
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-stimuli responsive heterotypic hydrogels based on nucleolipids show selective dye adsorption†
Ashok
Nuthanakanti
and
Seergazhi G.
Srivatsan
 *
*
Department of Chemistry, Indian Institute of Science Education and Research, Dr Homi Bhabha Road, Pashan, Pune 411008, India. E-mail: srivatsan@iiserpune.ac.in
First published on 13th July 2020
Abstract
Analogous to nucleic acids, the building blocks of nucleic acids and their derivatives are widely used to create supramolecular architectures for application mainly in the field of biomedicine. Here, we describe the construction of a multi-stimuli responsive and toxic dye adsorbing heterotypic hydrogel system formed using simple nucleoside–fatty acid conjugates. The nucleolipids are derived by coupling fatty acid chains of different lengths at the 5′ position of ribothymidine and uridine. The nucleolipids in the presence of a strong base (e.g. NaOH) undergo partial hydrolysis, which triggers the self-assembly of the hydrolysed components resulting in the formation of heterotypic hydrogels. Notably, the gels are formed specifically in the presence of Na+ ions as other ions such as Li+ and K+ did not support the hydrogelation process. Systematic analysis by microscopy, NMR, single crystal and powder X-ray diffraction and rheology indicated that the deprotonated nucleolipid and fatty acid salt interdigitate and provide necessary electrostatic interactions supported by Na+ ions to set the path for the hierarchical assembly process. Notably, the hydrogels are highly sensitive to external stimuli, wherein gel–sol transition can be reversibly controlled by using temperature, pH and host–guest interaction. One of the hydrogels made of 5′-O-myristate-conjugated ribothymidine was found to selectively adsorb cationic dyes such as methylene blue and rhodamine 6G in a recyclable fashion. Taken together, the easily scalable assembly, multi-stimuli responsiveness and ability to capture and release dyes highlight the potential of our nucleolipid hydrogel system in material applications and in the treatment of dye industry wastes.
Introduction
Nucleic acids serve as powerful supramolecular synthons in construction of unique and programmable nanoarchitectures.1 Many such DNA assemblies have been used for developing functional materials for various applications2 including multiplexed diagnosis,3 gene4 and drug delivery,5 and molecular patterning.6 Major impediments to nucleic acid based nanotechnology are the scalability and efficiency in reproducing the self-assembly process.7 As an alternative, synthons made of nucleic acid components have been highly useful in constructing architectures such as micelles, vesicles, fibres, and hydro- and organogels.8 In particular, nucleobases and nucleosides derivatized with lipophilic linkers or chains provide unique opportunities wherein predictable H-bonding interaction, stacking interaction, metal ion binding and amphiphilicity drive the hierarchical self-assembly process.9 For instance, guanosine and its derivatives can participate in H-bonding interactions through Watson–Crick and Hoogsteen faces, which facilitate self-assembly in the presence of monovalent alkali metal ions to produce unique aggregates such as ribbons, wires, quartets, ion channels and gels.10 Further, various hybrid nucleolipids and glyconucleolipids that form fibrous networks resulting in hydrogels and higher order structures have been used in biomedical and material applications.11Most hydrogels are made using single component gelators. Formation of such homotypic hydrogels is mainly driven by noncovalent interactions including H-bonding, stacking, electrostatic and hydrophobic interactions, and hence, the gelation process can be triggered and modulated by using an internal fuel12 or external stimuli such as temperature, pH, light, enzymes and ions.13 Alternatively, heterotypic hydrogels made of two or more supramolecular building blocks have certain advantages over homotypic hydrogels.14 Apart from the ease of synthesis of individual components, heterotypic systems offer a greater degree of tunablity of properties, and importantly, facilitate construction of multifunctional materials for sophisticated applications.15 Notably, many bioinspired hydrogels have been used as sensors and vehicles for sustainable delivery of drugs and tissue engineering.16
While development of nucleolipids for biomedical applications is a major theme of interest, creating nucleolipid-based smart multi-stimuli responsive materials and sensors with unique recognition features has been less explored. Our interest in this direction is to develop easily synthesizable and scalable supramolecular nucleolipid assemblies that support the formation of gels exhibiting reversible multi-stimuli responsiveness. We recently introduced nucleobase-modified fluorescent nucleolipid organogels, which exhibited aggregation induced enhanced emission and also their assembly process could be modulated using external stimuli.8b Similarly, by attaching fatty acid chains of different lengths to the sugar residue of nucleosides, we were able to generate gel surfaces which could be switched between highly hydrophilic and super hydrophobic states by using appropriate solvents during the gelation process.17 However, given the wide utility of hydrogels in medical and materials research, generating hydrogels out of neutral nucleoside–lipid conjugates is not straightforward due to the poor solubility of such synthons.
Here, we report a multi-stimuli responsive and selective dye adsorbing heterotypic hydrogel system obtained from the partial hydrolysis of 5′-O-fatty acid-substituted ribothymidine and uridine nucleolipids, which is specifically stabilized by Na+ ions (Fig. 1). Among the nucleolipids, ribothymidines (3a–3c) containing fatty acid alkyl chains of different lengths formed stable organogels, but did not support hydrogel formation by themselves. Intriguingly, upon addition of a strong base (NaOH) to the nucleolipids in water, the mixture formed hydrogels. Careful investigation by SEM, 1H NMR, PXRD and rheological analyses indicated a coordinated interplay of interactions between the nucleolipid, fatty acid carboxylate and specifically Na+ ions in the hydrogelation process. Further, the hydrogels exhibited reversible multi-stimuli responsiveness to changes in temperature, pH and host–guest interaction. We eventually used the recognition properties of the anionic heterotypic hydrogels in the selective capture and release of toxic water soluble cationic dyes in a recyclable fashion, a useful application, which could find utility in the treatment of dye effluents.
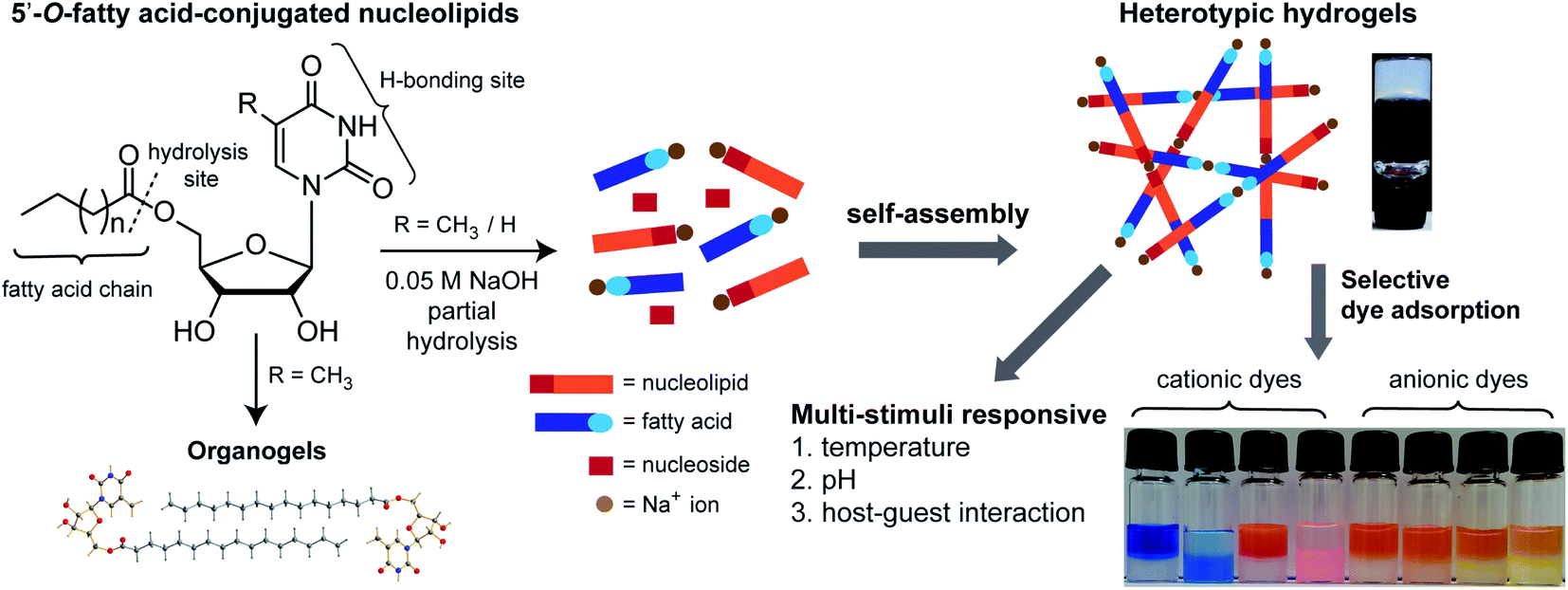 | ||
| Fig. 1 5′-O-fatty acid-substituted ribothymidine and uridine nucleolipids form heterotypic hydrogels due to partial hydrolysis and the gel network is specifically stabilized by Na+ ions. The hydrogels show multi-stimuli responsiveness and selectively adsorb cationic dyes. Vials from left to right indicate dyes suspended on the gel surface at zero time and 24 h (see Fig. 8 for details). Ribothymidine nucleolipids also supported the formation of organogels. | ||
Results and discussion
Synthesis of 5′-O-fatty acid-conjugated ribothymidine and uridine nucleolipids
5′-O-fatty acid-substituted nucleosides (3a–3c and 4a–4c) were synthesized by first protecting the 2′, 3′-OH groups in the form of an acetonide, followed by coupling reactions with fatty acids of different alkyl chain lengths in the presence of EDC (Scheme 1). The acetonide group was deprotected by using either p-toluenesulfonic acid or camphorsulfonic acid which then gave the desired products ribothymidine (3a–3c) and uridine (4a–4c) nucleolipids.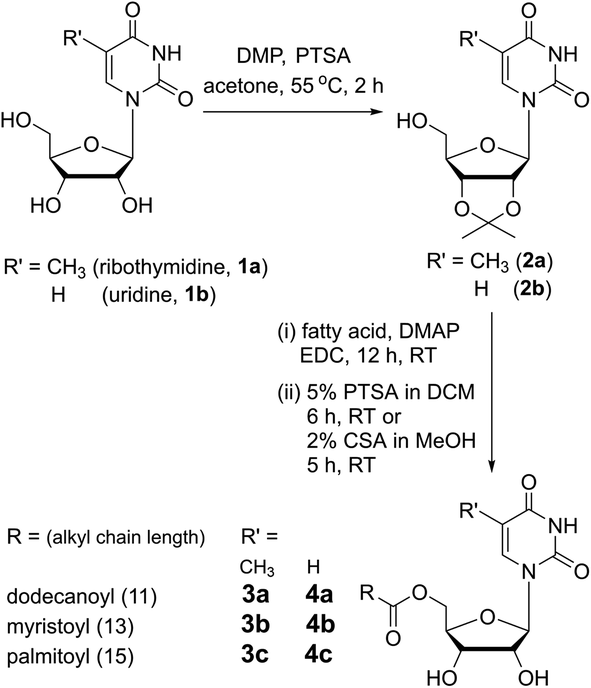 | ||
| Scheme 1 Synthesis of 5′-O-fatty acid-conjugated ribothymidine (3a–3c) and uridine (4a–4c) nucleolipids. DMP = 2,2-dimethoxypropane; PTSA = p-toluenesulfonic acid; DMAP = 4-dimethylaminopyridine; EDC = N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride; and CSA = camphorsulfonic acid. | ||
Ribothymidine and uridine nucleolipids form heterotypic hydrogels
Nucleolipids 3a–3c and 4a–4c are insoluble in water, but addition of a strong base such as LiOH, NaOH and KOH followed by heating solubilized the mixture in water. Intriguingly, a hot solution of nucleolipids containing myristoyl and palmitoyl chains (3b, 3c, 4b and 4c) in 0.05 M NaOH formed stable hydrogels upon cooling to room temperature (Fig. 2A). On the other hand, as high as 1 M LiOH or KOH did not induce hydrogelation. The gelation ability in terms of CGC depended on the fatty acid chain length (Table 1). Nucleolipids made of a shorter dodecanoyl chain 3a and 4a did not form hydrogels. As the chain length increased from myristoyl to palmitoyl chains, more weight percentage of the gelator was required to form the gel. From these results, it appears that this hydrogelation process depends on four parameters, namely the choice of the base, partial hydrolysis of nucleolipids (vide infra), specific cation interaction and fatty acid chain length. Since only NaOH induced the gelation process, we studied the influence of base strength by using milder bases such as Na2CO3 and NaHCO3 but without changing the cation. Both these bases did not solubilize the nucleolipids in water and also did not support gelation. Likewise, to study the role of cations, we tested the gelling ability of nucleolipids in LiCl, NaCl and KCl. In a control experiment, addition of even a 1 M aqueous solution of these salts did not solubilize the nucleolipids (data not shown). Interestingly, addition of 0.05 M NaCl to an aqueous solution of nucleolipids containing either KOH or LiOH resulted in efficient sol–gel transition, which was not observed when LiCl or KCl was added (Fig. S1†). It is to be noted that 50 mM NaOH or NaCl (in the case of a basic solution) is required for the gel formation. While lower amounts did not induce hydrogelation higher amounts did not affect gelation ability.18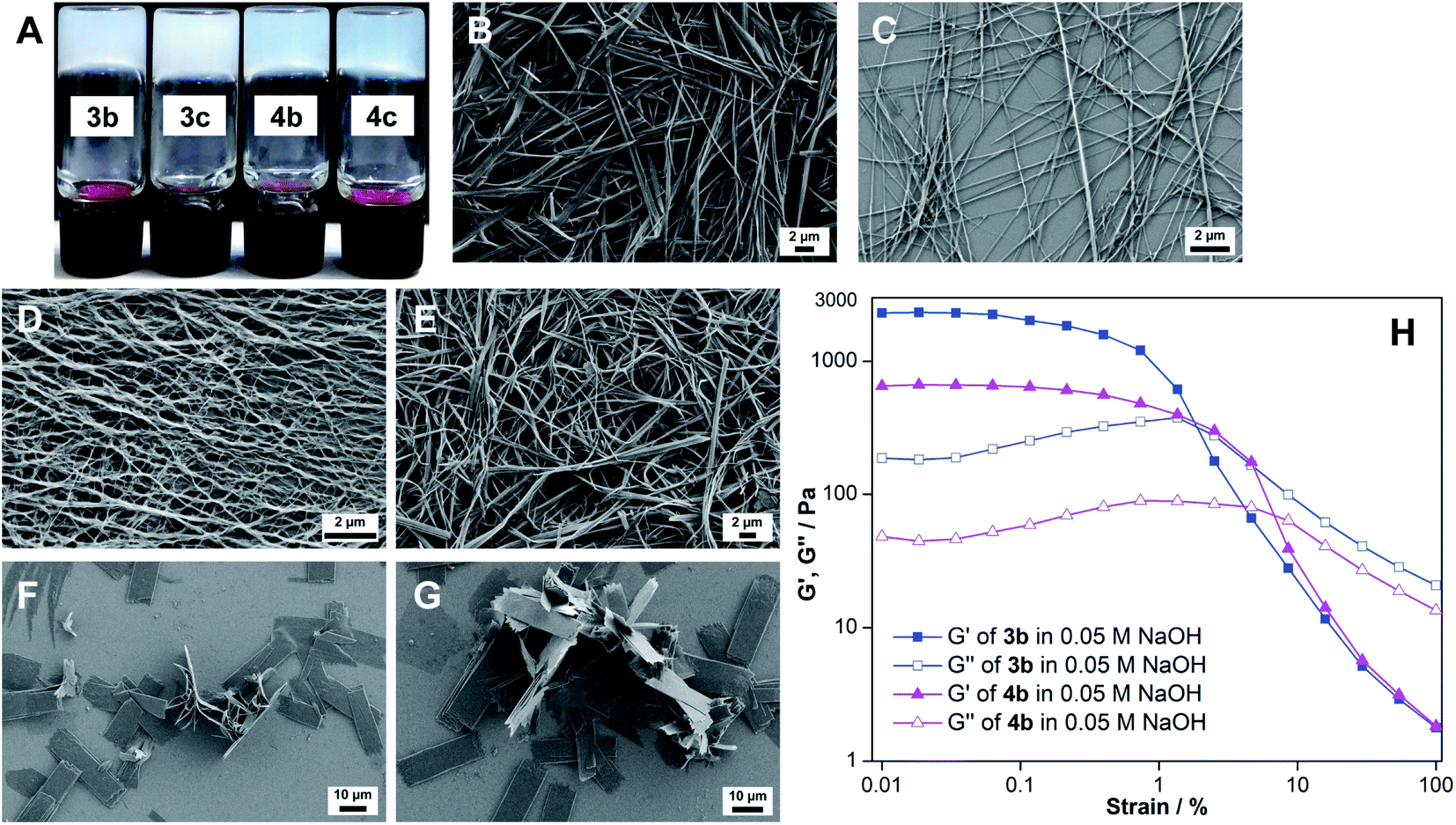 | ||
| Fig. 2 (A) Photograph of hydrogels formed using ribothymidine (3b and 3c) and uridine (4b and 4c) nucleolipids at the respective CGC in water containing 0.05 M NaOH. FESEM images of xerogels of nucleolipids (B) 3b, (C) 3c, (D) 4b and (E) 4c formed in the presence of 0.05 M NaOH. (F) and (G): FESEM images of nucleolipid 3b in LiOH and KOH, respectively. (H): Strain sweep rheological measurements of 3b and 4b hydrogels at constant oscillation frequency. | ||
| Solvent | 3a (w/v%) | 3b (w/v%) | 3c (w/v%) | 4a (w/v%) | 4b (w/v%) | 4c (w/v%) |
|---|---|---|---|---|---|---|
| a Hydrogels were formed in the presence of 0.05 M NaOH. I: insoluble, P: precipitate, G: gel, and S: sol. Critical gelation concentration (CGC in w/v %) is given in brackets. | ||||||
| Watera | I | G (0.3) | G (0.45) | I | G (0.3) | G (0.5) |
| CHCl3 | G (1.4) | G (1.2) | G (1.0) | P | I | I |
| Benzene | G (1.3) | G (0.9) | G (0.8) | P | S | S |
| Toluene | G (1.6) | G (1.0) | G (0.9) | S | S | S |
| 1,4-Dichlorobenzene | G (2.0) | G (1.8) | G (1.4) | S | S | S |
| Methanol | S | S | S | S | S | S |
| Acetone | S | S | S | S | S | S |
The NaOH-triggered gelation process was further confirmed by microscopy analysis. The FESEM images of the xerogels formed using the nucleolipids in the presence of NaOH revealed a highly entangled fibrous network, a morphology which typically assists in gelling solvents (Fig. 2B–E). However, in the presence of LiOH or KOH, the nucleolipids formed fragmented sheets, and hence, did not support gelation (Fig. 2F and G). Collectively, these results indicate that along with a strong basic environment, Na+ ions are specifically required to drive the hydrogelation of these nucleolipids.19
Next, we evaluated the mechanical properties of the hydrogels and for this purpose nucleolipids 3b and 4b having lower CGC values were used. The storage modulus (G′) and loss modulus (G′′) of the hydrogels were examined as a function of shear strain at a constant oscillation frequency (Fig. 2H). Based on the G′ value, ribothymidine nucleolipid 3b forms a discernibly stronger gel as compared to the uridine nucleolipid 4b. At lower strains, 3b and 4b hydrogels exhibited G′ values that were more than one order of magnitude greater than G′′, which revealed the elastic character of the gel. The crossover point of G′ and G′′ for 3b and 4b was observed at a 2.1% and 7.6% strain, respectively, where the gel transforms into a sol.
Nucleolipids form heterotypic hydrogels due to partial hydrolysis
We sought to understand the origin of base-induced hydrogelation exhibited by the nucleolipids. The pH of the nucleolipids containing 0.05 M LiOH, NaOH or KOH is around 12.8, and under these conditions the ester linkage between the nucleoside and fatty acid could potentially undergo hydrolysis. The pKa of the fatty acid (COOH), uracil imino hydrogen (N3–H) and ribose sugar secondary hydroxyl groups (2′- and 3′-OH) is around 4.9, 9.3 and 12.5, respectively.20 Hence, the carboxylic group of the fatty acid and 2′-OH, 3′-OH and N3–H of the nucleolipid should be deprotonated. To study the effect of the base, the 1H NMR experiment was first carried out using ribothymidine nucleolipid 3b in DMSO-d6 in the presence of LiOH, NaOH or KOH. N3–H, 2′-OH and 3′-OH signals almost completely disappeared indicating that these protons are deprotonated (Fig. S2†). Next, we recorded the 1H NMR spectrum of the three hydrogel samples formed using myristic acid (5), myristic acid–ribothymidine combination (5·1a) and nucleolipid 3b, all in the presence of 0.05 M NaOH in D2O at 40 °C (at RT we observed severe line broadening in the proton signal of the gels). These sets of experiments provided information on the components that are involved in the heterotypic hydrogel formation.15b Myristic acid by itself in NaOH formed a gel, and when its NMR spectrum was compared with the spectrum of a gel formed using the ribothymidine–myristic acid combination, the presence of individual components, namely ribothymidine and myristate, was observed (Fig. S3 and S4†). Interestingly, 1H NMR of the 3b gel in NaOH revealed 50% hydrolysis, which was inferred by comparing the integration values (Fig. S5A†). If 50% of 3b is hydrolyzed then the gel should contain 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 of ribothymidine, 3b and myristate. The integration values of individual proton signals corresponding to the myristoyl chain were found to be twice as much as those of the nucleoside proton signals, which suggests that the signals observed from the gel are due to a 1:1 mixture of 3b and myristate. The gel was stable and did not undergo further hydrolysis even after one week (Fig. S5B†). The hydrolyzed nucleoside precipitated and was possibly trapped in the gel network and immobilized, and hence was not visible in the spectrum. This was further ascertained by recording 1H NMR of the sol form of the 3b gel at 65 °C, which exhibited proton signals from the nucleolipid and fatty acid, but not for the precipitated nucleoside (Fig. S5C†).
:1:1 of ribothymidine, 3b and myristate. The integration values of individual proton signals corresponding to the myristoyl chain were found to be twice as much as those of the nucleoside proton signals, which suggests that the signals observed from the gel are due to a 1:1 mixture of 3b and myristate. The gel was stable and did not undergo further hydrolysis even after one week (Fig. S5B†). The hydrolyzed nucleoside precipitated and was possibly trapped in the gel network and immobilized, and hence was not visible in the spectrum. This was further ascertained by recording 1H NMR of the sol form of the 3b gel at 65 °C, which exhibited proton signals from the nucleolipid and fatty acid, but not for the precipitated nucleoside (Fig. S5C†).
Evidence for the heterotypic hydrogelation process was also ascertained by SEM and energy dispersive X-ray spectroscopy analyses (EDAX). The morphology of the 3b xerogel is markedly different from that of the xerogels formed using myristic acid (5) and myristic acid–ribothymidine (5·1a) under similar conditions (compare Fig. 2B with Fig. 3A and B). While the 3b gel shows a highly entangled fibrous network, the other two xerogels show a dendrimer like structure. Further, EDAX of fibers showed an elemental composition of carbon, nitrogen, oxygen and sodium, which corresponded to the gel network formed from a 1:1 ratio of the nucleolipid and myristate salt (Fig. S6†).15e We also carried out rheological measurements to demonstrate the co-assembly process. The mechanical properties of the gel of myristic acid and the two-component gel of myristic acid and ribothymidine in NaOH were noticeably weak as compared to those of the heterotypic nucleolipid 3b hydrogel (Fig. 3C). The reason for the mechanical reinforcement is probably due to formation of a highly entangled network due to the co-assembly of the nucleolipid and long-chain fatty acid salt.21 Collectively, these results indicate that partial hydrolysis of nucleolipids by NaOH generates heterotypic hydrogels, which are essentially assembled by electrostatic interactions between the deprotonated sites and Na+ ions and hydrophobic interactions between the alkyl chains.
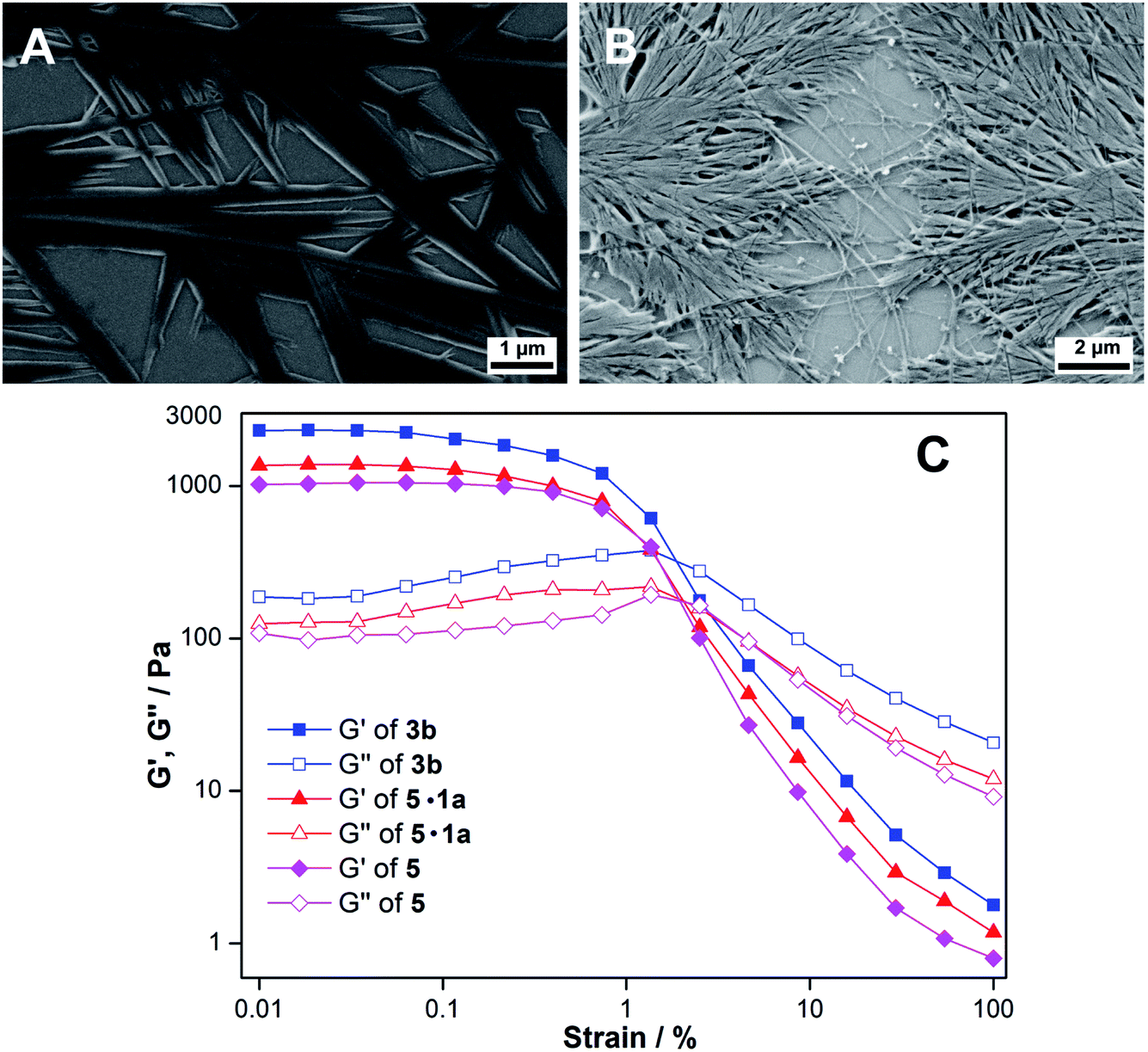 | ||
| Fig. 3 FESEM images of xerogels of (A) myristic acid 5 and (B) myristic acid–ribothymidine combination (5·1a) obtained in the presence of 0.05 M NaOH. (C) Strain sweep rheological profiles of 3b, 5 and 5·1a hydrogels at constant oscillation frequency. | ||
Ribothymidine nucleolipids form organogels
Ribothymidine nucleolipids (3a–3c) also formed opaque organogels in nonpolar solvents, whereas uridine nucleolipids (4a–4c) did not form organogels (Table 1). Notably, longer alkyl chain-conjugated nucleolipid 3c gelates efficiently at a lower CGC as compared to nucleolipid 3a containing a shorter alkyl chain.9f Microscopy analysis indicates that the gel network is formed from long-range tape-like structures (Fig. S7†).We obtained crystals of nucleolipids 3b, 3c and 4a by slow evaporation on nucleolipids dissolved in either methanol or acetone, which crystallized in a monoclinic system with the space group P21 (Fig. S8, Table S1–S3†). The torsion angle (C2–N1–C1′–O4′) in the range of 139–144° indicated an anti conformation about the glycosidic bond (Table S4†).22 Further, all the nucleolipids contained a C2′-endo puckered sugar as compared to a C3′-endo puckered sugar in native ribonucleosides, which could be due to conjugation of the long chain fatty acid at the 5′-OH position (Table S4†). The crystal structures of ribothymidine nucleolipids 3b and 3c show a complex H-bond network in which the N3H and O2 atoms of the nucleobase H-bond with the O2’ and O2′H atoms of two adjacent nucleosides, respectively (Fig. 4A and Fig S9†). O2′H and O3′H groups are involved in H-bonding interactions including the three-centered hydrogen bonded network. Further, ester carbonyl oxygen forms an intramolecular H-bond with the C6H of the base. The crystal packing of nucleolipids 3b and 3c revealed complete interdigitation of the alkyl chains as a result of head to head and tail to tail arrangements (Fig. S10 and S11†). Adjacent layers connected via H-bonds produced a 2D sheet-like structure, which was stabilized by a CH–π interaction between the C5-methyl hydrogen and nearby 5-methyluracil moiety (3.14(3) Å, Fig. S12†).
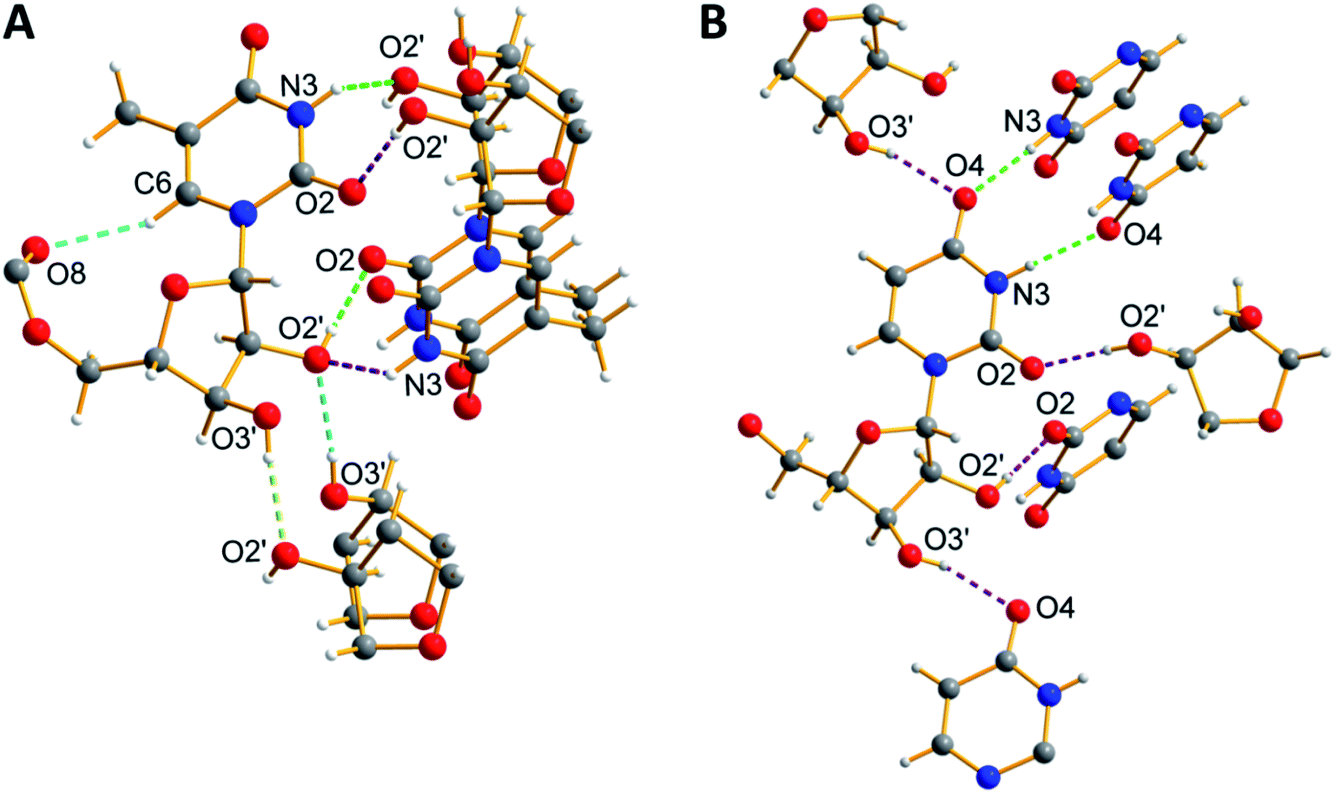 | ||
| Fig. 4 X-ray crystal structure of (A) 3b and (B) 4a showing a detailed view of the H-bonding interactions along the crystallographic b-axes. Hydrogen atoms other than in nucleosides are not shown for clarity. Atoms are coded as follows: off white, hydrogen; dark grey, carbon; blue, nitrogen; and red, oxygen. | ||
On the other hand, the structure of uridine nucleolipid 4a exhibited a different H-bonding pattern in which Watson–Crick H-bonding atoms N3H and C4 carbonyl oxygen base-paired with two adjacent uracil bases (Fig. 4B). In a complex network of H-bonding interactions, each nucleolipid interacts with six distinct molecules by utilizing both nucleobase and ribose sugar H-bonding sites. The packing structure revealed interdigitation of alkyl chains and strong interlayer interactions resulting in a 2D-sheet structure (Fig. S13†).
Probing the assembly process in gels by PXRD
The PXRD profile of the 3b hydrogel showed a low angle diffraction peak corresponding to an interlayer distance of 3.67 nm, which is shorter than twice the molecular length and larger than one molecular length (2.30 nm) obtained from a single crystal (Fig. 5A). Further, the diffraction pattern revealed peaks corresponding to a d spacing of 3.67, 1.83, 1.22, 0.91, and 0.73 nm, which are in a ratio of 1:1/2:1/3:1/4:1/5. Consistent with literature reports,23 these observations suggest that base-induced hydrogels are formed from a highly ordered lamellar structure with interdigitated bilayers using nucleolipids and fatty acid carboxylates as the basic unit (Fig. S14A†). As a result of partial hydrolysis of the nucleolipid 3b, ionic amphiphiles of the nucleolipid and fatty acid were generated, which were electrostatically linked by sodium ions to form bilayer units that subsequently assembled to form a fibrous network (Fig. S14†).19b,23 Similarly, the PXRD profile of the 3b organogel revealed a diffraction peak corresponding to a layer distance of 2.72 nm. This spacing is larger than one molecular length (2.30 nm) and nearly equal to the fully interdigitated molecule (2.77 nm, Fig. 5B and S14B†). Again the diffraction peaks (2.72, 1.36, 0.91, 0.68 and 0.54 nm) were in a ratio of 1:1/2:1/3:1/4:1/5 indicating the formation of gels from ordered lamellar aggregates made of interdigitated nucleolipids as the basic unit.17,24 The 3c organogel also showed similar PXRD patterns corresponding to an ordered lamellar arrangement made of interdigitated nucleolipids (Fig. S15†).
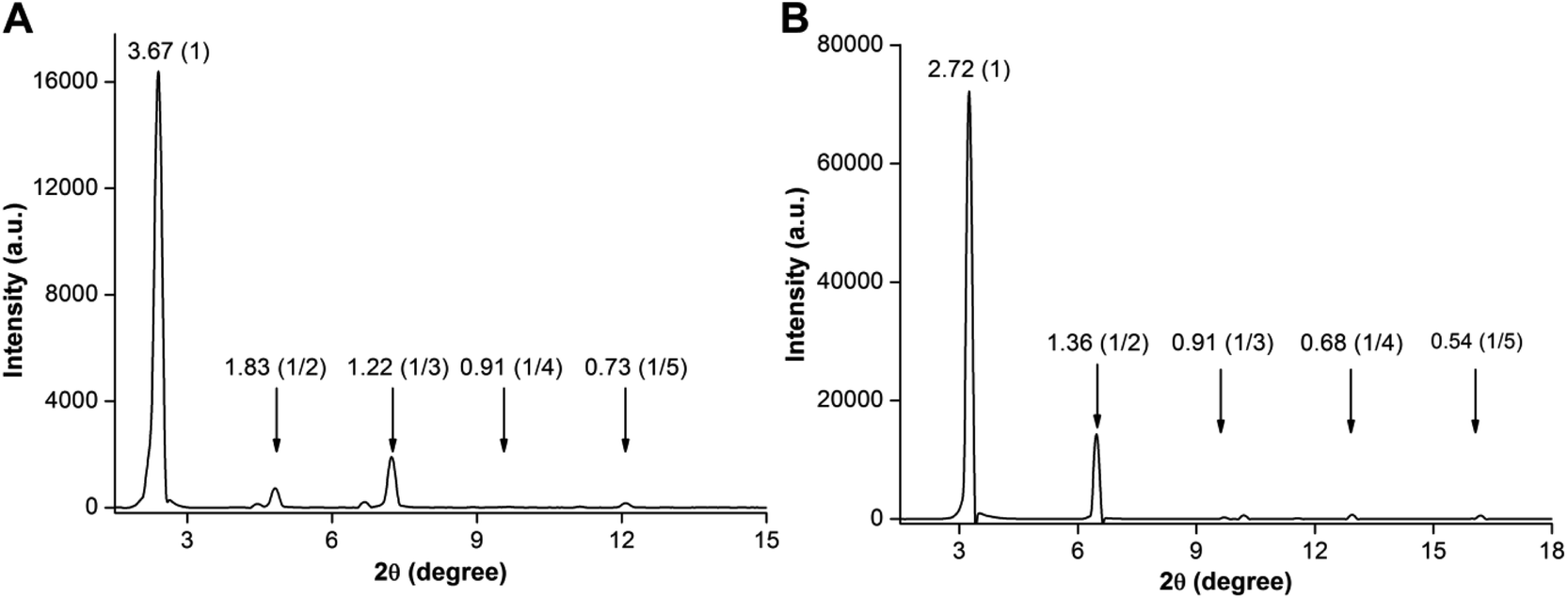 | ||
| Fig. 5 PXRD profiles of xerogels of (A) 3b hydrogel formed in the presence of NaOH and (B) 3b organogel formed in benzene. Layer spacing (nm) for prominent diffraction peaks and relative ratios are given in brackets. | ||
Nucleolipids 3a–3c formed stable organogels in nonpolar solvents but polar organic solvents such as methanol and acetone did not support gelation (Table 1). Nonpolar solvents potentially helped in H-bonding interactions between the nucleolipids and promoted self-assembly, but it is likely that the polar solvents competed for the H-bonding sites and disrupted the self-assembly, and hence, gelation. However, nucleolipid solutions in methanol or acetone upon slow evaporation crystallized due to the formation of multiple H-bonding interactions (Fig. 4, S8–S10†). Crystal packing revealed interdigitation of the hydrophobic alkyl chains. The PXRD profile of the xerogels also suggested the presence of ordered lamellar structures with interdigitated bilayers.
It is to be noted that the assembly process deduced from SEM, single crystal data and PXRD of xerogels need not be necessarily the same in the gel state. To test this, hydro and organogels formed using 3b (without drying) were visualized using a polarized light microscope. While the hydrogel in the gel state was largely amorphous with signs of crystallinity, the organogel formed in chloroform showed significant crystallization (Fig. S16†). The morphologies of both the gels, when dried were similar to the gel state under polarized light. From these results, it appears that the diffraction peaks observed in the PXRD spectrum of the xerogels likely emanate from the partially crystalline nature of the gels. Therefore, a combination of SEM, NMR, single crystal data and PXRD data suggests that the interdigitated bilayers could be the starting point for the hierarchical assembly process.
Nucleolipid hydrogel is responsive to multiple stimuli
Nucleolipid hydrogels are formed under basic conditions due to the electrostatic interactions between the deprotonated groups (carboxylic acid, N3H, 2′-OH and 3′-OH) and Na+ ions, and interdigitation of hydrophobic fatty acid chains. These interactions can be reversibly modulated using external stimuli such as heat and chemicals.25 To study multi-stimuli responsiveness, the ribothymidine nucleolipid 3b gel having a lower CGC value and higher gel strength was chosen (Fig. 6). The gel–sol transition was thermoreversible several times. A stable gel formed at basic pH (∼12.7) disintegrated when the pH was lowered to 6, but when the pH was increased by adding NaOH, a stable gel formed again. This is due to the protonation–deprotonation process, which changes the electrostatic interaction that supports the gelation process. Specific interaction of Na+ ions with the deprotonated species is very important for the gel formation, and hence, sequestering Na+ ions using an appropriate ion or a host could affect the gelling ability of the nucleolipid. Indeed, addition of even 0.2 equivalents of tetrabutyl ammonium chloride to 3b resulted in gel–sol conversion. However, addition of NaCl (0.2 equiv.) restored the gelling ability of the nucleolipid. To further prove the role of Na+ ions in the self-assembly process, 15-crown-5, a selective host for Na+ ions, was added. Gratifyingly, addition of 3 equivalents of 15-crown-5 resulted in gel–sol transition, and the gel was reversibly reconstructed by adding 3 equivalents of NaCl to the sol. | ||
| Fig. 6 Multi-stimuli responsiveness exhibited by the hydrogel obtained from 3b at its CGC. | ||
Nucleolipid hydrogel selectively adsorbs cationic dyes
Hydrogels have been used in the removal of pollutants and toxic substances via specific interactions, namely hydrophobic, electrostatic and hydrogen bonding interactions between the fibrous network and substances.26 Recently, a hydrazide-based system was utilized in generating a hydrogel that exhibited pH controlled adsorption–desorption of dyes.27b These reports prompted us to explore the usefulness of our nucleolipid hydrogels in the adsorption and removal of hazardous dyes that are a part of industrial effluents. The heterotypic hydrogel is made of a negatively charged fibrous network, and hence, we envisioned that cationic dyes would be selectively adsorbed in the gel network as compared to anionic dyes.27 For this study, cationic dyes methylene blue (MB) and rhodamine 6G (R6G) and anionic dyes eosin Y (EY) and methyl orange (MO) were used (Fig. 7). | ||
| Fig. 7 Chemical structures of the dyes used in this study. | ||
A hydrogel of 3b (0.3 w/v%, 6.4 mM) in 0.05 M NaOH was formed and an aqueous solution of individual dyes (0.25 mM) was placed on the gel surface. The adsorption of dyes by the gel network was visible to the naked eye, and was further quantified by measuring the absorbance of the dye remaining in the supernatant by UV-visible absorption spectroscopy. The heterotypic hydrogel showed very high adsorption capacity for cationic dyes MB and R6G as compared to anionic dyes EY and MO (Fig. 8A). The absorption profiles indicated a progressive decrease in the absorbance of MB and R6G in the supernatant, which was quite significant even after 6 h of incubation (Fig. 8B and C). A 24 h incubation time did not affect the gel and nearly 88% of the dye was removed, which increased to 93% over 48 h. However, EY and MO dyes were only marginally adsorbed after 6 h of incubation (Fig. 8D and E). When incubated for 24 h, the anionic dyes did show some reduction in absorbance possibly due to passive diffusion and hydrophobic interaction.26
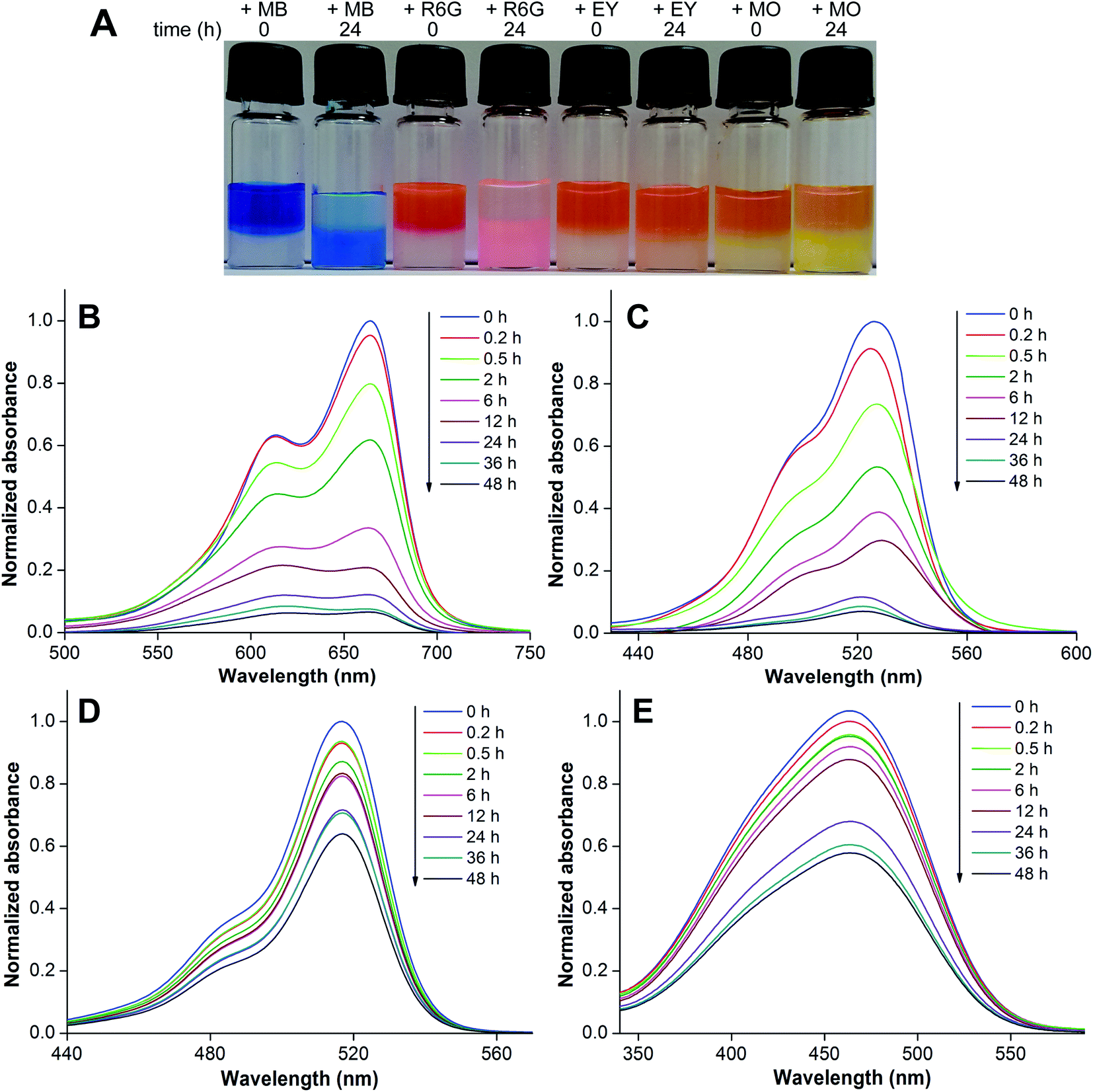 | ||
| Fig. 8 (A) Picture showing the effective adsorption of cationic dyes (MB and R6G) by the hydrogel formed using 3b. Anionic dyes (EY and MO) are discernibly less adsorbed. (B–E) Absorption spectra of dye solutions incubated with the gel at different time intervals. MB (B), R6G (C), EY (D) and MO (E). For details, see the Experimental section. | ||
The hydrogel was found to selectively remove cationic dyes from a solution containing a mixture of dyes. An aqueous solution containing equimolar concentrations of MB and EY or MB and MO was incubated with the gel. A plot of the normalized absorbance of the supernatant versus wavelength at zero time showed an intense band (∼670 nm) corresponding to MB and a weak band (∼515 nm) corresponding to EY (Fig. S17†). However, after 24 h incubation MB dye absorbance reduced significantly corresponding to ∼76% removal due to dye adsorption in the gel matrix. This was accompanied by an increase in the EY signal at 515 nm. Similarly, the gel was able to preferentially adsorb MB (∼60%) as compared to MO, which was also noticeably adsorbed (∼30%, Fig. S18†).
Recyclability of the hydrogel
A fresh gel formed using 3b was incubated with MB dye and after a 12 h interval the supernatant was removed, the gel surface was washed with water and a fresh solution of MB dye was suspended again on the gel surface (Fig. S19A†). This cycle was repeated 8 times and the percentage of dye adsorbed in each cycle was estimated by recording the absorbance of the dye left in the supernatant. The MB removal efficiency for the first 6 cycles was more than 75%, which subsequently dropped to 65% due to deformation of the gel (Fig. S19B†). The adsorbed dye after 8 cycles was recovered by reducing the pH using dilute HCl, which resulted in the precipitation of the gelator components (Fig. S19†). Simple filtration resulted in the removal of the dye. Addition of 50 mM NaOH to the precipitated gelator components gave the gel again, which could be used for the next round of dye extraction. Taken together, the heterotypic nucleolipid hydrogel provides a simple platform to extract cationic dyes, which can find utility in the treatment of dye industry waste.Conclusions
We have constructed heterotypic supramolecular hydrogels using simple nucleoside–fatty acid conjugates. Ribothymidine and uridine nucleolipids containing myristoyl and palmitoyl chains formed hydrogels specifically in the presence of NaOH. Systematic analysis by NMR, microscopy, X-ray diffraction and rheology provided insights into the mechanism by which partial hydrolysis of nucleolipids, deprotonation of nucleobase hydrogens and the ensuing electrostatic interaction specifically supported by Na+ ions and interdigitation of fatty acid alkyl chains drive the hierarchical assembly process to form heterotypic hydrogels. Notably, the hydrogels were found to be highly sensitive to external stimuli, wherein the gel–sol transition could be reversibly switched by changing temperature, pH and host–guest interaction. Further, we aptly utilized a hydrogel made of 3b in selectively adsorbing cationic dyes in a recyclable fashion. Taken together, the simple synthesis, easily scalable assembly, multi-stimuli responsiveness and ability to adsorb and desorb dyes underscore the potential utility of this nucleolipid hydrogel system in material applications and also in effluent treatment.Experimental section
General procedure for the synthesis of 5′-O-alkylated ribothymidine (3a–3c) and uridine (4a–4c) nucleolipids
Ribothymidine 2′,3′-isopropylidene (2a) and uridine 2′,3′-isopropylidene (2b) were synthesized by following a reported procedure.28 Compounds 2a or 2b (1.0 equiv.), fatty acids (dodecanoic acid, myristic acid or palmitic acid, 1.2 equiv.), EDC (1.2 equiv.) and DMAP (0.3 equiv.) were dissolved in dry dichloromethane (10 mL g−1 of 2a or 2b). The reaction mixture was stirred for 12 h at RT under a nitrogen atmosphere. After completion of the reaction, the crude product was diluted with dichloromethane and washed with saturated NH4Cl solution. The organic extract was dried over sodium sulfate. The resultant 2′,3′-O-acetonide-protected nucleolipids were used in the next step without further purification. In the case of compounds 3a–3c, deprotection was carried out using 5% p-toluenesulfonic acid (PTSA) in methylene chloride for 6 h at RT to remove the acetonide group. Similarly, compounds 4a–4c were treated with 2% camphorsulfonic acid (CSA) in methanol for 5 h at RT. The residues were worked up with saturated sodium bicarbonate and extracted with methylene chloride. The volatile solvents were evaporated and the crude residues were purified by silica gel column chromatography (5% methanol in methylene chloride) to afford the products. See the ESI† for characterization data and spectra of 3a–3c and 4a–4c.Gelation test by the inverted vial method
A weighed amount of the nucleolipid gelator molecules was taken in 1 mL of water and an appropriate amount of 0.05 M NaOH was added to adjust the solution pH to ∼12.8. The resultant aqueous suspension was heated until almost a clear solution was obtained and then cooled to RT. This set was repeated one more time. The samples were then kept for 12 h at RT to obtain stable hydrogels. In the case of organogels, the nucleolipids were dissolved in various organic solvents by heating. The samples were allowed to cool to RT and instantaneously stable opaque gels were observed. The gelation was confirmed by the inverted vial method. The minimum amount of gelator molecules required to form a stable gel is the critical gelation concentration (CGC). The hydrogels and organogels remained stable at RT for several months. Repeated heating and cooling steps confirmed the thermo-reversibility of the gelation process. All the experiments were performed at least in triplicate.Rheological studies
Rheological measurements were carried out on an Anton paar MCR 302 instrument by using a 15 mm diameter parallel plate at 25 °C. A strain sweep experiment in the range of 0.01–100% at a constant frequency (10 rad s−1) was performed to determine the linear viscoelastic region of the gel samples.FESEM analysis
The morphology of nucleolipid hydro and organogels was characterized by FESEM. Diluted gel samples were drop-cast on silicon wafers and dried in a vacuum desiccator for 6 h. To avoid sample charging, gold (Au) sputtering was performed on the sample surface before FESEM analysis.Crystallography
Single crystals of nucleolipids 3b, 3c and 4a were obtained from methanol, acetone and methanol, respectively, by a slow evaporation method. The diffraction data of the nucleolipids were collected using a Bruker KAPPA APEX II CCD Duo diffractometer (operated 1500 W, 50 kV and 30 mA) with graphite-monochromated MoKα (λ = 0.71073 Å) radiation. The structures were refined on F2 by the full-matrix least-squares technique using the SHELX program.29 Unless otherwise mentioned all non-hydrogen atoms were refined anisotropically. Hydrogen atoms were constrained in geometric positions with respect to their parent atoms. Crystallographic data are given in Tables S1–S3.† Crystallographic coordinates for the structures reported in this article were deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number (3b) 1554880, (3c) 1554881 and (4a) 1554882.Dye adsorption by the hydrogel
1 mL of the respective dye solution (0.25 mM) was slowly transferred to a vial containing the 3b hydrogel (0.3% w/v, 3 mg in 1 mL and 50 mM NaOH) and left to stand undisturbed at RT. A small aliquot of the supernatant at different time intervals was removed, diluted using water and the absorption spectrum was recorded from a 200 μL micro-cuvette. The experiment was performed in triplicate. The recyclability of the 3b hydrogel was investigated by incubating an aqueous solution of MB (1 mL, 0.25 mM) with the gel. After 12 h of incubation, the dye solution was removed carefully, the gel surface was washed with 2 mL of deionized water and was again incubated with MB (1 mL, 0.25 mM). The absorbance of all the supernatants was recorded.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Prof. M. Jayakannan for allowing us to use his laboratory to perform rheological analysis. A. N. is grateful to the University Grants Commission, India, for the graduate research fellowship. S. G. S. thanks CSIR, India (02-0086/12/EMR-II) and Wellcome Trust-DBT India Alliance (IA/S/16/1/502360) for the research grants.Notes and references
- (a) Y. Krishnan and F. C. Simmel, Angew. Chem., Int. Ed., 2011, 50, 3124–3156 CrossRef CAS; (b) F. Wang, C.-H. Lu and I. Willner, Chem. Rev., 2014, 114, 2881–2941 CrossRef CAS; (c) F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS; (d) M. Endo and H. Sugiyama, Molecules, 2018, 23, E1766 CrossRef.
- (a) Z.-G. Wang and B. Ding, Adv. Mater., 2013, 25, 3905–3914 CrossRef CAS; (b) J. B. Lee, S. Peng, D. Yang, Y. H. Roh, H. Funabashi, N. Park, E. J. Rice, L. Chen, R. Long, M. Wu and D. Luo, Nat. Nanotechnol., 2012, 7, 816–820 CrossRef CAS; (c) K. Leung, K. Chakraborty, A. Saminathan and Y. Krishnan, Nat. Nanotechnol., 2019, 14, 176–183 CrossRef CAS; (d) A. Buchberger, C. R. Simmons, N. E. Fahmi, R. Freeman and N. Stephanopoulos, J. Am. Chem. Soc., 2020, 142, 1406–1416 CrossRef CAS.
- J. B. Lee, Y. H. Roh, S. H. Um, H. Funabashi, W. Cheng, J. J. Cha, P. Kiatwuthinon, D. A. Muller and D. Luo, Nat. Nanotechnol., 2009, 4, 430–436 CrossRef CAS.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS.
- (a) E. Cheng, Y. Xing, P. Chen, Y. Yang, Y. Sun, D. Zhou, L. Xu, Q. Fan and D. Liu, Angew. Chem., Int. Ed., 2009, 48, 7660–7663 CrossRef CAS; (b) Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CAS.
- (a) D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS; (b) K. Hölz, E. Schaudy, J. Lietard and M. M. Somoza, Nat. Commun., 2019, 10, 3805 CrossRef.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS.
- (a) D. Berti, C. Montis and P. Baglioni, Soft Matter, 2011, 7, 7150–7158 RSC; (b) A. Nuthanakanti and S. G. Srivatsan, Nanoscale, 2016, 8, 3607–3619 RSC; (c) G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188–3206 RSC; (d) X. Liang, C. Gao, L. Cui, S. Wang, J. Wang and Z. Dai, Adv. Mater., 2017, 29, 1703135 CrossRef; (e) D. Zhang, Q. Liu, R. Visvanathan, M. R. Tuchband, G. H. Sheetah, B. D. Fairbanks, N. A. Clark, I. I. Smalyukh and C. N. Bowman, Soft Matter, 2018, 14, 7045–7051 RSC; (f) A. Nuthanakanti, M. B. Walunj, A. Torris, M. V. Badiger and S. G. Srivatsan, Nanoscale, 2019, 11, 11956–11966 RSC.
- (a) S. Sivakova and S. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC; (b) X. Li, Y. Kuang, H.-C. Lin, Y. Gao, J. Shi and B. Xu, Angew. Chem., Int. Ed., 2011, 50, 9365–9369 CrossRef CAS; (c) H. Zhao, X. Guo, S. He, X. Zeng, X. Zhou, C. Zhang, J. Hu, X. Wu, Z. Xing, L. Chu, Y. He and Q. Chen, Nat. Commun., 2014, 5, 3108 CrossRef; (d) O. Berger, L. Adler-Abramovich, M. Levy-Sakin, A. Grunwald, Y. Liebes-Peer, M. Bachar, L. Buzhansky, E. Mossou, V. T. Forsyth, T. Schwartz, Y. Ebenstein, F. Frolow, L. J. W. Shimon, F. Patolsky and E. Gazit, Nat. Nanotechnol., 2015, 10, 353–360 CrossRef CAS; (e) L. Latxague, M. A. Ramin, A. Appavoo, P. Berto, M. Maisani, C. Ehret, O. Chassande and P. Barthélémy, Angew. Chem., Int. Ed., 2015, 54, 4517–4521 CrossRef CAS; (f) A. Nuthanakanti, New J. Chem., 2019, 43, 13447–13456 RSC.
- (a) J. T. Davis and G. Spada, Chem. Soc. Rev., 2007, 36, 296–313 RSC; (b) J. M. Rivera and D. Silva-Brenes, Org. Lett., 2013, 15, 2350–2353 CrossRef CAS; (c) G. M. Peters, L. P. Skala, T. N. Plank, B. J. Hyman, G. N. M. Reddy, A. Marsh, S. P. Brown and J. T. Davis, J. Am. Chem. Soc., 2014, 136, 12596–12599 CrossRef CAS; (d) R. N. Das, Y. P. Kumar, O. M. Schütte, C. Steinem and J. Dash, J. Am. Chem. Soc., 2015, 137, 34–37 CrossRef CAS.
- J. Baillet, V. Desvergnes, A. Hamoud, L. Latxague and P. Barthélémy, Adv. Mater., 2018, 30, 1705078 CrossRef.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS.
- (a) Y. Gao, Y. Kuang, J.-F. Guo, Z. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS; (b) J. S. Park, S. Jeong, D. W. Chang, J. P. Kim, K. Kim, E.-K. Park and K.-W. Songe, Chem. Commun., 2011, 47, 4736–4738 RSC; (c) M. Ikeda, T. Tanida, T. Yoshii, K. Kurotani, S. Onogi, K. Urayama and I. Hamachi, Nat. Chem., 2014, 6, 511–518 CrossRef CAS; (d) M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS.
- (a) H. Wang and Z. Yang, Nanoscale, 2012, 4, 5259–5267 RSC; (b) D. Yuan and B. Xu, J. Mater. Chem. B, 2016, 4, 5638–5649 RSC.
- (a) L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC; (b) W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 5911–5920 CrossRef CAS; (c) K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. Ó Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2014, 4, 1480 CrossRef; (d) E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS; (e) E. R. Draper and D. J. Adams, Chem. Soc. Rev., 2018, 47, 3395–3405 RSC.
- (a) K. Thorat, S. Pandey, S. Chandrashekharappa, N. Vavilthota, A. A. Hiwale, P. Shah, S. Sreekumar, S. Upadhyay, T. Phuntsok, M. Mahato, K. K. Mudnakudu-Nagaraju, O. Sunnapu and P. K. Vemula, Sci. Adv., 2018, 4, 1780 CrossRef; (b) J. Liu, Y. Pang, S. Zhang, C. Cleveland, X. Yin, L. Booth, J. Lin, Y.-A. L. Lee, H. Mazdiyasni, S. Saxton, A. R. Kirtane, T. Erlach, J. Rogner, R. Langer and G. Traverso, Nat. Commun., 2017, 8, 124 CrossRef; (c) N. Latif, M. Asgari, H. Vali and L. Mongeau, Sci. Rep., 2018, 8, 1047 CrossRef.
- A. Nuthanakanti and S. G. Srivatsan, ACS Appl. Mater. Interfaces, 2017, 9, 22864–22874 CrossRef CAS.
- (a) B. Verdejo, F. Rodríguez-Llansola, B. Escuder, J. F. Miravet and P. Ballester, Chem. Commun., 2011, 47, 2017–2019 RSC; (b) F. Zhao, Y. Gao, J. Shi, H. M. Browdy and B. Xu, Langmuir, 2011, 27, 1510–1512 CrossRef CAS; (c) Q. Jin, J. Li, L. Zhang, S. Fanga and M. Liu, CrystEngComm, 2015, 17, 8058–8063 RSC.
- (a) H. Wang, W. Xu, S. Song, L. Feng, A. Song and J. Hao, J. Phys. Chem. B, 2014, 118, 4693–4701 CrossRef CAS; (b) M. A. Ramin, K. R. Sindhu, A. Appavoo, K. Oumzil, M. W. Grinstaff, O. Chassande and P. Barthélémy, Adv. Mater., 2017, 29, 1605227 CrossRef.
- (a) I. Velikyan, S. Acharya, A. Trifonova, A. Földesi and J. Chattopadhyaya, J. Am. Chem. Soc., 2001, 123, 2893–2894 CrossRef CAS; (b) T. Patra, A. Pal and J. Dey, Langmuir, 2010, 26, 7761–7767 CrossRef CAS.
- D. Li, Y. Shi and L. Wang, Chin. J. Chem., 2014, 32, 123–127 CrossRef CAS.
- IUPAC-IUB Commission on Biochemical Nomenclature, Abbreviations and Symbols for the Description of Conformations of Polynucleotide Chains, Pure Appl. Chem., 1983, 55, 1273–1280 Search PubMed.
- (a) Y. Kuang, Y. Gao, J. Shi, H.-C. Lin and B. Xu, Chem. Commun., 2011, 47, 8772–8774 RSC; (b) L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- (a) B. Adhikari, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC; (b) H. Kumari, S. E. Armitage, S. R. Kline, K. K. Damodaran, S. R. Kennedy, J. L. Atwood and J. W. Steed, Soft Matter, 2015, 11, 8471–8478 RSC.
- (a) Z. Qi, P. M. Molina, W. Jiang, Q. Wang, K. Nowosinski, A. Schulz, M. Gradzielski and C. A. Schalley, Chem. Sci., 2012, 3, 2073–2082 RSC; (b) S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356–2359 RSC.
- (a) M. Koyama, K. Bada, K. Sasaki, H. Tsuboi, A. Endou, M. Kubo, C. A. D. Carpio, E. Broclawik and A. Miyamoto, J. Phys. Chem. B, 2006, 110, 17872–17877 CrossRef CAS; (b) X. Dou, P. Li, D. Zhang and C.-L. Feng, Soft Matter, 2012, 8, 3231–3238 RSC.
- (a) F. Rodríguez-Llansola, B. Escuder, J. F. Miravet, D. Hermida-Merino, I. W. Hamley, C. J. Cardin and W. Hayes, Chem. Commun., 2010, 46, 7960–7962 RSC; (b) B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164–11166 RSC; (c) N. Cheng, Q. Hu, Y. Guo, Y. Wang and L. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 10258–10265 CrossRef CAS.
- R. Chung and K. S. Anderson, Tetrahedron Lett., 2006, 47, 8361–8363 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, nucleolipid characterization data, supplementary figures, tables, crystallographic data and NMR spectra. CCDC 1554880–1554882. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0na00509f |
| This journal is © The Royal Society of Chemistry 2020 |