
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Building heterogeneous nanostructures for photocatalytic ammonia decomposition
Shijie
Zhang
a,
Zuoli
He
 *a,
Xuan
Li
a,
Jing
Zhang
a,
Qianhao
Zang
b and
Shuguang
Wang
*a
*a,
Xuan
Li
a,
Jing
Zhang
a,
Qianhao
Zang
b and
Shuguang
Wang
*a
aShandong Key Laboratory of Water Pollution Control and Resource Reuse, School of Environmental Science and Engineering, Shandong University, Qingdao 266237, China. E-mail: zlhe@sdu.edu.cn; wsg@sdu.edu.cn
bSchool of Materials Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang 212003, China
First published on 10th July 2020
Abstract
Ammonia is an important chemical for human beings that is used in the synthesis of chemical fertilizers and products; meanwhile, it is also a hazardous compound which causes undesirable odors, several diseases, and environmental problems. Therefore, there is an urgent need to control and remove ammonia pollutants from water, air and soil. Hence, clean processes using photocatalysis to convert ammonia into H2 and N2 have been an important research topic in recent years. To date, only some metal-loaded common photocatalysts, such as TiO2, ZnO, C3N4, graphene and other carbon-based materials together with their hybrid materials, have been reported as active photocatalysts for the decomposition of aqueous ammonia solutions. In this review, we summarize the recent advances in heterogeneous nanostructures for photocatalytic ammonia decomposition. Particular emphasis is also given to metal-loading along with the resulting heterojunctions. Furthermore, the recent efforts toward the development of heterogeneous nanostructures for photocatalytic ammonia decomposition in this direction are discussed and appraised. Finally, perspectives and future opportunities regarding the challenges and future directions in the area of heterogeneous photocatalysts for ammonia decomposition are also provided.
 Shijie Zhang | Shijie Zhang received a B.S. in Environmental Engineering from the School of Environmental Science and Engineering of Shandong University (Qingdao, China) in 2019 and is currently pursuing a M.S. under the supervision of Prof. Zuoli He and Shuguang Wang at the same university. His research interests focus on heterogeneous nanostructures for photocatalytic nitrogen fixation, ammonia decomposition and other environmental oxidation or reduction processes. |
 Zuoli He | Zuoli He has been a research professor at the School of Environmental Science and Engineering in Shandong University since 2019. He received a Ph.D. in Electronic Science and Technology from Xi'an Jiaotong University in 2015. He spent one year at the University of Utah as a visiting researcher (2013–2014), two years at the Division of Environmental Science and Engineering, POSTECH as a postdoctoral researcher (2015–2017) and another two years at the Korea Institute of Materials Science (KIMS) as a senior researcher (2017–2019). His current research interests focus on functional nanostructured composite materials for photocatalytic solar energy conversion. |
 Xuan Li | Xuan Li is currently a B.S. student at the School of Environmental Science and Engineering in Shandong University (Qingdao, China). She will receive her B.S. in Environmental Engineering in June 2020. Then, she will pursue M.S. studies under the supervision of Prof. Zuoli He at the same university. Her research interests focus on 2D material-based heterojunctions for diverse photocatalytic applications, including nitrogen fixation, ammonia decomposition and other environmental oxidation or reduction processes. |
 Jing Zhang | Jing Zhang is currently a B.S. student at Yanshan University (Qinhuangdao, China) and a joint researcher in Prof Zuoli He's lab. She will start M.S. studies under the supervision of Prof. Zuoli He at Shandong University after receiving her B.S. in Environmental Engineering in June 2020. Her research interests focus on 1D heterogeneous nanostructures for diverse photocatalytic applications, including nitrogen fixation, ammonia decomposition and other environmental oxidation or reduction processes. |
 Qianhao Zang | Qianhao Zang is currently a lecturer at Jiangsu University of science and technology. He received his B.S. degree in Metallic Materials Engineering and his M.S. in Materials Engineering from Jiangsu University of Science and Technology in 2012 and 2015, respectively, and he received his Ph.D. in Materials Science and Engineering from Shandong University in 2019. He was a joint Ph.D. student at Korea Institute of Materials Science (KIMS) from 2016 to 2018. After obtaining his Ph.D. in 2019, he started his professorship at Jiangsu University of Science and Technology. His research interests focus on functional composites and nanomaterials. |
 Shuguang Wang | Shuguang Wang is currently a professor at the School of Environmental Science and Engineering at Shandong University. from obtained his B.S. and M.S. in Dalian Jiaotong University and pursued his Ph.D. at the Research Center for Eco-Environmental Sciences (RCEES), Chinese Academy of Sciences (CAS). After obtaining his Ph.D. in 2002, he started a professorship at Shandong University. His research interests cover eater pollution control and treatment, soil remediation and improvement and bioenergy of greenhouse gases. |
Introduction
In the past few decades, unavoidable consequences of fossil fuels and their excessive use have caused numerous negative effects on human health as well as on the environment, especially in developing countries.1–3 Therefore, there is a strong need to establish eco-friendly and sustainable alternatives to address environmental pollution while maintaining sustainable development of the global economy.4,5 Excess nitrogen compounds have been discharged from many types of industrial operations, such as chemical engineering and power plants as well as livestock industry facilities.6–11 For example, ammonia is mainly produced from the microbial decomposition of nitrogen-containing organic substances at very low oxygen concentrations and the decomposition of urea and nitrogen compounds in wastewater treatment processes. As a hazardous compound, it causes undesirable odors, several diseases and environmental problems; thus, there is an urgent need to control and remove ammonia pollutants from water, air and soil.12,13 It should be noted that ammonia is also a promising hydrogen carrier because it possesses an excellent energy density and storage capacity of H2 of 17.6 wt% (compared to other storage materials such as cyclohexane, ethanol and liquefied petroleum gas).14,15 Hydrogen production from ammonia decomposition is an eco-friendly and sustainable alternative to transform ammonia pollutants into resources.16–18 As shown in eqn (1)–(3), hydrogen production from ammonia decomposition is an uphill reaction from the thermodynamics point of view and requires energy for the reactions, and the Gibbs free energy of ammonia decomposition is far lower than that of water splitting; thus, a photocatalyst for water splitting can convert photon energy into chemical energy through this reaction.19,20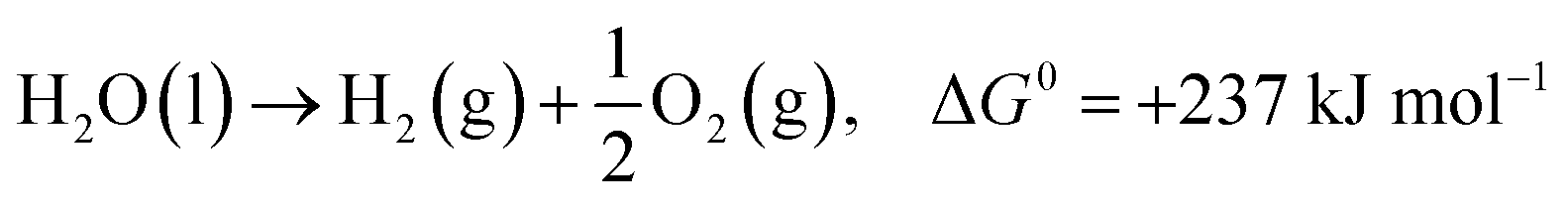 | (1) |
 | (2) |
 | (3) |
In the last ten years, many researchers working on the conversion of NH3 into non-toxic H2 and N2 (an uphill reaction, ΔG = 11 or 18 kJ mol−1) have made their best efforts to improve catalytic systems to better mimic COx-free hydrogen production performance.21 Therefore, this eco-friendly artificial synthetic reaction will be an important research topic. To date, three main catalytic approaches, namely thermal-catalysis,22–24 electro-catalysis25,26 and photo-catalysis,27–29 have been utilized for ammonia decomposition. Pertinent studies have proved that solar energy is an effective alternative among the various environmentally friendly energies, which is attributed to its great potential in electricity generation, chemical fuel production and pollutant degradation. Fujishima and Honda discovered the phenomenon of photocatalytic water splitting on titanium dioxide (TiO2) electrodes in 1972;30 this triggered the development of semiconductor photocatalysts for a wide range of environmental and energy applications, including degradation of organic pollutants, water splitting, nitrogen fixation, hydrogen production from ammonia, and carbon dioxide reduction. The efficiency of photocatalysis is related to its efficiency in five main steps during the reactions: (i) light absorption, (ii) formation of photogenerated electron–hole pairs, (iii) migration and separation of photogenerated electron–hole pairs, (iv) adsorption of reactants and desorption of products, and (v) occurrence of redox reactions on the semiconductor surface.31,32 Building heterogeneous nanostructures has been proved to enhance photocatalytic activity by increasing some of these five efficiencies; this often involves defect engineering, surface engineering, interface engineering and heterojunction engineering.
For uphill reactions such as water splitting, hydrogen production from ammonia, and carbon dioxide reduction, the slow surface multi-electron reaction kinetics leads to inevitable accumulation of photogenerated electrons and holes on the surface of photocatalysts, which accelerates the unexpected charge carrier recombination and the photocorrosion process of the photocatalysts themselves and consequently reduces their photoactivity.33 Photocatalytic decomposition of ammonia using sunlight, as an artificial uphill photosynthetic reaction, often takes place in alkaline conditions. When the irradiating light has higher energy than the band gap of the photocatalyst, electron–hole pairs will generally form (eqn (4)). The generated holes (h+) have been demonstrated to be powerful oxidants, whereas the conduction band electrons (e−) can act as strong reductants to reduce O2 for the formation of hydroxyl radicals (˙OH). The holes or ˙OH for the oxidation of ammonia to N2 or NOx are provided by the reactions shown in eqn (5)–(10).34 H2 production by the decomposition of an aqueous ammonia solution is an uphill reaction, as mentioned above. Thus, an uphill reaction to convert photon energy into chemical energy will be achieved using a heterogeneous photocatalyst driven by one-step photoexcitation. As shown in Fig. 1, electron/hole pairs can be generated by photons with higher energy than the corresponding band-gap energy of the photocatalyst; some of these electron/hole pairs can migrate to the surface or defects and initiate a series of chemical reactions with the adsorbed species on the surface of the catalyst, resulting in the degradation of ammonia. From the thermodynamic viewpoint, only when the ammonia reduction and oxidation potentials lie between the conduction band (CB) and the valence band (VB) potentials can such reactions be driven by photogenerated electrons and holes. During this process, different nitrogen-containing products (i.e., N2, NO2−, NO3−, and NOx) can be produced due to their similar redox potentials; the potentials of these typical reactions are given in Fig. 1.35 Ammonia can be decomposed into different amounts of various products under diverse reaction conditions (i.e., pH, temperature, initial ammonia or O2 concentration, trapping agent, sacrificial agent). To achieve the photocatalytic degradation of ammonia, the photogenerated electrons and holes on the surface of a semiconductor should have suitable reduction and oxidation ability to react with the adsorbed species (i.e., O2, NH4+, NO2−, NO3) on the surface of the catalyst and generate free radicals or different products.
| Photocatalyst + hν → e− + h+ | (4) |
| OH− + h+ → OH˙ | (5) |
| O2 + e− → O2−˙ | (6) |
| O2 + 2e− + 2H+ → H2O2 | (7) |
| 2O2−˙ + 2H+ → H2O2 + O2 | (8) |
| H2O2 + e− → OH− + OH˙ | (9) |
| H2O2 + O2−˙ → OH− + OH˙ + O2 | (10) |
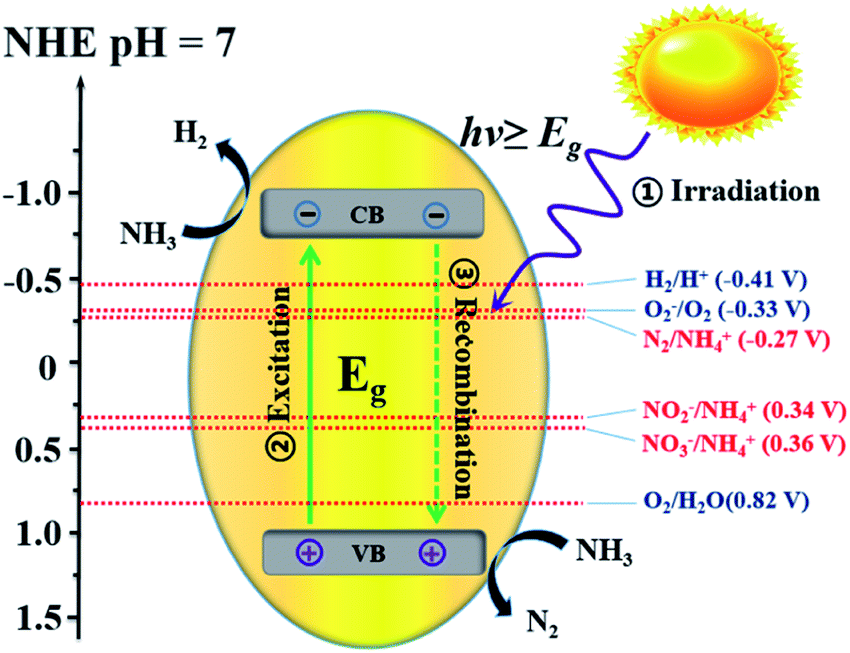 | ||
| Fig. 1 Illustration of the process of photocatalytic ammonia decomposition. | ||
Moreover, photocatalytic decomposition of ammonia usually stops under acidic conditions, which indicates that H+ ions inhibit the conversion of ammonia to NH2 free radicals. When the pH gradually increases, ammonia may react with surrounding oxygen to produce NO2−, NO3− and other nitrogen oxides (eqn (11)–(13)), which is detrimental to hydrogen production.36
| NH3 + 3O2 → 2NO2− + 2H+ + 2H2O, ΔG0 = +11 kJ mol−1 | (11) |
| 2NO2− + O2 → NO3−, ΔG0 = +11 kJ mol−1 | (12) |
| NH3 + 2O2 → NO3− + H+ + H2O, ΔG0 = +11 kJ mol−1 | (13) |
To date, only some metal-loaded common photocatalysts, such as TiO2, ZnO, C3N4, graphene and other carbon-based materials, together with their hybrid materials have been reported as active photocatalysts for the decomposition of an aqueous ammonia solution into H2 and N2, as presented in Table 1.19,25,37,38 In this review, particular emphasis will first be given to metal-loading along with the resulting heterojunctions; then, the recent developments of TiO2, ZnO, C3N4, graphene and other carbon-based materials in the application of ammonia decomposition will be respectively described. Finally, some concluding remarks and perspectives on the challenges and opportunities for exploring efficient heterogeneous photocatalysts for ammonia decomposition are presented. We hope that this review will assist and inspire further development of efficient heterogeneous materials for ammonia decomposition.
| Photocatalyst | Synthesis method | Band gap | Light source | Illumination | Initial ammonia concentration | Maximum decomposition ability | Ref. |
|---|---|---|---|---|---|---|---|
| Pt–TiO2 (0.5 wt% Pt) | Photochemical reduction | — | 450 W high pressure Hg lamp | 298 K, 24 h | 5 mM | Over 95% | 36 |
| Ni/TiO2 (0.5 wt% Ni) | Impregnation method | — | 500 W Xe lamp | 3 h | 5 mL, 0.59 mol L−1 | 131.7 μmol H2 per g-catalyst | 39 |
| Ce-doped TiO2 (0.6 wt% Ce) | Sol–gel method | 3.14 eV | 8 W Hg pen-ray lamp | 10 h | 100 mL, 0.8274 g L−1 | 810 mmol H2 per g-catalyst | 40 |
| Ce-doped TiO2 (1.2 wt% Ce) | Sol–gel method | 3.00 eV | 8 W Hg pen-ray lamp | 10 h | 100 mL, 0.8274 g L−1 | 1060 mmol H2 per g-catalyst | 40 |
| Ce-doped TiO2 (1.4 wt% Ce) | Sol–gel method | 2.97 eV | 8 W Hg pen-ray lamp | 10 h | 100 mL, 0.8274 g L−1 | 1010 mmol H2 per g-catalyst | 40 |
| Pt/Fe–TiO2 (0.5 wt% Pt; 1.0 wt% Fe) | Impregnation method | — | 500 W Xe lamp | 2 h | 5 mL, 0.59 mol L−1 | 27 μmol H2 per g-catalyst | 41 |
| Light expanded clay aggregate (LECA)-TiO2 nanoparticles | Thermal annealing | — | 80 W medium-pressure Hg lamp | 3 h | 0.05 M, 750 mL | 85% | 42 |
ZnO/Ag (molar ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
One-pot method | — | 300 W Xe lamp | 298 K, 2.5 h | 1.5 mg L−1 | Circa 90% | 43 |
| ZnO | Thermal annealing | 3.26 eV | 8 W Hg pen-ray lamp | 8 h | 0.8274 g L−1 | 1060 μmol H2 per g-catalyst | 19 |
| ZnO | Precipitation reaction + thermal annealing | 3.24 eV | 8 W Hg pen-ray lamp | 8 h | 0.8274 g L−1 | 670 μmol H2 per g-catalyst | 19 |
| ZnO | Precipitation reaction + UV irradiation + thermal annealing | 3.23 eV | 8 W Hg pen-ray lamp | 8 h | 0.8274 g L−1 | 860 μmol H2 per g-catalyst | 19 |
| TiO2–ZnO (molar ratio 1:2)/LECA (light expanded clay aggregate) |
Impregnation method | 3.22 eV | 125 W high pressure Hg lamp | 293 K, 3 h | 400 mg L−1 | 95.2% | 44 |
| ZnO/oak charcoal | Impregnation method | — | 125 W Hg lamp | 293 K, 4 h | 153.9 mg L−1 | About 80% | 34 |
| N–C@TiO2 (nitrogen-doped carbon framework) | hydrothermal method | — | 25 W UV lamp | 295 ± 1.5 K, 5 min | 100 μL aqueous ammonia (30%) | 100% | 45 |
| MoS2@TiO2 CNBs (carbon nanobelts) | Electrospinning followed by hydrothermal reaction method | — | 25 W UV lamp | 295 ± 1.5 K, 7 min | 100 μL aqueous ammonia (30%) | 91% | 46 |
| MoS2/N-doped graphene (5% of MoS2 mass) | Hydrothermal method | 0.80 eV | 300 W UV-visible lamp | 298 ± 2 K, 8 h | 100.0 mg L−1 | 99.6% | 47 |
| Nitrogen-doped rGO/TiO2 nanowires (NWs) membranes | Modified Hummers method + adsorption method | — | 8 W Hg pen-ray lamp | 12 h | 0.883 g L−1 | 208 μmol h−1 g−1 | 48 |
| SL g (single layer graphitic)-C3N4 | Thermal annealing + hydrothermal method | 3.0 eV | Xe lamp (195 mW cm−2) | 298 K, 6 h | 1.5 mg L−1 | Over 80% | 35 |
| GQDs (graphene quantum dots)/CN (g-C3N4) (0.5 wt% GQDs) | Thermal annealing + Hummer's method + hydrothermal method | 2.7 eV | 150 W Xe arc lamp | 7 h | 1.5 mg L−1 | 90% | 49 |
Building heterogeneous nanostructures
As mentioned above, building heterogeneous nanostructures has been proved to enhance photocatalytic performance by increasing some of the five efficiencies during photocatalysis reactions; this often involves defect engineering, surface engineering, interface engineering and heterojunction engineering. For ammonia decomposition, heterogeneous photocatalysts are often obtained by metal-loading and constructing heterojunctions with semiconductor photocatalysts for use in water splitting. As shown in Fig. 2, metal loading will introduce defect positions, high-speed charge transfer paths to increase the efficiencies of migration and separation of the photogenerated electron–hole pairs, adsorption of reactants and desorption of products, and occurrence of redox reactions on the semiconductor surface.50–57 Heterojunctions improve the activity of photocatalysts because of their suitable structures, which improve charge separation and transfer, promote optical absorption, optimize the bandgap position, and enhance the spatial separation of electron–hole pairs; this can increase some of the five efficiencies during photocatalysis, namely light absorption, formation of photogenerated electron–hole pairs, migration and separation of the photogenerated electron–hole pairs, adsorption of reactants and desorption of products, and occurrence of redox reactions on the semiconductor surface.50–52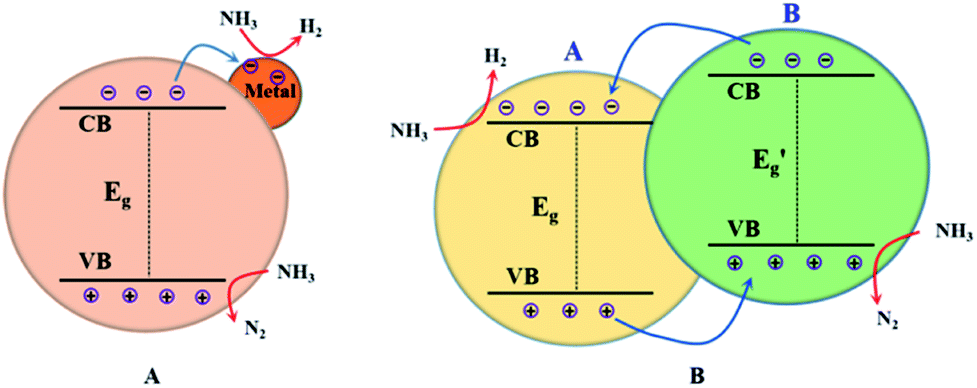 | ||
| Fig. 2 Illustrations of metal-loading (A) and heterojunction building (B) for enhanced photocatalytic ammonia decomposition performance. | ||
Taking metal-loaded TiO2 as an example to more deeply describe the advances in photocatalytic ammonia decomposition, metal loading can inhibit the recombination of electrons and holes on the surface of TiO2 to extend the carrier lifetimes by introducing defect positions or high-speed charge transfer paths.36,41,58–62 Furthermore, it can create active sites to increase the number of reaction locations and the adsorption capabilities of the reactants. Overall, metal loading will lead to enhanced photocatalytic performance and provide better chemical and environmental stability. Metal-loaded photocatalysts show higher activities than bare TiO2 to produce nitrogen and hydrogen from ammonia with an almost stoichiometric product ratio.62 As shown in Fig. 3A, among these metal-loaded TiO2 samples, metals with larger work functions are effective for gaseous ammonia decomposition. The most suitable cocatalyst for hydrogen evolution is Pt, which exhibits a high work function, low Fermi level and strong electron extracting capacity. In addition, it can be easily loaded by photo-deposition from a molecular precursor. In addition, the work function of the bulk metal and the logarithm of the hydrogen production rate have an almost linear relationship, as shown in Fig. 3B; this indicates that the photocatalytic hydrogen production activity in ammonia decomposition is mainly derived from the charge separation ability of the photogenerated hole and electron pairs rather than the original catalytic properties of the loaded metals for hydrogen production. In other words, the loaded metal with a larger work function, i.e., a lower Fermi level, will more easily accept the excited electrons from the conduction band (−0.16 V vs. NHE, Fig. 3C) to promote H2 production for ammonia decomposition. Although metal loading can effectively inhibit the recombination of electrons and holes on the catalyst surface, overloading of metal can also cause more recombination centers to appear and can affect the performance of the photocatalyst. Therefore, the optimal weight ratio of the metal loading to the main photocatalyst is mostly lower than 3 wt%.
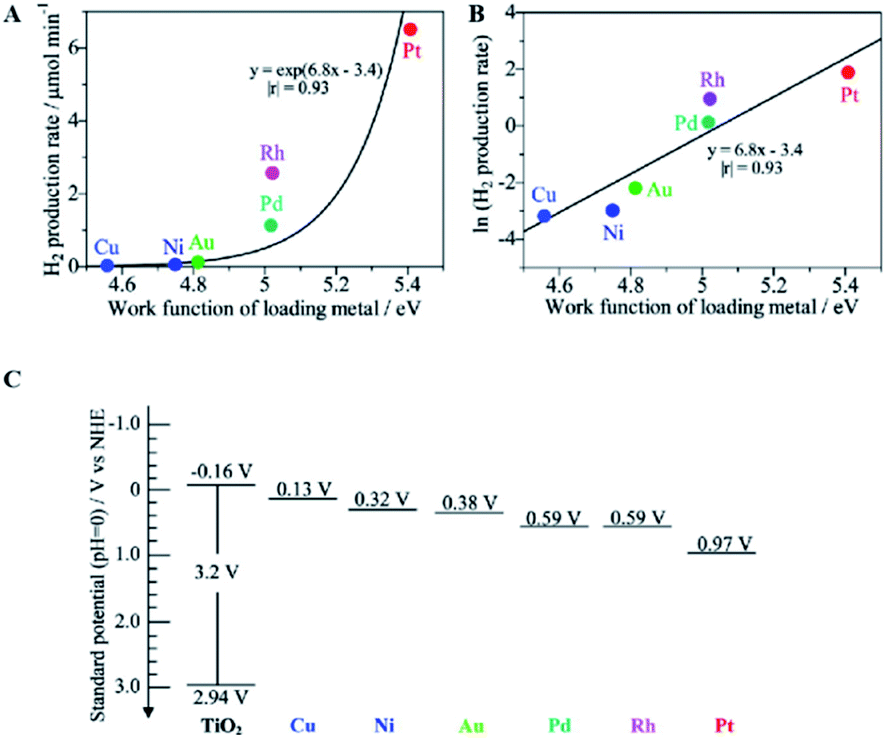 | ||
| Fig. 3 Relationship between the hydrogen production rate over the M(0.1)/TiO2 samples and the work function of the corresponding bulk metal (A), the logarithmic plot (B), and the standard potentials for the band edge positions of titanium oxide and the Fermi level of each loading metal (C). Reprinted with permission from ref. 62. Copyright 2012 American Chemical Society. | ||
In general, a heterojunction photocatalyst contains an interface between two different semiconductors with unequal band structures, which can result in suitable band alignments for photocatalytic reactions. For type-II heterojunction photocatalysts (see Fig. 2B), the CB and the VB levels of semiconductor B are higher than the corresponding levels of semiconductor A. Thus, the photogenerated electrons will transfer to semiconductor A while the photogenerated holes will migrate to semiconductor B, which will result in spatial separation of the electron–hole pairs.63 The redox ability of these heterojunction photocatalysts will also be reduced because the reduction reaction and the oxidation reaction take place on semiconductor A with lower reduction potential and on semiconductor B with lower oxidation potential, respectively.64 Generally, this type of heterojunction photocatalyst exhibits a smaller band-gap, good electron–hole separation, a wide light-absorption range, fast mass transfer and a larger surface area; this will increase the efficiency of some processes during photocatalysis, such as light absorption, formation of photogenerated electron–hole pairs, and migration and separation of the photogenerated electron–hole pairs. Additionally, metal-loading and heterojunction building have been applied together to design highly efficient ammonia decomposition photocatalysts. In the following sections, we provide a comprehensive overview of the ammonia decomposition photocatalysts, summarizing recent related studies in this area.
Metal-loaded TiO2 and its heterojunction photocatalysts
TiO2 is one of the most common photocatalysts; it has good performance in many photocatalytic applications, such as water splitting, hydrogen production, carbon dioxide reduction, nitrogen oxide reduction, nitrogen fixation, pollutant degradation, organic reactions, and ammonia synthesis and decomposition.31,42,65–74 Three crystal types of TiO2, including anatase type, rutile type and plate titanium type, exist in nature, with bandgap widths of 3.0, 3.2 and 3.25 eV, respectively.75 Bare TiO2 shows lower photocatalytic activity in ammonia decomposition due to the reactive energy barrier. Ammonia decomposition is an uphill reaction to convert photon energy into chemical energy; thus, it must be achieved by a heterogeneous photocatalyst driven by one-step photoexcitation. Additionally, the photocatalytic efficiency of TiO2 on the surface is relatively low due to the rapid recombination of photocarriers.76 Therefore, in some studies, TiO2 has been modified with loading metals and heterojunctions have been constructed with other semiconductors to overcome these limits.45,77Utsunomiya et al. compared the photocatalytic activities of ammonia decomposition of TiO2 loaded with various metals under UV irradiation at room temperature.39 As clearly shown in Table 2, Ni/TiO2 photocatalysts demonstrate the highest yields of H2. The NH2 radical was formed as a dominant intermediate during NH3 decomposition, which was confirmed by ESR measurements. H2 and H2N–NH2 as an intermediate were formed by the coupling of an NH2 radical and an NH3 molecule in the gas phase during the photodecomposition of NH3. XRD patterns proved that the crystal structure of Ni/TiO2 was stable, which further demonstrated that Ni has a good catalytic decomposition effect on ammonia. ESR measurements showed that NH2 radical is the dominant intermediate in the decomposition process. Ni0 was suggested to enhance the reaction pathways via H2N–NH2 because Ni0 was present in the catalyst that showed the highest catalytic activity. They also proposed three reaction pathways to investigate the mechanism of NH3 decomposition, as presented in Fig. 4. N2 and H2 were produced with the aid of an NH2 radical: route 1 involved the formation of NH radicals through extraction of one hydrogen atom from each of two NH2 radicals; route 2 involved the formation of NH2–NH2 by the coupling of adjacent NH2 radicals; and route 2′ involved the formation of NH2–NH2via formation of H2N–NH3. The activation energies for routes 1 and 2 were also calculated as 236 kcal mol−1 and 74.8 kcal mol−1, respectively, by density functional theory (DFT). Route 2 was found to be more energetically favorable than route 1. Possible reaction pathways by which N2 and H2 were formed through NH2–NH2 coupling were further split into route 2, involving the formation of H2N–NH2 by coupling of NH2 radicals; route 2′ involved the interaction of NH2 with one NH3 molecule in the gas phase. Their activation energies were estimated to be 74.4 kcal mol−1 and 59.2 kcal mol−1, respectively. Therefore, it is possible that NH3 decomposition proceeded by both routes 2 and 2′ via the formation of NH2–NH2.
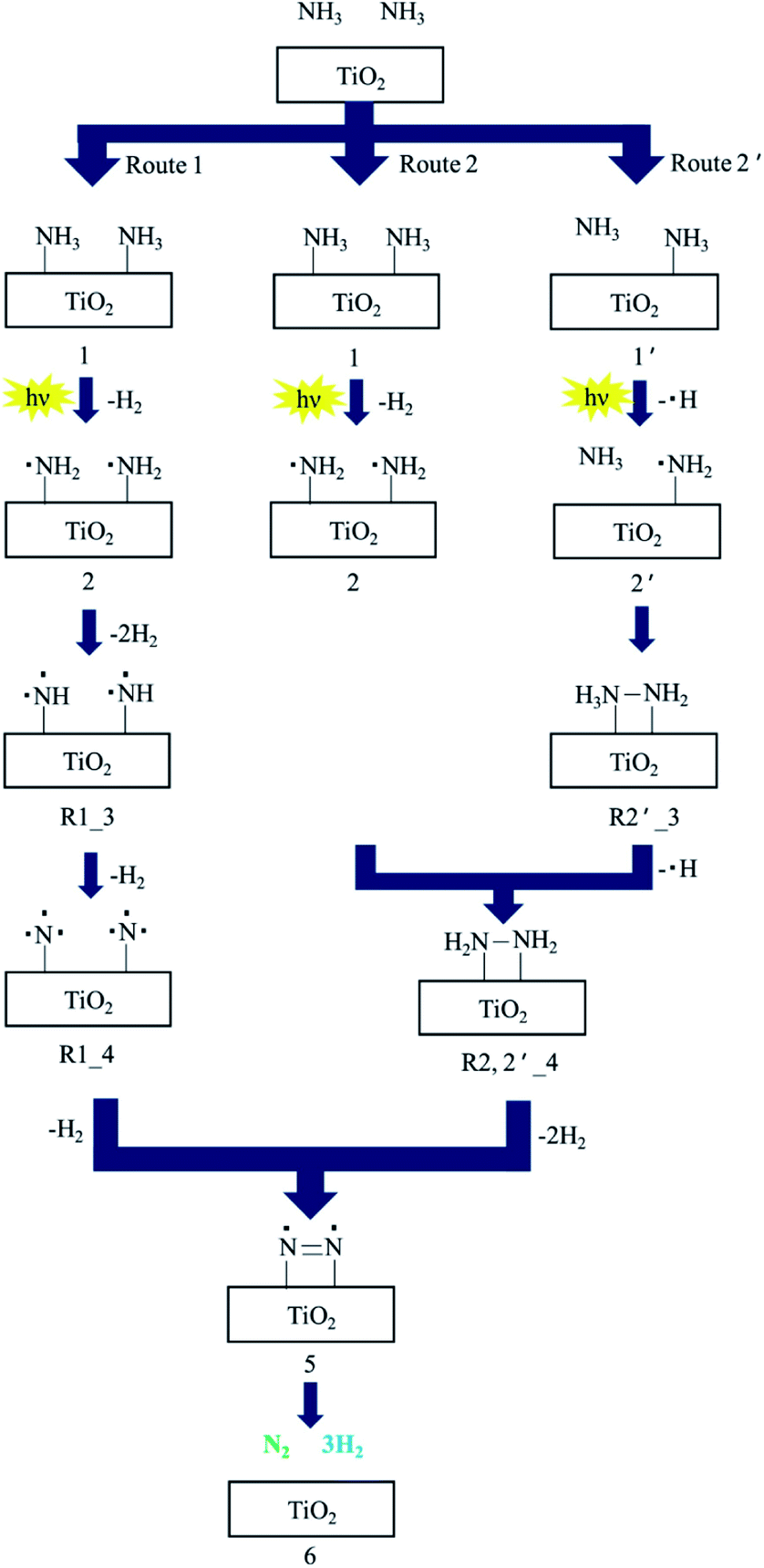 | ||
| Fig. 4 The suggested reaction mechanism for decomposition of NH3 to N2 and H2 over TiO2 photocatalysts. Reprinted from ref. 39, Copyright 2017, with permission from Elsevier. | ||
The influence of Ce loading on the performance of photocatalytic ammonia decomposition was also investigated by Reli et al.40 Cerium-doped TiO2 photocatalysts with different contents (0–1.4 wt%) were prepared within reverse micelles of nonionic surfactant Triton X-114 in cyclohexane. The anatase crystallite sizes decreased with increasing amount of cerium ions in TiO2, which corresponded to the increase of the specific surface area of the catalysts. The addition of cerium led to a decrease of the absorption edge from 3.19 eV to 2.97 eV. NH3 was decomposed to H2 and N2 stoichiometrically and side reactions such as deep oxidation to NO2− and NO3− did not occur when the reaction was carried out in their stirred batch annular reactor. This metal modification of TiO2 influenced the photocatalytic hydrogen formation. The direct correlation between the work functions, band gap energies and hydrogen yields of the catalysts was investigated in this research, as shown in Fig. 5. The optimal Ce content in anatase TiO2 will maximally lower the bandgap energy while maintaining sufficient electron and hole potentials for the photocatalytic reaction.
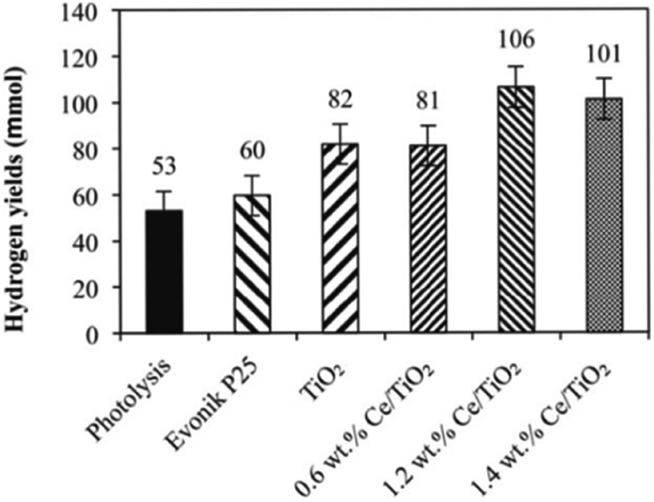 | ||
| Fig. 5 Hydrogen yields over each investigated photocatalyst and in the case of photolysis after 10 h irradiation. Conditions: 10 h of irradiation, 8 W Hg lamp (254 nm), 100 mL of NH4OH, 0.1 g of the photocatalyst. Reprinted from ref. 40. Copyright 2015, with permission from Elsevier. | ||
The reaction mechanism of ammonia decomposition to nitrogen and hydrogen over platinum-loaded titanium oxide photocatalysts has been investigated through various reaction tests, as well as ESR and FT-IR spectroscopy.62 The pH of the reaction suspension can strongly affect both the oxidation rate and selectivity, which was confirmed by Shibuya et al.36 They investigated the influence of pH and pH adjustment conditions on the photocatalytic oxidation of aqueous ammonia with Pt–TiO2 under airflow. The oxidation of ammonia could not proceed under acidic conditions and could only proceed under alkaline conditions, suggesting that molecular ammonia reacts with photogenerated holes. Both the maximum oxidation rate and the maximum amount of ammonia adsorbed on the photocatalyst are observed at ≈pH 10; increasing the adsorbed capability of molecular ammonia is important to increase the oxidation rate. The decrease in pH due to the formation of nitrite and nitrate from the photo-oxidation of ammonia and the cessation of the reaction at pH values below 7 indicated that for the oxidation of ammonia, the pH must be maintained in the alkaline region during the reaction. Moreover, the amount of ammonia adsorbed when the pH was adjusted with NaOH (aq) was higher than that achieved using the Na2CO3–NaHCO3 buffer solution at the same pH because carbonate/hydrocarbonate ions prevent the adsorption of ammonia and the photogenerated holes are consumed by the oxidation of nitrite in the presence of the buffer. A high initial concentration of ammonia and pH adjustment to 10 with NaOH (aq) promotes the oxidation of ammonia with low selectivity for the undesirable products nitrite and nitrate. Performing the reaction with a high density of N-containing species on the photocatalyst surface favorably increases the selectivity for dinitrogen.
Obata et al. analyzed the performance of Pt-supported metal-doped TiO2 (Pt/M-TiO2, M: dopant element) photocatalysts on decomposing NH3 to H2 and N2 by ultraviolet radiation at room temperature (Table 3).41 Fe-doped TiO2 photocatalyst (Pt/Fe–TiO2) obtained the highest yield of H2 production in ammonia photodecomposition. The Fe3+ replaced the Ti4+ sites in TiO2 crystal without changing the structure of TiO2, and the absorption edge of TiO2 was shifted from the ultraviolet to the visible light region by Fe substitution, as shown in Fig. 6. The resulting Fe–TiO2 catalyst material allows the effective utilization of irradiation light owing to the presence of an Fe impurity band, thereby leading to higher activity of decomposing NH3 aqueous solution and more effective hydrogen production.
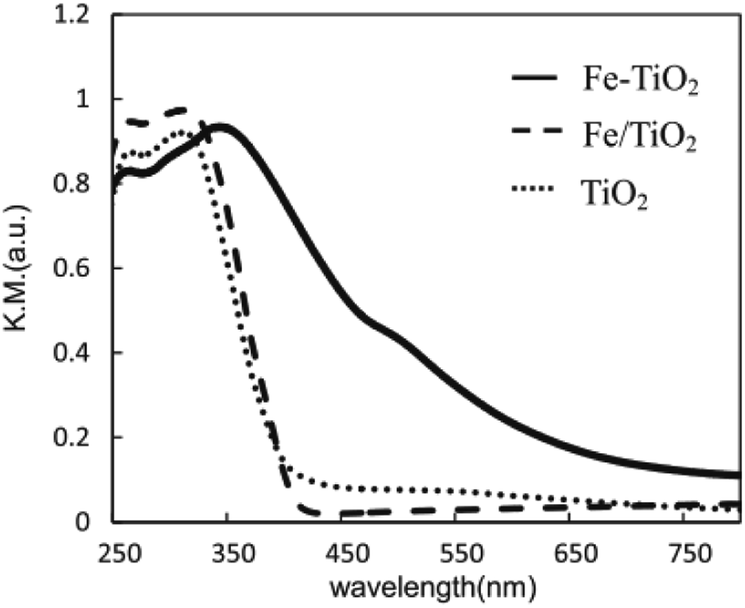 | ||
| Fig. 6 UV-vis-DR spectra of Fe–TiO2, Fe/TiO2 and TiO2 photocatalysts. Reprinted from ref. 41. Copyright 2015, with permission from Elsevier. | ||
In addition, TiO2 nanoparticles supported on light expanded clay aggregate granules (LECA) showed enhanced photocatalytic decomposition of ammonia.42 More than 85% of the ammonia was removed within 300 min under the optimum calcination temperature of 550 °C and pH 11.0. The kinetics of this photocatalytic reaction followed a pseudo-first-order model. By using floated TiO2/LECA as the photocatalyst in an aqueous solution of NH3, ammonia was photodegraded into N2 and H2 gases, while NO2− and NO3− were formed at very low concentrations. Furthermore, they reported a novel hybrid structure of TiO2/ZnO/LECA for efficient removal of ammonia from synthetic wastewater,44 which will be further discussed in the following section.
ZnO-based heterogeneous photocatalysts
Similar to TiO2, ZnO is one of the most widely used photocatalysts due to its high photocatalytic efficiency, low cost, and environmental sustainability.78–80 Its photocatalytic performance can be effectively improved by tailoring its crystallite size, surface area, and concentrations of active sites, oxygen defects, and facets.43,81–84 Moreover, metal-loaded ZnO and its heterojunctions have been used for photocatalytic decomposition of ammonia.Different synthetic procedures lead to the formation of different defects (oxygen vacancies and oxygen excess defects) in the ZnO lattice, which is a factor influencing the photocatalytic hydrogen production efficiency of decomposition of ammonia solution under ultraviolet light of 254 nm.19 The highest yields of hydrogen were observed in ZnO prepared by thermal annealing of zinc acetate in air, as shown in Table 4. This photocatalyst possesses the lowest concentration of oxygen vacancies, causing efficient trapping of electrons. Consequently, a higher number of electrons in the conduction band was available for photocatalytic decomposition of ammonia.
| Photocatalyst | Preparation method | L c (nm) | S BET (m2 g−1) | Band gap (eV) | H2 evolutiona (μmol g−1) | Rate of H2 production (μmol H2 per g per h) |
|---|---|---|---|---|---|---|
| a Values depict photocatalytic activities after 8 h irradiation. b Reprinted from ref. 19. Copyright 2015, with permission from Elsevier. | ||||||
| ZnO (1) | Thermal annealing | 36 | 15 | 3.26 | 1060 | 10.3 |
| ZnO (2) | Precipitation reaction + thermal annealing | 27 | 49 | 3.24 | 670 | 5.8 |
| ZnO (3) | Precipitation reaction + UV irradiation + thermal annealing | 15 | 43 | 3.23 | 860 | 6.8 |
| Evonik TiO2 P25 | Commercial | 26 | 50 | 3.39 | 800 | 7.3 |
As mentioned above, metal loading or heterojunction building has been proved to be an efficient approach to enhance the photocatalytic activity for ammonia decomposition. Guo et al. synthesized a porous Ag-loaded ZnO heterogeneous photocatalyst by a one-pot method, and the interface contact and the electron transfer capacity between ZnO and other materials were proved to be enhanced.43 The photocatalytic activity for decomposition of ammonia was significantly improved after Ag modification and retained high efficiency in the stability experiment; this can be attributed to inhibition of the recombination process of photogenerated electrons and holes by interconversion between Ag+ and Ag0 at the surface of ZnO. In addition, the effective separation of the photogenerated carriers can generate more active groups, which can promote the degradation of ammonia or organic dyes.
The experimental conditions of photocatalytic removal of ammonia from synthetic wastewater by a ZnO/oak charcoal photocatalyst were optimized using the Box–Behnken experimental design method.34 The contributions of different experimental conditions to the removal rate of ammonia were measured (Fig. 7), and the experimental data were fitted with a second-order polynomial regression model to derive the optimal experimental conditions, under which the maximum degradation efficiency of ammonia was about 80%. All three main variables of initial ammonia concentration, pH and catalyst dosage were effective in the ammonia decomposition. Among them, the initial ammonia concentration was found to be the most effective parameter, which is helpful for future research on ammonia decomposition. Based on their analysis of variance results, the significance order of the independent parameters was as follows: initial ammonia concentration > catalyst dosage > solution pH. In addition, the fabrication of ZnO-based heterojunctions is considered to be an effective approach to improve the photocatalytic activity of ZnO. The interface contact of heterojunctions can greatly affect the electron transfer capacity between ZnO and other materials. Mohammadi et al.44 immobilized TiO2/ZnO on a light-expanded clay aggregate (LECA) support to build heterostructures for removal of ammonia from synthetic wastewater. The capability of ammonia decomposition was significantly affected by the mobility and lifetime of the charge carriers generated in the TiO2/ZnO composite. Coupling of TiO2:ZnO with a molar ratio of 1:2 into a heterojunction photocatalyst showed the lowest photoluminescence emission intensity and maximum photocatalytic degradation activity of ammonia. The optimal pH, catalyst loading, and initial concentration of ammonia were found to be 11 g L−1, 25 g L−1, and 400 mg L−1, respectively. Also, 95.2% ammonia removal was achieved during 3 h of UV irradiation in these optimal conditions. More efforts will need to be focused on the improvement of the exploration of optimal experimental parameters.
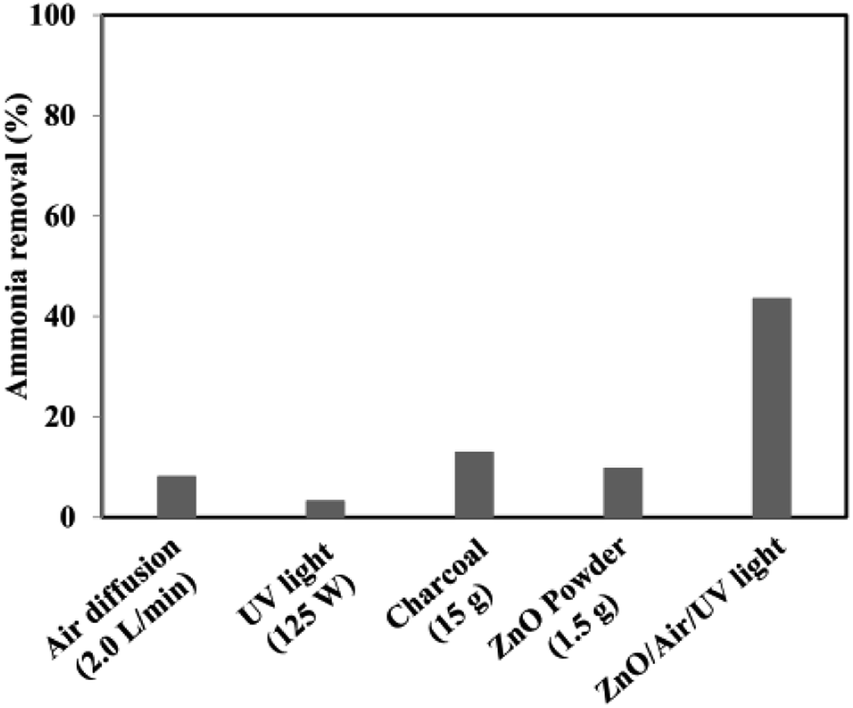 | ||
| Fig. 7 The results of reference tests to determine the proportion of adsorption and stripping as well as the effects of UV light and catalyst floating in the total removal of ammonia. Reprinted from ref. 34. Copyright 2017, with permission from Elsevier. | ||
Graphene-based photocatalysts
Graphene is a typical 2D material with a single atomic layer thickness composed of carbon atoms. Graphene is a zero band-gap material with dense honeycomb structures because its valence and conduction bands meet at the Dirac point.85 The low Fermi level of graphene also endows it with excellent electrical conductivity as a supporting material. Due to its high electron mobility and special optical, electrical, and mechanical properties, graphene has received extensive attention recently.86 When used for photocatalysis, graphene is often combined with other semiconductor materials because it cannot be directly utilized as a photoactive material because of its lack of a suitable bandgap.87–89 Graphene provides active sites, charge transfer tunnels or electron enrichment areas to increase the capture of visible light and promote photogenerated electron–hole pair transfer and separation. More active sites on the surface of photocatalysts, higher capacity and efficiency of decomposition will be obtained.Ammonia decomposition requires much less energy input to occur compared with water splitting; ammonia can spontaneously react with O2 to form N2O and gives off a large quantity of heat, which provides the energy for water splitting. N2O decomposition occurs synchronously under light irradiation. Therefore, ammonia may be able to produce hydrogen by using oxygen to drive the decomposition reaction of water, and the self-modification of nitrogen of the catalyst is beneficial to the production of H2. The individual layers of the nitrogen-doped TiO2 nanowires (NWs)-intercalated reduced graphene oxide (rGO) membrane are distinguishable, and the TiO2 NWs appear to be uniformly distributed between single rGO sheets.48 These nitrogen-doped rGO/TiO2 NWs hybrids were immersed in ammonia solution, and improved efficiency of hydrogen production from ammonia decomposition under ultraviolet irradiation was observed because rGO inhibited the recombination of the photogenerated carriers in TiO2. An interesting phenomenon was demonstrated: the yield of hydrogen production of these nitrogen-doped rGO/TiO2 NWs photocatalysts was 14 times higher than that of TiO2-P25 and 30-fold higher than that of the TiO2 NWs alone under the same conditions, which is probably due to nitrogen self-doping in rGO and the TiO2 NWs during the catalytic reaction.
Similar to this, Zhang et al. prepared a photocatalyst consisting of MoS2 and N-doped graphene (NG); this heterojunction responds to near-infrared (NIR) light, as shown in Fig. 8.47 MoS2 is a 2D graphene-like material with a similar layered structure to that of graphene. The bandgap of multilayer MoS2 is within 1.29 to 1.8 eV and is even as low as 1.0 eV depending on the layer number and defects, corresponding to 1240 nm NIR radiation; this indicates its possible full use of the NIR irradiation from the solar spectrum. A direct Z-scheme NIR-response photocatalytic system (MoS2/NG) was composed of two narrow band-gap MoS2 and NG semiconductors to harvest NIR irradiation and degrade ammonia under NIR light irradiation. As shown in Fig. 9A, the ammonia degradation efficiency of the MoS2/NG photocatalytic system under NIR irradiation could reach 99.6%, whereas the removal ratio of ammonia was only 64.0% when single MoS2 was used as a photocatalyst under similar conditions. This low degradation efficiency was attributed to the low valence band level of the narrow band gap MoS2, which was only equal to 0.40 eV, resulting in weak oxidisation. The degradation efficiency increased with the NG content in the initial stage from 1.0% to 5.0% at 8 h. Then, it reached a maximum value of 99.0% when the NG content was 5.0%, as presented in Fig. 9B, resulting from the effective separation of the photogenerated electron–hole pairs; this enhanced the oxidisation ability of the photocatalytic system via the combination of MoS2 with NG. Moreover, the overload of NG resulted in an electric short circuit, which resulted in decreased degradation efficiency. It should be noted here that the MoS2/NG catalyst is highly stable, with an average degradation efficiency of ammonia of over 90% (Fig. 9C) after the fifth cycle. As schematically shown in Fig. 9D, the valence band level (1.42 V) of the composite catalyst was higher than the potential level (−0.276 V vs. NHE) of E0(N2/NH4+), and the composite catalyst could oxidise NH4+ to N2 more effectively. At the same time, the more positive valence band level of the Z-scheme system mainly contributed to the enhanced photocatalytic activity for ammonia degradation.
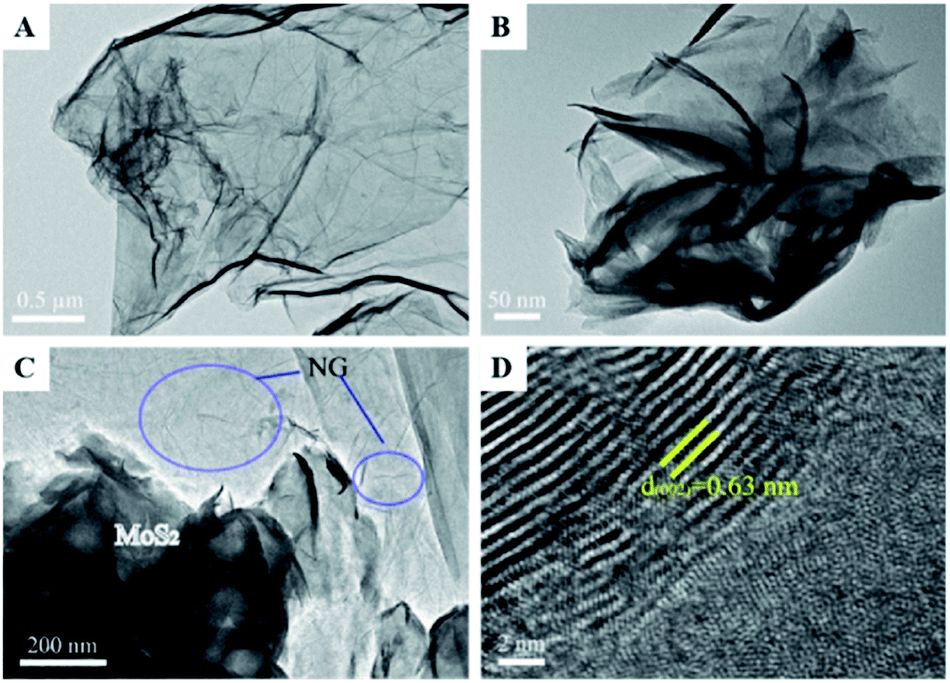 | ||
| Fig. 8 TEM images of the samples (A–D): (A), N-doped graphene (NG); (B), MoS2; (C), MoS2/NG; (D) high-resolution image of MoS2/NG. Reprinted from ref. 47. Copyright 2020, with permission from Elsevier. | ||
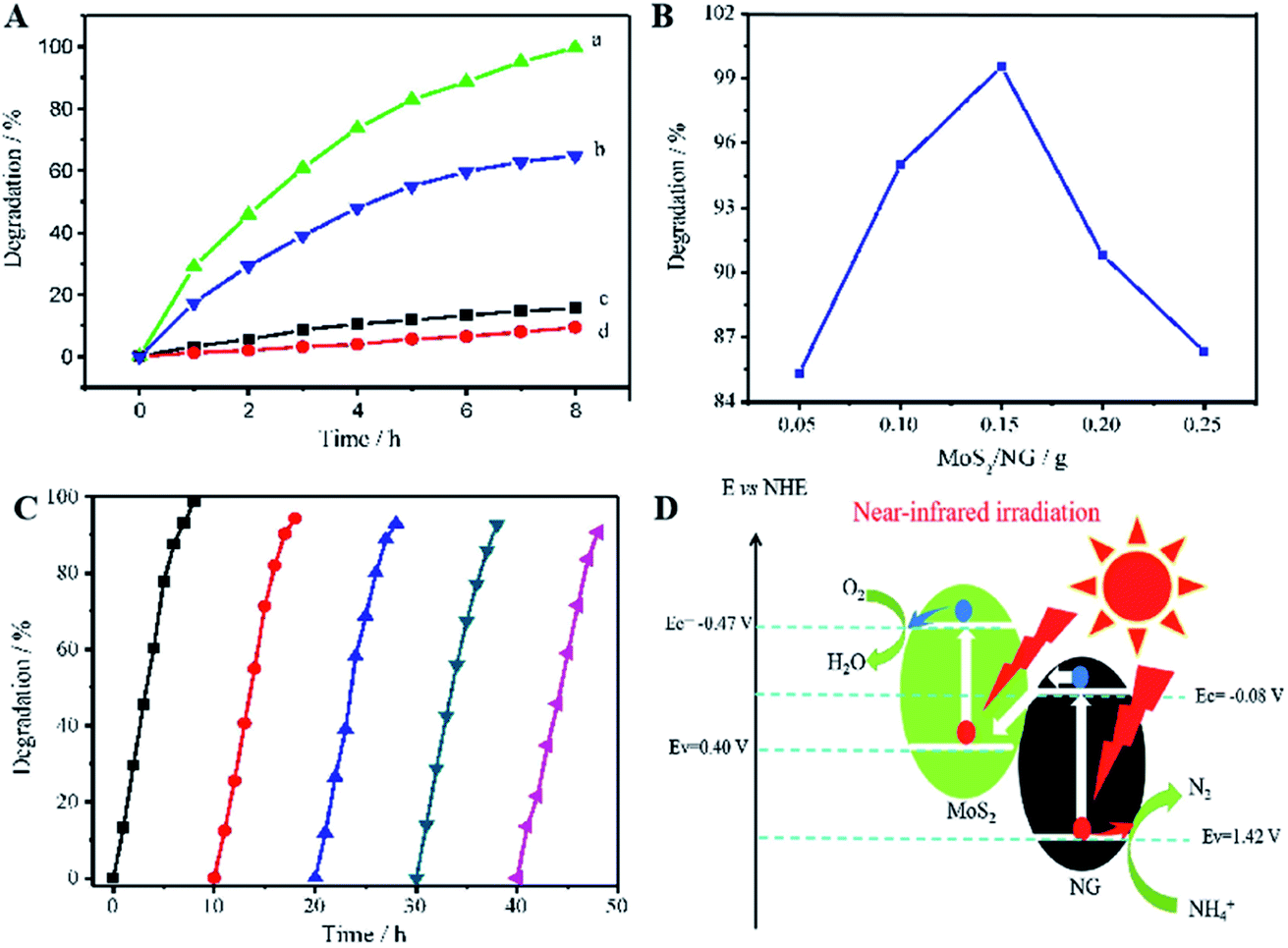 | ||
| Fig. 9 (A) Photocatalytic degradation of ammonia in 50 mL solutions containing NH3–N at 100 mg L−1 with pH 9.5. (a) 0.15 g of MoS2/NG catalyst under NIR irradiation; (b) 0.15 g of MoS2 catalyst under NIR irradiation; (c) 0.15 g of MoS2/NG in the dark and (d) under NIR irradiation without catalyst. (B) Effects of NG percentage on the ammonia degradation in 50 mL solutions containing 100 mg L−1 of NH3–N with pH 9.5 under NIR irradiation. (C) Five cyclic uses of the MoS2/NG catalyst to verify the stability. (D) The direct Z-scheme mechanism for ammonia degradation via the MoS2/NG photocatalyst under NIR irradiation. Reprinted from ref. 47. Copyright 2020, with permission from Elsevier. | ||
Graphitic-C3N4-based photocatalysts
Graphitic carbon nitride (g-C3N4), which is only composed of carbon and nitrogen, has been reported to be an attractive, inexpensive metal-free photocatalyst for hydrogen production from water splitting because its reduction and oxidation levels are both located inside the bandgap.29,90,91 In addition, g-C3N4 is fascinating because of its capability for potential applications, including the oxygen reduction reaction, selective organic synthesis, and organic pollutant degradation.29,92,93 A new type of atomic single layer graphitic-C3N4 (SL g-C3N4) has appealing applications as an inexpensive metal-free photocatalyst in aqueous ammonia treatment.35 SL g-C3N4 in alkaline solution demonstrated higher photocatalytic activity than in neutral or acidic solutions. Under the irradiation of a xenon lamp (195 mW cm−2), the TAN (initial concentration 1.50 mg L−1) removal rate in the presence of monolayer g-C3N4 within 6 h reached 80%. The investigation suggested that both photogenerated holes and hydroxyl radicals are involved in the TAN photocatalytic oxidation process and that the major oxidation product is NO3−–N. In addition, SL g-C3N4 exhibited good photocatalytic stability in aqueous solution.A graphene quantum dot (GQD)-modified g-C3N4 (GQDs/CN) heterogeneous photocatalyst demonstrated degradation properties for ammonia gas.49 Through GQD modification, the photon adsorption and electron transfer capability of the heterojunctions were effectively improved, leading to enhancing photocatalytic performance. As clearly shown in Fig. 10, the photocatalyst with 0.5 wt% GQD exhibited the best photocatalytic degradation performance of total ammonia nitrogen, which is approximately 3 times higher than that of pure g-C3N4. The increased photocatalytic properties can be attributed to the improvement of the photon adsorption ability and electron transfer capacity resulting from GQD modification. It should be noted here that a higher oxygen concentration and pH value are more favorable for the photocatalyst to degrade ammonia to produce NO3− (compared with NO2−).
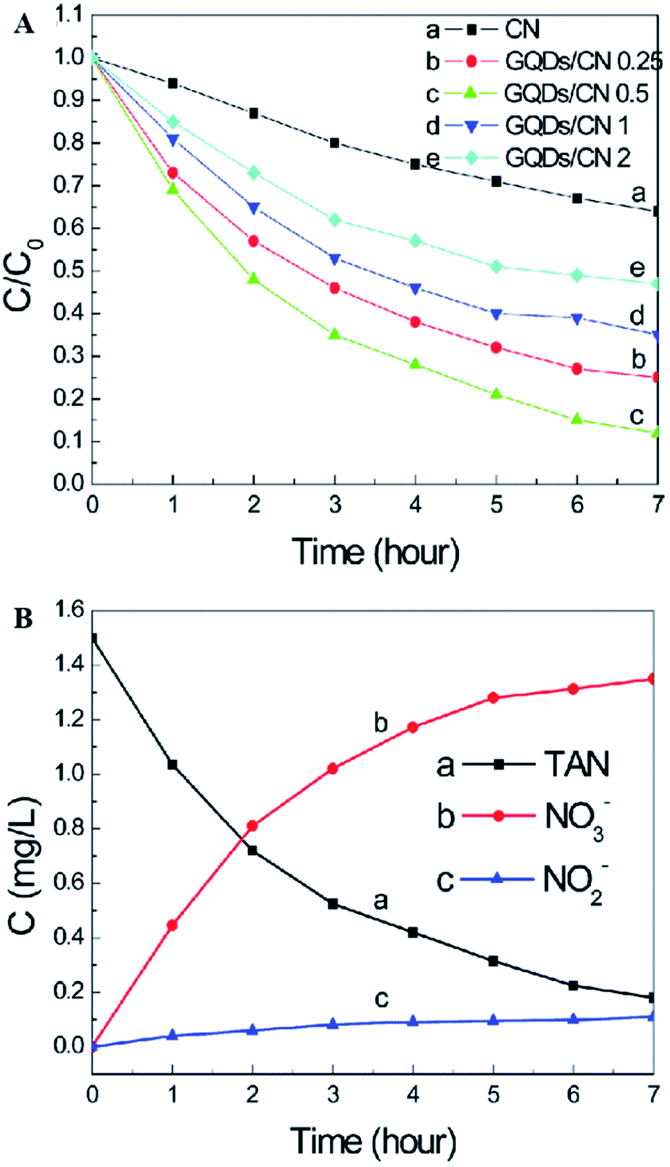 | ||
| Fig. 10 (A) Effects of the g-C3N4 and GQDs/CN composite dosage on the photocatalytic treatment of TAN solution. (B) Concentration changes of TAN, NO2− and NO3−. Reproduced from ref. 49 with permission from the Royal Society of Chemistry. | ||
Other C-based photocatalysts
In addition to graphene, other carbon-based materials (such as carbon frameworks, carbon nanotubes, and carbon nanobelts) have good electrical and optical properties that can be utilized by coupling the materials with other metal/non-metal materials to build heterogeneous photocatalysts. These heterogeneous structures show improved visible light absorption capacity, surface adsorption capacity, charge transmission, and separation capacities.45,46,94 Carbon-based composite photocatalysts (including activated carbon frameworks, carbon nanotubes, and carbon nanobelts) have been reported for the decomposition of ammonia or hydrogen production in the literature.45,46,94Ultrafine TiO2 encapsulated in a nitrogen-doped porous carbon framework was fabricated and used as a hierarchical-structured composite photocatalyst to degrade ammonia gas.45 After high-temperature calcination, the specific surface area and pore volume of the catalyst increased significantly, which may prompt pollutant molecules to penetrate into the holes and provide more active sites for photocatalysis. The as-prepared ultrafine TiO2 encapsulated in the nitrogen-doped porous carbon framework displayed excellent photocatalytic activity and degraded ammonia gas with 100% efficiency after only 5 min of light irradiation. This outstanding performance is related to the small size of TiO2, abundant porosity of the composite, and excellent light adsorption. In addition, the nitrogen-doped porous carbon framework in the composite efficiently accelerated the separation of photo-induced electrons and holes.
One-dimensional coaxial nanobelts of molybdenum disulfide grown on titanium dioxide-encapsulated carbon nanobelts (MoS2@TiO2 CNBs) by electrospinning followed by a hydrothermal reaction method were also prepared and investigated for the photocatalytic degradation of ammonia gas.46 The existence of Ti, O, Mo, S and C elements in the prepared TiO2 CNBs was confirmed by XRD, and the surface characteristics were observed by FESEM, HRTEM, TEM and EDS (Fig. 10). Under ultraviolet radiation, the MoS2@TiO2 CNBs heterojunction generates electron–hole pairs and participates in ammonia degradation (Fig. 11). The MoS2@TiO2 CNBs present excellent photocatalytic activity with an ammonia gas degradation rate of over 91% after 7 min due to the synergistic effect of the robust banding structure and chemical composition.
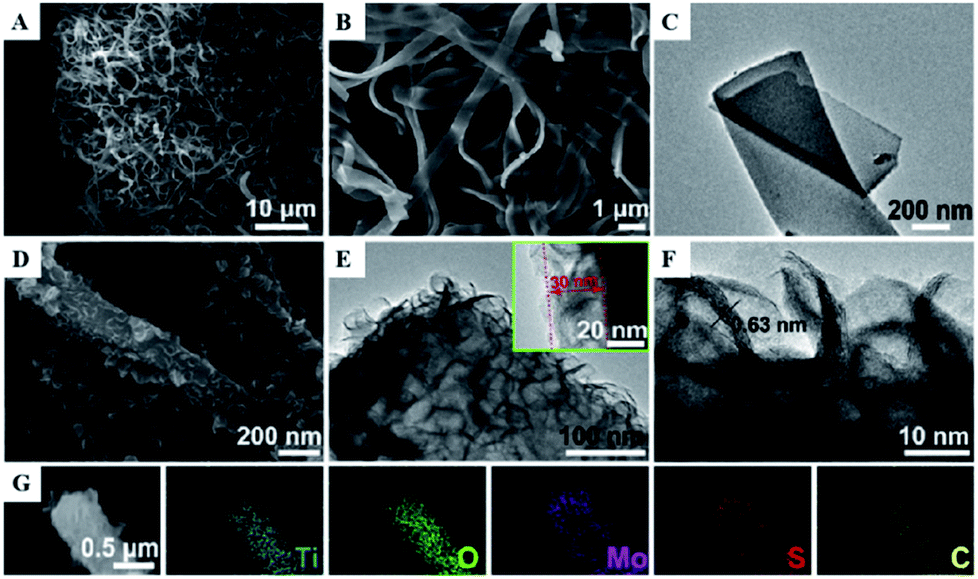 | ||
| Fig. 11 (A) Low, (B) high-resolution FESEM, and (C) TEM image of TiO2 CNBs. (D) FESEM, (E) TEM, and, (F) HRTEM images of MoS2@TiO2 CNBs. (G) EDS elemental maps of Ti, O, Mo, S, and C, respectively. Reprinted from ref. 46. Copyright 2017, with permission from Elsevier. | ||
Conclusions and outlook
Photocatalytic ammonia decomposition is a prospective technology for future industrial applications for the removal of ammonia and its conversion into harmless H2 and N2. It appears to be necessary to design heterogeneous structures combining metals or different semiconductors because many bare photocatalysts suffer from one or several limitations, such as poor activity, low stability, low surface area, complicated synthesis approaches, and larger band-gaps. Over the past five years, some significant strategies, such as the metal loading or heterojunction building listed above, have been successfully designed and applied in photocatalytic ammonia decomposition. Despite these significant advances, the photocatalytic efficiency is still too low for practical industrial applications. To address this challenge, future research opportunities and efforts are required in the following aspects:(1) More effort should be focused on building controllable heterogeneous structures by direct growth on base materials in future studies, which may significantly deepen our understanding of the photocatalytic enhancement mechanisms of ammonia decomposition. It is known that heterojunctions improve the activity to some degree in general; further enhancements will be obtained by controlling their morphologies or nanostructures. The designed morphologies or nanostructures show greater improvements (especially in charge separation and charge transfer capabilities between interfaces) than regular heterogeneous structures.
(2) Nitrogen oxides are easily generated during the photocatalytic decomposition of ammonia owing to the similar redox potentials of these nitrogen-containing products. Although current research has made great progress in the efficiency of photocatalytic decomposition of ammonia, much more research should be focused on controlling the decomposition products and improving the yield of hydrogen, which requires adjustment of the decomposition paths of ammonia to hydrogen by the design of new Z-scheme heterogeneous structures.
(3) Few available photocatalytic materials have been used for photocatalytic ammonia decomposition. The photocatalytic ammonia decomposition performance of more photocatalysts must be investigated in the future. We hope that the present review will stimulate scientific interest in photocatalytic ammonia decomposition using heterogeneous nanostructures as well as in other advanced environmental and energy-related photocatalytic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported under the framework of the Young Taishan Scholars Program of Shandong Province (No. tsqn. 201909026), Future Young Scholars Program of Shandong University (No. 61440089964189), and Youth Interdisciplinary Science and Innovative Research Groups of Shandong University (Grant No. 2020QNQT014).Notes and references
- C. J. Clarke, W. C. Tu, O. Levers, A. Brohl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- M. J. Foulkes, M. J. Hawkesford, P. B. Barraclough, M. J. Holdsworth, S. Kerr, S. Kightley and P. R. Shewry, Field Crops Res., 2009, 114, 329–342 CrossRef.
- L. Yuan and Y. J. Xu, Appl. Surf. Sci., 2015, 342, 154–167 CrossRef CAS.
- J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Chem. Soc. Rev., 2018, 47, 2322–2356 RSC.
- P. J. Landrigan, R. Fuller, N. J. R. Acosta, O. Adeyi, R. Arnold, N. Basu and A. B. Balde, et al. , Lancet, 2018, 391, 462–512 CrossRef.
- F. C. Meunier and J. R. H. Ross, Appl. Catal., B, 2000, 24, 23–32 CrossRef CAS.
- N. W. Cant and I. O. Y. Liu, Catal. Today, 2000, 63, 133–146 CrossRef CAS.
- F. C. Meunier, J. P. Breen, V. Zuzaniuk, M. Olsson and J. R. H. Ross, J. Catal., 1999, 187, 493–505 CrossRef CAS.
- M. Iwamoto, T. Zengyo, A. M. Hernandez and H. Araki, Appl. Catal., B, 1998, 17, 259–266 CrossRef CAS.
- M. Haneda, Y. Kintaichi, M. Inaba and H. Hamada, Catal. Today, 1998, 42, 127–135 CrossRef CAS.
- D. N. Thomas, R. J. Lara, H. Eicken, G. Kattner and A. Skoog, Polar Biol., 1995, 15, 477–483 CrossRef.
- F. Schuth, R. Palkovits, R. Schlogl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- M. Shaban, M. R. AbuKhadra, F. M. Nasief and H. M. Abd El-Salam, Water, Air, Soil Pollut., 2017, 228, 450 CrossRef.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- A. Yapicioglu and I. Dincer, Renewable Sustainable Energy Rev., 2019, 103, 96–108 CrossRef CAS.
- M. E. E. Abashar, J. King Saud Univ., Eng. Sci., 2018, 30, 2–11 Search PubMed.
- T. V. Choudhary, C. Sivadinarayana and D. W. Goodman, Catal. Lett., 2001, 72, 197–201 CrossRef CAS.
- S. Mukherjee, S. V. Devaguptapu, A. Sviripa, C. R. F. Lund and G. Wu, Appl. Catal., B, 2018, 226, 162–181 CrossRef CAS.
- M. Reli, M. Edelmannová, M. Šihor, P. Praus, L. Svoboda, K. K. Mamulová, H. Otoupalíková, L. Čapek, A. Hospodková, L. Obalová and K. Kočí, Int. J. Hydrogen Energy, 2015, 40, 8530–8538 CrossRef CAS.
- D. A. Hansgen, D. G. Vlachos and J. G. G. Chen, Nat. Chem., 2010, 2, 484–489 CrossRef CAS PubMed.
- R. Z. Sørensen, L. J. Nielsen, S. Jensen, O. Hansen, T. Johannessen, U. Quaade and C. H. Christensen, Catal. Commun., 2005, 6, 229–232 CrossRef.
- J. Ji, X. Duan, G. Qian, X. Zhou, G. Tong and W. Yuan, Int. J. Hydrogen Energy, 2014, 39, e12498 Search PubMed.
- K. Nagaoka, T. Eboshi, N. Abe, S. Miyahara, K. Honda and K. Sato, Int. J. Hydrogen Energy, 2014, 39, 20731–20735 CrossRef CAS.
- W. I. David, J. W. Makepeace, S. K. Callear, H. M. Hunter, J. D. Taylor, T. J. Wood and M. O. Jones, J. Am. Chem. Soc., 2014, 136, 13082–13085 CrossRef CAS PubMed.
- S. Podila, S. F. Zaman, H. Driss, Y. A. Alhamed, A. A. Al-Zahrani and L. A. Petrov, Catal. Sci. Technol., 2016, 6, 1496–1506 RSC.
- X. Duan, G. Qian, X. Zhou, Z. Sui, D. Chen and W. Yuan, Appl. Catal., B, 2011, 101, 189–196 CrossRef CAS.
- J. H. Carey, J. Lawrence and H. M. Tosine, Bull. Environ. Contam. Toxicol., 1976, 16, 697–701 CrossRef CAS PubMed.
- W. Zheng, J. Zhang, Q. Ge, H. Xu and W. Li, Appl. Catal., B, 2008, 80, 98–105 CrossRef CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- K. R. Reddy, M. Hassan and V. G. Gomes, Appl. Catal., A, 2015, 489, 1–16 CrossRef CAS.
- Z. He, C. Kim, L. Lin, T. H. Jeon, S. Lin, X. Wang and W. Choi, Nano Energy, 2017, 42, 58–68 CrossRef CAS.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- N. Tafreshi, S. Sharifnia, S. M. J. P. S. Dehaghi and E. Protection, Process Saf. Environ. Prot., 2017, 106, 203–210 CrossRef CAS.
- H. Wang, Y. Su, H. Zhao, H. Yu, S. Chen, Y. Zhang and X. Quan, Environ. Sci. Technol., 2014, 48, 11984–11990 CrossRef CAS PubMed.
- S. Shibuya, Y. Sekine and I. Mikami, Appl. Catal., A, 2015, 496, 73–78 CrossRef CAS.
- K. Nagaoka, T. Eboshi, Y. Takeishi, R. Tasaki, K. Honda, K. Imamura and K. Sato, Sci. Adv., 2017, 3, e1602747 CrossRef.
- T. Choudhary, C. Sivadinarayana and D. Goodman, Chem. Eng. J., 2003, 93, 69–80 CrossRef CAS.
- A. Utsunomiya, A. Okemoto, Y. Nishino, K. Kitagawa, H. Kobayashi, K. Taniya, Y. Ichihashi and S. Nishiyama, Appl. Catal., B, 2017, 206, 378–383 CrossRef CAS.
- M. Reli, N. Ambrozova, M. Sihor, L. Matejova, L. Capek, L. Obalova, Z. Matej, A. Kotarba and K. Koci, Appl. Catal., B, 2015, 178, 108–116 CrossRef CAS.
- K. Obata, K. Kishishita, A. Okemoto, K. Taniya, Y. Ichihashi and S. Nishiyama, Appl. Catal., B, 2014, 160, 200–203 CrossRef.
- M. Zendehzaban, S. Sharifnia and S. N. Hosseini, Korean J. Chem. Eng., 2013, 30, 574–579 CrossRef CAS.
- W. M. Guo and H. T. Liu, Chem. Res. Chin. Univ., 2017, 33, 129–134 CrossRef CAS.
- Z. Mohammadi, S. Sharifnia and Y. Shavisi, Mater. Chem. Phys., 2016, 184, 110–117 CrossRef CAS.
- Y. N. Li, Z. Y. Chen, S. J. Bao, M. Q. Wang, C. L. Song, S. H. Pu and D. B. Long, Chem. Eng. J., 2018, 331, 383–388 CrossRef CAS.
- S. H. Pu, D. B. Long, M. Q. Wang, S. J. Bao, Z. H. Liu, F. Y. Yang, H. Wang and Y. Q. Zeng, Mater. Lett., 2017, 209, 56–59 CrossRef CAS.
- H. Zhang, Q. Q. Gu, Y. W. Zhou, S. Q. Liu, W. X. Liu, L. Luo and Z. D. Meng, Appl. Surf. Sci., 2020, 504, 144065 CrossRef.
- Z. Wu, N. Ambrožová, E. Eftekhari, N. Aravindakshan, W. Wang, Q. Wang, S. Zhang, K. Kočí and Q. Li, Emergent Mater., 2019, 2, 303–311 CrossRef CAS.
- R. Wang, T. Xie, Z. Sun, T. Pu, W. Li and J. P. Ao, RSC Adv., 2017, 7, 51687–51694 RSC.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- Z. He and W. Que, Appl. Mater. Today, 2016, 3, 23–56 CrossRef.
- C. V. Reddy, I. N. Reddy, K. Ravindranadh, K. R. Reddy, N. P. Shetti, D. Kim, J. Shim and T. M. Aminabhavi, J. Environ. Manage., 2020, 260, 110088 CrossRef CAS PubMed.
- C. V. Reddy, I. N. Reddy, V. V. N. Harish, K. R. Reddy, N. P. Shetti, J. Shim and T. M. Aminabhavi, Chemosphere, 2020, 239, 124766 CrossRef CAS PubMed.
- N. R. Reddy, U. Bhargav, M. M. Kumari, K. K. Cheralathan, M. V. Shankar, K. R. Reddy, T. A. Saleh and T. M. Aminabhavi, J. Environ. Manage., 2020, 254, 109747 CrossRef CAS PubMed.
- S. B. Patil, P. S. Basavarajappa, N. Ganganagappa, M. S. Jyothi, A. V. Raghu and K. R. Reddy, Int. J. Hydrogen Energy, 2019, 44, 13022–13039 CrossRef CAS.
- C. Venkata Reddy, I. N. Reddy, B. Akkinepally, K. R. Reddy and J. Shim, J. Alloys Compd., 2020, 814, 152349 CrossRef CAS.
- T. Kamegawa and T. Nakaue, Chem. Commun., 2015, 51, 16802–16805 RSC.
- K. Fuku, T. Kamegawa, K. Mori and H. Yamashita, Chem.–Asian J., 2012, 7, 1366–1371 CrossRef CAS PubMed.
- M. V. Dozzi and E. Selli, J. Adv. Oxid. Technol., 2016, 19, 246–255 CAS.
- M. Sihor, K. Koci, L. Matejova, M. Reli, N. Ambrozova, J. Pavlovsky, L. Capek and L. Obalova, J. Nanosci. Nanotechnol., 2015, 15, 6833–6839 CrossRef CAS PubMed.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126–4136 CrossRef CAS.
- K. R. Reddy, C. V. Reddy, M. N. Nadagouda, N. P. Shetti, S. Jaesool and T. M. Aminabhavi, J. Environ. Manage., 2019, 238, 25–40 CrossRef CAS PubMed.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- V. Manh-Hiep, M. Sakar and D. Trong-On, Catal, 2018, 8, 621 CrossRef.
- Z. He, C. Kim, T. H. Jeon and W. Choi, Appl. Catal., B, 2018, 237, 772–782 CrossRef CAS.
- Z. He, W. Que, J. Chen, X. Yin, Y. He and J. Ren, ACS Appl. Mater. Interfaces, 2012, 4, 6816–6826 CrossRef CAS PubMed.
- Z. Zhang, H. Lu, X. Li, X. Li, S. Ran, Z. Chen, Y. Yang, X. Wu and L. Li, ACS Sustainable Chem. Eng., 2019, 7, 10299–10309 CrossRef CAS.
- G. Zhang, X. Yang, C. He, P. Zhang and H. Mi, J. Mater. Chem. A, 2020, 8, 334–341 RSC.
- Y. Wei, J. Wang, R. Yu, J. Wan and D. Wang, Angew. Chem., Int. Ed., 2019, 58, 1422–1426 CrossRef CAS PubMed.
- C. Tian, W. Sheng, H. Tan, H. Jiang and C. Xiong, ACS Appl. Mater. Interfaces, 2018, 10, 37453–37460 CrossRef CAS PubMed.
- P. Kar, S. Zeng, Y. Zhang, E. Vahidzadeh, A. Manuel, R. Kisslinger, K. M. Alam, U. K. Thakur, N. Mahdi, P. Kumar and K. Shankar, Appl. Catal., B, 2019, 243, 522–536 CrossRef CAS.
- C. Gao, T. Wei, Y. Zhang, X. Song, Y. Huan, H. Liu, M. Zhao, J. Yu and X. Chen, Adv. Mater., 2019, 31, 1806596 CrossRef PubMed.
- P. S. Basavarajappa, S. B. Patil, N. Ganganagappa, K. R. Reddy, A. V. Raghu and C. V. Reddy, Int. J. Hydrogen Energy, 2020, 45, 7764–7778 CrossRef CAS.
- Z. He, W. Que and H. Xie, Titanium Dioxide: Applications, Synthesis and Toxicity, Nova Science Publishers, Inc., 2013, pp. 1–34 Search PubMed.
- M. Guo, L. Li, H. Lin, Y. Zuo, X. Huang and G. Li, Chem. Commun., 2013, 49, 11752–11754 RSC.
- K. Mori, K. Miyawaki and H. Yamashita, ACS Catal., 2016, 6, 3128–3135 CrossRef CAS.
- V. Vaiano, M. Matarangolo, J. J. Murcia, H. Rojas, J. A. Navio and M. C. Hidalgo, Appl. Catal., B, 2018, 225, 197–206 CrossRef CAS.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- Z. He and W. Que, J. Nanoeng. Nanomanuf., 2012, 2, 17–21 CrossRef.
- J. Becker, K. R. Raghupathi, J. St. Pierre, D. Zhao and R. T. Koodali, J. Phys. Chem. C, 2011, 115, 13844–13850 CrossRef CAS.
- J. Wang, P. Liu, X. Fu, Z. Li, W. Han and X. Wang, Langmuir, 2009, 25, 1218–1223 CrossRef CAS PubMed.
- Z. R. Tian, J. A. Voigt, J. Liu, B. McKenzie, M. J. McDermott, M. A. Rodriguez, H. Konishi and H. Xu, Nat. Mater., 2003, 2, 821–826 CrossRef CAS PubMed.
- L. Zhang, H. Yang, J. Ma, L. Li, X. Wang, L. Zhang, S. Tian and X. Wang, Appl. Phys. A, 2010, 100, 1061–1067 CrossRef CAS.
- Y. Yan, J. Chen, N. Li, J. Tian, K. Li, J. Jiang, J. Liu, Q. Tian and P. Chen, ACS Nano, 2018, 12, 3523–3532 CrossRef CAS PubMed.
- J. W. Fu, J. G. Yu, C. J. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- H. J. Choi, S. M. Jung, J. M. Seo, D. W. Chang, L. Dai and J. B. Baek, Nano Energy, 2012, 1, 534–551 CrossRef CAS.
- G. Liao, S. Chen, X. Quan, H. Yu and H. Zhao, J. Mater. Chem., 2012, 22, 2721–2726 RSC.
- Q. Xiang and J. Yu, J. Phys. Chem. Lett., 2013, 4, 753–759 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868–12884 CrossRef CAS PubMed.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 6164–6175 CrossRef CAS.
- J. Fu, Y. Tian, B. Chang, F. Xi and X. Dong, J. Mater. Chem., 2012, 22, 21159–21166 RSC.
| This journal is © The Royal Society of Chemistry 2020 |