DOI:
10.1039/D0NA00301H
(Paper)
Nanoscale Adv., 2020,
2, 3872-3881
Amplification of oxidative stress via intracellular ROS production and antioxidant consumption by two natural drug-encapsulated nanoagents for efficient anticancer therapy†
Received
17th April 2020
, Accepted 13th July 2020
First published on 14th July 2020
Abstract
Cancer cells are commonly characterized by high cellular oxidative stress and thus have poor tolerance to oxidative insults. In this study, we developed a nano-formulation to elevate the level of reactive oxygen species (ROS) in cancer cells via promoting ROS production as well as weakening cellular anti-oxidizing systems. The nanoagent was fabricated by encapsulating two natural product molecules, cinnamaldehyde (CA) and diallyl trisulfide (DATS), in PLGA–PEG copolymer formulated nanoparticles. CA promotes ROS generation in cancer cells and DATS depletes cellular glutathione. CA and DATS exhibited a synergistic effect in amplifying the ROS levels in cancer cells and further in their combined killing of cancer cells. The in vivo experiments revealed that the CA and DATS-encapsulated nanoagent suppressed tumors more efficiently as compared with the single drug-loaded ones, and the tumor-targeted delivery further enhanced the therapeutic efficacy. This study suggests that the combined enhancement of oxidative stress by CA and DATS could be a promising strategy for cancer therapy.
Introduction
Reactive oxygen species (ROS) are a group of oxygen-containing chemical species with high reactivity, including the non-radical species of hydrogen peroxide and the free radicals such as superoxide and hydroxyl.1 In all the aerobic organisms, ROS serve as a second messenger in cellular signaling and play crucial roles in various biological processes such as cellular proliferation and differentiation, and the response to various cellular stresses.2–5 The imbalance of cellular ROS is generally involved in the promotion and progression of diverse diseases.6–9 For instance, cancer cells are characterized by high levels of oxidative stress due to the aberrant metabolism and signaling, and they have also adapted to the high oxidative stress by activating the non-enzymatic and/or enzymatic anti-oxidizing systems.10,11 In cancer cells, the high level of ROS production is exerted to promote proliferation, metastasis and other actions for tumor progression.12 However, high levels of ROS are generally detrimental to both normal and cancer cells.13 ROS can cause cellular damage of proteins, lipids and DNA, leading to apoptotic cell death.14 In this regard, ROS are also used to kill cancer cells.15–18 As evidence, all the non-surgical therapies including chemotherapy, radiotherapy and photodynamic therapy are found to share the mechanism of ROS-generation induced cell death.19–21
In comparison with normal cells, cancer cells have adapted to high levels of cellular oxidative stress, but have a low ROS tolerance and are, therefore, easily attacked by extra ROS induced by exogenous stimuli.22 Therefore, a new type of therapeutic strategy termed “oxidation therapy” has been recently developed, whereby the level of ROS in cancer cells is increased to cause ROS-mediated cancer cell death.23–26 In general, there are two ways to promote the levels of ROS in cancer cells. One way is to introduce agents to promote ROS generation, and the other is to disrupt the cellular anti-oxidizing systems.27 Based on this idea, the ROS generating agents like arsenic agents, curcumin and emodin have been utilized either alone or in combination with conventional therapeutics for the clinical treatment of diverse cancers.28–30 To enhance ROS production, disrupting the enzymatic (such as superoxide dismutase and thioredoxin) and/or the non-enzymatic (for instance, glutathione (GSH) and thiols) antioxidant systems can also increase the cellular oxidative stress to cause apoptosis of cancer cells.31–34 GSH plays a critical role in the maintenance of intracellular redox status and the defense against oxidative stress.35 It consumes the intracellular ROS directly as a free radical scavenger and/or as a substrate for GSH peroxidase.36 However, excessive amounts of ROS will exhaust GSH and cause cell apoptosis.37 Recently, a combination strategy has been reported, involving the construction of a hybrid anticancer molecule with dual functions of ROS generation and antioxidant depletion.38 This strategy has achieved the highly efficient suppression of tumors, but the design and synthesis of such a bifunctional anticancer molecule are commonly complicated and difficult. Therefore, a universal and efficient formulation should be developed for ROS-involved anticancer therapy.
In this study, we developed a nano-formulation to deliver two natural product molecules to promote ROS generation as well as interfere with cellular antioxidant systems for oxidation-associated cancer therapy. The nanoagent was fabricated by loading cinnamaldehyde (CA) and diallyl trisulfide (DATS) in poly(D,L-lactic-co-glycolic acid)-block-poly(ethylene glycol) (PLGA–PEG) nanoparticles. CA is a natural product molecule from the cinnamon tree and is able to elevate the cellular ROS production.39 DATS is also a natural constituent of allium vegetables, which can deplete GSH.40 Both CA and DATS can induce apoptosis in cancer cells due to the enhanced oxidative stress and have been used individually for anticancer therapy.41,42 We found that CA and DATS remarkably enhanced the oxidative stress in cancer cells in comparison with their individuals, and exhibited a synergistic effect on the efficiency of killing cancer cells in vitro. The CA and DATS-loaded PLGA–PEG nanoparticles (PP–CD) were fabricated for in vivo studies, and they could more efficiently kill cancer cells in vitro and suppress tumors in vivo as compared to the single drug-loaded nanoagents. Finally, the tumor-targeting peptide of cyclic Arg–Gly–Asp (RGD) was modified on PP–CD (PPR–CD) to improve the drug delivery and therapeutic efficiency.
Results and discussion
CA and DATS-induced ROS enhancement and cancer cell death
The anticancer effects of individual CA and DATS or their combination were first evaluated on human breast cancer MDA-MB-231 cells in vitro. The individual treatment using DATS at 40.0 μM showed little cytotoxicity (87.9% relative cell viability, Fig. 1a), based on which the concentrations of CA were tuned to determine the best synergistic effect for killing cancer cells. The data shown in Fig. 1a suggested that the combination treatment at a CA concentration of 37.5 μM and at a DATS concentration of 40.0 μM exhibited the best synergistic effect for killing cancer cells. The individual treatment of CA at 37.5 μM could only reduce the cell viability by 11.0%, and that of DATS at 40.0 μM by 12.1%. However, their combination decreased cell viability by 50.6%. Based on this, the typical CA concentration at 37.5 μM and DATS concentration at 40.0 μM were utilized in the subsequent treatment of MDA-MB-231 cells. To confirm the synergistic effect, we further evaluated CA and DATS-induced apoptosis individually or in combination on MDA-MB-231 cells. The individual treatment by CA only induced 16.3% apoptosis in cancer cells, and that by DATS induced 18.5% apoptosis (Fig. 1b). In comparison, the combination of CA and DATS amplified the killing efficiency, leading to 87.7% apoptosis of cancer cells (Fig. 1b). The data suggested that the combination treatment by CA and DATS could more efficiently kill cancer cells in vitro. We also evaluated the synergistic effect of CA and DATS on human breast cancer MCF-7 cells. The data showed that CA at 100 μM and DATS at 50 μM represented the best synergistic effect for killing MCF-7 cells (Fig. S1 and S2†). In order to explore the mechanism of killing cancer cells, we evaluated the ROS levels in cancer cells after different treatments. MDA-MB-231 and MCF-7 cells were stained with dichloro-dihydro-fluorescein diacetate (DCFH-DA), an agent for the quantitative assessment of oxidative stress. The fluorescence images and flow cytometry data showed that both CA and DATS could promote ROS production in cancer cells, and in comparison, their combination could remarkably elevate the ROS generation to a higher level (Fig. 1c, d and S3†). ROS-induced apoptosis is generally accompanied by the loss of mitochondrial membrane potential.43 The decrease in the red/green fluorescence intensity ratio in cancer cells stained by JC-1 dye indicates the mitochondria depolarization and thus the apoptosis of cells.44 The healthy MDA-MB-231 cells showed red fluorescence, and the cells treated individually by CA or DATS represented red plus yellow fluorescence, indicating little loss of the mitochondrial membrane potential (Fig. 1e). However, the cells treated by both CA and DATS represented a dominant green fluorescence intensity, indicating the major loss of the mitochondrial membrane potential (Fig. 1e). Similarly, the combination of CA and DATS on MCF-7 cells also induced a huge loss of the mitochondrial membrane potential (Fig. S4†). DATS could quickly consume GSH in vitro (Fig. S5†), which is consistent with the previous report.45 Therefore, DATS might act on the non-enzymatic system to elevate the ROS levels in cancer cells. To identify whether the ROS production responds to apoptosis, cancer cells were pretreated with GSH-reduced ethyl ester (GSHoet, an antioxidant agent) and then treated with CA and DATS. The data showed that GSHoet could reduce the anticancer efficiency of CA and DATS (Fig. 1f), which indicated that CA and DATS promoted ROS-induced apoptosis in cancer cells.
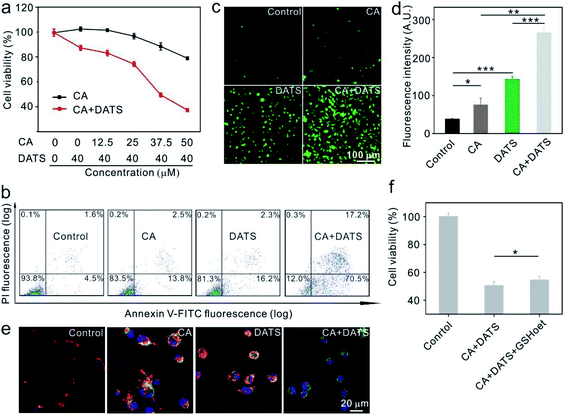 |
| | Fig. 1 The combination effect of CA and DATS on ROS promotion and cytotoxicity. (a) The cytotoxicity of CA, DATS, and their combination of CA + DATS on MDA-MB-231 cells after 48 h incubation. The concentration of DATS was fixed at 40.0 μM, and that of CA was varied in the range of 0–50 μM (n = 6). (b) Apoptosis of MDA-MB-231 cells treated with CA, DATS, and CA + DATS for 24 h. The cells were stained by Annexin V-FITC and PI and analyzed by flow cytometry (c and d) ROS detection via DCFH-DA staining. Fluorescence images (c) and the average fluorescence intensities (d) of MDA-MB-231 cells treated with CA, DATS, and CA + DATS for 12 h (n = 3). (e) The mitochondrial membrane potential changes of MDA-MB-231 cells treated with CA, DATS, and CA + DATS for 2 h. (f) Regression of ROS by GSHoet reduced the cytotoxicity of CA + DATS (n = 6). The concentration of CA was 37.5 μM, and that of DATS was 40.0 μM in the assays showed in b–f. *P < 0.05, **P < 0.01 and ***P < 0.001 were analyzed by Student's t-test. | |
Fabrication of the nanoagent of PP–CD
We fabricated the nano-formulation of PP–CD for the in vivo study. The PLGA–PEG copolymer was synthesized and employed as a carrier due to its excellent biocompatibility and biodegradability (Fig. S6†).46 The two agents CA and DATS were encapsulated in PLGA–PEG nanoparticles via a nanoprecipitation method (Fig. 2a). The concentrations of CA and DATS were determined by high-performance liquid chromatography (HPLC, Fig. S7†). The drug encapsulation efficiencies for CA and DATS were 1.0% and 20.0%, respectively, and the drug loading ratios for the two agents were 1.0% and 1.5%, respectively (Table S1†). The as-obtained PP–CD had a spherical shape and an average diameter of 123.5 ± 26.6 nm (Fig. 2b). The Fourier transform infrared (FT-IR) spectra of CA, DATS and PP–CD were recorded. The typical absorption peak for CA at 1764 cm−1 was assigned to the stretching vibration of the aldehyde group in its structure and the DATS peaks at 973 and 917 cm−1 were assigned to the bending vibration of the alkenyl group in its structure (Fig. 2c). The PP–CD spectrum consisted of both the typical absorption peak for CA at 1764 cm−1, and those for DATS at 973 and 917 cm−1 (Fig. 2c), which suggested that both CA and DATS were successfully encapsulated in the nanoparticles. The dynamic light-scattering analysis revealed that PP–CD had a hydrodynamic diameter of 145.0 nm (Fig. 2d), and had a negative zeta potential in deionized (DI) water (−21.5 mV, Fig. S8†). Their hydrodynamic sizes had negligible changes in phosphate-buffered saline (PBS) and in 10% fetal bovine serum (FBS) in PBS during a long incubation (Fig. 2e). The data suggested that PP–CD was very stable in physiological conditions. Moreover, PP–CD also had a negative zeta potential in PBS, which changed minimally after three days of incubation (Fig. 2f). The stability of the drugs in their formulation of PP–CD was further evaluated via measuring their ultraviolet-visible (UV-Vis) spectra at different time points. As shown in Fig. S9,† the absorption of both CA and DATS gradually decreased along with the time. In comparison, their absorption in the nanoparticles decreased slowly. The results indicated that the nanoparticle could improve the stability of both CA and DATS. Finally, the drug release behavior was evaluated. As shown in Fig. 2g, there was a burst release for both CA and DATS at the beginning, and then the slow release was continued until the assays were stopped. Finally, 71.1% CA and 63.3% DATS were released from PP–CD in 105 h (Fig. 2g).
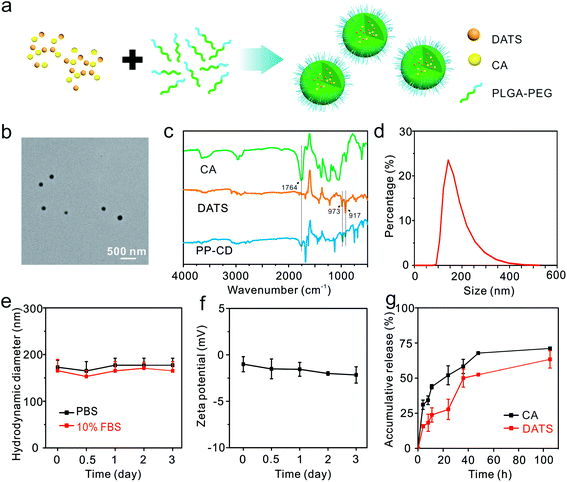 |
| | Fig. 2 Formation and characterization of PP–CD. (a) Schematic illustration of the formulation of PP–CD. CA and DATS were encapsulated in PLGA–PEG nanoparticles via nanoprecipitation. (b) TEM image of PP–CD. (c) FT-IR spectra of CA, DATS and PP–CD. (d) The hydrodynamic diameters of PP–CD in DI water. (e) The hydrodynamic diameter changes of PP–CD in PBS or PBS containing 10% FBS (n = 3). (f) Time-elapsed evolution of the zeta potential of PP–CD in PBS (n = 3). (g) The accumulative drug release of CA and DATS from PP–CD in PBS (pH = 7.4, n = 3). | |
PP–CD-associated ROS enhancement-induced cancer cell death
We further evaluated the efficiencies of the different nanoagents for killing cancer cells in vitro. The MDA-MB-231 cells were treated with PBS, CA-encapsulated PLGA–PEG nanoparticles (PP–C), DATS-encapsulated PLGA–PEG nanoparticles (PP–D) and PP–CD, respectively. The cellular ROS level was first determined by using DCFH-DA. As shown in Fig. 3a and S10,† the ROS levels were enhanced in the cells treated with PP–C and more obviously in the cells treated with PP–D 12 h post-incubation, while the cells treated with PP–CD showed the highest ROS enhancement. The cytotoxicity of these nanoagents was further evaluated. The data revealed that the individual treatment by using PP–C and PP–D had little cytotoxicity at both time points of 24 and 48 h post-treatment, as the cell viabilities were both at a relatively high level of around 80% (Fig. 3b and S11†). However, the cells treated with PP–CD retained 55.0% of their viability at 24 h, and 33.6% at 48 h (Fig. 3b and S11†). The cells treated with different nanoagents were further stained by acridine orange (AO)/ethidium bromide (EB) to detect apoptosis. As shown in Fig. 3c, few cells were stained with a red color 24 h after the treatment with PP–C and PP–D; however, in the cells treated with PP–CD, most of the cells died and thus only a few cells adhered in the culture media. Moreover, we employed flow cytometry to quantify the apoptotic cells. The data confirmed that PP–C and PP–D demonstrated little toxicity to the cells (14.1 apoptosis for PP–C and 16.3% apoptosis for PP–D), but PP–CD represented much greater toxicity than the individual formulation (78.2% apoptosis, Fig. 3d). We further decorated the tumor-targeting RGD peptides on the surface of PP–CD. The as-obtained PPR–CD had a hydrodynamic diameter of 164 nm (Fig. S12†) and was larger than that of PP–CD due to the modification of RGD (Fig. 2d). The zeta potential of PPR–CD was measured to be −19.0 mV, which was less negative than that of PP–CD (Fig. S8†). To quantitatively evaluate the cellular uptake in vitro, a hydrophobic fluorescein molecule coumarin-6 (Co6) was encapsulated in PLGA–PEG and PLGA–PEG–RGD (i.e. PP–Co6 and PPR–Co6). The flow cytometry data showed that more PPR–Co6 was taken by MDA-MB-231 cells than PP–Co6 (Fig. 3e). The DCFH-DA staining assay revealed that PPR–CD could more efficiently elevate the cellular ROS levels as compared to the PP–CD in MDA-MB-231 cells (Fig. S13†). Furthermore, the cell-killing efficiency of PPR–CD was evaluated. The AO/EB staining assay revealed that more cancer cells were killed on treatment with PPR–CD as compared with that by PP–CD (Fig. S14†). The apoptosis of MDA-MB-231 cells treated with PPR–CD was also quantified by the Annexin-V/propidium iodide (PI) staining assay. The data showed that PPR–CD caused more cell apoptosis than PP–CD (Fig. S15†). Finally, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay revealed that PPR–CD caused greater cytotoxicity than PP–CD in MDA-MB-231 cells (Fig. 3f). All these results suggested that RGD could enhance the cellular uptake of the nanoagents in cancer cells overexpressing αvβ3 integrin,47 and thus caused enhanced intracellular ROS and more cancer cell death.
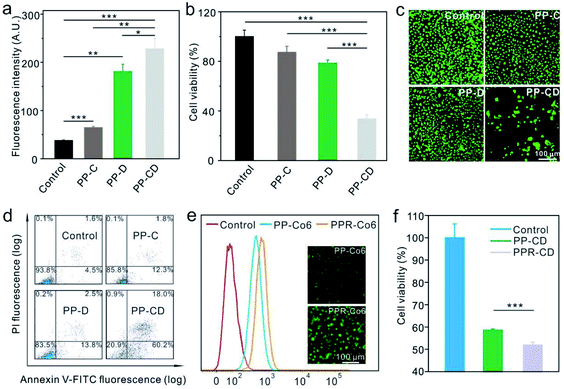 |
| | Fig. 3 ROS production and the cytotoxicity induced by different agents. (a) ROS generation in MDA-MB-231 cells treated with PP–C, PP–D and PP–CD for 12 h (n = 3). The cells were stained with DCFH-DA and analyzed by flow cytometry. (b) The cytotoxicity of PP–C, PP–D and PP–CD on MDA-MB-231 cells after 48 h incubation (n = 6). (c) AO/EB-stained MDA-MB-231 cells after incubation with PP–C, PP–D and P–CD for 24 h. (d) Apoptosis of MDA-MB-231 cells treated with PP–C, PP–D and PP–CD for 24 h. The cells were stained by Annexin V-FITC and PI and analyzed by flow cytometry. (e) The cellular uptake of PP–Co6 and PPR–Co6 analyzed by flow cytometry. The MDA-MB231 cells were incubated with PP–Co6 and PPR–Co6 for 1 h. Insets are the fluorescence images of the cells. (f) The cytotoxicity of PP–CD and PPR–CD on MDA-MB-231 cells after incubation for 18 h (n = 6). *P < 0.05, **P < 0.01 and ***P < 0.001 were analyzed by Student's t-test. | |
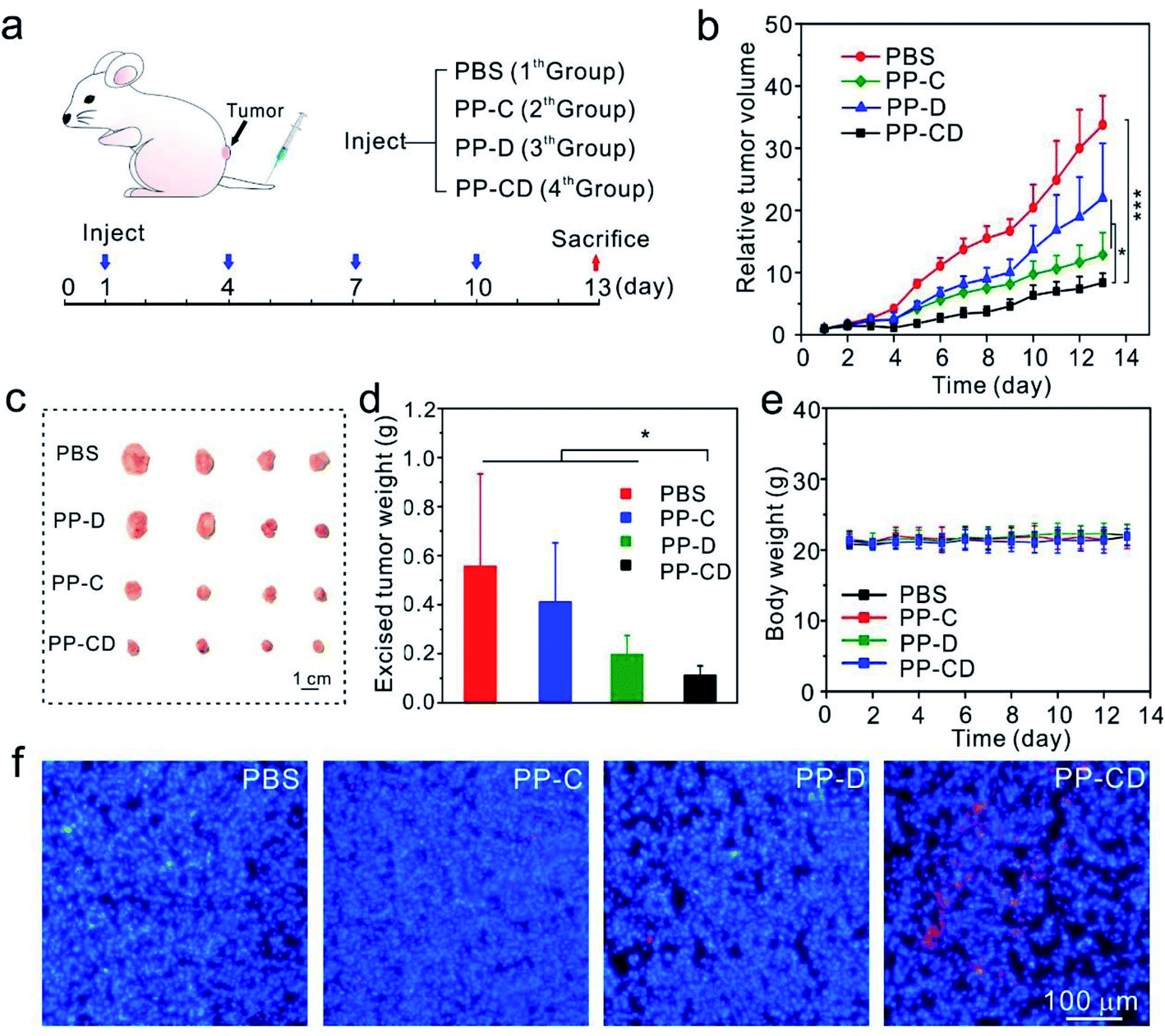
PP–CD-associated in vivo tumor treatment
The in vivo anticancer effect was assessed on a mice model bearing MDA-MB-231 tumors. The mice in four groups were intravenously administered with PBS, PP–C, PP–D and PP–CD, respectively (Fig. 4a). The average tumor sizes in the PP–C and PP–D groups were obviously reduced in comparison with that in the PBS group, which suggested that the two drugs had anticancer effects in their individual treatments (Fig. 4b). The combination treatment using PP–CD more efficiently caused regression of the tumor growth as compared to the individual treatments using PP–C or PP–D (Fig. 4b). The tumors were isolated from sacrificed mice after treatment. The tumors in the PP–CD group were small and had the lightest average weight (Fig. 4c and d). The terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay suggested that there were more apoptotic tumor cells in the PP–CD group as compared with PP–C and PP–D groups (Fig. 4f), which confirmed that PP–CD represented the highest anticancer efficiency. The body weights of all the mice changed negligibly during the treatment, and the main organs showed no detectable changes after treatment, which indicated that these nanoagents had non-detectable toxicity in vivo (Fig. 4e and S16†).
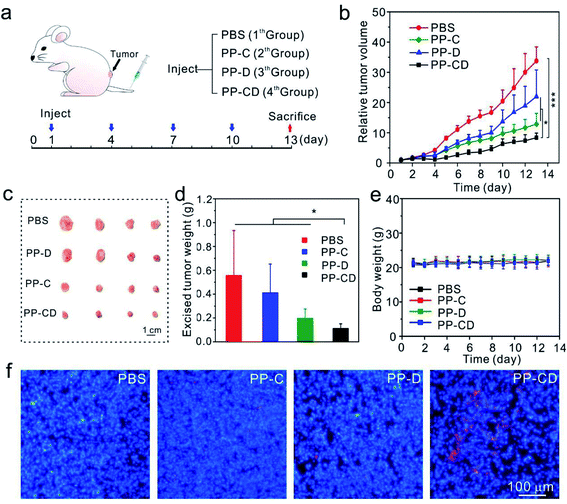 |
| | Fig. 4
In vivo anticancer treatment (a) the scheme depicts the experiment process. The mice bearing MDA-MB-231 tumors were intravenously injected with PBS, PP–C, PP–D and PP–CD, respectively. (b) The changes in tumor sizes during treatment. (c) Photograph of the tumors isolated from sacrificed mice after treatment. (d) The average weights of tumors after treatment (n = 5). (e) The changes in body weights during treatment. (f) Apoptosis (red) in tumor cells analyzed by a TUNEL assay. The cell nuclei were stained by Hoechst 33342. *P < 0.05 and ***P < 0.001 were analyzed by Student's t-test. | |
PPR–CD-associated targeted tumor treatment
To enhance the therapeutic efficacy, we conducted another in vivo assay for tumor-targeting therapy. The mice in three groups were injected with PBS, PP–CD and PPR–CD, respectively. It was observed that the tumors in the PPR–CD group grew more slowly as compared to those in the PP–CD group (Fig. 5a), and the isolated tumors in the PPR–CD group were smaller and lighter (Fig. 5b and c). The TUNEL assay revealed that there were more apoptotic tumor cells in the PPR–CD group than in the PP–CD group (Fig. 5d); the data confirmed that the targeted treatment enhanced the therapeutic efficacy. The body weights of the mice changed minimally during the treatment (Fig. 5e), and the main organs of the mice were all in healthy condition (Fig. S17†), which suggested that the nanoagents had no-detectable toxicity in vivo during the treatment.
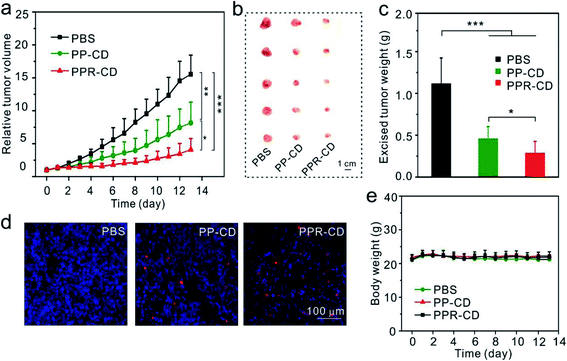 |
| | Fig. 5
In vivo anticancer effect of PPR–CD (a) the changes in tumor sizes during treatment (n = 5). (b) Photograph of the tumors isolated from sacrificed mice after treatment. (c) The average weights of tumors after treatment (n = 5). (d) Apoptosis (red) in tumor cells analyzed by a TUNEL assay. (e) The changes in body weights during treatment (n = 5). *P < 0.05, **P < 0.01 and ***P < 0.001 were analyzed by Student's t-test. | |
Conclusion
In summary, we fabricated a CA and DATS-encapsulated nanoagent formulated by PLGA–PEG copolymers for oxidation-mediated cancer therapy. CA and DATS are both natural product molecules. CA promoted cellular ROS production, and DATS interfered with the cellular antioxidant system. The combined use of CA and DATS could significantly enhance the ROS levels in cancer cells, and therefore caused the oxidation stress-induced apoptosis of cancer cells. The in vivo treatment demonstrated that the oxidation therapy associated with PP–CD could more efficiently suppress tumors than the individual treatment mediated by PP–C or PP–D, and the tumor-targeting PPR–CD formulation exhibited greater efficacy than PP–CD.
Materials and methods
Materials and chemicals
Carboxyl-terminated PLGA (PLGA–COOH, lactide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :glycolide = 50:50, molecular weight = 38000–54000 Da) was obtained from Aladdin (Shanghai, China). Amine–PEG–maleimide (NH2–PEG–Mal, molecular weight = 2000 Da) was bought from Ponsure Biotechnology (Shanghai, China). N-Hydroxysuccinimide (NHS) and GSHoet were obtained from Macklin Biochemical Co. Ltd (Shanghai, China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) was obtained from Shanghai Medpep Co. Ltd (Shanghai, China). CA was bought from J&K Scientific (Shanghai, China). DATS was obtained from Huaxia Reagent (Chengdu, China). Acetonitrile was bought from J. T. Baker (Phillipsburg, USA). Dimethyl sulfoxide (DMSO) was obtained from Sinopharm Chem (Shanghai, China). AO was purchased from Sangon Biotech (Shanghai, China). Annexin V-FITC/PI Apoptosis Detection Kit was obtained from BD Biosciences Pharmingen (San Diego, USA). EB was bought from Tiangen Biotech (Beijing, China). ROS Assay Kit and Mitochondrial Membrane Potential Assay Kit with JC-1 were bought from Beyotime Biotechnology (Shanghai, China). Hoechst 33342 and Co6 were obtained from Sigma-Aldrich (St Louis, USA). The TUNEL Apoptosis Assay Kit was obtained from Roche Diagnostics (Mannheim, Germany). All the chemicals were used as received without further purification.
:glycolide = 50:50, molecular weight = 38000–54000 Da) was obtained from Aladdin (Shanghai, China). Amine–PEG–maleimide (NH2–PEG–Mal, molecular weight = 2000 Da) was bought from Ponsure Biotechnology (Shanghai, China). N-Hydroxysuccinimide (NHS) and GSHoet were obtained from Macklin Biochemical Co. Ltd (Shanghai, China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) was obtained from Shanghai Medpep Co. Ltd (Shanghai, China). CA was bought from J&K Scientific (Shanghai, China). DATS was obtained from Huaxia Reagent (Chengdu, China). Acetonitrile was bought from J. T. Baker (Phillipsburg, USA). Dimethyl sulfoxide (DMSO) was obtained from Sinopharm Chem (Shanghai, China). AO was purchased from Sangon Biotech (Shanghai, China). Annexin V-FITC/PI Apoptosis Detection Kit was obtained from BD Biosciences Pharmingen (San Diego, USA). EB was bought from Tiangen Biotech (Beijing, China). ROS Assay Kit and Mitochondrial Membrane Potential Assay Kit with JC-1 were bought from Beyotime Biotechnology (Shanghai, China). Hoechst 33342 and Co6 were obtained from Sigma-Aldrich (St Louis, USA). The TUNEL Apoptosis Assay Kit was obtained from Roche Diagnostics (Mannheim, Germany). All the chemicals were used as received without further purification.
Synthesis of PLGA–PEG
In a standard synthesis, PLGA–COOH (100.00 mg) was dissolved in DMSO (2 mL), and then EDC (2.08 mg) was added. The reaction solution was stirred for 10 min. NHS (0.25 mg) was dissolved in DMSO (50 μL), and was then added to the reaction solution. The reaction was conducted under magnetic stirring for 2 h. After that, NH2–PEG–Mal (21.70 mg) was added to the reaction solution, and the reaction was conducted for 24 h. Finally, the product was dialyzed three times with DMSO and then ten times with DI water. The purified product was stored at 4 °C for use.
Preparation of PP–CD, PP–C and PP–D
PP–CD was prepared via a nanoprecipitation method. Typically, PLGA–PEG (10 mg), DATS (0.4 mg) and CA (6.6 mg) were dissolved in acetonitrile (1 mL), and then the solution was added dropwise to PBS (3 mL) under magnetic stirring. The reaction proceeded for 7 min. After that, the product was collected via centrifugation (11000 rpm, 15 min) and washed five times with PBS; the product was suspended in PBS for use. The PP–C and PP–D were synthesized and purified via the same procedure.
Synthesis of PPR–CD
PLGA–PEG–RGD was synthesized by a click reaction using PLGA–PEG–MAL and thiol-RGD. The PLGA–PEG–MAL was dissolved in DMSO, then thiol-RGD was added to the reaction solution. The reaction was conducted for 24 h. The product was dialyzed three times with DMSO and then ten times with DI water. The purified product was stored at 4 °C for use. PPR–CD was synthesized and purified via the same procedure as PP–CD.
Characterization
Transmission electron microscopy (TEM) images were taken on a transmission electron microscope (HT7700, HITACHI, Japan). The hydrodynamic diameter and zeta potential of nanoparticles were measured by dynamic light scattering (Zetasizer Nano ZS90, Malvern, UK). The nuclear magnetic resonance (NMR) spectra of polymers were collected using an NMR spectrometer (500 MHz, Bruker Avance, Germany). The FT-IR spectra of the drugs were collected using an FT-IR spectrometer (Nicolet iS50, Thermo Scientific).
Drug loading of PP–CD
The drug loading parameters of PP–CD were first determined. Typically, the PP–CD suspension in PBS (10 mg mL−1; 10 μL) was added to acetonitrile (90 μL) to disrupt the nanoparticles, and then DATS and CA in PP–CD were quantitatively determined on an Agilent 1100 HPLC. The standard curve for DATS and CA was determined in a concentration range of 0–0.2 mg mL−1 (Fig. S7†). To determine the drug-encapsulation efficiency and drug-loading ratio, the PP–CD suspension was lyophilized and weighed. The drug encapsulation efficiency and the drug loading ratios of PP–CD were calculated according to the following formulas:
| Drug encapsulation efficiency = the weight of the loaded drug/the weight of the drug in feed × 100% |
| Drug loading ratio = weight of the loaded drug/the weight of micelle × 100% |
Stability of PP–CD
The stability of PP–CD was also assessed by dynamic light scattering. PP–CD was incubated with PBS and PBS containing 10% FBS. The hydrodynamic diameters and the zeta potentials of the nanoparticles were recorded at different time points.
Drug stability in the nanoparticles
The stability of the drugs was assessed by measuring their UV-Vis spectra at different time points. The free drugs CA (3.75 μM) and DATS (4.0 μM) were dissolved in acetonitrile, and their nanoparticle formulation of PP–C (37.5 μM) and PP–D (40.0 μM) was incubated in DI water. At the time points of 0, 24, 72, 120, 168 h, 0.9 mL of CA and DATS in acetonitrile were taken out, and then 0.1 mL DI water was added to each sample. At the same time points, 0.1 mL PP–C and PP–D were taken out and diluted with 0.9 mL acetonitrile. The UV-Vis spectra of these samples were recorded on an Agilent Cary 60 UV-Vis spectrophotometer (Agilent Technologies, USA). The absorption of CA was measured at the wavelength of 291 nm, and that of DATS was done at 254 nm.
In vitro drug release from PP–CD
In a typical assay, twenty-one samples of PP–CD (10 mg mL−1, 10 μL in PBS) in EP tubes were prepared and incubated at room temperature. At each time point, three tubes were picked out, and the nanoparticles in each were collected via centrifugation (15000 rpm, 30 min), dissolved in acetonitrile and analyzed using HPLC.
Cell culture
MDA-MB-231 cells (a human breast adenocarcinoma cell line, ATCC) were cultured in MEM medium. MCF-7 (a human breast adenocarcinoma cell line, ATCC) cells were cultured in DMEM medium. The cell culture media were supplemented with 10% FBS, 100 μg mL−1 streptomycin, and 100 μg mL−1 penicillin. All the cells were cultured at 37 °C under 5% CO2.
Cytotoxicity assay
MDA-MB-231 cells (1 × 104) were cultured in 96-well plates for 24 h. The cells were co-incubated with DATS at a concentration of 40.0 μM and CA at different concentrations (0, 12.5, 25, 37.5, 50 μM) for 48 h. After that, a standard MTT assay was carried out to evaluate the cell viability. The cytotoxicity of CA and DATS was also determined on MCF-7 cells. The MCF-7 cells (1 × 104) were cultured in 96-well plates for 24 h. The cells were co-incubated with DATS at a concentration of 50 μM and CA at different concentrations (0, 25, 50, 100, 200 μM) for 48 h. A standard MTT assay was carried out to evaluate the cell viability.
The cytotoxicities of PP–C (CA, 37.5 μM), PP–D (DATS, 40.0 μM) and PP–CD (CA 37.5 μM, DATS 40.0 μM) were analyzed on MDA-MB-231 cells by using a standard MTT assay after 24 and 48 h incubation.
Cytotoxicity of PP–CD on GSHoet pre-treated cells
MDA-MB-231 cells (1 × 104) were cultured in 96-well plates for 24 h. The cells were incubated with GSHoet (1 mM) for 2 h and then CA (37.5 μM), DATS (40.0 μM) and CA (37.5 μM) + DATS (40.0 μM) were added. After 48 h incubation, the cell viability was examined by a standard MTT assay.
Cytotoxicity of PPR–CD
MDA-MB-231 cells (2 × 105) were cultured in a 24-well culture plate for 24 h and treated with PBS, PP–CD (CA 37.5 μM, DATS 40.0 μM) and PPR–CD (CA 37.5 μM, DATS 40.0 μM). After 18 h, the cell viability was assessed via a standard MTT assay.
AO/EB staining assay
The cytotoxicities of PP–C, PP–D and PP–CD were also evaluated by the AO/EB staining assay. MDA-MB-231 cells were treated as above, and then the cells were stained by AO (5 μg mL−1) and EB (5 μg mL−1) in PBS for 10 min. Finally, the cells were washed with PBS and imaged by a fluorescence microscope (Moticam 5000, Motic Instruments Inc.). The cell death induced by PP–CD (CA 37.5 μM, DATS 40.0 μM) and PPR–CD (CA 37.5 μM, DATS 40.0 μM) were also assessed by the same method.
Apoptosis detection
The Annexin V-FITC/PI kit was used to detect cell apoptosis. MDA-MB-231 cells (2 × 105) were incubated in 24-well plates for 24 h. The cells were then incubated with CA (37.5 μM), DATS (40.0 μM) and CA (37.5 μM) + DATS (40.0 μM) for another 24 h. For the analysis of apoptosis, the cells were stained with Annexin V-FITC (5 μL) and PI (5 μL) at room temperature for 15 min. Finally, the cells were analyzed by flow cytometry (FACS Caliber, Becton Dickinson, San Jose, CA). The nanoparticles of PP–D (DATS, 40.0 μM), PP–C (CA, 37.5 μM) and PP–CD (CA 37.5 μM, DATS 40.0 μM)-induced apoptosis was assessed by using the same method. The comparison of PP–CD (CA 25.0 μM, DATS 26.7 μM) and PPR–CD (CA 25.0 μM, DATS 26.7 μM)-induced apoptosis was also evaluated by the same method, except that a lower concentration of the agents was added.
ROS detection
MDA-MB-231 cells (2 × 105) were cultured in a 24-well culture plate for 24 h, and then were treated with CA (37.5 μM), DATS (40.0 μM) and CA (37.5 μM) + DATS (40.0 μM) for 12 h. After that, the cells were washed twice with cold PBS and were added to diluted DCFH-DA (1:1000), followed by incubation for 20 min at 37 °C in the dark. Finally, the cells were imaged by a fluorescence microscope and analyzed by a flow cytometer. The ROS promotion capability of PP–C (CA, 37.5 μM), PP–D (DATS, 40.0 μM) and PP–CD (CA 37.5 μM, DATS 40.0 μM) was also evaluated via the same method. The comparison of PP–CD (CA 25.0 μM, DATS 26.7 μM) and PPR–CD (CA 25.0 μM, DATS 26.7 μM)-induced ROS was also evaluated by the same method.
Mitochondrial membrane potential detection
MDA-MB-231 cells (1 × 106) were cultured for 24 h and treated with CA (37.5 μM), DATS (40.0 μM) and CA (37.5 μM) + DATS (40.0 μM) for 2 h. The cells were then dyed with JC-1 and Hoechst 33342 according to the instructions of the kit. Finally, the cells were observed by a laser scanning confocal microscope (Olympus FV1000).
Cellular uptake of the RGD-modified nanoagent
MDA-MB-231 cells (2 × 105) were cultured in a 24-well culture plate for 24 h and then incubated with PP–Co6 and PPR–Co6 for 1 h at 37 °C in the dark. Next, the cells were washed with PBS three times and imaged by an inverted fluorescence microscope (Olympus); they were then collected via trypsin-digestion and analyzed by a flow cytometer (BD, FACSCalibur, USA).
In vivo treatment of tumors
All the animal experiments in this study were carried out according to the National Institutes of Health guidelines for the care and use of laboratory animals and approved by the ethics committee of the East China Normal University (ECNU). The animals were housed under specific-pathogen-free conditions within the animal care facility at the ECNU. BALB/c nude female mice (4 weeks old, ∼18 g) were used to establish the tumor model. MDA-MB-231 cells (∼106 cells) suspended in 150 μL PBS were subcutaneously injected in the lower back of the nude mice. After two weeks of feeding, the tumor-bearing mice were randomly grouped, with five mice in each group, and were intravenously injected with one of the following: PBS, PP–C (CA, 0.55 mg kg−1), PP–D (DATS, 0.77 mg kg−1) and PP–CD (CA 0.55 mg kg−1, DATS 0.77 mg kg−1). The treatments were repeated every three days for four injections. The tumor volumes and the body weights of the mice were recorded every day. The anticancer capability of PPR–CD was further evaluated as above.
Histological examination
After the mice were sacrificed, the fresh heart, liver, spleen, lungs, and kidneys were collected and fixed with 4% paraformaldehyde. After dehydration, the tissues embedded in paraffin were cut into slices of 4 μm thickness on a microtome. After that, the tissue sections were stained with hematoxylin and eosin and were observed using a fluorescence microscope.
TUNEL assay
The tumor tissues harvested from sacrificed mice were fixed in 4% formalin solution, embedded in paraffin, and sectioned into slices with a thickness of 4 μm. The tumor sections were then incubated with proteinase K, the TUNEL reaction mixture and Hoechst 33342 according to the protocols of the in situ apoptosis detection kit. Apoptotic cells in the sections were imaged by a fluorescence microscope.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (31871010). The authors acknowledge East China Normal University (ECNU) Electron Microscopy Center for the nanoparticle characterization and ECNU Multifunctional Platform for Innovation (011) for the animal experiments.
References
- C. Gorrini, I. S. Harris and T. W. Mak, Nat. Rev. Drug Discovery, 2013, 12, 931–947 CrossRef CAS PubMed.
- B. C. Dickinson and C. J. Chang, Nat. Chem. Biol., 2011, 7, 504–511 CrossRef CAS PubMed.
- P. D. Ray, B. W. Huang and Y. Tsuji, Cell Signal, 2012, 24, 981–990 CrossRef CAS PubMed.
- S. J. Dixon and B. R. Stockwell, Nat. Chem. Biol., 2014, 10, 9–17 CrossRef CAS PubMed.
- K. Ito, A. Hirao, F. Arai, K. Takubo, S. Matsuoka, K. Miyamoto, M. Ohmura, K. Naka, K. Hosokawa, Y. Ikeda and T. Suda, Nat. Med., 2006, 12, 446–451 CrossRef CAS PubMed.
- P. T. Schumacker, Cancer Cell, 2006, 10, 175–176 CrossRef CAS PubMed.
- D. A. Callaway and J. X. Jiang, J. Bone Miner. Metab., 2015, 33, 359–370 CrossRef CAS PubMed.
- S. Manoharan, G. J. Guillemin, R. S. Abiramasundari, M. M. Essa, M. Akbar and M. D. Akbar, Oxid. Med. Cell Longev., 2016, 2016, 8590578 Search PubMed.
- N. Panth, K. R. Paudel and K. Parajuli, Adv. Med., 2016, 2016, 9152732 Search PubMed.
- S. Galadari, A. Rahman, S. Pallichankandy and F. Thayyullathil, Free Radic. Biol. Med., 2017, 104, 144–164 CrossRef CAS PubMed.
- P. T. Schumacker, Cancer Cell, 2015, 27, 156–157 CrossRef CAS PubMed.
- V. Nogueira and N. Hay, Clin. Cancer Res., 2013, 19, 4309–4314 CrossRef CAS PubMed.
- S. E. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- M. Schieber and N. S. Chandel, Curr. Biol., 2014, 24, R453–R462 CrossRef CAS PubMed.
- A. T. Shaw, M. M. Winslow, M. Magendantz, C. Ouyang, J. Dowdle, A. Subramanian, T. A. Lewis, R. L. Maglathin, N. Tolliday and T. Jacks, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8773–8778 CrossRef CAS PubMed.
- L. Tong, C. C. Chuang, S. Wu and L. Zuo, Cancer Lett, 2015, 367, 18–25 CrossRef CAS PubMed.
- L. Raj, T. Ide, A. U. Gurkar, M. Foley, M. Schenone, X. Li, N. J. Tolliday, T. R. Golub, S. A. Carr, A. F. Shamji, A. M. Stern, A. Mandinova, S. L. Schreiber and S. W. Lee, Nature, 2015, 526, 596 CrossRef CAS PubMed.
- J. Wang and J. Yi, Cancer Biol. Ther., 2008, 7, 1875–1884 CrossRef CAS PubMed.
- P. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu and J. Lin, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- M. Misawa and J. Takahashi, Nanomedicine, 2011, 7, 604–614 CrossRef CAS PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- J. Fang, T. Seki and H. Maeda, Adv. Drug Deliv. Rev., 2009, 61, 290–302 CrossRef CAS PubMed.
- B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS PubMed.
- L. S. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H. H. Yang and X. Chen, Angew. Chem., Int. Ed. Engl., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- G. Huang, H. Chen, Y. Dong, X. Luo, H. Yu, Z. Moore, E. A. Bey, D. A. Boothman and J. Gao, Theranostics, 2013, 3, 116–126 CrossRef CAS PubMed.
- A. M. Durantini, L. E. Greene, R. Lincoln, S. R. Martinez and G. Cosa, J. Am. Chem. Soc., 2016, 138, 1215–1225 CAS.
- K. S. Kim, D. Lee, C. G. Song and P. M. Kang, Nanomedicine, 2015, 10, 2709–2723 CrossRef CAS PubMed.
- W. C. Chou, C. Jie, A. A. Kenedy, R. J. Jones, M. A. Trush and C. V. Dang, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4578–4583 CrossRef CAS PubMed.
- M. A. Khan, S. Gahlot and S. Majumdar, Mol. Cancer Ther., 2012, 11, 1873–1883 CrossRef CAS PubMed.
- J. Ma, J. Yang, C. Wang, N. Zhang, Y. Dong, C. Wang, Y. Wang and X. Lin, Biomed. Res. Int., 2014, 2014, 107671 Search PubMed.
- K. Davison, S. Cote, S. Mader and W. H. Miller, Leukemia, 2003, 17, 931–940 CrossRef CAS PubMed.
- J. E. Biaglow and R. A. Miller, Cancer Biol. Ther., 2005, 4, 6–13 CAS.
- D. Trachootham, Y. Zhou, H. Zhang, Y. Demizu, Z. Chen, H. Pelicano, P. J. Chiao, G. Achanta, R. B. Arlinghaus, J. Liu and P. Huang, Cancer Cell, 2006, 10, 241–252 CrossRef CAS PubMed.
- J. Fang, T. Sawa, T. Akaike, T. Akuta, S. K. Sahoo, G. Khaled, A. Hamada and H. Maeda, Cancer Res., 2003, 63, 3567–3574 CAS.
- M. Khan, T. Li, M. K. A. Khan, A. Rasul, F. Nawaz, M. Sun, Y. Zheng and T. Ma, Biomed. Res. Int., 2013, 2013, 719858 Search PubMed.
- L. Chen, S. Li, X. Guo, P. Xie and J. Chen, Environ. Toxicol., 2016, 31, 552–560 CAS.
- Y. Sun, Y. Zheng, C. Wang and Y. Liu, Cell Death. Dis., 2018, 9, 753 CrossRef PubMed.
- J. Noh, B. Kwon, E. Han, M. Park, W. Yang, W. Cho, W. Yoo, G. Khang and D. Lee, Nat. Commun., 2015, 6, 6907 CrossRef CAS PubMed.
- H. Ka, H. J. Park, H. J. Jung, J. W. Choi, K. S. Cho, J. Ha and K. T. Lee, Cancer Lett, 2003, 196, 143–152 CrossRef CAS PubMed.
- T. Seki, T. Hosono, T. Hosono-Fukao, K. Inada, R. Tanaka, J. Ogihara and T. Ariga, Asia Pac J. Clin. Nutr., 2008, 17, 249–252 CAS.
- C. M. Cabello, W. B. Bair III, S. D. Lamore, S. Ley, A. S. Bause, S. Azimian and G. T. Wondrak, Free Radic. Biol. Med., 2009, 46, 220–231 CrossRef CAS PubMed.
- H. K. Na, E. H. Kim, M. A. Choi, J. M. Park, D. H. Kim and Y. J. Surh, Biochem. Pharmacol., 2012, 84, 1241–1250 CrossRef CAS PubMed.
- T. Satoh, Y. Enokido, H. Aoshima, Y. Uchiyama and H. Hatanaka, J. Neurosci. Res., 1997, 50, 413–420 CrossRef CAS PubMed.
- J. R. Wu, J. Wang, S. K. Zhou, L. Yang, J. L. Yin, J. P. Cao and Y. B. Cheng, Neural. Regen. Res., 2015, 10, 1120–1124 CrossRef CAS PubMed.
- D. Liang, H. Wu, M. W. Wong and D. Huang, Org. Lett., 2015, 17, 4196–4199 CrossRef CAS PubMed.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS PubMed.
- J. Morlieras, S. Dufort, L. Sancey, C. Truillet, A. Mignot, F. Rossetti, M. Dentamaro, S. Laurent, L. Vander Elst, R. N. Muller, R. Antoine, P. Dugourd, S. Roux, P. Perriat, F. Lux, J. L. Coll and O. Tillement, Bioconjug. Chem., 2013, 24, 1584–1597 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00301h |
| ‡ The authors (Y. Liu and H. Liu) contributed equally on this manuscript. |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a