
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of inorganic nanoparticle degradation in cancer therapy
Christy
Maksoudian
 ,
Neshat
Saffarzadeh
,
Evelien
Hesemans
,
Nora
Dekoning
,
Kiana
Buttiens
and
Stefaan J.
Soenen
*
,
Neshat
Saffarzadeh
,
Evelien
Hesemans
,
Nora
Dekoning
,
Kiana
Buttiens
and
Stefaan J.
Soenen
*
Department of Imaging and Pathology, KU Leuven, NanoHealth and Optical Imaging Group, Herestraat 49, B3000, Belgium. E-mail: s.soenen@kuleuven.be; Tel: +32 16 330034
First published on 27th July 2020
Abstract
Nanomaterials are currently widely exploited for their potential in the development of novel cancer therapies, and so far, mainly nanoparticles (NPs) consisting of liposomes and polymers have made their way into the clinic. However, major bottlenecks for the clinical translation of other types of NPs (i.e. inorganic) are the lack of knowledge concerning their long-term distribution in vivo and their potential toxicity. To counter this, various research groups have worked on soluble NPs, such as zinc oxide (ZnO), copper oxide (CuO), and silver (Ag), which tend to dissolve spontaneously into their ionic form, releasing toxic metal ions and leading to reactive oxygen species (ROS) generation when exposed to cellular environments. By fine-tuning the dissolution kinetics of these NPs, it is possible to control the level of ROS production and thus cytotoxicity to selectively destroy tumor tissue. Specifically, cancer cells tend to exhibit a higher basal level of oxidative stress compared to normal cells due to their higher metabolic rates, and therefore, by engineering NPs that generate sufficient ROS that barely exceed toxic thresholds in cancer cells, normal cells will only experience reversible transient damage. This review focuses on the use of these soluble inorganic NPs for selective cancer therapy and on the various in vitro and in vivo studies that have aimed to control the dissolution kinetics of these NPs, either through particle doping or surface modifications.
 Christy Maksoudian | Christy Maksoudian obtained her BSc in Biomedical Sciences from New York University and MSc in Drug Discovery and Development from Imperial College London, during which she worked extensively on the use of cell-penetrating peptides and gold nanoparticles in cancer therapy, respectively. She is currently pursuing her PhD at KU Leuven, with her work mainly focusing on the optimization of nanoparticle delivery to solid tumors using a variety of biological methods, particularly cell-based therapy. |
 Neshat Saffarzadeh | Neshat Saffarzadeh obtained her Masters' degree in Microbial Biotechnology from the University of Tehran in Iran in 2017 and is currently pursuing her PhD in Biomedical Science at KU Leuven under the supervision of Prof. Stefaan Soenen. Her research involves the use of various doped metal oxide nanoparticles for the treatment of cancer as well as bacterial and fungal infections. |
 Evelien Hesemans | Evelien Hesemans obtained a Masters' degree in Pharmaceutical Sciences in 2017, with a specialization in Drug Development, followed by a Master in Management, both at KU Leuven. At the moment she is a PhD candidate in the NanoHealth and Optical Imaging lab supervised by Prof. Soenen. Her PhD project focuses on the development and validation of optical imaging tools for advanced preclinical cancer research. She strives to design an imaging platform to visualize small cell numbers including therapy-resistant cells, cells undergoing EMT and cancer stem cells. |
 Nora Dekoning | Nora Dekoning graduated from the University of Antwerp with a Master's degree in Biochemical Engineering Technology in 2018. She joined the lab of Prof. Soenen at KU Leuven in 2019 as a PhD student in Biomedical Sciences in the cancer doctoral school. The main focus of her PhD is developing biomimetic nanoparticles for the optimization of innovative optical imaging techniques. |
 Kiana Buttiens | Kiana Buttiens is a PhD candidate in the Doctoral School of Biomedical Sciences, within the domain of Cancer. After receiving her Masters' degree in Biochemistry and Biotechnology from KU Leuven in 2018, she joined Prof. Soenen's NanoHealth and Optical Imaging Lab. Her research focuses on the use of nanoparticles in cancer therapy, mainly exploring their effect on extracellular vesicle-mediated cellular communication. |
 Stefaan J. Soenen | Stefaan J. Soenen heads the NanoHealth and Optical Imaging Group at KU Leuven, Belgium since January 2018. His main research interests lie in the use of nanomaterials for therapeutic and diagnostic purposes and on developing novel preclinical optical imaging methods. He has published over 75 manuscript with an h-index of 34 and received an ERC Starting Grant for his work on nanotherapeutics for cancer therapy. |
1. Introduction
Despite the long use of nanomaterials for biomedical applications, the field of nanomedicine only truly established itself as a separate field from nanotechnology in 2004 and involves the use of nanoparticles (NPs) that range from one to hundreds of nanometers for medical purposes.1 At this small scale, the surface area is significantly increased, altering the physical and chemical characteristics of the particles and giving rise to their various unique properties that render them suitable for a wide range of biomedical applications.2–4 For instance, NPs with varying compositions, sizes, and shapes can be used to enhance the solubility, biodistribution, and magnetic and optical properties of formulations, while their surfaces can be functionalized with various coatings and ligands to improve tissue selectivity and uptake into targeted cells.5,6 A wide range of different materials including liposomes, polymers, micelles, metal oxides (MOs) and other inorganic components can be employed for NP synthesis. These NPs can be subdivided in several ways based on their various characteristics. Two classifications that are often employed include chemical composition (organic or inorganic) and use (as delivery vehicle or direct physical/chemical therapy).The focus of nanomedicine in the clinic so far has been in the treatment of cancer (i.e. Doxil©, Abraxane©, Onivyde MM-398©, DaunoXome©), anemia and Fe deficiency (i.e. Cosmofer©, Feraheme©), and infections (i.e. AmbiSome©), and in their use as contrast agents for non-invasive imaging (i.e. Ferumoxtran-10©, SonoVue©) and in vaccinations (i.e. Epaxal©, Inflexal©). Specifically, the anticancer regimens Doxil© and DaunoXome© consist of liposomes loaded with doxorubicin (DOX) and daunorubicin, respectively, and are approved for the treatment of HIV-associated Kaposi's sarcoma. Additional indications for Doxil© include ovarian cancer and multiple myeloma. Moreover, the approval of no less than three new nanomedicines, VYXEOS© (FDA-approved), Patisiran© (FDA-approved) and Hensify© (CE-Mark approved), in the last couple of years clearly shows that the field of nanomedicine is being actively investigated in by companies and academic and research institutions worldwide.7 VYXEOS© consists of liposomes loaded with daunorubicin and cytarabine and was approved for the treatment of acute myeloid leukemia cancer in 2017. Hensify©, on the other hand, is a hafnium oxide NP-based aqueous suspension that is used for enhanced radiotherapy treatment and was recently approved in 2019. Patisiran©, the first siRNA/RNAi therapeutic, is also encapsulated within liposomes and has received FDA-approval in 2018 for the treatment of rare amyloidogenic polyneuropathy. Currently, there are a few thousand ongoing studies that use NPs mainly focusing on cancer and gene therapy.7 These studies mostly involve the so-called ‘soft’ NPs, consisting of liposomes or polymers, which predominantly aim to obtain a more efficient delivery of traditional anticancer agents (Fig. 1).
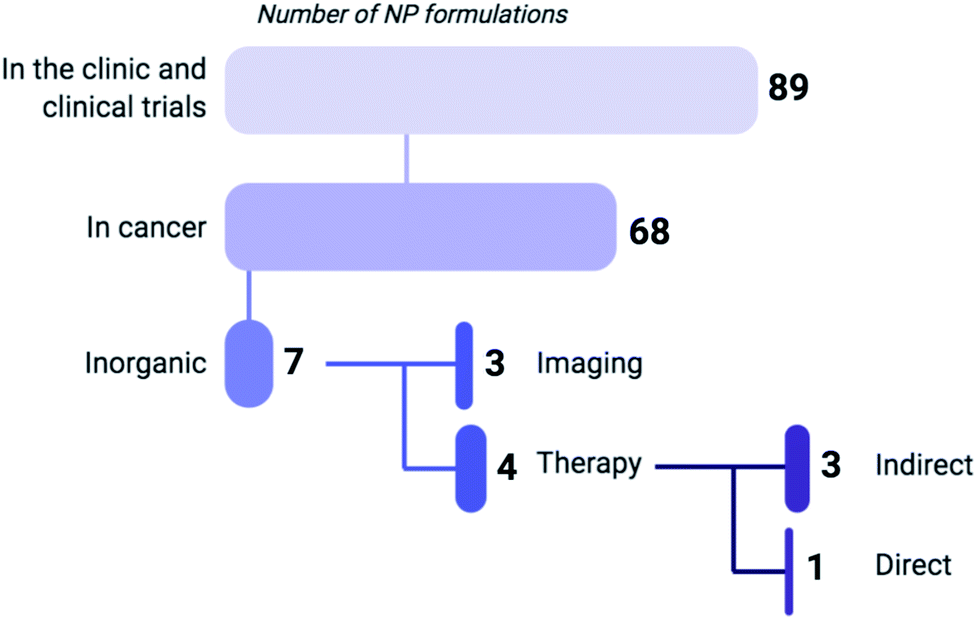 | ||
| Fig. 1 Based on a recent review by Anselmo and Mitragotri (2019) listing 89 NP formulations that are currently approved for use in the clinic or undergoing clinical trials, 75% of these regimens are indicated for cancer treatment (including gene therapy) or diagnosis, of which 90% are composed of organic materials such as liposomes, polymers and micelles.7 Six of the inorganic NP formulations that are clinically approved are used for imaging, while only one (NU-0129) functions as a direct therapeutic. As of 2019, NU-0129, consisting of Au NPs surface-covered with nucleic acids, is being tested in phase I clinical trials for the treatment of glioblastoma. The indirect-acting NPs exert their anticancer activity through radiation or thermal ablation by an external source. | ||
Harder NPs, often consisting of inorganic metals or MOs, have lower translational use, although a lot of progress has been made over the last years. For example, Fe oxide (IO) NPs are already clinically approved as hyperthermia agents for the treatment of multiforme glioblastoma (MagForce©) and are currently being tested on other types of tumors such as prostate cancer. Gold (Au) NPs are also being investigated in diverse cancer studies as agents for photothermal therapy (Aurolase©). In both cases, the NPs are used as indirect therapeutic agents, where external stimuli such as alternating magnetic fields and laser lights are applied upon administration of MagForce© and Aurolase©, respectively, to produce local heat, leading to cancer cell (CC) death. The lower translation of such NPs in the clinic has mainly been attributed to the lack of knowledge concerning their safety due to their potential to induce high levels of toxicity in biological systems compared to their bulk counterparts.8 This can be explained by the greater number of reactive sites on the NP surface associated with their higher surface-to-volume ratio as well as the high chemical reactivity that results from the structural modifications of their surface electronic properties.9,10 Concurrently, their nano size enables them to circumvent clearance by the reticuloendothelial system (RES), remain longer in blood circulation,11 passively traverse across the leaky tumor vasculature and penetrate deep into tumor tissue – a phenomenon known as the enhanced permeability and retention (EPR) effect.12,13 Put together, the larger exposure of the surface reactivity of NPs and their EPR effect lead to a higher level of particle interaction with their immediate surroundings, and therefore, greater potential for toxicity. However, this may not be the case for all types of materials, and even within a single type, the degree of toxicity is dependent upon multiple factors such as size, shape, coating and other surface modifications, as well as the nature of the exposed cell type.6,14–16
Therefore, there is a strong need to control NP-induced toxicity for effective tumor therapy. Alongside unaccounted-for toxicity, another factor that restricts the use of NPs in the clinic is the limited knowledge concerning their final destination, especially in the long-term. For example, Au NPs remain in the body for a long time and it is not yet entirely clear where these NPs will go after the therapy has been completed and what their long-term effects are, mainly in terms of the immune system, liver, spleen and kidneys.17–19 It has been suggested that such NPs remain intact within the tumor environment as a result of the thick extracellular matrix, poor lymphatic drainage, and eventual particle agglomeration. To counter this, various research groups have worked on soluble NPs, such as zinc oxide (ZnO), copper oxide (CuO), and silver (Ag). These NPs tend to dissolve spontaneously into their ionic form, releasing toxic metal ions (Mz+) when exposed to biotic/abiotic environments.20,21 In the context of the cell, the NPs are mainly taken up by endocytosis, during which their Mz+ release induces reactive oxygen species (ROS)-mediated toxicity in tumor tissues. This review will discuss the mechanisms by which these inorganic NPs degrade and exert their cytotoxicity, and how controlling the degree of Mz+ release from NPs can be harnessed to achieve selective tumor therapy. Although IONPs are the one of the most commonly used type of inorganic NPs for biomedical applications to date, their intracellular degradation has thus far not been linked to a cancer-selective toxicity (to our knowledge) particularly since the release of ferric ions is easily incorporated into the natural metabolism of the cell to be used in various biological functions and is thus tolerated at high levels.22 Therefore, this review will mainly focus on ZnO, CuO, and Ag NPs.
2. Inorganic NPs and their degradation
For all engineered NPs to be used for biomedical applications, their potential toxicity and removal from the body must be studied. In general, for organic materials, most commonly applied materials are biodegradable, resulting in the generation of biologically relevant components (e.g. polylactic-co-glycolic acid (PLGA) breaks down into lactic and glycolic acid, which are metabolites that can be processed in cellular Krebs cycle23). While the effect of these additional metabolites on cell growth and metabolism may not be negligible in all conditions, the toxicity thereof will be minimal. For inorganic NPs, this is typically a far more difficult story. Some NPs, such as Au NPs tend not to degrade and can persist very long inside the body.17 Other NPs can degrade, upon which they release Mz+. Any toxicity associated with this depends on multiple factors, including the chemical nature of the ions released and the rate of degradation. It is important to note that while some ions are physiologically relevant (e.g. Zn, Fe), their presence may affect other metabolites or disturb cellular homeostasis and hereby cause secondary toxicity. In the sections below, we aim to provide an overview of NP degradation (also referred to as “dissolution” throughout the text) with respect to how this can affect cellular homeostasis and how the process of NP degradation can be more controlled to selectively destroy CCs. Specifically, given that CCs exhibit a higher basal level of ROS compared to normal cells (NCs) due to their higher metabolic rates, NPs can be engineered to release Mz+ and generate ROS at finely-tuned levels that exceed toxic thresholds in CCs but only cause transient cellular damage in healthy tissue and allow gradual recovery.2.1. Oxidative stress due to Mz+ release from inorganic NPs as main cause of toxicity
The mechanism by which metallic NPs exert their cytotoxic effects has been mainly attributed to the generation of ROS or ROS-induced damage (Fig. 2).4,24 These species can be classified as non-radicals (i.e. H2O2, HOCl, O3) or free radical-containing (i.e.1O2, O2˙−, HO˙, HO2˙).25,26 Under normal conditions, ROS are produced as intermediates by the cell's natural oxidative metabolism and serve as key mediators in processes such as cell survival and signaling, and production of inflammatory factors.27,28 Their intracellular levels are strongly maintained by antioxidants (i.e. glutathione GSH), and detoxifying enzymes, including superoxide dismutases (SODs), GSH peroxidase (GPx), and catalase (CAT).29 SOD serves as a catalyst for the conversion of O2− to O2 and H2O2, and CAT further reduces H2O2 to H2O. The cellular compartments that are mainly associated with ROS production are the mitochondria, endoplasmic reticula (ER), peroxisomes, and NADPH oxidase (NOX) complexes located on the cell membranes.29–31 At the early stages of the ROS production process, increased levels of cytoplasmic calcium (Ca2+) trigger the activation of the mitochondrial electron transport chain, leading to the production of ATP and therefore, ROS as by-products.32,33 Disruption in the redox balance that regulates ROS homeostasis leads to the oxidative damage of various biomolecules and consequently, interference with different cellular functions.34,35 Among the alterations that ROS generation exert is the peroxidation of lipids leading to cell membrane damage, oxidation of proteins which interferes with enzymatic function and membrane permeability, and nucleic acid oxidation that causes genotoxicity.4,36 ROS-induced oxidative stress has been delineated as a three-tier model.35,37 Tier 1 is characterized by a defensive response wherein detoxifying enzymes including HO1, NAD(P)H, SOD and CAT aim to restore the redox balance and minimize damage through the activation of the phase II genes by Nrf2.38,39 Tier 2 results from ROS-induced stress that surpasses the enzymes' defensive capacity, leading to the production of pro-inflammatory cytokines and chemokines and thus, activation of inflammatory cells such as macrophages and neutrophils.40,41 Among the pro-inflammatory responses is the activation of mitogen-activated protein kinase (MAPK) and nuclear factor κB (NFκB).42,43 Tier 3 involves perturbation of the mitochondrial membranes and their potential through the formation of transient pores and increased influx of intracellular calcium, resulting in morphological changes or cell death by apoptosis, autophagy or necrosis.35 When NPs are internalized by the cells, they are taken up into the endosomes where the acidic environment and cellular biomolecules stimulate metal ion release from the NPs causing an imbalance in the local metal ion homeostasis and producing a strong but gradual increase in ROS that reaches optimum/Tier 3 levels after 24–48 h. Specifically, transition metals located at the surface of NPs, such as Cu on CuO NPs, have been shown to react with H2O2 to produce HO˙ and oxidized Cu ions via Fenton-type reactions.44,45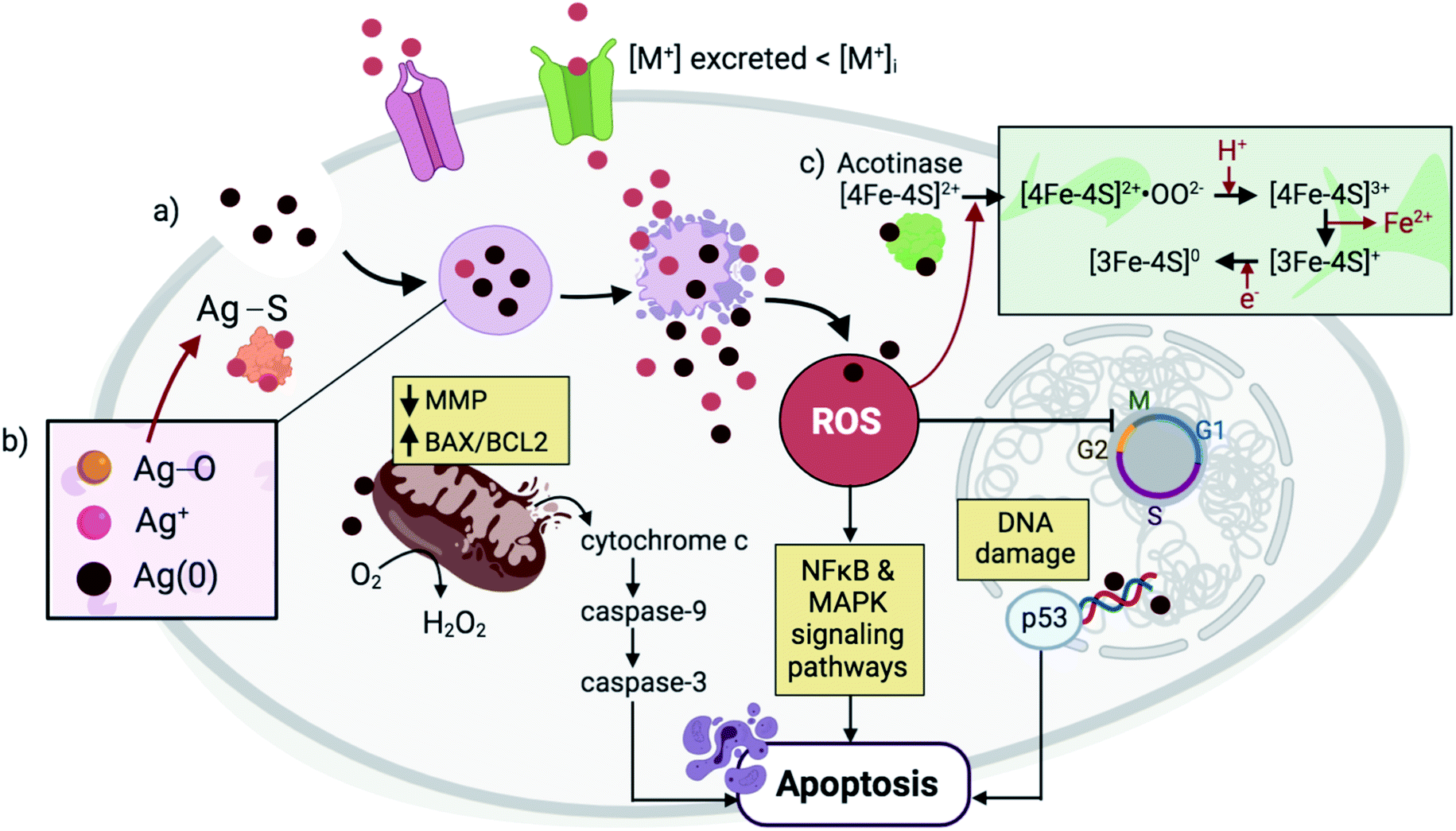 | ||
| Fig. 2 NPs are endocytosed and are contained within lysosomal compartments, during which they are exposed to low pH environments that accelerate their dissolution. (a) The high concentration of local Mz+ release (pink) exceeds the metal transporters' capacity to excrete ions, leading to ROS generation and DNA damage. In response, p53 triggers cell cycle arrest with the aim of restoring oxidative balance. Further elevation in ROS levels causes a decrease in the MMP and an increase in the BAX/BCL2 ratio, triggering cytochrome c release and activation of a caspase signaling pathway that leads to apoptosis. Other signaling pathways such as NFκB and MAPK are also activated during NP-mediated apoptosis. (b) In the case of Ag NPs, three forms of Ag exist simultaneously in the lysosomes, including solid Ag(0), Ag+, and Ag2O.65 Subsequent interaction of Ag2O intermediates with cysteine residues results in Ag–S formation that cause disruptions in protein secondary structures. (c) Meanwhile, in the case of ZnO NPs, Mz+ release and ROS generation cause alterations in the Fe–S clusters that are found abundantly in several protein families including aconitase, leading to protein inactivation.42 | ||
As with CuO and ZnO NPs, there have been several debates on the cytotoxicity induced by Ag NPs and their metal salts. In a study by Kim et al. (2009), the authors demonstrate comparable levels of cytotoxicity between Ag NPs and AgNO3, although the mechanism of toxicity differed for both.62 In cells treated with Ag NPs, no particular effect on the expression of MTs could be detected, which contrasts with the increased expression observed in AgNO3-exposed cells. When coupled with studies reporting that Ag NPs only partially dissolve in solution and thus release less free Ag+ compared with Ag salt, these findings suggest that the Ag NPs-induced toxicity is a product of oxidative stress rather than Mz+ release.63 Given that the mechanism of Ag+ release is shown to be via oxidative solution – during which surface metallic Ag reacts with protons and dissolved O2 to yield Ag2O, which is subsequently fully solubilized into Ag+ – the partial dissolution of Ag NPs has been attributed to the Ag+ readsorption onto the NP surface.64,65 In fact, Liu and Hurt (2010) demonstrate that three forms of Ag are found in colloidal suspensions of Ag NPs, including solid Ag(0), Ag+ that are available or have formed complexes with nutrients in the surrounding medium, and surface-adsorbed Ag+.65 The buildup of surface Ag+, in turn, blocks the O2 and proton access to metallic Ag, and this effect has been stated to be due to various NP surface coatings or particle agglomerations.65,66 However, others have reported the full dissolution of Ag NPs, and theoretical calculations suggest that Ag NPs are fully soluble in solutions with pH ranging from 4 to 12.65 At the same time, various studies have revealed that Ag NPs exhibit higher toxicity than Ag+.67,68 Specifically, a study by Eom and Choi (2010) indicates a time-dependent cytotoxic effect of Ag NPs,69 an observation that is similar to one made by Llop et al. (2014) on ZnO NPs.57 They show that, within 24 h of cellular incubation with Ag NPs and Ag+, the levels of oxidative stress were similar for both species. After 24 h, Ag NPs exhibit significantly higher toxicity compared to free Ag+, suggesting that they may serve as a long-term source of constant ion release. This data supports the notion that free Mz+ disperse themselves throughout the cells, while NPs tend to accumulate within endosomal compartments and release their Mz+ locally at high concentrations, exceeding toxic thresholds.
Similarly, the homeostasis of Cu2+ is maintained by Cu importers such as CTR1 and ATPase that respectively increase or decrease the flux of Cu2+ within the cell.78,79 When the level of intracellular Cu2+ is increased beyond normal conditions, CTR1 is internalized while ATPase is transferred to the cell membrane to eliminate the excess ions. However, during cellular uptake and subsequent dissolution of CuO NPs, the release of Cu2+ is significantly higher and more rapid than the capacity of ATPase to excrete the excess ions, disrupting the redox balance and inducing ROS generation and oxidative stress. The cellular homeostasis of Ag is also tightly regulated by metal transport genes and proteins. In a study by Wang et al. (2015), exposure of cells to Ag NPs led to an increase in mRNA expression levels of genes responsible for the extracellular release or sequestration of Mz+ and metal-bound complexes into lysosomes, including ATP-binding cassette transporter subfamily C member C1 (ABCC1), divalent metal transporter 1 (DMT1) and Cu-transporting ATPase 1 (ATP7A).80 The presence of Ag NPs within the lysosomal compartments also led to the disruption of the organelle membrane, followed by a significant reduction in MMPs after 24 h.
2.2. Factors influencing NP dissolution
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, as confirmed using single-crystal X-ray diffraction (SC-XRD).79 These findings are in line with previously reported structural formations between Cu2+ and glutamine.112 In fact, several early works have described the crystal structure that results from Cu2+ complexation with various amino acids, such as asparagine,113 glutamic acid114 and aspartic acid.115 Records of electron paramagnetic resonance (EPR) spectra in growth medium also indicate that all the Cu2+ released immediately bind with the nutrients present and precipitate to form crystals.79 Additionally, Gilbert et al. (2012) used high-resolution X-ray spectromicroscopy and high elemental sensitivity X-ray microprobe analyses to reveal that the full intracellular ZnO NP dissolution generated Zn2+ that immediately formed complexes with molecular ligands.116 This NP–protein interaction is able to cause protein misfolding, fibrillation, thiol crosslinking and thus loss of function.117–119 Interestingly, in a study by Pokhrel et al. (2013), the authors developed a model explaining that Zn2+ released from ZnO NP dissolution interact with rhombic [2Fe–2S], cuboidal [3Fe–4S], and cubane [4Fe–4S] FeS clusters found in various intracellular proteins, causing changes in cellular responses as a result of the reduced enzymatic activity.42,120,121 Specifically, NP-induced ROS production is able to damage the highly oxidation-sensitive [4Fe–4S]2+ cluster found in the aconitase protein family,122 generating the intermediate [4Fe–4S]2+˙OO2−. This resulting species reacts with free protons and releases Fe2+ to produce [3Fe–4S]3+, which is then rendered inactive ([3Fe–4S]0) upon univalent reduction.123,124 Concurrently, Ag+ have been shown to complex with Cl−, Br− and thiol-containing biomolecules leading to further damage to cellular functionality.125–127 Using SR-XANES, Wang et al. (2015) revealed the chemical transformation of Ag NPs – as a result of surface oxidation by ROS and dissolved O2 – from Ag(0) to Ag2O, which then interacts with organic acid molecules to form AgS.80 Major sources of intracellular thiol include cysteine and methionine-containing peptides, proteins and antioxidants (i.e. MTs), which complex with Ag NPs to form more stable entities.63,128,129 In fact, the presence of Ag NPs caused a 5-fold and 10-fold increase in the mRNA expression of MTs after 12 h and 24 h, respectively, while circular dichroism spectra revealed alterations in the secondary structures of these antioxidants.63,80
:2, as confirmed using single-crystal X-ray diffraction (SC-XRD).79 These findings are in line with previously reported structural formations between Cu2+ and glutamine.112 In fact, several early works have described the crystal structure that results from Cu2+ complexation with various amino acids, such as asparagine,113 glutamic acid114 and aspartic acid.115 Records of electron paramagnetic resonance (EPR) spectra in growth medium also indicate that all the Cu2+ released immediately bind with the nutrients present and precipitate to form crystals.79 Additionally, Gilbert et al. (2012) used high-resolution X-ray spectromicroscopy and high elemental sensitivity X-ray microprobe analyses to reveal that the full intracellular ZnO NP dissolution generated Zn2+ that immediately formed complexes with molecular ligands.116 This NP–protein interaction is able to cause protein misfolding, fibrillation, thiol crosslinking and thus loss of function.117–119 Interestingly, in a study by Pokhrel et al. (2013), the authors developed a model explaining that Zn2+ released from ZnO NP dissolution interact with rhombic [2Fe–2S], cuboidal [3Fe–4S], and cubane [4Fe–4S] FeS clusters found in various intracellular proteins, causing changes in cellular responses as a result of the reduced enzymatic activity.42,120,121 Specifically, NP-induced ROS production is able to damage the highly oxidation-sensitive [4Fe–4S]2+ cluster found in the aconitase protein family,122 generating the intermediate [4Fe–4S]2+˙OO2−. This resulting species reacts with free protons and releases Fe2+ to produce [3Fe–4S]3+, which is then rendered inactive ([3Fe–4S]0) upon univalent reduction.123,124 Concurrently, Ag+ have been shown to complex with Cl−, Br− and thiol-containing biomolecules leading to further damage to cellular functionality.125–127 Using SR-XANES, Wang et al. (2015) revealed the chemical transformation of Ag NPs – as a result of surface oxidation by ROS and dissolved O2 – from Ag(0) to Ag2O, which then interacts with organic acid molecules to form AgS.80 Major sources of intracellular thiol include cysteine and methionine-containing peptides, proteins and antioxidants (i.e. MTs), which complex with Ag NPs to form more stable entities.63,128,129 In fact, the presence of Ag NPs caused a 5-fold and 10-fold increase in the mRNA expression of MTs after 12 h and 24 h, respectively, while circular dichroism spectra revealed alterations in the secondary structures of these antioxidants.63,80
3. Controlling NP dissolution for selective tumor therapy
Selective tumor therapy can thus be achieved by engineering NPs that are responsive to the low pH surrounding tumor tissue and that generate sufficient levels of ROS to induce apoptotic responses in CCs with high metabolic rates, while the additional ROS experienced by normal peripheral cells with higher physiological pH and lower metabolic rates should not reach apoptotic thresholds and only cause temporary reversible damage (Fig. 3). Several methodologies have already been explored to tune the toxicity profiles of the NPs by controlling their distribution, dissolution and oxidative capacity. For instance, Le et al. (2016) conducted a multiparametric evaluation of ZnO NPs with varying physicochemical properties such as size (proportional to calcination temperature), aspect ratio (spheres, grains, rods, or needles), dopants with different concentrations (Fe, Mn, or Co), and coatings (polymethyl-methacrylate PMMA, silica, oleic acid OA, or serum protein) and assessed their effect on human umbilical vein endothelial cells (HUVECs) and hepatocellular liver carcinoma (HepG2) cells using three toxicity endpoints, including viability, membrane integrity and oxidative stress.136 The objective of this study was to determine the parameters that have the greatest impact on toxicity and subsequently develop computational models that would be able to predict NP behavior. The descriptors used for the doping elements were similar although not identical to those described above by Simeone et al. (2019), and include IP, conduction band energy (EC), and reduction potential (RP). With respect to cell viability, the parameters that had the greatest impact include NP concentration and size, extent of NP shielding from surrounding (solubility and coating type), and the resulting change in IP and EC associated with doping, particularly for Mn-doped oxides which had the largest cytotoxic effect, in contrast to serum-coated NPs that enhanced cell viability. Next, membrane damage was measured by the amount of lactate dehydrogenase (LDH) released, and was shown to be significantly affected by IP, EC, and RP, as well as NP shape, size, solubility and zeta potential. Specifically, in line with previously reported studies, positively-charged NPs exhibited higher LDH release compared to their negatively-charged counterparts, possibly due to the greater interaction of the cationic NPs with the abundant negative residues on cell membranes. Finally, antioxidant response elements (AREs) were used as markers for oxidative stress and were shown to be most affected by the dopant descriptors IP, EC and RP, particularly as a result of Mn- and Co-doping. However, in the context of selective cancer therapy using ZnO and CuO NPs, given that these particles in their bare form already exert the toxicity required for apoptosis in most cell lines as will be discussed in Section 4, doping them with elements such as Mn and Co that further induce cellular toxicity does not serve the purpose of reducing the impact on NCs. In contrast, doping these NPs with elements such as Fe reduces their dissolution kinetics and associated toxicity, which provides an opportunity to maintain the viability of normal tissue while only exceeding toxic thresholds in the highly metabolic CCs.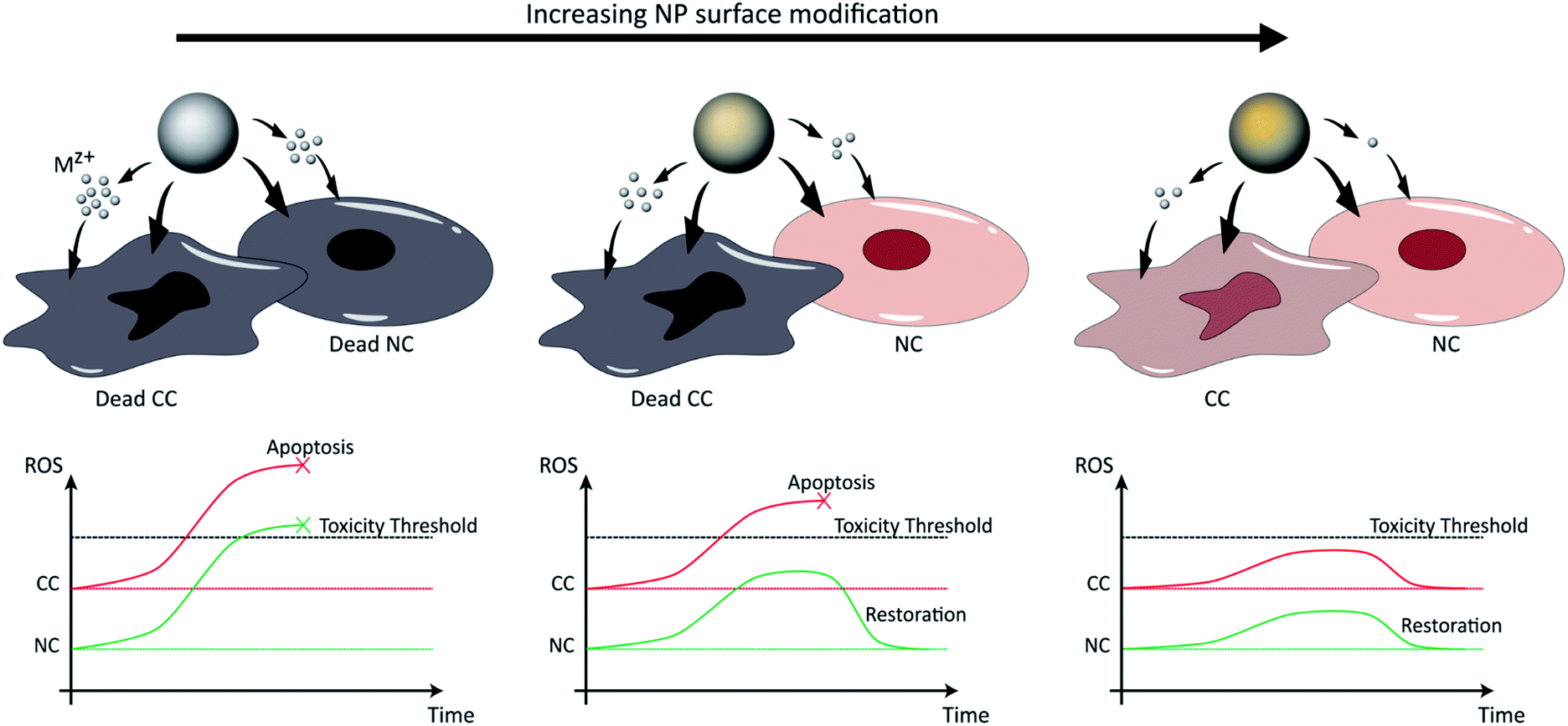 | ||
| Fig. 3 NP dissolution kinetics and associated toxicity can be tuned by surface modification, including doping and coating, and are increased in the low pH tumor microenvironment. In the first scenario (left), pure NPs release high concentrations of ions that induce ROS generation at levels exceeding toxic thresholds for both CCs and NCs. In the second scenario (right), the excess surface modification of NPs causes a slow release of Mz+ at non-toxic amounts, preserving the viability of both cell types. Finally, in the third scenario (middle), the Mz+ release associated with specific extents of NP surface modification is sufficient to exceed toxic thresholds in CCs that possess high metabolism (higher basal levels of ROS), but low enough to exert only transient damage in NCs with low metabolism and to restore oxidative balance. | ||
3.1. The effect of NP doping on dissolution properties
Doping the NPs with various elements leads to modifications in their electronic state density, magnetic moments, charge distribution, and lattice distortions, altering their reactivity, stability and various other properties. Given that kinetic factors hinder the incorporation of dopants into the NP structure, effective doping is only achieved when the growth rates of the host and dopant are balanced.137 In this way, the dopant properly substitutes the host core atoms rather than simply adsorb onto the NP surface. In an early work by Bilecka et al. (2011), ZnO NPs were doped with Fe, Mn, Ni, Co and V, and the greatest doping efficiency was found for Fe and Co due to their comparable reactivity and size of their divalent ions with Zn.138 Vanadium, however, had the least effective doping (<3% of dopant precursor content) as it has a larger ionic radius and is a much harder Lewis acid compared with Zn.As a result, Fe-doping has been heavily explored for ZnO and CuO NPs, and has been shown to significantly reduce particle dissolution and cytotoxicity at levels proportional to the extent of doping,92 although the toxicity reduction mechanisms for both NP types differ. Within the CuO NP structure, Cu resides among two and four oxygen ligands in the apical and planar positions respectively, which result in Jahn–Teller distortions upon Fe-doping as well as CuFe2O4 spinal formation.139 The resulting increased structural stability and surface spinal formation hinders excess Cu2+ release. In Fe-doped ZnO NPs, the increased stability compared with pure ZnO is due to the stronger structural binding of Fe than Zn to O, while the Fe atoms simultaneously serve as kinetic constraints for Zn release.140,141 It has also been suggested that the additional cytoplasmic Fe2+ availability originating from the Fe-doped ZnO NPs is able to reduce the previously mentioned oxidized FeS clusters to reverse the protein inactivation process by regenerating the active [4Fe–4S]2+ cluster.42 These reports were further supported by an experiment conducted in E. coli that reveal that unstable [4Fe–4S]2+ clusters tend to form inactive [4Fe–4S]+, and subsequent exposure to Fe ions and dithiothreitol leads to protein reassembly with a 60% gain in protein activity within the span of a few minutes.142 Therefore, this mechanism of protein function salvaging may also contribute to the reduced cytotoxicity observed with Fe-doped NPs compared to their pure counterparts. Additionally, doping MOs results in the narrowing or expansion of the NP band structure and changes in the positioning of the conduction band edges, altering the levels of ROS generation.99
In a recent study from our group investigating the effects of varying Fe doping levels on CuO NP toxicity and cancer therapy, we found that the rapid release of Cu2+ from pure CuO causes cell death in both normal and tumor cells, whereas a too slow release leaves both cell types mostly unaffected.79 By adjusting the Fe-doping levels to fit the exact desired release kinetics of the Mz+, it was possible to selectively target CCs. The most beneficial response was achieved at Fe doping levels of 6%. Although the release rate of 6% Fe-doped CuO NPs was initially rapid and overlapped with that of pure CuO, it significantly diminished with time, leading to a slow long-term release (Fig. 4a and b). This decrease in dissolution rate is due to the strong Jahn–Teller distortion that adjusts the planar and apical Cu–O bond lengths. In fact, a 10% Fe-doping of CuO has been previously associated with a 6.4% plane elongation and 4.1% distortion compared to pure CuO.139 Our EPR spectra showed a Cu release of ∼65% from pure CuO and only 8% from 10% Fe-doped CuO in the first 10 minutes of NP exposure to cell culture medium. Using powder diffraction, Rietveld analysis and EDX spectroscopy, we suggested that dissolution causes a transformation from CuO → CuFe2O4 → Fe3O4 as well as the formation of Fe–O (Fig. 4e and f). Taking into account several assumptions, such as a dissolution rate that is proportional to the surface Cu concentration which itself is proportional to the active surface area for instance, we developed a two-step model to describe the dissolution kinetics of Fe-doped CuO (Fig. 4g). The first step involves the repeated Fe redistribution on the NP surface during Cu2+ release and thus a continuous increase in the Fe/Cu surface ratio until all the surface Cu is released, forming an Fe shell. This step is followed by the slow solid-state diffusion of the core Cu to the surface until all the Cu has been released. The remaining Fe particles are then degraded into their ionic form (i.e. via Fenton reactions, enzymes44), which are then incorporated into the natural Fe metabolism of the cell with the help of ferritin. In fact, this Fe integration has been evidenced through the radioactive labeling of magnetic FeO NPs and the subsequent localization of labeled Fe within the hemoglobin of newly-developed erythrocytes in vivo.22 Therefore, given the safety and success of Fe doping in controlling Mz+ release and reducing NP cytotoxicity, efforts have been made to test various core-dopant combinations for selective tumor therapy, such as Zn-doped CuO NPs, which have shown to have significant anticancer properties in vitro and in vivo. These findings will be discussed further in Sections 4 and 5.
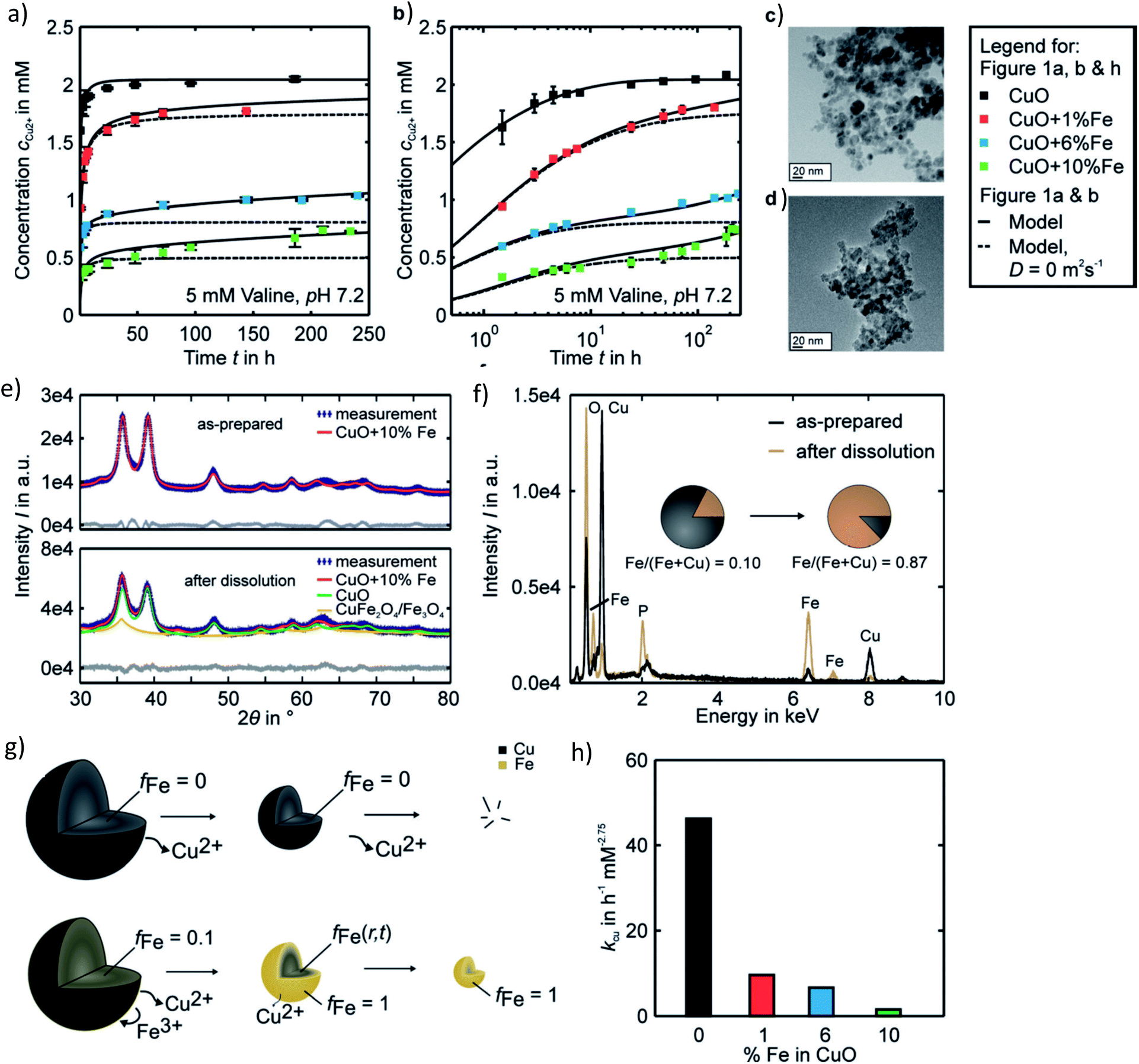 | ||
| Fig. 4 Pharmacokinetics of Fe-doped CuO NPs as described by Naatz et al.79 (a and b) Cu2+ release profiles showing a slow long-term release in the case of the higher doped NPs in the logarithmic time scale. (c and d) TEM images of 10% Fe-doped NPs before and after dissolution in the span of 4 weeks. (e and f) Powder diffraction patterns and EDX spectra depicting the change in particle composition before and after dissolution of the 10%-doped CuO NPs. (g) Proposed two-step model of Fe-doped CuO NP dissolution wherein all the surface available Cu is first released until an Fe shell forms, followed by the solid-state diffusion of core Cu to the surface until all the particle Cu is removed. (h) Decrease in the dissolution rate constants with increasing Fe doping. This image has been reproduced with permission from Naatz et al.,79 © Wiley-VCH, 2020. | ||
3.2. The effect of NP coating on dissolution properties
Besides doping, surface modification of NPs using a wide variety of coatings have also been heavily investigated for their potential in controlling the dissolution kinetics of NPs. For instance, given that silica is highly stable in neutral and acidic environments, a silica coating has been developed around ZnO NPs with the aim of reducing NP degradation and toxicity.143 This strong hydrophilic coating turns the previously positively-charged surface of the ZnO NPs negative, and the absolute increase in the zeta potential enhances NP stability in water. Optimizing the amount of silica coating could thus result in ZnO NPs with specific degradation profiles. In two separate studies, other types of surface coatings were assessed on either CuO144 or ZnO145 NPs with the aim of completely reducing the surface reactivity and Mz+ release from the particles. Osmond-McLeod et al. (2013) demonstrate almost full reduction of ZnO NP toxicity as a result of surface coating them with dimethoxy-diphenylsilane/triethoxy-caprylylsilane crosspolymer (MAX) compared to pure ZnO.145 However, coating the NPs with triethoxycaprylylsilane (HP1) resulted in moderate cytotoxic response. Given that the aim of the study was to fully reduce NP dissolution as a safety measure and as a result, the HP1-coated NPs were not further investigated, these findings suggest the need to further test the HP1-coated NPs for the purposes of cancer therapy by tuning their dissolution kinetics. Similar conclusions can be derived from the study by Cai et al. (2017), who show that after coating with ethylenediamine tetra (methylene phosphonic acid) (EDTMP), the extent of CuO dissolution was less than 0.2% and almost no ROS generation and toxicity were observed in THP-1 and BEAS-2B cells.144 They also report that the coating does not disrupt the CuO crystal structure and morphology. Meanwhile, they tested the impact of coating NPs with citrate and PVP (polyvinylpyrrolidone), which had limited and moderate protective effects, respectively. Similarly to the case of HP1-coated ZnO NPs, optimizing NP dissolutions profiles may be achieved by investigating varying degrees of NP coating with PVP. A follow-up study by Líbalová et al. (2018) revealed that varying NP surface modifications (anionic sodium citrate CIT, sodium ascorbate ASC, neutral PVP, cationic polyethylenimine PEI) only moderately affected Cu2+ release and that the toxicity observed in a murine macrophage cell line was mainly determined by the NP coating agent and Cu bioavailability rather than the intracellular Cu burden, with the PEI-coated NPs being the most toxic.474. Cellular studies on cancer-specific NP toxicity in vitro
Although a few types of doping and coatings were introduced in the previous section, there exists a variety of other surface materials that have been experimented with in order to fine tune the dissolution kinetics of NPs. In this next part, we initially discuss the various studies that have tested the anticancer activity of pure ZnO, CuO, and Ag NPs in vitro, followed by the efforts that have been made over the years to further enhance CC selectivity using the assorted doping and coating materials and their cellular responses. Please note that a wide range of different cell types have been used in the various studies, which will not be described in detail as this would be outside of the scope of the current manuscript. For clarity purposes, we have listed all cell types described with their abbreviation and full name in Table 1. A summary of all the discussed studies is also provided in Tables 2–4.| Cell line | Type | Cell line | Type |
|---|---|---|---|
| A172 | Human glioblastoma | KCL22 | Human chronic myeloid leukemia |
| A549 | Human lung adenocarcinoma | KLN205 | Murine lung carcinoma |
| AsPC-1 | Human pancreatic tumor | L02 | Human hepatic |
| B16 | Murine melanoma | L5178Y-R | Murine lymphoma |
| BEAS-2B | Human lung epithelial | LS174T | Human colon cancer |
| BeL7402 | Human hepatocellular carcinoma | MCF-7 | Human breast cancer |
| BJ | Human normal fibroblasts | MCF-7/ADR | MCF-7 adriamycin-resistant (ADR) |
| BxPC-3 | Human pancreatic cancer | MDA-MB-231 | Human breast cancer |
| C6 | Rat glioma | MG-63 | Human osteosarcoma |
| CHO | Chinese hamster ovary epithelial | MIA Paca-2 | Human pancreatic cancer |
| COLO 205 | Human colon adenocarcinoma | MSC | Mesenchymal stem cells |
| CT26 | Murine colorectal carcinoma | N417 | Human small cell lung cancer |
| DAL | Canine hemangiosarcoma | NCI-H460 | Human non-small lung cancer |
| EAC | Murine Ehrlich Ascites carcinoma | NIH3T3 | Mouse embryonic fibroblasts |
| ESC | Embryonic stem cells | OUS-11 | Human lung normal |
| H1299 | Human non-small lung cancer | PANC1 | Human pancreatic cancer |
| H187 | Human small cell lung carcinoma | Panc28 | Human pancreatic adenocarcinoma |
| H82 | Human small cell lung carcinoma | PBMC | Peripheral blood mononuclear cells |
| HCC | Human hepatocellular carcinoma | PC3 | Human prostatic cancer |
| HCT116 | Human colorectal carcinoma | RAW264.7 | Murine macrophage |
| HeLa | Human Henrietta Lacks immortal cells | SHSY5Y | Human neuroblastoma |
| HepG2 | Human liver hepatocellular carcinoma | SK-MES-1 | Human lung cancer |
| HT-29 | Human colon cancer | SKBR3 | Human breast cancer |
| HT1080 | Human fibrosarcoma | SUM159 | Mesenchymal TNBC |
| hTERT-HPNE | Immortalized human pancreatic duct | TNBC | Triple-negative breast cancer |
| HUVEC | Human umbilical vein endothelial | U251 | Human glioblastoma |
| Jurkat | Human immortalized T lymphocytes | U87 | Human primary glioblastoma |
| K562 | Human myelogenous leukemia | WI38 | Human lung fibroblasts |
| K562/A02 | Human leukemic | WISH | Human amnion |
| Surface mod. | Size (in nm) | Main finding | VT | VV | Model systems | Dose/concentration | Cellular/tumoral effects | CCS | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ccore size, HDhydrodynamic size, VT in vitro, VV in vivo, CCS cancer-cell specific. | ||||||||||
| Pure | 21C, 131HD | ZnO NPs selectively induce apoptosis in CCs via ROS, mediated by p53, bax/bcl-2 and caspase pathways | ● | HepG2, A549, BEAS-2B, rat astrocytes & hepatocytes | 5–15 μg mL−1 | Dose-dependent ↓ cell viability | ● | ↑ p53 and bax and ↓ bcl2 | 89 | |
| Pure | 5C, 6C | Newly synthesized ZnO QDs with promising optical properties show cytotoxic effects in CC lines | ● | K562, K562/A02, HepG2 | 1.25–100 μg mL−1 | Dose-dependent ↓ cell viability | 146 | |||
| Pure | 35C | ZnO NPs restore oxidative balance, hepatocyte integrity and tumor markers to normal levels in HCC | ● | ● | VT: HepG2, PC3, A549; VV: HCC | VT: 0–1000 μmol L; VV: 10 μg kg−1 per week | Dose-dependent ↓ cell viability | Restoration of oxidant and antioxidant activity (MDA, GSH, GPx, GSR, SOD, CAT), hepatocyte integrity (ALT, AST, LDH) and tumor biomarkers (AFP, AFU) to normal levels in CCs | 147 | |
| Pure | 95HD | ZnO NPs exert toxicity in normal and CCs through ROS generation, whereas CeO2 and TiO2 NPs do not | ● | A549, NCI-H460, SK-MES-1, HeLa, Jurkat, AT II | 2–6 μg cm−2 | ↓ cell viability | Complete particle dissolution and ↑ ROS | 57 | ||
| Pure | 100HD | ZnO and Ag NPs evoke stress-induced autophagy in pulmonary and hepatic cells while TiO2 NPs do not | ● | A549, HepG2 | 1–500 μg mL−1 | Dose-dependent ↓ cell viability | Autophagy induction (↑ LC3B, atg4b, p62 and ↓ atg12, atg5) in a time-dependent manner, and apoptotic cell death via caspase-3 | 148 | ||
| Pure | 40C | Apoptosis and oxidative stress as relevant mechanisms of antitumor activity and genotoxicity of ZnO-NPs | ● | ESC | 50, 300 and 500 mg kg−1 per body weight | Oxidative stress (↑ MDA and ↓ CAT, GST) and DNA damage. Apoptosis (↑ Bax and p53, ↓ Bcl2). NAC restores oxidative balance in the liver and kidney without ↓ the antitumor efficacy of NPs | 184 | |||
| Pure | 20HD, 70HD | Genotoxic anticancer effects of ZnO NPs via ROS, leading to non-apoptotic cell death in an orthotopic mouse model of human small-cell lung cancer | ● | ● | VT: H82, H187, BEAS-2B, MCF-7, OUS-11, LS174T, N417; VV: N417 | VT: 0–20 μg mL−1; VV: 0.04–0.25 mg kg−1 | Dose-dependent ↓ cell viability. ↓ tumor density | ● | ↑ ROS and DNA leakage from nuclei. Phos. of CHK2 in N417 and LS174T cells. ↑ cleaved PARP in LS174T cells. Q-VD-OPh (inhibitor of caspase-3, 1, 8, 9, slightly ↑ viability by inhibiting apoptosis, but did not block ZnO toxicity) | 185 |
| Fe-doped (0–10%) | 20.2C–8.3C (0–10%) | Fe doping reduces ZnO toxicity in animals due to decreased NP dissolution rates and associated toxicological responses | ● | ● | VT: RAW264.7, BEAS-2B; VV: rat & mouse lung, zebrafish embryo | VT: 12.5 μg mL−1; VV: 0–50–150 μg mL−1 | ↓ toxicity with ↑ Fe doping | ↑ Fe doping leads to ↓ inhibitory effect of Zn2+ in zebrafish embryo hatching, ↓ PMN cell counts in the BAL fluid and IL-6 mRNA and ↑ heme oxygenase 1 in mouse lung, and ↓ BAL PMN cell counts, LDH, and albumin in rat lung | 163 | |
| Fe-doped (0–10%) | 11C, 5.5C (0, 10%) | 2% Fe-doped ZnO NPs are found to be optimal to cause selective CC death and reduce metastasis formation | ● | ● | VT: mMSC, Beas-2B, HeLa, KLN205; VV: KLN 205 | VT: 0–35 μg mL−1; VV: 125 μg per animal | ↓ cell viability. ↓ toxicity with ↑ Fe doping. ↓ tumor growth and metastasis (2, 10%) | ● | ↑ ROS, membrane and mitochondrial damage and autophagy. ↓ toxicological response with ↑ Fe doping due to resulting ↓ in cellular levels of Zn2+. VV: pure NPs led to weight loss and premature death of mice | 77 |
| La-doped (0–5%) | 33C, 29C (0, 5%) | Doping ZnO NPs with La increases NP photocatalytic activity and cytotoxicity | ● | MDA-MB-231, KCL22, HeLa | 6–500 μg mL−1 | Dose-dependent ↓ viability. ↑ toxicity with La doping | 164 | |||
| Sm-, Eu-, Gd-doped | >100C | Sm-doped ZnO NPs displays the most significant antitumor activity compared to other lanthanide-doped ZnO NPs | ● | ● | EAC | VT: 0–0.05 mol; VV: 150–350 mg kg−1 | Dose-dependent ↓ cell viability for Sm3+:ZnO NPs |
Compared to pure NPs, Sm-doping ↓ tumor size, ↓ PI3K, Akt and mTOR, ↓ CXCR4 and P450, ↓ Bcl2:Bax ratio, ↓ liver function enzymes (AST, ALT) and induce G2 cell cycle arrest |
165 | |
| PEG or starch coated | 40–1200HD | Toxicity towards osteoblast CCs is dependent on NP size, aspect ratio and coating. PEG-capped NPs exhibit higher toxicity than starch-capped NPs | ● | MG-63 | 1 μM to 7 mM | Dose-dependent ↓ cell viability. ↓ toxicity with ↑ NP size | 167 | |||
| Surface mod. | Size (in nm) | Main finding | VT | VV | Model systems | Dose/concentration | Cellular/tumoral effects | CCS | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ccore size, HDhydrodynamic size, VT in vitro, VV in vivo, CCS cancer-cell specific. | ||||||||||
| Pure | 7C, 127HD | CuO NPs inhibit pancreatic tumor growth primarily by targeting TICs via ROS and mitochondrial pathway | ● | ● | PANC1 | VT: 0–50 μg mL−1; VV: 0–12.5 mg kg−1 | Dose- and time-dependent ↓ cell viability. Tumor growth inhibition | ● | ↑ ROS, ↓ MMP, apoptosis of TICs (arrest in sub G1 phase) | 149 |
| Pure | 22C, 167HD | CuO NPs induce mitochondria-mediated apoptosis in human hepatocarcinoma cells | ● | HepG2 | 0–50 μg mL−1 | Dose-dependent ↓ cell viability | Oxidative stress (↑ MDA, ↓ GSH) ↑ ROS, DNA damage. Mitochondria-mediated apoptosis (↓ MMP, ↑ P53, ↑ BAX/BCL2 and caspase-3) | 150 | ||
| Pure | 20C | CuO NPs induce cytotoxicity via mitochondrial pathway | ● | K562, PBMC | 0–25 mg mL−1 | Dose-dependent ↓ cell viability | ● | ↑ ROS, mitochondria-mediated pathway, ↑ P53 and Bax/Bcl2 | 151 | |
| Pure | 30C, 235HD | Autophagy is the main mechanism of CuO NP-induced cell death, while apoptosis is only triggered secondarily | ● | MCF7 | 0–12 μg mL−1 | Dose- and time-dependent ↓ cell viability | Autophagy: ↑ MAP-LC3-II, Beclin1 and ATG5. 3 MA inhibits autophagy, and further Beclin1 KD leads to apoptosis (↑ PARP-cleavage, BAD dephosphorylation and caspase-3) | 95 | ||
| Pure | 12C | CuO NPs synthesized from Eucalyptus globulus induce apoptosis in breast carcinoma | ● | MCF7 | 0–100 μg mL−1 | Dose-dependent ↓ cell viability | Impaired MMP, ↑ ROS, ↑ p53, bax, caspase-3, and caspase-9, cell cycle arrest in G1, S and G2/M phases | 90 | ||
| Fe-doped (0–10%) | 11.8C–10.7C (0–10%) | 6% Fe-doped CuO NPs induce inhibition of tumor growth and complete tumor remission when combined with immunotherapy | ● | ● | VT: MSC, Beas-2B, HeLa, KLN205; VV: KLN205 | VT: 0–35 μg mL; VV: 0–225 μg kg−1 bw (6%) | ↓ toxicity with ↑ Fe doping. ↓ tumor growth (6%-doped) | ● | ↑ membrane damage, ROS, autophagy. ↓ toxicity with ↑ Fe doping. VV: ↑ local antitumor immune response (activation of CD8+ and NK cells). Complete tumor remission due to treatment with 6%-doped NPs and EPAC | 79 |
| Fe-doped (10%) | 235HD, 247HD (0, 10%) | Fe-doping of CuO NPs lowers their toxic potential on glioblastoma cells by slowing down Cu release | ● | C6 | 0–1000 μM | Dose- and time-dependent ↓ cell viability. ↓ toxicity with ↑ Fe doping | ↑ ROS and oxidative stress. Cu chelators can prevent Cu-induced toxicity | 168 | ||
| Zn-doped | 30C | Zn–CuO NPs exert selective antitumor activity (inhibition of glioblastoma growth) and reverse temozolomide resistance in glioblastoma by inhibiting AKT and ERK1/2 | ● | ● | VT: Panc28, HCT116, U87, C6, HELA, BeL7402, U251, A172, HUVEC, NIH3T3; VV: U87 | VT: 5.0–20.0 μg mL−1; VV: 0–100 mg kg−1 | Dose-dependent ↓ GBM cell proliferation. ↓ tumor growth, cell migration and invasion | ● | ↑ ROS, apoptosis (↑ procaspase-9, procaspase-3 and ↓ bcl-2/bax ratio), inhibition of AKT and ERK1/2 | 169 |
| Zn-doped | 30C | Zn–CuO NPs inhibit pancreatic cancer growth by inducing autophagy through AMPK/mTOR pathway | ● | ● | VT: AsPC1, MIA Paca2, HepG2, BxPC3, PANC1, HT29; VV: AsPC1 | VT: 0–160 μg mL−1; VV: 5 and 10 mg kg−1 | Dose-dependent ↓ cell viability. Inhibition of tumor growth | ↑ ROS. Autophagy induced via AMPK/mTOR pathway (↑ p-AMPK, p-ULK1, Beclin-1 and LC3-II/LC3-I ratio, ↓ mTOR phosphorylation) | 170 | |
| Zn-doped | 2–10C | Zn–CuO NPs inhibit tumor growth by NF-κB pathway. NAC restores the balance disrupted by autophagy and apoptosis | ● | ● | VT: HepG2, Panc28; VV: Panc28 | VT: 0–40 μg mL−1; VV: 5 and 10 mg kg−1 | Dose-dependent ↑ cell proliferation inhibitory rates | NF-κB signaling involved in ROS-induced apoptosis (↑ Bax and caspase 3, Bcl-2 ↓) and autophagy (↑ LC3B and LC3 B/A). DNA, ER & Golgi damage. All effects restored with NAC | 172 | |
| Zn-doped | 3C | Zn–CuO NPs inhibit human CC growth through ROS-mediated NFκB activations | ● | HepG2, Bel7402, A549, Panc28, HT1080, Hela, HUVEC, L02 | 0–60 μg mL−1 | Dose-dependent ↑ cell proliferation inhibitory rates | ● | ↑ ROS and NF-κB pathway activation (↑ p-IKKα/β and nucleus p-NF-κB p65, ↓ IKKα, IKKβ, IκBα and nucleus NF-κB p65 expression). Induction of G2/M cell cycle arrest | 171 | |
| Carbon (C)-coated | 10.4–19.4C (CuO–C/Cu) | C-coat decreases cytotoxicity of CuO NPs due to reduced solubility, and CuO NPs induce greater toxicity than Cu2+ | ● | CHO, HeLa | 30 ppm C/Cu, 34 ppm CuO | Dose-dependent ↓ cell viability. ↓ toxicity with C-coating | ● | 103 | ||
| Protein coating (DMSA) | 141HD (pure), 167HD (coated) | pCuO-NP-induce cell death in glioblastoma cells to a lesser extent than pure CuO NPs, due to reduced Cu ion release | ● | C6, primary astrocytes | 0–1000 μM | Dose- and time-dependent ↓ cell viability. ↓ toxicity with protein coat | ↓ LDH and ↓ MTT reduction capacity. Cu chelators and low temperature ↓ toxicity | 173 | ||
| Surface mod. | Size (in nm) | Main finding | VT | VV | Model systems | Dose/concentration | Cellular/tumoral effects | CCS | Mechanism | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ccore size, HDhydrodynamic size, VT in vitro, VV in vivo, CCS cancer-cell specific. | ||||||||||
| Pure | 8–22C | Ag NPs synthesized from plant extracts induce anticancer activity in lung cancer models | ● | ● | H1299 | VT: 2–30 μg mL−1; VV: 10 μg g−1 | Dose-dependent ↓ cell viability. ↓ tumor size & growth | Morphological alterations, ↓ NF-κB transcriptional activity and Bcl2, ↑ caspase-3 and survivin | 152 | |
| Pure | 5–35C | Bio-synthesized Ag NPs exhibit anticancer, antioxidant and anti-angiogenic activity with no adverse effect on liver and kidney | ● | DAL | VT: 0–100 μg mL; VV: 50 μg mL−1 | Dose-dependent ↓ cell viability ex vivo | 187 | |||
| Pure | 9.4–25.9C | Ag NPs synthesized from Nostoc linckia extracts exert anticancer activity in breast cancer models | ● | ● | VT: MCF-7, WI38, WISH; VV: EAC | VT: 1.56–100 μg mL−1; VV: 5 mg kg−1 | Dose-dependent ↓ cell viability. ↓ cell volume, cell count and weight of tumors | ● | 153 | |
| Pure | 31.8C | Ag NPs synthesized from Acorus calamus show antitumor activity with complete NP elimination 89 days post-treatment | ● | ● | VT: Hep2, COLO 205, SH-SY5Y | 0–200 μg mL−1 | ↓ cell viability | ↑ ROS, ↓ MMP, ↑ caspases-8, 9 and 3, ↑ lamin and PARP, ↑ MDA, ↓ SOD, GPx and CAT | 154 | |
| Pure | 8–20C | Ag NPs synthesized from Agaricus bisporus cause apoptosis and anti-angiogenesis in combination with gamma radiation | ● | ● | VT: MCF-7; VV: EAC | VV: 0.1–10 μg kg−1 | Dose-dependent ↓ cell viability | ↑ caspase-3, ↑ nitric oxide and MDA, ↑ ROS, DNA damage | 155 | |
| Pure | 2.6C, 18C | Ag NPs induce size-dependent cell death in chemo-resistant pancreatic CCs via apoptosis, autophagy, necroptosis and mitotic catastrophe | ● | PANC-1, hTERT-HPNE | 0–5μg mL−1 (2.6 nm), 0–100 μg mL−1 (18 nm) | Dose-dependent ↓ cell viability. ↓ toxicity with ↑ NP size | ● | Apoptosis: ↑ bax, p53, ↓ bcl-2. Necroptosis: ↑ MLKL, RIP1, RIP3. Autophagy: ↑ LC3-II | 91 | |
| Citrate-coated | 5C | Ag NPs initiate antitumor effects and trigger the activation of a tumor cell-specific immune response | ● | ● | VT: HeLa, A549, KLN205; VV: KLN205, CT26 | VT: 5–50 μg mL−1; VV: 100 μg | Dose-dependent ↓ cell viability. Tumor inflammation and ↓ tumor size | ↑ ROS, mitochondrial damage, autophagy, immunomodulatory effects (↑ NFκB pathway and ↑ IL-1α) | 175 | |
| PVP-coated | 30–50 | Ag NPs induce toxicity to CCs via necrosis and increase survival of mice | ● | ● | L5178Y-R | VT: 9–579 nM; VV: 20 mg kg−1 | Dose-dependent ↓ cell viability | 97 | ||
| PVP-coated | 5–75C | Ag NPs show selective cytotoxicity against TNBC cells regardless of size, shape or coating | ● | ● | VT: TNBC, MDA-MB-231, SUM159; VV: TNBC | VT: 0–60 μg mL−1; VV: 6 mg kg−1 | ↓ proliferation. ↓ tumor growth rate, ↑ survival rate | ● | Impairment of cellular redox balance, ↑ ER stress, UPR activation, ↑ CHOP, DNA damage | 178 |
| EPS-coated | 11C | Ag NPs biogenerated by Klebsiella oxytoca DSM 29614 show anticancer activity mainly by induction of autophagy | ● | SKBR3 | 5 and 50 μg mL−1 | Dose- and time-dependent ↓ colony-forming ability | ↑ ROS. Autophagy: ↑ ATG5, ATG7, LC3-II and Beclin-1, ↓ AKT, p-AKT, p62 and HSP90 | 177 | ||
| Ag+-R, Ag0-R | <2C | Compared to Ag+-R NCs, Ag0-R nanoclusters (NCs) exhibit greater release of Ag species | ● | BJ | 62.5, 250 and 1000 mM | Toxicity by Ag0-R NCs > Ag+-R NCs | Activation of p53 | 176 | ||
| TAT-coated | 8C | Antitumor activity in both multidrug resistant and non-resistant CCs is greater with TAT functionalization | ● | ● | VT: HeLa, MCF-7(/ADR), B16; VV: B16 | VV: 1 nmol kg−1 | ↓ cell viability. ↓ tumor growth at lower doses than DOX | 191 | ||
4.1. CC-specific toxicity of pure NPs
Similarly to the previously reported findings concerning the cytotoxic effects of ZnO NPs, CuO NPs were found to have a dose-dependent (DD) toxic effect on HepG2 (ref. 150) and K562 (leukemia)151 cells in two separate studies, and were associated with the upregulation of p53, increased ratio of BAX/BCL2, and decrease in MMP, suggesting that a mitochondria-mediated pathway is involved in CuO NP-induced apoptosis. Expression levels of caspase-3 were also measured in the HepG2 cell line and were found to be elevated. Multiple studies have also tested the effects of CuO NPs in human breast CCs. In the MCF7 cell line, CuO NPs induced dose- and time-dependent autophagy as a possible defence mechanism against CuO-induced toxicity.95 Apoptosis-related cell death was indicated by PARP cleavage, bcl2-associated death promoter (BAD) de-phosphorylation and increased caspase 3 cleavage. A more recent in vitro study on MCF7 cells by Ali et al. (2020) used CuO NPs synthesized from Eucalyptus globulus leaf extract (ELE) with anticancer and antifungal activity.90 They demonstrated a considerable reduction in cell survival via an impairment of MMP and elevation in intracellular ROS. Cell cycle analysis indicated disruption/arrest in different phases (G1, S and G2/M phases) upon CuO NP administration, leading to apoptosis. This data was further confirmed by the upregulated expression levels of p53, bax, caspase-3, and caspase-9 genes.
With the aim of circumventing the use of toxic and expensive chemicals, Nakkala et al. (2018) synthesized Ag NPs using a green pathway (rhizome extract of Acorus calamus), and these NPs significantly impacted the viabilities of different CC lines, including Hep2 (human epidermoid carcinoma), COLO 205 (human colon adenocarcinoma) and SH-SY5Y (neuroblastoma).154 Hep2 cells, which clearly showed lower IC50 values after 24 h, displayed characteristics of late apoptosis such as the rounding up of nuclei, cell shrinkage, nuclear condensation and fragmentation, and loss of MMP. Furthermore, an increase of ∼66% in ROS levels and 1.2-fold in MDA levels (marker of oxidative stress), as well as a decrease in SOD, GPx and CAT antioxidant activity were detected. It was thereby concluded that both intrinsic and extrinsic apoptotic pathways are involved in the cell death of Ag NP-treated cells. Other biologically-synthetized Ag NPs were also shown to induce a DD reduction in viability in MCF-7 cells.155 The selectivity of Ag towards CCs was further highlighted in a study by Zielinska et al. (2018), wherein 2.6 and 18 nm Ag NPs exerted higher cytotoxicity against PANC-1 CCs compared to non-tumor cells of the same tissue (hTERT-HPNE cells).91 Both Ag NPs induced, in a size- and concentration-dependent manner, a significant decrease in proliferation and cell death after 24 h, which appeared to be regulated by apoptosis, necroptosis, autophagy and/or mitotic catastrophe as a result of alterations in the protein levels associated with each mechanism. The role of apoptosis in Ag NPs-induced PANC-1 cell death was confirmed by significant increases in the levels of BAX and P53 and very low levels of BCL2. Evidence for necroptosis and autophagy was shown by a size- and concentration-dependent increase in MLKL, RIP1 and RIP3 protein levels and an increase in LC3-II, respectively. Finally, the role of mitotic catastrophe in Ag NPs-induced cell death was confirmed by the observed increase in cell size combined with multinucleation.
IONPs have been shown to induce some interesting effects, linked to their degradation, which are unique among the other NPs. Firstly, when relatively low levels of IONPs are used, the released ferric ions can promote cell proliferation, as demonstrated for mesenchymal stem cells, as Fe is a vital component in cell cycle progression.159 Secondly, upon degradation of IONPs, it has been shown that they keep evolving with time. Initially, they undergo a rapid burst of degradation, after which ferric ions are transported to ferritin for long-term storage under its ferrihydrite form ref. 160 and 161. It has been shown that stem cells can however resynthesize novel biogenic IONPs with a magnetite structure.162
4.2. Effect of NP doping and surface modification on cellular toxicity in vitro
Other metals were also tested as potential dopants of ZnO NPs. For instance, in a study by Shakir et al. (2016), pure and different lanthanum-doped ZnO NPs (1%, 3% and 5% La/ZnO) were synthesized and their cytotoxicity was examined using various concentrations (6–500 μg mL−1) in different CC lines, including MDA-MB-231, KCL22 (chronic myeloid leukemia) and HeLa cells.164 Unlike Fe-doped NPs, La-doped ZnO NPs were shown to be more potent at inducing cytotoxicity than pure ZnO NPs under the same experimental conditions. These findings suggest that in cases where specific cell types are resistant to ZnO NPs, doping them with La may enhance the NP dissolution kinetics and potentially serve as a means of enhancing toxicity. Recently, another study was conducted evaluating the cytotoxic effects of ZnO NPs doped with various lanthanides, including samarium (Sm), europium (Eu) and gadolinium (Gd).165 They found that, in the Ehrlich ascites carcinoma (EAC) cell line, Sm3+:ZnO displayed the most significant antitumor activity with 95% cell death, while both Eu3+ and Gd3+ caused only 10% and 5% tumor cell death, respectively. Pure ZnO NPs also showed weak antitumor activity, possibly due to the micro-sized particles utilized throughout the study. These findings are in line with the previously reported selective anticancer effects of nanocomplexes composed of Sm3+ and green tea polyphenols on the melanoma (B16F10) cell line.166
Finally, a study investigating the effects of varying NP surface modification was conducted using nano- and micro-particles of ZnO with sizes ranging from 40 nm to 1.2 μm.167 These particles were either bare or coated with polyethylene glycol (PEG) or starch and were tested at various concentrations (1 μM to 7 mM). In human osteoblast CCs (MG-63), PEG-capped particles exerted higher toxicity with decreasing size and increasing concentration. In contrast, starch-capped ZnO exerted the lowest toxicity compared with the other particles, highlighting the importance of surface capping on ZnO toxicity towards CCs.
Another type of metal-doping of CuO NPs was performed using Zn with the aim of inducing selective anticancer activity in glioma cells both in vitro and in vivo (Fig. 5).169 Treatment with nZn-doped CuO NPs has shown to not only inhibit cell proliferation in a DD manner, but also trigger ROS generation and the intrinsic apoptotic pathway. Specifically, it was found that nZn–CuO NPs disturb cell growth by downregulating the gene expression of bcl-2 and inhibiting AKT and ERK1/2. In a similar study, the anticancer activity of these particles was tested on human pancreatic CCs (AsPC-1 and MIA Paca-2).170 Here, the authors highlight that the AMPK/mTOR pathway plays an important role in the inhibition of tumor growth induced by Zn–CuO NPs, as they increased both the protein levels and gene expression of p-AMPK, p-ULK1, Beclin-1 and the ratio of LC3-II/LC3-I, while NP exposure downregulated the phosphorylation of mTOR. The specificity of Zn–CuO NPs towards CCs was further tested in multiple cell lines.171 In a DD manner, Zn–CuO NPs exerted anti-proliferative effects on CCs, including HepG2, Bel7402 (human hepatoma), A549, Panc28 (human pancreatic), HT1080 (human cervical), and Hela. However, in NC lines such as HUVEC and L02 (human hepatocyte) cells, these anti-proliferative effects were significantly weaker compared to the impact on CC lines. It was further shown that Zn–CuO NPs affect HepG2 and Panc28 CCs through ROS generation, apoptosis and the induction of G2/M cell cycle arrest. Molecular data revealed an increase in the expression of p-IKKα/β and nucleus p-NF-κB p65 and a decrease of IKKα, IKKβ, IκBα and nucleus NF-κB p65 expression in both HepG2 and Panc28 CC lines. In a parallel study that also revealed the importance of the NF-κB pathway, further treatment with NAC restored all of the above effects on protein expression.172 Therefore, in addition to the activation of the AMPK/mTOR pathway, Zn–CuO NPs trigger the ROS-mediated NF-κB pathway during the selective inhibition of CC growth.
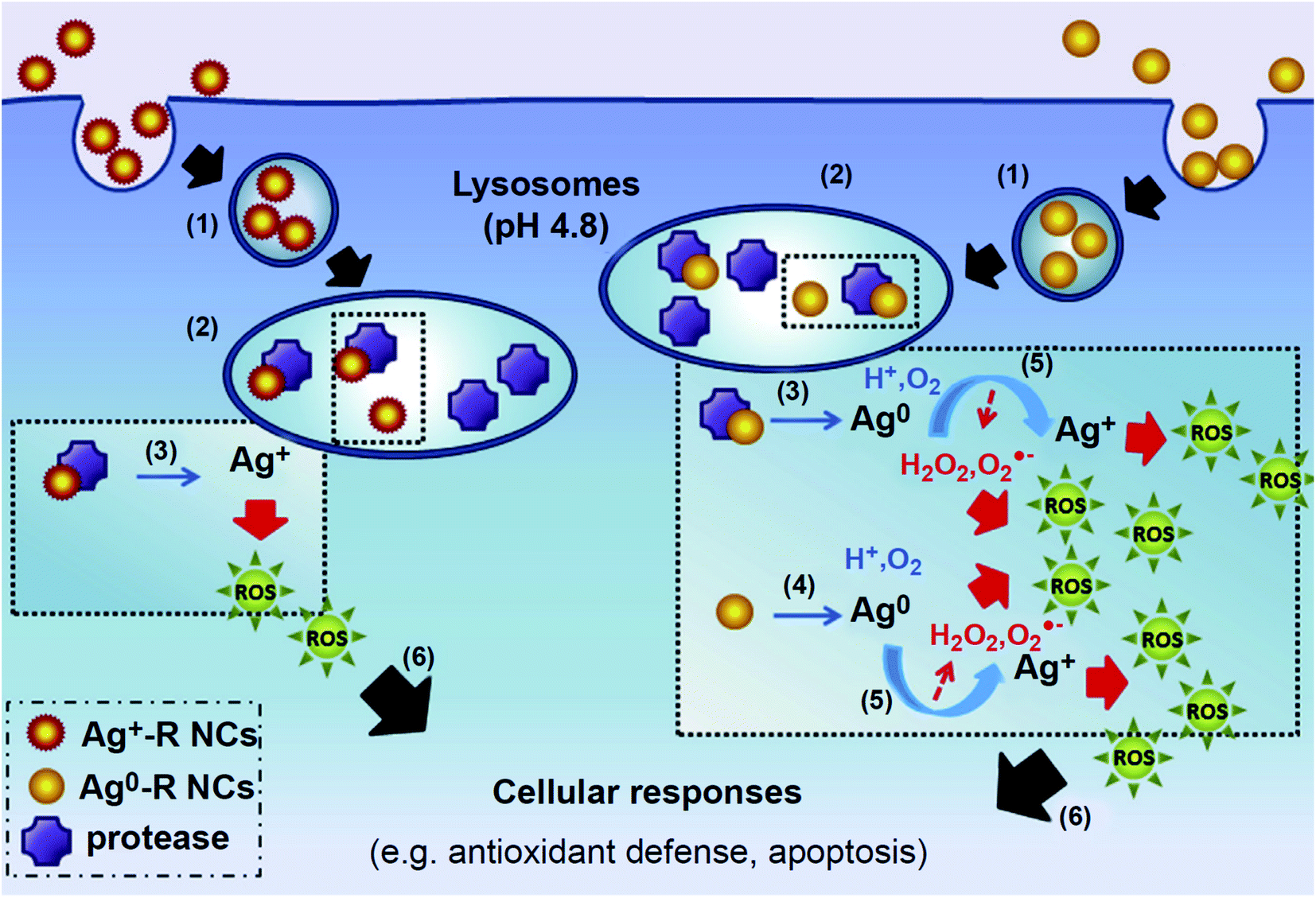 | ||
| Fig. 5 Proposed mechanism of cellular responses induced by GSH-Ag NCs. GSH-Ag NCs were taken up by the cell via an endocytotic pathway (1) and send to the lysosome (2),where stable GSH-Agþ-R NCs were enzymatically degraded by the lysosomal protease (3) to form reactive Agþ. In contrast, GSH-Ag0-R NCs decomposition originated not only from enzymatic degradation of the clusters (3) but also caused by their instability (4) in lysosome environment. The result of this decomposition is the formation of Ag0, which then undergone oxidative dissolution (5), resulting in Ag+ ion formation with ROS as the byproducts. Ag+ ions being release in turn affects the cell's respiratory chain, leading to increase of ROS which then triggers cellular responses (6), such as activation of anti-oxidant defense and programmed cell death. Reproduced with permission from Setyawati et al.,176 © 2014, Elsevier Publishing. | ||
Besides metal doping, CuO NPs have been synthesized with a variety of coatings with the aim of controlling NP dissolution. Studer et al. (2010) reported that coating CuO NPs with a stabilizing carbon layer not only reduced their toxicity in CCs (HeLa) but also altered the release and cellular internalization of Cu2+.103 Protein coats have also been applied to CuO NPs to tune their toxicity towards C6 glioma cells, including dimercaptosuccinic acid (DMSA) and bovine serum albumin (BSA),173 which are able to improve the colloidal stability of CuO NPs in physiological media.174 Within these cells, DMSA protein-coated NPs (pCuO-NPs) resulted in Cu accumulation and severe toxicity in a time-, concentration- and temperature-dependent manner. Exposure of the cells to pCuO-NPs for 30 minutes caused severe loss in enzyme activity, including cellular lactate dehydrogenase (LDH) activity and MTT reduction capacity, which are markers for membrane integrity and cell viability, respectively. This effect was only detected in C6 glioma cells when the Cu content exceeded 20 nmol mg−1. Once again, the addition of Cu chelators (TTM or BCS) led to lower intracellular Cu content and protected glioma cells against toxicity induced by pCuO-NPs.
In a separate study, the toxicity of Ag nanoclusters (Ag NCs) with two different core surface speciation (Ag+-rich NCs (Ag+-R NCs) and Ag0-rich NCs (Ag0-R NCs)) of the same size (<2 nm core) was investigated on human neonatal foreskin fibroblast cells (BJ).176 Ag0-R NCs induced higher cellular toxicity in a dose- and time-dependent manner, as well as higher ROS generation compared to the Ag+-R NCs. As the Ag0-R bond is weaker than Ag+-R, making it more potent to release Ag in acidic environments, this difference in toxicity may be due to the faster release of Ag from Ag0-R NCs which then oxidizes immediately to Ag+ within the lysosomes. After 24 and 48 h of exposure to Ag0-R NCs, only 47% and 15% of the viable population remained, respectively, while Ag+-R NCs did not induce cell death in BJ cells. Activation of p53 was increased by 3.3-fold by Ag0-R NCs after 24 h, while only a 2-fold increase was observed for Ag+-R NCs (Fig. 5).
Ag NPs embedded in microbial exopolysaccharides (EPS) and biogenerated by Klebsiella oxytoca DSM 29614 under aerobic (Ag NPs-EPSaer) conditions were also investigated for their cytotoxic effects on SKBR3 cells.177 Significant inhibition of cellular proliferation was observed, and the colony-forming ability, which was used to assess cell viability, showed a dose- and time-dependent reduction after Ag NPs-EPSaer treatment. The selectivity of Ag NPs-EPSaer for SKBR3 cells was confirmed by calculating the selectivity index using DOX as a positive control and the non-tumoral mammary cell line HB2. Morphological assessment revealed apoptotic-like changes such as cell shrinkage, loss of contact between adjacent cells, membrane blebbing and nuclear chromatin condensation. Additionally, a concentration-dependent elevation of intracellular ROS was observed after 24 h of exposure. They found that autophagic cell death seemed to be the main mechanism in Ag NPs-EPSaer-induced cell death within the first 24 h of treatment, as supported by the evaluation in the expression levels of multiple autophagic markers. Specifically, the upregulation of ATG5, ATG7, LC3-II and Beclin-1 and downregulation of AKT, p-AKT, p62 and HSP90 were detected. It was also shown that DNA was not a primary target within the first 24 h of treatment but in the long-term, treatment with Ag NPs-EPSaer could increase and/or stabilise the interaction of Ag with DNA and thereby induce cell death. Together these results suggest that the cytotoxicity of Ag NPs-EPSaer consists of a direct effect caused by cellular uptake and induction of intracellular ROS as well as an indirect effect caused by the release of Ag+ – initially in the mitochondria and later on in the nuclei to interact with DNA – and is regulated by the induction of autophagy, followed by apoptosis.
Furthermore, Swanner et al. (2019) found that triple-negative breast cancer (TNBC) cells possess specific vulnerability to Ag NPs which can be exploited to avoid toxicity induction in normal tissue.178 They found that internalization of the NPs is essential to exert any TNBC-selective cytotoxic effects and that selective inhibition of TNBC cell growth takes place regardless of NP size, shape or coating (including PVP). However, they show that TNBC cells and their normal counterparts are comparably sensitive to Ag+, highlighting the importance of the NP itself in exerting a tumor selective response. Next, they followed the uptake and intracellular distribution of Ag NPs and reported that after 1 h of incubation with cells, Ag NPs were localized within the endosomes and exhibited clear degradation in MDA-MB-231 cells while remaining intact in non-malignant breast epithelial cells (MCF-10A). Interestingly, after 6 h, MCF-10A cells still displayed intact particles even upon endosomal fusion with lysosomes. The same observations were made when comparing the effects on iMEC and SUM159 cells, a non-malignant breast epithelial and TNBC cell line respectively. It was also shown that Ag NPs impaired the cellular redox balance in MDA-MB-231 cells and induced ER stress, UPR activation, and an increased expression of CHOP, a pro-apoptotic protein in TNBC cells. This effect was not observed in the MCF-10A cell line. Finally, MDA-MB-231 and non-neoplastic S1 mammary epithelial cells (S1 cells) were grown in 3D tumor organoid cultures, and in line with the previous findings, Ag NPs induced apoptosis and DNA damage only in MDA-MB-231 cells.
5. Cancer-specific NP toxicity in vivo: an overview of preclinical results obtained for NP degradation
Despite the multitude of in vitro studies that have been conducted over the years exploiting the Mz+ release and ROS generation of ZnO, CuO and Ag NPs to selectively target CCs, only a few have made their way to the preclinical setting. Specifically, the lack of in vivo studies on pure ZnO and CuO NPs in the literature can be attributed to the high toxicity of these particles, which tend to cause major weight loss in animals and often premature death shortly after treatment. Thus, the termination of such experiments occurs due to the underlying ethical reasons associated with NP administration.77 In this next section, we discuss the few in vivo experiments associated with pure ZnO, CuO, and Ag NP, as well as the progress that has been made over the recent years in rendering such studies safer via doping and coating of these NPs. A summary of all the discussed studies is provided in Tables 2–4.5.1. Effects of pure ZnO, CuO, Ag and IONPs in mouse tumor models
Among the few studies that have tested the effects of ZnO NPs in vivo, El-Shorbagy (2019) administered ZnO NPs alone or in combination with 100 mg kg−1 body weight N-acetyl cysteine (NAC) – a GSH precursor that has shown to lower intracellular ROS levels182,183 – to Ehrlich solid carcinoma (ESC)-bearing mice for a period of 7 days.184 During the treatment, three different doses of ZnO NPs including 50, 300 and 500 mg kg−1 were administered through oral gavage. It was shown that treatment with NPs inhibited the tumor growth by 31.5% and 46% for the 300 and 500 mg kg−1 doses, respectively. In line with respective in vitro studies, a significant increase in p53 and bax and a decrease in bcl2 expression levels were observed along with significant reductions of glutathione-S-transferase (GST) and CAT. Their results suggest that ZnO NPs have a significant effect on the inhibition of tumor growth through induction of oxidative stress, DNA damage and activation of the p53-mediated pathway, leading to apoptosis. Additionally, pre-treatment with NAC was shown to protect normal tissue including the liver and kidney without interfering with the antitumor activity of the ZnO NPs. In the latest study, ZnO NPs were used to treat lung cancer in mice and were found to exert anticancer activity when a minimum dose of 0.25 mg kg−1 was administered.185 However, contradicting results have been published by Hassan et al. (2017), wherein they observed an antioxidant effect of ZnO NPs, characterized by a decrease in rat serum α-fetoprotein (AFP) and α-L-fucosidase (AFU) levels and restoration of caspase-3 normal levels upon treatment of hepatocellular carcinoma (HCC)-bearing mice with ZnO NPs.147 It must be noted that AFP and AFU have been shown to be common biomarkers of HCC tumors.186The cytotoxic effects of pure CuO NPs have been investigated in mice bearing pancreatic tumors (Fig. 6) and were shown to delay tumor growth and significantly increase the number of apoptotic TICs, which is in line with previously discussed in vitro studies.149
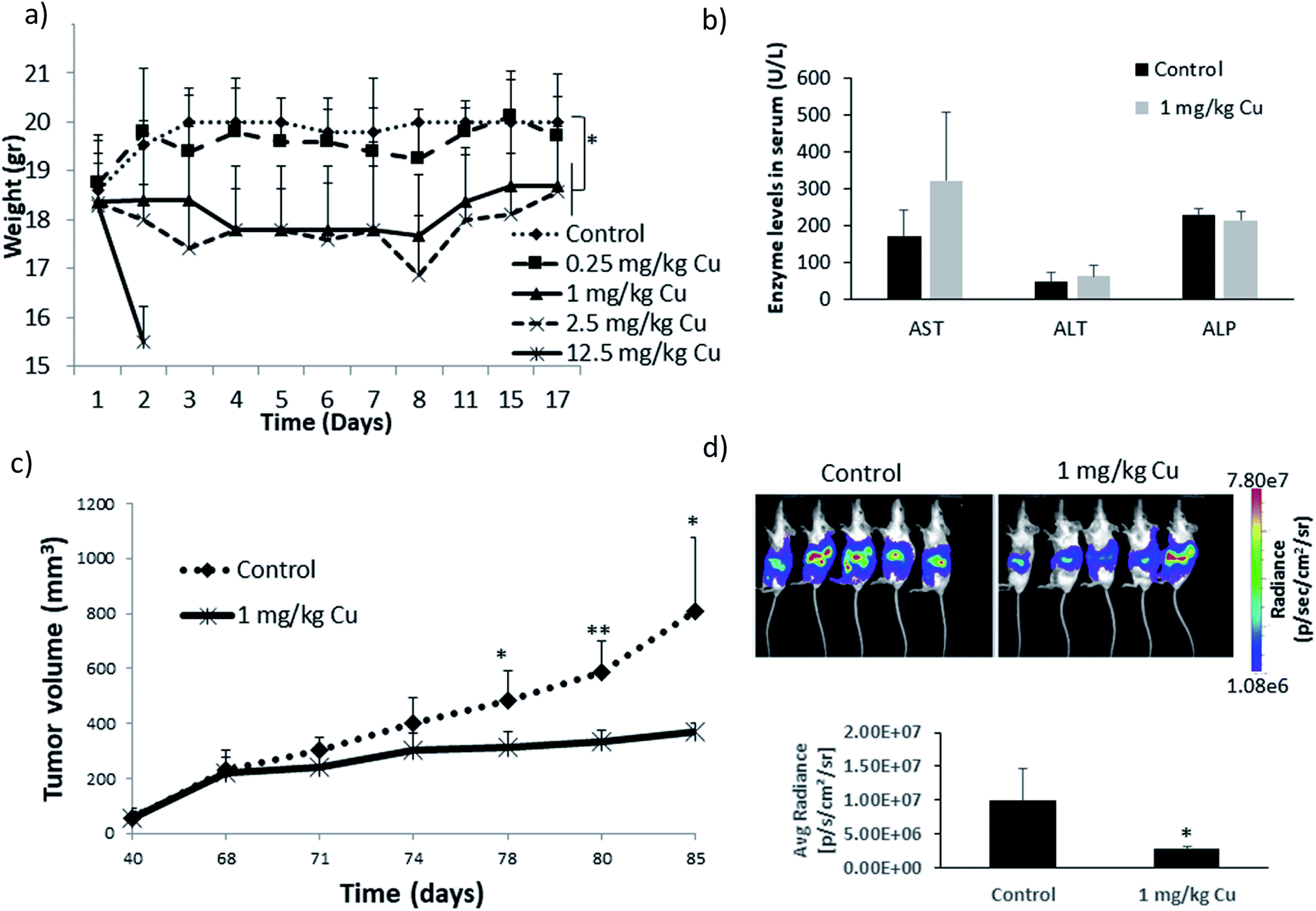 | ||
| Fig. 6 In vivo studies of pure CuO NPs as reported by Benguigui et al.149 (a) Effect of different doses of NPs on the weight of mice with no tumors over time. (b) Changes in the biomarkers of liver damage (AST, ALT, ALP) as a result of NP treatment in mice with no tumors. (c and d) Effect of daily CuO NP treatment (total 7 days) on tumor size in mice bearing subcutaneous PANC-1 tumors (c) and on the bioluminescence signal in orthotopically-implanted PANC-1 (luciferase-tagged) tumors after 5 weeks (d). Reproduced with permission from Benguigui et al.149 © Nature Publishing Group, 2019. | ||
With respect to Ag NPs, their effect on the progression and growth of human lung CCs (H1299) has been evaluated in immunodeficient SCID mice.152 The Ag NPs which were administered intraperitoneally were able to slow down the development of tumors and significantly reduce their size. Another study assessed the antitumor activity of pure Ag NPs in EAC-bearing mice and showed that a dose of 5 mg kg−1 could significantly inhibit the cell volume, cell count and weight of the tumors, as compared to the non-treated control group.153 A rapid increase in ascitic tumor volume and a decrease in body weight was observed in the control and treated mice, respectively. The body weight was correlated with the ascites fluid volume, which served as a direct nutritional source for tumor cells. These results are in agreement with a previous report on an Ehrlich ascites tumor model in mice, in which the antitumor activity of biologically synthetized Ag NPs was detected.155 In this study, increases in nitric oxide, MDA and ROS levels, and DNA damage were reported, and were associated with higher numbers of necrotic and apoptotic cells as shown on photomicrographs of H&E sections. Histopathological observations and cell cycle analysis also revealed an antiangiogenic effect of Ag NPs. In another similar study that employs biologically-synthesized particles, Ficus religiosa-derived Ag NPs were examined for their anticancer activity in a mouse model of Dalton's ascites lymphoma (DAL).187 As was the case in the previous study, NP-treated mice experienced loss of body weight (in line with reduced ascites fluid volume) as well as increased survival. These Ag NPs were shown to induce CC apoptosis via DNA damage and exert antioxidant (restoration of MDA, SOD and CAT levels) and antiangiogenic effects without affecting kidney and liver function.
For IONPs, no cancer-selective toxicity has been observed, but clinically approved dextran-coated IONPs have been shown to activate macrophages to a pro-inflammatory M1-type status. This then further resulted in a reduction in the growth of subcutaneous adenocarcinoma, due to a more aggressive immune microenvironment.188 This indirect effect could play an important role in macrophage-related cancer immunotherapies.
5.2. In vivo effects of doped and coated NPs
Another recent work in 2020 was carried out in which Sm-doped ZnO NPs were tested in Swiss albino female mice after the LD50 was determined.165 The selected dose was 45 mg kg−1 (20% of LD50) given that it exhibited no mortality and was administered by intramuscular injection to the Ehrlich solid tumor-bearing mice. While the average tumor size in the control group increased over time, the treated mice experienced reduced tumor size and a downregulation in CXCR4 and cytochrome P450 expression, which are markers of different aspects of cancer.189,190
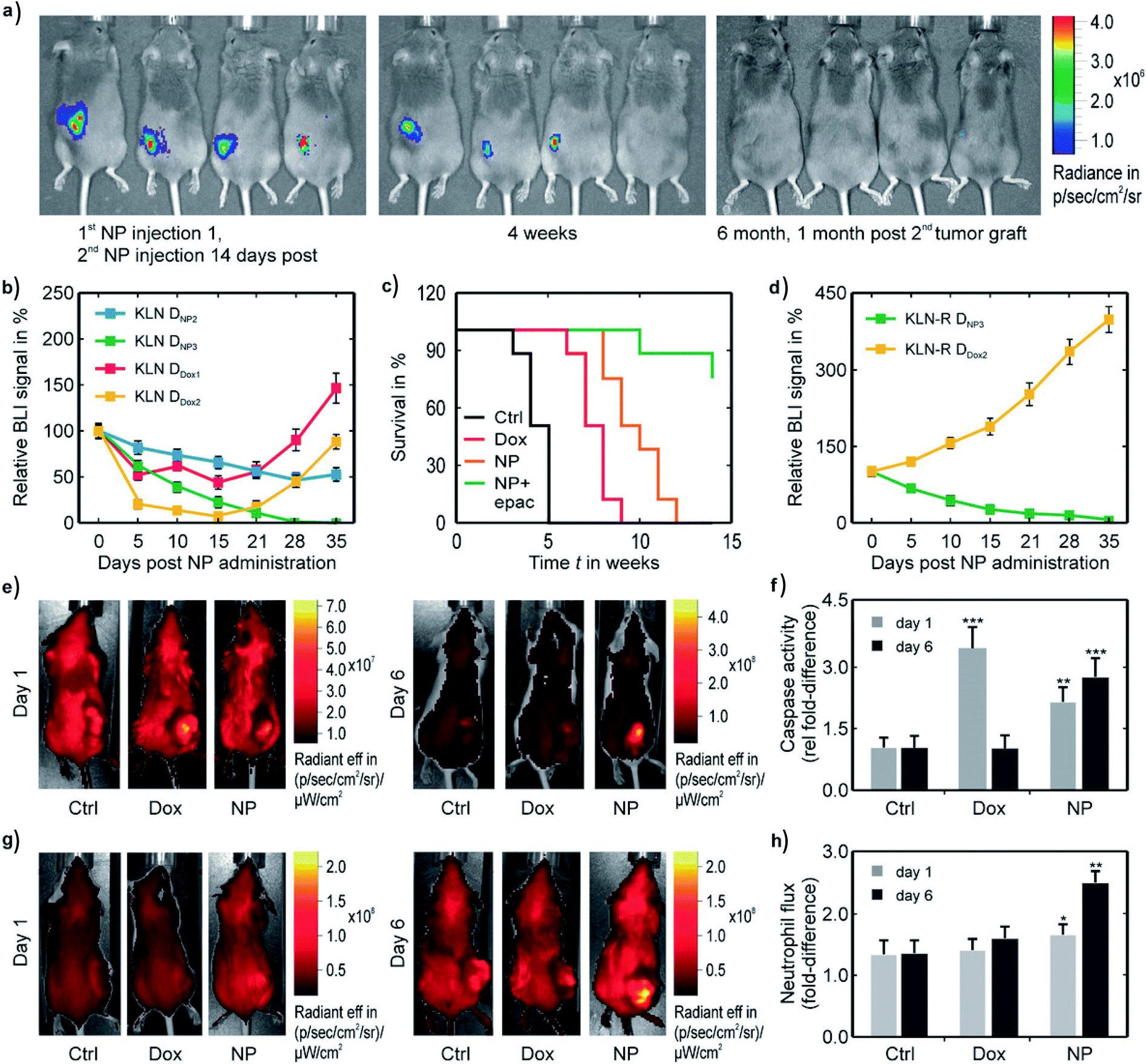 | ||
| Fig. 7 Synergistic therapeutic effect of treatment combinations with 6% Fe-doped CuO NPs and EPAC in mice bearing firefly-expressing KLN205 (a–d) or CT26 (e–h) tumors, as assessed by Naatz et al.79 (a) Luminescence images of tumors after treatment over time. (b and d) Bioluminescence imaging (BLI) signals comparing combination treatments of EPAC with either NPs or DOX in normal (b) and DOX-resistant cells (d). (c) Survival curves of mice exposed to different treatments. (e–h) Fluorescence images of mice treated with a fluorescent pan-caspase probe (e) and neutrophil-specific peptide (g) and their respective signal quantifications. The DOX and NP-treated groups also received EPAC. This image has been reproduced with permission from Naatz et al.,79 © Wiley-VCH, 2020. | ||
At the same time, the potential of Zn-doped CuO nanocomposites to selectively target CCs has been investigated in multiple studies in vivo. In a glioblastoma mouse model established by U87 cell transplantation, significant antitumor effects of Zn–CuO NPs have been demonstrated.169 Specifically, increased procaspase-9 and procaspase-3 levels and a decreased BCL2/BAX ratio indicated that NP-mediated cell death occurs through the mitochondrial apoptotic pathway. Histological studies confirmed a DD decrease in Ki-67, a marker for cell proliferation, as well as no toxicity in the main organs. Furthermore, this study demonstrated that temozolomide (TMZ)-resistance of cells can be reversed upon Zn–CuO NP exposure via inhibition of AKT/ERK1/2 activation. Similar observations on tumor growth inhibition have been made in mice bearing pancreatic cancer tumors (Panc28 (ref. 172) and AsPC-1 (ref. 170)) upon treatment with Zn–CuO NPs.
To investigate the contribution of the immune system to the effects of citrate-coated Ag NPs on the treatment of cancer, Manshian et al. (2017) administered Ag NPs to immune-deficient as well as immune-competent mice bearing KLN 205 CCs.175 In a first experiment, the pro-inflammatory effects of the Ag NPs were tested. The relative fluorescence intensity – originating from an inflammation-activatable probe given to the animals 5 days post saline or NP administration – exhibited a higher signal in the NP-treated immune-competent mice, indicating tumor inflammation and thus activation of the immune system. In a parallel experiment using the same animal groups, the therapeutic efficacy of the NPs was evaluated, during which a clear reduction in tumor size was shown upon NP treatment. Ten days post treatment, tumors regrew due to the dilution of cellular NP levels that results from continuous cell division. At the same time, for the treated immune-deficient mice, the tumor growth rate was nearly identical to that observed in the untreated group, highlighting the importance of the complementary role of the immune system during Ag NP cancer treatment.
6. Hypothetical mechanism summary
While all the NPs described above have redox potential and can induce ROS while undergoing degradation in the endo- or lysosomal environment, the true mechanism behind cancer-selective toxicity remains somewhat unclear, and the lack of effect for IONPs compared to other NPs also remains unanswered. From our perspective, and based on the literature review, we suggest a hypothetical mechanism for both activities.First, the ability of CuO, ZnO or Ag NPs to induce cancer cell selective toxicity may be due to the fact that many cancer cells have a high metabolic rate and intrinsically have higher levels of (mitochondrial) ROS. Any additional ROS that is generated, such as that generated by the NPs, can therefore result in exaggerated toxic effects compared to non-cancerous counterparts. For every cell, cellular defense mechanisms against ROS will protect the cell against certain levels of ROS generation.192 However, when this threshold is exceeded, oxidative stress occurs, which can be lethal to the cells. It is therefore important to finely tune the kinetics of NP degradation and associated ROS generation to be able to induce sufficient ROS to exceed toxic limits in cancerous cells, while not inducing too many that they would also affect non-cancerous cells. Furthermore, the effects observed may not be generalized to every type of cancerous or normal cell. If the cancer cells have a high level of oxidative defense mechanisms, then they will not be easily targeted by this form of therapy. Cancer cells that contain mutations that limit their oxidative stress defenses would however be excellently suited to this type of therapy.
Secondly, the ability of some NPs to induce cancer-selective toxicity while others cannot (e.g. IONP), can simply be due to the chemical nature of the metal ion and how these are normally processed under typical physiological conditions. For Fe, any Fe to be taken up by cells will be bound to transferrin, and cellular uptake involves the transferrin molecule binding to the cell surface-located transferrin receptor. This triggers an internalization pathway, where the transferrin receptor/transferrin complex is shuttled inside early endosomes and at lower pH, Fe is released. The transferrin receptor can then be recycled back to the cell membrane, while Fe can be transferred to late endosomes or lysosomes. It can then bind to small molecules, such as citrate, which can shuttle it into the cytoplasm, where it can become part of the labile Fe pool.193 When cellular Fe levels increase, they can be stored long-term in Fe storage proteins (ferritin), the level of which depends on the amount of cytoplasmic Fe. For IONPs, this would essentially be a similar story, where internalization of IONPs results in endo- or lysosomal sequestration of the NPs followed by their degradation and transport of free Fe ions by means of citrate to become part of the labile Fe pool.160 Excessive amounts of Fe can be relatively well tolerated by increasing the level of ferritin storage complexes. Any toxicity would be linked to excessive amounts of Fe in the endo- or lysosomes, linked to high levels of Fenton reactions that exceed local toxicity thresholds.
For Ag, Zn or Cu, the physiological handling of these ions is far different. For Ag, which is extremely rare in our body, there are no specific transport pathways or predetermined routes of cellular processing. Ag ions will mainly be linked to calcium channels, and monovalent metal transporters.194 For Zn ions, a highly specific pathway is present. In short, for eukaryotic cells, Zn ions are transported from the extracellular space into the cell cytoplasm by the so-called Zrt-, Irt-like protein (ZIP, SLC39) while the cation diffusion facilitator (CDF, SLC30) work the opposite way and transport Zn from the cytoplasm to the extracellular space. Once inside the cytoplasm, Zn can be transported to the Golgi apparatus, or mitochondria, where it can be used in Zn-dependent proteins or matrices.195 For Cu, the process is similar. Here, Cu enters the cells mainly via the Cu transporter 1 (CTR1), and is immediately linked to its chaperones, such as Cu chaperone for SOD (CCS) or an enzyme involved in the synthesis of cytochrome c oxidase (Sco1).196 Cu can be then stored at low levels in MTs. For Ag, Zn or Cu, it is therefore important to note that transport of these ions from endo- and lysosomes towards the cytoplasm is very limited as it does not occur naturally. For metal-containing NPs, this thus poses an entirely different scenario, where the cell is not naturally equipped to deal with excess level of the Mz+ in its endosomal compartments. ZnO, CuO or Ag NP, all of which typically dissolve rather quickly, result in free Mz+ already present in cell medium due to premature degradation, and cellular uptake of excess Mz+ that can lead to cell death.197 Alternatively, leakage of Mz+ from endosomes can also lead to further oxidative stress and cytotoxicity, while the intraendosomal presence of these Mz+ in itself can generate additional ROS.
7. Conclusion and future outlook
Here, we have summarized recent advances and key mechanisms behind the degradation of degradable metal and MO NPs. We highlight the various types of doping strategies and surface coatings that have been used to control NP dissolution and achieve selective tumor targeting, as well as certain computational models that have been developed to predict the impact of various physicochemical alterations on cytotoxicity. Besides the composition of the surface modifications, the toxicity profiles of these NPs have been optimized by changing the size, aspect ratio, and concentration of dopants and surface coatings. We then discussed several recent in vitro and in vivo advances that utilized these variously modified NPs to highlight their great potential in selective cancer therapy.Studies assessing the toxicity of pure ZnO, CuO and Ag NPs on a wide variety of cell lines showed a dose-dependent toxicity, as well as a time-dependent mechanism. In addition, cellular studies on Fe- and lanthanide-doped ZnO NPs showed the potential fine-tuning capacities of the doping approach to control their cytotoxicity. Similar results were found for carbon-coated, protein-coated and Fe-doped CuO NPs, and for citrate- and EPS-coated Ag NPs. At the same time, pure ZnO, CuO and Ag NPs have not been extensively investigated in vivo, due to their high cytotoxicity levels that cause premature death in animals shortly after treatment. Nevertheless, in vivo research on their coated and doped counterparts have gained tremendous interest over the recent years, although the NPs remain in the early phases of development. Specifically, doping of ZnO NPs with Fe and samarium, as well as coating Ag with citrate for instance, have provided a first glimpse of the potential use of these particles in cancer therapy. For CuO NPs, doping with Fe and Zn have exhibited promising results in vivo, especially in combination with epacadostat, which achieved complete tumor remission.79
Therefore, the latest advances on controlling NP dissolution provide a positive outlook in the direction of selective tumor targeting, indicating a promising future for the use of nanotechnology in cancer therapy. Still, several aspects require further investigation to improve the clinical translation of such NMs, including the inconsistencies in in vitro research concerning the setup, NP synthesis methods and evaluated parameters, making it hard to reproducibly come to the same results in follow-up studies. In particular, a GMP-certified synthesis method that can produce the levels of NPs required for clinical use is essential and more work is needed to set up such GMP-certified synthesis methods. Another aspect involves a further understanding of their underlying biological pathways. Several studies already suggest mechanisms and pathways involved in NP-induced cancer cell death, yet more understanding is needed to make firm conclusions. This in particular addresses the reason why some cancer cell types are more sensitive to this type of NP-mediated therapy than others, irrespective of the NP internalization levels. If any biomarkers could be found that can predict whether a particular tumor type would be sensitive to this of treatment or not would be a major breakthrough. Another point of attention would be the ability of the NPs to induce immunogenic cell death and hereby sensitize the tumors to immunotherapy. A broader spectrum of studies must be performed to understand whether this is true for all NPs or which NP-related parameters can enhance this. Additionally, also here the question of tumor-type specificity comes up, where the biological outcome of NP treatment will likely also depend on the nature of the tumor cells and its immune microenvironment. Lastly, while some promising preclinical studies have been performed, this must be repeated on a broader range of tumor types, and also include more relevant tumor models, such as patient-derived xenografts, and by combining different immunotherapy-NP combinations in view of the tumoral immune landscape.
Conflicts of interest
There are no conflicts to declare.References
- Lancet, 2003, 362, 673 Search PubMed.
- S. C. Baetke, T. Lammers and F. Kiessling, Br. J. Radiol., 2015, 88, 20150207 CrossRef CAS PubMed.
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1217 CrossRef CAS PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- L. E. Euliss, J. A. DuPont, S. Gratton and J. DeSimone, Chem. Soc. Rev., 2006, 35, 1095–1104 RSC.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958 CrossRef CAS PubMed.
- A. C. Anselmo and S. Mitragotri, Bioeng. Transl. Med., 2019, 4, e10143 Search PubMed.
- C. Buzea, I. I. Pacheco and K. Robbie, Biointerphases, 2007, 2, MR17–MR71 CrossRef PubMed.
- C. Sioutas, R. J. Delfino and M. Singh, Environ. Health Perspect., 2005, 113, 947–955 CrossRef PubMed.
- G. Oberdörster, A. Maynard, K. Donaldson, V. Castranova, J. Fitzpatrick, K. Ausman, J. Carter, B. Karn, W. Kreyling and D. Lai, Part. Fibre Toxicol., 2005, 2, 8 CrossRef PubMed.
- Y. Wei, L. Quan, C. Zhou and Q. Zhan, Nanomedicine, 2018, 13, 1495–1512 CrossRef CAS PubMed.
- S. K. Golombek, J.-N. May, B. Theek, L. Appold, N. Drude, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2018, 130, 17–38 CrossRef CAS PubMed.
- H. Maeda, G. Bharate and J. Daruwalla, Eur. J. Pharm. Biopharm., 2009, 71, 409–419 CrossRef CAS PubMed.
- H. L. Karlsson, J. Gustafsson, P. Cronholm and L. Möller, Toxicol. Lett., 2009, 188, 112–118 CrossRef CAS PubMed.
- C. Maksoudian, S. J. Soenen, K. Susumu, E. Oh, I. L. Medintz and B. B. Manshian, Bioconjugate Chem., 2020, 31, 1077–1087 CrossRef CAS PubMed.
- M. J. Akhtar, M. Ahamed and H. A. Alhadlaq, Clin. Chim. Acta, 2018, 487, 186–196 CrossRef CAS PubMed.
- S. Fraga, A. Brandão, M. E. Soares, T. Morais, J. A. Duarte, L. Pereira, L. Soares, C. Neves, E. Pereira and M. de Lourdes Bastos, Nanomedicine, 2014, 10, 1757–1766 CrossRef CAS PubMed.
- J.-Y. Wang, J. Chen, J. Yang, H. Wang, X. Shen, Y.-M. Sun, M. Guo and X.-D. Zhang, Int. J. Nanomed., 2016, 11, 3475 CrossRef CAS PubMed.
- W. Poon, Y.-N. Zhang, B. Ouyang, B. R. Kingston, J. L. Wu, S. Wilhelm and W. C. Chan, ACS Nano, 2019, 13, 5785–5798 CrossRef CAS PubMed.
- S. Sabella, R. P. Carney, V. Brunetti, M. A. Malvindi, N. Al-Juffali, G. Vecchio, S. M. Janes, O. M. Bakr, R. Cingolani and F. Stellacci, Nanoscale, 2014, 6, 7052–7061 RSC.
- K. Midander, P. Cronholm, H. L. Karlsson, K. Elihn, L. Möller, C. Leygraf and I. O. Wallinder, Small, 2009, 5, 389–399 CrossRef CAS PubMed.
- R. a. Weissleder, D. D. Stark, B. L. Engelstad, B. R. Bacon, C. C. Compton, D. L. White, P. Jacobs and J. Lewis, Am. J. Roentgenol., 1989, 152, 167–173 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- A. Abdal Dayem, M. K. Hossain, S. B. Lee, K. Kim, S. K. Saha, G.-M. Yang, H. Y. Choi and S.-G. Cho, Int. J. Mol. Sci., 2017, 18, 120 CrossRef PubMed.
- O. Augusto and S. Miyamoto, Principles of free radical biomedicine, 2011, vol. 1, pp. 19–42 Search PubMed.
- B. Halliwell, Plant Physiol., 2006, 141, 312–322 CrossRef CAS PubMed.
- C. F. Mueller, K. Laude, J. S. McNally and D. G. Harrison, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 274–278 CrossRef CAS PubMed.
- M. J. Akhtar, M. Ahamed, H. A. Alhadlaq and A. Alshamsan, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 802–813 CrossRef CAS PubMed.
- Y. R. Li, Free Radical Biomedicine: Principles, Clinical Correlations, and Methodologies, Bentham Science Publishers, 2012 Search PubMed.
- V. J. Thannickal and B. L. Fanburg, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2000, 279, L1005–L1028 CrossRef CAS PubMed.
- M. J. Akhtar, M. Ahamed and H. A. Alhadlaq, Clin. Chim. Acta, 2017, 469, 53–62 CrossRef CAS PubMed.
- T. Finkel, J. Biol. Chem., 2012, 287, 4434–4440 CrossRef CAS PubMed.
- P. S. Brookes, Y. Yoon, J. L. Robotham, M. Anders and S.-S. Sheu, Am. J. Physiol.: Cell Physiol., 2004, 287, C817–C833 CrossRef CAS PubMed.
- C. Tapeinos and A. Pandit, Adv. Mater., 2016, 28, 5553–5585 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, L. Madler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121–2134 CrossRef CAS PubMed.
- P. D. Ray, B.-W. Huang and Y. Tsuji, Cell. Signalling, 2012, 24, 981–990 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, J. Brant, M. Hotze, J. Sempf, T. Oberley, C. Sioutas, J. I. Yeh, M. R. Wiesner and A. E. Nel, Nano Lett., 2006, 6, 1794–1807 CrossRef CAS PubMed.
- G. G. Xiao, M. Wang, N. Li, J. A. Loo and A. E. Nel, J. Biol. Chem., 2003, 278, 50781–50790 CrossRef CAS PubMed.
- P. Talalay, A. T. Dinkova-Kostova and W. D. Holtzclaw, Adv. Enzyme Regul., 2003, 43, 121–134 CrossRef CAS PubMed.
- I. M. Kennedy, D. Wilson and A. I. Barakat, Research report, Health Effects Institute, 2009, pp. 3–32 Search PubMed.
- Z. Zhang, A. Berg, H. Levanon, R. W. Fessenden and D. Meisel, J. Am. Chem. Soc., 2003, 125, 7959–7963 CrossRef CAS PubMed.
- S. Pokhrel, A. E. Nel and L. Mädler, Acc. Chem. Res., 2013, 46, 632–641 CrossRef CAS PubMed.
- H.-M. Lee, D.-M. Shin, H.-M. Song, J.-M. Yuk, Z.-W. Lee, S.-H. Lee, S. M. Hwang, J.-M. Kim, C.-S. Lee and E.-K. Jo, Toxicol. Appl. Pharmacol., 2009, 238, 160–169 CrossRef CAS PubMed.
- H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- Y. Wei and M. Guo, Angew. Chem., Int. Ed., 2007, 46, 4722–4725 CrossRef CAS PubMed.
- B. M. Strauch, R. K. Niemand, N. L. Winkelbeiner and A. Hartwig, Part. Fibre Toxicol., 2017, 14, 28 CrossRef PubMed.
- H. Líbalová, P. M. Costa, M. Olsson, L. Farcal, S. Ortelli, M. Blosi, J. Topinka, A. L. Costa and B. Fadeel, Chemosphere, 2018, 196, 482–493 CrossRef PubMed.
- N. M. Franklin, N. J. Rogers, S. C. Apte, G. E. Batley, G. E. Gadd and P. S. Casey, Environ. Sci. Technol., 2007, 41, 8484–8490 CrossRef CAS PubMed.
- C. Beer, R. Foldbjerg, Y. Hayashi, D. S. Sutherland and H. Autrup, Toxicol. Lett., 2012, 208, 286–292 CrossRef CAS PubMed.
- P. Cronholm, H. L. Karlsson, J. Hedberg, T. A. Lowe, L. Winnberg, K. Elihn, I. O. Wallinder and L. Möller, Small, 2013, 9, 970–982 CrossRef CAS PubMed.
- A. R. Gliga, S. Skoglund, I. O. Wallinder, B. Fadeel and H. L. Karlsson, Part. Fibre Toxicol., 2014, 11, 11 CrossRef PubMed.
- M. Heinlaan, A. Ivask, I. Blinova, H.-C. Dubourguier and A. Kahru, Chemosphere, 2008, 71, 1308–1316 CrossRef CAS PubMed.
- N. M. Franklin, N. J. Rogers, S. C. Apte, G. E. Batley, G. E. Gadd and P. S. Casey, Environ. Sci. Technol., 2007, 41, 8484–8490 CrossRef CAS PubMed.
- S. W. Wong, P. T. Leung, A. Djurišić and K. M. Leung, Anal. Bioanal. Chem., 2010, 396, 609–618 CrossRef CAS.
- T. W. Turney, M. B. Duriska, V. Jayaratne, A. Elbaz, S. J. O'Keefe, A. S. Hastings, T. J. Piva, P. F. Wright and B. N. Feltis, Chem. Res. Toxicol., 2012, 25, 2057–2066 Search PubMed.
- A. Ivask, K. G. Scheckel, P. Kapruwan, V. Stone, H. Yin, N. H. Voelcker and E. Lombi, Nanotoxicology, 2017, 11, 150–156 CrossRef CAS PubMed.
- J. Llop, I. Estrela-Lopis, R. F. Ziolo, A. González, J. Fleddermann, M. Dorn, V. G. Vallejo, R. Simon-Vazquez, E. Donath and Z. Mao, Part. Part. Syst. Charact., 2014, 31, 24–35 CrossRef CAS.
- S. K. Misra, S. Nuseibeh, A. Dybowska, D. Berhanu, T. D. Tetley and E. Valsami-Jones, Nanotoxicology, 2014, 8, 422–432 CrossRef CAS PubMed.
- A. L. Neal, N. Kabengi, A. Grider and P. M. Bertsch, Nanotoxicology, 2012, 6, 371–380 CrossRef CAS PubMed.
- P. Asharani, M. P. Hande and S. Valiyaveettil, BMC Cell Biol., 2009, 10, 65 CrossRef CAS PubMed.
- S. J. Soenen, U. Himmelreich, N. Nuytten, T. R. Pisanic, A. Ferrari and M. De Cuyper, Small, 2010, 6, 2136–2145 CrossRef CAS PubMed.
- S. Kim, J. E. Choi, J. Choi, K.-H. Chung, K. Park, J. Yi and D.-Y. Ryu, Toxicol. In Vitro, 2009, 23, 1076–1084 CrossRef CAS PubMed.
- E. M. Luther, M. M. Schmidt, J. Diendorf, M. Epple and R. Dringen, Neurochem. Res., 2012, 37, 1639–1648 CrossRef CAS PubMed.
- T. S. Peretyazhko, Q. Zhang and V. L. Colvin, Environ. Sci. Technol., 2014, 48, 11954–11961 CrossRef CAS PubMed.
- J. Liu and R. H. Hurt, Environ. Sci. Technol., 2010, 44, 2169–2175 CrossRef CAS PubMed.
- R. Ma, C. Levard, S. M. Marinakos, Y. Cheng, J. Liu, F. M. Michel, G. E. Brown Jr and G. V. Lowry, Environ. Sci. Technol., 2012, 46, 752–759 CrossRef CAS PubMed.
- K. Kawata, M. Osawa and S. Okabe, Environ. Sci. Technol., 2009, 43, 6046–6051 CrossRef CAS PubMed.
- J. Fabrega, S. R. Fawcett, J. C. Renshaw and J. R. Lead, Environ. Sci. Technol., 2009, 43, 7285–7290 CrossRef CAS PubMed.
- H.-J. Eom and J. Choi, Environ. Sci. Technol., 2010, 44, 8337–8342 CrossRef CAS.
- V. Frazzini, E. Rockabrand, E. Mocchegiani and S. Sensi, Biogerontology, 2006, 7, 307–314 CrossRef CAS PubMed.
- J. M. Falcón-Pérez and E. C. Dell'Angelica, Exp. Cell Res., 2007, 313, 1473–1483 CrossRef PubMed.
- A. Okado-Matsumoto and I. Fridovich, J. Biol. Chem., 2001, 276, 38388–38393 CrossRef CAS PubMed.
- I. G. Gazaryan, I. P. Krasinskaya, B. S. Kristal and A. M. Brown, J. Biol. Chem., 2007, 282, 24373–24380 CrossRef CAS PubMed.
- A. M. Brown, B. S. Kristal, M. S. Effron, A. I. Shestopalov, P. A. Ullucci, K.-F. R. Sheu, J. P. Blass and A. J. Cooper, J. Biol. Chem., 2000, 275, 13441–13447 CrossRef CAS PubMed.
- S. L. Sensi, H. Z. Yin, S. G. Carriedo, S. S. Rao and J. H. Weiss, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2414–2419 CrossRef CAS PubMed.
- K. E. Dineley, T. V. Votyakova and I. J. Reynolds, J. Neurochem., 2003, 85, 563–570 CrossRef CAS PubMed.
- B. B. Manshian, S. Pokhrel, U. Himmelreich, K. Tämm, L. Sikk, A. Fernández, R. Rallo, T. Tamm, L. Mädler and S. J. Soenen, Adv. Healthcare Mater., 2017, 6, 1601379 CrossRef PubMed.
- D. Denoyer, S. Masaldan, S. La Fontaine and M. A. Cater, Metallomics, 2015, 7, 1459–1476 RSC.
- H. Naatz, B. B. Manshian, C. Rios Luci, V. Tsikourkitoudi, Y. Deligiannakis, J. Birkenstock, S. Pokhrel, L. Madler and S. J. Soenen, Angew. Chem., Int. Ed., 2020, 59, 1828–1836 CrossRef CAS PubMed.
- L. Wang, T. Zhang, P. Li, W. Huang, J. Tang, P. Wang, J. Liu, Q. Yuan, R. Bai and B. Li, ACS Nano, 2015, 9, 6532–6547 CrossRef CAS PubMed.
- Y. Toduka, T. Toyooka and Y. Ibuki, Environ. Sci. Technol., 2012, 46, 7629–7636 CrossRef CAS PubMed.
- H. Xie, M. M. Mason and J. P. Wise, Rev. Environ. Health, 2011, 26, 251–268 CAS.
- M. Farnebo, V. J. Bykov and K. G. Wiman, Biochem. Biophys. Res. Commun., 2010, 396, 85–89 CrossRef CAS PubMed.
- B. Antonsson, Cell Tissue Res., 2001, 306, 347–361 CrossRef CAS PubMed.
- J. M. Brown and B. G. Wouters, Cancer Res., 1999, 59, 1391–1399 CAS.
- D. Jiang, P. G. Sullivan, S. L. Sensi, O. Steward and J. H. Weiss, J. Biol. Chem., 2001, 276, 47524–47529 CrossRef CAS PubMed.
- M. M. Compton, Cancer Metastasis Rev., 1992, 11, 105–119 CrossRef CAS PubMed.
- Y. Sánchez-Pérez, Y. I. Chirino, Á. R. Osornio-Vargas, R. Morales-Bárcenas, C. Gutiérrez-Ruíz, I. Vázquez-López and C. M. García-Cuellar, Cancer Lett., 2009, 278, 192–200 CrossRef PubMed.
- M. J. Akhtar, M. Ahamed, S. Kumar, M. M. Khan, J. Ahmad and S. A. Alrokayan, Int. J. Nanomed., 2012, 7, 845 CAS.
- K. Ali, Q. Saquib, B. Ahmed, M. A. Siddiqui, J. Ahmad, M. Al-Shaeri, A. A. Al-Khedhairy and J. Musarrat, Process Biochem., 2020, 91, 387–397 CrossRef CAS.
- E. Zielinska, A. Zauszkiewicz-Pawlak, M. Wojcik and I. Inkielewicz-Stepniak, Oncotarget, 2018, 9, 4675 CrossRef PubMed.
- S. George, S. Pokhrel, T. Xia, B. Gilbert, Z. Ji, M. Schowalter, A. Rosenauer, R. Damoiseaux, K. A. Bradley and L. Mädler, ACS Nano, 2010, 4, 15–29 CrossRef CAS PubMed.
- N. Mizushima, B. Levine, A. M. Cuervo and D. J. Klionsky, Nature, 2008, 451, 1069–1075 CrossRef CAS PubMed.
- M. J. Akhtar, H. A. Alhadlaq, S. Kumar, S. A. Alrokayan and M. Ahamed, Arch. Toxicol., 2015, 89, 1895–1907 CrossRef CAS PubMed.
- D. Laha, A. Pramanik, J. Maity, A. Mukherjee, P. Pramanik, A. Laskar and P. Karmakar, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 1–9 CrossRef CAS PubMed.
- P. Vandenabeele, T. V. Berghe and N. Festjens, Science's STKE, 2006, 2006, pe44 CrossRef PubMed.
- J. H. Lara-González, R. Gomez-Flores, P. Tamez-Guerra, E. Monreal-Cuevas, R. Tamez-Guerra and C. Rodríguez-Padilla, J. Adv. Med. Med. Res., 2013, 1308–1316 CrossRef.
- P. Vandenabeele, L. Galluzzi, T. V. Berghe and G. Kroemer, Nat. Rev. Mol. Cell Biol., 2010, 11, 700–714 CrossRef CAS PubMed.
- N. B. Saleh, D. J. Milliron, N. Aich, L. E. Katz, H. M. Liljestrand and M. J. Kirisits, Sci. Total Environ., 2016, 568, 926–932 CrossRef CAS PubMed.
- E. Burello and A. P. Worth, Nanotoxicology, 2011, 5, 228–235 CrossRef CAS PubMed.
- N. Odzak, D. Kistler, R. Behra and L. Sigg, Environ. Pollut., 2014, 191, 132–138 CrossRef CAS PubMed.
- M.-L. Avramescu, P. Rasmussen, M. Chénier and H. Gardner, Environ. Sci. Pollut. Res., 2017, 24, 1553–1564 CrossRef CAS PubMed.
- A. M. Studer, L. K. Limbach, L. Van Duc, F. Krumeich, E. K. Athanassiou, L. C. Gerber, H. Moch and W. J. Stark, Toxicol. Lett., 2010, 197, 169–174 CrossRef CAS PubMed.
- W. Zhang, Y. Yao, N. Sullivan and Y. Chen, Environ. Sci. Technol., 2011, 45, 4422–4428 CrossRef CAS PubMed.
- S. Elzey and V. H. Grassian, J. Nanopart. Res., 2010, 12, 1945–1958 CrossRef CAS.
- G. M. Cooper and D. Ganem, Nat. Med., 1997, 3, 1042 CrossRef.
- S. Müller, J. Dennemärker and T. Reinheckel, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 34–43 CrossRef PubMed.
- V. Sée, P. Free, Y. Cesbron, P. Nativo, U. Shaheen, D. J. Rigden, D. G. Spiller, D. G. Fernig, M. R. White and I. A. Prior, ACS Nano, 2009, 3, 2461–2468 CrossRef PubMed.
- X. Jiang, T. Miclăuş, L. Wang, R. Foldbjerg, D. S. Sutherland, H. Autrup, C. Chen and C. Beer, Nanotoxicology, 2015, 9, 181–189 CrossRef CAS PubMed.
- W. G. Kreyling, A. M. Abdelmonem, Z. Ali, F. Alves, M. Geiser, N. Haberl, R. Hartmann, S. Hirn, D. J. De Aberasturi and K. Kantner, Nat. Nanotechnol., 2015, 10, 619 CrossRef CAS PubMed.
- J. W. Moreau, P. K. Weber, M. C. Martin, B. Gilbert, I. D. Hutcheon and J. F. Banfield, Science, 2007, 316, 1600–1603 CrossRef CAS PubMed.
- J. M. Schveigkardt, A. C. Rizzi, O. E. Piro, E. E. Castellano, R. C. d. Santana, R. Calvo and C. D. Brondino, Eur. J. Inorg. Chem., 2002, 2002, 2913–2919 CrossRef.
- F. Stephens, R. Vagg and P. Williams, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 841–845 CrossRef.
- L. Antolini, Inorg. Chem., 1985, 24, 3621–3626 CrossRef CAS.
- R. Calvo, C. Steren, O. Piro, T. Rojo, F. Zuniga and E. E. Castellano, Inorg. Chem., 1993, 32, 6016–6022 CrossRef CAS.
- B. Gilbert, S. C. Fakra, T. Xia, S. Pokhrel, L. Mädler and A. E. Nel, ACS Nano, 2012, 6, 4921–4930 CrossRef CAS PubMed.
- M. Chen and A. von Mikecz, Exp. Cell Res., 2005, 305, 51–62 CrossRef CAS PubMed.
- S. Linse, C. Cabaleiro-Lago, W. F. Xue, I. Lynch, S. Lindman, E. Thulin, S. E. Radford and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8691–8696 CrossRef CAS PubMed.
- A. A. Vertegel, R. W. Siegel and J. S. Dordick, Langmuir, 2004, 20, 6800–6807 CrossRef CAS PubMed.
- S. S. Mansy and J. Cowan, Acc. Chem. Res., 2004, 37, 719–725 CrossRef CAS PubMed.
- R. H. Holm, Acc. Chem. Res., 1977, 10, 427–434 CrossRef CAS.
- D. H. Flint, J. Tuminello and M. Emptage, J. Biol. Chem., 1993, 268, 22369–22376 CAS.
- S. S. Leal and C. M. Gomes, Biol. Chem., 2005, 386, 1295–1300 CAS.
- P. R. Gardner, I. Raineri, L. B. Epstein and C. W. White, J. Biol. Chem., 1995, 270, 13399–13405 CrossRef CAS PubMed.
- C. Levard, E. M. Hotze, B. P. Colman, A. L. Dale, L. Truong, X. Yang, A. J. Bone, G. E. Brown Jr, R. L. Tanguay and R. T. Di Giulio, Environ. Sci. Technol., 2013, 47, 13440–13448 CrossRef CAS PubMed.
- C. m. Levard, S. Mitra, T. Yang, A. D. Jew, A. R. Badireddy, G. V. Lowry and G. E. Brown Jr, Environ. Sci. Technol., 2013, 47, 5738–5745 CrossRef CAS PubMed.
- Q. L. Feng, J. Wu, G. Chen, F. Cui, T. Kim and J. Kim, J. Biomed. Mater. Res., 2000, 52, 662–668 CrossRef CAS PubMed.
- G. V. Lowry, K. B. Gregory, S. C. Apte and J. R. Lead, Environ. Sci. Technol., 2012, 46, 6893–6899 CrossRef CAS PubMed.
- J. M. Zook, S. E. Long, D. Cleveland, C. L. A. Geronimo and R. I. MacCuspie, Anal. Bioanal. Chem., 2011, 401, 1993 CrossRef CAS PubMed.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H.-M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175 CrossRef CAS PubMed.
- C. Kaweeteerawat, A. Ivask, R. Liu, H. Zhang, C. H. Chang, C. Low-Kam, H. Fischer, Z. Ji, S. Pokhrel and Y. Cohen, Environ. Sci. Technol., 2015, 49, 1105–1112 CrossRef CAS PubMed.
- Q. Mu, G. Jiang, L. Chen, H. Zhou, D. Fourches, A. Tropsha and B. Yan, Chem. Rev., 2014, 114, 7740–7781 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- F. C. Simeone and A. L. Costa, Environ. Sci.: Nano, 2019, 6, 3102–3112 RSC.
- M. V. Ganduglia-Pirovano, A. Hofmann and J. Sauer, Surf. Sci. Rep., 2007, 62, 219–270 CrossRef CAS.
- T. C. Le, H. Yin, R. Chen, Y. Chen, L. Zhao, P. S. Casey, C. Chen and D. A. Winkler, Small, 2016, 12, 3568–3577 CrossRef CAS PubMed.
- R. Buonsanti and D. J. Milliron, Chem. Mater., 2013, 25, 1305–1317 CrossRef CAS.
- I. Bilecka, L. Luo, I. Djerdj, M. D. Rossell, M. Jagodic, Z. Jaglicic, Y. Masubuchi, S. Kikkawa and M. Niederberger, J. Phys. Chem. C, 2011, 115, 1484–1495 CrossRef CAS.
- H. Naatz, S. Lin, R. Li, W. Jiang, Z. Ji, C. H. Chang, J. Köser, J. Thöming, T. Xia and A. E. Nel, ACS Nano, 2017, 11, 501–515 CrossRef CAS PubMed.
- R. Wang, A. W. Sleight and M. Subramanian, J. Solid State Chem., 1996, 125, 224–227 CrossRef CAS.
- J. Xiao, A. Kuc, S. Pokhrel, M. Schowalter, S. Parlapalli, A. Rosenauer, T. Frauenheim, L. Mädler, L. G. Pettersson and T. Heine, Small, 2011, 7, 2879–2886 CrossRef CAS PubMed.
- S. Jang and J. A. Imlay, J. Biol. Chem., 2007, 282, 929–937 CrossRef CAS PubMed.
- S. L. Chia and D. T. Leong, Heliyon, 2016, 2, e00177 CrossRef PubMed.
- X. Cai, A. Lee, Z. Ji, C. Huang, C. H. Chang, X. Wang, Y. P. Liao, T. Xia and R. Li, Part. Fibre Toxicol., 2017, 14, 13 CrossRef PubMed.
- M. J. Osmond-McLeod, R. I. Osmond, Y. Oytam, M. J. McCall, B. Feltis, A. Mackay-Sim, S. A. Wood and A. L. Cook, Part. Fibre Toxicol., 2013, 10, 54 CrossRef PubMed.
- Y. Zhang, H. Wang, H. Jiang and X. Wang, Nanoscale, 2012, 4, 3530–3535 RSC.
- H. F. Hassan, A. M. Mansour, A. M. Abo-Youssef, B. E. Elsadek and B. A. Messiha, Clin. Exp. Pharmacol. Physiol., 2017, 44, 235–243 CrossRef CAS PubMed.
- A. Kermanizadeh, K. Jantzen, M. B. Ward, J. A. Durhuus, L. Juel Rasmussen, S. Loft and P. Moller, Nanotoxicology, 2017, 11, 184–200 CrossRef CAS PubMed.
- M. Benguigui, I. S. Weitz, M. Timaner, T. Kan, D. Shechter, O. Perlman, S. Sivan, Z. Raviv, H. Azhari and Y. Shaked, Sci. Rep., 2019, 9, 1–10 CrossRef CAS PubMed.
- M. A. Siddiqui, H. A. Alhadlaq, J. Ahmad, A. A. Al-Khedhairy, J. Musarrat and M. Ahamed, PLoS One, 2013, 8, e69534 CrossRef CAS PubMed.
- M. Shafagh, F. Rahmani and N. Delirezh, Iran. J. Basic Med. Sci., 2015, 18, 993 Search PubMed.
- Y. He, Z. Du, S. Ma, Y. Liu, D. Li, H. Huang, S. Jiang, S. Cheng, W. Wu and K. Zhang, Int. J. Nanomed., 2016, 11, 1879 CrossRef CAS PubMed.
- N. E.-A. El-Naggar, M. H. Hussein and A. A. El-Sawah, Sci. Rep., 2017, 7, 1–20 CrossRef CAS PubMed.
- J. R. Nakkala, R. Mata, K. Raja, V. K. Chandra and S. R. Sadras, Mater. Sci. Eng., C, 2018, 91, 372–381 CrossRef CAS PubMed.
- S. El-Sonbaty, Cancer Nanotechnol., 2013, 4, 73 CrossRef CAS PubMed.
- S. M. Dadfar, K. Roemhild, N. I. Drude, S. von Stillfried, R. Knüchel, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2019, 138, 302–325 CrossRef CAS PubMed.
- P. Aisen, C. Enns and M. Wessling-Resnick, Int. J. Biochem. Cell Biol., 2001, 33, 940–959 CrossRef CAS PubMed.
- T. R. Pisanic II, J. D. Blackwell, V. I. Shubayev, R. R. Fiñones and S. Jin, Biomaterials, 2007, 28, 2572–2581 CrossRef PubMed.
- D.-M. Huang, J.-K. Hsiao, Y.-C. Chen, L.-Y. Chien, M. Yao, Y.-K. Chen, B.-S. Ko, S.-C. Hsu, L.-A. Tai and H.-Y. Cheng, Biomaterials, 2009, 30, 3645–3651 CrossRef CAS PubMed.
- S. J. Soenen, U. Himmelreich, N. Nuytten and M. De Cuyper, Biomaterials, 2011, 32, 195–205 CrossRef CAS PubMed.
- H. Arami, A. Khandhar, D. Liggitt and K. M. Krishnan, Chem. Soc. Rev., 2015, 44, 8576–8607 RSC.
- A. Van de Walle, A. P. Sangnier, A. Abou-Hassan, A. Curcio, M. Hemadi, N. Menguy, Y. Lalatonne, N. Luciani and C. Wilhelm, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4044–4053 CrossRef CAS PubMed.
- T. Xia, Y. Zhao, T. Sager, S. George, S. Pokhrel, N. Li, D. Schoenfeld, H. Meng, S. Lin and X. Wang, ACS Nano, 2011, 5, 1223–1235 CrossRef CAS PubMed.
- M. Shakir, M. Faraz, M. A. Sherwani and S. I. Al-Resayes, J. Lumin., 2016, 176, 159–167 CrossRef CAS.
- A. I. Nabeel, Tumor Biol., 2020, 42 DOI:10.1177/1010428320909999.
- K. Li, G. Xiao, J. J. Richardson, B. L. Tardy, H. Ejima, W. Huang, J. Guo, X. Liao and B. Shi, Adv. Sci., 2019, 6, 1801688 CrossRef PubMed.
- S. Nair, A. Sasidharan, V. D. Rani, D. Menon, S. Nair, K. Manzoor and S. Raina, J. Mater. Sci.: Mater. Med., 2009, 20, 235 CrossRef PubMed.
- A. Joshi, H. Naatz, K. Faber, S. Pokhrel and R. Dringen, Neurochem. Res., 2020, 1–16 Search PubMed.
- N. Wu, C. Zhang, C. Wang, L. Song, W. Yao, A. Gedanken, X. Lin and D. Shi, Nanomedicine, 2018, 13, 1303–1318 CrossRef CAS PubMed.
- X. Li, H. Xu, C. Li, G. Qiao, A. A. Farooqi, A. Gedanken, X. Liu and X. Lin, Front. Pharmacol., 2019, 10, 319 CrossRef CAS PubMed.
- R. Yuan, H. Xu, X. Liu, Y. Tian, C. Li, X. Chen, S. Su, I. Perelshtein, A. Gedanken and X. Lin, ACS Appl. Mater. Interfaces, 2016, 8, 31806–31812 CrossRef CAS PubMed.
- H. Xu, R. Yuan, X. Liu, X. Li, G. Qiao, C. Li, A. Gedanken and X. Lin, Nanomedicine, 2019, 14, 131–149 CrossRef CAS PubMed.
- A. Joshi, W. Rastedt, K. Faber, A. G. Schultz, F. Bulcke and R. Dringen, Neurochem. Res., 2016, 41, 3004–3019 CrossRef CAS PubMed.
- A.-K. Ostermeyer, C. Kostigen Mumuper, L. Semprini and T. Radniecki, Environ. Sci. Technol., 2013, 47, 14403–14410 CrossRef CAS PubMed.
- B. B. Manshian, J. Jimenez, U. Himmelreich and S. J. Soenen, Adv. Healthcare Mater., 2017, 6, 1601099 CrossRef PubMed.
- M. I. Setyawati, X. Yuan, J. Xie and D. T. Leong, Biomaterials, 2014, 35, 6707–6715 CrossRef CAS PubMed.
- M. Buttacavoli, N. N. Albanese, G. Di Cara, R. Alduina, C. Faleri, M. Gallo, G. Pizzolanti, G. Gallo, S. Feo and F. Baldi, Oncotarget, 2018, 9, 9685 CrossRef PubMed.
- J. Swanner, C. D. Fahrenholtz, I. Tenvooren, B. W. Bernish, J. J. Sears, A. Hooker, C. M. Furdui, E. Alli, W. Li, G. L. Donati, K. L. Cook, P. A. Vidi and R. Singh, FASEB BioAdv., 2019, 1, 639–660 CrossRef CAS PubMed.
- Z. Shaterabadi, G. Nabiyouni and M. Soleymani, Mater. Sci. Eng., C, 2017, 75, 947–956 CrossRef CAS PubMed.
- M. A. Malvindi, V. De Matteis, A. Galeone, V. Brunetti, G. C. Anyfantis, A. Athanassiou, R. Cingolani and P. P. Pompa, PLoS One, 2014, 9, e85835 CrossRef PubMed.
- Y. Javed, L. Lartigue, P. Hugounenq, Q. L. Vuong, Y. Gossuin, R. Bazzi, C. Wilhelm, C. Ricolleau, F. Gazeau and D. Alloyeau, Small, 2014, 10, 3325–3337 CrossRef CAS PubMed.
- G. Spagnuolo, V. D'Antò, C. Cosentino, G. Schmalz, H. Schweikl and S. Rengo, Biomaterials, 2006, 27, 1803–1809 CrossRef CAS PubMed.
- G. Fukami, K. Hashimoto, K. Koike, N. Okamura, E. Shimizu and M. Iyo, Brain Res., 2004, 1016, 90–95 CrossRef CAS PubMed.
- H. M. El-Shorbagy, S. M. Eissa, S. Sabet and A. A. El-Ghor, Int. J. Nanomed., 2019, 14, 3911 CrossRef CAS PubMed.
- R. Tanino, Y. Amano, X. Tong, R. Sun, Y. Tsubata, M. Harada, Y. Fujita and T. Isobe, Mol. Cancer Ther., 2020, 19, 502–512 CrossRef CAS PubMed.
- E. Waidely, A.-R. O. Al-Yuobi, A. Bashammakh, M. S. El-Shahawi and R. M. Leblanc, Analyst, 2016, 141, 36–44 RSC.
- J. J. Antony, M. A. A. Sithika, T. A. Joseph, U. Suriyakalaa, A. Sankarganesh, D. Siva, S. Kalaiselvi and S. Achiraman, Colloids Surf., B, 2013, 108, 185–190 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman and M. Moseley, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- C. Rodriguez-Antona and M. Ingelman-Sundberg, Oncogene, 2006, 25, 1679–1691 CrossRef CAS PubMed.
- K. Nobutani, Y. Shimono, K. Mizutani, Y. Ueda, T. Suzuki, M. Kitayama, A. Minami, K. Momose, K. Miyawaki and K. Akashi, PLoS One, 2015, 10, e0130032 CrossRef PubMed.
- J. Liu, Y. Zhao, Q. Guo, Z. Wang, H. Wang, Y. Yang and Y. Huang, Biomaterials, 2012, 33, 6155–6161 CrossRef CAS PubMed.
- A. Manke, L. Wang and Y. Rojanasakul, BioMed Res. Int., 2013, 2013, e942916 Search PubMed.
- T. Kurz, A. Terman, B. Gustafsson and U. T. Brunk, Histochem. Cell Biol., 2008, 129, 389–406 CrossRef CAS PubMed.
- P. Klíma, M. Laňková, F. Vandenbussche, D. Van Der Straeten and J. Petrášek, Plant Cell Rep., 2018, 37, 809–818 CrossRef PubMed.
- D. J. Eide, Biochim. Biophys. Acta, Mol. Cell Res., 2006, 1763, 711–722 CrossRef CAS PubMed.
- J. H. Kaplan and E. B. Maryon, Biophys. J., 2016, 110, 7–13 CrossRef CAS PubMed.
- S. Liu, Y. Liu, B. Pan, Y. He, B. Li, D. Zhou, Y. Xiao, H. Qiu, M. G. Vijver and W. J. Peijnenburg, Chemosphere, 2020, 245, 125612 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |