
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInducing endoplasmic reticulum stress in cancer cells using graphene oxide-based nanoparticles†
Shalini
Pandey
a,
Aditi
Nandi
a,
Sudipta
Basu
 *b and
Nirmalya
Ballav
*a
*b and
Nirmalya
Ballav
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Pune, Dr Homi Bhabha Road, Pashan, Pune, Maharashtra 411008, India. E-mail: nballav@iiserpune.ac.in
bDiscipline of Chemistry, Indian Institute of Technology (IIT) Gandhinagar, Palaj, Gandhinagar, Gujarat 382355, India. E-mail: sudipta.basu@iitgn.ac.in
First published on 19th August 2020
Abstract
The endoplasmic reticulum is one of the vital organelles primarily involved in protein synthesis, folding, and transport and lipid biosynthesis. However, in cancer cells its functions are dysregulated leading to ER stress. ER stress is now found to be closely associated with hallmarks of cancer and has subsequently emerged as an alluring target in cancer therapy. However, specific targeting of the ER in a cancer cell milieu remains a challenge. To address this, in this report we have engineered ER-targeted self-assembled 3D spherical graphene oxide nanoparticles (ER-GO-NPs) encompassing dual ER stress inducers, doxorubicin and cisplatin. DLS, FESEM and AFM techniques revealed that the nanoparticles were spherical in shape with a sub 200 nm diameter. Confocal microscopy confirmed the specific homing of these ER-GO-NPs into the subcellular ER within 3 h. A combination of gel electrophoresis, confocal microscopy and flow cytometry studies revealed that these ER-GO-NPs induced ER stress mediated apoptosis in HeLa cells. Interestingly, the nanoparticles also activated autophagy which was inhibited through the cocktail treatment with ER-GO-NPs and chloroquine (CQ). At the same time these ER-GO-NPs were found to be efficient in prompting ER stress associated apoptosis in breast, lung and drug resistant triple negative breast cancer cell lines as well. We envision that these ER specific self-assembled graphene oxide nanoparticles can serve as a platform to exploit ER stress and its associated unfolded protein response (UPR) as a target resulting in promising therapeutic outcomes in cancer therapy.
Introduction
Cancer cells endure both oncogenic and environmental stresses while they relentlessly proliferate. These stresses act as potential growth inhibiting factors for tumorigenesis.1–3 To counter them, cancer cells exploit an innate adaptive mechanism of the unfolded protein response (UPR) launched by the endoplasmic reticulum (ER) of cells.4,5 The ER is a major regulator of various metabolic processes including protein synthesis and folding.6,7 A high proliferation rate of cancer cells requires increased protein demands leading to ER stress, a condition characterized by accumulation of unfolded or misfolded proteins in the ER lumen.8,9 In response to such stresses, the cytoprotective UPR mechanism is triggered and cells work in a coordinated fashion to establish ER homeostasis.9–11The baseline activity level of the ER stress response system in normal and cancer cells differs.12 Cancer cells display chronically elevated ER stress levels hinting that they are already struggling to survive. The burden on the ER stress response system when aggravated would lose its pro-survival feature. On the other hand, its proapoptotic module is triggered.13 The persistent activity of chronic ER stress in tumor cells may constitute an Achilles heel and provide a window of opportunity for development of therapeutic regimens. Growing interest in the UPR as a therapeutic target in cancer has led to the development of several pharmacological agents that induce cancer cell death by impairing the UPR.14–16 Bortezomib, 17-AAG, and brefeldin-A are to name a few that have recently been added to the list of promising UPR inhibitors.17,18 Interestingly, several in-clinic chemotherapeutics such as doxorubicin and cisplatin have been explored to establish new targets in the ER. Doxorubicin has been shown to inhibit the IRE1-α arm of the UPR.19 On the other hand, cisplatin binds to proteins such as calreticulin and protein disulphide isomerase (PDI) residing in the ER and induces stress.20–23 In fact, various cancer cell lines have shown increased sensitivity to cisplatin when it is used in combination with other ER stress inducers.16 However, doxorubicin and cisplatin exert off-target cytotoxicity especially damaging topoisomerases and nuclear DNA respectively.24,25 Hence selectively delivering these stress inducers directly into the subcellular ER in cancer cells is a daunting task due to limited chemical tools present.
In recent years, nanoscale materials have been used to address this challenge. Lipidic and polymeric nanoparticles, small molecule based supramolecular self-assemblies have been developed to specifically navigate to the ER, induce stress and impair the UPR.26,27 Graphene oxide (GO) based nanoplatforms have also emerged as a promising candidate owing to the panoply of features they possess.28,29 Apart from being biocompatible and biodegradable, their unique aromatic π–π system and surface modalities present allow for stacking of drugs and conjugation of targeting moieties.30–33 Nonetheless, there is a serious lack of effective nanoscale tools for impairment of the adaptive UPR and induction of ER stress mediated apoptosis in cancer cells.
To address this, in this manuscript we report easy and robust engineering of self-assembled graphene oxide nanoparticles (ER-GO-NPs), decorated with dansyl groups (for ER localization), encompassing ER stress inducers (doxorubicin and cisplatin) (Fig. 1a). These ER-GO-NPs were spherical in shape with a sub-200 nm diameter confirmed by light scattering (DLS) and electron microscopy (SEM and AFM). These ER-GO-NPs specifically localized into the subcellular ER of HeLa cervical cancer cells within 3 h and released their payload causing UPR impairment as visualised by confocal laser scanning microscopy (CLSM) and gel electrophoresis (Fig. 1b). This nanoparticle-mediated ER stress led to the activation of autophagy which was inhibited by autophagy inhibitor chloroquine in a combination treatment.
 | ||
| Fig. 1 (a) Synthetic scheme for engineering ER targeted GO-nanoparticles (ER-GO-NPs). (b) Schematic representation of ER stress induction of ER-GO-NPs in cancer cells leading to autophagy and apoptosis. | ||
Experimental
Materials
Graphene oxide, dansyl chloride, cisplatin, Nunc® Lab-Tek® II chambered coverglass, sodium dodecyl sulfate (SDS) and silicon wafers for FESEM were purchased from Sigma Aldrich. Doxorubicin was bought from Selleck Chemicals. Analytical thin-layer chromatography (TLC) was performed using 60 F254 pre-coated silica gel aluminium sheets bought from EMD Millipore Laboratories. UV-Visible spectra was recorded on Shimadzu. DMEM and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from HiMedia. MitoTracker® Deep Red, ER Tracker Green and LysoTracker Green DND-26 were purchased from Invitrogen. An Annexin-V-FITC staining kit was purchased from Roche. Flow cytometry analysis was performed on a BD-Accuri. Western blot analysis was performed on a Las ImageQuant 400.Synthesis of N-(2-aminoethyl)-5-(dimethylamino) naphthalene-1-sulfonamide (2)
A dansyl–ethylene diamine conjugate (2) was prepared according to a previously reported procedure.26Synthesis of graphene-oxide-dansyl-doxorubicin-cisplatin nanoparticles (ER-GO-NPs)
ER-GO-NPs were synthesised according to a previously reported procedure.34 Briefly, graphene oxide (4 mg mL−1) was dispersed in distilled water (1 mL) and the dansyl–ethylene diamine conjugate (2) was added in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 weight ratio in the presence of EDC as a coupling agent to obtain a GO–Dan conjugate (3). Doxorubicin (1 mg) dissolved in distilled water (1 mL) was then added to this GO–Dan conjugate (3) at room temperature. The reaction mixture was then allowed to stir for 24 h. To remove unreacted doxorubicin, the reaction mixture was dialyzed against distilled water through a dialysis membrane (MWCO = 1000 Dalton) for 6 h. Aquated cisplatin (5 mg mL−1) was then added to the GO–Dan–Dox conjugate (4) and stirred at room temperature for 24 h. After the reaction was completed, the mixture was dialyzed again to remove excess aquated cisplatin to obtain ER-GO-NPs. The ER-GO-NPs were stored at 4 °C for further use.
:5 weight ratio in the presence of EDC as a coupling agent to obtain a GO–Dan conjugate (3). Doxorubicin (1 mg) dissolved in distilled water (1 mL) was then added to this GO–Dan conjugate (3) at room temperature. The reaction mixture was then allowed to stir for 24 h. To remove unreacted doxorubicin, the reaction mixture was dialyzed against distilled water through a dialysis membrane (MWCO = 1000 Dalton) for 6 h. Aquated cisplatin (5 mg mL−1) was then added to the GO–Dan–Dox conjugate (4) and stirred at room temperature for 24 h. After the reaction was completed, the mixture was dialyzed again to remove excess aquated cisplatin to obtain ER-GO-NPs. The ER-GO-NPs were stored at 4 °C for further use.
Determination of the shape, size and morphology
The shape, size and morphology of the ER-GO-NPs were determined using dynamic light scattering (DLS) and electron microscopy techniques such as field-emission scanning electron microscopy (FESEM) and atomic force microscopy (AFM). The samples were prepared according to previously reported procedures.34Quantification of the drug loading in the nanoparticles
The loading of doxorubicin and cisplatin in the ER-GO-NPs was determined at λmax= 488 nm and 706 nm by UV-Vis spectroscopy using a previously reported method.31 The drug loading efficiency was calculated using the formula:| Drug loading efficiency (%) = (amount of drug loaded in nanoparticles) × 100/(amount of drug used) |
In vitro studies
Confocal microscopy, cell viability assay, flow cytometry analysis and western blot analysis were performed by previously published methods.26,27,34Results and discussion
Engineering ER specific GO-based nanoparticles
To specifically navigate the GO-based NPs to the subcellular ER, we planned to conjugate the dansyl moiety to the GO surface via the ethylene diamine linker. We have chosen the dansyl moiety for ER targeting (a) as it has the necessary sulfonamide functionality to interact with the sulphonamide receptors present on the ER surface and (b) due to its fluorescent nature for sub-cellular ER trafficking of the nanoparticles in cancer cells.26,27,35–37 We have capitalized upon the combination of doxorubicin and cisplatin, clinically used to treat several malignancies, to inhibit the UPR in cancer cells. Moreover, fluorescent Dox will also help us to visualize the localization of the nanoparticles into the sub-cellular ER through fluorescence microscopy. The dansyl moiety was conjugated to the free carboxylic acid (–COOH) groups of 2D GO sheets (1) using the ethylene diamine linker via amide coupling by reacting GO and the dansyl ethylene diamine conjugate (2) in a 1:5 weight ratio in the presence of EDC as a coupling reagent in water for 24 h to obtain the GO–dansyl conjugate (GO–Dan) (3) (Fig. 1a). Doxorubicin was then stacked onto the two-dimensional GO–Dan (3) surface by aromatic π–π interactions by incubating Dox and GO–Dan in a 1:1 weight ratio in water for 24 h to obtain GO–Dan–Dox (4). The presence of the dansyl moiety and Dox over GO was confirmed by UV-Vis spectroscopy (Fig. S1a and b, ESI†). The characteristic peaks at λmax = 350 nm and λmax = 480 nm validated the presence of dansyl and doxorubicin respectively. Furthermore, stacking of Dox on GO–Dan–Dox (4) was confirmed by fluorescence microscopy. Drastic reduction in the fluorescence emission intensity of Dox stacked on GO compared to free Dox at λmax = 590 nm confirmed the incorporation of doxorubicin over the GO surface (Fig. S1c, ESI†). GO–Dan–Dox (4) was further reacted with [(OH2)2Pt(NH3)2]2+ in a 1:5 ratio in water for another 24 h followed by dialysis to afford the GO–Dan–Dox–CDDP conjugate (6). Conjugate 6 was found to hierarchically self-assemble into spherical nanoparticles (ER-GO-NPs) which was in complete agreement with our previous study.33,34 The hydrodynamic diameter of the ER-GO-NPs was found to be 145 nm by dynamic light scattering (DLS) (Fig. 2a). The spherical morphology of ER-GO-NPs was visualized by field emission-scanning electron microscopy (FESEM) and atomic force microscopy (AFM) (Fig. 2b and c). These shape and size are suitable for the accumulation of ER-GO-NPs into tumor tissues through the enhanced permeability and retention (EPR) effect.38,39 The presence of cisplatin in ER-GO-NPs was confirmed by elemental mapping of Pt using FESEM based energy dispersive X-ray spectroscopy (EDAX) (Fig. S2, ESI†). The characteristic D and G bands in the single particle Raman spectra confirmed the presence of GO in ER-GO-NPs (Fig. 2d). We finally evaluated the loading of doxorubicin and cisplatin in ER-GO-NPs using UV-Vis spectroscopy. It was estimated that the ER-GO-NPs contained 196 μg mL−1 and 100 μg mL−1 doxorubicin and cisplatin with 78% and 80% drug loading efficiency respectively (Fig. S3, ESI†).
 | ||
| Fig. 2 (a–c) DLS, FESEM and AFM images of ER-GO-NPs. (d) Single particle Raman spectra of ER-GO-NPs showing the presence of GO. | ||
ER homing
We hypothesized that the dansyl group will facilitate the internalization of the ER-GO-NPs into the sub-cellular ER due to the presence of sulphonamide receptors. To validate our hypothesis, we treated HeLa cervical cancer cells with ER-GO-NPs at different time points (3 h, 6 h, 12 h and 24 h) and co-stained them with ER-Tracker Green. We then visualised the live cells using confocal laser scanning microscopy (CLSM). The fluorescence microscopy images revealed that the ER-GO-NPs started internalizing in the ER within 3 h and the accumulation gradually increased with time (Fig. 3). Further quantification by CLSM using Pearson's and Mander's coefficients confirmed that the ER-GO-NPs localized into the ER with Pearson's coefficient of 0.72 and 0.86 and Mander's coefficients of 0.92 and 0.97 at 3 h and 6 h respectively (Table S1, ESI†). The confocal images of incubation at higher time points (12 h and 24 h) revealed that the ER-GO-NPs were retained in the ER with Pearson's coefficient of 0.73 and 0.86 and Mander's coefficients of 0.972 and 0.97 respectively.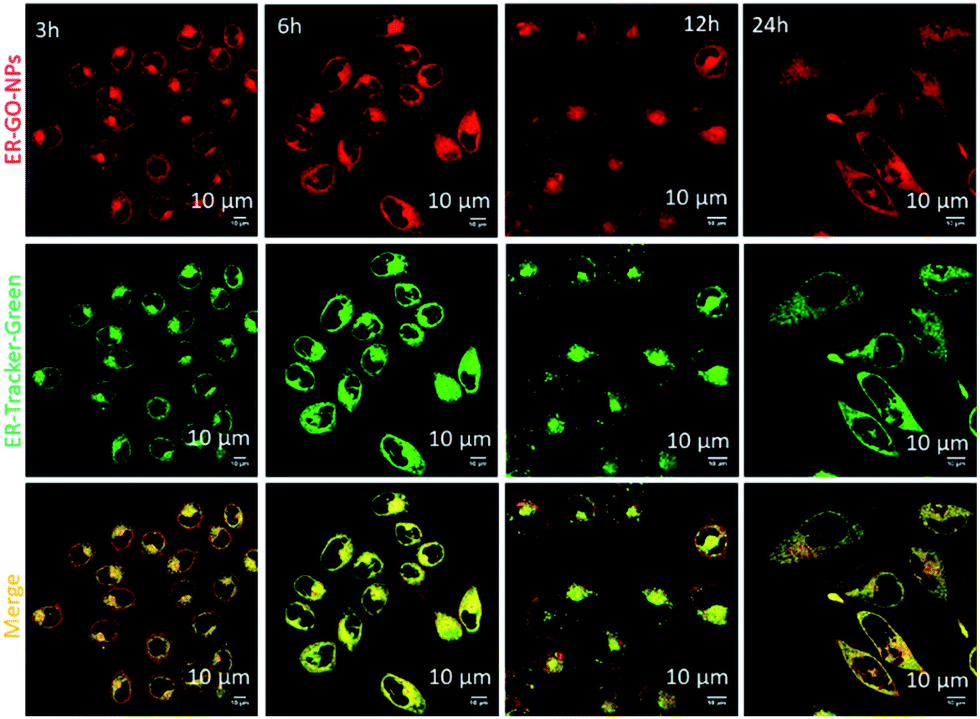 | ||
| Fig. 3 Confocal laser scanning microscopy images of HeLa cells treated with ER-GO-NPs (red fluorescent) at 3 h, 6 h, 12 h and 24 h. The cells were counter stained with ER-Tracker-Green dye. Scale bar = 10 μm. | ||
To evaluate the specific localization of ER-GO-NPs into the ER compared to the other organelles, we also visualized the localization of ER-GO-NPs in mitochondria. We treated the HeLa cells with ER-GO-NPs for 12 h and 24 h followed by staining mitochondria with MitoTracker Deep Red dye. The cells were then visualized by confocal microscopy. From the confocal images, we can clearly observe that even at 12 h and 24 h, very less yellow regions were visible by overlapping green and red fluorescence from ER-GO-NPs and MitoTracker Deep Red respectively (Fig. S4, ESI†). We also confirmed the lower accumulation of ER-GO-NPs in mitochondria at 12 h and 24 h from the lower value of Pearson's and Mander's coefficients (Table S2, ESI†).
Our previous experience showed that GO nanoparticles initially localize into lysosomes in cancer cells through clathrin mediated endocytosis.34,40 To evaluate the homing into lysosomal compartments, we treated HeLa cells with ER-GO-NPs at 1 h, 3 h and 6 h time points and co-stained the lysosomes with Lyso Tracker Green dye. The ER-GO-NPs showed inherent red fluorescence due to the presence of doxorubicin. We visualized the cells by confocal microscopy. We observed a considerable number of yellow signals at 3 h (Fig. S5, ESI†) which showed that the ER-GO-NPs localized into the lysosomes within 3 h. However, at 6 h, we hardly visualized any colocalization. This observation was also confirmed from the high value of Pearson's and Mander's coefficients at 3 h with very low values at both 1 h and 6 h time points (Table S3, ESI†). It was previously observed that dansyl-coated nanoparticles showed a positive surface charge in an acidic milieu (in lysosomes) which we expected to be the reason behind the quick escape of dansyl-coated ER-GO-NPs from lysosomes through electrostatic repulsion.26 Furthermore, from our earlier report, we found out that doxorubicin and cisplatin loaded self-assembled GO nanoparticles released less than 10% of their payload in an acidic medium of pH = 5.5 at 6 h.34 This observation clearly indicated that ER-GO-NPs would lose a minimum amount of ER stress inducers in lysosomes due to their quick escape from the lysosomes within 3 h. Hence, from these confocal microscopy studies, it was evident that ER-GO-NPs homed into the ER within 3 h and hardly localized into mitochondria. These ER-GO-NPs localized initially into lysosomes within 3 h, but slowly escaped them to home into the ER.
Induction of ER stress and DNA damage
Once ER-GO-NPs internalized into the ER of the HeLa cells, we anticipated that the carboxylesterases present in the ER41 will cleave the coordinate bond between Pt and the carboxylic acid of GO to release cisplatin followed by disintegration of the self-assembled ER-GO-NPs. This carboxylesterase mediated fragmentation of the nanoparticle will also release GO-stacked doxorubicin.34 This simultaneous release of cisplatin and doxorubicin would further induce ER stress impairing the UPR. The onset of ER stress leads to increased expression of CHOP, a marker for ER stress induced apoptosis.42,43 We visualized the increase in CHOP expression by immunofluorescence assay. We incubated HeLa cells with ER-GO-NPs for 24 h followed by treatment with CHOP specific primary antibody. The primary antibody was further detected using an Alexa Fluor 594 tagged secondary antibody (red fluorescent). The nuclei of the cells were stained with blue fluorescent DAPI and then visualized using confocal microscopy. The confocal images showed that the cells treated with ER-GO-NPs showed a significant increase in red fluorescence intensity as compared to the control (Fig. 4a). Confocal microscopy-based quantification showed that ER-GO-NP treatment increased the expression of CHOP by 3.8 fold compared to that in the control non-treated cells (Fig. S6, ESI†). Induction of ER stress leads to increase in the levels of CHOP as well as its accumulation in the nucleus. The overlap of red fluorescence signals with blue fluorescence signals of DAPI leading to purple fluorescence confirmed that CHOP accumulated in the nucleus upon ER stress induction by ER-GO-NPs, which was absent in the control cells. | ||
| Fig. 4 (a) Fluorescence confocal images of HeLa cells treated with ER-GO-NPs for 24 h followed by treatment with CHOP-primary antibody and Alexa Fluor 594-tagged secondary antibody (red). Nuclei were stained with DAPI (blue). Scale bar = 10 μm. (b and c) The western blot images of HeLa cells after treatment with ER-GO-NPs for 24 h to evaluate the expression of CHOP and GRP78 as ER stress markers respectively. | ||
We also evaluated the expression of CHOP by gel electrophoresis. We treated HeLa cells with ER-GO-NPs for 24 h followed by their lysis and then separated the proteins to perform western blot analysis. We observed an increase in CHOP expression by 1.6 fold compared to that in the non-nanoparticle treated control cells (Fig. 4b and S7a†). We expected this lower increase in CHOP expression as doxorubicin inhibits XBP1 protein which prevents upregulation of CHOP.44 To further estimate the ER stress, we evaluated the expression level of GRP78, one of the key markers for triggering of ER stress induction by western blot analysis.45–47 It was observed that while the control cells hardly showed any expression of GRP78, the ER-GO-NP treated cells showed increased levels of GRP78 (1.5 fold) indicating the onset of ER stress (Fig. 4c and S7b†).
The proto-oncogene CHOP also known as growth arrest and DNA damage 153 (GADD 153) encodes for DNA damage as well.48,49 Therefore, we expect that induction of ER stress by ER-GO-NPs will lead to damage of nuclear DNA. We validated the DNA damage by ER-GO-NPs by immunofluorescence assay. We incubated HeLa cells with ER-GO-NPs for 24 h and then treated the cells with γH2AX specific primary antibody followed by Alexa Fluor 594 tagged secondary antibody (red fluorescence). We also stained the nucleus of the HeLa cells with blue fluorescent DAPI. We then visualized the cells by confocal microscopy and observed an enhanced red fluorescence in the treated cells as compared to the control (Fig. 5a). Furthermore, purple fluorescence observed due to the overlap of red and blue fluorescence signals confirmed that the ER-GO-NPs damaged the nuclear DNA. We also evaluated the ER-GO-NP-mediated DNA damage by western blot analysis. We treated HeLa cells with ER-GO-NPs for 24 h, extracted the proteins and performed gel electrophoresis. From the western blot image, it was clear that the control cells showed negligible expression of γH2AX. However, the ER-GO-NP treated cells showed a significant increase (3 fold) in the expression of γH2AX (Fig. 5b and S7c, ESI†). These confocal imaging and western blot analyses revealed that ER-GO-NPs induced ER stress and DNA damage in HeLa cells.
 | ||
| Fig. 5 (a) Confocal images of the HeLa cells treated with ER-GO-NPs for 24 h and stained with γH2AX selective primary antibody followed by Alexa Fluor 594-tagged secondary antibody (red). Nuclei were stained with DAPI (blue). Scale bar = 10 μm. (b) western blot analysis after treating HeLa cells with ER-GO-NPs for 24 h to show the expression of γH2AX as a DNA damage marker. | ||
Autophagy induction
ER stress is also a potent trigger for autophagy, a self-degradative process with adaptive functions.50,51 To validate if autophagy was activated upon treatment with ER-GO-NPs, we evaluated the expression level of LC3B as an autophagy marker.52 We validated the expression of LC3B using immunofluorescence assay. HeLa cells were treated with ER-GO-NPs for 24 h and incubated with LC3B primary antibody followed by secondary antibody tagged with Alexa Fluor 488 dye (green). We stained the nucleus with DAPI (blue). We observed the formation of puncta (autophagosomes) in the ER-GO-NP-treated cells as compared to the control cells (Fig. 6a). This formation of autophagosomes is a hallmark of autophagy.53 We further validated the expression of LC3B by western blot analysis. We treated HeLa cells with ER-GO-NPs for 24 h and the cellular proteins were subjected to gel electrophoresis. From the western blot image, we observed that ER-GO-NPs increased the expression of LC3B in HeLa cells compared to the control cells (Fig. 6b). Further quantification also confirmed that ER-GO-NPs increased the expression of LC3B by 11 fold compared to that in the control cells (Fig. S7d, ESI†). We also evaluated the level of Beclin, another autophagy marker via immunofluorescence assay.54 We incubated HeLa cells with ER-GO-NPs for 24 h. We then treated HeLa cells with Beclin specific primary antibody followed by incubation with Alexa Fluor 594 tagged secondary antibody (red). We also counter stained the nucleus of the cells with DAPI (blue). We then visualized the cells using confocal microscopy. The increased red fluorescence in the treated cells as compared to the control indicates that Beclin expression has indeed increased to trigger autophagy (Fig. 6c). These immunofluorescence assays and western blot analysis thus showed that ER-GO-NPs induced ER stress in HeLa cells. Prolonged ER stress triggered autophagy as an adaptive response to it. Interestingly, to induce autophagy the ER-GO-NPs needed to escape the lysosomes quickly and localize into the ER to trigger ER stress. Prolonged lysosomal homing of ER-GO-NPs would release the ER stress inducers prematurely into the cytosol leading to non-specific effects in other organelles such as the nucleus towards direct apoptosis. We anticipate that this induction of autophagy could be due to specific degradation of the endoplasmic reticulum through ER-phagy which is currently an elusive field of study.55,56 | ||
| Fig. 6 (a) Confocal images of HeLa cells after treatment with ER-GO-NPs for 24 h and stained with primary antibody specific for LC3B, followed by Alexa Fluor 488 tagged secondary antibody (green). (b) Western blot analysis of LC3B after incubation of HeLa cells with ER-GO-NPs for 24 h. (c) Confocal images of the HeLa cells after treatment with ER-GO-NPs for 24 h. The cells were stained with primary antibody specific for Beclin followed by Alexa Fluor 594 tagged secondary antibody (red). Scale bar = 10 μm. | ||
Apoptosis and cell death
Finally, we expected the ER-GO-NPs to induce ER stress mediated apoptosis in HeLa cells. We estimated the apoptosis induced by ER-GO-NPs by flow cytometry analysis. We treated HeLa cells with ER-GO-NPs and then stained the apoptotic and necrotic cells with Annexin V-FITC (green) and propidium iodide (PI) (red) followed by flow cytometry. We found that ER-GO-NPs triggered early and late apoptosis in 61.3% and 35.7% of the cells respectively (Fig. 7a). Since autophagy is triggered upon treatment with ER-GO-NPs, we anticipated it to help the cells escape apoptosis. Hence, we performed the flow cytometry study in combination with chloroquine (CQ). We treated HeLa cells with ER-GO-NPs and 50 μM CQ and incubated them for 24 h. The flow cytometry data revealed that co-treatment with ER-GO-NPs and CQ indeed resulted in 27.5% and 60.5% of the cells in early and late apoptotic stages respectively (Fig. 7a). These flow cytometry data confirmed that ER-GO-NPs triggered early apoptosis and combination treatment with the autophagy inhibitor induced late apoptosis. | ||
| Fig. 7 (a) Flow cytometry analysis of ER-GO-NPs and CQ in HeLa cells to evaluate the induction of apoptosis. (b) Dose dependent viability of HeLa, MDA-MB-231, MCF-7 and A549 cells after treatment with ER-GO-NPs, 24 h post incubation. (c) Cell viability of HeLa cells after co-treatment with ER-GO-NPs and 4-PBA, 24 h post incubation. | ||
Finally, to evaluate the effect of apoptosis induced by ER-GO-NPs on cancer cells, we treated HeLa cells with ER-GO-NPs at different concentrations for 24 h and quantified the cell viability by MTT assay. It was observed that on treatment with ER-GO-NPs, HeLa cells exhibited cell death with IC50 = 5 μM (Fig. 7b). We also treated HeLa cells with a combination of ER-GO-NPs in a dose dependent manner with CQ at 50 μM concentration for 24 h. The cell viability assay revealed that the combination of ER-GO-NPs and CQ showed much lower IC50 = 1.25 μM (Fig. 7b). Inspired by these data, we also validated the effect of the ER-GO-NPs and CQ combination on other cancer cells such as A549 (human lung carcinoma), MCF-7 (human breast cancer) and MDA-MB-231 (drug resistant triple negative breast cancer) by MTT assay. At 24 h post incubation, it was found that ER-GO-NPs demonstrated IC50 = 5 μM, 0.8 μM and 4 μM in A549, MCF-7 and MDA-MB-231 cells respectively (Fig. 7b). Interestingly, in the combination treatment with CQ, the IC50 value reduced to 1 μM in A549 and MDA-MB-231 cells. However, the combination treatment did not change the IC50 value in MCF-7 cells. It is intriguing to note that ER-GO-NPs and combination treatment with CQ lead to nearly 25% cell viability in all the cancer cell lines tested at the highest concentration (50 μM). We anticipate that the cell killing ability of the ER-GO-NPs alone or in combination could be improved by either increasing the exposure time to 48 h or using a more potent autophagy inhibitor bafilomycin A1.57 Moreover, without the ER targeting dansyl moiety, doxorubicin and cisplatin containing self-assembled GO nanoparticles would localize mainly into lysosomes and release the dual drugs at acidic pH followed by nuclear DNA damage leading to direct apoptosis and improved cell killing (IC50 = 2 μM) in HeLa cells without autophagy.34
To ascertain our hypothesis that the apoptosis induced in HeLa cells by ER-GO-NPs was ER stress mediated, we treated HeLa cells with ER-GO-NPs and 4-phenyl butyric acid (4-PBA), a well-known chemical chaperone to alleviate both toxicity and proteomic alterations induced by an ER stress inducer, by aiding in protein folding in the ER.58,59 We co-treated HeLa cells with ER-GO-NPs along with 15 μM PBA for 24 h and then performed the cell viability assay. We observed that co-treatment with 4-PBA drastically reduced the cell death as compared to treatment with ER-GO-NPs alone (Fig. 7c). 4-PBA augmented protein folding and thereby helped HeLa cells survive. This proved that apoptosis induced by ER-GO-NPs in HeLa cells was indeed ER stress mediated.
Conclusions
In conclusion, we have engineered dansyl (ER targeting moiety) decorated graphene oxide (GO) based nanoparticles simultaneously encompassing ER stress inducers doxorubicin and cisplatin through π–π stacking and coordinate linkage, respectively. We demonstrated specific homing of ER-GO-NPs into the subcellular ER of the HeLa cells, released their payload and induced ER stress as well as autophagy as a cellular defence mechanism. Further, ER-GO-NPs were employed to induce ER stress associated apoptosis in cervical cancer cells. Finally, these ER-GO-NPs alone and in combination with CQ showed remarkable cell killing efficacy in breast, lung and drug resistant triple negative breast cancer cells. We envision that these graphene oxide-based ER specific nanoparticles can be used as an effective tool in simultaneously impairing multiple targets in the unfolded protein response (UPR) signalling pathway in the ER thereby leading to better cancer therapeutics in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SB and NB acknowledge the Department of Biotechnology (DBT) (BT/PR14724/NNT/28/831/2015) and Department of Science and Technology (DST) [SB/NM/NB-1083/2017 (G) and DST/TMD/MES/2k18/50], Govt. of India for financial support. SP and AN thank IISER Pune for the doctoral fellowship. We also thank Dr Mayurika Lahiri (IISER Pune) for helping with the cell culture experiments.References
- E. Madden, S. E. Logue, S. J. Healy, S. Manie and A. Samali, Biol. Cell., 2019, 111, 1–17 CrossRef.
- J. R. Cubillos-Ruiz, S. E. Bettigole and L. H. Glimcher, Cell, 2017, 168, 692–706 CrossRef.
- M. Wang and R. J. Kaufman, Nat. Rev. Cancer, 2014, 14, 581–597 CrossRef.
- S. S. Cao and R. J. Kaufman, Antioxid. Redox Signaling, 2014, 21, 396–413 CrossRef.
- J. Miles, R. Scherz-Shouval and P. van Oosten-Hawle, Trends Biochem. Sci., 2019, 44, 927–942 CrossRef.
- D. Ron and H. P. Harding, Cold Spring Harbor Perspect. Biol., 2012, 4, a013177 Search PubMed.
- J. Lee and U. Ozcan, J. Biol. Chem., 2014, 289, 1203–1211 CrossRef CAS.
- H. J. Clarke, J. E. Chambers, E. Liniker and S. J. Marciniak, Cancer Cell, 2014, 25, 563–573 CrossRef CAS.
- R. Clarke, K. L. Cook, R. Hu, C. O. B. Facey, I. Tavassoly, J. L. Schwartz, W. T. Baumann, J. J. Tyson, J. Xuan, Y. Wang, A. Wӓrri and A. N. Shajahan, Cancer Res., 2012, 72, 1321–1331 CAS.
- A. Walczak, K. Gradzik, J. Kabzinski, K. Przybylowska-Sygut and I. Majsterek, Oxid. Med. Cell. Longevity, 2019, 2019, 5729710 Search PubMed.
- A. Almanza, A. Carlesso, C. Chintha, S. Creedican, D. Doultsinos, B. Leuzzi, A. Luἰs, N. McCarthy, L. Montibeller, S. More, A. Papaioannou, F. Püschel, M. L. Sassano, J. Skoko, P. Agostinis, J. de Belleroche, L. A. Eriksson, S. Fulda, A. M. Gorman, S. Healy, A. Kozlov, C. Muňoz-Pinedo, M. Rehm, E. Chevet and A. Samali, FEBS J., 2019, 286, 241–278 CrossRef CAS.
- A. H. Schönthal, Scientifica, 2012, 2012, 857516 CrossRef.
- A. D. Garg, H. Maes, A. R. van Vliet and P. Agostinis, Mol. Cell. Oncol., 2015, 2, e975089 CrossRef.
- R. K. Yadav, S. W. Chae, H. R. Kim and H. J. Chae, J Cancer Prev, 2014, 19, 75–88 CrossRef.
- K. Shen, D. W. Johnson, D. A. Vesey, M. A. McGuckin and G. C. Gobe, Cell Stress Chaperones, 2018, 23, 317–334 CrossRef CAS.
- D. Xu, S. Q. Liang, H. Yang, U. Lüthi, C. Riether, S. Berezowska, T. M. Marti, S. R. R. Hall, R. Bruggmann, G. J. Kocher, R. A. Schmid and R. W. Peng, Br. J. Cancer, 2018, 119, 65–75 CrossRef CAS.
- D. H. Suh, M. K. Kim, H. S. Kim, H. H. Chung and Y. S. Song, Ann. N.Y. Acad. Sci., 2012, 1271, 20–32 CrossRef CAS.
- A. M. Martelli, F. Paganelli, F. Chiarini, C. Evangelisti and J. A. McCubrey, Cancers, 2020, 12, 333 CrossRef CAS.
- D. Jiang, C. Lynch, B. C. Medeiros, M. Liedtke, R. Bam, A. B. Tam, Z. Yang, M. Alagappan, P. Abidi, Q. T. Le, A. J. Giaccia, N. C. Denko, M. Niwa and A. C. Koong, Sci. Rep., 2016, 6, 33353 CrossRef CAS.
- R. M. Cunningham and V. J. DeRose, ACS Chem. Biol., 2017, 12, 2737–2745 CrossRef CAS.
- Y. Xu, C. Wang and Z. Li, Mol. Clin. Oncol., 2014, 2, 3–7 CrossRef CAS.
- T. Karasawal, M. Sibrian-Vazquez, R. M. Strongin and P. S. Steyger, Plos One, 2013, 8, e66220 CrossRef.
- A. Mandic, J. Hansson, S. Linder and M. C. Shoshan, J. Biol. Chem., 2003, 278, 9100–9106 CrossRef CAS.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 338–350 CrossRef CAS.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef.
- C. Ghosh, A. Nandi and S. Basu, Nanoscale, 2019, 11, 3326–3335 RSC.
- C. Ghosh, A. Nandi and S. Basu, ACS Appl. Bio Mater., 2019, 2, 3992–4001 CrossRef.
- S. A. Sydlik, S. Jhunjhunwala, M. J. Webber, D. G. Anderson and R. Langer, ACS Nano, 2015, 9, 3866–3874 CrossRef.
- S. Shi, F. Chen, E. B. Ehlerding and W. Cai, Bioconjugate Chem., 2014, 25, 1609–1619 CrossRef.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef.
- H. Hong, Y. Zhang, J. W. Engle, T. R. Nayak, C. P. Theuer, R. J. Nickles, T. E. Barnhart and W. Cai, Biomaterials, 2012, 33, 4147–4156 CrossRef.
- A. Mallick, A. Nandi and S. Basu, ACS Appl. Bio Mater., 2019, 2, 14–19 CrossRef.
- A. Nandi, A. Mallick, P. More, P. Sengupta, N. Ballav and S. Basu, Chem. Commun., 2017, 53, 1409–1412 RSC.
- H. Xiao, C. Wu, P. Li, W. Gao, W. Zhang, W. Zhang, L. Tong and B. Tang, Chem. Sci., 2017, 8, 7025–7030 RSC.
- Y. Liu, C. N. Lok, B. C. Ko, T. Y. Shum, M. K. Wong and C. M. Che, Org. Lett., 2010, 12, 1420–1423 CrossRef.
- P. Gao, W. Pan, N. Li and B. Tang, Chem. Sci., 2019, 10, 6035–6071 RSC.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 Search PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Control. Release, 2000, 65, 271–284 CrossRef.
- A. Nandi, C. Ghosh and S. Basu, Nanoscale Adv, 2019, 1, 4965–4971 RSC.
- W. Hakamata, S. Tamura, T. Hirano and T. Nishio, ACS Med. Chem. Lett., 2014, 5, 321–325 CrossRef.
- S. Oyadomari and M. Mori, Cell Death Differ., 2004, 11, 381–389 CrossRef.
- H. Nishitoh, J. Biochem., 2012, 151, 217–219 CrossRef.
- H. Hu, M. Tian, C. Ding and S. Yu, Front. Immunol., 2019, 9, 3083 CrossRef.
- M. Wang, S. Wey, Y. Zhang, R. Ye and A. S. Lee, Antioxid. Redox Signaling, 2009, 11, 2307–2316 CrossRef.
- J. B. Gifford, W. Huang, A. E. Zeleniak, A. Hindoyan, H. Wu, T. R. Donahue and R. Hill, Mol. Cancer Ther., 2016, 15, 1043–1052 CrossRef.
- A. S. Lee, Nat. Rev. Cancer, 2014, 14, 263–276 CrossRef CAS.
- Y. Yang, L. Liu, I. Naik, Z. Braunstein, J. Zhong and B. Ren, Front. Immunol., 2017, 8, 1612 CrossRef.
- A. Forus, V. A. Flørenes, G. M. Maelandsmo, Ø. Fodstad and O. Myklebost, Cancer Genetics Cytogenet, 1994, 78, 165–171 CrossRef CAS.
- W. S. Lee, W. H. Yoo and H. J. Chae, Curr. Mol. Med., 2015, 15, 735–745 CrossRef CAS.
- V. Sica, L. Galluzzi, J. M. Bravo-San Pedro, V. Izzo, M. C. Maiuri and G. Kroemer, Mol. Cell, 2015, 59, 522–539 CrossRef CAS.
- I. Tanida, T. Ueno and E. Kominami, Methods Mol. Biol., 2008, 445, 77–88 CrossRef CAS.
- Z. Xie and D. J. Kilonsky, Nat. Cell Biol., 2007, 9, 1102–1109 CrossRef CAS.
- Y. Cao and D. J. Klionsky, Cell Res, 2007, 17, 839–849 CrossRef CAS.
- H. Chino and N. Mizushima, Trends Cell Biol, 2020, 30, 384–398 CrossRef CAS.
- S. Wilkinson, FEBS J., 2019, 286, 2645–2663 CAS.
- L. Galluzzi, J. M. Bravo-San Pedro, B. Levine, D. R. Green and G. Kroemer, Nat. Rev. Drug Discovery, 2017, 16, 487–511 CrossRef CAS.
- B. Kaur, A. Bhat, R. Chakraborty, K. Adlakha, S. Sengupta, S. Roy and K. Chakraborty, Mol. Omics, 2018, 14, 53–63 RSC.
- A. U. Nissar, L. Sharma, M. A. Mudasir, L. A. Nazir, S. A. Umar, P. R. Sharma, R. A. Vishwakarma and S. A. Tasduq, J. Lipid Res., 2017, 58, 1588–1868 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: UV-Vis and fluorescence spectra, EDAX, drug loading, confocal microscopy, western blot and confocal microscopy quantification. See DOI: 10.1039/d0na00338g |
| This journal is © The Royal Society of Chemistry 2020 |