
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomedical nanomaterials for immunological applications: ongoing research and clinical trials
Vincent
Lenders†
 ,
Xanthippi
Koutsoumpou†
,
Ara
Sargsian
and
Bella B.
Manshian
*
,
Xanthippi
Koutsoumpou†
,
Ara
Sargsian
and
Bella B.
Manshian
*
NanoHealth and Optical Imaging Group, Department of Imaging and Pathology, KU Leuven, B-3000 Leuven, Belgium. E-mail: bella.manshian@kuleuven.be
First published on 24th August 2020
Abstract
Research efforts on nanomaterial-based therapies for the treatment of autoimmune diseases and cancer have spiked and have made rapid progress over the past years. Nanomedicine has been shown to contribute significantly to overcome current therapeutic limitations, exhibiting advantages compared to conventional therapeutics, such as sustained drug release, delayed drug degradation and site-specific drug delivery. Multiple nanodrugs have reached the clinic, but translation is often hampered by either low targeting efficiency or undesired side effects. Nanomaterials, and especially inorganic nanoparticles, have gained criticism due to their potential toxic effects, including immunological alterations. However, many strategies have been attempted to improve the therapeutic efficacy of nanoparticles and exploit their unique properties for the treatment of inflammation and associated diseases. In this review, we elaborate on the immunomodulatory effects of nanomaterials, with a strong focus on the underlying mechanisms that lead to these specific immune responses. Nanomaterials to be discussed include inorganic nanoparticles such as gold, silica and silver, as well as organic nanomaterials such as polymer-, dendrimer-, liposomal- and protein-based nanoparticles. Furthermore, various approaches for tuning nanomaterials in order to enhance their efficacy and attenuate their immune stimulation or suppression, with respect to the therapeutic application, are described. Additionally, we illustrate how the acquired insights have been used to design immunotherapeutic strategies for a variety of diseases. The potential of nanomedicine-based therapeutic strategies in immunotherapy is further illustrated by an up to date overview of current clinical trials. Finally, recent efforts into enhancing immunogenic cell death through the use of nanoparticles are discussed.
 Vincent Lenders | Vincent Lenders is currently a Ph.D. researcher at the Nanohealth and Optical Imaging group at KU Leuven. His research interests include nanomedicine and advanced drug delivery systems for immunological and perinatal applications. He obtained his M.Sc. in Bioscience Engineering from KU Leuven in 2019. |
 Xanthippi Koutsoumpou | Xanthippi Koutsoumpou is a Ph.D. researcher at the Nanohealth and Optical Imaging group, KU Leuven. Her research interests include nanomedicine, magnetic hyperthermia and drug delivery systems for cancer treatment. She obtained her M.Sc. in Nanosciences and Nanotechnologies from Aristotle University of Thessaloniki in 2018. |
 Ara Sargsian | Ara Sargsian finished his Masters thesis successfully at the Nanohealth and Optical Imaging group at KU Leuven and from 1/09/2020 he will be a part of the group as a Ph.D. researcher. His research interests include Developing ex vivo cultures of animal and human explants for toxicity screening and efficacy testing of new drugs and nanomedical formulations. He obtained his M.Sc. in Biomedical science (specialisation: Biomedical Basic and Translational Research) from KU Leuven in 2020. |
 Bella Manshian | Bella Manshian is a group leader at the Nanohealth and Optical Imaging group at KU Leuven. Bella's research focuses on nanotoxicology and the manipulation of nanomaterials for in vivo theranostic applications. Bella has been using multimodal and multiparametric in vivo imaging, of mainly preclinical animal models, to assess disease mechanisms and therapy accompanied with screening of individual nanoparticle toxicity, the mechanisms involved and cellular–nanoparticle interaction kinetics. Bella's main interest is in developing non-invasive methods for the dual function of visualization and tracking of specific cell types with a strong focus on tumor cell, stem cells, immune cells and beta cells while simultaneously delivering therapeutic agents. |
1. Introduction
Our immune system has a crucial role in protecting us from various diseases. In the presence of danger signals, such as infection, tissue damage and cancer, an immune response is initiated that surveilles and eliminates foreign intruders and cancerous cells.1 However, dysregulation of self-tolerance can occur either by autoantigen triggering or by cross-reaction between self and foreign antigens. This subsequently leads to an immune reactivity against self-antigens and eventually results in an autoimmune disease.2 Additionally, cancerous cells have the ability to avoid recognition by the immune system by tumour-induced tolerance, leading to cancer growth and evasion.3 Therefore, there is a high need for the development of immune suppressive therapies for the treatment of autoimmune diseases such as; rheumatoid arthritis (RA), diabetes, inflammatory bowel disease (IBD) and multiple sclerosis (MS). Multiple immunomodulatory agents are used for the treatment of inflammation in autoimmune diseases, but limitations, such as side effects caused by low solubility and adherence of drugs, impede their therapeutic efficiency.4 Contrarily, cancer immunotherapy involves immunostimulatory therapies in order to overcome the immunosuppressive activity of cancer cells. Several immunotherapeutic strategies have shown promising results in cancer treatment, such as tumour-antigen vaccination, immune checkpoint blockade and adoptive cell therapy.5 However, undesired side effects and the immunosuppressive tumour microenvironment hinder the effectiveness of these strategies.6Some of these therapeutic limitations can be mitigated by the emerging field of nanomedicine, as nanomaterials can facilitate the targeted delivery of immunosuppressive agents, protecting them from degradation, improving drug absorption and preventing unwanted accumulation at off-target organs and tissues.7 The gained bioavailability of the drugs, for example by reducing the degradation in the gastrointestinal tract for oral delivery,8 significantly improves therapeutic efficacy. The extensiveness of the increase in drug availability can be controlled through modifications applied to the surface of nanoparticles, as they can be steered towards specific tissues or exhibit a stimuli-responsive behaviour for targeted and controlled drug release.9 Additionally, nanoparticles demonstrate potential advantages for immune stimulation. They have shown promising results as nanovaccines, for co-delivery of antigens and adjuvants, specifically to antigen presenting cells (APCs). Due to their unique properties, nanoparticles can protect antigens from premature degradation, improve their stability, enhance the cell uptake by APCs and stimulate immune response, as they exhibit good adjuvant properties themselves.10 Furthermore, nanoparticles can improve the therapeutic efficacy of cancer immunotherapy, by the targeted co-delivery of various immunomodulatory cytokines and immune checkpoint inhibitors to the tumour microenvironment (TME) in order to reprogram it and resume immune surveillance.11 Moreover, nanoparticles can co-deliver anti-cancer drugs and immunomodulatory compounds resulting in a synergistic anti-tumour therapeutic effect.5
However, in the process of designing nanomaterial-based therapies, it is key to understand the different mechanisms by which these nanomaterials interact and interfere with the immune system. Therefore, we summarize in this review the current understanding of interaction mechanisms of different nanomaterials with the immune system, focusing both on how nanomaterials can inflict stress and the different signaling pathways involved in the onset of the immune response. The mechanisms of the most important inorganic nanomaterials (gold, silica and silver) and organic nanomaterials (polymers, dendrimers, liposomes and proteins) will be discussed in detail. Additionally, strategies leveraging the improved understanding of the immunomodulatory behavior of nanoparticles, are elaborated on for a variety of immunological applications. The major potential of nanoparticles in immunotherapy is further illustrated by an up to date overview of current clinical trials.
2. Nanoparticle-induced mechanisms for immunological alterations
Although the field of nanomedicine has made major advances, nanomaterials continue to cause concern due to their potential toxic effects following human exposure. In the past years, major research efforts have been undertaken to understand these toxic effects, and certain pathways, mechanisms and key influential parameters have been implicated as a consequence of cellular exposure to nanomaterials. These nanotoxicology evaluations have also shed light on the immunological alterations caused by these nanomaterials. In order to control the immunomodulatory effect of nanomaterials in applications, the mechanism by which they induce immunological reactions needs to be understood. Therefore, we discuss the main pathways that can lead to NP-induced immune alterations (Table 1). Additionally, we discuss how these mechanisms can be influenced by e.g. adjusting physicochemical parameters.| Mechanism | NP-associated initiating effect | Cellular response | Immunological alteration | Examples |
|---|---|---|---|---|
| Stress mechanisms | ||||
| Oxidative stress | Intracellular release of reactive metal ions | ROS generation | MAPK and inflammasome activation | Silver; ZnO; CuO; TiO2 |
| Lysosomal membrane permeabilization (LMP) | Increased cationic charge in lysosome | Proton sponge effect; lysosomal enlargement and pore formation | Inflammasome activation | Polystyrene; crystalline silica; MWCNTs; titanium nanobelts |
| Lysosomal alkalization | Dissociation of ATPase V1 protein complex from membrane-associated ion conductance V0 protein complex | Dysregulation of vacuolar H+-(V)-ATPase; alkalization of the lysosome | No direct effect | Gold |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Signalling pathways | ||||
| Toll-like receptors (TLR) | Binding of NP on TLR through e.g. hydrophobic effects | NA | TLR activation; increase or decrease of cytokines | CNTs; ZnO; silver; gold |
| Mitogen-activated protein kinases (MAPK) | ROS generation | NA | MAPK stimulates NF-κB; increase of pro-inflammatory cytokines | Silica (colloidal and mesoporous) |
| Inflammasomes (NLRP3) | ROS generation; lysosomal cathepsin release | NA | NLRP3 activates caspase 1; production of IL-1β and IL-18 | Silver; AlO; MWCNT; TiO2 |
| Pyroptosis | NLRP3 and caspase 1 activation; Cleavage of gasdermin D | Pore formation in cell membrane; cell death | Immunogenic cell death; release of intracellular contents e.g. IL-1β | Mesoporous silica; La2O3 and Gd2O3 |
| Necroptosis | Upregulation of ZBP1; activation of RIPK3 | Pore formation in plasma membrane; cell death | Immunogenic cell death | Selenium; silica |
| Ferroptosis | Increased iron availability | Lipid peroxidation and glutathione depletion; cell death | Immunogenic cell death | Doxorubicin-loaded iron-saturated ferritin |
2.1. NP-induced stress
It is important to note that depending on the intrinsic material reactiveness and possible surface modifications, the induced oxidative stress varies for different nanomaterials. Redox-active nanomaterials for example, can either be antioxidant or prooxidant. Iron oxide is an example of a prooxidant nanomaterial and is heavily researched, especially superparamagnetic iron oxide nanoparticles (SPIONs), for their use in chemotherapy.14 Trough Fenton and Haber-Weiss reactions, Fe2+ and Fe3+ ions can catalyze ROS generating reactions.15 When exposed to ultraviolet radiation, titanium oxide (TiO2) is also prooxidant. UV exposure will lead to the formation of electron–hole pairs in the nanomaterial, which can react with oxygen to form ROS. This mechanism can be exploited in the photodynamic treatment of cancer.16 Cerium oxide (CeO) is an example of an antioxidative nanomaterial, that scavenges ROS through the Ce3+ and Ce4+ on the surface of the particles.17 An interesting study, performed by Dowding et al., showed that the synthesis method of redox-reactive nanomaterials like CeO, can greatly influence the redox state of the material and thereby the anti- or prooxidative property of the material. This is a possible explanation for contradictory findings on toxicity in literature.18
Another way NPs can generate ROS is through oxidation of the nanomaterial itself, which subsequently leads to the release of (toxic) metal ions in a process called NP dissolution. A few, easily ionized nanomaterials (e.g. Ag, ZnO, CuO) have been described to release ions, and especially silver nanoparticles have been studied in detail, because of their popular use in consumer products leveraging their antimicrobial activity.19 However, AgNPs have been shown to induce toxicity upon in vivo exposure.20 Multiple reports have indicated that this toxicity effect is mainly due to the release of ions during NP dissolution, although some reports indicate that NP-associated effects contribute to the toxicity of the particles as well.21 However, the most accepted mechanism for easily ionized metal nanoparticles is based on the so-called Trojan-horse mechanism.22 This mechanism was studied in more detail by Setyawati et al. and showed that particles are taken up via endocytosis by the cell and subsequently degraded in the lysosome, because the lower pH level accelerates NP dissolution. Ag0 is then oxidized to Ag+, in a conversion step that includes ROS byproducts. The Ag+ ions themselves interfere with the respiratory chain of the cell and lead to additional ROS formation.23
ROS, either generated by redox-active materials or through NP dissolution, can elicit a variety of cellular processes with detrimental outcomes (Fig. 1). NP-induced ROS generation has, for example, been linked to mutagenic and carcinogenic effects and to induce lung fibrosis.24 Additionally, ROS plays an important role in the induction of inflammation through the activation of oxidant-dependent mitogen-activated protein kinases (MAPKs) or the activation of the inflammasome. Murphy et al. investigated the pro-inflammatory effect of AgNP in THP-1 cells and results showed an increased gene expression of IL-1, IL-6 and TNF-α. Additionally, a higher release of IL-1β indicates the activation of the inflammasome.25
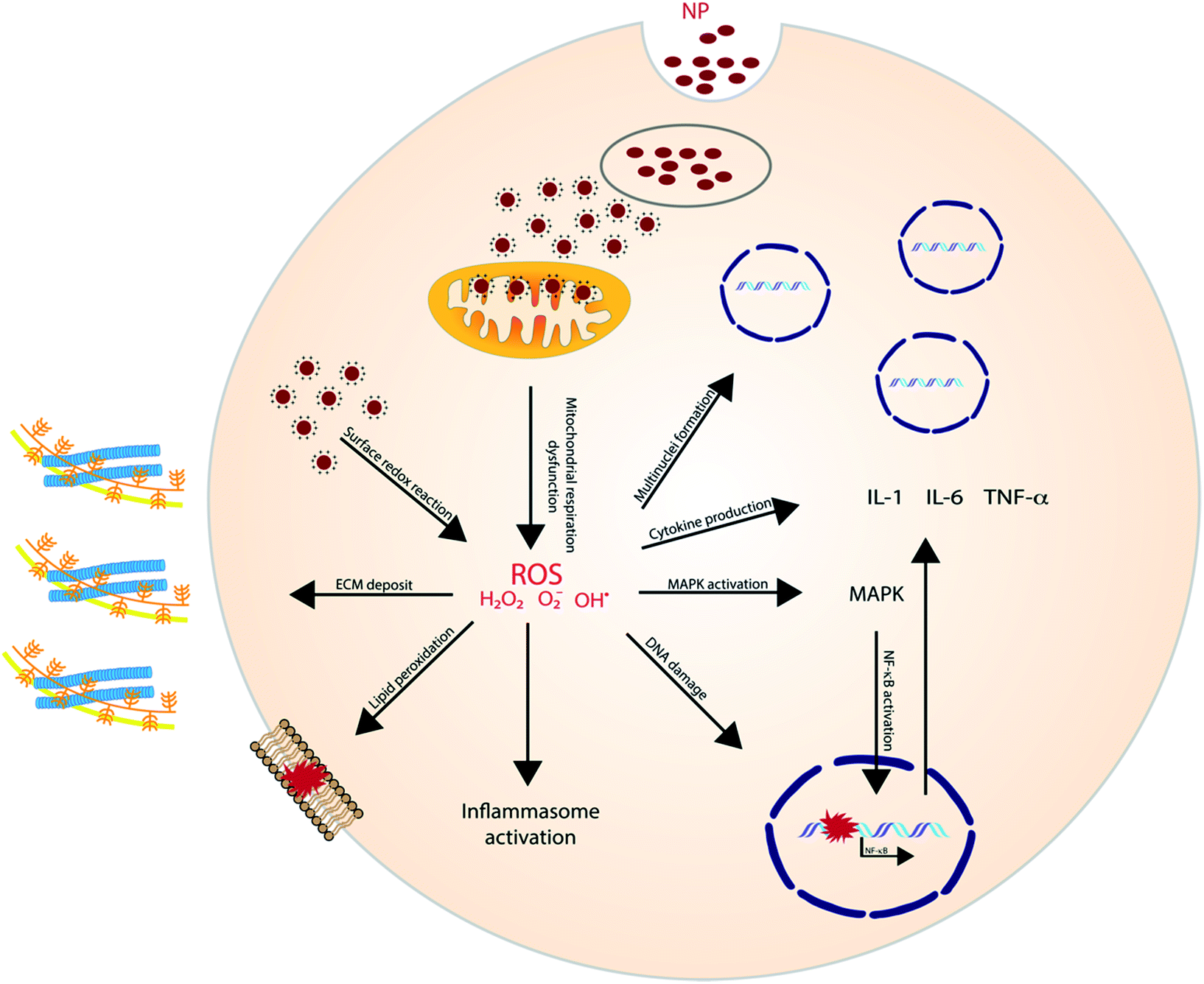 | ||
| Fig. 1 ROS generated oxidative stress elicit a variety of adverse, cellular processes. | ||
Other metal NPs have shown to have a similar effect as well. In Brzicova et al., it was shown that zinc oxide (ZnO) dissolution, and thus the release of Zn2+, leads to the release of pro-inflammatory cytokines in THP-1 cells. Additionally, an enhanced expression of ICAM-1 and VCAM-1 was observed, which plays an important role in leukocyte adhesion.26 The same inflammatory effect of ZnO was later shown in vivo and was proven to be regulated by the ROS-triggered activation of MAPKs.27,28 Similarly, Cu+ ions were illustrated to contribute to ROS generation after copper oxide (CuO) NP dissolution,29 and have shown to induce oxidative stress and trigger apoptosis in lymphocytes. ROS triggered lipid peroxidation, membrane potential collapse and lysosomal membrane leakage were all shown to contribute to the observed lymphocyte cell death.30
Several parameters can influence oxidative stress and, therefore, the immunomodulatory effect of nanomaterials, e.g. by influencing their dissolution rate. Specific NP properties include composition of the metal core, size and surface coating. Size influences the dissolution rate, as more oxidation will take place on the larger reactive surface area of smaller NP.31 Park et al., showed that 4 nm AgNPs induce a higher release of chemokines (IL-8) in comparison to larger AgNPs (20 and 70 nm) after in vitro exposure to macrophages.32 A similar size-dependent inflammatory effect was observed after in vivo pulmonary exposure to AgNPs with sizes 15 and 410 nm. Exposure to the smallest AgNPs led to a 5-fold increase in pro-inflammatory cytokines (IL-1beta and MCP-1) and a 175-fold increase influx of neutrophils to the lungs.33
A second way to influence dissolution and toxicity is the use of adequate coatings. Alcaron et al., showed that type 1 collagen coated AgNPs are very stable and elicit no toxic effects in human fibroblasts and keratinocytes.34 Similarly, Manshian et al. evaluated the effect of 3 different coatings on AgNP toxicity: mercaptoundecanoic acid (MUA), dodecylamine-modified poly(isobutylene-alt-maleic anhydride) (PMA) and polyethylene glycol (PEG). The different coatings were shown to have little effect on the intrinsic level of NP dissolution but did influence cellular NP uptake and thereby the level of Ag+ in the cell. Additionally, different toxicity levels were observed for the different NPs, initiated by different toxicity pathways: PMA-coated NPs affected the cells through autophagy and cytoskeletal deformations, while MUA-NPs induce membrane damage because of agglomerate sedimentation. The difference in toxicity between the 3 different NPs indicates the importance of surface chemistry and proves that besides NP dissolution, also NP-associated effects contribute to ROS formation.21 The effect of surface chemistry was also illustrated for CuO NPs by Ilves et al., in vivo. It was shown that inhalation of unmodified CuO lead to an exacerbation of allergic airway inflammation through an increased neutrophil influx. However, by coating the particles with PEG, this immune effect was suppressed.35 The PEGylation effect on immune avoidance is commonly known. PEG-coating increases the hydrophilicity and neutralizes the surface charge on the particles. Without PEGylation, hydrophobic and cationic polymers show increased opsonization by serum proteins and as a result, a higher uptake by phagocytic cells.36
 | ||
| Fig. 2 Lysosomal membrane permeabilization mechanism. Reproduced from ref. 38 with permission from The Royal Society of Chemistry. | ||
LMP has been reported to play a key role in NP-induced inflammation, more specifically, NLRP3 inflammasome activation. Jessop et al. illustrated that crystalline silica, multiwalled carbon nanotubes (MWCNTs) and titanium nanobelts induce acidification of the phagolysosome, which appeared to be critical in the eventually caused LMP in the exposed macrophages. This was proven by comparing silica exposure with or without Bafilomycin A1, a vATPase inhibitor which impedes proton influx. Results indicated that Bafilomycin A1 treatment inhibited lysosomal acidification and LMP.39 Also, the cationic polymer polyerthylenimine (PEI), which is mostly used as gene transfer vehicle, has been linked to induce LMP.40 For example; Sang-Hyun Park and colleagues demonstrated that treating HeLa cells with lysosomal membrane stabilization protein inhibitors resulted in a reduction in lysosomal Cl− concentrations and the induction of LMP.41
2.2. Signaling pathways
Stress, generated by the exposure to nanoparticles, initiates (multiple) cell signaling pathways that eventually leads to immunological alterations. Several signaling molecules and pathways have been proposed to be involved and proven to be of importance for the different nanomaterials.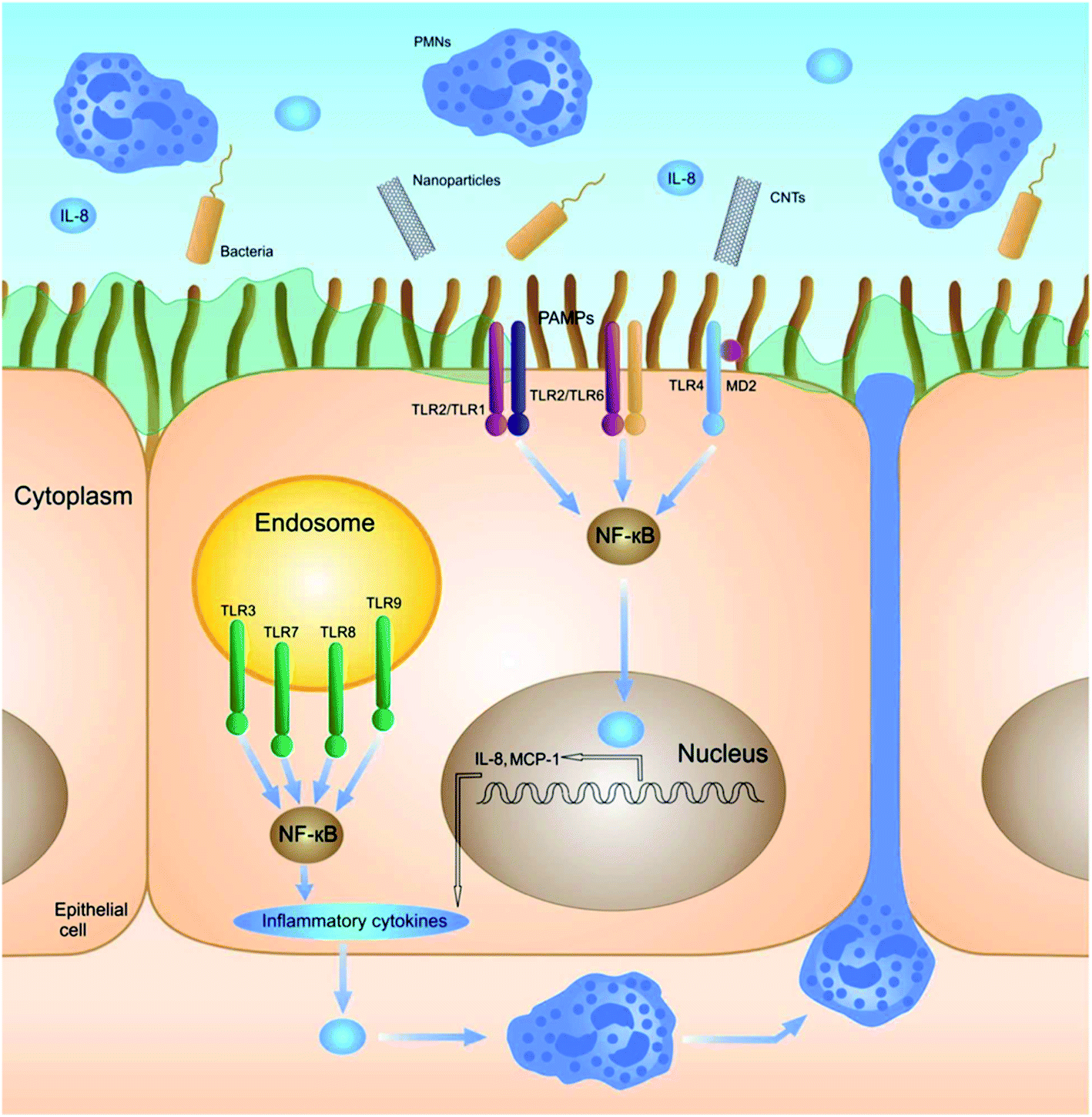 | ||
| Fig. 3 Schematic illustration of the nanoparticle-induced signaling pathway regulated by TLRs. Reproduced from ref. 46 with permission of The Royal Society of Chemistry. | ||
Interestingly, NP interaction with TLRs can also lead to an immunosuppressive effect. Gliga et al. recently showed that Ag nanoparticles reduce the LPS-induced release of pro-inflammatory cytokines in THP-1 cells (IL-1β, TNF-α and IL-6). It was shown that co-treatment of LPS and AgNPs resulted in a dose-dependent inhibition of TLR2, which was suggested to be caused by the release of silver ions.49 Tsai et al. illustrated that AuNPs can also lead to TLR-regulated immunosuppression. They showed that small AuNPs (4 nm) can inhibit TLR9 and, therefore, inhibit the production of pro-inflammatory cytokines.50
The role of NLRP3 is repeatedly shown in inflammatory responses to high aspect ratio nanomaterials. Manshian et al. illustrated the effect of aspect ratio for aluminum oxide (AlO) NPs in four different cell lines (KLN205, HeLa, A549 and SKOV3). The study showed that rod-like AIO NP resulted in higher toxicity, due to a higher cellular uptake, while wire-like particles showed higher activation of the NLRP3 inflammasome.44 Hamilton et al. showed similar effects for TiO2 by comparing nanospheres, 5 μm and 20 μm nanobelts in mouse alveolar macrophages. The longest nanobelts induced a significantly higher amount of pro-inflammatory cytokines (IL-1β and IL-18).56 Also, MWCNTs linked to inducing lung fibrosis, induce inflammation through the activation of NLRP3. Interestingly, Sun et al. showed the contribution of NADPH oxidase, able to generate an oxidative burst, for the MWCNT-induced inflammasome activation.57
Pyroptosis can be stimulated by multiple microbial infections, like Salmonella and Legionella, but also a few nanomaterials have been shown to induce pyroptotic cell death. Reisetter et al. showed that exposure of human alveolar macrophages to black carbon nanoparticles leads to the activation of the inflammasome and caspase 1, increased IL-1β production and eventually pyroptosis. The distinction between pyroptosis and apoptosis was proven by the protective effect of treatment with YVAD, a capsase-1 inhibitor, and glycine, a pyroptosis inhibitor.59 Furthermore, Zang et al. linked MSN to liver inflammation and hepatotoxicity. MSNs were shown to increase ROS levels, activate NLRP3 and subsequently initiate caspase 1-dependent pyroptosis in hepatocytes, both, in vitro and in vivo. The crucial role of NLRP3 was proven by the mitigation following NPRP3 knockdown or treatment with MCC950, a selective inhibitor of NLRP3.60 A similar liver inflammation was observed in an interesting study evaluating the effect of 29 metal oxides. Of these metal oxides, only the rare-earth oxide (REO) NPs, e.g. La2O3 and Gd2O3, induced pyroptosis. However, pyroptosis was only observed in Kupffer cells, contrary to the observed apoptosis in hepatocytes. Lysosomal membrane permeabilization, induced by the lysosomal accumulation of REO NPs, was shown to play a key role in the activation of caspase 1. Caspase 1 subsequently cleaves Pro-IL-1β and gasdermin D (GDDMD), resulting in N-GSDMD, which, after oligomerization in the cell membrane, leads to the formation of pores and the release of intracellular contents61 (Fig. 4).
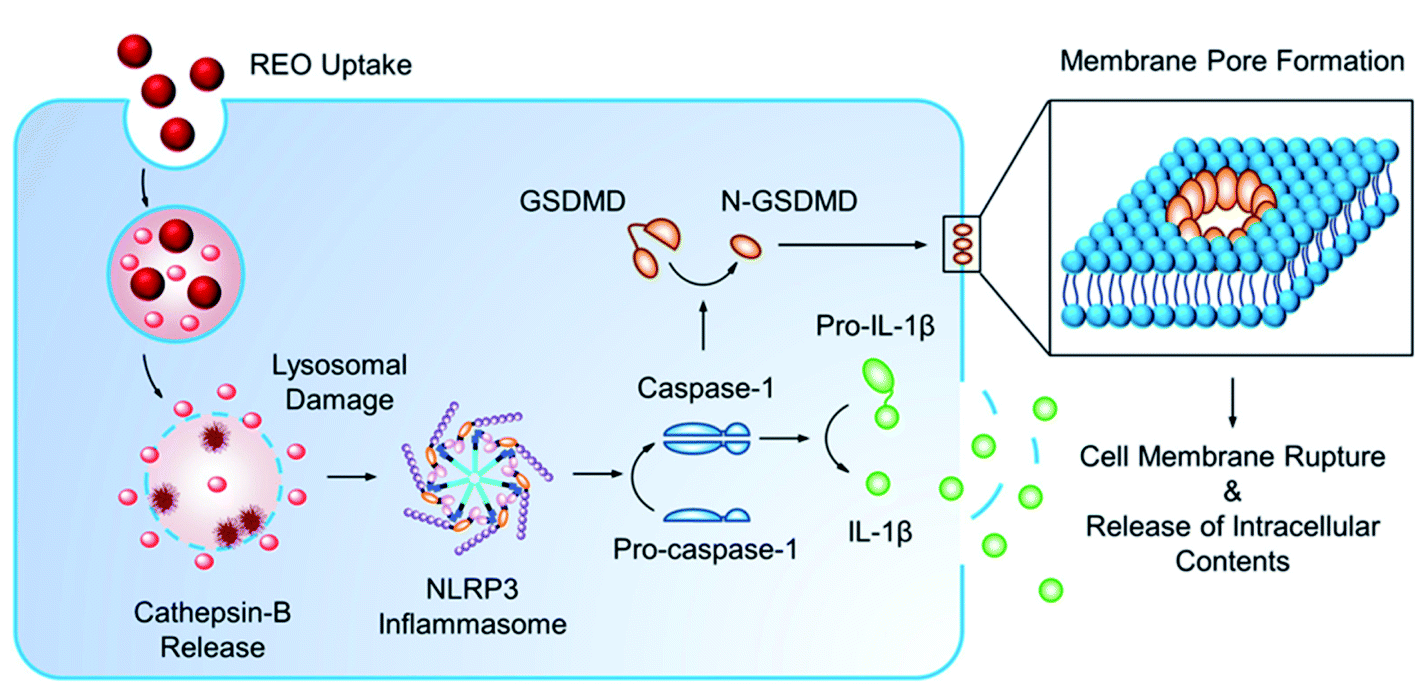 | ||
| Fig. 4 Mechanism of pyroptosis after uptake of nanoparticles. Reprinted with permission from ref. 61. Copyright (2018) American Chemical Society. | ||
Necroptosis is another immunogenic cell death mechanism, regulated by 2 key components: (1) receptor-interacting serine–threonine kinase 3 (RIPK3) and (2) mixed lineage kinase domain-mike (MLKL). These 2 components are crucial in the characteristic plasma membrane permeabilization. RIPK3 is responsible for phosphorylation of serine and threonine residues on MLKL, which facilitates MLKL oligomerization, eventually leading to pore formation in the plasma membrane. Additionally, both RIPK3 and MLKL have been linked to the activation of the inflammasome, which leads to the proinflammatory potential associated with necroptosis.62 A few nanomaterials have been linked to necroptosis, for example, silica nanoparticles have been shown to induce necroptosis in hepatocellular carcinoma cells. Interestingly, it was shown that Z-DNA binding protein 1 (ZBP1), an RIPK3 activating protein, was upregulated after silica exposure and played a crucial role in necroptosis.63 Additionally, Selenium nanoparticles were shown to induce necroptosis in PC-3 cancer cells.64
A third immunogenic cell death pathway is ferroptosis. This regulated cell death mechanism is mediated by lipid peroxidation and iron availability and induced by glutathione depletion.65 This type of cell death has been first proposed by Dixon et al. in 2012 and therefore nanomaterials linked to ferroptosis are limited at present.66 Yang et al. designed doxorubicin loaded iron saturated ferritin nanoparticles that efficiently led to cell death of HT29 cells via ferroptosis. However, this was only seen for the drug loaded nanomaterials. Unloaded ferritin nanoparticles showed almost no cytotoxicity to HT29 and thus were unable to initiate ferroptosis.67
3. Inorganic nanomaterials in immunological applications
The insights discussed above have allowed researchers to tune these nanomaterials in applications where an enhanced activation of the immune response is required. The most important applications where this is the case are nanovaccines. Nanoparticles can serve 3 purposes in vaccines: they can serve as adjuvant, as an antigen carrier and as an adjuvant carrier. Beside applications in which NP-induced immune responses are desired, nanoparticles can also be used as delivery vehicles for the targeted delivery of compounds that interact with the immune system. In these applications, the goal of using nanomaterials is to enhance the efficiency of the active compound, while the toxic or immunogenic effect of the material is undesirable. At present, inorganic nanomaterials qualifying these criteria and used for immunological applications, are scarce.3.1. Gold
Gold NPs are among the most popular nanoparticles, because they are considered bioinert, are easily synthesized and modified and have proven to be successful in a wide range of applications such as; biosensors, drug delivery and optical imaging applications.68 In addition, their use as nanovaccines has shown to have potential in cancer therapy,69 Influenza A70 and Malaria,71 where they can serve as adjuvant, as antigen carrier, and adjuvant carrier. This was illustrated by Almeida and colleagues, who studied the use of AuNPs in peptide vaccines in anti-tumour immunotherapy. They showed that OVA-coated AuNPs, induce a higher systemic antigen-specific response compared to free OVA due to a facilitated delivery. This immune response translated in tumour growth reduction in a B16-OVA tumour mouse model. Also, the use of AuNPs as adjuvant carrier was shown by evaluating coupled CpG:AuNPs. Results show that a higher immune response is generated after administration of free OVA in combination with CpG:AuNPs compared to both free OVA and CpG. Interestingly, comparing the immune response to either OVA:AuNPs or to OVA:AuNPs in combination with CpG:AuNPs, was not significantly different.69 However, the adjuvant effect of AuNP does not imply the uselessness of other adjuvants. Wang et al. showed, indeed, that the combination of influenza A hemagglutinin bound AuNPs (AuNP-HA) with flaggelin (Flic)-bound AuNPs (Flic-AuNP) was essential for the effectiveness of the vaccine. Without the adjuvant Flic-AuNP, the AuNP-HA induced a similar antigen-specific immune response, but was unable to effectively induce a favorable IgG1/IgG2a ratio and, therefore, the promotion of cellular immunity.72Optimizing AuNPs as a vaccine delivery platform is possible by, for example, increasing the amount of peptides per particle, as illustrated by the bottom-up, self-assembling method proposed by Lin et al. (Fig. 5). By first coating the AuNPs with PEG-SH and subsequently conjugating the peptides using EDC/sulfo-NHS chemistry, multiple repeats of the peptide can be bound to the AuNP. This can enhance the efficiency of nanovaccines and deliver larger doses, resulting in stronger immunogenicity.73 Another optimization method is choosing peptides that conjugate in a highly ordered, densely packed manner. Higher order organization of epitopes, similar to highly efficient surfaces of viruses, provokes a strong immune response. The coating order is influenced by the presence of the hydrophobic chains in the peptides.74
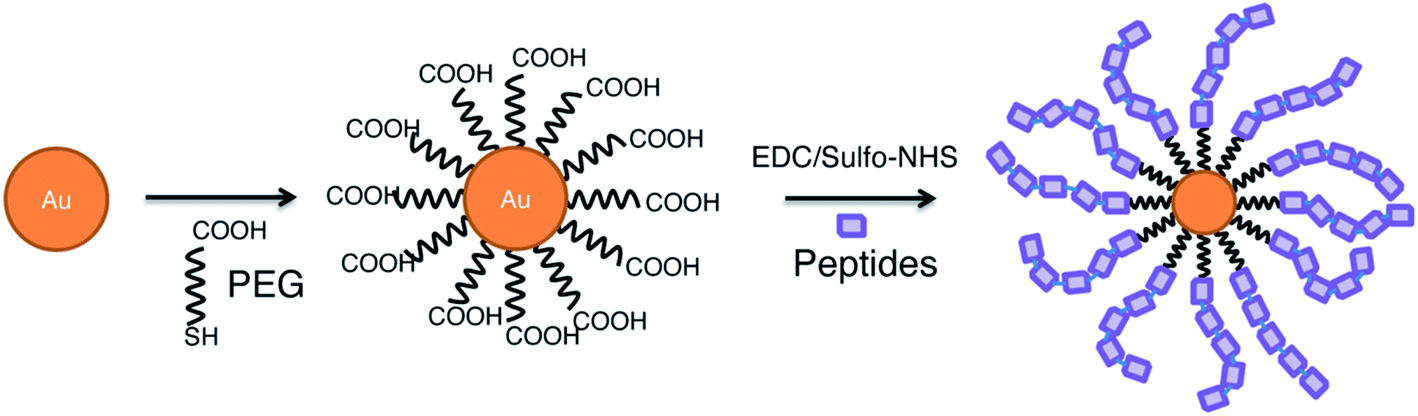 | ||
| Fig. 5 Self-assembly of peptides on gold nanoparticle surface.73 | ||
Given the immunomodulatory effect of AuNPs, they have also been researched as potential anti-virals. The anti-viral effect of gold has been proven in vivo for respiratory syncytial virus (RSV) in Bawage et al. By stimulating the innate immune system through AuNP mediated TLR, NOD-like and RIG-1-like receptor signaling pathways, the production of cytokines and chemokines increases leading to an enforced defense mechanism and subsequently, a reduced replication of RSV.75
3.2. Silica
Mesoporous silica nanoparticles have been heavily researched for their potential in nanovaccines.76 The use of these hard, inorganic nanomaterials is especially interesting for oral vaccines. Despite the advantages of oral vaccination (ease of administration and increased patient compliance), poses the harsh gastrointestinal environment an enormous challenge for effective vaccination due to drug inactivation or degradation. For the purpose of developing novel oral vaccine adjuvants, Wang et al. investigated the potential use of mesoporous silica nanoparticles, serving as both the antigen carrier and as adjuvant. They designed 3 different particles with different particle sizes and pore diameter/geometry, which were loaded with BSA. Results of the systemic and humoral immune response in mice showed that the silica loaded nanoparticles induced a significant higher IgG production in comparison with the free BSA. Within different particle designs, a higher antibody titer can be obtained if the release of the antigen is prolonged. A larger pore diameter and particle size were shown to possibly prolong the release of antigens. However, an optimal size range around 500 nm was reported, balancing the antigen release rate and the cellular uptake of the particles. Besides its clear antigen delivery potential, results showed that both the Th1 (cell-mediated) and Th2 (humoral) immune responses were induced upon administration of porous silica.77 The self-adjuvant effect of MSN was strongly illustrated by Mahony et al., were the adjuvant performance of AVO-loaded MSN was compared to the widely used adjuvant QuilA. Immunization results in mice showed that an MSN formulation with 10 μg of OVA resulted in a strong antibody and cell-mediated response, which was only slightly smaller than the OVA-QuilA formulation. The high immune response with a lower amount of antigen, together with the observation that MSN did not induce any toxic events, shows the potential of MSN as a self-adjuvant vaccine delivery platform.78 Efforts have been made to optimize this platform further, for example by tuning the adjuvant potency by changing the surface chemistry of the MSN particles. Yang et al., showed that a hydrophobic –C18 modification, in comparison to –OH and –NH2-groups, significantly enhances antigen uptake by APCs, macrophage maturation and antibody response in mice. Additionally, the positively charged –NH2 particles showed a higher antibody response in comparison to the negatively charged –OH particles.79Mesoporous silica is also one of the most interesting inorganic nanomaterials for drug delivery. The potential of MSN-based drug delivery is based on its large internal volume and high drug loading capacity. Additionally, as explained previously, physical properties of MSN can be tuned so that the immune response is limited. In Heidegger et al., a pro-inflammatory drug (synthetic TLR7/8 ligand R848) was loaded in MSN particles and was shown, in contrast to empty MSN, to provoke a strong immune response in mice.80 Additionally, anti-inflammatory agents can be loaded in MSN for drug delivery. Braz et al. loaded MSN with the anti-inflammatory drug indomethacin and showed that the MSN-drug complex significantly lowered the in vitro cytotoxicity compared to the free form of the drug.81
These MSN-based systems can be further improved, for example by using coatings such as ethylcellulose, which prolongs the drug release.82 Additionally, it can be modified for stimuli-responsive drug release by e.g. adding a lipid layer, which resolves in the reductive intracellular environment and subsequently sets the encapsulated drug free.83 The drug loading capacity and entrapment efficiency can be improved by using hollow MSNs, but these have the disadvantage that all pores are connected to one large reservoir and the drug can be released by any of the interconnected pores.84,85
3.3. Silver
In the process of wound healing, the inflammatory stage is essential for a rapid clean-up of the tissue and the neutralization of invading agents.86 Studies have shown that AgNPs induce an acute immune response, which normalizes over time.33 This short-term inflammation has been found to be beneficial in wound healing. The initial rapid inflammation speeds up the wound healing process, eventually decreasing inflammation faster.87 Tian et al., demonstrated the beneficial effect of AgNPs in a thermal injury mouse model. By applying AgNP-coated dressings on the burn wound, an upregulation in mRNA levels of VEGF, IL-10 and IFN-γ was obtained, contributing to a fast wound healing process.88 Additionally, Kwan et al. showed that using AgNPs for wound healing also leads to better restored functionality of the healed skin.893.4. Aluminium
Aluminium salts (Alum) are widely used as adjuvants but have been shown not to be effective for several vaccine targets such as influenza, HPV and HBV, because alum mainly induces Th2 type immunity.90 Orr et al. showed in their study that engineering the properties of Alhydrogel nanoparticles (nanoalum) can greatly influences their adjuvant properties. Results showed that, while the unbound formulation of poly(acryl) acid (PAA) and nanoalum promoted a Th2 immune response, PAA-coated nanoalum strongly promoted a Th1 immune response.91 The engineering of these nanoparticles has thus led to a possible new adjuvant class that can be used for diseases where Th1 immunity is important. In a follow-up study, Khandar et al. showed that the oxidative state of the core of the nanoalum influences its adjuvant capability. Nanoalum made from AlO(OH) did induce CD4+ T cells in mice, while the γ-Al2O3 derived particles did not.903.5. Silicon
Porous silicon is a biodegradable and biocompatible material with a large drug loading capacity. In Gu et al., porous silicon nanoparticles (SiNPs) were incorporated with multiple copies of the FGK45 antibody, an agonistic antibody to the APC receptor CD40. In vitro stimulation of mice B cells revealed 30–40-fold activation level FGK-SiNP in comparison with free FGK45, while empty SiNP showed no B cell activation, indicating the immunological inertness of the nanomaterials. The increased activation was shown to be due to an increased potency of the conjugated FGK45 and not because of multivalency.923.6. Others
Some inorganic nanomaterials have proven to be effective in interacting with microbes and viruses and can offer potential in therapeutics for infectious diseases. In this way, inorganic nanoparticles can impact indirectly the induced immune response. The most researched nanomaterial for this is silver, that has proven to act, both, as anti-microbial93 and anti-viral.94 Additionally, given the previously explained lysosomal alkalization effect of gold nanoparticles, AuNPs might be of interest as less toxic variant for chloroquine in reducing replication of ACE2 receptor dependent viruses, like the coronaviruses SARS-Cov, NL63 and SARS-Cov-2.95 A higher pH in lysosomes (and endosomes) reduces the rupture of the virus particle and therefore prevents the release of infectious viral nucleic acids.96 Additionally, alkalization affects glycolisation of the ACE2 receptor and, therefore, the viral binding affinity of the receptor.97 A detailed discussion of nanomaterials in infectious diseases falls outside the scope of this review. Finally, inorganic nanomaterials can be used in combination therapy, which will be discussed in a later chapter.4. Organic nanoparticles in immunological applications
A plethora of organic nanocarriers have been reported as promising drug delivery systems including polymer-based nanoparticles, liposomes, dendrimers and protein nanoparticles (Fig. 6).98,99 Their advantages include: (1) the improved bioavailability of insoluble drugs and reduced side effects, (2) the protection of their cargo from degradation and rapid clearance, (3) specific tissue targeting using surface-coupled ligands, (4) controlled drug release and (5) co-delivery of agents for a synergistic therapeutic effect. Due to their preferable properties, such as their increased biodegradability, biocompatibility and stability,98 organic nanomaterials have been extensively investigated as potential delivery vehicles of anti-inflammatory agents to inflamed tissues (Table 2), while they have also been explored as a cargo for vaccines and vaccine adjuvants (Table 3). Many studies have shown that the physicochemical characteristics and surface modifications of organic nanoparticles moderate their immunological response. As has been well illustrated in the review of M. A. Dobrovolskaia and S. E. McNeil, different parameters, including size, shape, hydrophobicity, surface chemistry and functionalization as well as their route of administration have an essential role on the immunomodulatory effects of nanoparticles and consequently on their successive outcome, depending on the application used.100 In this section, we focused on organic nanoparticle-based treatments for inflammatory diseases as well as on their role as vaccine delivery systems and vaccine adjuvants.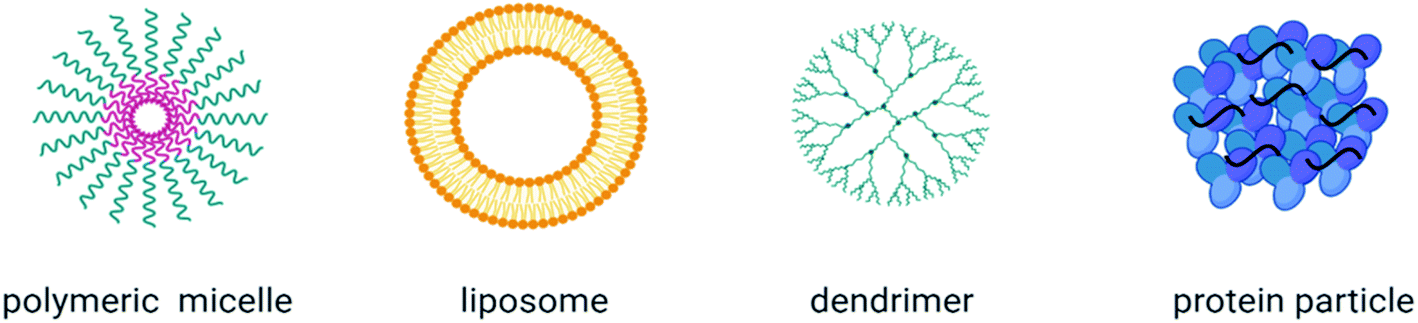 | ||
| Fig. 6 Classification of organic nanoparticles. Created with Biorender.com. | ||
| Nanoparticle | Loaded anti-inflammatory agent | Modification | Application | System |
|---|---|---|---|---|
| Polymeric | ||||
| PLA-PEG105 | TNFa-siRNA | Covalent surface Fab’ attachment | Inflammatory bowel disease (IBD) treatment | Mouse model of colitis |
| PLGA-PEG-PLGA110 | Etoricoxib (NSAID) | NA | Osteoarthritis (OA) treatment | Intra-articular injection in rat OA model |
| PLA-PEG111 | Curcumin | NA | Liver inflammation treatment | STZ-induced diabetic rats |
| PLA-PEG115 | NA | Surface coupled mAbs to E-selectin, VCAM-1, and ICAM-1 | Inflamed endothelium targeting | In vitro: adhesion assay (HUVEC), in vivo: adhesion assay post TNF-a injection (mice) |
| PLGA-PEG116 | Genistein (protein tyrosine kinase inhibitor) | Surface coupled islet-homing peptide (CHVLWSTRC; Pep I) | Insulitis therapy | Islet CE cells [leukocyte−endothelial cell adhesion assay] |
| PLGA118 | NA | Surface coupled a2,8 N-acetylneuraminic acid (NANA) | Sepsis treatment | In vitro: LPS stimulated mice peritoneal macrophages, primary human monocyte derived macrophages and monocytes (MDMs), in vivo: mouse model of systemic inflammation, mouse model of lung injury, ex vivo: human lung perfusion model |
| PLGA119 | NA | Macrophage membrane coating | Sepsis treatment | In vitro: mouse TLR4 reporter cells, HUVECs, in vivo: mouse bacteraemia model, mouse endotoxemia model |
| PLGA120 | NA | Human neutrophil membrane coating/mouse neutrophil membrane coating | Rheumatoid arthritis (RA) treatment | In vitro: human chondrocytes, HUVECs, in vivo: injured cartilage mouse model, inflammaed cartilage mouse model, collagen-induced arthritis (CIA) mouse model, human transgenic mouse model of inflammatory arthritis |
| APN micelle121 | Prednisolone (PD) [glucocorticoid] | NA | Rheumatoid arthritis (RA) treatment | Mouse model of rheumatoid arthritis |
| PPS-PNIPAm (ROS-temperature responsive copolymer)122 | DOX | NA | Inflammation and tumour targeting | MCF-7 cell line |
| Polyketal (PK)123 | Superoxide dismutase (SOD) | NA | Interstitial lung diseases (ILD) treatment | Bleomycin mouse model of lung fibrosis |
| HPOX125 | Biodegradable hydroxybenzyl alcohol (HBA) | NA | Airway inflammatory diseases treatment | Mouse model of allergic asthma |
|
||||
| Liposomes | ||||
| DPPC/DPPG/Chol, 50/10/40 mol% (ref. 139) | Dexamethasone phosphate (DXM-P) (glycocorticoid) | NA | Rheumatoid arthritis (RA) treatment | Antigen-induced RA rat model |
| (NH+)-DOPC/DOPE/DOTAP140 | Guanosine 5′-diphosphate (GDP) | NA | Anaemia of inflammation (AI) treatment | In vitro: U937 monocytic cells and co-culture model consisting of HepG2 and Caco2 cells, in vivo: acute and chronic AI mouse model |
| LEC, F70101C-AL, FormuMax Scientific, Sunnyvale, CA, USA141 | Clodronate (LEC) | NA | Acute inflammation treatment | Carrageenan-induced inflammation model |
| DPPC, PEG-DSPE (1.85:0.15:1.0)148 |
Prednisolone phosphate (PP) | NA | Rheumatoid arthritis (RA) treatment | Adjuvant induced arthritis rat model |
| DPPC, PEG-(2000)-DSPE, NBD-PE, cholesterol (1.85:0.15:0:1)150 |
Dexamethasone phosphate | NA | Hepatitis and liver fibrosis treatment | Mouse models of acute concanavalin A (ConA)-based hepatitis and chronic toxic carbon tetrachloride (CCl4)-based liver injury |
| DSPE-PEG2000 (ref. 151) | Dexamethasone (Dex) | DC8,9 PC molecules crosslinked in the bilayer by UV irradiation | Rheumatoid arthritis (RA) treatment | In vitro: raw264.7 cell line, in vivo: adjuvant induced arthritis rat model |
| LipoCardium155 | Cyclopentenone prostaglandin (PGA2) | Surface coupled anti-VCAM-1 antibodies | Atherosclerosis treatment | In vitro: U937pro-monocytic cell line, in vivo: atherosclerosis induced mouse model |
| DPPC 53% cholesterol 45%, DSPE–PEG2000 2%156 | NA | Surface coupled anti-ICAM-1 antibodies | Intra-arterial drug delivery of CNS disorders | Brain inflammation mouse model induced by microinjection of TNF |
| DSPC:CH: SA (7.5:2.5:0.5) (inner liposome)158 |
Prednisolone (PRD) with methotrexate (MTX) | Folate-PEG-DSPE (outer liposome) | Rheumatoid arthritis (RA) treatment | Collagen-induced arthritis (CIA) rat model |
| Leukosome159 | Dexamethasone (Dex) | Constituted by proteins derived from the leukocytes' plasmalemma integrated into a synthetic phospholipid bilayer | Targeting of inflamed endothelia | In vitro: HUVECs, in vivo: LPS induced murine ear inflammation |
|
||||
| Dendrimers | ||||
| Polyamidoamine (PAMAM) (G2–G4)176 | Ketoprofen, Ibuprofen, Diflunisal, Naproxen [NSAIDs] | NA | Improvement of NSAIDs solubility | NA |
| G4-NH2-PAMAM177 | NA | NA | Inflammation treatment | In vitro: rat peritoneal macrophages, in vivo: adjuvant-induced arthritis rat model, Carrageenan-induced edema rat model |
| 3.5 polyamidoamine (PAMAM)178 | NA | Glucosamine and glucosamine 6-sulfate conjugation | Prevention of scar tissue formation after surgery | PBMCs, HUVECs rabbit glaucoma filtration surgery model of excessive scar tissue formation |
| Polyglycerol sulfates (dPGS)185 | NA | Surface coupled E-, P- and L-selectin ligands | Blockade of leukocytes recruitment for therapeutic intervention in inflammatory disorders | Surface plasmon resonance (SPR)-based binding assay |
|
||||
| Protein | ||||
| HSA194 | Methotrexate (MTX) | Chlorin e6 (Ce6) conjugation | SPARC targeting for rheumatoid arthritis (RA) treatment | Collagen-induced arthritis (CIA) mouse model |
| HSA195 | 5-Aminosalicylic acid (5-ASA) | Crosslinking with glutaraldehyde | MPO targeting for inflammatory bowel disease (IBD) treatment | DDS induced colitis mouse model |
| BSA196 | Piceatannol (Syk inhibitor) | Crosslinking with glutaraldehyde | Inactivation of neutrophil transmigration and vascular inflammation mitigation | In vitro: isolated mouse neutrophils, in vivo: LPS-induced acute lung injury mouse model |
| Gelatin203 | Ibuprofen | PEGylation and CaCl2 crosslinking | Enhanced pharmacokinetics and bioavailability of Ibuprofen for rheumatoid arthritis and chronic arthropathies treatment | In vitro: platelet rich plasma (PRP), platelet poor plasma (PPP) human peripheral blood mononuclear cells (PBMCs), RAW264.7 cells, in vivo: 5 week-old Sprague Dawley WT rats |
| Gelatin204 | Epigallocatechin gallate (EGCG) | Surface coupled hyaluronic acid (HA) | Dry-eye syndrome (DES) treatment | In vitro: HCECs, in vivo: DES induced rabbit model, Wistar rats |
| Lipoprotein-mimicking peptide-phospholipid scaffold (HPPS)206 | Curcumin | NA | Autoimmune encephalomyelitis (EAE) treatment | In vitro: mouse isolated monocytes and neutophils, in vivo: EAE mouse model |
| Elastin like particle (ELP)207 | Stromal cell-derived factor1 (SDF1) | Fusion of ELP with SDF1 | Wound healing | Diabetic mice wound assay |
| Nanoparticle | Loaded antigen | Properties | Application | System |
|---|---|---|---|---|
| Polymeric | ||||
| Delta-inulin (Advax™)132 | NA | Complement activation | Improvement of recombinant hepatitis B virus (HBV) immunogenicity [vaccine adjuvant] | Mice and guinea pigs |
| Carboxylated polysterene133 | Ovalbumin (OVA) | Shape and size related immune response modulation (smaller spherical NPs produce a greater Th1 biased cell mediated immune response, while larger rod-shaped NPs a Th2 biased humoral immune response) | Vaccine | In vitro: mouse dendritic cell line (DC2.4), in vivo: female balb/C mice |
| Amphiphilic γ-PGA128 | Ovalbumin (OVA) or recombinant human immuno-deficiency virus (HIV)-1 | Enhanced protein delivery to iDCs | Vaccine/vaccine adjuvant | In vitro: murine iDCs, in vivo: female BALB/c mice |
| Cationicalginate-PEI nanogels129 | Ovalbumin (OVA) | Enhanced MHC I presentation and IFN-γ production | Vaccine/vaccine adjuvant | In vitro: mouse splenocytes, mouse bone marrow dendritic cells (BMDCs), raw 264.7 mouse macrophages, in vivo: female C57BL/6 mice |
| CD205 and CD11c Ab functionalised poly (N -vinylpyrrolidone) (PVPON)127 | NA | CD11c-NPs: Successfull internalisation by DCs DC205-NPs: Unsuccesfull internalization by DCs | Vaccine/vaccine adjuvant | Mouse spleen derived dendritic cells (DCs) |
| Anti-human DEC-205 functionalised PLGA126 | MART-1peptide | DEC-205 receptor-mediated targeting of tumor Ag to DCs | Cancer vaccine | Human monocyte-derived DCs |
|
||||
| Liposomes | ||||
| Mannosylated phosphatidylethanolamine (Man-PE)162 | Neisseria meningitidis type B antigen PorA | Recognised by the DC surface molecule mannose receptor (MR), enhanced uptake by human DCs compared to unmannosylated liposomes | Vaccine/vaccine adjuvant | Human monocyte-derived DCs (MoDCs) and murine bone marrow-derived DCs (BMDCs) |
| Positively charged MLVs (PC/Chol/SA: molar ratio 4:5:1)163 |
Ovalbumin (OVA) | Efficient vectors to APC and antigen-depots | Vaccine/vaccine adjuvant | In vitro: C57BL/6 T lymphoma EG7 cell line, OVA-transfected clone of EL4 cell line, in vivo: male C57BL/6 mice, female BALB/c mice |
| Cationic PEGylated DDA: TDB164 | Ag85B-ESAT-6 | Size and % PEGylation controls depot formation and Th1/Th2 balance | Vaccine/vaccine adjuvant | Female Balb/C mine, female C57BL/6 mice |
| ALF- and ALFQ167 | NA | Enhanced anti-polysaccharide antibody responses | Vaccine adjuvants | C57BL6/J mice |
| PLFE-based archaeosomes168 | Ovalbumin (OVA) | Antigen specific both systemic immune response and mucosal immune response by oral administration | Oral vaccination | Female BALB/c mice |
|
||||
| Dendrimers | ||||
| Leb-conjugated poly(amido amine) (PAMAM)186 | Ovalbumin (OVA) | G3 and G4: optimal size and multivalency to achieve the most efficient DC-SIGN targeting | Antigen delivery to DCs | Human monocyte-derived dendritic cells |
| Dendrimeric FMDV peptide188 | NA | T-cell activation that efficiently contributes to FMDV protection | Vaccination against foot-and-mouth disease virus (FMDV) | Pigs |
|
||||
| Protein | ||||
| Virus like particles (VLPs)208 | NA | Presentation of viral Env spikes in their natural conformation, efficient internalisation by APCs and strong humoral and cellular immune responses stimulation | Vaccination against hepatitis B, hepatitis E, HPV | Commercially available for humans |
4.1. Polymer-based nanoparticles
4.1.1.1. PEGylation for escaping immune system recognition. Hydrophobic and cationic polymers present increased opsonization by serum proteins and as a result a higher uptake by phagocytic cells.100 The uptake of nanoparticles usually increases with increasing the zeta potential values while excess of positive charge can induce toxicity and stimulate immune reactions.112 When the goal is to avoid the immune system recognition, coating of NPs surface with polyethylene glycol (PEG) is thought to be a good strategy in order to increase the hydrophilicity and neutralise the surface charge of the particles.113 PEG generates a hydrated volume around the NPs, due to its hydrophilic nature, precluding NPs from steric interactions with other NPs and blood components. There are many factors that influence the interactions and circulation of PEGylated NPs in the blood, including PEG molecular weight, surface density and the physicochemical properties of NP core which should be taken into consideration for the optimal engineering of NP vehicles, in association with the targeting tissues, therapeutic application, loaded cargo and administration route.114
4.1.1.2. Ligand-targeted polymeric nanoparticles. Furthermore, polymeric NPs can be functionalized in order to target specific tissues by active targeting or escape the MPS. Several targeting ligands are used for drug delivery such as antibodies, peptides and lectins.115–118 Sakhalkar H. et al., functionalized PLA-PEG NPs with monoclonal antibodies in order to target E-selectin, P- selectin, VCAM-1, and ICAM-1 on inflamed endothelium tissue. The targeted particles showed an increased adhesion to inflamed HUVEC cells (up to 33-fold) compared to non-inflamed and increased adhesion to cytokine (up to 6-fold) and trauma induced inflamed endothelium (up to 10-fold) compared to untreated endothelium.115 Apart from antibodies, peptides could also be used as targeting ligands to inflamed tissues. Ghosh K. et al., coated PLGA-PEG NPs with an islet-homing peptide (CHVLWSTRC-Pep I) in order to target the endothelium of insulin-producing pancreatic islet β cells that are progressively destroyed by the body's immune cells in type 1 diabetes. The autoimmune destruction of islet β cells in insulitis initiate with the adhesion of blood leukocytes to the inflamed islet vascular endothelium, followed by extravasation of the immune cells into the islet parenchyma where they attack the islet β cells. The Pep I functionalized PLGA-PEG NPs loaded with genistein (Gen), a protein tyrosine kinase inhibitor that is known to impair leukocyte binding to TNF- stimulated endothelial cells. The Pep I-Gen-PLGA-PEG NPs exhibited a 3-fold increased binding capability to islet endothelial cells in vitro compared to controls and a 200-fold inhibition of leukocyte adhesion to islet endothelial cells compared to free drug.116 Sialic acid-binding immunoglobulin-like lectins (Siglecs) are type I transmembrane proteins and are considered to be self-associated molecular patterns found on hemopoietic and immune cells which regulate immunity.117 Spence S. et al., demonstrated that PLGA NPs conjugated with N-acetylneuraminic acid (NANA), a murine Siglec-E ligand, were able to target Singlec-E receptors and exhibited a significant anti-inflammatory effect in murine and human inflammatory models.118
4.1.1.3. Biomimetic functionalization of polymeric nanoparticles. Another promising strategy seems to be the coating of nanoparticles with leukocytes' membranes in order to recapitulate their biological properties. For example, Thamphiwatana S. et al., exploited the capability of macrophages to bind to endotoxins and pro-inflammatory cytokines in order to find a therapeutic effect against sepsis. They coated PLGA NPs with macrophage membranes which maintained their binding properties without retaining the inflammatory signals of macrophages. This resulted in the sequestering of bacterial endotoxin and inflammatory factors in a sepsis in vivo mouse model with a significant therapeutic potential.119 In a similar study, Zhang Q. et al., used the membrane of neutrophils to coat PLGA NPs in order to target cytokines activated by chondrocytes in a rheumatoid arthritis mouse model which showed promising anti-arthritis effects.120
4.1.1.4. Responsive polymeric micelles. Polymeric micelles are core–shell nanostructures that are formed by the self-assemble of amphiphilic block copolymers and can solubilize hydrophobic drug molecules that are not well dissolved in water.98 Polymeric micelles with pH-, thermal and reactive oxygen species (ROS) responsiveness have also attracted a lot of attention due to their capabilities to target inflammation. Li C. et al., synthesized a pH-sensitive amphiphilic NP by conjugating the glucocorticoid drug prednisolone (PD) with the polymer APN which is a branched derivative of PEG. The APN micelles were able to target the inflamed tissue of a rheumatoid arthritis mouse model, while the acidic environment at the sites of inflammation led to a targeted drug release, resulting in better therapeutic results than the free drug.121 In another study, Tang M. et al., created a diblock copolymer combining poly(propylene sulfide) (PPS) which is a ROS-sensitive polymer with PNIPAm, a thermo-sensitive polymer, in order to engineer a drug delivery vehicle which will release its cargo specifically in inflamed and cancerous tissues, characterized by increased ROS generation and higher temperatures.122
4.1.1.5. Polymeric nanoparticles for pulmonary administration. The route of administration influences the toxicity profiles of NPs and should thus be tested for each administration route separately. For instance, although PLGA has been approved by United States-Food and Drug Administration (US-FDA) for biomedical and pharmaceutical applications, the use of PLGA NPs for pulmonary administration is not optional due to its acidic products and its slow degradation which results to an airway inflammatory response.123 Springer S. et al., instilled PLGA microspheres in the trachea of hamsters in order to investigate whether they are easily detectable within alveolar macrophages, which would constitute them as a possible marker for the diagnosis of respiratory lung disease. They noticed an acute inflammatory response on the instillation day of PLGA particles, demonstrated by an increase in neutrophils in whole-lung lavage and lung parenchyma of hamsters, that disappeared thereafter.124 V. F. Fiore et al. compared the inflammatory response of polyketal (PK3) with PLGA microparticles after intratracheal injection in mice in order to investigate their potential use as drug delivery vehicles for interstitial lung diseases (ILD). Their results showed that PLGA microparticles induced a significant macrophage infiltration starting at day 4 and continuing through day 7 and a transient lymphocyte infiltration at day 4, while PK3 particles did not exhibit any airway inflammation.123 In addition, Yoo D. et al., demonstrated the advantage of HPOX as a polymeric prodrug of HBA, as a potential therapeutic for treating airway inflammatory diseases compared to PLGA. The biocompatibility of HPOX in the lungs may be due to its rapid degradation rate (half-life of ∼12 h) and the excellent antioxidant and anti-inflammatory activity of its degradation product, HBA.125
 | ||
| Fig. 7 Schematic representation of intracellular trafficking of nanoparticles with different morphological features.135 | ||
4.2. Liposomes
Liposomes have been extensively investigated since 1965, when introduced first by Bangham et al., for drug delivery applications.134 Their unique properties such as their biocompatibility and biodegradability as well as their chemical structure that enable them to incorporate both hydrophilic and hydrophobic compounds make them suitable as drug delivery platforms.135 Many liposomal particles have been approved for clinical practice such as Doxil®, Ambisome®, DepoDur™ etc., while a lot of liposome formulations are currently in clinical trials for various therapeutic applications.1364.2.1.1. Conventional liposomes. Liposomes are composed of phospholipid bilayers and are formed due to hydrophobic interactions which drive their hydrophobic carbon tails to cluster together while their polar groups interact with the aqueous media.137 Liposomes have been extensively investigated as drug delivery systems of glucocorticoids (GC) in order to be specifically distributed in the inflamed tissues and reduce the out of target side effects of GC. Liposomal glucocorticoids have been evaluated for their therapeutic efficacy for various inflammatory diseases such as asthma, rheumatoid arthritis, multiple sclerosis and cancer and have been shown increased anti-inflammatory activity and/or prolonged therapeutic duration which make them promising agents for the treatment of inflammatory diseases.138 As an example, Data C. et al., demonstrated that liposomes (DPPC/DPPG/Chol, 50/10/40 mol%, size: 295 (SD 15) nm) loaded with glucocorticoid dexamethasone phosphate (DXM-P) showed an increased suppression of joint destruction compared to the free drug in rat antigen-induced arthritis.139
Encapsulation of other anti-inflammatory strategies, using liposomes have also been investigated. Angmo S. et al., developed cationic phospholipid mixture 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-Dioleoyl-sn-glycero-3 phosphoethanolamine (DOPE) and DOTAP, with single positive surface charge (NH+) with the encapsulation of GDP, a natural compound which inhibits hepcidin action. Hepcidin is an hepatic peptide hormone, which present abnormally high levels in inflammation and inhibits iron transport by binding to the iron export channel ferroportin located on the basolateral surface of gut enterocytes and the plasma membrane of reticuloendothelial cells (macrophages). NH+-GPD liposomes demonstrated decreased hepcidin levels in hepatocytes and macrophages, indicating to be promising therapeutic for anaemia of inflammation (AI).140 Macrophages are important key players in initiation and progression of inflammation and their depletion is used as a strategy to manage inflammation. Clodronate (LEC)-encapsulated liposomes have been reported for the specific depletion of macrophages.141 Clodronate (LEC) belongs to the drug family of bisphosphonates and is used for the treatment of osteolytic bone disease and post-menopausal osteoporosis because of its ability to inhibit osteoclast function.142 Liposomes are undergone phagocytosis by macrophages (MΦ), while LEC is released into their cytosol causing their apoptosis. Mert et al., investigated the effects of MΦ depletion using LEC-liposomes on a carrageenan-induced rat inflammation model and their results showed suppressive effects on inflammatory markers at the inflammation site and spinal cord level.143
4.2.1.2. Stealth liposomes. One obstacle for the desired clinical efficacy of liposomes is thought to be their rapid clearance from the mononuclear phagocyte system (MPS). Blood circulating proteins can be absorbed to the surface of liposomes resulting to the formation of a protein corona which can lead to complement activation and phagocytic clearance.144 It has been observed that PEGylation of liposomes results in decreased phagocytosis and enhanced circulation half-life.145 Prolonged circulation time of liposomes when administered systemically can improve the pharmacokinetics of the encapsulated drugs and accumulate in the tumour tissues and sites of inflammation due to the “Enhanced Permeability and Retention Effect” (EPR).146 Several studies have shown the advantage of PEGylated liposomes in glucocorticoid delivery.138,146,147 Several animal studies have shown that prednisolone containing long-circulating PEG-liposomes have higher therapeutic efficacy compared to the free drug administration.148,149 Bartneck M. et al., evaluated PEG-liposomes loaded with dexamethasone for the treatment of experimental liver injury models, demonstrating the depletion of hepatic and systemic T cells, as well as polarization of macrophages towards an anti-inflammatory phenotype, which could be promising for the treatment of acute and chronic liver injury.150
In addition, conventional liposomes present stability issues under physiological conditions which lead to the off-target leakage of encapsulated drug. An effective strategy for increasing liposomes' structural integrity is the polymerization of lipids in their lipid bilayers. Recently, Wang Q. et al., prepared polymerized stealth liposomes composed of 1,2-bis(10,12-tricosadiynoyl)-sn-glycero-3-phospho- choline (DC8,9PC) and 1,2-distearoyl-sn-glycero-3-phospho-ethanol-amine-poly(ethyleneglycol) (DSPE-PEG2000) by a thin-film hydration method, in which DC8,9PC molecules were crosslinked in the bilayer by UV irradiation and PEG chains provided a stealth layer to enhance their blood circulation time. The polymerized stealth liposomes showed an increased blood circulation time compared to nonpolymerized stealth liposomes, while when loaded with dexamethasone (Dex) showed a significant suppression of proinflammatory cytokines (TNF-a and IL-1b) in joint tissues and reduction of inflamed joints' swelling, compared to free Dex, in RA induced rats.151
Liposomes have weak immunogenic response, however, there have been reported some unwanted immune reactions in several studies. It has been shown that exposure to liposomes can lead to complement activation which may result in some cases to a serious hypersensitivity reaction called complement activation-related pseudoallergy (CARPA).152 Although PEGylation reduces the RES clearance and increase the circulation lifetime, it may also lead to hypersensitivity responses.153 It has been demonstrated that repeated injections of PEG-PE liposomes can generate anti-PEG antibodies which may lead to accelerated blood clearance (ABC) after prolonged administration.154
4.2.1.3. Ligand targeted liposomes for active targeting of inflammation. Active targeting of liposomes to the inflamed tissues or inflammatory mediated cells can be facilitated by surface functionalization with specific ligands such as proteins, peptides, antibodies and antibody fragments.155 Functionalization of liposomes with antibodies in order to target adhesion molecules of inflamed endothelium, such as ICAM-1, VCAM-1, E-selectin and P-selectin has been explored in liposomal technology as well, in order to deliver therapeutics to the inflamed sites. Homem P. et al., targeted cyclopentenone prostaglandin (CP-PG) loaded liposomes using anti-VCAM antibodies to endothelial cells and foamy macrophages in atherosclerotic lesions of mice. CP-PGs inhibit the nuclear factor-κB (NF-κB) transcription factor, the activation of which leads to a cascade of inflammatory responses.155 Their formulation resulted in enhanced targeting of endothelial cells and foamy macrophages in atherosclerotic lesions and indicated significant anti-inflammatory effects which rescued treated mice from death by myocardium infarction or stroke, compared to control ones. In a recent study, Marcos-Contreras O. et al., showed that intraarterial administration of anti-ICAM functionalised liposomes exhibited superior targeting to inflamed brain vasculature compared to untargeted liposomes, implying their clinical potential in the treatment of cerebrovascular diseases.156
Activated macrophages, which have an essential role in the development and maintenance of inflammatory diseases, have the folate receptor beta (FR-b) upregulated. Functionalized folate-linked PEG liposomes were demonstrated to specifically target sites of inflammation in colitis and atherosclerosis mouse models and deliver their anti-inflammatory cargo there (betamethasone) which leaded to the reduction of activated macrophages.157 In a similar study, Verma A. et al., developed functionalized PEGylated double liposomes (DSPE and DSPC) with the vitamin folic acid (FA), which exhibits good affinity with the folate receptor. FA-PEG liposomes were loaded with the anti-inflammatory agent prednisolone (PRD) and methotrexate (MTX) (a disease modifying anti-rheumatoid agent, DMARDs) and their therapeutic effects evaluated in collagen-induced arthritis (CIA) rats. Their results indicated significantly higher edema inhibition compared to non-FA functionalized liposomes and free drugs.158
4.2.1.4. Biomimetic functionalization of liposomes. Bioinspired ligands on NPs' surface have been extensively investigated in order to mimic the functions of immune cells. The incorporation of cell membranes as coatings is a potential alternative in order to optimize targeting and therapeutic effects of liposomes. Molinaro R. et al., first developed the so called “leukosomes”, a novel vesicle constituted by cholesterol and phospholipids enriched with proteins derived from leukocytes' membranes. Leukosomes exhibited increased targeting efficiency of activated human endothelial cells in vitro as well as an enhanced accumulation in inflamed murine tissue in vivo compared to conventional liposomes. In vivo investigation of leukosomes in an ear inflamed mouse model showed increased accumulation of the leukosomes in the inflamed ears compared with control liposomes.159
Although cationic liposomes have shown an increased antigen presentation when administered intramuscularly, it was reported that their positive charge blocks DNA vaccine activity after intradermal administration. This was explained by the immobilization of the positively charged nanoparticles in the negatively charged extracellular matrix (ECM), preventing the particles to reach their target. PEGylation of these particles resulted in shielding of their surface charge and a significant 55-fold increase in vivo antigen expression.165 Therefore, the administration route and desired immune responses should be taken into consideration when PEG coated cationic liposomes engineered as vaccines adjuvants. Combination of antigens with various immunostimulators, as ligands for pattern recognition receptors (PRRs) of APCs, have been investigated as cargos of cationic liposomes vaccine adjuvants. Differences in the signaling pathways induced by the different receptors can lead towards Th1 or Th2 biased immune responses. Therefore, the choice and combination of immunostimulators play an important role in controlling the type of immune response whereas different strategies can be employed for their engineering depending on the type of molecules and purpose of their use (Fig. 8). For example, PRRs recognizing ligands of bacterial origin often induce a Th1 response, which is suitable for fighting certain bacterial infections, while (ds)RNA derived from virus induces cytotoxic T-lymphocyte (CTL) immune responses, capable of combatting a virus infection.166 Ramakrishnan A. et al., developed two novel liposome-based adjuvant systems, the Army Liposome Formulation (ALF), containing synthetic monophosphoryl lipid A, and ALF plus QS-21 (ALFQ), in order to enhance immonogeneicity of capsule conjugate vaccine against C. jejuni strain 81–176 (CPS-CRM). Both liposome adjuvants exhibited enhanced immunity in mice and nonhuman primates compared to alum, providing promising evidence that these adjuvant formulations may enhance immunogenicity in humans as well.167 In the case of hydrophobic immunostimulators, they can be incorporated in the liposomal bilayer. For example, the use of archaeal lipids, can increase the immunogeneicity of liposomes (“archeosomes”) and was demonstrated by Li Z. et al., that were significantly more potent than liposomes made with Egg phosphatidylcholine (EPC)/Chol at inducing ovalbumin- (OVA-) specific IgG and IgA antibodies following oral administration in a mouse model.168
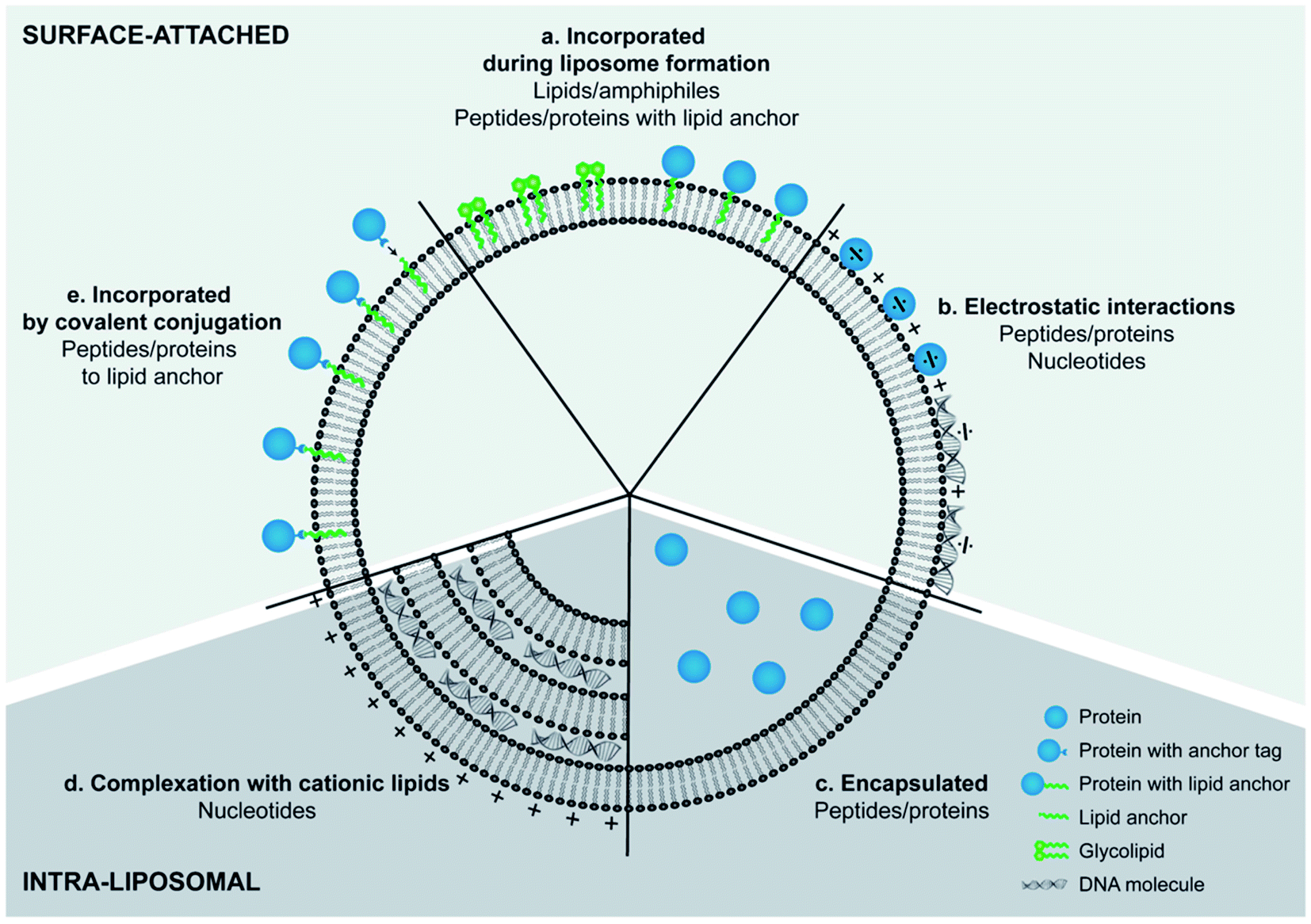 | ||
| Fig. 8 Different strategies for incorporating antigens and immunostimulators into liposomes.166 | ||
4.3. Dendrimers
Dendrimers belong to another category of nanoformulations with promising biomedical applications, due to their unique properties of low polydispersity, low viscosity, multi-functionality and biocompatibility.169 They are hyperbranched molecules which are generally prepared using either a divergent method or a convergent one. In the divergent method, dendrimers are built up from a multifunctional core molecule on which successive series of branches are attached with repeated reactions on activated end groups, while in the convergent method the synthesis starts from exterior branches and ends to the core.170 The total number of series of branches determines the generation of the dendrimer (Fig. 9).171 Their molecular weight and number of terminal groups are determined by the dendrimer generation. Conformational changes occur when dendrimers reach a specific generation, with low generation dendrimers (G0–G3) presenting ellipsoidal shapes without an interior cavity, while high-generation dendrimers (G4–G10) have spherical shapes and well-defined interior cavities.172 It is shown that higher generation and positive charged dendrimers result in higher cytotoxicity and immunogenicity and for that reason different chemical groups are attached on their surface in order to increase their biocompatibility173 The end groups on their outer shell could be also functionalized in order to enhance their targeting capability.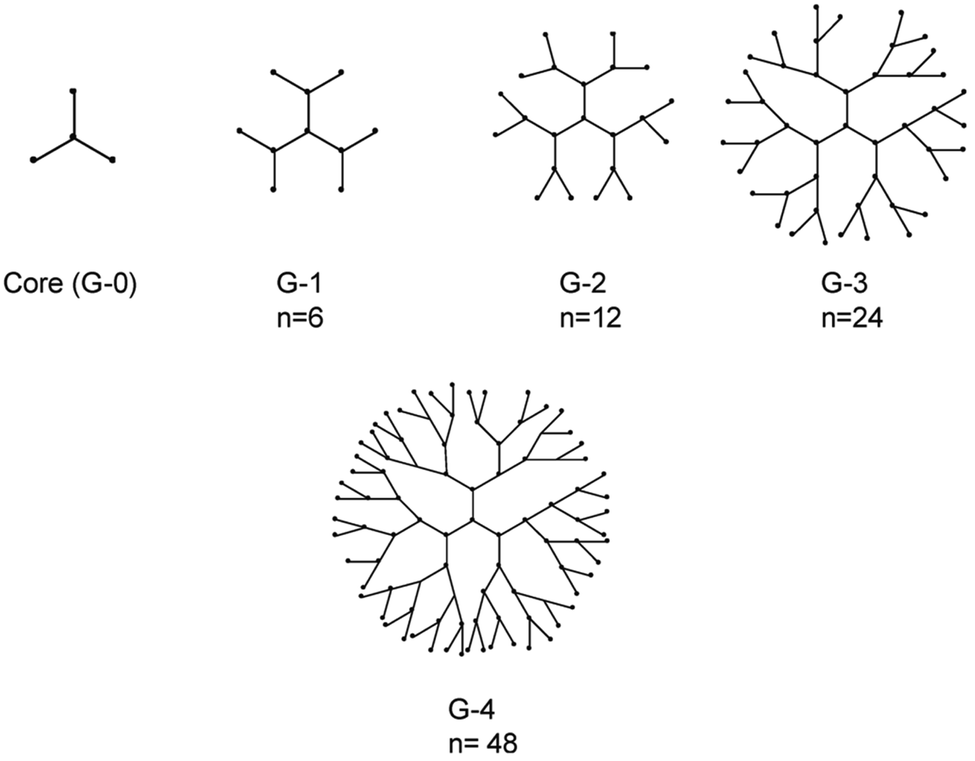 | ||
| Fig. 9 Representation of dendrimers of generations 1–4. The n denotes number of terminal functional groups.174 | ||
In addition to PAMAM dendrimers, other types of dendrimers have also been reported to possess anti-inflammatory properties. Azabisphosphonate (capped by amino-bisphosphonate groups) (ABP) anionic dendrimers have shown to increase the production of the immunomodulatory cytokine IL10 via the activation of different immune cells, such as monocytes, macrophages and CD4+ T lymphocytes. In addition, they seem to selectively amplify Natural Killer (NK) cells, which could lead to promising applications in cancer immunotherapy.181,182 Molecular dynamic simulations have shown that ABP dendrimers with low flexibility and more directional surface groups leads to enhanced anti-inflammatory properties.183,184 Adhesive interactions of leukocytes and endothelial cells initiate leukocyte migration to inflamed tissue which can lead to acute and chronic inflammatory diseases. Selectin receptors on endothelial cells facilitate the adhesion of leukocytes and it is proposed that pharmacological inhibitors of those molecules, such as heparin, could lead to an effective treatment for inflammatory diseases. Dernedde J. et al., investigated the role of synthetic dendritic polyglycerol sulphates (dPGs) as inhibitors of the cell adhesion molecules E-, L- and P-selectin. Using Surface Plasmon Resonance (SPR) in order to evaluate the selectin binding potential of dPGS in vitro, they showed that L- and P-selectin were efficiently inhibited, with IC50 values of 30 and 90 nM, whereas E-selectin binding was not affected. The number of sulphate groups per molecule and the core size of dPGs determined the selectin inhibition efficacy. Their results indicated that the larger is the core of the dendritic NPs the more effectively shields the selectin binding sites.185
Multiple antigenic peptides (MAPS) with a dendrimer-structure are able to present multiple copies of an antigen or multiple antigens to the immune system at the same time and are thought as promising formulations in vaccine development.187 Mimicking protective responses using synthetic peptides has attracted a lot of attention due to their advantages such as ease of production, thermal stability and safety. However, the complexity of the interactions between pathogens and host immune responses have limited the development of successful peptide vaccines. The multivalency of dendrimers can be exploited for the presentation of multiple peptides through the enhancement of peptide immunogenicity. Cubillos C. et al., demonstrated the increased efficiency of peptide dendrimeric vaccines against foot-and-mouth disease virus (FMDV) in pigs, containing T-cell and B-cell epitopes for an enhanced immune reactivity. The pigs did not develop significant clinical signs upon FMDV challenge, while a potent anti-FMDV immunoglobulin A response (local and systemic) was observed, despite the parenteral administration of the peptide, which constitute MAPs dendrimers as promising vaccine candidates.188
4.4. Protein nanoparticles
4.4.1.1. Albumin. Human serum albumin (HSA) is the most abundant protein in blood plasma which has a critical role as a pH and osmotic pressure determinant while it facilitates the transport of many substances (endogenous or exogenous) inside the body due to its high affinity to many ligands.191 SPARC (secreted protein acidic and rich in cysteine) is an extracellular glycoprotein involved in tissue development, wound healing and angiogenesis. It has been observed that various tumour tissues present an overexpression of SPARC which is associated with metastasis and poor prognosis.192 Albumin has a high affinity for SPARC which has leaded to the success of Abraxane® to enhance the targeting of the chemotherapeutic drug PTX to metastatic tumour tissues.193 Bottini N. et al., showed that SPARC is overexpressed in the synovial fluid and joint tissues in patients with RA as well as RA induced mice. Therefore, they aimed to target the antirheumatic drug methotrexate (MTX) to the arthritic joins of RA induced mice using MTX-loaded HSA nanoparticles, exploiting the high affinity of albumin for SPARC. In addition, arthritic joints seemed to metabolize more albumin than healthy tissues which enhanced the delivery of albumin NPs to these tissues. They demonstrated that the MTX-albumin bound NPs increased the therapeutic effects of MTX with no significant side effects compared to free MTX due to the greater accumulation of MTX-albumin NPs to the arthritic joints of mice.194 In another innovative study, Iwao Y. et al., exploited the affinity of albumin to myeloperoxidase (MPO) which is reported to be elevated and cause damage to the sites of inflammation, in IBD patients. In order to deliver the anti-inflammatory drug, 5-aminosalicylic acid (5-ASA), to the inflamed colon, they prepared 5-ASA-HSA NPs by a desolvation technique in which the active site for enzymatic degradation of HSA was internalized inside the NPs in order to protect it from the proteolytic enzymes in the gastrointestinal tract. 5-ASA-HSA NPs were shown to have a significant therapeutic effect in a mouse model of ulcerative colitis (UC) with the delivery of a lower drug dose needed, compared to the dose of the free drug, in order to cause similar therapeutic effects.195 Wang Z. et al., demonstrated that bovine serum albumin (BSA) NPS are preferentially internalized by activated neutrophils in vivo, through FcgR signaling. As neutrophil infiltration and activation at the vessel wall is the primary cause of vascular inflammation which leads to various diseases such as acute lung injury and ischaemic tissue injury, the targeting of neutrophils in order to block their adhesion to endothelial cells seems to be a promising approach. In this study, BSA NPs were prepared using a desolvation technique and loaded with piceatannol, a tyrosine kinase inhibitor which blocks outside-in integrin signaling of neutrophils. Piceatannol loaded BSA NPs showed a significantly reduced neutrophil sequestration in acute lung injury induced mice compared to piceatannol alone, confirming the specific targeting of albumin NPs to activated neutrophils.196
4.4.1.2. Gelatin. Gelatin is another biocompatible and biodegradable protein which can be used for the fabrication of nanomaterials. It is obtained by controlled hydrolysis of collagen, which is a major component of the skin, bones, and connective tissues.197 There are two different types of gelatin, A and B, which can be produced following either acid or base hydrolysis, resulting in proteins with different isoelectric point (pI), molecular weight, amino acid composition, and viscosity.198 Gelatin NPs can be prepared by several different techniques, including desolvation, coacervation-phase separation, emulsification-solvent evaporation, reverse phase microemulsion, and nanoprecipitation.199 Various cross-linking agents such as glutaraldehyde, glyceraldehyde etc. could be used for the cross-linking of gelatin in order to provide stability and enhance the circulation lifetime of nanoparticles (Fig. 10).197,198 Gelatin particles have been used effectively as drug delivery vehicles as they demonstrate a controlled drug release profile by the manipulation of their size, cross-linker density, and PIs of the particles.200 Wang E. et al., showed that the crosslinking density of gelatin nanoparticles affects the degradability of the gelatin polymers and their drug release.201 Gelatin nanoparticles are easily functionalized with ligands in order to enhance their targeting and have been used for the targeted delivery of inflammatory drugs, siRNAs and DNA. IBS, the sodium salt of Ibuprofen, is a NSAID drug used for the treatment of rheumatoid arthritis and chronic arthropathies, but its short plasma half-life demands multiple administrations in order to maintain a therapeutic dose.
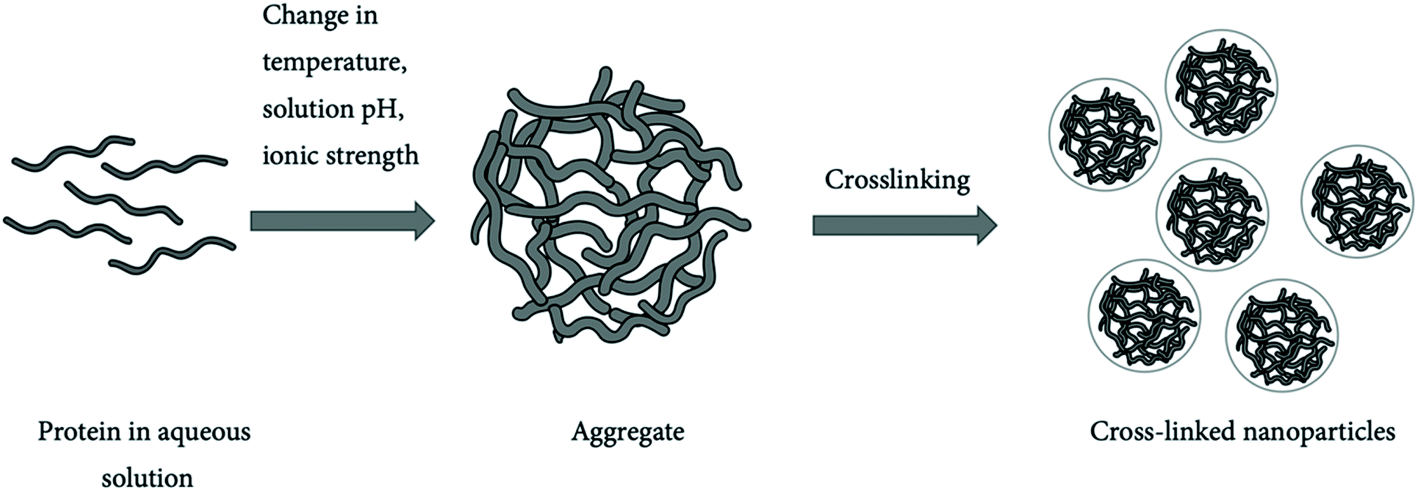 | ||
| Fig. 10 Protein nanoparticles formed by crosslinking.198 | ||
In general, positively charged nanoparticles more readily interact with cells due to the negatively charged cell membranes, resulting in their rapid clearance by the reticuloendothelial system upon intravenous administration.202 Narayanan D. et al., showed that PEGylated gelatin nanoparticles loaded with IBS, improved the in vivo pharmacokinetics of the drug, exhibiting a sustained drug release and prolonged blood circulation, whereas gelatin nanocarriers did not exhibit any significant immunotoxicity even at high doses.203 In another study, Huang et al.,204 functionalized gelatin nanoparticles with the mucoadhesive hyaluronic acid (HA) in order to increase the interaction of particles on the corneal surface for the treatment of dry-eye syndrome (DES), which is characterized by inflammation of the ocular surface and lacrimal glands.205 Epigallocatechin gallate (EGCG), a polyphenol of green tea with anti-inflammatory properties, encapsulated inside the HA coated gelatin particles (GEH NPs) as an ocular drug delivery system and was administered to DES induced rabbits. GEH NPs exhibited an enhanced therapeutic outcome compared to EGCG alone and gelatin nanoparticles without HA coating, due to their slow drug release and prolonged ocular retention.204
4.4.1.3. Recombinant proteins. The biomimetic characteristics of recombinant proteins could also be exploited for engineering of NP vehicles when the availability of the natural proteins is limited or has undesired immunogenic response. In a recent study, Lu L. et al., developed high-density lipoprotein-mimicking peptide-phospholipid scaffold (HPPS) nanoparticles loaded with curcumin, as an anti-inflammatory agent, in order to target monocytes, which are thought to play an important role in the initiation and development of multiple sclerosis (MS). Thus, the group has designed HPPS to target high-density lipoprotein (HDL) receptor of monocytes. Curcumin loaded HPPS NPs were specifically taken up by inflammatory monocytes hindering their crossing of the blood brain barrier (BBB). The internalised curcumin NPs inhibited NF-κB activation which leaded to the downregulation of intercellular adhesion molecules 1 (ICAM-1) and macrophage-1 antigen (MAC-1) expression in the monocytes with a significant therapeutic outcome in mice with autoimmune encephalomyelitis (EAE).206
Elastin like peptides (ELPs) are recombinant peptides with structural and functional properties similar to elastin, which is a component of extracellular matrix and promotes tissue repair. Above a transition temperature ELPs form self-assemble nanoparticles which can been used as drug delivery vehicles.197 ELP fusion proteins, with a nanoparticulate form, have been shown to protect biomolecules from proteolysis. Yeboah A. et al., engineered stromal cell-derived factor1 (SDF1)-ELP fusion protein NPs, in order to increase stability of SDF1, a key mediator of the wound healing response. The SDF1-ELP NPs showed accelerated wound healing compared to SDF1 alone after treatment of diabetic mice with skin wounds, which was attributed to the increased stability and half-life of SDF1-ELP NPs.207
5. Combination therapy: immunotherapy combined with nanoparticle treatment
5.1. Cancer immunotherapy
Immunotherapeutic agents for the treatment of cancer have provided limited evidence of clinical success until recently. In 2010, FDA approved Provenge (sipuleucel-T), a cell-based immunotherapy for advanced prostate cancer. It followed the approval of ipilimumab in 2011, an anti-cytotoxic T lymphocyte-associated protein 4 (CTLA-4) antibody, the programmed cell death protein 1 (PD-1) antibodies pembrolizumab and nivolumab in 2014 for the treatment of melanoma, and the PD-1 ligand (PD-L1) antibody atezolizumab in 2015 for the treatment of bladder cancer.211 Cancer immunotherapy can be classified into the active and passive type. Active immunotherapy stimulates endogenous immune system using prophylactic or therapeutic vaccines, immune adjuvants or cytokines, while passive immunotherapy uses engineered cells or antibodies which are administered to patients in order to enhance immunity against cancer.212Although promising, Immunotherapies can be problematic due to undesired side effects that can lead to autoimmune diseases. In addition, they seem to be more effective against lymphoma than solid tumours while the immunosuppressive tumour microenvironment hinders their effectiveness.211 The emerging field of nanomedicine seems to be promising for overcoming the current shortcomings of cancer immunotherapy. Nanoparticles could have different roles in cancer immunotherapy (Fig. 16) which are in-detailed discussed in the following sections.
5.2. NPs for checkpoint blockade immunotherapy and immune-suppressive TME modulation
The effectiveness of cancer immunotherapy is challenging, due to the immunosuppressive properties of cancerous cells. Tumours express ligands that bind to inhibitory receptors of tumour-specific T cells and downregulate their immune response. Such examples are the CTLA-4 receptor, an inhibitory receptor that down-regulates the initial stages of T cell activation, and PD1 receptor, which bounds to PD-L1 and PD-L2 ligands expressed by tumours and leads to the inhibition of T-cell proliferation, cytokine release and cytotoxicity.212 Cancer immunotherapeutic agents, using immune checkpoint inhibitors, such as CTLA-4 antibody (Ab) (ipilimumab), PD-L1 Ab (atezolizumab), and two PD-1 Abs (nivolumab and pembrolizumab) have been FDA approved, but seem to be effective only in a minority of patients while are associated with adverse side effects.212 Nanoparticles can be used for the delivery of immune checkpoint inhibitors in order to enhance their efficiency. Zhang X. et al., generated membrane derived nanovesicles (NVs) with PD-1 receptors tagged with DsRed protein. The membrane of transfected HEK 293T cells was used for the engineering of PD1-NVs (Fig. 11a), while 1-methyl-tryptophan (1-MT), an inhibitor of indoleamine 2,3-dioxygenase IDO, which is an immunosuppressive molecule overexpressed by tumour and DC cells (IDO + DCs), was encapsulated inside the NVs. The PD-1 NVs could bind to the surface of tumour cells and achieve PD-L1 blockade while the encapsulated 1-MT inhibited IDO which is induced by Treg cells (Fig. 11b and c). The 1-MT loaded PD1-NVs significantly suppressed the tumour growth in melanoma bearing mice compared to the treatment with 1-MT or PD-1 NVs alone, by the disruption of the dual immune tolerance mechanisms in the tumours.213 | ||
| Fig. 11 (a) Schematic representation of the preparation of PD-1 NVs loaded with 1-MT. (i) Engineering of HEK 293T cell line stably expressing mouse PD-1 receptors on the cell membranes. (ii) Harvesting of the cell membrane expressing PD-1 receptors. (iii) Preparation of PD-1 NVs through extrusion. (iv) Loading 1-MT into PD-1 NVs. (b) PD-L1 downregulates CD8+ T cells by interacting with their PD-1 receptors. The expression of IDO is induced by Treg cells, which inhibits the activity of CD8+ T cells. (c) PD-L1 blockade by PD-1 NVs lead to the activation of CD8+ cells to attack tumour cells. The inhibition of IDO by the 1-MT inhibitor also reverts the immunosuppressive activity of the DCs to CD8+ T cells. © 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. Reproduced from ref. 213 with permission from John Wiley and Sons. | ||
Furthermore, tumour microenvironment (TME) infiltrates cells with an immunosuppressive activity. Regulatory T-cells (Tregs) are chemoattracted to the TME and suppress immune functions through various mechanisms such as CTLA-4-mediated suppression of APC function, consumption of IL-2, production of immunosuppressive cytokines, and immune suppressive metabolites.214 Tumour associated macrophages (TMA) are frequently found in the TME exhibiting two opposing phenotypes, either tumour-supportive (M2-like cells) or tumouricidal (M1-like cells). In most tumours the M2 phenotype dominates, producing high levels of IL-10 and TGF-β immunoregulatory cytokines, inhibiting immune cell activation, maturation, and differentiation. In addition, myeloid derived suppressor cells (MDSCs) are tumour infiltrated cells which release IL-10, ARG1, NOS2, and IDO activating Tregs and suppress other immune cells.212 Nanoparticles that can target these immunosuppressive cells in TME and inhibit their action or re-educate them are promising agents for cancer immunotherapy. Rodell C. et al., engineered β-Cyclodextrin nanoparticles (CDNPs) with R848 entrapment in order to re-educate TAMs. R848 is an inhibitor of the macrophage colony-stimulating factor 1 receptor (CSF1R), the activation of which leads to M2-like polarization. Colorectal cancer (MC38) bearing mice treated with CDNP-R848 demonstrated nanoparticle accumulation in TAMs and a significant decrease in the tumour growth rates compared to R848 and CDNP alone and a vehicle control.215 In another study, Ou W. et al., demonstrated Treg cell targeting using PLGA NPs coated with polyethylene glycol-distearoyl-phosphatidylethanolamine (PEG-DSPE) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC). NPs were loaded with imatinib (IMT), a tyrosine kinase inhibitor that blocks STAT3 and STAT5 signalling and impairs their immunosuppressive functions. In addition, the surface of the NPs functionalized with tLyp1 peptide for targeting the Nrp1 receptor on Treg cells and an anti-CTLA4 immuno-checkpoint inhibitor for a combined immune-suppressive effect (imatinib loaded tLyp1-hNPs). Their results indicated a strong immune response against B16/BL6 melanoma tumours in mice by down-regulating immunosuppressive Treg cells while activating CD8+ T cells. The antitumour capability of IMT was enhanced by tLyp1 and anti-CTLA4 antibody functionalization of the NPs inhibiting the tumour cell proliferation and decrease angiogenesis.216
5.3. Targeting immune cells resident in lymph nodes
Nanoparticles are exploited as vehicles of tumour-specific antigens (TSAs) in order to APCs and specifically DCs. Administration of free antigens does not lead to an effective immune response due to their enzymatic degradation, low cellular uptake by APCs and insufficient delivery to the lymph nodes. Nanoparticles can be functionalized with ligands in order to target DCs resident in lymph nodes, such as; mannose, DEC-205, and CD40. In addition, the size of NPs is also an important factor for their accumulation in the lymph nodes. Smaller NPs, in a size range of 10–50 nm exhibit higher accumulation in the lymph nodes, whereas larger particles are taken up by macrophages.217 DCs exhibit pattern recognition receptors (PRRs) in order to scavenge pathogens, such as Toll-like receptors (TLRs). Using ligands for TLRs as adjuvants, could lead to the activation of DCs and upregulation of co-stimulatory factors such as CD40, CD80, and CD86.219 Adjuvants could be loaded in the NPs, linked to their surface or co-administered with them. Rosalia R. et al., developed PLGA NPs loaded with the ovalbumin tumour associated antigen and the adjuvants Pam3Csk4 (TLR2L) and poly(I:C) (TLR3L), for DCs' TLR receptors activation. Furthermore, NPs were surface functionalised with DC40 monoclonal antibodies for the specific targeting of DCs. CD40 is a highly expressed surface receptor on DCs which plays a crucial role in the maturation of DCs and T-cell signalling. The multi-compound nanoparticulate vaccine resulted in selective delivery to DCs via CD40 targeting and efficient T-response through the synergistic effect of TLR2-and TLR3-adjuvants.220 In another study, Kakwere H. et al., synthesized amphiphilic polymeric NPs, conjugated with the tumour-associated antigen peptide derived from tyrosinase-related protein 2 (TRP2) and functionalized with mannose for the specific targeting of DCs (Fig. 12). In vitro immunogenicity assay of bone marrow derived mouse DCs treated with the antigen conjugated NPs showed poor immunogenicity indicating the necessity of adjuvant co-administration. Treatment of mice with melanoma (B16-F10) tumours, using CpG, a TLR9 agonist that can stimulate the immune system, as an adjuvant, showed that antigen conjugated NPs in combination with an adjuvant resulted in an improved survival (up to 24 days) compared to the untreated group (14 days).221 | ||
| Fig. 12 Schematic representation of the antigen-conjugated nanoparticles and their immunogenic action. Reprinted from ref. 221. Copyright 2017 American Chemical Society. | ||
Using another approach, Noh Y. et al., generated immunostimulatory liposomes, named “tumosomes”, by the reconstitution of tumour cell membranes containing tumour-associated antigens and the incorporation of two immunomodulatory adjuvants, 3-O-desacyl-4′-monophosphoryl lipid A (MPLA) and dimethyldioctadecylammonium bromide (DDA) (Fig. 13). After an intranodal injection of the tumosomes in B16-F10 melanoma tumour-bearing mice, the results indicated a significant inhibition of tumour growth compared to injections of antigen or liposomes alone. The treatment with tumosomes increased the proliferation of immune cells from lymph nodes and spleen, while lymph nodes cells and splenocytes produced a higher amount of IFN-γ levels resulting in an enhanced antitumour immunogenicity.218 The cytosolic nucleotide sensor stimulator of IFN genes (STING), has attracted a lot of attention as a target of potential adjuvants. Cyclic dinucleotides (CDNs) are ubiquitous small molecule second messengers synthesized by bacteria which activate innate immunity by binding to the endoplasmic reticulum-resident receptor STING, activating a signalling pathway that induces the expression of interferon-β (IFN-β).222 Hanson M. et al., used a PEGgylated liposomal carrier to transfer chemically synthesized CDNs, called cdGMP, to lymphoid tissues in order to enhance their potency as immune stimulators and decrease their side effects. Mice immunization with cdGMP-liposomes demonstrated enhanced CD4+ and CD8+ T cell responses, with a 30-fold higher potency and 15-fold increased accumulation in the lymph nodes compared to free CDNs.223
 | ||
| Fig. 13 Schematic representation of the generation of the antigenic liposomes “tumosomes” as cancer immunotherapeutic agents. Reproduced from ref. 218. Copyright WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
5.4. Immunogenic cell death induction
There is evidence that tumour cell death promotes an antitumour immune response known as immunogenic cell death (ICD). Dying cells release or express molecules, which are referred to as damage-associated molecular patterns (DAMPs) and can act as danger signals for the innate immune system. Some known DAMPs are ATP, high mobility group protein B1 (HMGB, also known as amphoterin), calreticulin (CRT) and heat shock protein 90 (HSP90).224 Consequently, anticancer agents which cause toxicity and cell death in cancerous cells could be used as a combination treatment with immunotherapy, acting as adjuvants and enhancing the therapeutic outcome.However, effectively inducing ICD is challenging and so nanomaterials are attractive modalities to promote ICD, as they specifically concentrate cytotoxic agents in tumour cells, thereby reducing common systemic side effects.225 They can be designed to directly interact with external energy sources, allowing for targeted radiotherapy, magnetic hyperthermia, photothermal and photodynamic therapy, or deliver a toxic therapeutic agent combined with immunotherapeutic strategies. Additionally, as previously explained in the section on nanoparticle-induced mechanisms, ICD can be induced by the intrinsic properties of nanomaterials.
PTT can also be combined with chemotherapy which can lead to enhanced anti-cancer effects exploiting ICD triggering. Wen et al. co-delivered the chemotherapeutic agent Doxorubicin (Dox) with photothermal reactive palladium nanoparticles (PDNPs) in amphiphile triglycerol monostearates (TGMs). Chemotherapy and PTT proved to effectively kill mouse colon cancer (CT26) cells, but additionally promoted the release of ‘eat-me’ signals like calreticulin (CRT) and adenosine triphosphate (ATP). This improved immunogenicity led to an improved T-lymphocyte infiltration and eventually better treatment efficacy.227 Ma et al. achieved similar effects with gold nanoparticle irradiation using the second near-infrared bio window NIR(II), with wavelengths between 950 and 1350 nm. This window is more preferred as it reduces photon scattering, has a low tissue background and most importantly, allows a deeper penetration depth in comparison to NIR(I). This was proven by measuring the different levels of calreticulin (CALR) in mouse breast cancer (4T1) tumours after intratumoural injection of Au-DOPC (gold nanoparticles absorbed on liposomes) and treatment with 3 different laser irradiations. NIR(I) irradiation led to ICD up until 3 mm into the tumour, while NIR(II) reached significantly further up until 6 mm (Fig. 14). Additionally, the researchers illustrated that using this technique after administration of erythrocyte membrane-coated 2D polypyrrole nanosheets, an anti-tumour immunity can be elicited that prevents tumour metastasis.228
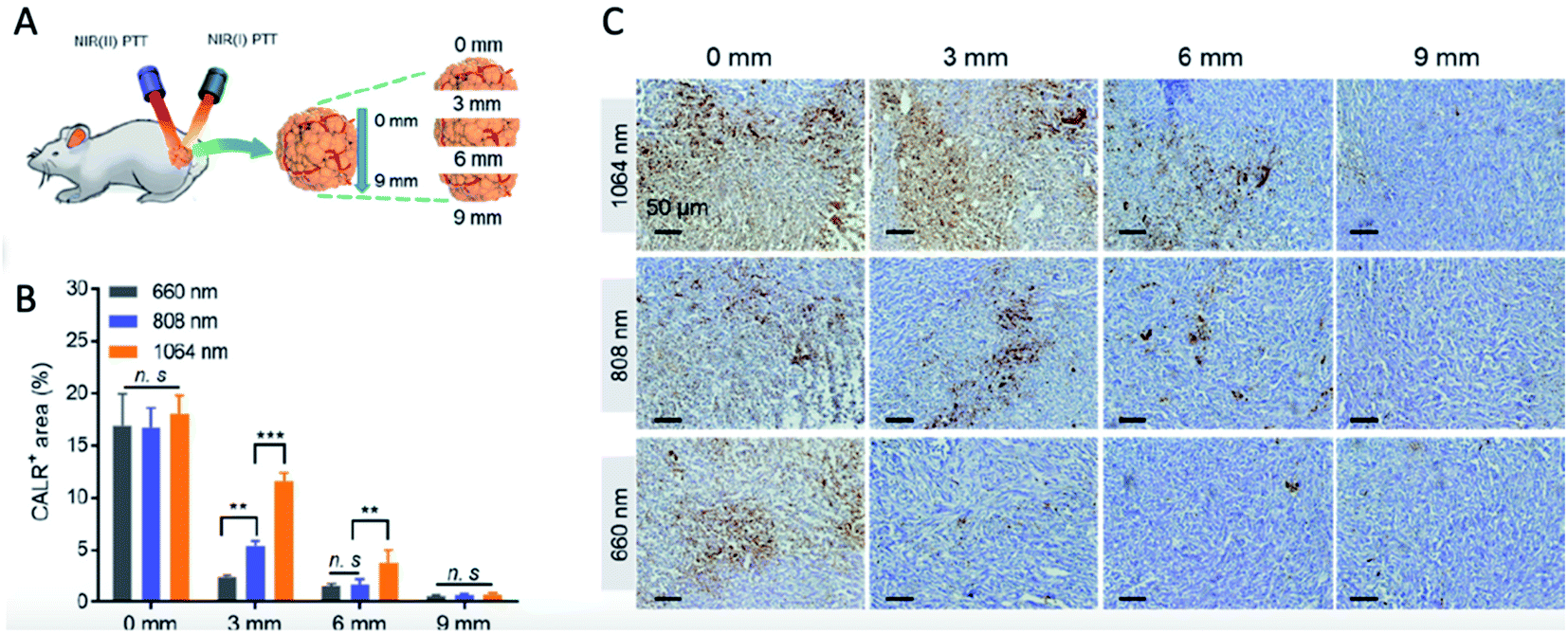 | ||
| Fig. 14 Calreticulin (CALR) exposure at different depths using different laser irradiation wavelengths. (A) Schematic representation of the experiment. (B) Percentage of CALR positive areas at different depths in dissected 4T tumours (C) immunohistochemical staining of CALR exposure in tumours. Reprinted from ref. 228. Copyright 2019 American Chemical Society. | ||
Further optimization research for PTT was performed by Sweeney et al., which revealed the importance of the correct thermal dose that is generated. Results indicated that a thermal window between 50 and 60 °C is optimal for the immunogenicity of the tumour cell death, and thus, critical for generating an anti-tumour immunity.229 Therefore, optimization of appropriate nanoparticle dose as well as radiation intensity, has to be performed on an individual application level.
Recently, Ploetz E. et al., demonstrated that hybrid metal–organic framework (MOF) nanoparticles loaded in liposomes (Lip-MOFs) could create a controllable platform for intracellular iron delivery. The high amount of realized ions by the internalized particles induced pyroptosis, a highly inflammatory form of programmed cell death, which occurred only in slightly acidic extracellular environments. This unique property of Lip-MOFs and similar nanostructures could be exploited in cancer immunotherapy, as the particles could result in the cell death of targeted cells in the acidic tumour microenvironment, via pyroptosis, inducing a significant immune response at the same time.236
Finally, activation of the stimulator of interferon gene (STING) pathway leads to increased IFN-I production and induction of ICD. Activator molecules are cyclin dinucleotides (CDNs) such as cGAMP.237 Nanoparticles can be used for the optimal and effective delivery of CDNs. Cheng et al. showed that the encapsulation of cGAMP in liposomal nanoparticles significantly upregulates IFN-I levels and suppress tumour growth, compared to free cGAMP.238 A similar approach was taken by Shae et al. using polymersomes as delivery vehicles.239 In another study, Hoang M. et al. investigated the antigen production from human B-lymphocytic (U266) cells using bPEI-SPIONs combined with ultraviolet B (UVB) irradiation. Superparamagnetic iron oxide nanoparticles (SPIONs) are known to produce high amounts of ROS and induce an immunogenic response. Apoptotic U266 cells can be used as antigens in order to induce antitumour induction of DCs to T-cells. Two hours post irradiation U266 cells with b-PEI-SPIONs demonstrated increased ROS production, extracellular release of surface Heat Shock Proteins 70 and 90 (Hsp70, Hsp90), and high mobility group box 1 protein (HMGB1). DCs were loaded with the antigenic cells which showed an enhanced expression of IFN-γ, a Th-1 polarizing cytokine promoting Th1 polarization of T-cells.240
5.5. Adoptive cell therapy (ACT)
Adoptive cell therapy (ACT) is a promising strategy for cancer immunotherapy. Tumour-specific lymphocytes are isolated from patients' blood, tumour draining lymph nodes, or tumour tissues, and expanded ex vivo before reinfusion, in order to express high reactivity against tumour antigens and overcome the immunosuppressive tumour microenvironment. T-cells can be engineered in order to express antitumour receptors. The first engineered T-cells were designed to express T-cell receptors (TCRs) with affinity to a tumour antigen. TCRs were activated only when they recognized peptides bound to major histocompatibility complex (MHC) on tumours and antigen presenting cells. However, many tumours exhibit a downregulation of the MHC constituting TCR-T cells ineffective. In order to overcome this limitation, artificial chimeric antigen receptors (CARs) have been engineered which combine the high affinity of antibody binding, without the need for antigen expression through MHC, with the intracellular signalling domains of TCRs.241 CAR-T cell-based agents were FDA approved in 2017 exhibiting significant antitumour activity against CD19-positive B-cell leukaemia and lymphoma in humans.242 However, the therapeutic efficacy of CAR-T therapies in the treatment of solid tumours is low, mainly, due to the immunosuppressive tumour microenvironment.243 Nanoparticles can be loaded with supporting drugs or chemokines and conjugated with the adoptively transferred cells (cellular backpacks) or functionalised with ligands in order to target adoptive T-cells in vivo enhancing ACT effects. For example, Zheng et al., studied the effect of TGF-β inhibitor loaded liposomes on ACT T-cell therapy. They investigated whether T-cells preloaded ex vivo with liposomes targeting an internalizing receptor (CD90, or Thy1) versus a non-internalizing receptor (CD45) were more efficient compared to antibody-functionalized liposomes that target TGF-β directly to T-cells in vivo (Fig. 15). They showed that in vitro loaded T-cells with CD45 functionalized liposomes exhibited an enhanced tumour infiltration and therapeutic efficacy, while anti-Thy1.1 internalizing liposomes, targeting T-cells in vivo, demonstrated a better therapeutic outcome. These results indicated that Thy1 exhibits an increased specificity to the donor T-cells compared to CD45. Targeted liposomes to T-cells in vivo could have a preferable clinical application, as liposomes can be administered repeatedly compared to the limitations exhibited in T-cell infusions.244 | ||
| Fig. 15 Schematic representation of the fate of Anti-Thy1.1 and Anti-CD45 targeted liposomes after T-cell targeting. Reprinted from ref. 244. Copyright 2017 American Chemical Society. | ||
 | ||
| Fig. 16 Schematic representation of the different roles of nanoparticles used in cancer immunotherapy. Created with BioRender.com. | ||
The targeted delivery of supporting cytokines and tumour-microenvironment-modulating factors using nanoparticles conjugated to adoptive T-cells (backpacks) has been explored in order to enhance ACT effectiveness and avoid systemic side effects. However, low drug loading capacity and leakiness of the drug from the backpacks before reaching their target limit their therapeutic effect. Tang L. et al., addressed these challenges using nanogels (NGs), as adoptive T-cell backpacks, synthesized with a disulphide cross-linker which was designed to be cleaved in response to reducing conditions at the T cell surface, allowing TCR activation in vivo. NGs were loaded with a human interleukin-15 super-agonist (IL-15Sa), a potent cytokine that increases CD8+ T and NK cell numbers and function.245 The NGs were functionalized with anti-CD45 antibodies in order to be conjugated to T-cell surfaces, avoiding their internalization inside the cell cytoplasm. Their results showed that T cells backpacked with TCR-responsive NGs expanded 16-fold more in B16F10 mouse melanoma tumours compared to T cells supported by systemic cytokine injections. Furthermore, they evaluated whether NG-backpacked CAR-T cells targeting epidermal growth factor receptor (EGFR) in a human glioblastoma model in immunodeficient mice could enhance the impact of CAR-T cell therapy. Treatment with NG-backpacked CAR-T cells lead to an increased therapeutic outcome, with the complete eradication of tumours in 4 or 5 mice, compared to CAR-T cells alone or CAR-T cells with an equivalent systemic dose of free IL-15Sa.246 Nanoparticles can be used as DNA delivery vehicles in order to introduce leukaemia-targeting CAR genes into circulating T cells in vivo. Smith et al., synthesized poly(β-amino ester) polymeric NPs, functionalized with T-cell-targeting anti-CD3e f(ab')2 fragments which enabled their receptor-mediated endocytosis by lymphocytes and peptides containing microtubule-associated sequences (MTAS) and nuclear localization signals (NLS) in order to import their DNA cargo into T-cell nuclei via the microtubule transport machinery. NPs were loaded with a plasmid DNA, encoding the leukaemia-specific 194-1BBz CAR and they exhibited selective targeting and reprogramming of T-cells in vivo with leukaemia regression in mice at efficacies comparable to conventional adoptive transfer of laboratory-manufactured CAR T-cells.247
Despite the advantages of CAR-T therapy, the elaborate procedures needed to isolate, engineer, selectively expand and reinfuse patient derived T-lymphocytes can only be performed at a few specialized centres worldwide, preventing their use as standard-of-care treatment for cancer.
5.6. The role of nanoparticles for the treatment of allergy and auto-immune diseases
Immunotherapy for the treatment of cause-oriented allergy involves the gradually increased dose administration of allergen natural extracts. Administration of high-dose of allergen leads to a shift from allergic Th2 inflammation toward Th1 inflammation and the generation of suppressive “regulatory” immune cells with the decrease of allergen-specific IgE and an increase of protective IgG antibodies. In addition, the infiltration and activation of eosinophils, mast cells, and basophilic leukocytes is reduced.248,249 Despite the promising therapeutic outcome of allergen-specific immunotherapy (AIT), it has some limitations, including potential systemic side-effects, long-term duration of the treatment needed and, in some cases, limited treatment effect. Nanoparticles can be used as allergen delivery vehicles, with potential adjuvant effects, in order to target their cargo, specifically, to the site of action avoiding their recognition by IgEs on the surface of mast cells or basophils, which would lead to adverse systemic side effects (Fig. 17).249 | ||
| Fig. 17 Schematic representation of allergen-specific immunotherapy using (A) soluble allergen and (B) allergen encapsulated in nanoparticles (NPs). Reproduced from ref. 249. Copyright 2017 EAACI and John Wiley and Sons A/S. Published by John Wiley and Sons Ltd. | ||
Various types of NPs have been investigated as antigen delivery vehicles with potential therapeutic use.245 For example, Smarr C. et al., demonstrated that PLGA NPs loaded with the OVA antigen [PLG(Ag) NPs] effectively inhibited Th2 responses and airway inflammation in an allergic airway inflammation mouse model without inducing adverse side effects.250 Allergen-specific immunotherapy (AIT) using NPs could be a potential treatment strategy for food allergies, which currently remains challenging due to high rates of allergic side effects and no evidence of long-term efficacy.249 Kawakita A. et al., investigated whether intranasal administration of oligomannose-coated liposomes (OML), loaded with the OVA food allergen, could induce immune tolerance in OVA-sensitized mice. Their results indicated that OVA-OML induced regulatory CD8+ T cells and antigen-specific secretory IgA in localized tissues of OVA-sensitized mice and inhibited the development of allergic diarrhea.251 Furthermore, inorganic nanoparticles and specifically gold nanoparticles exhibiting immunomodulatory effects have been investigated for their impact on allergic diseases. Baretto E. et al., demonstrated that intranasal installation of gold nanoparticles inhibited allergen-induced airway hyperreactivity in asthma mouse models. The treatment with gold nanoparticles blocked the Th2 pro-inflammatory cytokine production and reduced ROS and MDA production, resulting in protective effects against asthma allergen sensitization.252
It is known that autoimmune diseases result from an attack of the immune system against specific tissues, compromising their function. In the context of an autoimmune disease, self-peptide antigens displayed by APCs activate T-cells, instead of promoting their tolerance, resulting in auto-immune response.253 Currently, the treatment of autoimmune diseases relies mostly on total immunosuppression, which, in the long term, leads to adverse side effects. Therefore, new approaches need to be developed that could target only the disease mediating cells. Altered peptide ligands (APLs) of autoantigens have been used to induce antigen-specific tolerance in animal models of autoimmune diseases exploiting the specificity of the TCR–pMHC interaction in order to modulate the function of autoreactive T-cells. However, the immune response to a specific APL cannot be predicted and the suppression of the autoreactive T-cells without affecting the systemic immunity is not currently possible. Nanoparticle based approaches are investigated for the treatment of autoimmune diseases, in order to achieve specific targeting and avoid systemic side effects. One approach is the use of NPs as delivery vehicles of autoantigens to tolerogenic APCs.254 Getts D. et al., showed that polystyrene microparticles coupled to encephalitogenic myelin epitopes induced T-cell tolerance in multiple sclerosis mouse models, alleviating the clinical symptoms of experimental autoimmune encephalomyelitis (EAE). They demonstrated that the beneficial effect of the particles required expression of the ‘macrophage receptor with collagenous structure’ (MARCO), a scavenger receptor expressed on splenic marginal zone macrophages that contributes to the uptake and clearance of particulate debris.255 Another approach involves the delivery of tolerogenic substances to T-cells. Naive CD4+ T lymphocytes in the periphery, recognize self-peptides on APCs with the concurrent stimulatory or inhibitory signals in the microenvironment playing an important role to their transformation to effector T-cells (Teff) or regulatory T-cells (Treg), respectively. Park J. et al., demonstrated that CD4-targeted PLGA nanoparticles loaded with the inflammatory cytokine interleukin-6 (IL-6) leaded to inappropriate Th17 cell activity enhancing RORγT expression and inflammatory activity. On the contrary, NPs loaded with leukemia inhibitory factor (LIF), a tolerogenic cytokine, enhanced Foxp3+ expression which leaded to Treg lineage development and immune tolerance (Fig. 18).256
 | ||
| Fig. 18 (A) Schematic model of the LIF and IL-6 cytokine loaded antibody (anti-CD4)-targeted PLGA nanoparticle and their modulation of CD4+ T cell differentiation. Nanoparticles encapsulating LIF (LIF-nano) were found to enhance Foxp3 expression leading to Treg lineage development, while nanoparticles encapsulating IL-6 (IL-6-nano) enhanced RORγT expression leading to Teff lineage development. (B) Schematic representation of the PLGA anti-CD4 PLGA NPS used. Reprinted from ref. 256. Copyright 2011 American Chemical Society. | ||
Adoptive transfer of polyclonal FOXP3+CD4+CD25+ regulatory T (Treg) cells has been proposed as a therapeutic approach for autoimmune diseases. However, there are currently no effective strategies for expanding antigen specific Treg cells in vitro. Clemente-Casares X. et al., discovered a new strategy for the expansion of Treg cells in vivo. They showed that systemic therapy with nanoparticles coated with autoimmune-disease-relevant peptides bound to major histocompatibility complex class II (pMHCII) molecules triggered the generation and expansion of antigen-specific regulatory CD4+ T cells in different autoimmune disease mouse models in vivo, leading to the resolution of inflammation. Their data suggests that the use of any single pMHC, involved in a given autoimmune disease, could promote the differentiation of disease autoreactive T cells into Treg which, in turn, suppress autoantigen-loaded APCs and lead to the differentiation of B cells into disease-suppressing regulatory B cells. Therefore, pMHC-based nanomedicines represent a new class of therapeutics in the treatment of autoimmune diseases, without compromising systemic immunity.257
6. Clinical trials
The current understanding of the immune alterations induced by nanomaterials, together with optimized strategies for their use in drug delivery or vaccines, allowed multiple products to reach clinical trials. In Table 4 we review clinical trials of nanoparticles in immunotherapy that have a start and/or end date in the past 10 years. Not surprisingly, more trials are found using organic based NP-strategies, in contrast to only 3 trials using inorganic NPs. This trend is in accordance with current approved nanoparticle-based therapeutics, with iron oxide applications, e.g. as contrast agents for MRI, one of the few inorganic examples.258 The hafnium oxide nanoparticle-based strategy (NBTXR3) of Nanobiotix is another example and has been approved (CE mark) for its radio enhancing effect for the treatment of locally advanced soft tissue sarcoma.259 Additionally, promising preclinical in vivo results for its use in combination with anti-PD-1 immunotherapy, show potential for the treatment of lung cancer and has led to the initiation of a new clinical trial (NCT03589339).260| Name | Nanoparticle | Intervention | Immunotherapeutic indication | Disease application | Phase | Start date | Status | Clinical trials.gov identifier |
|---|---|---|---|---|---|---|---|---|
| a PIK3 (phosphoinositide 3-kinases); PD-L1 (programmed death-ligand 1); VEGFR2 (vascular endothelial growth factor 2); AHSCT (autologous hematopoietic stem cell transplantation). | ||||||||
| Inorganic | ||||||||
| NBTXR3 activated by radiotherapy for patients with advanced cancers treated with an anti-PD-1 therapy | Hafnium oxide (HfO2) nanoparticle activated by radiotherapy | Combination of NBTXR3 with anti-PD-1 therapy | Combination with immune checkpoint inhibitors to convert checkpoint inhibitor non-responders into responders | Locoregional recurrent (LRR) or recurrent and metastatic (R/M) head and neck squamous cell carcinoma (HNSCC) and lung and liver metastases from any primary cancer eligible for anti-PD-1 therapy | 1 | 16/01/2019 | Recruiting | NCT03589339 |
| Enhanced epidermal antigen specific immunotherapy trial-1 (EE-ASI-1) | Human C19A3 proinsulin derived peptide coupled to gold nanoparticles | Intradermal micro-injectable solution of C19-A3 GNP | Induce peptide-specific IL-10 secreting T cells to modify the autoimmune response and the switching off the white blood cells causing damage to the pancreatic islet cells | Diabetes type 1 | 1A | 29/09/2016 | Active | NCT02837094 |
| Study to assess the impact of inhaled silver nanoparticles on lung cell immune response | Silver nanoparticles | Inhalation of nanosilver | Inhaled nanosilver can affect the lungs immune system and enter the bloodstream | Toxicity screening on healthy adults | 0 | 03/04/2015 | Withdrawn | NCT02408874 |
|
||||||||
| Organic | ||||||||
| Nanoparticles as adjuvants | ||||||||
| Safety and efficacy trial of DNA vaccines to treat Genital Herpes in Adults | Liposome (Vaxfectin) | VCL-HB01: Plasmid DNA vaccine encoding two HSV-2 proteins formulated with Vaxfectin |
Cationic lipid-based adjuvant Vaxfectin to improve vaccine efficiency | Genital Herpes Simplex type 2 | 1/2A | December 2013 | Completed (February 2016) | NCT02030301 |
| 2 | September 2016 | Completed (December 2018) | NCT02837575 | |||||
| Nanoparticles as delivery vehicle for immunotherapy | ||||||||
| RNA-immunotherapy of IVAC_W_bre1_uID and IVAC_M_uID (TNBC-MERIT) | Liposomes containing RNA for patient specific tumour-associated antigens and p53 RNA | Indivudualized cancer immunotherapy with immunogenic RNA vaccines IVAC_W_bre1_uID vaccination | Induction of T-cell memory against patient specific tumour-associated antigens | Triple negative breast cancer | 1 | October 2016 | Active | NCT02316457 |
| JVRS-100 for the treatment of patients with relapsed or refractory leukemia | Cationic liposome loaded with plasmid DNA complex | JVRS-100 therapy | Immune stimulation | Relapsed or refractory leukemia | 1 | 01/03/2009 | Completed (February 2017) | NCT00860522 |
| Intravenous administration of RGI-2001 in patient undergoing allogenic hematopoietic stem cell transplantation (AHSCT) | Liposomal formulation of α-galactosylceramide | IV-injection of RGI-2001 after completion of transplant | Mitigation of graft versus host disease by increasing immune tolerance | Patients undergoing allogeneic hematopoietic stem cell transplantation with or without radiation myeloablative preparative treatment | 1/2 | September 2011 | Completed (April 2017) | NCT01379209 |
| The effects of oral nanocurcumin on expression levels of microRNAs and Treg cells and Th17 cells development factors in ankylosing spondylitis patients | Curcumin in nanomicelles | Oral nano-curcumin application in patients with ankylosing spondylitis (AS) | Curcumin induces down-regulation of various inflammatory cytokines including TNF-α and IL 1 | Ankylosing spondylitis | 2 | 29/04/2017 | Completed (January 2018) | NCT03140657 |
| Combination therapy: immunotherapy in combination with nanoparticle treatment | ||||||||
| Nab-paclitaxel and durvalumab with or without neoantigen vaccine in treating patients with metastatic triple negative breast cancer | Nab-paclitaxel | Combination of nab-paclitaxel and durvalumab with or without personalized synthetic long peptide vaccine (neoantigen vaccine) | Durvalumab is a monoclonal antibody that blocks the interaction of PD-L1 with PD-1 leading to immune cell activation. Vaccines made from peptides may help to build an effective immune response to kill tumor cells | Metastasized triple negative breast cancer | 2 | 15/12/2018 | Not yet recruiting | NCT03606967 |
| Nab-paclitaxel and atezolizumab before surgery in treating patients with triple negative breast cancer | Nab-paclitaxel | Atezolizumab and Nab-paclitaxel | Combination of anti-PD-L1 antibody with nab-paclitaxel to reduce tumour size before surgery | Primary invasive adenocarcinoma of triple negative breast cancer with no prior treatment | 2 | 04/02/2016 | Recruiting | NCT02530489 |
| Safety, tolerability and immunogenicity of EGFR(V)-EDV-Dox in subjects with recurrent glioblastoma multiforme (GBM) (CerebralEDV) | EGFR(V)-EDV-Dox using EnGeneIC EDV™ technology | EGFR(V)-EDV-Dox is a bacterially derived minicell with encapsulated doxorubicin, which targets specific cancer cells using bispecific antibodies (BsAb) | The residual EDVs that do not make it into the tumor microenvironment, are engulfed by cells of the immune system and since the EDVs are derived from bacteria, they carry potent immuno-stimulating components which appear to bypass the immuno-suppression caused by the tumor | Recurrent glioblastoma multiforme | 1 | 25/10/2016 | Recruiting | NCT02766699 |
| Atezolizumab with chemotherapy in treating patients with anaplastic or poorly differentiated thyroid cancer | Nab-paclitaxel | Combination of atezolizumab with chemotherapy (Cobimetinib, nab-paclitaxel, paclitaxel, vemurafenib) | Atezolizumab is a monoclonal antibody which will bind with PD-L1 and lead to immune cell activation. Combining it with chemotherapy may improve how immune cells respond and attack tumor cells | Anaplastic or poorly differentiated thyroid cancer | 2 | 27/07/2017 | Recruiting | NCT03181100 |
| A study to evaluate safety/tolerability of immunotherapy combinations in participants with triple-negative breast cancer and gynecologic malignancies | Nab-paclitaxel pegylated liposomal doxorubicin (PLD) | AB928 in combination with PLD with or without IPI-549, or AB928 in combination with nanoparticle albumin-bound-paclitaxel | Enhancing the tumour-infiltrating lymphocytes and myeloid cells in combination with anthracycline topoisomerase II inhibitor with or without PIK3 inhibitor or enhancing the tumour-infiltrating lymphocytes and myeloid cells in combination with microtubules stabilizers | Triple negative breast cancer or ovarian cancer | 1/1B | 15/10/2018 | Recruiting | NCT03719326 |
| Dose escalation study of mRNA-2752 for intrtumoral injection to patients with advanced malignancies | Lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ | Intratumoral injections of mRNA-2752 alone and in combination with intravenously administered durvalumab (immune checkpoint blockade therapy) | mRNA-2752 in combination with durvalumab, enhances the immune response at the tumour site | Relapsed/refractory solid tumor malignancies or lymphoma | 1 | 27/11/2018 | Recruiting | NCT03739931 |
| Nab-paclitaxel/rituximab-coated nanoparticle AR160 in treating patients with relapsed or refractory B-cell non-Hodgkin lymphoma | Nab-paclitaxel/rituximab-coated nanoparticle AR160 | Combination treatment of paclitaxel albumin-stabilized nanoparticle formulation and rituximab | Ritixumab is a monoclonal antibody which binds to the cell surface protein CD20 of B cells forming a cap which triggers natural killer cells to destroy these B-cells | Non-Hodgkin lymphoma | 1 | 25/4/2019 | Recruiting | NCT03003546 |
| Durvalumab in combination with chemotherapy in treating patients with advanced solid tumors, (DURVA + study) | Nab-paclitaxel-pegylated liposomal doxorubicin hydrichloride l | Combination of durvalumab with chemotherapy (capecitabine, nab-paclitaxel, paclitaxel, carboplatin, gemcitabine hydrochloride, pegylated liposomal doxorubicin hydrichloride) | Durvalumab is a monoclonal antibody which will bind with PD-1 and lead to immune cell activation. Combining it with chemotherapy may improve how immune cells respond and attack tumor cells | Advanced solid tumors (spread to other places in the body) | 2 | 25/06/2019 | Recruiting | NCT03907475 |
| Evaluation of IPI-549 combined with front-line treatments in Pts. With triple-negative breast cancer or renal cell carcinoma (MARIO-3) | Nab-paclitaxel | Combination of IPI-549 with atezolizumab and nab-paclitaxel | IPI-549 targets immune-suppressive tumor-associated myeloid cells through selective inhibition of phosphoinositide-3-kinase (PI3K)-gamma. In combination with immune therapy, it may enhance immune cell activation against the tumour cells | Triple negative breast cancer and renal cell cancer | 2 | 17/12/2019 | Recruiting | NCT03961698 |
| Cryoablation, Atezolizumab/Nab-paclitaxel for locally advanced or metastatic triple negative breast cancer | Nab-paclitaxel | Atezolizumab (humanized monoclonal antibody against PD-L1),Cryosurgery (cryoablation of the primary tumour), Nab-paclitaxel | Cryoablation in combination with anti-PD-L1 antibody and Nab- paclitaxel | PD-L1 positive triple negative metastatic breast cancer or breast cancer spread into neighboring tissue or lymph nodes | Early 1 | 23/01/2020 | Recruiting | NCT04249167 |
| A single-arm, phase II study of neoadjuvant MPDL3280A, Nab-paclitaxel and carboplatin (MAC) in resectable non-small cell lung cancer (NSCLC) | Nab-paclitaxel | Combination of chemotherapeutic agents with immune-based therapy | MPDL3280A (atezolizumab) is an immune checkpoint inhibitor which will bind to PD-1 and activate the immune cells | Non-small cell lung cancer | 2 | 07/06/2016 | Active | NCT02716038 |
| Neoadjuvant pembrolizumab (Pbr)/Nab-paclitaxel followed by Pbr/epirubicin/cyclophosphamide in TNBC (NIB) | Nab-paclitaxel | Pembrolizumab in combination with Nab-paclitaxel | Combination of anti-PD-L1 antibody with nab-paclitaxel to reduce tumour size before surgery | Triple negative breast cancer | 2 | 23/03/2018 | Active | NCT03289819 |
| S0800, Nab-paclitaxel, doxorubicin, cyclophosphamide, and pegfilgrastim with or without bevacizumab in treating women with inflammatory or locally advanced breast cancer | Nab-paclitaxel | IV injection of nab-paclitaxel with or without intravenous injection of bevacizumab | Comparison of treating efficiency of | HER2/NEU negative breast cancer | 2 | 04/2010 | Completed (December 2015) | NCT00856492 |
| Nab-paclitaxel and bevacizumab or ipilimumab as first-line therapy in treating patients with stage IV melanoma that cannot be removed by surgery | Nab-paclitaxel | Combination of Nab-paclitaxel with bevacizumab in patients with stage IV melanoma | Bevacizumab is a monoclonal antibody that can bind to vascular endothelial growth factor and prevent the growth of new blood vessels that tumours need to grow | Stage IV melanoma | 2 | 18/10/2013 | Completed (October 2019) | NCT02158520 |
| Safety and feasibility study of AGS-003-LNG for the treatment of stage 3 non small cell lung cancer | Nab-paclitaxel | AGS-003-LNG dosing initiated concurrently or subsequent to 3rd cycle of nano-particle albumin-bound paclitaxel chemotherapy & radiation therapy | Effects of combination treatment of autologous dendritic cell immunotherapy (AGS-003-LNG) and nanoparticlealbumin-bound paclitaxel in patients with resectable non-small cell lung cancer |
Resectable non-small cell lung cancer | 2 | March 2016 | Withdrawn | NCT02662634 |
| MM-398 and ramucirumab in treating patients with gastric cancer or gastroesophageal junction adenocarcinoma | Encapsulating irinotecan within long-circulating liposome-based nanoparticles | MM-398: interrupts cell reproduction ramucirumab: anti-VEGFR2 | Treating patients with ramucirumab to enhance body's immune system to attack cancer cells and the combination with MM-398 for treating patients by interrupting cell reproduction | Gastric cancer or gastroesophageal junction adenocarcinoma | 1/2 | 06/04/2020 | Withdrawn (PI left) | NCT03739801 |
For organic nanoparticles, we divided the trials based on their type of application; nanoparticles as adjuvants, nanoparticles as immunotherapeutic delivery vehicles or nanoparticles in combination therapy. Within the first category, adjuvants, the clinical trials (NCT02030301 and NCT02837575) for VCL-HB01, a plasmid DNA vaccine formulated with Vaxfectin (cationic lipid-based adjuvant), were completed. Although promising results from the phase 1/2A showed significant lowering of lesion rates after 9 months, Vical completely shut down the HSV-2 program following unsatisfactory results in the phase 2 trial. The disappointing results were allocated to a limited recurrence rate in the placebo group.261
Within the second category, nanoparticles as immunotherapeutic delivery vehicles, the clinical trial for RGI-2001 (NCT01379209) was finalized in April 2017. RGI-2001 is a liposomal formulation of KRN700, an α-galactosylceramide, and is intended to prevent Graft-versus-Host-Disease (GVHD) after allogeneic bone marrow transplantation. In pre-clinical studies in a murine model, RGI-2001 was shown to prevent GVHD, due to an increased number of naturally occurring CD4+Foxp3+ Tregs, while retaining the intended Graft-versus-Leukemia effect of the transplantation.262 In the phase 2A clinical trial, 29 patients were enrolled, of which 14 patients received a dose of 1 μg kg−1 and 15 patients a dose of 100 μg kg−1 on day 0 of transplantation. Results show that the treatment was well tolerated and led to an increase in Tregs in 8 out of 29 patients. Of these 8 responders, 6 had received sirolimus, an immunosuppressant, and appeared to result in a synergetic effect on the total Treg number. Additionally, the incidence of grade 2 to 4 and of grade 3 or 4 of GVHD was respectively, 12.5% and 0% in the responder group, contrary to 52.4% and 9.5% in non-responders. Although results are promising, it should be noted that no control group was included in the study, and no definitive conclusion can be made regarding the potential of RGI-2001.263 Further studies are needed to clarify the precise effect of RGI-2001 and to understand the lack of effect in non-responders. In another completed clinical trial (NCT03140657), the use of nanocurcumin (curcumin encapsulated in biodegradable polymer nanoparticles) for the treatment of ankylosing spondylitis, a chronic autoimmune disease, was evaluated. In this phase 2 trial, 12 patients were given a daily dose of 80 mg kg−1 nanocurcumin for 4 months. The study showed a significant increase of Treg cells and higher expression levels of FoxP3, TGF-β and IL-10, while the expression of IL-6 was significantly suppressed. Therefore, nanocurcumin shows potential as therapeutic agent for ankylosing spondylitis.264
After the successful use of liposome and albumin nanoparticles in current chemotherapies, like Doxil® and Abraxane®, similar strategies have been used in combination treatment with immunotherapy, the third category of clinical trials. Abraxane® (albumin-bound paclitaxel) is a very promising strategy with, currently, 10 ongoing clinical trials evaluating its potential for treating Triple Negative Breast Cancer (NCT03606967, NCT02530489, NCT03961698, NCT03719326, NCT04249167 and NCT03289819), thyroid cancer (NCT03181100), Non-Hodgkin lymphoma (NCT03003546), Non-small cell lung cancer (NCT02716038) and advanced solid tumours (NCT03907475). It was also used in the completed SWOG S0800 clinical trial, in which the addition of bevacizumab to a neoadjuvant chemotherapy (Nab-paclitaxel and dose-dense doxorubicin/cyclophosphamide) was shown to enhance the cytotoxicity of the therapy.265
7. Future prospects and conclusions
Nanoparticles have gained increasing interest over the years and are being explored for enhancing therapies of a variety of diseases and disorders. Here, we have provided an overview of the current research and understanding of the immunological alterations induced by nanomaterials and how this has allowed researchers to design nanomaterials suitable for drug delivery, immunostimulation and immunotherapy. Inorganic nanomaterials, like gold and mesoporous silica, were shown to have excellent adjuvant properties, and thus, could be used as self-adjuvant vaccine carriers. Additionally, porous inorganic materials can leverage their high internal volumes for enhanced drug delivery.Organic nanomaterials exhibit considerable potential as delivery vehicles of anti-inflammatory agents, genetic material like DNA or RNA, antibodies and antigens, as they can protect their cargo from degradation and target specifically the inflamed tissues without inducing adverse side effects. Their physicochemical characteristics have significant effects on their therapeutic efficacy and should be modulated accordingly, taking into consideration their intended application. For example, cationic liposomes are recommended to be used for antigen presentation due to their immunostimulatory effects, whereas neutral and anionic liposomes would be preferable as drug delivery vehicles due to their prolonged circulation lifetime. Furthermore, surface functionalization of the NPs has a critical role on their site-specific targeting. Various ligands have been used for targeting enhancement while their specificity to the diseased site is essential for engineering safe and efficient nanotherapeutics. As discussed in this review, nanomaterial-based immunotherapy has shown great promise in the treatment of cancer, allergies and autoimmune diseases. Several approaches have been proposed in order to overcome the limitations of conventional immunotherapy strategies. Nanomaterials are able to deliver antigens specifically to antigen presenting cells (APC) in the lymph nodes, regulate Tregs, and TAMs, activate T cells and enhance adoptive cell therapies. In addition, nanoparticles could be harnessed as immunogenic cell death (ICD) inducers, in combination with immunotherapy for an enhanced therapeutic efficacy.
The findings, discussed in this review, illustrate that research efforts on nanomaterial-based therapies have spiked and have made rapid progress. Progress in this field has largely been pushed by research for cancer therapies and, although translation is still relatively low,266 more and more nanoparticle strategies reach clinical trials. Enhancing translation ability of nanomedicine research will be one of the key challenges in the coming years. Upcoming strategies, such as cell-based nanoparticle delivery, functionalization with elaborating ligands with specific microenvironment responsiveness, and co-delivery of agents with synergistic immunomodulatory effects, are promising ideas for tackling this challenge.
The second major challenge is to assure safety of nanomaterials in the human body. Although multiple mechanisms have been suggested and indicated to contribute to the toxicological and immunological effect of nanomaterials, the exact pathways are still not fully unraveled. Additionally, tuning influential physicochemical parameters requires a time consuming, systematic experimental optimization. Computational methods are upcoming in bionanotechnological research and might facilitate optimization steps. For example, artificial neural network modelling has been used to fully evaluate the effect of PEG chain length, chitosan/PEG ratio and pH of the solution on the size and zeta potential of PEGylated Chitosan NPs.267 Additionally, computational fluid dynamics (CFD) has been used to predict the distribution and targeting efficiencies of nanoparticles.268 Further integration of computational models in bionanotechnology could accelerate nanoparticle designs.
Despite the major accomplishments over the past years, it is clear that we are only at the beginning of exploiting the potential of nanotechnology to its fullest and that a deeper understanding of the interactions of nanomaterials within the human body will lead to safer and more efficient immunological applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Commission under the Horizon 2020 framework for ERC (ERC StG 750973), FWO research project (G0B2919N) and KU Leuven Internal Funds C2 (C24/18/101).References
- G. P. Dunn, A. T. Bruce, H. Ikeda, L. J. Old and R. D. Schreiber, Nat. Immunol., 2002, 3, 991, DOI:10.1038/ni1102-991.
- P. J. Delves, in Encyclopedia of Immunology, 2nd edn, 1998 Search PubMed.
- C. G. Drake, E. Jaffee and D. M. Pardoll, Adv. Immunol., 2006, 90, 51–81 CAS.
- A. N. Ilinskaya and M. A. Dobrovolskaia, Br. J. Pharmacol., 2014, 171, 3988–4000 CrossRef CAS.
- S. Farkona, E. P. Diamandis and I. M. Blasutig, BMC Med., 2016, 14, 1–18 CrossRef.
- X. Feng, W. Xu, Z. Li, W. Song, J. Ding and X. Chen, Adv. Sci., 2019, 6 DOI:10.1002/advs.201900101.
- S. Musetti and L. Huang, ACS Nano, 2018, 12, 11740–11755 CrossRef CAS.
- A. K. Jain, K. Thanki and S. Jain, Mol. Pharmaceutics, 2013, 10, 3459–3474 CrossRef CAS.
- P. I. Siafaka, N. Üstündag Okur, E. Karavas and D. N. Bikiaris, Int. J. Mol. Sci., 2016, 17, 1440, DOI:10.3390/ijms17091440.
- R. Pati, M. Shevtsov and A. Sonowane, Front. Immunol., 2018, 9, 2224, DOI:10.3389/fimmu.2018.02224.
- M. Saeed, J. Gao, Y. Shi, T. Lammers and H. Yu, Theranostics, 2019, 9, 7981–8000 CrossRef CAS.
- M. Poljak-Blaži, M. Jaganjac and N. Žarković, Cell Oxidative Stress: Risk of Metal Nanoparticles, CRC Press Taylor, London, UK; New York, NY, USA, 2010 Search PubMed.
- I. Pujalte, I. Passagne, B. Brouillaud, M. Treguer, E. Durand, C. Ohayon-Courtes and B. L'Azou, Part. Fibre Toxicol., 2011, 8, 10 CrossRef CAS.
- M. Licciardi, C. Scialabba, R. Puleio, G. Cassata, L. Cicero, G. Cavallaro and G. Giammona, Int. J. Pharm., 2019, 556, 57–67 CrossRef CAS.
- R. J. Wydra, C. E. Oliver, K. W. Anderson, T. D. Dziubla and J. Z. Hilt, RSC Adv., 2015, 5, 18888–18893 RSC.
- Z. Hou, Y. Zhang, K. Deng, Y. Chen, X. Li, X. Deng, Z. Cheng, H. Lian, C. Li and J. Lin, ACS Nano, 2015, 9, 2584–2599 CrossRef CAS.
- F. Caputo, M. De Nicola, A. Sienkiewicz, A. Giovanetti, I. Bejarano, S. Licoccia, E. Traversa and L. Ghibelli, Nanoscale, 2015, 7, 15643–15656 RSC.
- J. M. Dowding, S. Das, A. Kumar, T. Dosani, R. McCormack, A. Gupta, T. X. T. Sayle, D. C. Sayle, L. Von Kalm, S. Seal and W. T. Self, ACS Nano, 2013, 7, 4855–4868 CrossRef CAS.
- G. Benetti, E. Cavaliere, R. Brescia, S. Salassi, R. Ferrando, A. Vantomme, L. Pallecchi, S. Pollini, S. Boncompagni, B. Fortuni, M. J. Van Bael, F. Banfi and L. Gavioli, Nanoscale, 2019, 11, 1626–1635 RSC.
- N. Leynen, F. G. A. J. Van Belleghem, A. Wouters, H. Bove, J.-P. Ploem, E. Thijssen, S. A. S. Langie, R. Carleer, M. Ameloot, T. Artois and K. Smeets, Nanotoxicology, 2019, 13, 476–491 CrossRef CAS.
- B. B. Manshian, C. Pfeiffer, B. Pelaz, T. Heimeri, M. Gallego, M. Möller, P. del Pino, U. Himmelreich, W. J. Parak and S. J. Soenen, ACS Nano, 2015, 9, 10431–10444 CrossRef CAS.
- S. Sabella, R. P. Carney, V. Brunetti, M. A. Malvindi, N. Al-Juffali, G. Vecchio, S. M. Janes, O. M. Bakr, R. Cingolani, F. Stellacci and P. P. Pompa, Nanoscale, 2014, 6, 7052–7061 RSC.
- M. I. Setyawati, X. Yuan, J. Xie and D. T. Leong, Biomaterials, 2014, 35, 6707–6715 CrossRef CAS.
- X. Lai, H. Zhao, Y. Zhang, K. Guo, Y. Xu, S. Chen and J. Zhang, Sci. Rep., 2018, 8, 4499 CrossRef.
- A. Murphy, A. Casey, G. Byrne, G. Chambers and O. Howe, J. Appl. Toxicol., 2016, 36, 1311–1320 CrossRef CAS.
- T. Brzicova, E. Javorkova, K. Vrbova, A. Zajicova, V. Holan, D. Pinkas, V. Philimonenko, J. Sikorova, J. Klema, J. Topinka and P. Rossner, Nanomaterials, 2019, 9, 687, DOI:10.3390/nano9050687.
- A. Pal, S. Alam, L. K. S. Chauhan, P. N. Saxena, M. Kumar, G. N. Ansari, D. Singh and K. M. Ansari, Toxicol. Res., 2016, 5, 1066–1077 CrossRef CAS.
- H. Attia, H. Nounou and M. Shalaby, Toxics, 2018, 6(2), 29, DOI:10.3390/toxics6020029.
- C. Angelé-Martínez, K. V. T. Nguyen, F. S. Ameer, J. N. Anker and J. L. Brumaghim, Nanotoxicology, 2017, 11, 278–288 CrossRef.
- E. Assadian, M. Zarei, A. Gilani, M. Farshin, H. Degampanah and J. Pourahmad, Biol. Trace Elem. Res., 2018, 184, 350–357 CrossRef CAS.
- J. Y. Ma, H. Zhao, R. R. Mercer, M. Barger, M. Rao, T. Meighan, D. Schwegler-Berry, V. Castranova and J. K. Ma, Nanotoxicology, 2011, 5, 312–325 CrossRef CAS.
- J. Park, D.-h. Lim, H.-j. Lim, T. Kwon, J.-s. Choi, S. Jeong, I.-h. Choi and J. Cheon, Chem. Commun., 2011, 47, 4382–4384 RSC.
- H. M. Braakhuis, I. Gosens, P. Krystek, J. A. F. Boere, F. R. Cassee, P. H. B. Fokkens, J. A. Post, H. van Loveren and M. V. D. Z. Park, Part. Fibre Toxicol., 2014, 11, 1743–8977 Search PubMed.
- E. I. Alarcon, K. Udekwu, M. Skog, N. L. Pacioni, K. G. Stamplecoskie, M. González-Béjar, N. Polisetti, A. Wickham, A. Richter-Dahlfors, M. Griffith and J. C. Scaiano, Biomaterials, 2012, 33, 4947–4956 CrossRef CAS.
- M. Ilves, P. A. S. Kinaret, J. Ndika, P. Karisola, V. Marwah, V. Fortino, Y. Fedutik, M. Correia, N. Ehrlich, K. Loeschner, A. Besinis, J. Vassallo, R. D. Handy, H. Wolff, K. Savolainen, D. Greco and H. Alenius, Part. Fibre Toxicol., 2019, 16(28), 1–21 CAS.
- S. Haque, V. M. McLeod, S. Jones, S. Fung, M. Whittaker, M. McIntosh, C. Pouton, D. J. Owen, C. J. H. Porter and L. M. Kaminskas, Eur. J. Pharm. Biopharm., 2017, 119, 408–418 CrossRef CAS.
- J.-P. Behr, CHIMIA, 1997, 51, 34–36 CAS.
- F. Wang, M. G. Bexiga, S. Anguissola, P. Boya, J. C. Simpson, A. Salvati and K. A. Dawson, Nanoscale, 2013, 5, 10868–10876 RSC.
- F. Jessop, R. F. Hamilton, J. F. Rhoderick, P. Fletcher and A. Holian, Toxicol. Appl. Pharmacol., 2017, 318, 58–68 CrossRef CAS.
- R. Benjaminsen, M. A. Mettebjerg, J. Henriksen, S. M. Moghimi and T. Andresen, Mol. Ther., 2013, 21, 149–157 CrossRef CAS.
- S.-H. Park, J. Y. Hyun and I. Shin, Chem. Sci., 2018, 10, 56–66 RSC.
- X. Le Guével, M. Perez Perrino, T. D. Fernández, F. Palomares, M.-J. Torres, M. Blanca, J. Rojo and C. Mayorga, ACS Appl. Mater. Interfaces, 2015, 7, 20945–20956 CrossRef.
- J. Y. Ma, H. Zhao, R. R. Mercer, M. Barger, M. Rao, T. Meighan, D. Schwegler-Berry, V. Castranova and J. K. Ma, Nanotoxicology, 2011, 5, 312–325 CrossRef CAS.
- B. B. Manshian, J. Poelmans, S. Saini, S. Pokhrel, J. J. Grez, U. Himmelreich, L. Mädler and S. J. Soenen, Acta Biomater., 2018, 68, 99–112 CrossRef CAS.
- Y.-H. Luo, L. W. Chang and P. Lin, BioMed Res. Int., 2015, 2015, 143720, DOI:10.1155/2015/143720.
- M. Turabekova, B. Rasulev, M. Theodore, J. Jackman, D. Leszczynska and J. Leszczynski, Nanoscale, 2014, 6, 3488–3495 RSC.
- H. Chang, C.-C. Ho, C. S. Yang, W.-H. Chang, M.-H. Tsai, H.-T. Tsai and P. Lin, Exp. Toxicol. Pathol., 2013, 65, 887–896 CrossRef CAS.
- C.-C. Ho, Y.-H. Luo, T.-H. Chaung, C.-S. Yang and P. Lin, Toxicology, 2013, 308, 1–9 CrossRef CAS.
- A. R. Gliga, J. De Loma, S. Di Bucchianico, S. Skoglund, S. Keshavan, I. O. Wallinder, H. L. Karlsson and B. Fadeel, Nanoscale Adv., 2020, 2, 648–658 RSC.
- C. Y. Tsai, S. L. Lu, C. W. Hu, C. S. Yeh, G. B. Lee and H. Y. Lei, J. Immunol., 2012, 188, 68–76 CrossRef CAS.
- S. Lee, H.-S. Yun and S.-H. Kim, Biomaterials, 2011, 32, 9434–9443 CrossRef CAS.
- A. Gallud, O. Bondarenko, N. Feliu, N. Kupferschmidt, R. Atluri, A. Garcia-Bennett and B. Fadeel, Biomaterials, 2017, 121, 28–40 CrossRef CAS.
- O. Kepp, L. Galluzzi and G. Kroemer, Nat. Immunol., 2011, 12, 199–200 CrossRef CAS.
- R. Zhou, A. S. Yazdi, P. Menu and J. Tschopp, Nature, 2011, 469, 211–215 CrossRef.
- C. S. Yang, D. M. Shin and E. Jo, Int. Neurourol. J., 2012, 16, 2–12 CrossRef.
- R. F. Hamilton, N. Wu, D. Porter, M. Buford, M. Wolfarth and A. Holian, Part. Fibre Toxicol., 2009, 6, 35 CrossRef.
- B. Sun, X. Wang, Z. Ji, M. Wang, Y. P. Liao, C. H. Chang, R. Li, H. Zhang, A. E. Nel and T. Xia, Small, 2015, 11, 2087–2097 CrossRef CAS.
- S. L. Fink and B. T. Cookson, Cell. Microbiol., 2006, 8, 1812–1825 CrossRef CAS.
- A. Reisetter, L. V. Stebounova, J. Baltrusaitis, L. Powers, A. Gupta, V. Grassian and M. M. Monick, J. Biol. Chem., 2011, 286, 21844–21852 CrossRef CAS.
- X. Zhang, J. Luan, W. Chen, J. Fan, Y. Nan, Y. Wang, Y. Liang, G. Meng and D. Ju, Nanoscale, 2018, 10, 9141–9152 RSC.
- V. Mirshafiee, B. Sun, C. H. Chang, Y.-P. Liao, W. Jiang, J. Jiang, X. Liu, X. Wang, T. Xia and A. E. Nel, ACS Nano, 2018, 12, 3836–3852 CrossRef CAS.
- Q. Remijsen, V. Goossens, S. Grootjans, C. V. D. Haute, N. Vanlangenakker, Y. Dondelinger, R. Roelandt, I. Bruggeman, A. Goncalves, M. J. M. Bertrand, V. Baekelandt, N. Takahashi, T. V. Berghe and P. Vandenabeele, Cell Death Dis., 2014, 5, e1004, DOI:10.1038/cddis.2013.531.
- Y. Niu, E. Tang and Q. Zhang, Toxicol. Res., 2019, 8, 1042–1049 CrossRef CAS.
- P. Sonkusre and S. S. Cameotra, J. Nanobiotechnol., 2017, 15, 43, DOI:10.1186/s12951-017-0276-3.
- T. T. Mai, A. Hamaï, A. Hienzsch, T. Caneque, S. Müller, J. Wicinski, O. Cabaud, C. Leroy, A. David, V. Acevedo, A. Ryo, C. Ginestier, D. Birnbaum, E. Charafe-Jauffret, P. Codogno, M. Mehrpour and R. Rodriguez, Nat. Chem., 2017, 9, 1025–1033 CrossRef CAS.
- S. Dixon, K. Lemberg, M. Lamprecht, R. Skouta, E. Zaitsev, C. Gleason, D. Patel, A. Bauer, A. Cantley, W. Yang and B. Morrison, Cell, 2012, 149, 1060–1072 CrossRef CAS.
- R. Yang, Y. Li, X. Wang, J. Yan, D. Pan, Y. Xu, L. Wang and M. Yang, RSC Adv., 2019, 9, 28548–28553 RSC.
- H.-H. Jeong, E. Choi, E. Ellis and T.-C. Lee, J. Mater. Chem. B, 2019, 7, 3480–3496 RSC.
- J. P. M. Almeida, A. Y. Lin, E. R. Figueroa, A. E. Foster and R. A. Drezek, Small, 2015, 11, 1453–1459 CrossRef CAS.
- W. Tao, K. S. Ziemer and H. S. Gill, Nanomedicine, 2014, 9, 237–251 CrossRef CAS.
- R. Kumar, P. C. Ray, D. Datta, G. P. Bansal, E. Angov and N. Kumar, Vaccine, 2015, 33, 5064–5071 CrossRef CAS.
- C. Wang, W. Zhu, Y. Luo and B.-Z. Wang, Nanotechnol. Biol. Med., 2018, 14, 1349–1360 CrossRef CAS.
- A. Y. Lin, J. Lunsford, A. S. Bear, J. K. Young, P. Eckels, L. Luo, A. E. Foster and R. A. Drezek, Nanoscale Res. Lett., 2013, 8, 72 CrossRef.
- N. G. Bastús, E. Sánchez-Tilló, S. Pujals, C. Farrera, C. López, E. Giralt, A. Celada, J. Lloberas and V. Puntes, ACS Nano, 2009, 3, 1335–1344 CrossRef.
- S. S. Bawage, P. M. Tiwari, A. Singh, S. Dixit, S. R. Pillai, V. A. Dennis and S. R. Singh, Nanotechnol. Biol. Med., 2016, 12, 2299–2310 CrossRef CAS.
- K. T. Mody, A. Popat, D. Mahony, A. S. Cavallaro, C. Yu and N. Mitter, Nanoscale, 2013, 5, 5167–5179 RSC.
- T. Wang, H. Jiang, Q. Zhao, S. Wang, M. Zou and G. Cheng, Int. J. Pharm., 2012, 436, 351–358 CrossRef CAS.
- D. Mahony, A. S. Cavallaro, F. Stahr, T. J. Mahony, S. Z. Qiao and N. Mitter, Small, 2013, 9, 3138–3146 CrossRef CAS.
- Y. Yang, M. Jambhrunkar, P. L. Abbaraju, M. Yu, M. Zhang and C. Yu, Adv. Healthcare Mater., 6, 1700466, DOI:10.1002/adhm.201700466.
- S. Heidegger, D. Gl, A. Schmidt, S. Niedermayer, C. Argyo, S. Endres, T. Bein and C. Bourquin, Nanoscale, 2015, 8, 938–948 RSC.
- W. R. Braz, N. L. Rocha, E. H. de Faria, M. L. A. e. Silva, K. J. Ciuffi, D. C. Tavares, R. A. Furtado, L. A. Rocha and E. J. Nassar, Nanotechnology, 2016, 27, 385103, DOI:10.1088/0957-4484/27/38/385103.
- Y. C. Hacene, A. Singh and G. Van Den Mooter, Int. J. Pharm., 2016, 506, 138–147 CrossRef CAS.
- N. Han, Q. Zhao, L. Wan, Y. Wang, Y. Gao, P. Wang, Z. Wang, J. Zhang, T. Jiang and S. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 3342–3351 CrossRef CAS.
- B. G. Trewyn, S. Giri, I. I. Slowing and V. S. y. Lin, Chem. Commun., 2007, 3236–3245 RSC.
- H. Geng, Y. Zhao, J. Liu, Y. Cui, Y. Wang, Q. Zhao and S. Wang, Int. J. Pharm. Res., 2016, 1, 184–194 Search PubMed.
- S.-K. Min, S.-C. Lee, S.-D. Hong, C.-P. Chung, W. H. Park and B.-M. Min, Biomaterials, 2010, 31, 4725–4730 CrossRef CAS.
- S. J. Cameron, F. Hosseinian and W. G. Willmore, Int. J. Mol. Sci., 2018, 19, 2030, DOI:10.3390/ijms19072030.
- J. Tian, K. K. Y. Wong, C. M. Ho, C. N. Lok, W. Y. Yu, C. M. Che, J. F. Chiu and P. K. H. Tam, ChemMedChem, 2007, 2, 129–136 CrossRef CAS.
- K. H. L. Kwan, X. Liu, M. K. T. To, K. W. K. Yeung, C.-M. Ho and K. K. Y. Wong, Nanotechnol. Biol. Med., 2011, 7, 497–504 CrossRef CAS.
- A. P. Khandhar, H. Liang, A. C. Simpson, S. G. Reed, D. Carter, C. B. Fox and M. T. Orr, Nanoscale, 2020, 12, 2515–2523 RSC.
- M. T. Orr, A. P. Khandhar, E. Seydoux, H. Liang, E. Gage, T. Mikasa, E. L. Beebe, N. D. Rintala, K. H. Persson, A. Ahniyaz, D. Carter, S. G. Reed and C. B. Fox, npj Vaccines, 2019, 4, 1 CrossRef CAS.
- L. Gu, L. E. Ruff, Z. Qin, M. Corr, S. M. Hedrick and M. J. Sailor, Adv. Mater., 2012, 24, 3981–3987 CrossRef CAS.
- U. Ikoba, H. Peng, H. Li, C. Miller, C. Yu and Q. Wang, Nanoscale, 2015, 7, 4291–4305 RSC.
- J. L. Elechiguerra, J. L. Burt, J. R. Morones, A. Camacho-Bragado, X. Gao, H. H. Lara and M. J. Yacaman, J. Nanobiotechnol., 2005, 3, 6, DOI:10.1186/1477-3155-3-6.
- H. P. Jia, D. C. Look, L. Shi, M. Hickey, L. Pewe, J. Netland, M. Farzan, C. Wohlford-Lenane, S. Perlman and B. McGray, J. Virol., 2005, 23, 14614–14621 CrossRef.
- C. Devaux, J.-M. Rolain, P. Colson and D. Raoult, Int. J. Antimicrob. Agents, 2020, 105938, DOI:10.1016/j.ijantimicag.2020.105938.
- M. A. A. Al-Bari, Pharmacol. Res. Perspect., 2017, 5(1), e00293, DOI:10.1002/prp2.293.
- M. J. R. Virlan, D. Miricescu, R. Radulescu, C. M. Sabliov, A. Totan, B. Calenic and M. Greabu, Molecules, 2016, 21, 207, DOI:10.3390/molecules21020207.
- R. Kumar and S. Lal, J. Nanomater. Mol. Nanotechnol., 2014, 3(4), 1000150, DOI:10.4172/2324-8777.1000150.
- M. A. Dobrovolskaia and S. E. McNeil, Nat. Nanotechnol., 2007, 2, 469–478 CrossRef CAS.
- N. Kamaly, Z. Xiao, P. M. Valencia, A. F. Radovic-Morenob and O. C. Farokhzad, Chem. Soc. Rev., 2012, 41, 2971–3010 RSC.
- H. K. S. Yadav, A. A. Almokdad, S. I. M. Shaluf and M. S. Debe in Nanocarriers for Drug Delivery: Polymer-Based Nanomaterials for Drug-Delivery Carriers, ed. S. S. Mohapatra, S. Ranjan, N. Dasgupta, R. K. Mishra and S. Thomas, Elsevier Inc., 2019, pp. 531–556 Search PubMed.
- D. Dupeyrón, M. Kawakami, A. M. Ferreira, P. R. Cáceres-Vélez, J. Rieumont, R. B. Azevedo and J. C. T. Carvalho, Int. J. Nanomed., 2013, 8, 467–3477 Search PubMed.
- K. Tatiparti, S. Sau, S. K. Kashaw and A. K. Iyer, Nanomaterials, 2017, 7, 77, DOI:10.3390/nano7040077.
- H. Laroui, E. Viennois, B. Xiao, B. S. B. Canup, D. Geem, T. L. Denning and D. Merlin, J. Controlled Release, 2014, 186, 41–53 CrossRef CAS.
- N. Parameswaran and S. Patial, Crit. Rev. Eukaryotic Gene Expression, 2010, 20(2), 87–103 CrossRef CAS.
- A. M. Aldayel, H. L. O'Mary, S. A. Valdes, X. Li, S. G. Thakkar, B. E. Mustafa and Z. Cui, J. Controlled Release, 2018, 283, 280–289 CrossRef CAS.
- M. A. A. Jansen, L. H. Klausen, K. Thanki, J. Lyngsø, J. S. Pedersen, H. Franzyk, H. M. Nielsen, W. van Eden, M. Dong, F. Broere, C. Foged and X. Zeng, Eur. J. Pharm. Biopharm., 2019, 142, 38–48 CrossRef CAS.
- S. J. Lee, A. Lee, S. R. Hwang, J. Park, J. Jang, M. S. Huh, D. Jo, S. Yoon, Y. Byun, S. H. Kim, I. C. Kwon, I. Youn and K. Kim, Mol. Ther., 2014, 22(2), 397–408 CrossRef CAS.
- P. Liu, L. Gu, L. Ren, J. Chen, T. Li, X. Wang, J. Yang, C. Chen and L. Sun, Am. J. Transl. Res., 2019, 11(11), 6775–6789 CAS.
- M. E. El-Naggar, F. Al-Joufi, M. Anwar, M. F. Attia and M. A. El-Bana, Colloids Surf., B, 2019, 177, 389–398 CrossRef CAS.
- M. Elsabahy and K. L. Wooley, Chem. Soc. Rev., 2012, 41, 2545–2561 RSC.
- M. Üner and G. Yener, Int. J. Nanomed., 2007, 2(3), 289–300 Search PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS.
- H. S. Sakhalkar, M. K. Dalal, A. K. Salem, R. Ansari, J. Fu, M. F. Kiani, D. T. Kurjiaka, J. Hanes, K. M. Shakesheff and D. J. Goetz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(26), 15895–15900 CrossRef CAS.
- K. Ghosh, M. Kanapathipillai, N. Korin, J. R. McCarthy and D. E. Ingber, Nano Lett., 2012, 12, 203–208 CrossRef CAS.
- H. Läubli and A. Varki, Cell. Mol. Life Sci., 2020, 77, 593–605 CrossRef.
- S. Spence, M. K. Greene, F. Fay, E. Hams, S. P. Saunders, U. Hamid, M. Fitzgerald, J. Beck, B. K. Bains, P. Smyth, E. Themistou, D. M. Small, D. Schmid, C. M. O'Kane, D. C. Fitzgerald, S. M. Abdelghany, J. A. Johnston, P. G. Fallon, J. F. Burrows, D. F. McAuley, A. Kissenpfennig and C. J. Scott, Sci. Transl. Med., 2015, 7, 303, DOI:10.1126/scitranslmed.aab3459.
- S. Thamphiwatana, P. Angsantikul, T. Escajadillo, Q. Zhang, J. Olson, B. T. Luk, S. Zhang, R. H. Fang, W. Gao, V. Nizet and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(43), 11488–11493 CrossRef CAS.
- Q. Zhang, D. Dehaini, Y. Zhang, J. Zhou, X. Chen, L. Zhang, R. H. Fang, W. Gao and L. Zhang, Nat. Nanotechnol., 2018, 13, 1182–1190 CrossRef CAS.
- C. Li, H. Li, Q. Wang, M. Zhou, M. Li, T. Gong, Z. Zhang and X. Sun, J. Controlled Release, 2017, 246, 133–141 CrossRef CAS.
- M. Tang, P. Hu, Q. Zheng, N. Tirelli, X. Yang, Z. Wang and Y. Wang, J. Nanobiotechnol., 2017, 1–11 Search PubMed.
- V. F. Fiore, M. C. Lofton, S. Roser-Page, S. C. Yang, J. Roman, N. Murthy and T. H. Barker, Biomaterials, 2010, 31, 810–817 CrossRef CAS.
- C. Springer, S. Benita, Y. Sherman, N. Gursoy and D. Gilhar, Pediatr. Res., 2005, 58(3), 537–541 CrossRef CAS.
- D. Yoo, K. Guk, H. Kim, G. Khang, D. Wu and D. Lee, Int. J. Pharm., 2013, 450, 87–94 CrossRef CAS.
- D. J. Hanlon, F. A. Sharp, D. Khalil, E. Robinson, R. Tigelaar and T. M. Fahmy, 2014, 5231–5246.
- J. D. Mintern, C. Percival, M. M. J. Kamphuis, W. J. Chin, F. Caruso and A. P. R. Johnston, Adv. Healthcare Mater., 2013, 2, 940–944 CrossRef CAS.
- T. Akagi, X. Wang, T. Uto, M. Baba and M. Akashi, Biomaterials, 2007, 28, 3427–3436 CrossRef CAS.
- P. Li, Z. Luo, P. Liu, N. Gao, Y. Zhang, H. Pan, L. Liu, C. Wang, L. Cai and Y. Ma, J. Controlled Release, 2013, 168, 271–279 CrossRef CAS.
- M. Diwan, P. Elamanchili, M. Cao and J. Samuel, Curr. Drug Delivery, 2004, 1(4), 405–412 CrossRef CAS.
- P. J. Tacken, I. S. Zeelenberg, L. J. Cruz, M. A. van Hout-Kuijer, G. van de Glind, R. G. Fokkink, A. J. Lambeck and C. G. Figdor, Blood, 2011, 118(26), 6836–6844 CrossRef CAS.
- N. P. F. Saade, Y. Honda-Okubo and S. Trec, Vaccine, 2013, 31(15), 1999–2007 CrossRef.
- S. Kumar, A. C. Anselmo, A. Banerjee, M. Zakrewsky and S. Mitragotri, J. Controlled Release, 2015, 220, 141–148 CrossRef CAS.
- A. D. Bangham, M. M. Standish and J. C. Watkins, J. Mol. Biol., 1965, 13, 238–252 CrossRef CAS.
- Z. Chu, S. Zhang, B. Zhang, C. Zhang, C. Fang, I. Rehor, P. Cigler, H. Chang, G. Lin, R. Liu and Q. Li, Sci. Rep., 2014, 4, 4495, DOI:10.1038/srep04495.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12, DOI:10.3390/pharmaceutics9020012.
- B. Ozbakir, B. J. Crielaard, J. M. Metselaar, G. Storm and T. Lammers, J. Controlled Release, 2014, 190, 624–636 CrossRef CAS.
- C. Data, Ann. Rheum. Dis., 2009, 68, 12, DOI:10.1136/ard.2009.108985.
- S. Angmo, S. Rana, K. Yadav, R. Sandhir and N. K. Singhal, Sci. Rep., 2018, 8, 17684, DOI:10.1038/s41598-018-35992-2.
- M. Ponzoni and F. Pastorino, Int. J. Mol. Sci., 2018, 19, 1953, DOI:10.3390/ijms19071953.
- T. M. Drake, L. B. Clarke and S. Khosla, Mayo Clin. Proc., 2008, 83, 1032–1045 CrossRef.
- T. Mert, M. Sahin, E. Sahin and S. Yaman, Inflammopharmacology, 2019, 27, 603–612 CrossRef CAS.
- N. M. La-Beck, X. Liu and L. M. Wood, Front. Pharmacol., 2019, 10, 220, DOI:10.3389/fphar.2019.00220.
- R. Nisini, N. Poerio, S. Mariotti, F. De Santis and M. Fraziano, Front. Immunol., 2018, 9, 155, DOI:10.3389/fimmu.2018.00155.
- R. M. Schiffelers, M. Banciu, J. M. Metselaar, G. Storm, R. M. Schiffelers, M. Banciu and J. M. Metselaar, J. Liposome Res., 2006, 16(3), 185–194 CrossRef CAS.
- A. Gauthier, A. Fisch, K. Seuwen, B. Baumgarten, H. Ruffner, A. Aebi, M. Rausch, F. Kiessling, M. Bartneck, R. Weiskirchen, F. Tacke, G. Storm, T. Lammers and M. Ludwig, Biomaterials, 2018, 178, 481–495 CrossRef CAS.
- J. M. Metselaar, W. B. van den Berg, A. Wagenaar-hilbers, O. C. Boerman and G. Storm, Arthritis Rheum., 2003, 48(7), 2059–2066 CrossRef CAS.
- J. M. Metselaar, W. B. van den Berg, A. E. Holthuysen, M. H. Wauben, G. Storm and P. L. van Lent, Ann. Rheum. Dis., 2004, 63(4), 348–353 CrossRef CAS.
- M. Bartneck, K. M. Scheyda, K. T. Warzecha, L. Y. Rizzo, K. Hittatiya, T. Luedde, G. Storm, C. Trautwein, T. Lammers and F. Tacke, Biomaterials, 2015, 37, 367–382 CrossRef CAS.
- Q. Wang and J. Fang, J. Mater. Chem. B, 2020, 8(9), 1773–1982 RSC.
- J. Szebeni and G. Storm, Biochem. Biophys. Res. Commun., 2015, 468, 490–497 CrossRef CAS.
- E. Wenande and L. H. Garvey, Clin. Exp. Allergy, 2016, 907–922 CrossRef CAS.
- J. J. F. Verhoef and T. J. Anchordoquy, Drug Delivery Transl. Res., 2013, 3(6), 499–503 CrossRef CAS.
- P. I. Homem de Bittencourt Jr, D. J. Lagranha, A. Maslinkiewicz, S. M. Senna, A. M. V. Tavares, L. P. Baldissera, D. R. Janner, J. S. Peralta, P. M. Bock, L. L. P. Gutierrez, G. Scola, T. G. Heck, M. S. Krause, L. A. Cruz, D. S. P. Abdalla, C. J. Lagranha, T. Lima and R. Curi, Atherosclerosis, 2007, 193, 245–258 CrossRef.
- O. A. Marcos-contreras, J. S. Brenner, R. Y. Kiseleva, V. Zuluaga-ramirez, C. F. Greineder, C. H. Villa, E. D. Hood, J. W. Myerson, S. Muro, Y. Persidsky and V. R. Muzykantov, J. Control Release, 2019, 301, 54–61 CrossRef CAS.
- S. Poh, V. Chelvam, W. Ayala-lópez, K. S. Putt and P. S. Low, Nanomedicine, 2018, 14(3), 1033–1043 CrossRef CAS.
- A. Verma, A. Jain, A. Tiwari, S. Saraf and P. K. Panda, Pharm. Res., 2019, 36, 123, DOI:10.1007/s11095-019-2653-0.
- R. Molinaro, C. Corbo, J. O. Martinez, F. Taraballi, M. Evangelopoulos, S. Minardi and I. K. Yazdi, Nat. Mater., 2016, 15, 1037–1047 CrossRef CAS.
- M. Henriksen-lacey, K. S. Korsholm, P. Andersen, Y. Perrie and D. Christensen, Expert Opin. Drug Delivery, 2011, 8(4), 505–519 CrossRef CAS.
- C. Foged, C. Arigita, A. Sundblad, W. Jiskoot, G. Storm and S. Frokjaer, Vaccine, 2004, 22, 1903–1913 CrossRef CAS.
- M. Henriksen-lacey, D. Christensen, V. W. Bramwell, T. Lindenstrøm, E. Marie, P. Andersen and Y. Perrie, J. Controlled Release, 2010, 145, 102–108 CrossRef CAS.
- T. Nakanishi, J. Kunisawa, A. Hayashi, Y. Tsutsumi, K. Kubo, S. Nakagawa, H. Fujiwara, T. Hamaoka and T. Mayumi, Biochem. Biophys. Res. Commun., 1997, 797, 793–797 CrossRef.
- R. Kaur, V. W. Bramwell, D. J. Kirby and Y. Perrie, J. Controlled Release, 2012, 164, 331–337 CrossRef CAS.
- J. H. van den Berg, K. Oosterhuis, W. E. Hennink, G. Storm, L. J. van der Aa, J. F. J. Engbersen, J. B. A. G. Haanen, J. H. Beijnen, T. N. Schumacher and B. Nuijen, J. Controlled Release, 2010, 141, 234–240 CrossRef CAS.
- S. T. Schmidt, C. Foged, K. S. Korsholm, T. Rades and D. Christensen, Pharmaceutics, 2016, 8, 7, DOI:10.3390/pharmaceutics8010007.
- A. Ramakrishnan, N. M. Schumack, C. L. Gariepy, H. Eggleston, G. Nunez, N. Espinoza, M. Nieto, R. Castillo, R. J. Rojas, A. J. McCoy, Z. Beck, G. R. Matyas, C. R. Alving, P. Guerry, F. Poly and R. M. Laird, mSphere, 2019, 4(3), e00101 CAS.
- Z. Li, L. Zhang, W. Sun, Q. Ding, Y. Hou and Y. Xu, Vaccine, 2011, 29, 5260–5266 CrossRef CAS.
- R. H. J. Khandare, M. Calderon and N. M. Dagia, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- R. K. Kesrevani and A. K. Sharma, in Nanoarchitectonics for Smart Delivery and Drug Targeting: Present Status and Future Prospects in Drug Delivery, ed A. M. Holban and A. M. Grumezesku, William Andrew, Oxford, Uk, 2016 Search PubMed.
- S. Fruchon and R. Poupot, Nanomaterials, 2017, 7, 251, DOI:10.3390/nano7090251.
- Y. Cheng, J. Wang, T. Rao, X. He and T. Xu, Front. Biosci., 2008, 13, 1447–1471 CrossRef CAS.
- A. Janaszewska, J. Lazniewska, P. Trzepiński, M. Marcinkowska and B. Klajnert-Maculewicz, Biomolecules, 2019, 9, 330 CrossRef CAS.
- S. Biswas and V. P. Torchilin, Pharmaceuticals, 2013, 6, 161–183 CrossRef CAS.
- H. Yang, Nanomedicine, 2016, 12, 309–316 CrossRef CAS.
- C. Yiyun and X. Tongwen, Eur. J. Med. Chem., 2005, 40(12), 1384–1389 CrossRef.
- A. S. Chauhan, P. V. Diwan, N. K. Jain and D. A. Tomalia, Biomacromolecules, 2009, 10, 1195–1202 CrossRef CAS.
- S. Shaunak, S. Thomas, E. Gianasi, A. Godwin, E. Jones, I. Teo, K. Mireskandari, P. Luthert, R. Duncan, S. Patterson, P. Khaw and S. Brocchini, Nat. Biotechnol., 2004, 22, 977–984 CrossRef CAS.
- E. R. Gillies, J. M. J. Fréchet and F. C. Szoka, Mol. Pharm., 2005, 2, 129–138 CrossRef CAS.
- I. Durocher and D. Girard, Inflammation Res., 2016, 65, 745–755 CrossRef CAS.
- L. Griffe, M. Poupot, P. Marchand, A. Maraval, C. Turrin, O. Rolland, P. Møtivier, G. Bacquet, J. Fourniø, A. Caminade, R. Poupot and J. Majoral, Angew. Chem., Int. Ed., 2007, 46, 2523–2526 CrossRef CAS.
- S. Fruchon and R. Poupot, Molecules, 2018, 23(6), 1272, DOI:10.3390/molecules23061272.
- L. Griffe, M. Poupot, P. Marchand, A. Maraval, C. Turrin, O. Rolland, P. Møtivier, G. Bacquet, J. Fourniø, A. Caminade, R. Poupot and J. Majoral, Angew. Chem., Int. Ed., 2007, 46, 2523–2526 CrossRef CAS.
- G. M. Hayder, M. Garzoni, D. Bochicchio, A. Caminade, F. Couderc, V. Ong-Meang, J. L. Davignon and C. Turrin, Biomacromolecules, 2018, 19, 712–720 CrossRef.
- J. Dernedde, A. Rausch, M. Weinhart, S. Enders, R. Tauber, K. Licha, M. Schirner, U. Zügel, A. Von Bonin and R. Haag, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19679–19684 CrossRef CAS.
- J. J. García-Vallejo, M. Ambrosini, A. Overbeek, W. E. van Riel, K. Bloem, W. W. J. Unger, F. Chiodo, J. G. Bolscher, K. Nazmi, H. Kalay and Y. van Kooyk, Mol. Immunol., 2013, 53, 387–397 CrossRef.
- F. S. Tabatabaei Mirakabad, M. S. Khoramgah, K. F. Keshavarz, M. Tabarzad and J. Ranjbari, Life Sci., 2019, 233, 116754 CrossRef CAS.
- C. Cubillos, G. Beatriz, A. Jakab, G. Clementi, E. Borrás, J. Bárcena, D. Andreu, F. Sobrino and E. Blanco, J. Virol., 2008, 82(14), 7223–7230 CrossRef CAS.
- D. Verma, N. Gulati, S. Kaul, S. Mukherjee and U. Nagaich, J. Pharm., 2018, 2018, 9285854, DOI:10.1155/2018/9285854.
- A. Sparreboom, C. D. Scripture, V. Trieu, P. J. Williams, T. De, A. Yang, B. Beals, W. D. Figg, M. Hawkins and N. Desai, Clin. Cancer Res., 2005, 11, 4136–4144 CrossRef CAS.
- G. Fanali, V. Trezza, M. Marino, M. Fasano and P. Ascenzi, Mol. Aspects Med., 2012, 33, 209–290 CrossRef CAS.
- F. Zhang, Y. Zhang, J. Da, Z. Jia, H. Wu and K. Gu, J. Cancer, 2020, 11, 414–420 CrossRef CAS.
- N. Desai, V. Trieu, Z. Yao, L. Louie, S. Ci, A. Yang, C. Tao, T. De, B. Beals, D. Dykes, P. Noker, R. Yao, E. Labao, M. Hawkins and P. Soon-shiong, Clin. Cancer Res., 2006, 12, 1317–1325 CrossRef CAS.
- L. Liu, F. Hu, X. Wu, A. S. Eltahan, S. Stephanie, N. Bottini, H. Xiao, M. Bottini, W. Guo and X. Liang, ACS Nano, 2019, 13, 5036–5048 CrossRef CAS.
- Y. Iwao, I. Tomiguchi, A. Domura, Y. Mantaira and A. Minami, Eur. J. Pharm. Biopharm., 2018, 125, 141–147 CrossRef CAS.
- Z. Wang, J. Li, J. Cho and A. B. Malik, Nat. Nanotechnol., 2014, 9, 204–210 CrossRef CAS.
- A. Verma, A. Jain, A. Tiwari, S. Saraf and P. K. Panda, ACS Biomater. Sci. Eng., 2018, 4, 3939–3961 CrossRef.
- W. Lohcharoenkal, L. Wang, Y. C. Chen and Y. Rojanasakul, BioMed Res. Int., 2014, 2014, 180549, DOI:10.1155/2014/180549.
- A. O. Elzoghby, J. Controlled Release, 2013, 172, 1075–1091 CrossRef CAS.
- M. Santoro, A. M. Tatara and A. G. Mikos, J. Controlled Release, 2014, 190, 210–218 CrossRef CAS.
- H. Wang, Q. Zou, O. C. Boerman, A. W. G. Nijhuis, J. A. Jansen, Y. Li and S. C. G. Leeuwenburgh, J. Controlled Release, 2013, 166, 172–181 CrossRef CAS.
- M. Bartneck, K. T. Warzecha and F. Tacke, Hepatobiliary Surg Nutr, 2014, 3, 364–376 Search PubMed.
- D. Narayanan, M. G. Geena, H. Lakshmi, M. Koyakutty, S. Nair and D. Menon, Nanomedicine, 2013, 9, 818–828 CrossRef CAS.
- H. Y. Huang, M. C. Wang, Z. Y. Chen, W. Y. Chiu, K. H. Chen, I. C. Lin, W. C. V. Yang, C. C. Wu and C. L. Tseng, Int. J. Nanomed., 2018, 13, 7251–7273 CrossRef CAS.
- M. Javadi and S. Feizi, J. Ophthalmic Vision Res., 2011, 6, 192–198 Search PubMed.
- L. Lu, S. Qi, Y. Chen, H. Luo, S. Huang, X. Yu, Q. Luo and Z. Zhang, Biomaterials, 2020, 245, 119987, DOI:10.1016/j.biomaterials.2020.119987.
- A. Yeboah, R. I. Cohen, R. Faulknor, R. Schloss, M. L. Yarmush and F. Berthiaume, J. Controlled Release, 2016, 232, 238–247 CrossRef CAS.
- Y. Gao, C. Wijewardhana and J. F. S. Mann, Front. Immunol., 2018, 9, 1–18 CrossRef.
- J. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 521–535 CrossRef CAS.
- Y. Seow and M. J. Wood, Mol. Ther., 2009, 17, 767–777 CrossRef CAS.
- M. A. Buabeid, E. S. A. Arafa and G. Murtaza, J. Immunol. Res., 2020, 2020, 9624532, DOI:10.1155/2020/9624532.
- S. Farkona, E. P. Diamandis and I. M. Blasutig, BMC Med., 2016, 14, 73, DOI:10.1186/s12916-016-0623-5.
- X. Zhang, C. Wang, J. Wang, Q. Hu, B. Langworthy, Y. Ye, W. Sun, J. Lin, T. Wang, J. Fine, H. Cheng, G. Dotti, P. Huang and Z. Gu, Adv. Mater., 2018, 30, 1707112, DOI:10.1002/adma.201707112.
- Y. Ohue and H. Nishikawa, Cancer Sci., 2019, 110, 2080–2089 CrossRef CAS.
- C. B. Rodell, S. P. Arlauckas, M. F. Cuccarese, C. S. Garris, R. Li, M. S. Ahmed, R. H. Kohler, M. J. Pittet and R. Weissleder, Nat. Biomed. Eng., 2018, 2, 578–588 CrossRef CAS.
- W. Ou, R. Kumar, L. Jiang, Z. Chi, M. Gautam and J. Chang, J. Controlled Release, 2018, 281, 84–96 CrossRef CAS.
- H. Zhang, M. T. Silvestrini, J. S. Lewis and K. W. Ferrara, Bioconjugate Chem., 2018, 28, 2756–2771 Search PubMed.
- Y. W. Noh, S. Y. Kim, J. E. Kim, S. Kim, J. Ryu, I. Kim, E. Lee, S. H. Um and Y. T. Lim, Adv. Funct. Mater., 2017, 27, 1605398, DOI:10.1002/adfm.201605398.
- N. I. Ho, L. G. M. Huis, T. K. Raaijmakers and G. J. Adema, Front. Immunol., 2018, 9, 2874, DOI:10.3389/fimmu.2018.02874.
- R. A. Rosalia, L. J. Cruz, S. Van Duikeren, A. T. Tromp, J. Oostendorp, A. L. Silva, W. Jiskoot, T. De Gruijl, L. Clemens, S. H. Van Der Burg and F. Ossendorp, Biomaterials, 2015, 40, 88–97 CrossRef CAS.
- H. Kakwere, E. S. Ingham, R. Allen, L. M. Mahakian, S. M. Tam, H. Zhang, M. T. Silvestrini, J. S. Lewis and K. W. Ferrara, Bioconjugate Chem., 2017, 28, 2756–2771 CrossRef CAS.
- T. W. Dubensky, D. B. Kanne and M. L. Leong, Ther. Adv., 2013, 1, 131–143 Search PubMed.
- M. C. Hanson, M. P. Crespo, W. Abraham, K. D. Moynihan, G. L. Szeto, S. H. Chen, M. B. Melo, S. Mueller and D. J. Irvine, J. Clin. Invest., 2015, 125, 2532–2546 CrossRef.
- D. J. Irvine and E. L. Dane, Nat. Rev. Immunol., 2020, 20, 321–334 CrossRef CAS.
- D. V Krysko, A. D. Garg, A. Kaczmarek and O. Krysko, Nat. Rev. Cancer, 2012, 12, 860–875 CrossRef.
- A. S. Bear, L. C. Kennedy, J. K. Young, S. K. Perna, J. P. Mattos, A. Y. Lin, P. C. Eckels, R. A. Drezek and A. E. Foster, PLoS One, 2013, 8, 69073, DOI:10.1371/journal.pone.0069073.
- Y. Wen, X. Chen, X. Zhu, Y. Gong, G. Yuan, X. Qin and J. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 43393–43408 CrossRef CAS.
- Y. Ma, Y. Zhang, X. Li, Y. Zhao, M. Li, W. Jiang, X. Tang, J. Dou, L. Lu, F. Wang and Y. Wang, ACS Nano, 2019, 13, 11967–11980 CrossRef CAS.
- E. E. Sweeney, J. Cano-Mejia and R. Fernandes, Small, 2018, 14(20), 1800678, DOI:10.1002/smll.201800678.
- W. Li, J. Yang, L. Luo, M. Jiang, B. Qin, H. Yin, C. Zhu, X. Yuan, J. Zhang, Z. Luo, Y. Du, Q. Li, Y. Lou, Y. Qiu and J. You, Nat. Commun., 2019, 10, 3349, DOI:10.1038/s41467-019-11269-8.
- D. Liu, B. Chen, Y. Mo, Z. Wang, T. Qi, Q. Zhang and Y. Wang, Nano Lett., 2019, 19, 6964–6976 CrossRef CAS.
- L. Raes, C. Van Hecke, J. Michiels, S. Stremersch, J. C. Fraire, T. Brans, R. Xiong, S. De Smedt, L. Vandekerckhove, K. Raemdonck and K. Braeckmans, Crystals, 2019, 9, 411, DOI:10.3390/cryst9080411.
- L. Van Hoecke, L. Raes, S. Stremersch, T. Brans, J. C. Fraire, R. Roelandt, W. Declercq, P. Vandenabeele, K. Raemdonck, K. Braeckmans and X. Saelens, Int. J. Mol. Sci., 2019, 20, e4254, DOI:10.3390/ijms20174254.
- X. Duan, C. Chan, W. Han, N. Guo, R. R. Weichselbaum and W. Lin, Nat. Commun., 2019, 10, 1899, DOI:10.1038/s41467-019-09221-x.
- H. Naatz, B. B. Manshian, C. R. Luci, V. Tsikourkitoudi, Y. Deligiannakis, J. Birkenstock, S. Pokhrel, M. Lutz and S. J. Soenen, Angew. Chem., Int. Ed., 2020, 59, 1828–1836 CrossRef CAS.
- E. Ploetz, A. Zimpel, V. Cauda, D. Bauer, D. C. Lamb, C. Haisch, S. Zahler, A. M. Vollmar, S. Wuttke and H. Engelke, Adv. Mater., 2020, 1907267, DOI:10.1002/adma.201907267.
- L. W. Bishop, M. Wehbe, D. Shae, J. James, B. C. Hacker, K. Garland, P. P. Chistov, M. Rafat, J. M. Balko and J. T. Wilson, J. Imunotherapy Cancer, 2020, 8(1), e000282, DOI:10.1136/jitc-2019-000282.
- N. Cheng, R. Watkins-Schulz, R. D. Junkins, C. N. David, B. M. Johnson, S. A. Montgomery, K. J. Peine, D. B. Darr, H. Yuan, K. P. McKinnon, Q. Liu, L. Miao, L. Huang, E. M. Bachelder, K. M. Ainslie and J. P. Y. Ting, JCI Insight, 2018, 3(22), e120638, DOI:10.1172/jci.insight.120638.
- D. Shae, K. W. Becker, P. Christov, D. S. Yun, A. K. R. Lytton-Jean, S. Sevimli, M. Ascano, M. Kelley, D. B. Johnson, J. M. Balko and J. T. Wilson, Nat. Nanotechnol., 2019, 14, 269–278 CrossRef CAS.
- M. Hoang, H. Lee, H. Lee, S. Jung, N. Choi, M. Vo, H. Kim, I. Park and J. Lee, J. Immunol. Res., 2015, 2015, 706379, DOI:10.1155/2015/706379.
- T. R. Abreu, N. A. Fonseca, N. Gonçalves and J. N. Moreira, J. Controlled Release, 2020, 319, 246–261 CrossRef CAS.
- J. N. Kochenderfer, W. H. Wilson, J. E. Janik, M. E. Dudley, M. Stetler-Stevenson, S. A. Feldman, I. Maric, M. Raffeld, D. A. N. Nathan, B. J. Lanier, R. A. Morgan and S. A. Rosenberg, Blood, 2010, 116, 4099–4102 CrossRef CAS.
- K. Newick, E. Moon and S. M. Albelda, Mol. Ther.--Oncolytics, 2016, 3, 16006, DOI:10.1038/mto.2016.6.
- Y. Zheng, L. Tang, L. Mabardi, S. Kumari and D. J. Irvine, ACS Nano, 2017, 11, 3089–3100 CrossRef CAS.
- L. Tang, Y. Zheng, M. B. Melo, L. Mabardi, A. P. Castaño, Y. Xie, N. Li, S. B. Kudchodkar, H. C. Wong, E. K. Jeng, M. V Maus and D. J. Irvine, Nat. Biotechnol., 2018, 36(8), 707–716, DOI:10.1038/nbt.4181.
- C. P. Bailey, T. Budak-Alpdogan, C. T. Sauter, M. M. Panis, C. Buyukgoz, E. K. Jeng, H. C. Wong, N. Flomenberg and O. Alpdogan, Oncotarget, 2017, 8, 44366–44378 CrossRef.
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. Mcknight, W. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, Nat. Nanotechnol., 2017, 12, 813–822 CrossRef CAS.
- N. C. Gunawardana and S. R. Durham, Ann. Allergy, Asthma, Immunol., 2018, 121, 293–305 CrossRef CAS.
- H. Pohlit, I. Bellinghausen, H. Frey and J. Saloga, Allergy, 2017, 72, 1461–1474 CrossRef CAS.
- C. B. Smarr, W. Teck, T. P. Neef, R. M. Pearson, Z. N. Hunter, I. Ifergan and D. R. Getts, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5059–5064 CrossRef CAS.
- A. Kawakita, H. Shirasaki, M. Yasutomi, S. Tokuriki, M. Mayumi, H. Naiki and Y. Ohshima, Allergy, 2012, 67, 371–379 CrossRef CAS.
- E. Barreto, M. F. Serra, R. V. Dos Santos, C. E. A. Dos Santos, J. Hickmann, A. C. Cotias, C. R. Rodrigues Pão, S. G. Trindade, V. Schimidt, C. Giacomelli, V. F. Carvalho, P. M. Rodrigues E Silva, R. S. Balão Cordeiro and M. A. Martins, J. Biomed. Nanotechnol., 2015, 11, 1038–1050 CrossRef CAS.
- H. C. Probst, S. Muth and H. Schild, Eur. J. Immunol., 2014, 44, 927–933 CrossRef CAS.
- P. Serra and P. Santamaria, Clin. Immunol., 2015, 160(1), 3–13 CrossRef CAS.
- D. R. Getts, A. J. Martin, D. P. Mccarthy, R. L. Terry, Z. N. Hunter, W. T. Yap, M. T. Getts, M. Pleiss, X. Luo, N. J. C. King, L. D. Shea and S. D. Miller, Nat. Biotechnol., 2012, 30, 1217–1224 CrossRef CAS.
- J. Park J, W. Gao W, R. Whiston R, T. B. Strom TB, S. Metcalfe S and T. M. Fahmy, Mol. Pharm., 2011, 8(1), 143–152 CrossRef.
- X. Clemente-Casares, J. Blanco, P. Ambalavanan, J. Yamanouchi, S. Singha, C. Fandos, S. Tsai, J. Wang, N. Garabatos, C. Izquierdo, S. Agrawal, M. B. Keough, V. W. Yong, E. James, A. Moore, Y. Yang, T. Stratmann, P. Serra and P. Santamaria, Nature, 2016, 530, 434–440 CrossRef CAS.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2016, 1, 10–29 CrossRef.
- Nanobiotix, https://www.nanobiotix.com/wp-content/uploads/2019/06/PR_Nanobiotix_marquage_CE_04042019_VF-1.pdf, accessed May 28, 2020.
- Nanobiotix, https://www.nanobiotix.com/wp-content/uploads/2019/06/PR_Nanobiotix_AACR_Results_Announcement.pdf, accessed May 28, 2020.
- Globes news wire, https://www.globenewswire.com/news-release/2018/06/11/1519433/0/en/Vical-Reports-Phase-2-Trial-of-HSV-2-Therapeutic-Vaccine-Did-Not-Meet-Primary-Endpoint.html, accessed May 28, 2020.
- O. Duramad, A. Laysang, J. Li, Y. Ishii and R. Namikawa, Biol. Blood Marrow Transplant., 2011, 17, 1154–1168 CrossRef CAS.
- Y.-B. Chen, Y. A. Efebera, L. Johnston, E. D. Ball, D. Avigan, L. J. Lekakis, C. R. Bachier, P. Martin, O. Duramad, Y. Ishii, S. Han, Y.-J. Jung, D. Lee, L. Kunkel, R. S. Negrin and J. D. Bui, Biol. Blood Marrow Transplant., 2017, 23, 625–634 CrossRef CAS.
- M. Ahmadi, M. Hajialilo, S. Dolati, S. Eghbal-Fard, H. Heydarlou, M. Ghaebi, A. Ghassembaglou, L. Aghebati-Maleki, H. S. Kafil, A. Kamrani, B. Rahnama, R. Rikhtegar and M. Yousefi, J. Cell. Biochem., 2019, 121, 103–110 CrossRef.
- Z. A. Nahleh, W. E. Barlow, D. F. Hayes, A. F. Schott, J. R. Gralow, W. M. Sikov, E. A. Perez, S. Chennuru, H. R. Mirshahidi, S. W. Corso, D. L. Lew, L. Pusztai, R. B. Livingston and G. N. Hortobagyi, Breast Cancer Res. Treat., 2016, 158, 485–495 CrossRef CAS.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014, DOI:10.1038/natrevmats.2016.14.
- U. Bozuyuk, N. O. Dogan and S. Kizilel, ACS Appl. Mater. Interfaces, 2018, 10, 33945–33955 CrossRef CAS.
- G. Fullstone, J. Wood, M. Holcombe and G. Battaglia, Sci. Rep., 2015, 5, 10649, DOI:10.1038/srep10649.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |