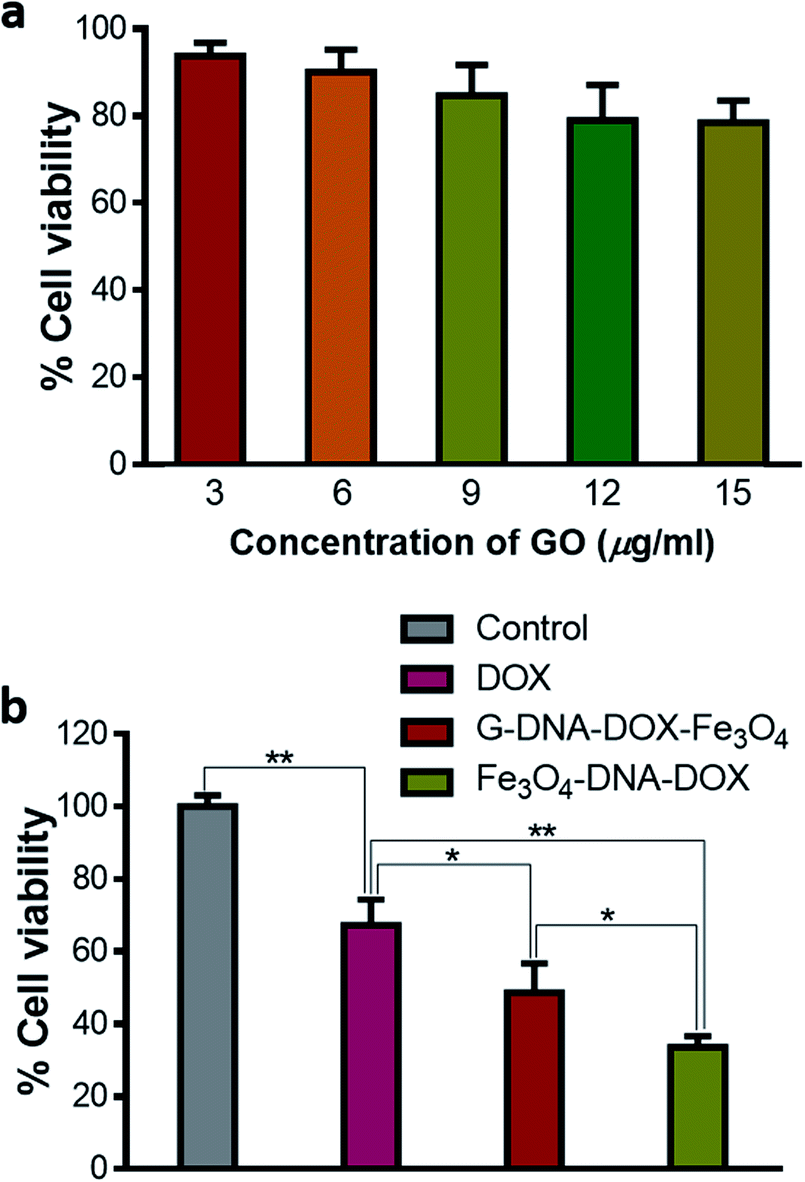 | ||
| Fig. 7 MTT assay. (a) Effect of GO (3, 6, 9, 12, and 15 μg mL−1) on the cell viability of HeLa cells. (b) Cell viability assay showed Fe3O4–DNA–DOX (33.62 ± 3.03% cell viability) to be the most toxic, followed by G–DNA–DOX–Fe3O4 showing 48.75 ± 8.03% cell viability. Both the synthesized nanosystems were more toxic than free DOX (67.29 ± 7.04% cell viability). The values were statistically analyzed using the unpaired Student t-test, ** represents p ≤ 0.01 and * represents p ≤ 0.05. | ||
Thus, G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX were successfully synthesized and covalent conjugation was confirmed from FTIR. Particles were in the nano-size range and TEM showed the presence of Fe3O4 NPs on the GO sheet. DOX intercalation with DNA was confirmed from the fluorescence spectral shift to a higher wavelength in λem of DOX. BSA adsorption on G–DNA–DOX–Fe3O4 was greater compared to Fe3O4–DNA–DOX measured after BSA interaction for 30 min. After 24 h incubation of HeLa cells with free DOX, DOX accumulated in the nucleus with no significant traces in the cytoplasm. G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX preferably localized in the lysosome with gradual release of DOX into the nucleus. Morphological alterations were maximum in free DOX, with a drastic decrease in CSA and NSA with increased N/C ratio and cell roundness. Cytotoxicity to HeLa cells was computed in the order DOX < G–DNA–DOX–Fe3O4 < Fe3O4–DNA–DOX.
Discussion
The flexibility in incorporation of multifunctional NPs of varied dimensions to build the nanosystems helped regulate their activity and cytotoxic strategies within the cells. For instance, in both synthesized nanosystems, the double stranded DNA strongly intercalated with DOX. Further addition of a planar component such as a GO sheet drastically modified the cell uptake, cytotoxicity and residence time of DOX in various cell organelles.In the course of NP fabrication, covalent bonding between DNA and GO is crucial to maintain DNA stability, owing to electrostatic repulsion between DNA and GO sheets.31 Shielding of DNA within the nanosystems serves to prevent occurrence of enzymatic degradation when internalised into the cells.32,33 Hence, the synthetic schemes for both Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 were designed such that DNA would be sandwiched between the nanosystem components, GO or Fe3O4via covalent bonding. Intercalation of DOX within DNA rendered the DOX release from the nanosystems dependent on the degradation of DNA within the NPs. Release studies were conducted at pH 7.4 and pH 5.5 for G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX, mimicking physiological and lysosomal pH conditions (Nandi et al., unpublished data). At both pH states, poor DOX release was observed which implied the pH stability of the DNA–DOX complex. However, DOX release was observed within the cells which can be majorly attributed to the intracellular/lysosomal enzymes such as endonucleases that cleave the DNA strands, thereby releasing DOX within cell compartments containing the NPs.
A comparison between the drug loading capacities of the present G–DNA–DOX–Fe3O4 nanosystem with our previously described DOX bearing nanosystem, G–Cys–Fe3O4–DOX reveals interesting differences (Table S2†). G–DNA–DOX–Fe3O4, with DOX bound to DNA, had ∼20% higher DOX loading than G–Cys–Fe3O4–DOX, which had DOX directly bound to the GO sheets. DNA intercalation with the anthracycline nucleus of DOX occurs between a guanine–cytosine nucleotide pair and consequently, multiple DOX molecules can be bound to the ample guanine–cytosine sites within a single DNA molecule.34 DNA therefore proved to be an efficient drug carrier which increased the loading capacity of DOX compared to the DOX carrying capacity for GO alone.
Quenching of BSA–tryptophan fluorescence was utilized as a validated probing tool that allows quantification of DOX–nanosystems interactions with serum blood and cell media proteins represented here by BSA. The fluorescence intensity (λem) of BSA in the presence of DNA, DOX, GO, Fe3O4 NPs, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 (Fig. 3) was determined at incremental concentrations. BSA in the presence of (free) DNA, DOX and GO showed a linearly decreasing fluorescence varying indirectly with the incremental nanomaterial content. The progressive reduction in the λem intensity of BSA was an outcome of adsorption of the NPs and DOX onto BSA, arising from the greater accessibility of the tryptophan residue to the nanosystems leading to stronger association between the NPs and BSA. However, in the case of Fe3O4, Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4, the BSA-binding plots were non-linear, suggestive of poor BSA: Fe3O4–DNA–DOX affinity. An unbound NP fraction implied by the above result indicated the greater availability of Fe3O4–DNA–DOX, as compared to G–DNA–DOX–Fe3O4 NPs for targeting HeLa cells; which corresponds with the greater cytotoxic effect of Fe3O4–DNA–DOX on cancer cells.
Despite the apparent BSA-binding capacity, G–DNA–DOX–Fe3O4 had higher cytotoxicity (48.75 ± 8.03% cell viability) compared to free DOX (67.29 ± 7.04% cell viability), although lower than the cytotoxicity indicated for Fe3O4–DNA–DOX (Fig. 7). It is likely that the GO terminal of the nanosystem is able to demonstrate significant protein binding comparable to that of free GO (Fig. 3), however it is conceivable that the transient nature of the binding35 may allow a reversible binding of the BSA:nanosystem pair. Such a reversible mechanism may allow G–DNA–DOX–Fe3O4 to be stabilized in the circulation system and serve as a reservoir for dynamic nanosystem blood content.
Comparison of the DOX-laden DNA nanosystems provided insights into the mechanism of the nanosystem–HeLa cell interaction. Upon incubating the cells with the DOX–nanosystems for 24 h, the DOX intensity was significantly increased in the nucleus for both Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4. The observed nuclear accumulation was likely due to the cleavage of the amide bond in Fe3O4–DNA–DOX and the ester bonds in G–DNA–DOX–Fe3O4 NPs due to the dual effect of the acidic digestion within the lysosomes and the presence of proteolytic lysosomal enzymes.36,37 The resulting exposure of the DNA–DOX intercalated complex within the cell's microenvironment likely enabled subsequent endonuclease mediated DNA degradation, yielding free DOX.38 In this context, it may be inferred that the lower cytotoxicity of G–DNA–DOX–Fe3O4 compared to Fe3O4–DNA–DOX may likely arise from its relatively lower nuclear and cytosolic accumulation (Fig. 5). Fig. S8† shows the DNA:DOX signal of G–DNA–DOX–Fe3O4 retained in the lysosomal region after 48 h; in contrast, cells appear drastically shrunken and round when treated with Fe3O4–DNA–DOX with appreciable nuclear accumulation of DOX. Taken together, the results suggest a role of GO in providing a spatially stabilized microenvironment to protect the DNA–DOX complex against endonuclease mediated DNA degradation. Fe3O4–DNA–DOX, on the other hand, lacked the steric protection afforded by GO and subsequently bore greater susceptibility to enzymatic digestion of DNA.39 It is conceivable that the latter's limited protection from endonuclease degradation resulted in greater liberation of DOX from DNA which further underwent rapid nuclear diffusion, shown in Fig. S8a.†
RFI measurements of specific regions within a single cell demonstrated Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 to largely localize in the perinuclear compartments, specifically in the lysosomal compartment (Fig. 5). RFI denoted the presence of DOX fluorescence in the cytosol, with higher cytosol RFI measured for G–DNA–DOX–Fe3O4 compared to Fe3O4–DNA–DOX. In the case of free DOX, which rapidly diffused into the nucleus and was retained therein, inhibition of topoisomerase-II has been indicated as the primary mode of inducing cytotoxic activity.20 Under this mechanism of cell death, DOX causes nuclear bulging which was evident from the increased NSA in the first few hours of DOX exposure (Fig. S9b†). DOX also interferes and disrupts the nuclear membrane leading to loss of nucleus integrity which leads to shrinkage of the nuclear area (observed for NSA of DOX at 24 h) leading towards cell death.40 We have shown earlier14,18 that DOX is liberated from (non-DNA-conjugated) nanosystems upon their lysosomal processing; consequently, a greater cellular accumulation of DOX–nanosystems would be responsible for releasing a greater amount of DOX intracellularly (DOX-dumping) which would dramatically improve the nuclear uptake of the drug as compared to free DOX as well as the G–DOX.
Inclusion of DNA in the nanosystems distinctly altered their cellular compartmentalization. As seen in Fig. 4, the DOX fluorescence, attributable to DOX-laden nanosystems was observed to be concentrated in the lysosomal regions (distinctly lacking a diffuse DOX signal throughout the cell), while the DOX signal was significantly low in the nucleus. Specifically, the GO-lacking Fe3O4–DNA–DOX demonstrated enhanced DOX loading, faster internalization and significant cytotoxicity in treated HeLa cells. Fe3O4–DNA–DOX showed a low concentration of DOX in the nucleus (Fig. 5) suggesting a slow liberation of DOX from the lysosomal compartment, also indicated by a low but detectable DOX content in the cytosol. The low nuclear DOX accumulation, in the context of superior cytotoxicity of the NPs suggests an alternate pathway of cell death. Lower DOX concentrations are capable of destabilizing the nuclear lamina leading to NSA changes, evident in the present study from the changes in the nuclear morphology of Fe3O4–DNA–DOX.40 DOX is implicated in altered mitochondrial functions consistent with enhanced production of mitochondrial ROS. DOX is reduced to a semiquinone radical by the oxidoreductases present within the cell, which further undergoes spontaneous reoxidation producing ROS, ultimately leading to cell death.20 Dual-modality DOX activity via nuclear and extra-nuclear pathways may potentiate the lethality of DOX (via Fe3O4–DNA–DOX).
Conversely, the present study indicates that G–DNA–DOX–Fe3O4 had lower nuclear DOX accumulation than its counterpart but significantly higher cytosolic DOX content suggesting that the toxicity borne by G–DNA–DOX–Fe3O4 was primarily elicited due to cytosolic DOX. The resulting lethal effects of cytosolic DOX were also evident from the terminal decrease in CSA for both Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 treatments. Further, the sharply reduced nuclear DOX levels for both nanosystems, juxtapositioned with their cytotoxic effect (∼48% and 33% cell viability for G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX respectively), in comparison with free drug administration, suggested a time dependent DOX reservoir effect which allowed DOX to be slowly liberated from the nanosystems over time leading to their cytotoxic outcome. The appearance of membrane blebbing was another important feature to infer the apoptotic effect attributable to the nanosystem (Fig. S10†).
We have previously reported G–DOX and G–Cys–Fe3O4–DOX (without DNA) to demonstrate a marked increase in nuclear accumulation within 4 h of nanosystem incubation in HeLa cells.14 Here, incorporation of nucleic acid into the NP design was correlated to lysosomal retention of the DNA-bound DOX. Fe3O4–DNA–DOX was the most toxic to HeLa cells and also demonstrated higher cytoplasmic as well as nuclear DOX intensity over G–DNA–DOX–Fe3O4, implying improved cellular internalization of Fe3O4–DNA–DOX, and its comparatively superior ability to release DOX from the nanosystem. Additionally, improved internalization of Fe3O4–DNA–DOX could be attributed to its evidently smaller size range (41.2 ± 11.6 nm) compared to G–DNA–DOX–Fe3O4.
Taken together, the current work and our previously published results yield interesting insights into the localization control of the nanosystems and the roles of individual biomaterial components in cellular internalization and pharmacological efficiency such as the controlled release of DOX from its nanoconjugate systems, improved nanosystem biocompatibility and increased cytotoxicity.14,18 The results illustrate the sub-cellular localization and prolonged retention of DNA-incorporating nanosystems, whereas non-nucleic acid NPs can demonstrate nuclear localization of DOX in about 4 h, depending on the cell type.
It is likely that protein binding may retard the access of GO-based nanosystems to target cells, subsequently leading to lower cellular accumulation of nanosystems. We have also reported earlier that higher anticancer drug levels (≥IC50) may be crucial in controlling the proliferation of cancer cells.41 The results suggest molecular tunability of NPs to alter cell entry kinetics, cellular localization depending on the nature of the drug and residence time to suit the pharmacological need.
Conclusions
Fe3O4–DNA–DOX and G–DNA–DOX–Fe3O4 conjugates were synthesized for multifunctional roles via formation of amide and ester bonds respectively. FTIR spectra, fluorescence spectroscopy and TEM images confirmed successful conjugation and the presence of all nanocarrier components. The hydrodynamic size of the synthesized nanocarriers was in the nano-size range which was appropriate for further cell kinetics studies. The synthesized nanosystems were superior to free DOX in terms of improved cellular retention and greater cytotoxicity. G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX demonstrated tunability in terms of DOX loading, BSA affinity, cytoplasmic/nuclear retention and cellular uptake rate in treated HeLa cells. Both NPs reduced the binding capacity of plasma proteins to DOX and greater unbound concentration was available for interaction with HeLa cells. G–DNA–DOX–Fe3O4 and Fe3O4–DNA–DOX altered DOX PK and were demonstrated to be efficient organelle-selective drug delivery systems that influenced the characteristic morphological adaptations in HeLa cells. The above multimodal approach applied to synthesize nanosystems presents an incremental strategy that provides greater control over regulation of specific cellular compartmentalisation of these nanosystems, and enhances the anticancer drug efficacy and reduces undesired effects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge the financial support of DBT-Nano-Biotechnology, DST-FIST and DST-Nano Mission, Government of India.Notes and references
- S. Baek, R. K. Singh, D. Khanal, K. D. Patel, E.-J. Lee, K. W. Leong, W. Chrzanowski and H.-W. Kim, Nanoscale, 2015, 7, 14191–14216 RSC.
- K. Xiao, T.-y. Lin, K. S. Lam and Y. Li, Nanoscale, 2017, 9, 7765–7770 RSC.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- D. Maysinger, Org. Biomol. Chem., 2007, 5, 2335–2342 RSC.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS.
- J. Khandare, M. Calderón, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469–4476 CrossRef CAS.
- C. Peng, W. Hu, Y. Zhou, C. Fan and Q. Huang, Small, 2010, 6, 1686–1692 CrossRef CAS.
- E. Campbell, M. T. Hasan, C. Pho, K. Callaghan, G. Akkaraju and A. Naumov, Sci. Rep., 2019, 9, 1–9 CrossRef CAS.
- M. Balcioglu, M. Rana and M. V. Yigit, J. Mater. Chem. B, 2013, 1, 6187–6193 RSC.
- R. Gonzalez-Rodriguez, E. Campbell and A. Naumov, PLoS One, 2019, 14, e0217072 CrossRef CAS.
- P. S. Wate, S. S. Banerjee, A. Jalota-Badhwar, R. R. Mascarenhas, K. R. Zope, J. Khandare and R. D. K. Misra, Nanotechnology, 2012, 23, 415101 CrossRef.
- S. Nandi, N. R. Kale, V. Takale, G. P. Chate, M. Bhave, S. Banerjee and J. Khandare, J. Mater. Chem. B, 2020, 8, 1852–1862 RSC.
- J. Khandare, A. Mohr, M. Calderon, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268 CrossRef CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- P. Kolhe, J. Khandare, O. Pillai, S. Kannan, M. Lieh-Lai and R. M. Kannan, Biomaterials, 2006, 27, 660–669 CrossRef CAS.
- S. S. Andhari, R. D. Wavhale, K. D. Dhobale, B. V. Tawade, G. P. Chate, Y. N. Patil, J. J. Khandare and S. S. Banerjee, Sci. Rep., 2020, 10, 1–16 CrossRef.
- N. R. Kale, S. Nandi, A. Patil, Y. N. Patil, S. Banerjee and J. Khandare, Biomater. Sci., 2020, 5729–5738 RSC.
- M. A. Mitry and J. G. Edwards, IJC Heart & Vasculature, 2016, 10, 17–24 Search PubMed.
- D. R. Dreyer, A. D. Todd and C. W. Bielawski, Chem. Soc. Rev., 2014, 43, 5288–5301 RSC.
- S. Kayal and R. Ramanujan, Mater. Sci. Eng., C, 2010, 30, 484–490 CrossRef CAS.
- L. Shen, B. Li and Y. Qiao, Materials, 2018, 11, 324 CrossRef.
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2018, 119, 6459–6506 Search PubMed.
- H. Bai, G. M. S. Lester, L. C. Petishnok and D. A. Dean, Biosci. Rep., 2017, 37, BSR20160616 CrossRef CAS.
- S. S. Banerjee and D.-H. Chen, J. Hazard. Mater., 2007, 147, 792–799 CrossRef CAS.
- N. Luque, P. Velez, K. Potting and E. Santos, Langmuir, 2012, 28, 8084–8099 CrossRef CAS.
- B. Jawad, L. Poudel, R. Podgornik, N. F. Steinmetz and W.-Y. Ching, Phys. Chem. Chem. Phys., 2019, 21, 3877–3893 RSC.
- D. Agudelo, P. Bourassa, J. Bruneau, G. Berube, É. Asselin and H.-A. Tajmir-Riahi, PLoS One, 2012, 7, e43814 CrossRef CAS.
- M. Anjomshoa, S. J. Fatemi, M. Torkzadeh-Mahani and H. Hadadzadeh, Spectrochim. Acta, Part A, 2014, 127, 511–520 CrossRef CAS.
- M. Wu, R. Kempaiah, P.-J. J. Huang, V. Maheshwari and J. Liu, Langmuir, 2011, 27, 2731–2738 CrossRef CAS.
- R. Di Santo, L. Digiacomo, S. Palchetti, V. Palmieri, G. Perini, D. Pozzi, M. Papi and G. Caracciolo, Nanoscale, 2019, 11, 2733–2741 RSC.
- C.-H. Lu, C.-L. Zhu, J. Li, J.-J. Liu, X. Chen and H.-H. Yang, Chem. Commun., 2010, 46, 3116–3118 RSC.
- C. Pérez-Arnaiz, N. Busto, J. M. Leal and B. a. García, J. Phys. Chem. B, 2014, 118, 1288–1295 CrossRef.
- J. Kuchlyan, N. Kundu, D. Banik, A. Roy and N. Sarkar, Langmuir, 2015, 31, 13793–13801 CrossRef CAS.
- S. Jayant, J. J. Khandare, Y. Wang, A. P. Singh, N. Vorsa and T. Minko, Pharm. Res., 2007, 24, 2120–2130 CrossRef CAS.
- S. Mahesh, K.-C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef.
- O. Igoucheva, V. Alexeev and K. Yoon, Gene Ther., 2006, 13, 266–275 CrossRef CAS.
- H. Lei, L. Mi, X. Zhou, J. Chen, J. Hu, S. Guo and Y. Zhang, Nanoscale, 2011, 3, 3888–3892 RSC.
- V. A. Sardão, P. J. Oliveira, J. Holy, C. R. Oliveira and K. B. Wallace, Cell Biol. Toxicol., 2009, 25, 227–243 CrossRef.
- C. D. Bobade, S. Nandi, N. R. Kale, S. Banerjee, Y. N. Patil and J. Khandare, Nanoscale Adv., 2020, 2, 2315–2325 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 to S10, Tables 1 and 2. See DOI: 10.1039/d0na00575d |
| This journal is © The Royal Society of Chemistry 2020 |