
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold(I)–silver(I)-calix[8]arene complexes, precursors of bimetallic alloyed Au–Ag nanoparticles
Marie
Clément
ab,
Ibrahim
Abdellah
 b,
Cyril
Martini
b,
Frédéric
Fossard
c,
Diana
Dragoe
b,
Hynd
Remita
a,
Vincent
Huc
b and
Isabelle
Lampre
*a
b,
Cyril
Martini
b,
Frédéric
Fossard
c,
Diana
Dragoe
b,
Hynd
Remita
a,
Vincent
Huc
b and
Isabelle
Lampre
*a
aUniversité Paris-Saclay, CNRS, Institut de Chimie Physique, UMR 8000, 91405 Orsay, France. E-mail: Isabelle.lampre@universite-paris-saclay.fr
bUniversité Paris-Saclay, CNRS, Institut de Chimie Moléculaire et des Matériaux d’Orsay, UMR 8182, 91405 Orsay, France
cUniversité Paris-Saclay, ONERA, CNRS, Laboratoire d'Etude des Microstructures, 92322 Châtillon, France
First published on 6th May 2020
Abstract
In this paper, we report the first synthesis and characterisations of bimetallic gold(I)–silver(I) calix[8]arene complexes. We show that the radiolytic reduction of these complexes leads to the formation of small bimetallic nanoparticles with an alloyed structure, as evidenced by XPS, HR-TEM and STEM/HAADF-EDX measurements.
Introduction
In the last decades, research involving metal nanoparticles (NP) has seeped into many domains: material science, analytical chemistry, biochemistry… Indeed, due to their unique properties, metal NPs find applications in different fields, for instance catalysis, optics, sensing or medicine. However, their properties depend on their size, shape and morphology, parameters which need to be controlled. Towards that goal, the use of ligands, capping agents or supports is often required. But, the NP surface and its interactions with the environment and/or the ligands play an important role for applications. Among the ligands, macrocycles such as calixarenes have attracted attention due their conformational behaviour, functionalisation tunability, host-guest properties as well as non-toxicity, biological mimics and surface accessibility. Calixarenes-based NPs have already been the subject of several reviews.1–4Lately, we reported the radiolytic synthesis of mono and bimetallic gold–silver nanoparticles (NPs) stabilised by octa(hydroxyl)-octa(mercaptobutoxy)calix[8]arenes (C8, Chart 1).5 For a metal/calixarene ratio of 10, the reduction of metallic salts, AgClO4 or HAuCl4, in the presence of C8 in ethanolic solution leads to the formation of small spherical NP, homogeneous in size (diameter <5 nm). In the case of the reduction of ethanolic solution containing both Au(III) and Ag(I) salts in the presence of C8, alloyed Au–Ag NPs. were obtained with a mean size of 3.5 nm. However, the proportions of gold and silver were not constant from one NP to another and the NP structure appears non-homogeneous with domains containing more gold atoms and others more silver atoms. Such non-homogeneous structure might come from aggregation of small clusters with different compositions. The variations in the composition might result from different initial complexations between Au(III), Ag(I) and calix[8]arenes. In order to get a better control of the initial complexation between metallic ions and calixarenes, we undertook the synthesis of calix[8]arene-based metallic complexes. First, trimethylphosphine Au(I)-appended calix[8]arene containing eight and sixteen equivalents of gold (Au(I)-C8 and 2Au(I)-C8, Chart 1) were synthesised and characterised by 1H and 31P NMR spectroscopy.6 We also showed that the radiolytic reduction of these complexes leads to the formation of small Au NPs homogeneous in size. This has prompted us to prepare bimetallic complexes with eight equivalents of gold and eight equivalents of silver, and to reduce them to produce alloyed bimetallic NPs. Even if several metallo calixarenes have already been reported in the literature,7–14 few have been used as precursors of NPs or clusters, and to our knowledge none corresponds to bimetallic complex and NP. For instance, Chen et al. used Co16-calix[4]arenes to promote the nucleation and growth of Co NPs under solvothermal conditions.14 A. Katz and co-workers synthesised gold clusters via NaBH4 reduction of different Au(I)-calix[4]arene complexes bearing one or two metallic centres.10,11 They showed the formation of small clusters (<1.6 nm in diameter) with a small influence of the calixarene conformation and lower rim substituents and they also quantified the accessibility of the gold surface by steady-state fluorescence measurements.
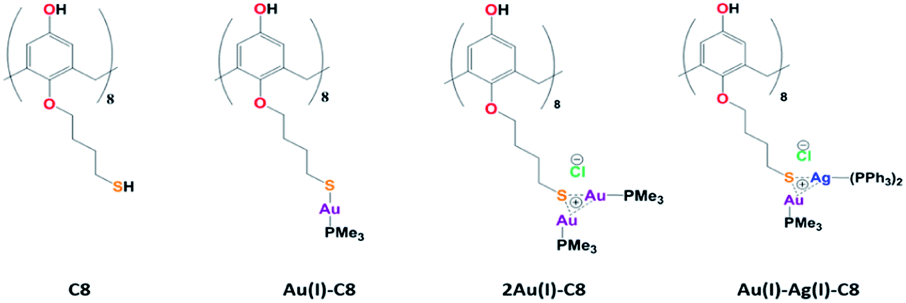 | ||
| Chart 1 Structures of the calix[8]arenes involved in this study. | ||
Herein, we report the first synthesis of bimetallic Au(I)–Ag(I)-calix[8]arene complexes with eight equivalents of each metal based on our recent study on Au(I)-calix[8]arene complexes. Then, we investigate the radiolytic reduction of these complexes in ethanolic solution. The formed NPs are characterised by high-resolution transmission electron microscopy (HR-TEM), X-ray photoelectron spectroscopy (XPS) and scanning transmission electron microscopy/energy dispersive X-ray spectroscopy (STEM/EDX). We show that the reduction of Au(I)–Ag(I)-calix[8]arene complexes generates homogeneously-alloyed Au–Ag NPs.
Results and discussion
Synthesis of Au(I)–Ag(I)-calix[8]arene complexe
The Au(I)–Ag(I)-calix[8]arene complexe (Au(I)-Ag(I)-C8, Chart 1) was synthesised directly in the NMR tube using two routes starting from either C8 or Au(I)-C8 (Scheme 1). In the first route R1, to a DMSO-d6 solution containing C8, were added eight equivalents of Cl–Au-PMe3 per calixarene molecule and eight equivalents of Cl–Ag-(PPh3)2. Then, thiethylamine (Et3N) was introduced to deprotonate the thiol group to facilitate the metal coordination. In the second route R2, eight equivalents of Cl–Ag-(PPh3)2 were added to a DMSO-d6 solution of the already synthesised monometallic Au(I)-calixarene complex, Au(I)-C8. It is to note that a third route starting from Ag(I)-appended calix[8]arene, Ag(I)-C8, was impossible as attempts to synthesize and isolate Ag(I)-C8 from Cl–Ag-(PPh3)2 and C8 were unsuccessful, no clear evidence of coordination between the two compounds being found.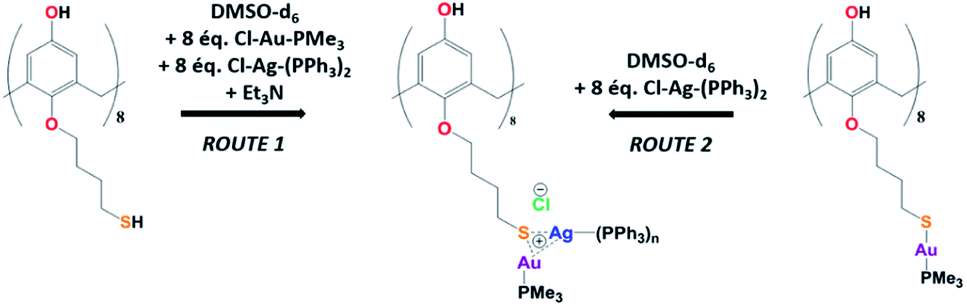 | ||
| Scheme 1 Synthetic routes of Au(I)–Ag(I) calix[8]arene complexes. | ||
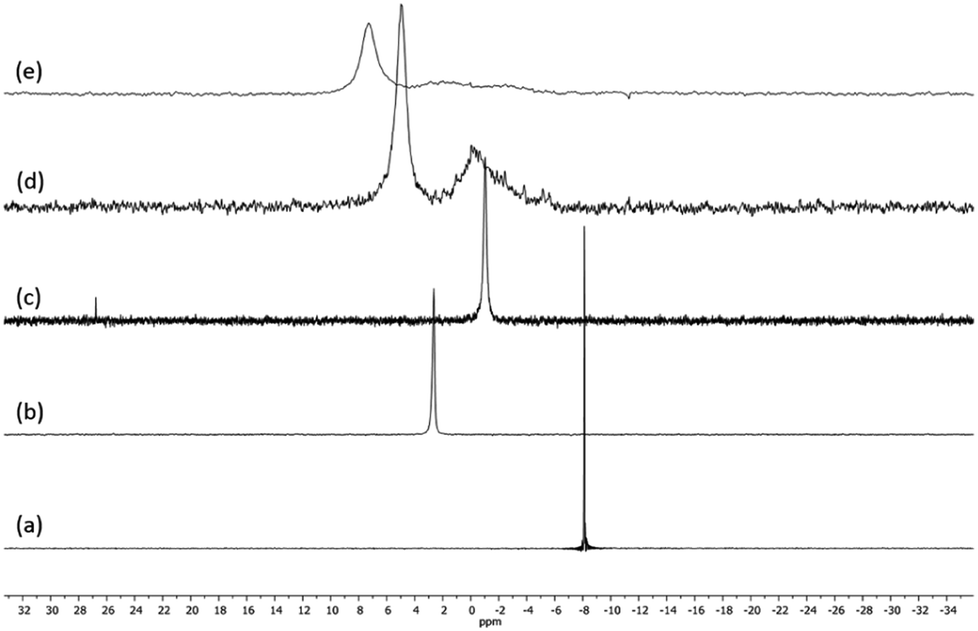
Fig. 1 presents the 1H NMR spectra of Au(I)–Ag(I)-calix[8]arene complexes obtained by the two synthetic routes Au(I)-Ag(I)-C8_R1 and Au(I)-Ag(I)-C8_R2 (Fig. 1d and e) as well as those of the initial calixarenes C8 and Au(I)-C8 (Fig. 1a and c) and silver complex Cl–Ag-(PPhe3)2 (Fig. 1b). Whatever the synthetic procedures, the spectra of the Au(I)-Ag(I)-C8 are complex but close as they present broad peaks at similar chemical shifts. The comparison with the initial compounds allow to retrieve the characteristic peaks of the calix[8]arene structure and triphenylphosphine groups. For the metallic complexes, Au(I)-C8 and Au(I)-Ag(I)-C8, the signal due to thiol group (δ = 2.08 ppm) is absent, confirming the coordination of the metallic centres to the sulphur atoms. The addition of the metal centres induces a broadening of the peaks indicating a loss in the flexibility of the molecule. The main aromatic resonances observed around 6.3 ppm for Au(I)-C8 disappear in favour of two broad signals around 5.9 and 6.5 with the addition of Ag(I)-(PPh3)2 revealing a lowering of the symmetry of the formed Au(I)Ag(I)-C8 complexes, as already noted in the case of the previously synthesised 2Au(I)-C8 complexes.6 Moreover, in the case of Au(I)-Ag(I)-C8_R1, the spectrum shows more resolved peaks compared to Au(I)-Ag(I)-C8_R2 suggesting more rigid conformers. The presence of Et3N in excess in the case of the route R1 might account for such results possibly by hydrogen bonding between the acidic [H–N(Et3)]+ and the free hydroxyl groups of the calixarene. The formation of inclusion complexes with the calixarenic cavity may also be invoked. Moreover, the presence of Et3N has also an effect on the 31P NMR spectra of the compounds (Fig. 2). The 31P spectra of both Au(I)-Ag(I)-C8_R1 and Au(I)-Ag(I)-C8_R2 differ from those of the metallic precursors, Cl–Au-PMe3 and Cl–Ag-(PPhe3)2, corroborating the complexation and the absence of free precursors in solution (Fig. 2). The spectra of Au(I)-Ag(I)-C8_R1 and Au(I)-Ag(I)-C8_R2 also differ slightly from each other, with the main peaks observed at 5 and 7.5 ppm for Au(I)-Ag(I)-C8_R1 and Au(I)-Ag(I)-C8_R2, respectively. The possibility of an exchange between a phosphine linked to the metal and triethylamine as a co-ligand (Scheme 2) as well as the formation of triethyl ammonium chlorohydrate could lead to a change in the solvation sphere of the calixarene complexes and account for different 31P spectra.
 | ||
| Fig. 1 1H NMR spectra of the initial compounds C8 (a), Cl–Ag(PPhe3)2 (b) and Au(I)-C8 (c) and those of the gold–silver calix[8]arene complexes Au(I)-Ag(I)-C8_R1 (d) and Au(I)-Ag(I)-C8_R2 (e) obtained by two different synthetic routes (Scheme 1); Δ indicates the peaks corresponding to trimethylamine and * marks CHCl3 and CH2Cl2 impurities. | ||
 | ||
| Fig. 2 31P NMR spectra of the precursor compounds Cl–Au-PMe3 (a), Cl–Ag-(PPhe3)2 (b) and Au(I)-C8 (c) and those of the gold–silver calix[8]arene complexes Au(I)-Ag(I)-C8_R1 (d) and Au(I)-Ag(I)-C8_R2 (e), obtained by two different synthetic routes (Scheme 1). | ||
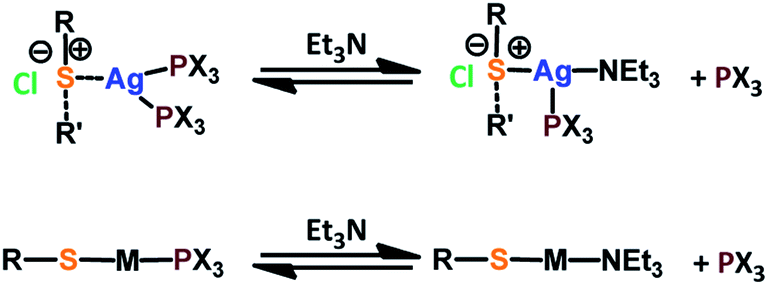 | ||
| Scheme 2 Possible exchanges between the phosphine and the thiethylamine as metal ligands. | ||
For the formation of 2Au(I)-C8 complexes,6 the tendency of gold thiolates to associate and form multinuclear complexes due to the high affinity of Au(I) to thiol(ate) groups and aurophilic interactions was evoked.15 Here, the strong metallophilic interactions between Ag(I) and Au(I), both d10 transition metal systems,16 can account for the formation of the bimetallic complexes, Au(I)-Ag(I)-C8. Indeed, several self-assembled Au(I)–Ag(I) systems with various metallophoric Au–Ag arrangements revealed by X-ray diffraction studies have already been reported.17–19 In these systems, the metal atoms are separated by distances around 3 angströms shorter than the sum of their van der Waals radii.17–19
Reduction of Au(I)–Ag(I) calix[8]arene complexes
The Au(I)-Ag(I)-C8 synthesised in situ in DMSO-d6 were diluted in ethanol and then reduced by gamma-irradiation. Indeed, radiolysis is a powerful technique to synthesise metallic NPs and nanomaterials of controlled size, shape and structure, without the addition of a chemical reductant.20–23 Solvated electrons and alcohol radicals produced by solvent (ethanol here) radiolysis induce homogeneous reduction and nucleation leading to metallic NPs. After centrifugation, the NPs were analysed by TEM. In Fig. 3, small, well-dispersed, spherical NPs are observed with a narrow distribution in size (3.5 ± 0.6 nm) whatever the initial complexes, Au(I)-Ag(I)-C8_R1 or Au(I)-Ag(I)-C8_R2. While the mean size is similar to that previously reported in the case of bimetallic NPs synthesised from metallic salts (HAuCl4 and AgClO4) and stabilised by C8,5 the size distribution is narrower here, indicating a better control of the size with the use of a metallic complex. But, as expected due to the absence of observable colour in the solution after irradiation and as already observed for the bimetallic NPs synthesized from the metallic salts,5 UV-visible spectra of the NPs formed here from the bimetallic complexes present no characteristic surface plasmon resonance (SPR) band. Such result is in agreement with the formation of small spherical NP with a mean size of 3.5 nm.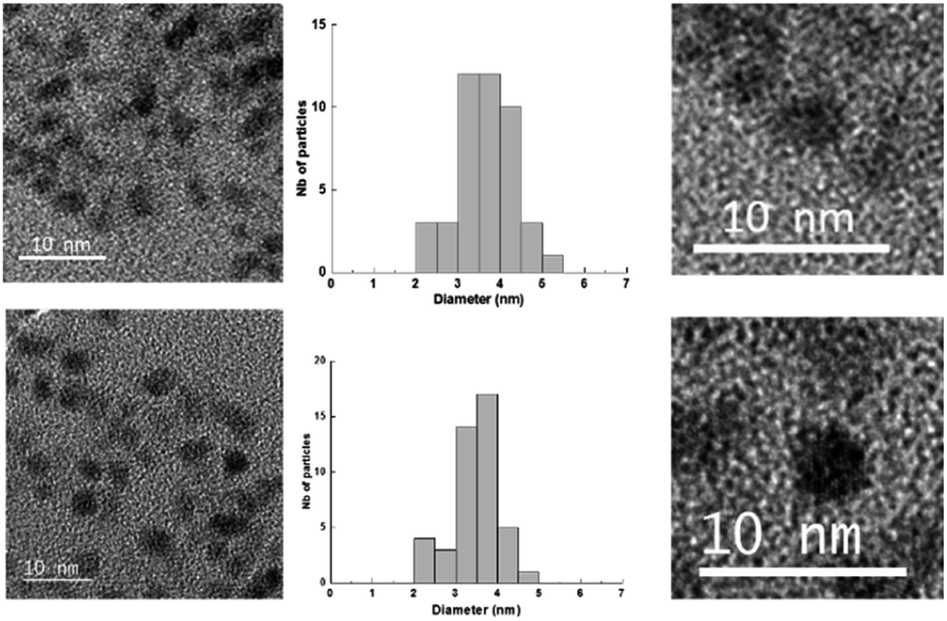 | ||
| Fig. 3 TEM images and size distribution of the nanoparticles obtained by radiolytic reduction of Au(I)-Ag(I)-C8_R1 complexes (bottom) and Au(I)-Ag(I)-C8_R2 complexes (top). | ||
XPS characterisations allow to get information on the composition of the NPs and their surface. The wide-scan spectra recorded for NPs obtained from Au(I)-Ag(I)-C8_R1 (Fig. 4) or Au(I)-Ag(I)-C8_R2 (not shown) are similar and show the presence of the expected elements: carbon, oxygen, sulphur, silver and gold. It is to note that, due to a high contribution of oxygen from the SiO2 support, the XPS spectrum of the O1s core-level was not analysed. The XPS spectra of the C1s, S2p, Ag3d and Au4f core-levels are presented in Fig. 5. The C1s signal corresponds to an asymmetric peak at 285.2 eV, and can be related to three contributions as referred in the literature: C–C bonding in the phenyl group (sp2 C) at 284.8 eV, C–C bonding in aliphatic chain (sp3 C) at 285.4 eV and C–O/S bonding at 286.3 eV.24 It is to note that the ratio of the area under the first two peaks is equal to 1.09, close to the ratio of the number of sp3 (40) and sp2 (32) carbon atoms in C–C bonds in the C8 ligand. The S2p signal appears as a broad asymmetric peak with the contributions of S2p3/2 and S2p1/2 at 162.5 and 163.3 eV, respectively. This S2p doublet can be attributed to sulphur bound to metal (Au or Ag) as already referred in the literature.25–27 The spin–orbit doublet of Ag3d corresponds to two well-defined peaks at 368.3 and 374.3 eV for the contributions of Ag3d5/2 and Ag3d3/2, respectively. This signal can be assigned to zero-valent silver.28,29 However, a small contribution of more-oxidised silver cannot be excluded as the binding energies reported for Ag2S (E(Ag3d5/2) = 368.2 eV)30 and Ag2O (E(Ag3d5/2) = 368.6 eV)28 are very close in energy. The signal of Au4f is also a spin–orbit doublet but here the two observed peaks at 85.3 and 89 eV are asymmetric and not well-separated. The fitting procedure requires the presence of two doublets. The former with Au4f7/2 and Au4f5/2 contributions at 84.2 and 87.9 eV, respectively, is attributed to metallic gold (Au0). The second doublet with components at 85.5 eV (Au4f7/2) and 89.2 eV (Au4f5/2) corresponds to more oxidised gold and is related to gold atoms bound to sulphur on the NP surface.29,31–33 That also suggests that the sulphur atoms from the calixarenic ligand tend to specifically bind to gold atoms on the NP surface. On the whole, the XPS characterisations attest the formation of bimetallic Au–Ag NPs with calix[8]arene ligand grafted on the surface. This result is confirmed by STEM analysis.
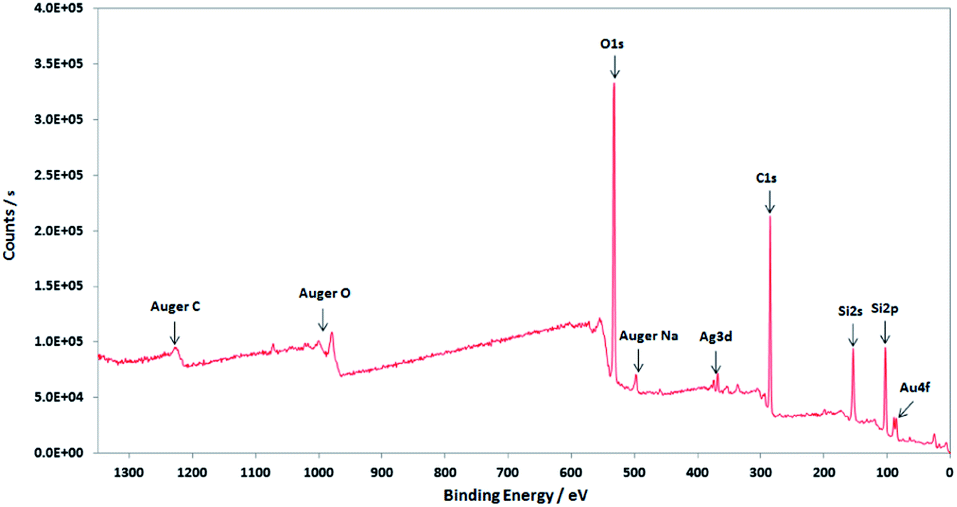 | ||
| Fig. 4 Wide-scan XPS spectrum of the nanoparticles obtained by radiolytic reduction of Au(I)-Ag(I)-C8_R1 complexes. | ||
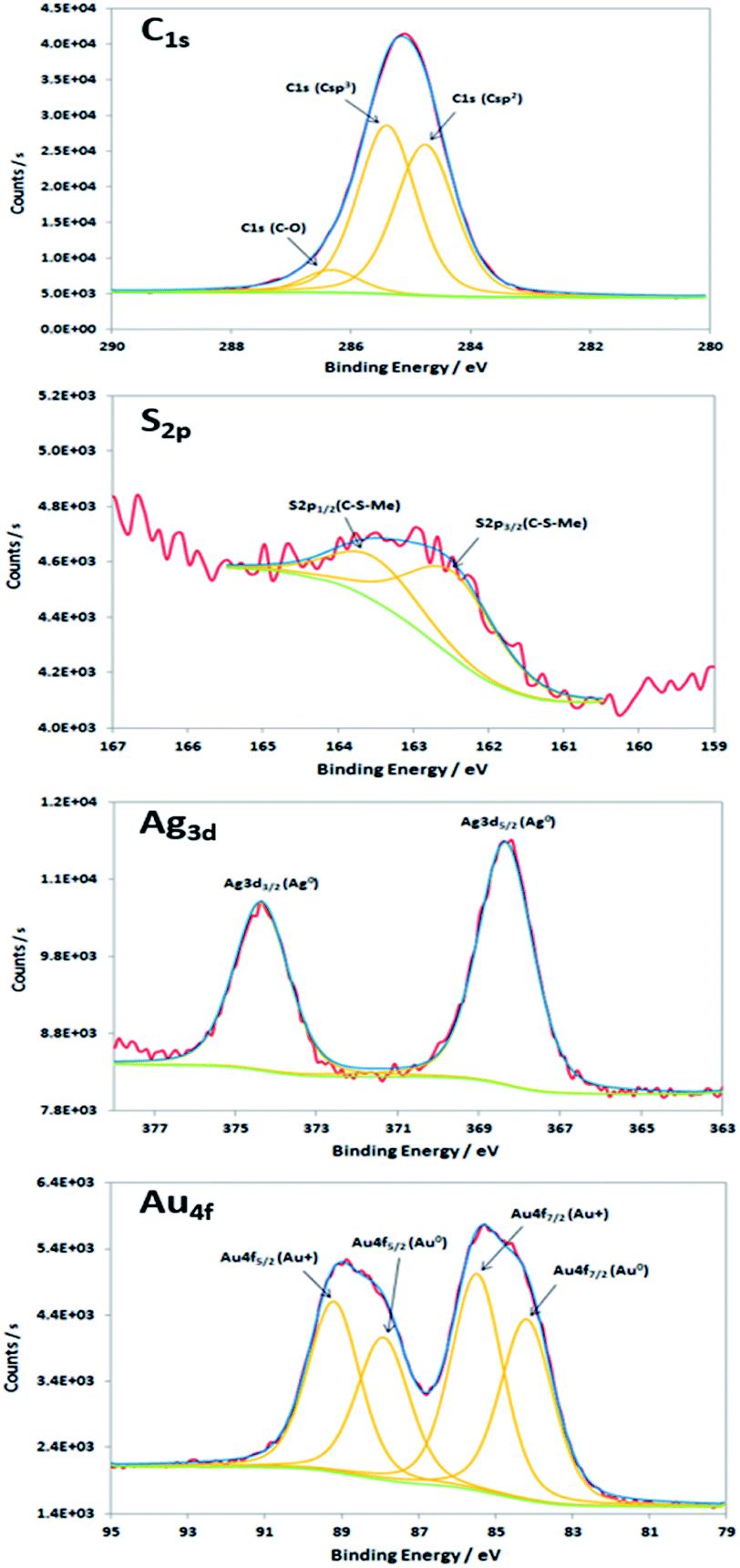 | ||
| Fig. 5 XPS spectra of the C1s, S2p, Ag3d and Au4f core-levels of the nanoparticles produced by radiolytic reduction of Au(I)-Ag(I)-C8_R1 complexes. | ||
Fig. 6 presents the high resolution STEM image in High Angle Annular Dark Field (HAADF) mode. In addition to the expected NPs with sizes around 3.5 nm, the HAADF images reveal the presence of small clusters (<1 nm in diameter). However, although NPs larger than 4 nm are rare, we select one of them for clarity reasons. Indeed, on the chosen HAADF image, the crystallographic planes of the NP are easily seen (Fig. 6). The EDX mapping of two NPs is shown in Fig. 7. The superposition of the EDX maps for gold and silver reveals the presence of both metals inside the NPs with a quite homogeneous distribution. The elemental quantification indicates a similar amount of gold (49 ± 3%) and silver (51 ± 3%) atoms, whatever the NP. The EDX map for sulphur also confirm the presence of the calix[8]arene ligands at the surface. Therefore, the reduction of the Au(I)-Ag(I)-C8 complexes leads to the formation of alloyed bimetallic Ag–Au NPs stabilised by calix[8]arenes.
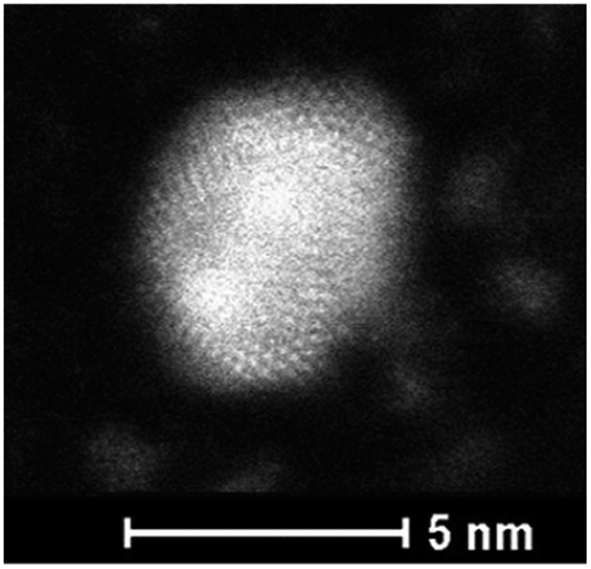 | ||
| Fig. 6 HAADF-STEM image of nanoparticles produced by reduction of Au(I)–Ag(I)–C8_R1 complexes. | ||
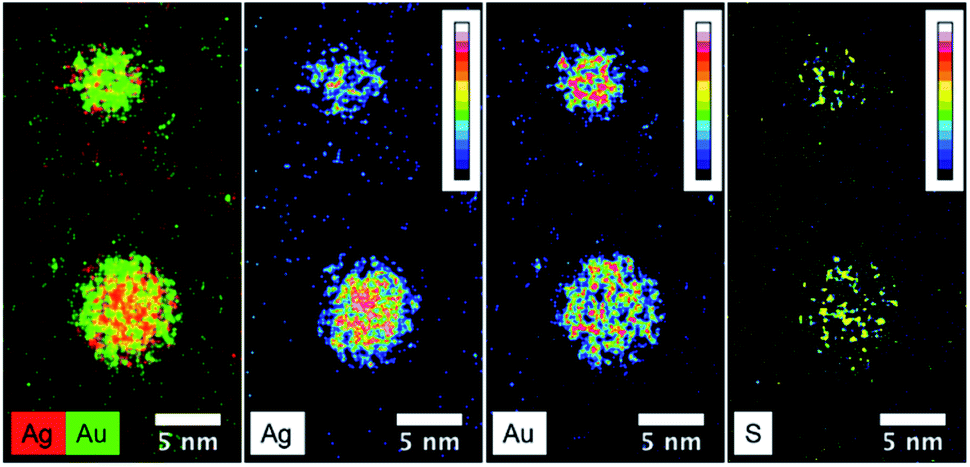 | ||
| Fig. 7 STEM-EDX maps of silver, gold and sulphur for the nanoparticles produced by reduction of Au(I)-Ag(I)-C8_R1 complexes, as well as the superposition of silver(red) and gold (green) maps. | ||
Conclusions
We have synthesised and characterised by 1H and 31P NMR a bimetallic gold(I)–silver(I) calix[8]arene complexe. For the first time a fully metallated calix[8]arene with 8 equivalents of Au(I) and 8 equivalents of Ag(I) is reported. The reduction of this bimetallic complex leads to the formation of small spherical bimetallic Au–Ag NPs with an alloyed structure as shown by STEM analysis. It is to note that starting from well-defined bimetallic complexes allows to keep the ratio of the metals for all the produced NPs ([Ag]/[Au] = 1.00 ± 0.12) and to get quite homogeneous structures, in contrast to what was previously observed in the case of the reduction of Au(III) and Ag(I) salts in the presence of calix[8]arenes.5 The possibility to prepare and reduce calixarene-complexes with others metals allows considering the production of various bimetallic alloyed nanostructures. This can be useful for applications, for instance in catalysis or electrocatalysis by taking advantage of the synergetic effect of different metals and surface accessibility offered by calixarene ligands.Experimental
Materials
All compounds (silver chloride, AgCl; trimethylamine, Et3N; triphenylphosphine, PPh3) and solvents (chloroform, CHCl3; pentane, C5H10; ethanol, EtOH; deuterated dimethylsulfoxide, DMSO-d6) were purchased with the highest available purity from commercial sources (Sigma-Aldrich and Strem chemicals) and were used without further purification. Argon (Ar, U grade, purity 99%) and dinitrogen (N2, U grade, purity 99.999%) gases were purchased from Air Liquide.Synthetic procedure
The synthesis of p-octa(hydroxy)-octa(mercaptobutoxy)-calix[8]arene C8 and p-octa(hydroxy)-octa(mercaptobutoxy)-octa(trimethylphosphine)gold(I)-calix[8]arene (Au(I)-C8) have already been described in details elsewhere.5,6Route R1. In a NMR tube, to a solution of C8 (8 mg, 0.005 mmol, 1 equiv.) in DMSO-d6 (0.3 mL) Cl–Au-PMe3 (12 mg, 0.04 mmol, 8 equiv.), Cl–Ag-(PPh3)2 (25 mg, 0.04 mmol, 8 equiv.) and then dry Et3N (7.7 μL, 0.06 mmol, 12 equiv.) were added under argon. The mixture was sonicated for 2 minutes and heated with a gun for 15 seconds.
Route R2. In a NMR tube, to a solution of Au(I)-C8 (8 mg, 0.002 mmol, 1 equiv.) in DMSO-d6 (0.3 mL), Cl–Ag-(PPh3)2 (11 mg, 0.017 mmol, 8 equiv.) was added under argon. The mixture was sonicated for 2 minutes and heated with a gun for 15 seconds.
Methods and instrumentation
1H and 31P NMR spectra were recorded on Brüker Avance spectrometers at 298 K.XPS spectra were recorded on a K Alpha (Thermo Fisher) spectrometer, equipped with a monochromatic Aluminum source (Al, Kα = 1486.6 eV, beam size: 200 μm). Wafers were 300 nm thermal SiO2 coated silicon wafers purchased from SiMat. Samples were introduced, without prior surface cleaning. Analysis chamber pressure was 2 × 10−9 mbar. Hemispherical analyzer was used in Constant Analyzer Energy (CAE) mode. Pass energies were 200 eV for the surveys acquisition and 50 eV for the narrow scans. Energies were recorded with a 1 eV path for the survey and 0.1 eV for narrow scans. Charge neutralization is performed by irradiation of the surface with low energy electrons (5 eV maximum).
High-resolution transmission electron microscopy (HR-TEM) images were recorded on a FEI TECNAI F30 microscope operating at an accelerating voltage of 300 kV. The irradiated ethanolic solutions were centrifuged to collect the formed NPs, which were dispersed in propan-2-ol. Droplets of the NPs solution were then deposited onto copper grids coated with an amorphous carbon membrane and dried at room temperature for 20 minutes.
High-angle annular dark field (HAADF) images and energy dispersive spectroscopy (EDX) were performed on a FEI Titan G2 probe-corrected scanning transmission electron microscope (STEM) operating at 200 kV.
The gamma-irradiation were carried out using a panoramic 60Co source facility. The dose rate, determined by the Fricke method in water solution, was 3.7 kGy h−1. The absorbed dose (576 Gy) was then calculated taking into account the relative electronic density factor of the used solvent (0.8 for ethanol) and adjusted in order to have a total reduction of the metallic complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank J.-L. Rodriguez-Lopez and H. Silva at IPiCYT, San Luis Potosi, Mexico, for TEM measurements. HRSTEM-EDX study was carried out within the MATMECA consortium, supported by the ANR-10-EQPX-37 contract and has benefited from the facilities of the Laboratory MSSMat, UMR 8579 CNRS, CentraleSupelec, Université Paris-Saclay.Notes and references
- A. Wei, Chem. Commun., 2006, 1581–1591 RSC.
- A. Acharya, K. Samanta and C. P. Rao, Coord. Chem. Rev., 2012, 256, 2096–2125 CrossRef CAS.
- V. Montes-Garcia, J. Pérez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzan, Chem. - Eur J., 2014, 20, 10874–10883 CrossRef CAS PubMed.
- A. R. Kongor, V. A. Mehta, K. M. Modi, M. K. Panchal, S. A. Dey, U. S. Panchal and V. K. Jain, Top. Curr. Chem., 2016, 374, 28 CrossRef PubMed.
- P. Ray, M. Clément, C. Martini, I. Abdellah, P. Beaunier, J. L. Rodriguez-Lopez, V. Huc, H. Remita and I. Lampre, New J. Chem., 2018, 42, 14128–14137 RSC.
- M. Clément, I. Abdellah, P. Ray, C. Martini, Y. Coppel, H. Remita, I. Lampre and V. Huc, Inorg. Chem. Front., 2020, 7, 953–960 RSC.
- D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed.
- D. Mendoza-Espinosa and T. A. Hanna, Dalton Trans., 2009, 5211–5225 RSC.
- D. Mendoza-Espinosa, A. L. Rheingold and T. A. Hanna, Dalton Trans., 2009, 5226–5238 RSC.
- N. de Silva, J.-M. Ha, A. Solovyov, M. M. Nigra, I. Ogino, S. W. Yeh, K. A. Durkin and A. Katz, Nat. Chem., 2010, 2, 1062–1068 CrossRef CAS PubMed.
- M. M. Nigra, A. J. Yeh, A. Okrut, A. G. DiPasquale, S. W. Yeh, A. Solovyov and A. Katz, Dalton Trans., 2013, 42, 12762–12771 RSC.
- C. Redshaw, Dalton Trans., 2016, 45, 9018–9030 RSC.
- B. Ourri, O. Tillement, T. Tu, E. Jeanneau, U. Darbost and I. Bonnamour, New J. Chem., 2016, 40, 9477–9485 RSC.
- Z. Chen, J. Liu, A. J. Evans, L. Alberch and A. Wei, Chem. Mater., 2014, 26, 941–950 CrossRef CAS PubMed.
- A. Sladek, W. Schneider, K. Angermaier, A. Bauer and H. Schmidbaur, Z. Naturforsch. B Chem. Sci., 1996, 51, 765–772 CAS.
- V. Wing-Wah Yam, V. Ka-Man Au and S. Yu-Lut Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef PubMed.
- O. Schuster, U. Monkowius, H. Schmidbaur, R. S. Ray, S. Krüger and N. Rösch, Organomettalics, 2006, 25, 1004–1011 CrossRef CAS.
- A. Laguna, T. Lasanta, J. M. Lopez-de-Luzuriaga, M. Monge, P. Naumov and M. E. Olmos, J. Am. Chem. Soc., 2010, 132, 456–457 CrossRef CAS PubMed.
- M. Gil-Moles, M. Conception Gimero, J. M. Lopez-de-Luzuriaga, M. Monge and M. Elena Olmos, Dalton Trans., 2019, 48, 5149–5155 RSC.
- J. Belloni, M. Mostafavi, H. Remita, J.-L. Marignier and M.-O. Delcourt, New J. Chem., 1998, 22, 1239–1255 RSC.
- H. Remita, I. Lampre, M. Mostafavi, E. Balanzat and S. Bouffard, Radiat. Phys. Chem., 2005, 72, 575–586 CrossRef CAS.
- W. Abidi and H. Remita, Recent Pat. Eng., 2010, 4, 170–188 CrossRef CAS.
- A. Abedini, A. A. A. Bakar, F. Larki, P. S. Menon, M. S. Islam and S. Shaari, Nanoscale Res. Lett., 2016, 11, 287 CrossRef PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray photoelectron spectroscopy, Perkin-Elmer Corporation, USA, 1992 Search PubMed.
- G. Xue, M. Ma, J. Zhang, Y. Lu and K. Carron, J. Colloid Interf. Sci., 1992, 150, 1–6 CrossRef CAS.
- A. J. Leavitt and T. P. Beebe, Surf. Sci., 1994, 314, 23–33 CrossRef CAS.
- H. Peisert, T. Chassé, P. Streubel, A. Meisel and R. Szargan, J. Electron Spectrosc. Relat. Phenom., 1994, 68, 321–328 CrossRef CAS.
- A. M. Ferraria, A. P. Carapeto and A. M. Botelho de Rogo, Vacuum, 2012, 86, 1988–1991 CrossRef CAS.
- L. Carlini, C. Fasolato, P. Postorino, I. Fratoddi, I. Venditti, G. Testa and C. Battocchio, Colloids Surf., A, 2017, 532, 183–188 CrossRef CAS.
- M. Romand, M. Roubin and J. P. Deloume, J. Electron Spectrosc. Relat. Phenom., 1978, 13, 229–242 CrossRef CAS.
- A. McNeillie, D. H. Brown, W. E. Smith, M. Gibson and L. Watson, J. Chem. Soc., Dalton Trans., 1980, 767–770 RSC.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
- E. Bedford, V. Humblot, C. Méthivier, C.-M. Pradier, F. Gu, F. Tielens and S. Boujday, Chem. - Eur J., 2015, 21, 14555–14561 CrossRef CAS PubMed.
- D. V. Sanghani, P. J. Smith, D. W. Allen and B. F. Taylor, Inorg. Chim. Acta, 1982, 59, 203–206 CrossRef CAS.
- J. C. Russell and G. R. Freeman, J. Phys. Chem., 1967, 71, 755–762 CrossRef CAS.
- J. J. J. Myron and G. R. Freeman, Can. J. Chem., 1965, 43, 381–394 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |